

Abstract
Gastrin Releasing Peptide (GRP), an evolutionarily-conserved neuropeptide, significantly contributes to influenza-induced lethality and inflammation in rodent models. Since GRP is produced by pulmonary neuroendocrine cells (PNEC) in response to γ-aminobutyric acid (GABA), we hypothesized that influenza infection promotes GABA release from innervated PNECs that activate GABAB receptors on PNEC to secrete GRP. Oxidative stress was increased in lungs of influenza A/PR/8/34 (PR8)-infected mice, as well as serum glutamate decarboxylase 1 (GAD1), the enzyme that converts L-glutamic acid into GABA. Therapeutic administration of saclofen, a GABAB receptor antagonist, protected PR8-infected mice, reduced lung proinflammatory gene expression of CCR2, CD68, and TLR4, and decreased levels of GRP and HMGB1 in sera. Conversely, baclofen, a GABAB receptor agonist, significantly increased lethality and inflammatory responses. The GRP antagonist, NSC77427, as well as the GABAB antagonist, saclofen, blunted PR8-induced monocyte infiltration into the lung. Together, these data provide the first report of neuroregulatory control of influenza-induced disease.
Keywords: influenza, GABA, GRP, TLR4, ALI, HMGB1
INTRODUCTION
The crosstalk between the innate immune system and the peripheral nervous system plays an integral protective role in the host response by detecting and responding to both external threats such as pathogens as well as internal signals such as danger associated molecular patterns (DAMP)s and other inflammatory mediators1,2. The lungs are highly innervated with both autonomic and sensory neurons3,4. The sensory neurons interact with the immune system at mucosal surfaces such as in the lungs. Pulmonary neuroendocrine cells (PNEC) are rare epithelial cells that represent less than 1% of the lung epithelium5. PNECs function as chemosensors in the lungs by responding to changes in oxygen levels and chemical stimuli6 and are a source of neuropeptides and neurotransmitters responsible for eliciting both physiological and aberrant immune responses7.
Gastrin releasing peptide (GRP) is a 27 amino acid peptide first isolated from porcine intestine8,9. GRP is highly conserved evolutionarily: it shares 9 of 10 C-terminal amino acids with bombesin, an amphibian neuropeptide, and was first described for its ability to mediate gastric acid secretion in the mammalian gut10,11. GRP is translated as an inactive “pre-proform” that, upon cleavage and amidation of the C-terminal methionine, becomes a mature, active, secreted peptide10. GRP interacts primarily with a 7-transmembrane, G-protein coupled, high affinity receptor, GRPR/BB2, to stimulate many signaling pathways including cAMP, MAPK, PI3K, and AKT, directly or indirectly through the transactivation of other ligand/receptor systems10,12. GRP stimulates fetal lung growth/maturation, but has also been associated with pulmonary inflammatory diseases and malignancies including bronchopulmonary dysplasia, chronic obstructive pulmonary disease, emphysema, fibrosis, small cell lung cancer, and non-small lung cancer10,13,14.
Influenza is a highly contagious respiratory illness. The WHO estimate of influenza-associated deaths globally is ~300,000–650,00015, and The Centers for Disease Control and Prevention has estimated ~8–13 million influenza-associated illnesses with 5,000–14,000 deaths in the U.S.A. between October 2021 and June 202216. With new antigenic variants emerging annually, the ability to predict the influenza strains to be incorporated into vaccines can be difficult and has resulted in reduced efficacy due to an unanticipated strain predominance17,18. The role of the neuroimmune response to influenza has not been closely studied. We previously reported that GRP plays a contributory role in influenza-mediated disease19. Herein, we have further investigated the mechanism(s) by which GRP is induced in response to influenza infection and contributes to significant inflammatory disease. Influenza induces oxidative stress as well as production of γ-aminobutyric acid (GABA), which acts on GABAB receptors on PNEC to release GRP20,21. GRP, in turn, mediates monocytic infiltration into the lung. Therapeutic administration of GABAB receptor agonist (baclofen) or antagonist (saclofen) enhance and depress the inflammatory response, respectively, with a corresponding effect on survival and the proinflammatory response to infection. This study represents the first direct evidence for neuroendocrine cross-talk in the host inflammatory response to virus infection.
RESULTS
Influenza infection induces oxidative stress and GABA in mice
Influenza induces oxidative stress through production of a variety of products22–25. To confirm this, wild type (WT) C57BL/6J mice were infected intranasally (i.n.) with an ~LD90 of a mouse-adapted influenza strain, PR826. Mice were euthanized on day 6 post-infection (p.i.) and the sera collected for measurement of markers of oxidative stress. Malondialdehyde (MDA), a lipid peroxidation marker, and 8-hydroxy 2-deoxyguanasine (8-OHdG), one of the predominant forms of free radical-induced oxidative stress, were measured27,28. Levels of both MDA and 8-OHdG were significantly increased in the lungs of PR8-infected mice (Figure 1A), consistent with previous studies29–32.
Figure 1. Oxidative stress induced by influenza PR8 infection is accompanied by the release of GAD1 and GABA.
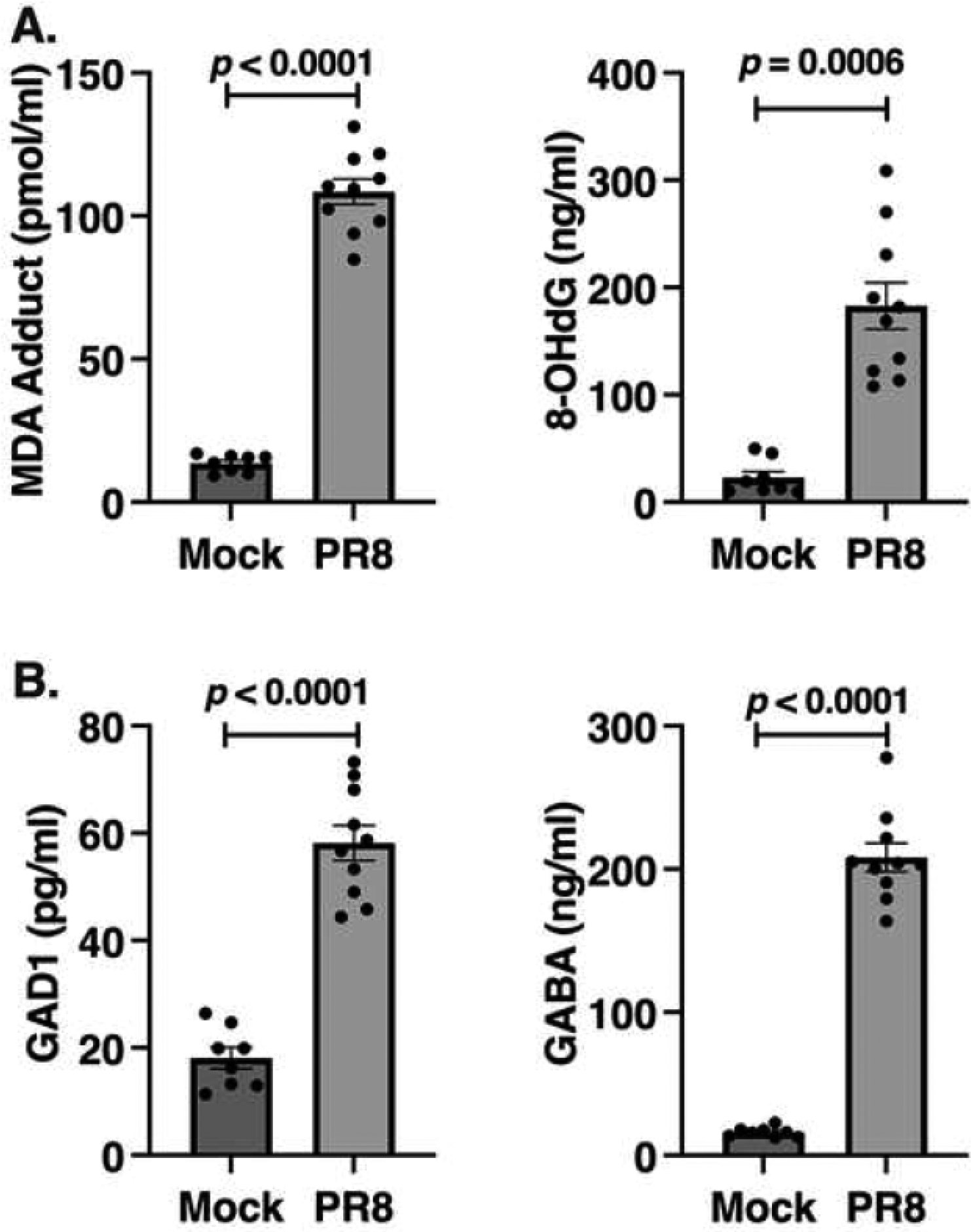
WT C57BL/6J mice were infected with and LD90 dose of mouse-adapted influenza strain PR8. On day 6 post-infection, protein levels were measured by ELISA in sera for oxidative stress markers, MDA and 8-OHdG (A) and GAD1 and GABA (B). Data shown are combined results of 2 separate experiments (N = 4–5 mice/group/experiment).
The lung has been characterized as an endocrine organ that produces immunoregulatory peptides such as GRP9,13. Rare cells of the bronchiolar columnar epithelium, PNEC33, cluster in response to noxious stimuli to form “neuroepithelial bodies” (NEBs)34–36. These specialized cells secrete GRP in response to environmental agents that lead to oxidative stress, including hypoxia, hyperoxia, ozone, smoking, and others13,34. Many such oxidative stressors have been associated with influenza infection23,24. The PNECs/NEBs are the only innervated epithelial cells in the lung37. When PNEC/NEBs are activated by the neurotransmitter γ-aminobutyric acid (GABA), they secrete GRP20,21. We previously reported that GRP is detectable in lung homogenates of influenza-infected mice and in the sera of influenza-infected cotton rats19. Therapeutic treatment of mice with a well-characterized small molecule GRP inhibitor, NSC7742738–40, a monoclonal anti-GRP antibody (2A11)41, and a GRPR/BB2 small molecule receptor antagonist (BW2258U89)14 resulted in significant survival after infection as well as decreased numbers of GRP-producing PNECs19.
Multiple groups have reported the effects of oxidative stress on GABA with regard to traumatic brain injury and related diseases42–44, however, its role in influenza infection has not been studied. Since the PNECs are the only cell type in the lung known to be in close juxtaposition to neurons45, we hypothesized that influenza-induced oxidative stress would stimulate neurons to produce GABA by producing glutamate decarboxylase-1 (GAD1). GAD1 is a pyridoxal 5’-phosphate-dependent enzyme that catalyzes the conversion of the L-glutamate to the neurotransmitter GABA46. Protein levels of both GAD1 and GABA were significantly upregulated in the sera of mice following influenza PR8 infection (Figure 1B).
Effect of antagonizing or stimulating the GABAB receptor during influenza infection in mice
There are three classes of GABA receptors, GABAA, GABAB, and GABAC47. GABAA and GABAC receptors are ionotropic. GABAB is a metabotropic G-protein coupled receptor that is often found pre-synaptically in the nervous system where it inhibits the release of other transmitters47. Therefore, we hypothesized that blocking or stimulating the GABAB receptor on PNEC/NEBs would, in turn, modulate secretion of GRP and its subsequent role in influenza-induced disease. To assess the role of the GABAB receptor during influenza infection, WT C57BL/6J mice were infected intranasally (i.n.) with an ~LD90 of PR8. On days 2 through 4 post-infection (p.i.), mice were treated once daily with either vehicle (saline) or with saclofen, a competitive antagonist for the GABAB receptor48. Mice treated with saclofen were significantly protected from influenza PR8-induced lethality (~67% survival; p = 0.0022) compared to infected mice treated with vehicle (Figure 2A). Saclofen treatment of influenza PR8-infected mice also showed ameliorated lung pathology compared to vehicle-treated, PR8-infected mice at day 6 p.i. (Figure 2B). Therapeutic treatment of influenza PR8-infected mice with saclofen also led to reduced levels of inflammatory cytokines: 6 days p.i, lungs of mice treated therapeutically with saclofen produced significantly lower mRNA levels of proinflammatory genes Tnfa, Il1b, Ifnb, and Ccl5 compared to mice infected with PR8 and treated with vehicle (Figure 2C). This reduction in gene induction was also paralleled at the protein level for TNF-α, IL-1β, IFNβ, CCL5, and CCL2 (Supplemental Figure 1). Additionally, the saclofen-treated PR8-infected mice produced significantly lower protein levels of GRP and the danger-associated molecular pattern (DAMP), High Mobility Group Box 1 (HMGB1), in their sera than measured in PR8-infected, vehicle-treated control mice (Figure 2D). We have previously reported that influenza-induced HMGB1 elicits TLR4-mediated influenza-induced disease49.
Figure 2. Effect of antagonizing the GABAB receptor during influenza PR8 infection.

(A) Mice were infected with and LD90 of mouse-adapted influenza strain PR8. Mice received vehicle (saline) or GABAB receptor antagonist (saclofen) daily from day 2 to day 4 p.i. Survival was monitored for 14 days. Data shown are combined results of 3 separate experiments (5 mice/treatment group/experiment). (B,C,D) Mice were infected and treated as described in (A). Lungs were harvested on day 6 p.i. for histopathology (B), lung gene expression by qRT-PCR (C), and serum levels of GRP and HMGB1 (D). Data represent the combined results of 2 separate experiments N = 4–5 mice/treatment group/mock; N = 7–10 mice/treatment group. *** p = 0.0002; **** p < 0.0001.
While antagonizing the GABAB receptor resulted in increased protection from influenza disease, we tested whether stimulating GABAB receptor after infection would worsen influenza-induced disease. Baclofen is a well characterized GABAB receptor agonist that is used clinically for muscle spasticity50. To test this hypothesis, WT C57BL/6J mice were infected with a low dose of influenza PR8 (~LD10)51 and subsequently administered either vehicle or baclofen once daily from days 2 to 4 p.i. Survival was significantly greater (~79%) in vehicle-treated, PR8-infected mice compared to baclofen-treated, PR8-infected mice (p < 0.0001) with ~94% of the baclofen-treated mice succumbing to infection (Figure 3A). Baclofen treatment alone did not affect survival of mice (data not shown). Baclofen treatment of PR8-infected mice also led to a significant increase in lung pathology (Figure 3B). Post-infection stimulation of the GABAB receptor with baclofen also significantly increased the inflammatory response to low dose PR8 infection: at day 6 p.i., baclofen-treated mice produced significantly higher levels of Tnfa, Il1b, Ifnb, and Ccl5 mRNA and protein levels (Figure 3C; Supplemental Figure 2), as well as increased protein levels of GRP and HMGB1 in their sera (Figure 3D), compared to vehicle-treated, infected mice, consistent with their increased sensitivity to low dose influenza PR8 infection.
Figure 3. Effect of stimulating GABAB receptor during influenza PR8 infection.

(A) Mice were infected with an LD10 of mouse-adapted influenza strain PR8. Mice received vehicle (saline) or GABAB agonist (baclofen) daily from day 2 to day 4 p.i. Survival was monitored for 14 days. Data shown are combined results of 2 separate experiments N = 4–5 mice/treatment group/mock; N = 7–10 mice/treatment group/influenza). (B,C,D) WT C57BL/6J mice were infected with mouse-adapted influenza strain PR8. Mice received vehicle (saline) or GABAB agonist (baclofen) daily from day 2 to day 4 p.i. Lungs were harvested on day 6 p.i. for histopathology (B), lung gene expression by qRT-PCR (C), and levels of GRP and HMGB1 (D). Data represent the combined results of 2 separate experiments N = 4–5 mice/treatment group/mock; N = 7–10 mice/treatment group. *** p = 0.0002; **** p < 0.0001.
Effect of GABA on GRP-mediated infiltration of lung macrophages during influenza infection in mice
Previous studies suggested that acute lung injury (ALI) induced by oxidative stress leads to enhanced GRP expression and this, in turn, acts as a chemotactic factor for neutrophils and monocytes via stimulation of the high affinity GRP receptor, GRPR/BB213,52,53. It has been widely reported that in response to influenza, the infiltrating cells are largely monocytic54–57. Since influenza infection induced oxidative stress (Figure 1), as well as increased the number of PNEC/NEBs producing GRP19, we examined the effect of the GRP small molecule antagonist, NSC7742719,39, on the infiltration of monocytic cells into the lungs of mice in response to PR8 infection. Expression of CD68 (a pan-monocyte marker), CCR2 (the infiltrating monocyte chemokine receptor for CCL2), and TLR4 (the primary target of HMGB158), were first measured in the lungs of mice infected with influenza PR8 and treated therapeutically with vehicle or with NSC77427, the small molecule GRP antagonist that we previously reported to be protective during PR8 infection19. Mice infected with influenza PR8 and treated with vehicle exhibited significantly increased mRNA levels of all three monocytic markers, while therapeutic treatment of PR8-infected mice with NSC77427 on days 2–6 p.i. significantly decreased expression of all three genes (Figure 4). Immunohistochemical staining of lung sections from these same mice was carried out for CD68 and TLR4. Blinded quantification of CD68- and TLR4-positive cells in multiple sections per slide confirmed the mRNA data that influenza infection induced a significant infiltration of monocytes, and that treatment of influenza-infected mice with NSC77427 significantly decreased the number of infiltrating CD68+, TLR4+ monocytic cells (Table I). Collectively, this data indicates that blocking GRP during influenza infection results in a reduction of TLR4+ monocytes infiltrating into the lungs.
Figure 4. GRP mediates infiltration of CD68+, TLR4+ CCR2+ monocytic cells.

WT C57BL/6J mice were infected with and LD90 of mouse-adapted influenza strain PR8 (LD90; ~7500 TCID50) and treated with vehicle (0.0096% DMSO in saline; i.v.) or NSC77427 (20 mM; i.v.) daily from days 2 to 6 p.i. Lungs were harvested on day 7 post-infection for mRNA gene expression by qRT-PCR. Data represent the combined results of 2 separate experiments. N = 4–5 mice/treatment group/mock; N = 5 mice/treatment group. *p < 0.01; **p < 0.007.
Table I.
Blocking GRP decreases infiltrating TLR4+ monocytes
| CD68+ cells/20X field | SEM | TLR4+ cells/20X field | SEM | |
|---|---|---|---|---|
| PR8+Vehicle | 2.6 | 0.11 | 3.1 | 0.23 |
| PR8+NSC77427 | 1.5 | 0.34 | 1.19 | 0.31 |
| Statistics | p = 0.04 | p = 0.0002 |
The data presented in Figures 2D and 3D indicate that treating influenza PR8-infected mice with either saclofen or baclofen resulted in the decreased or increased production of GRP, respectively. We hypothesized that if less GRP were produced, or its ability to interact with its receptor is blocked, then fewer TLR4+ inflammatory monocytes would be recruited to the lungs. Conversely, if more GRP were produced, then more TLR4+ inflammatory cells would be recruited, resulting in more severe disease. To test whether antagonizing (saclofen) or stimulating (baclofen) the GABAB receptor also affects the recruitment of TLR4+ inflammatory monocytic cells, we examined induction of Cd68 and Tlr4 mRNA in the lung samples of influenza PR8-infected mice that were treated with vehicle only, saclofen, or baclofen as shown in Figures 2C and 3C. Lungs from mice infected with influenza PR8 and treated with saclofen expressed significantly less Cd68 and Tlr4 mRNA compared to untreated PR8-infected mice (Figure 5A), while mice that were infected and treated with baclofen showed a significant increase in both Cd68 and Tlr4 mRNA compared to infection alone (Figure 5B). Immunohistochemical staining for the monocytic marker, CD68, in saclofen-treated, PR8-infected mice confirmed a marked reduction in the number of CD68+ cells compared to vehicle-treated PR8-infected mice (Figure 6). Panels A and B from mock-infected mice show typical lung architecture with one normal CD68+ macrophage visible in an alveolar space as shown by an arrow in panel B. Mice infected with influenza PR8 and treated with vehicle, as shown in panels B and C, show severe acute pneumonia with multiple small veins surrounded by CD68+ monocytes and macrophages crossing through the vascular wall as indicated with arrows. Importantly, mice infected with influenza PR8 and treated with saclofen showed very few CD68+ macrophages as pointed out by arrows in panel F. Taken together, our data support the conclusion that influenza infection leads to the release of GABA that, in turn, elicits production of GRP by PNEC/NEBs. GRP then recruits CD68+ monocytic cells to the lung that mediate the TLR4-dependent inflammation associated with influenza infection.
Figure 5. Antagonizing or stimulating GABAB receptor during influenza PR8 infection alters gene expression of markers of monocytic infiltration and GRP.

(A) Mice were infected with an LD90 of mouse-adapted influenza strain PR8. Mice received vehicle (saline) or GABAB receptor antagonist (saclofen) daily from day 2 to day 4 post-infection. Lungs were harvested on day 6 p.i. and gene expression analyzed by qRT-PCR. Data represent the combined results of 2 separate experiments. N = 4–5 mice/treatment group/mock; N = 7–10 mice/treatment group. (B) Mice were infected with an LD10 of mouse-adapted influenza strain PR8. Mice received vehicle (saline) or GABAB agonist (baclofen) daily from day 2 to day 4 p.i. Lungs were harvested on day 6 p.i. and gene expression was analyzed by qRT-PCR. Data represent the combined results of 2 separate experiments N = 4–5 mice/treatment group/mock; N = 7–10 mice/treatment group. *** p = 0.0002; **** p < 0.0001.
Figure 6. IHC staining for CD68 in mouse lungs infected with influenza.

Lung sections from Figure 2 were stained in mock-infected (panels A and B), PR8 + vehicle treatment (panels C and D), and PR8 + saclofen treatment (panels E and F). Representative images shown are results of 2 separate experiments N = 4–5 mice/treatment group/mock; N = 7–10 mice/treatment group. Scale bars for panels A, C, and E at 100X are 200 μm and at 400X are 50 μm for panels B, D, and F. Arrows point to CD68+ cells. Abbreviations: Alv, alveoli; Art, artery; Aw, airway; V, vein.
DISCUSSION
The lungs and respiratory tract act as a key innate mechanical barrier against respiratory pathogens. The lung, which has been classified as an endocrine organ, is highly innervated throughout the airway by neurons that are able to respond and work in conjunction with the immune response to a variety of chemical and microbial insults59. While much work has been done on a neuro-immune response during allergic inflammation59, the neuro-immune response during the innate immune response to viral infections is less understood with the preponderance of studies focused on mechanisms by which viral infections elicit airway hyperreactivity and sympathetic reflex responses (i.e., cough and sneezing)60 and not on the initial response to infection. Our previous study focused on the role of GRP during influenza infection19. We were the first to show that influenza infection triggered GRP production in mice and cotton rats, a unique rodent species that is susceptible to non-adapted human respiratory virus strains61, and showed that inhibiting either GRP or its receptor greatly increased survival in mice, reduced the inflammatory response, and improved lung histology19.
PNEC/NEBs are the only lung epithelial cell that is innervated37. GABA is a potent neurotransmitter and neuromodulator that has been assumed to be produced by neurons that act on PNEC/NEB to produce GRP20,21. However, a study by Barrios et al.62 provided convincing evidence that PNECs are the major source of GABA in the lungs in primate and human models of Th2-mediated allergic inflammation induced by exposure to ovalbumin and LPS; however, their study does not exclude the possibility that in a more Th1-driven model of inflammation, such as that induced by influenza infection, neurons and/or PNECs are a source of GABA. Future studies will be required to determine the cellular source of GABA in response to influenza infection. Regardless of the cellular source of the GABA, stimulation of PNEC/NEBs in an autocrine/paracrine manner would result in enhanced GRP production as we have shown.
In this study, we focused on the production of GABA in response to influenza infection and how its modulation influences the outcome of infection, specifically on the neuroendocrine response and resulting monocytes coming into the tissue after infection. While GABA is known as an inhibitory neurotransmitter in the brain47, it can also be involved in migration and cellular proliferation in neuronal and non-neuronal cells63–65. We specifically focused on modulation of the metabotropic GABAB receptor on PNEC/NEBs due to our previous study showing release of GRP during influenza infection. GABAB receptor-specific agonists, such as baclofen, have been shown to decrease the airway responses by modulating acetylcholine release to bronchoconstricting agents66,67. However, baclofen has also been shown to induce more severe respiratory responses in asthma studies following administration of methacholine68. These studies were again focused on Th2 models, thus, the role of modulating GABAB during a severe Th1 model such as influenza had not been reported. In our current study, inhibiting the GABAB receptor with a specific inhibitor improved the response to a lethal influenza infection, while administration of a GABAB receptor-specific agonist worsened the response to an otherwise low dose infection and resulted in increased inflammatory mediator production and more severe lung pathology.
In conclusion, the interplay of GABA and the host innate immune response plays a pivotal role in the response to influenza-induce ALI. In Figure 7, we provide a hypothetical model that encompasses our previous and new data. We posit that influenza-infected epithelial cells lead to inflammatory and oxidative stress responses resulting in cell death and the release of HMGB1. Our data presented herein shows that influenza-induced release of GABA acts on the PNEC/NEBs through the GABAB receptor to produce GRP that, in turn, acts as a chemotactic factor to recruit TLR4+ monocytic cells into the lung that respond to HMGB1-mediated TLR4/MD-2 signaling to release inflammatory mediators of lung damage. Blocking GRP with a small molecule inhibitor or monoclonal antibody or blocking the GRPR/BB2 reduced lung damage and lethality19. Blocking GABAB signaling also blocked GRP production and release, and the recruitment of TLR4+ monocytic cells that amplify TLR4-dependent, HMGB1-mediated signaling and lung damage. Conversely, stimulating GABAB signaling leads to further damage and sustained inflammation. To our knowledge, our findings represent the first report of a novel neuroendocrine pathway leading to the induction of inflammation and lung pathology during influenza-induced ALI.
Figure 7. Hypothetical model for involvement of GABA, GRP, and TLR4 during influenza infection.

Influenza-infected epithelial cells lead to inflammatory and stress responses resulting in HMGB1 release and the production of secretion of GABA by neurons and NEBs. HMGB1 signals through TLR4/MD2, leading to further oxidative stress, cytokine release, tissue damage, and lethality. GABA production by neurons and/or PNECs/NEBs triggers GRP production, which recruits TLR4+ monocytes into the lung that are the targets of HMGB1-mediated TLR4/MD-2 signaling, leading to proinflammatory mediator production and subsequent lung damage. Blocking GRP reduces lung damage and lethality as previously reported. Blocking GABAB signaling blocks GRP production and release, thereby preventing further recruitment of TLR4+ monocytes that amplify signaling and damage. Stimulating GABAB signaling leads to further damage and sustained inflammation.
Methods
Reagents:
GRP Enzyme ImmunoAssay (EIA), that is specific for the mature form of GRP (amide 1–27), was purchased from Phoenix Pharmaceuticals, Inc. (Catalog # EK-027–07; Burlingame, CA). GABA ELISA kit was purchased from Aviva Systems Biology (Catalog # OKEH02564; San Diego, CA). HMGB1 ELISA kit was purchased from IBL International (Catalog # ST51011; Toronto, Ontario, Canada). The 8-hydroxy 2 deoxyguanosine (8-OHdG) ELISA kit was purchased from Abcam (Catalog # ab201734; Branford, CT). GAD ELISA was purchased from Lifeome Biolabs (Catalog # EL009159MO-96; Oceanside, CA). The ELISA kit for IFN-β was purchased from PBL Assay Science (Catalog # 42400; Piscataway, NJ). The ELISA kits for TNF-α (Catalog # MTA00B), IL-1β (Catalog # MLB00C), CCL5/RANTES (Catalog # MMR00), and CCL2/MCP-1 (Catalog # MJE008) were purchased from R&D Systems (Minneapolis, MN). The GRP inhibitor, NSC77427, was made by the Small Molecule Library Reagent Program (National Cancer Institute, Division of Cancer Treatment & Diagnosis/Developmental Therapeutics Program, NIH). Saclofen and baclofen were purchased from Tocris Bioscience (Minneapolis, MN). The TLR4 antibody was purchased from Invitrogen (Catalog # 14–991782; Waltham, MA) and the CD68 antibody purchased from Biolegend (San Diego, CA).
Mice:
Six-week old, WT C57BL/6J mice were purchased from the Jackson Laboratory (Bar Harbor, ME). All animal experiments were conducted with institutional IACUC approvals from University of Maryland, Baltimore.
Viruses:
Mouse-adapted H1N1 influenza A/PR/8/34 virus (“PR8”) (ATCC, Manassas, VA) was grown in the allantoic fluid of 10-day old embryonated chicken eggs as described69 and was kindly provided by Dr. Donna Farber (Columbia University).
Virus challenge and treatment:
For survival experiments, WT C57BL/6J mice were infected with mouse-adapted influenza virus, strain A/PR/8/34 (PR8; ~7500 TCID50i.n., 25 ml/nares), a dose of PR8 that kills ~90% of infected mice26,49 for saclofen experiments. For baclofen experiments, WT C57BL/6J mice were infected with mouse-adapted influenza virus, strain A/PR/8/34 (PR8; ~1500 TCID50 i.n., 25 μl/nares), a dose of PR8 that kills ~10% of infected mice51. Two days after PR8 infection, mice received either vehicle (indicated in the figure legend), saclofen (20 μg; 100 ml i.p.), or baclofen 100 μg; 100 ml i.p.) daily for 3 consecutive days (day 2 until day 4). For tissue analysis, mice were treated as above and the sera and lungs harvested on day 6 p.i. for histology, gene expression, and protein levels.
Histology and staining:
Lungs were inflated and perfused and fixed with 4% PFA. Fixed sections (5 μm) of paraffin-embedded lungs were stained with hematoxylin and eosin (H&E). Four inflammatory parameters were scored independently from 0 to 4 for each section: peribronchiolitis (inflammatory cells, primarily lymphocytes, surrounding a bronchiole), perivasculitis (inflammatory cells, primarily lymphocytes, surrounding a blood vessel), alveolitis (inflammatory cells within alveolar spaces), and interstitial pneumonitis (increased thickness of alveolar walls associated with inflammatory cells). Slides were randomized, read blindly, and scored for each parameter. Data is shown as a cumulation of the four parameters measured.
Immunohistochemistry:
IHC staining for CD68 for imaging in Figure 6 was carried out as follows: Paraformaldehyde-fixed, paraffin-embedded lung sections were treated for 10 min with Triton X-100 (0.3% in PBS), then normal serum blocking. Slides were incubated with anti-CD68 antibody diluted 1:100 overnight at 4°C, followed by PBS washes and incubation for 2 h at 4° C with 1:200 dilution of biotinylated secondary antibody, then ABC-alkaline phosphatase complex (Vector #AK-5000), and ImmPACT Vector Red Substrate kit for alkaline phosphatase (AP; Vector #SK-5105). Slides were counterstained with hematoxylin. IHC staining for CD68 and TLR4 was carried out as follows for quantitation shown in Table I: Paraformaldehyde-fixed, paraffin-embedded lung sections were treated for 10 min with Triton X-100 (0.3% in PBS), then normal serum blocking. Diluted primary antibodies were added to serial sections overnight at 4°C, followed by PBS washes and incubation for 2 h at 4° C with 1:250 dilution of biotinylated secondary antibodies. After blocking with 3% H2O2 in methanol, and slides were incubated with ready to use avidin-biotin complex (peroxidase ready; Vectastain), all slides were then washed and incubated with diaminobebzidine (DAB) solution then counterstained with 2.5% aqueous methyl green. All slides were finally blinded for semi-quantitative analysis by a board-certified pathologist. Results are expressed as the mean numbers of cells positive for CD68 or TLR4 per mm2 lung tissue section (Table I). Statistical analysis was carried out using a one-tailed Student’s t test.
Quantitative real-time PCR (qRT-PCR):
Total RNA isolation and qRT-PCR were performed as previously described70,71. Levels of mRNA for specific genes were normalized to the level of the housekeeping gene, HPRT, in the same samples and are expressed as “fold-increase” over the relative gene expression measured in mock-infected lungs.
Protein levels in serum and lung homogenates:
Protein levels in sera for GABA, GRP, HMGB1, 8-OHdG, MDA, and GAD were measured by ELISA according to the manufacturers’ protocols in the serum of mice infected with influenza as described above. Protein levels in lung tissue homogenates for TNF-α, IL-1β, IFN-β, CCL5/RANTES, and CCL2/MCP-1 were measured by ELISA according to the manufacturers’ protocols.
Statistics:
Statistical differences between two groups were determined using an unpaired, one-tailed Student’s t test with significance set at p < 0.05. For comparisons between ≥3 groups, analysis was done by one-way ANOVA followed by a Tukey’s multiple comparison post-hoc test with significance determined at p < 0.05. For survival studies, a Log-Rank (Mantel-Cox) test was used.
Supplementary Material
Acknowledgements:
This work was supported by the following grants: NIH AI159507 (KAS) and NIH AI123371 (SV). We thank Professor Margaret McCarthy (UMB) for thoughtful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests: The authors declare no competing interests.
References Cited
- 1.Baral P, Udit S, & Chiu IM Pain and immunity: implications for host defense. Nat Rev Immunol. 19, 433–447 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Szallasi A, Cortright DN, Blum CA, & Eid SR The vanilloid receptor TRPV1: 10 years from channel cloning to antagonist proof-on-concept. Nat Rev Drug Discov. 6, 357–372 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Blake KJ, Jiang XR, & Chiu IM Neuronal regulation of immunity in the skin and lungs. Trends Neurosci. 42, 537–551 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chu C, Artis D, & Chiu IM Neuro-immune interactions in the tissues. Immunity 52, 464–474 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boers JE, den Brok JL, Koudstall J, Arends JW, & Thunnissen FB Number and proliferation of neuroendocrine cells in normal human airway epithelium. Am J Resp Crit Care Med. 154, 758–763 (1996). [DOI] [PubMed] [Google Scholar]
- 6.Branchfield K, Nantie L, Verheyden JM, Sui P, Weinhold MD, & Sun X Pulmonary neuroendocrine cells function as airway sensors to control lung immune responses. Science 351, 707–710 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hewitt RJ & Lloyd CM Regulation of immune responses by airway epithelial cell landscape. Nat Rev Immunol. 21,347–362 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McDonald T, Nilsson G, Vagne M, Ghatei M, Bloom SR, & Mutt V A gastrin releasing peptide from the porcine nonantral gastric tissue. Gut 19:767–774 (1978). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McDonald TJ et al. Characterization of a gastrin releasing peptide from porcine nonantral gastric tissue. Biochem Biophysical Res Comm. 90: 227–233 (1979). [DOI] [PubMed] [Google Scholar]
- 10.Jensen RT, Battey JF, Spindel ER, & Benya RV International union of pharmacology. LXVIII. Mammalian bombesin receptors: nomenclature, distribution, pharmacology, signaling, and functions in normal and disease states. Pharmacol. Rev 60:1–41 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ramos-Álvarez I, Moreno P, Mantey SA, Nakamura T, Nuche-Berenguer B, Moody TW, Coy DH, Jensen RT. 2015. Insights into bombesin receptors and ligands: highlighting recent advances. Peptides 72:128–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jaeger N, Czepielewski RS, Bagatini M, Porto BN, & Bonorino C Neuropeptide gastrin-releasing peptide induces PI3K/reactive oxygen species-dependent migration in lung adenocarcinoma cells. Tumor Biol. 39:1–11 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Sunday MW Oxygen, gastrin-releasing peptide, and pediatric lung disease: life in the balance. Front. Pediatr Jul 18; 72. doi: 10.3389/fped.2014.00072 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moody TW, Venugopal R, Zia F, Patierno S, Leban JJ, & McDermed J BW2258U89: a GRP receptor antagonist which inhibits small cell lung cancer growth. Life Sci. 56:521–529 (1995). [DOI] [PubMed] [Google Scholar]
- 15.World Health Organizstion. Influenza (seasonal). DATE. Retrieved 10/5/2022 from: https://www.who.int/news-room/fact-sheets/detail/influenza-(seasonal).
- 16.Centers for Disease Control and Prevention. Retrieved 10/5/2022 from: https://www.cdc.gov/flu/about/burden/preliminary-in-season-estimates.html.
- 17.Interim adjusted estimates of seasonal influenza vaccine effectiveness - United States, February 2013. MMWR Morb Mortal Wkly Rep (2013) 62:119–123. [PMC free article] [PubMed] [Google Scholar]
- 18.Emergence of Avian Influenza A (H7N9) virus causing severe human illness – China, February – April 2013. MMWR Morb Mortal Wkly Rep (2013) 62:366–371. [PMC free article] [PubMed] [Google Scholar]
- 19.Shirey KA et al. Novel role of gastrin releasing peptide-mediated signaling in the host response to influenza infection. Mucosal Immunol. 12, 223–231 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weigert N, Schepp W, Haller A, & Schusdziarra V Regulation of gastrin, somatostatin, and bombesin release from the isolated rat stomach by exogenous and endogenous gamma-aminobutyric acid. Digestion 59, 16–25 (1998). [DOI] [PubMed] [Google Scholar]
- 21.Solorzano SR et al. GABA promotes gastrin-releasing peptide secretion in NE/NE-like cells: contribution to prostrate cancer progression. Sci Reports. 8:10272. Doi: 10.1038/s41598-018-28538-z (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Imai Y et al. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell 133, 235–249 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu M, Chen F, Liu T, Chen F, Liu S, & Yang J The role of oxidative stress in influenza infection. Microbes Infect. 19:580–586 (2017). [DOI] [PubMed] [Google Scholar]
- 24.Sgarbanti R et al. , Intracellular redox state as a target for anti-influenza therapy: are antioxidants always effective? Curr Top Med Chem. 14:2529–2541 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shirey KA, Blanco JCG, & Vogel SN Targeting TLR4 signaling to blunt viral-mediated acute lung injury. Front. Immunol 2021 Jul 2;12:705080.doi: 10.3389/fimmu.2022.705080 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shirey KA et al. The TLR4 antagonist Eritoran protects mice from lethal influenza infection. Nature. 497, 498–502 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cui X et al. Relationship between free and total malondialdehyde, a well-established marker of oxidative stress, in various types of human biospecimens. J Thorac Dis. 10, 3088–3097 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elkabany ZA et al. , Oxidative stress markers in neonatal respiratory distress syndrome: advanced oxidative protein product and 8-hydroxy-2-deoxyguanosine in relation to disease severity. Pediatric Research 87, 74–80 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang R et al. Kaempferol ameliorates H9N2 swine influenza virus-induced acute lung injury by inactivation of TLR4/MyD88-mediated NF-kB and MAPK signaling pathways. Biomedicine and Pharmacotherapy 89, 660–672 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Gonzalez-Juarbe N et al. Influenza-induced oxidative stress sensitizes lung cells to bacterial-toxin-mediated necroptosis. Cell Reports Aug 25;32(8):108062. doi: 10.1016/j.celrep.2020.108062 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tanaka R et al. Therapeutic impact of human serum albumin-thioredoxin fusion protein on influenza virus-induced lung injury mice. Front. Immunol May 5; 10.3389/fimmu.2014.00561 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kash JC et al. Treatment with the reactive oxygen species scavenger EUK-207 reduces lung damage and increases survival during 1918 influenza virus infection in mice. Free Radical Biology and Med. 67, 235–247 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cutz E, Chan W, Wong V, & Conen PE Ultrastructure and fluorescence histochemistry of endocrine (APUD-type) cells in tracheal mucosa of human and various animal species. Cell. Tissue Res 158:425–437 (1975). [DOI] [PubMed] [Google Scholar]
- 34.Cutz E Neuroendocrine cells of the lung and overview of morphologic characteristics and development. Experimental Lung Res. 3:3–4:185–208 (1982). [DOI] [PubMed] [Google Scholar]
- 35.Cutz E, Yeger H, & Pan J Pulmonary neuroendocrine cell system in pediatric lung disease-recent advances. Pediatric and Developmental Path. 10:419–435 (2007). [DOI] [PubMed] [Google Scholar]
- 36.Van Lommel A Pulmonary neuroendocrine cells (PNEC) and neuroepithelial bodies (NEB): chemoreceptors and regulators of lung development. Paediatric Resp. Rev 2:171–176 (2001). [DOI] [PubMed] [Google Scholar]
- 37.Barrios J et al. Early life allergen-induced mucus overproduction requires augmented neural stimulation of pulmonary neuroendocrine cell secretion. FASEB J. 31:4117–4128 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martínez A, Julián M, Bregonzio C, Notari L, Moody TW, & Cuttitta F Identification of vasoactive nonpeptide positive and negative modulators of adrenomedulin using a neutralizing antibody-based screening strategy. Endocrinology 145, 3858–3865 (2004). [DOI] [PubMed] [Google Scholar]
- 39.Martínez A Zudaire E, Julián M, Moody TW, & Cuttitta F Gastrin-releasing peptide (GRP) induces angiogenesis and the specific GRP blocker 77427 inhibits tumor growth in vitro and in vivo. Oncogene 24, 4106–4113 (2005). [DOI] [PubMed] [Google Scholar]
- 40.Tighe RM et al. Immediate release of gastrin-releasing peptide mediates delayed radiation-induced pulmonary fibrosis. Am J Pathol. 189, 1029–1040 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cuttitta F et al. Bombesin-like peptides can function as autocrine growth factors in human small-cell lung cancer. Nature 316, 823–826 (1985). [DOI] [PubMed] [Google Scholar]
- 42.Cao Y et al. Glutamate carboxypeptidase II gene knockout attenuates oxidative stress and cortical apoptosis after traumatic brain injury. BMC Neuroscience 17, 15. 10.1186/s12868-016-0251-1 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.MacMullin P et al. Increase in seizure susceptibility after repetitive concussion results from oxidative stress, parvalbumin-positive interneuron dysfunction and biphasic increases in glutamate/GABA ratio. Cerebral Cortex 30, 6108–6120 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Abruzzo PM, Panisi C, & Marini M The alteration of chloride homeostasis/GABAergic signaling in brain disorders: could oxidative stress play a role? Antioxidants 10, 1316. Doi:org/ 10.3390/antiox10081316 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Noguchi M, Furukawana KT, & Morimoto M Pulmonary neuroendocrine cells: physiology, tissue homeostasis and disease. Dis Model Mech. Dec 1; 13;12:dmm046920. Doi: 10.1242/dmm.046920:10.1242/dmm.046920 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee S-E, Lee Y, & Lee GH The regulation of glutamic acid decarboxylases in GABA neurotransmission in the brain. Archives Of Pharmacal Research 42, 1031–1039 (2019). [DOI] [PubMed] [Google Scholar]
- 47.Wu C & Sun D GABA receptor in brain development, function, and injury. Metab Brain Dis. 30, 367–379 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Drew CA, Johnston GAR, Kerr DIB, & Ong J Inhibition of baclofen binding to rat cerebellar membranes by phaclofen, saclofen, 3-aminopropylphosphonic acid and related GABAB receptor antagonists. Neuroscience Letters 113, 107–110 (1990). [DOI] [PubMed] [Google Scholar]
- 49.Shirey KA et al. Novel strategies for targeting innate immune responses to influenza. Mucosal Immunol. 9, 1173–1182 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Krach LE Pharmacotherapy of spacity: oral medication and intrathecal baclofen. J Child Neurol. 16, 31–36 (2001). [DOI] [PubMed] [Google Scholar]
- 51.Shirey KA et al. Influenza “trains” the host for enhanced susceptibility to secondary bacterial infection. mBio. 2019 May 7;10(3). Pii:e00810–19. Doi: 10.1128/mBio.00810-19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aguayo SM Determinants of susceptibility to cigarette smoke. Potential roles for neuroendocrine cells and neuropeptides in airway inflammation, airway wall remodeling, and chronic airflow obstruction. Am. J. Crit. Care Med 149, 1692–1698 (1994). [DOI] [PubMed] [Google Scholar]
- 53.Czepieleski RS et al. Gastrin-releasing peptide receptor (GRPR) mediates chemotaxis in neutrophils. Proc. Natl. Acad. Sci. U.S.A 109, 547–552 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lin KL, Suzuki Y, Nakano H, Ramsburg E, & Gunn MD CCR2+ monocyte-derived dendritic cells and exudate macrophages produce influenza-induced pulmonary immune pathology and mortality. J. Immunol 180, 2562–2572 (2008). [DOI] [PubMed] [Google Scholar]
- 55.Lin S-j. et al. The pathological effects of CCR2+ inflammatory monocytes are amplified by an IFNAR1-triggered chemokine feedback loop in highly pathogenic influenza infection. J. Biomed. Sci 21:99. Doi: 10.1186/s12929-014-0099-6 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huang S et al. PPAR-g in macrophages limits pulmonary inflammation and promotes host recovery following respiratory viral infection. J. Virol 93:e00030–19. Doi: 10.1128/JVI.00030-19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Guo K et al. Cellular heterogeneity and molecular reprogramming of the host response during influenza acute lung injury. J. Virol 96:e0124622. Doi: 10.1128/jvi.01246-22 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang H et al. MD-2 is required for disulfide HMGB1-dependent TLR4 signaling. J Exp Med. 212, 5–14 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jean EE, Good O, Inclan Rico JM, Rossi HL, & Herbert DR Neuroimmune regulatory networks of the airway mucosa in allergic inflammatory disease. J Leukoc Biol. 111, 209–221 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zaccone EJ & Undem BJ Airway vagal neuroplasticity associated with repiratory viral infections. Lung 194, 25–29 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ottolini MG et al. The cotton rat provides a useful small-animal model for the study of influenza virus pathogenesis. J Gen Virol. 86, 2823–30. [DOI] [PubMed] [Google Scholar]
- 62.Barrios J et al. Pulmonary neuroendocrine cells secrete g-aminobutyric acid to induce goblet cell hyperplasia in primate models. Am J Cell Mol Biol. 60, 687–694 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fujimori S, Hinoi E, & Yoneda Y Functional GABAB receptors expressed in cultured calvarial osteoblasts. Biochem Biophys Res Commun. 293, 1145–1452 (2002). [DOI] [PubMed] [Google Scholar]
- 64.Cunningham MD & Enna EJ Evidence for pharmacologically distinct GABAB receptor associated with cAMP production in rat brain. Brain Res. 720, 220–224 (1996). [DOI] [PubMed] [Google Scholar]
- 65.Couve A, Moss SJ, & Pangolos MN GABAB receptors: a new paradigm in G protein signalling. Mol Cell Neurosci. 16, 296–312 (2000). [DOI] [PubMed] [Google Scholar]
- 66.Tohda Y, Ohkawa K, Kubo H, Muraki M, Fukuoka M, & Nakajima S Role of GABA receptors in the bronchial response: studies in sensitized guinea-pigs. Clin Exp Allergy 28, 772–777 (1998). [DOI] [PubMed] [Google Scholar]
- 67.Chapman RW, Danko G, Rizzo C, Egan RW, Mauser PJ, & Kreutner W Prejunctional GABA-B inhibition of cholinergic, neurally-mediated airway contractions in guinea-pigs. Pulm Pharmacol 4, 218–224 (1991). [DOI] [PubMed] [Google Scholar]
- 68.Dicpinigaitis PV Effect of the GABA-agonist baclofen on bronchial responsiveness in asthmatics. Pulm Pharmacol Ther 12,257–260 (1999). [DOI] [PubMed] [Google Scholar]
- 69.Teijaro JR et al. Costimulation modulation uncouples protection from immunopathology in memory T cell response to influenza virus. J. Immunol 182, 6834–43 (2009). [DOI] [PubMed] [Google Scholar]
- 70.Shirey KA, Cole LE, Keegan AD, & Vogel SN Francisella tularensis live vaccine strain induces macrophage alternative activation as a survival mechanism. J. Immunol 181, 4159–4167 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cole LE et al. Immunological consequences of Francisella tularensis live vaccine strain infection: role of the innate immune response in infection and immunity. J. Immunol 176, 68888–99 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.