

Summary
In early life, susceptibility to invasive infection skews towards a small subset of microbes, while other pathogens associated with disease later in life, including Streptococcus pneumoniae (Spn), are uncommon among neonates. To delineate mechanisms behind age-dependent susceptibility, we compared age-specific mouse models of invasive Spn infection. We show enhanced CD11b-dependent opsonophagocytosis by neonatal neutrophils improved protection against Spn during early life. Augmented function of neonatal neutrophils was mediated by higher CD11b surface expression at the population level due to dampened efferocytosis, which also resulted in more CD11bhi ‘aged’ neutrophils in peripheral blood. Dampened efferocytosis during early life could be attributed to lack of CD169+ macrophages in neonates, and reduced systemic expression of multiple efferocytic mediators, including MerTK. Upon experimentally impairing efferocytosis later in life, CD11bhi neutrophils increased and protection against Spn improved. Our findings reveal how age-dependent differences in efferocytosis determine infection outcome through modulation of CD11b-driven opsonophagocytosis and immunity.
Graphical Abstract
eTOC Blurb
Many pathogens rarely cause invasive disease during neonatal life. Bee et al. delineate an immunologic determinant of this phenomenon. During early life, developmental impairments in macrophage function (efferocytosis) alter neutrophil homeostasis to augment CD11b-dependent opsonophagocytosis. This results in increased protection against certain pathogens, and accounts for age-related patterns of susceptibility.
Introduction
While newborns are generally considered more susceptible to infection, this trend is predominantly seen with a small subset of microbes, clinically defined as neonatal pathogens. Many microbes known to cause systemic infection at later stages in life are infrequently problematic during the neonatal period1. While it has been assumed that this protection is due to maternally-acquired passive immunity, active mechanisms in neonates may also protect against invasive infection by these ‘non-neonatal pathogens’. This epidemiological observation is at odds with the idea of hypertolerogenic responses thought to characterize neonatal immunity or a weak immune system, as seen in elderly or immunocompromised individuals. To delineate this dichotomy of susceptibility versus resistance in early life, a better understanding of immune mechanisms that characterize the neonatal period is needed. Much of our understanding of host defenses are founded on evidence relying solely on adult mouse models, which do not account for environmental and developmental challenges faced by neonates, or human cord blood leukocytes, which may not be representative of immune cells during immediate postnatal life2.
This dichotomy between neonatal and ‘non-neonatal pathogens’ is demonstrated by the major pathogenic streptococci. Streptococcus agalactiae (GBS) is a leading cause of invasive neonatal infection, while Streptococcus pneumoniae (Spn) and Streptococcus pyogenes (GAS) invasive infections are uncommon during this period3-7. In the case of Spn, rates of nasopharyngeal carriage peak during early childhood8, and therefore the rarity of invasive infection among neonates cannot be attributed to a lack of environmental exposure during early life9,10. Spn carriage and invasive infection following colonization can be modeled in mice where maternal immunity from prior exposure is not a confounding factor. We have previously demonstrated that for a strain of Spn that causes invasive disease in adults11, mice are protected during the first two weeks of life regardless of the postnatal age by which they were nasally challenged12. Using Spn as a model ‘non-neonatal pathogen,’ we sought to investigate immune mechanisms that facilitate systemic resistance to pathogens during early life. We then extended our findings to other invasive streptococcal species. Herein, we show that dampened efferocytosis in early life alters neutrophil homeostasis to augment CD11b-mediated responses, whereby the extent of dependence on CD11b effector activity dictates invasive infection outcome during the neonatal versus non-neonatal period of life.
Results
Neonatal mice are resistant to systemic infection by Spn, a ‘non-neonatal pathogen’
To model age-dependent susceptibility to invasive infection by a ‘non-neonatal pathogen’, we adopted an intraperitoneal (IP) model of Spn infection to induce systemic challenge in neonatal (6 - 9 days of life) and adult (6 - 14 weeks of life) mice, with adjustment of dose based on relative weight of the animal. The survival rate of neonatal mice upon IP challenge was significantly higher than that of adult mice (Fig 1A), correlating with our previous observation of a protective window in neonates following intranasal challenge with an invasive Spn strain12. At early time points of 24 and 32 hours post-Spn IP challenge, neonatal mice had significantly lower bacterial burden in the blood and spleen compared to adult mice (Fig 1B-C). To exclude the possibility that improved protection in neonates was solely attributed to adjusted doses based on body weight, we infected neonatal and juvenile (21 - 28 days of life) mice with an equivalent dose of Spn. Even when challenged with the same dose, neonates had higher survival rates than the larger juveniles (Fig 1A), and we continued to observe significantly lower bacterial burden in both blood (Fig 1D) and spleen (Fig 1E) of neonates compared to juveniles. Together, these results suggested that control of Spn dissemination is more efficient in neonatal (early in life) compared to juvenile and adult mice (later in life).
Figure 1: Neonates are resistant to systemic infection by Streptococcus pneumoniae, a ‘non-neonatal pathogen’.

[A] Survival of WT neonatal (NNT, solid circle black line, n = 26), juvenile (JUV, half solid circle grey spotted line, n = 25) and adult mice (AD, open circle grey line, n = 18) following intraperitoneal (IP) challenge with P2406, an Spn strain with invasive potential in humans adapted in mice, at 5 x 101 – 1 x 102 colony forming units (CFU) per gram of body weight.
[B] Recoverable live Spn in blood of NNT and AD to assess systemic dissemination at two early time points, 24 hours (n = 16 for NNT, n = 11 for AD) and 32 hours (n = 19 for NNT, n = 13 for AD) post-infection, following IP challenge with Spn at 5 x 101 – 1 x 102 CFU per gram of body weight.
[C] Similar as described in [B], but for spleen at 24 hours (n = 16 for NNT, n = 11 for AD) and 32 hours post-infection (n = 17 for NNT, n = 11 for AD); BW, body weight.
[D] Recoverable live Spn in blood of NNT (n = 10) and JUV (n = 13) to assess systemic dissemination at 32 hours post-infection, following IP challenge with Spn at 5 x 101 – 1 x 102 CFU per gram of neonatal body weight.
[E] Similar as described in [D] but for spleen (n = 10 for NNT, n = 13 for JUV).
[F] Blood neutrophil counts from mice treated with an IgG isotype control clone 2A3 (n = 6) or neutrophil-specific depletion antibody clone 1A8 (n = 6) following IP challenge with Spn at 5 x 101 – 1 x 102 CFU per gram body weight at 32 hours post-infection.
[G] Recoverable live bacteria in spleen from mice described in [F].
[H] Blood monocyte counts from mice that were either wild-type, Ccr2+/+ (n = 15), or CCR2-deficient, Ccr2−/− (n = 9), following IP challenge with Spn at 5 x 101 – 1 x 102 CFU per gram body weight at 32 hours post-infection.
[I] Recoverable live bacteria in spleen from mice described in [H]. Each data point represents an individual mouse, and data are representative of experiments repeated at least twice. Data with error bars are presented as mean ± SEM. N.S., not significant.
Neutrophils facilitate resistance against invasive Spn infection in neonatal mice
Next, we asked what cellular mediator is directly responsible for the improved protection in neonates. The quick onset of protection in neonates as early as 24 hours post-IP challenge, combined with absence of pre-existing Spn-specific antibodies (as dams were not exposed to Spn), suggested the importance of innate, rather than adaptive, immunity in this process. We turned our attention to phagocytic cells, specifically neutrophils, given their established importance as the initial line of defense against invading extracellular microbes, including Spn13,14. While the role of neutrophils in controlling Spn dissemination is established in non-neonatal stages of life15,16, we questioned if this holds true in the neonatal period. We tested this hypothesis by depleting neutrophils in neonates with the 1A8-clone monoclonal antibody (which specifically recognize Ly6G, a pan-neutrophil marker), followed by IP challenge with Spn. In the 1A8-treated neonates, an approximate 50% reduction in blood neutrophil numbers was associated with a significant increase in splenic bacterial load compared to isotype control, 2A3-treated neonates (Fig 1F-G), supporting a role for neutrophils in conferring immune protection against Spn in early life. To further ascertain the role of neutrophils as the primary cellular mediator of this protection, we infected neonatal mice that were deficient in CCR2, which have impaired monocyte trafficking17. As expected, monocyte counts were lower in Ccr2−/− neonates compared to wild-type (Ccr2+/+) neonates (Fig 1H). We did not observe differences in bacterial burden between Ccr2−/− and Ccr2+/+ neonates (Fig 1I), indicating that neutrophils were the primary circulating myeloid cellular mediator conferring protection against Spn during the neonatal period.
Increased CD11b expression on early life neutrophils drives systemic immunity against Spn
Considering the importance of neutrophils in controlling bacterial infection in both neonatal and non-neonatal stages of life, we sought to distinguish the features of neonatal neutrophils that accounted for improved protection in early life. We utilized flow cytometry to phenotype neutrophils in the bone marrow (BM) of mice at different ages. Gating strategy is listed in Fig S1A.
We found that surface expression of CD11b on BM neutrophils was higher in neonatal mice, and exhibited an age-dependent decay (Fig 2A). Higher CD11b surface expression was also observed on Ly6C-expressing neonatal monocytes (Fig S1B). In juvenile mice, surface expression of CD11b on myeloid cells were similar to that of adults (Fig 2A & Fig S1B). This correlated with the increased susceptibility to Spn systemic infection in juveniles compared to neonates, despite receiving similar infectious doses. We did not observe differences in CD11b mRNA transcript amounts in neutrophils from neonatal versus adult BM, indicating that increased CD11b surface expression on neonatal neutrophils is driven by post-transcriptional mechanisms (Fig 2B). The early life increase in CD11b on neutrophils did not reflect a transient phase in development as neutrophils from peripheral blood of neonatal mice also exhibited a similar phenotype (Fig 2C). Our observations in mice were recapitulated in humans. Mass cytometry analysis of peripheral blood neutrophils from human neonates during the first week of life (1 – 5 days of life) also exhibited higher CD11b surface expression than those from the immediate post-neonatal period (31 – 80 days of life), although this was not observed in monocytes (Fig 2D & Fig S1C). These results underscore in both mice and humans that surface expression of CD11b is increased on neutrophils during neonatal life.
Figure 2: Increased CD11b expression in early life enhances neutrophil function and drives systemic immunity against Spn.

[A] Flow cytometry analysis of CD11b surface expression via relative median fluorescence intensity (MFI) on bone marrow neutrophils (gated on Singlets+ CD45+ CD11b+ Ly6G+ Ly6Cmid) from mice at different ages [3 days old (3d), n = 4; 6 days old (6d), n = 5; 9 days old (9d), n = 5; JUV, n = 5; AD, n= 10].
[B] RT-PCR analysis of CD11b (Itgam) mRNA transcript in NNT (n = 15) versus AD (n = 11) bone marrow neutrophils isolated via density centrifugation.
[C] MFI of CD11b on peripheral blood neutrophils from NNT (n = 10) versus AD mice (n = 7).
[D] CD11b marker expression of neutrophils from mass cytometry experiment comparing newborns aged 0 – 5 days (n = 75) and 31 – 81 days after birth (n = 31), mean expression from binned days used for coloring.
[E] Opsonophagocytic uptake of mouse sera opsonized GFP-fluorescent Spn (P2531) by neutrophils from bone marrow suspensions of NNT versus AD mice at multiplicity of infection (MOI) of 25 (n = 8 for both groups).
[F] MFI of GFP on GFP+ neutrophils from [E] (n = 8 for both groups).
[G] Recoverable live bacteria in blood (left) and spleen (right) of WT (Itgam+/+) and CD11b-deficient (Itgam−/−) NNT mice challenged IP with Spn at 32 hours post-infection (n = 8 for Itgam+/+ n = 10 for Itgam−/−).
[H] MFI of CD11b on neutrophils from Itgam+/+ NNT (n = 3), CD11b-heterozygous (Itgam+/−) NNT (n = 6), and Itgam+/+ AD (n = 6). Itgam−/− AD is included as negative control (n = 2).
[I] Survival of Itgam+/+ NNT (solid circle black line; n = 20), Itgam+/− NNT (solid circle orange line; n = 17), Itgam−/− NNT (solid circle green line; n = 17) and Itgam−/− AD (open circle muted green line; n = 12) mice challenged IP with Spn at 5 x 101 – 1 x 102 CFU/g body weight.
[J] Neutrophil counts in peripheral blood from Itgam+/+ (n = 10), Itgam+/− (n = 10) and Itgam−/− (n = 9) NNT mice.
[K] Survival of Itgam+/+(solid circle dotted black line, n = 9) and Itgam−/− (solid circle dotted green line, n = 9) NNT mice treated with 25μg cobra venom factor at 24 hours prior infection, followed by IP challenge with Spn at 5 x 101 – 1 x 102 CFU/g body weight.
[L] Survival of WT NNT (solid circle black line; n = 18) versus AD mice (open circle grey line; n = 20), and both groups of mice treated with 25μg cobra venom factor (WT NNT, solid circle dotted black line, n = 9; WT AD, open circle dotted grey line, n = 11) following IP challenge with complement sensitive capsule switch mutant, P2453, at 5 x 101 – 1 x 102 CFU/g body weight.
[M] Recoverable live bacteria in blood of Itgam−/− neonate recipients receiving either Itgam+/+ neonatal neutrophils (solid circle, n = 9), Itgam+/− neonatal neutrophils (solid orange circles, n = 8) or Itgam−/− neonatal neutrophils (solid green circle, n = 8) relative to that of recipients receiving Itgam+/+ adult neutrophils (open circle; n = 9, n = 9, n = 7, respectively) at 20 hours post infection with Spn via IP. Each data point represents a biological replicate (refer to STAR Methods for number of mice pooled), and data are representative of experiments repeated at least twice. Data with error bars are presented as mean ± SEM. N.S., not significant. See also Figure S1.
Given the importance of the microbiota in regulating immune system development, we postulated that microbial cues in early life enhance CD11b surface expression on neonatal neutrophils. Contrary to our prediction, CD11b expression was similar between BM neutrophils from neonatal mice housed in germ-free and specific pathogen free conditions (Fig S1D). This suggests that the increase in CD11b expression on neonatal neutrophils was driven by developmentally-intrinsic mechanisms that are independent of microbial cues.
CD11b forms part of a heterodimeric β2 integrin complex that recognizes complement protein C3 deposited on bacterial surfaces, leading to neutrophil phagocytosis18; in humans, recognition of C3-tagged Spn by CD11b on neutrophils is important for control of Spn19-21. We hypothesized that increased CD11b surface expression on neonatal neutrophils enhances C3-dependent phagocytic capacity. To test this, we utilized an ex vivo phagocytosis assay using GFP-expressing Spn opsonized with mouse sera, and quantified bacterial uptake using flow cytometry. We observed that co-localization of neutrophils with a GFP signal was significantly higher in neonatal over adult neutrophils (Fig 2E). This difference was abrogated when Spn was incubated with heat-inactivated sera, in which complement proteins are degraded, supporting a critical role for C3-CD11b interactions in this process. For neutrophils with a GFP signal (co-localized with Spn), median fluorescence intensity (MFI) of GFP did not differ between adult and neonatal mouse neutrophils, suggesting that there was no difference in events following surface binding of Spn (Fig 2F). These trends were similarly observed with mouse monocytes (Fig S1E-F). In contrast to neutrophil CD11b expression, sera from neonates and adults deposited similar amounts of C3 on the surface of Spn (Fig S1G). Combined, these observations demonstrate that on the population level, more neonatal neutrophils are able to take up Spn, an observation that correlates with increased CD11b expression on early life myeloid cells.
Next, we tested whether the age-dependent increase in CD11b drives systemic immune resistance to Spn in early life in vivo. We first confirmed that CD11b-deficient (Itgam−/−) neonates had significantly higher bacterial burden than wild-type (Itgam+/+) neonates at 32 hours post-IP challenge with Spn (Fig 2G). To modulate expression of CD11b on neutrophils, we generated heterozygous CD11b (Itgam+/−) mice. Surface expression of CD11b on neutrophils from Itgam+/− neonates significantly decreased compared to Itgam+/+ neonates and phenocopied that of Itgam+/+ adults (Fig 2H). As predicted, upon IP Spn challenge both Itgam−/− and Itgam+/− neonates were more susceptible compared to WT neonates (Fig 2I). While Itgam+/− mice appear to have a survival advantage over the Itgam−/− mice during the first 72 hours post-infection, these mice ultimately succumb to the infection similarly. We also did not observe a difference in survival between Itgam−/− neonatal and Itgam−/− adult mice (Fig 2I). The effect of CD11b surface expression on bacterial density and neonate survival was not attributable to differences in neutrophil numbers (Fig 2J).
To confirm the protective effect of CD11b is dependent on interactions with complement C3, we first showed that Itgam+/+ and Itgam−/− neonates treated with cobra venom factor to deplete C3 were equally susceptible to Spn (Fig 2K). The polysaccharide capsule of Spn inhibits the deposition of C3 on the bacterial surface22,23. To further resolve C3-dependent CD11b phagocytic interactions as the critical mechanism driving age-dependent differences in resistance to Spn, we performed infection experiments using a serotype switch Spn mutant strain, whereby the locus for capsule expression was switched while preserving the genomic background of the strain. To this end, we switched the native serotype 4 capsule for the serotype 6A capsule, the latter of which has been demonstrated to have higher deposition of C3 on its surface24. We predicted that the increase in C3 deposition on surface of Spn overcomes the CD11b-C3 interactions bottleneck imposed during infection in adults, and thus, result in improved protection. In support of this hypothesis, we found that neonates and adults now become equally protected following infection with this capsule switch Spn strain (Fig 2L). This protection in both neonates and adults is lost following cobra venom factor pre-treatment to deplete C3 (Fig 2L).
Finally, we demonstrated that elevated CD11b expression on neutrophils specifically drove resistance to Spn in vivo during the neonatal period. To do so, we performed experiments whereby neutrophils isolated from either neonatal or adult donor bone marrow were adoptively transferred into Itgam−/− neonate recipients infected with Spn. Transfer of neutrophils from Itgam+/+ neonate donors resulted in improved bacterial control compared to that of Itgam+/+ adult donors, but not recipients receiving neutrophils from either Itgam+/− neonates or Itgam−/− neonates (Fig 2M & Fig S1H). As predicted, recipients receiving Itgam+/− neonatal neutrophils had similar bacterial burden to those receiving Itgam+/+ adult neutrophils; this confirms that other age-dependent but CD11b-independent phenotypic differences in neutrophils are not responsible for improved neonatal protection (Fig 2M & Fig S1H). Collectively, these in vivo observations illustrate the protective benefit against Spn of enhanced neutrophil CD11b surface expression in neonates.
Circulating neutrophils in neonatal mice bias towards an ‘aged’ phenotype
Similar to BM neutrophils, we also observed increased CD11b surface expression on early life blood neutrophils compared to those from later in life (Fig 2C). This suggested that increased CD11b expression did not merely reflect a transient phase in development within the BM and that circulating neutrophils are capable of enhanced CD11b-dependent effector function. This observation prompted us to ask about other characteristics of circulating neutrophils in early life.
There is increasing appreciation that neutrophils can specialize and adapt to different environments, a feature that can lead to generation of functional diversity. For example, neutrophils which have circulated in blood extensively and nearing the end of their lifespan, known as ‘aged’ neutrophils, have increased pro-inflammatory potential25,26. ‘Aged’ neutrophils are canonically marked by gradual loss of CD62L expression, which limits their migration into lymphatics, and a concomitant increase in CXCR4 expression, which promotes their migration into CXCL12-expressing tissues. These features help ‘aged’ neutrophils infiltrate tissues for degradation27. Since increased expression of CD11b is a characteristic feature of ‘aged’ neutrophils, we determined whether neonatal blood neutrophils have an ‘aged’ phenotype.
Expression of CD62L was significantly decreased on neonatal compared to adult mouse peripheral neutrophils (Fig 3A). By gating for CD62Llo (‘aged’) versus CD62Lhi (‘non-aged’) neutrophils, we found that neonates had a significantly higher proportion of CD62Llo neutrophils compared to adults (Fig 3B). As expected, expression of CXCR4 and CD11b were higher in the CD62Llo compared to the CD62Lhi population of neonatal neutrophils (Fig 3C-D). Other known phenotypic features of ‘aged’ neutrophils were observed in the CD62Llo population in neonates, including higher CD45 expression and lower Ly6G expression (Fig 3E-F), as well as lower side and forward scatters, suggesting reduced granularity and cell size, respectively (Fig 3G-H). As additional validation of our gating scheme, we observed a similar expression profile in the CD62Llo versus CD62Lhi populations of adult circulating neutrophils (Fig S2A-G). These results demonstrate that a higher proportion of ‘aged’ neutrophils exist in neonatal blood.
Figure 3: Circulating neutrophils in neonatal mice bias towards an ‘aged’ phenotype.

[A] MFI of CD62L on peripheral blood neutrophils from NNT (n = 10) versus AD mice (n = 7).
[B] Frequency of CD62Llo ‘aged’ neutrophils from peripheral blood of NNT (n = 10) versus AD mice (n = 7) as proportion of all hematopoietic cells [Live, CD45+ cells] (left) and of all neutrophils [Ly6G+ cells] (right); FMO, fluorescence-minus-one control.
[C-H] MFI analysis of known markers between CD62Llo ‘aged’ (red dots) and CD62Lhi ‘non-aged’ (blue dots) neutrophils in neonatal blood (n = 10). Each data point represents a biological replicate (refer to STAR Methods for number of mice pooled), and data are representative of experiments repeated three times. Data with error bars are presented as mean ± SEM. N.S., not significant. See also Figure S2.
Impaired efferocytosis in early life increases ‘aged’ neutrophil numbers in blood
Recently, a mechanism involving myeloperoxidase and T-cell death in influencing neutrophil lifespan has been described28. However, these parameters do not exhibit an age-dependent component (Fig S3A-B), suggesting this mechanism does not explain the ‘aged’ phenotype bias during neonatal life. Further, mediators of known neutrophil-intrinsic pathways, CXCR2- and CXCR4-dependent signaling29, involved in the ‘aging’ program did not appear to exhibit age-dependent differences (Fig S3C-D). Expression of CD11b itself also did not determine the proportion of ‘aged’ neutrophils present in blood (Fig S3E).
In addition to phenotypic differences of peripheral blood neutrophils between neonates and adults, we also observed that neonates have higher numbers of neutrophils in blood (Fig 4A). This raised the question of whether mechanisms to remove ‘aged’ neutrophils are impaired in neonates. Degradation of ‘aged’ cells, or efferocytosis, is mediated by macrophages including those of hematopoietic origin that express CD16930. While a previous study noted the absence of CD169+ macrophages in neonatal spleens31, the functional consequences of this observation on neutrophil homeostasis in early life was not explored.
Figure 4: Impaired efferocytosis in early life increases ‘aged’ neutrophil numbers in blood.

[A] Neutrophil counts in peripheral blood from NNT (6d old, n = 7; 9d old, n = 10), JUV (n = 6) and AD (n = 11) mice.
[B-F] Transcriptional analysis of whole spleen from naïve NNT versus AD for [B] Cd169, [C] Lxra, [D] Mertk, [E] Abca1 and [F] Gas6. Expression was calculated as fold change in NNT relative to AD (n = 5 for both groups).
[G] Confocal images of naive NNT (n = 5) and AD (n = 4) lungs immunostained for CD169 (blue), MerTK (red), F4/80 (yellow), Ly6G (green) and CD3 (cyan). Scale bar is at 50μm. Quantification of densitometry values are adjacent to the images. Each data point represents an individual mouse, and data are representative of experiments repeated at least twice. Data with error bars are presented as mean ± SEM. N.S., not significant. See also Figure S1, S3-S4.
Besides the spleen32, there are other sites for efferocytosis of’aged’ neutrophils, such as the lungs33 and bone marrow27. Because expression of Cd169 is restricted to macrophages in mice34, we assessed tissue expression of Cd169 in spleens, lungs and BM from neonate and adult mice. We found significant downregulation of Cd169 across all three neonatal tissues, which suggests decreased numbers of CD169+ macrophages at these sites in the neonatal period (Fig 4B & Fig S4A). Expression of Lxra, which regulates development of CD169+ macrophages in the spleen31 and facilitates efferocytosis32,35, was similarly downregulated across neonatal tissues (Fig 4C & Fig S4B). This trend was also observed with genes both facilitating and induced upon efferocytosis, Mertk32,35,36 and Abca137-38 (Fig 4D-E & Fig S4C-D). Expression of Gas6, a bridging molecule which facilitates recognition of phosphatidylserine on ‘aged’ cells by MerTK, induced during efferocytosis through LXRα-independent pathways32,39 was decreased in lungs and spleens of neonates, but not BM (Fig 4F & Fig S4E). Collectively, our transcriptional analyses demonstrate that expression of mediators driving efferocytosis is dampened in the neonatal period. We also performed confocal imaging of immunostained spleen sections. Firstly, we observed significantly decreased CD169 staining in neonatal compared to adult spleens (Fig 4G). This validated our transcriptional analysis and supports a reduced density of CD169+ macrophages in neonates. We also found that Ly6C-expressing monocytes, which can seed tissues to develop into CD169+ macrophages31, were increased in blood circulation and within the spleen (Fig S1I-H). This suggests that absence of CD169+ macrophages was not due to defective seeding by progenitors; rather, the developmental signals (i.e. induction of LXRα-signaling pathways) to promote their functional maturation were lacking (Fig 4C). Moreover, LXRα expression in the spleen has been shown to increase over the first weeks of life31. We also observed that the defined localization of CD169 staining in the marginal zone of adult spleens was not observed in neonatal spleens. This disorganized architecture in conjunction with decreased numbers of CD169+ macrophages in neonatal spleens potentially contribute to impaired efferocytosis. Secondly, while there was no difference in staining of macrophages by F4/80 in neonatal versus adult spleens, neonatal spleens exhibited significantly reduced MerTK co-staining. Even though numbers of F4/80+ macrophages may be similar to that in adults, this suggests that other tissue resident non-CD169 expressing macrophages in early life, such as those in the red pulp, may also exhibit a defect in efferocytosis. Combined, these observations support a dampening of efferocytic processes in early life, which contributes to increased ‘aged’ neutrophil numbers in blood of neonates.
Impairing efferocytosis later in life confers systemic protection against Spn dependent on CD11b-effector ability
To test if we could improve protection against invasive Spn infection in adult mice, we utilized CD169-diphteria toxin receptor (DTR) mice to transiently impair efferocytosis30, and predicted this would increase the generation of ‘aged’ neutrophils. First, we confirmed loss of CD169 staining cells in spleens of diphtheria toxin (DT)-treated CD169-DTR mice (Fig S5A). In line with previous work25, we found that DT-treated CD169-DTR mice had higher proportions and absolute counts of ‘aged’ neutrophils than their WT counterparts (Fig 5A-B). Importantly, these ‘aged’ neutrophils (CD62Llo) in the DT-treated CD169-DTR mice also exhibited increased CD11b expression over those of their ‘non-aged’ (CD62Lhi) counterparts (Fig 5C). We also observed that neutrophils from the BM of DT-treated CD169-DTR mice had higher CD11b surface expression than those from WT mice (Fig 5D). This was similarly observed with Ly6C-expressing monocytes (Fig S5B-C).
Figure 5: Impairing efferocytosis later in life protects against Spn in a CD11b-effector activity dependent manner.

[A] Frequency of CD62Llo ‘aged’ neutrophils from peripheral blood of WT versus CD169-DTR adult mice per all Live, CD45+ cells (n = 9 for both groups).
[B] Absolute counts of CD62Llo ‘aged’ neutrophils in peripheral blood of WT versus CD169-DTR adult mice (n = 9 for both groups).
[C] MFI of CD11b surface expression on CD62Llo ‘aged’ and CD62Lhi ‘non-aged’ neutrophils in blood of WT versus CD169-DTR adults (n = 9 for both groups).
[D] MFI of CD11b surface expression on neutrophils in bone marrow of WT (n = 11) versus CD169-DTR adults (n = 8).
[E] Recoverable live Spn in blood (left) or spleen (right) of WT and CD169-DTR adults to assess systemic dissemination at 32 hpi (n = 9 for both groups) following IP Spn challenge.
[F] Survival of WT (open circle grey line, n = 12), CD169-DTR (open circle pink line, n = 13) and both groups of mice treated with 25μg of cobra venom factor (WT, open circle dotted grey line, n = 9; CD169-DTR, open circle dotted pink line, n = 9) following IP Spn challenge.
[G] Frequency of CD62Llo ‘aged’ neutrophils from peripheral blood of Mertkfl/fl (n = 6) versus Lyz2Cre/+ Mertkfl/fl (n = 9) adult mice per all Live, CD45+ cells.
[H] Recoverable life Spn in blood (left) or spleen (right) of Mertkfl/fl (n = 4) versus Lyz2Cre/+ Mertkfl/fl (n = 9) adult mice to assess systemic dissemination at 32hpi following IP Spn challenge. Each data point represents an individual mouse, and data are representative of experiments repeated at least twice. Data with error bars are presented as mean ± SEM. N.S., not significant. See also Figure S5-S6.
Following IP challenge of Spn in DT-treated CD169-DTR and WT adult mice, CD169-DTR adults were more protected than WT adults. CD169-DTR adults had significantly lower bacterial burden in both blood and spleen at 32 hours post infection (Fig 5E), and improved survival over WT adults (Fig 5F). Upon depletion of C3 using cobra venom factor, we found that the protective benefit conferred by depleting CD169+ macrophages was abrogated, and both WT and CD169-DTR mice were equally susceptible. This indicated that CD11b recognition of C3 by myeloid cells, such as neutrophils, was crucial for improved protection in CD169-DTR mice. To eliminate the possibility that the CD169 receptor itself was responsible for differences observed between WT and CD169-DTR adults, we infected Cd169−/− adults (lacks the CD169 molecule but retains this subset of macrophages) and compared them to WT adults. We observed that Cd169−/− mice were similarly susceptible to WT mice, and had similar circulating myeloid cell counts (Fig S5D-H).
MerTK is a receptor which facilitates macrophage efferocytosis of ‘aged’ neutrophils32, of which we observed reduced expression during neonatal life (Fig 4D, 4G & S4C). As additional validation on the role of efferocytosis in augmenting CD11b-driven immunity, we generated mice with floxed Mertk alleles expressing Cre recombinase driven by the Lyz2 promoter (Lyz2Cre/+ Mertkfl/fl) to genetically ablate MerTK from lysozyme-expressing macrophages. Despite (1) targeting only one of many receptors that facilitate efferocytosis and (2) an incomplete deficiency of MerTK from all macrophages in Lyz2Cre/+ Mertkfl/fl adult mice (Fig S6), we still observed an increase in CD11bhi ‘aged’ neutrophils in these mice over Cre-negative (Mertkfl/fl) adult mice (Fig 5G) and improved systemic control of Spn (Fig 5H). Thus, our genetic and pharmacologic approaches to impair macrophage-dependent efferocytosis demonstrate its role in modulating CD11b-driven immunity and consequently, generation of protective immunity against Spn.
Dependence on CD11b-driven immunity predicts streptococcal infection outcome in early life
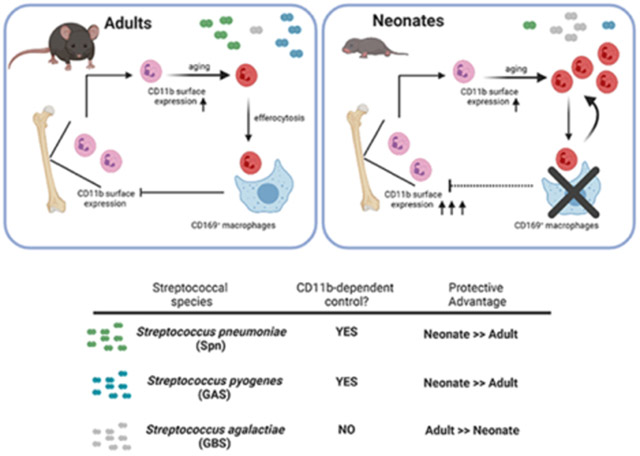
Our data suggested that neonates adopt enhanced CD11b-dependent immunity as a strategy for systemic protection against potential pathogens. We infected neonatal and adult mice with Streptococcus pyogenes (GAS), another ‘non-neonatal pathogen’. Following IP challenge with GAS, neonatal mice were more protected compared to adult mice, and protection was CD11b dependent (Fig 6A). As predicted, the protection seen in Itgam+/+ neonates over the Itgam−/− neonates was abrogated if C3 is depleted using cobra venom factor prior infection with GAS (Fig 6A), supporting the importance of CD11b-C3 interactions as shown with Spn. This suggested our model where enhanced CD11b functionality drives early life microbial immunity was relevant to a broader array of ‘non-neonatal pathogens’. In contrast to the streptococcal species examined above, Streptococcus agalactiae (GBS) is the leading cause of invasive bacterial infection in neonates. We postulated that systemic defense against GBS is independent of CD11b-mediated responses and that might in part explain increased susceptibility to GBS in neonates, as mechanisms in place to enhance CD11b-mediated responses are no longer advantageous to the host. To test this, we infected neonatal and adult mice with GBS via an IP route. In contrast to our observations with Spn and GAS, we found neonates to be significantly more susceptible to GBS compared to adults. Importantly, in adults, the more protected group over neonates, Itgam−/− adult mice were equally protected as Itgam+/+ counterparts (Fig 6B).
Figure 6: Dependence on CD11b-driven immunity predicts streptococcal infection outcome in early life.

[A] Survival of WT NNT (solid circle black line, n = 17), Itgam−/− NNT (solid circle green line, n = 10) and both groups of mice treated with 25μg of cobra venom factor (WT NNT, solid circle dotted black line, n = 12; Itgam−/− NNT, solid circle dotted green line, n = 11) and WT AD mice (open circle grey line, n = 11) following IP challenge with GAS5, a Streptococccus pyogenes strain with necrotizing fasciitis potential in humans, at 5 – 50 CFU per gram of body weight.
[B] Survival of WT NNT (solid circle black line, n = 13), WT AD (open circle grey line, n = 7) and Itgam−/− (open circle muted green line, n = 8) mice following IP challenge with GBS5, a Streptococcus agalactiae strain at 3 x 105 CFU per gram of body weight. Each data point represents an individual mouse, and data are representative of experiments repeated at least twice. N.S., not significant.
Discussion
Microbial colonization of neonates begins immediately after exit from the sterile environment of the womb. To accommodate this transition, the neonatal immune system is typically thought of as hypertolerant. Many have proposed that both strict and opportunistic pathogens exploit this feature to cause disease. However, this hypothesis fails to explain the lack of association between numerous common or leading adult pathogens and neonatal sepsis, which implies that mechanisms exist in early life to protect against such ‘non-neonatal pathogens,’ such as Spn.
In the case of Spn, opsonophagocytosis driven by complement component C3–integrin CD11b interactions on myeloid cells, such as neutrophils, plays a pivotal role in host immunity and is often the limiting factor in generating a protective response40,41. Since neonatal mice were more resistant than adult mice in our systemic Spn infection model, we hypothesized that this aspect of immune function might be augmented during early life. By profiling cells in the bone marrow and blood, we discovered that neonatal neutrophils have enhanced CD11b-dependent opsonophagocytic function. Our data shows that impaired efferocytosis in early life, as mediated by CD169+ macrophages, increased relative CD11b surface expression on neutrophils during development in BM and promoted retention of CD11bhi expressing ‘aged’ neutrophils in blood. Consequently, these drove a CD11b-dependent systemic immune advantage against Spn in neonates. We found this advantage to be lost by weaning age (~21 days of life), as juvenile mice were more susceptible to Spn than neonatal mice. This correlated with the observation where CD11b surface expression on myeloid cells from juvenile mice phenocopied those of adult mice, and CD169+ macrophages have populated tissues such as the spleen31. The importance of this augmented CD11b-dependent immunity was demonstrated in our experiments where partially impairing efferocytosis in DT-treated CD169-DTR adult mice rescued protection against Spn in a C3-dependent manner.
Our results also provide a mechanistic explanation for age-dependent biases underscoring etiology of invasive streptococcal infections. We extrapolated on our observations with Spn and speculated that augmented CD11b immunity in neonates improves protection against microbes requiring CD11b for efficient control, but not against microbes that did not. We infected mice with GAS, which is rarely associated with human neonatal sepsis, and showed that neonatal mice are more protected over adult mice in a CD11b-dependent manner, similar to Spn. However, with GBS, a leading cause of human neonatal sepsis, we found neonatal mice to be more susceptible compared to adult mice. Contrary to Spn and GAS, we showed protection against GBS systemic infection was independent of CD11b, suggesting the augmented CD11b responses in neonates was inessential during systemic GBS infection. Protection in the case of GBS could instead require direct activity of the siglec CD169, which recognizes sialic acid42; sialic acid is a surface feature of the GBS polysaccharide capsule43. The age-dependent developmental delay in CD169 expression was thus disadvantageous for immunity against GBS, as previously proposed44,45. We expanded on this model and propose that reduced CD169+ macrophages in early life is not wholly detrimental, but akin to a double-edged sword. Decreased CD169 expression would impair control of sialic acid coated bacteria, such as GBS. On the other hand, decreased efferocytosis enhances CD11b-effector ability, and improves bactericidal control against Spn and GAS, which both lack cell surface sialic acid46. It is tempting to speculate that dependence on CD11b-versus-CD169 functionality can also predict susceptibility to other non-streptococcal microbes during early life. Another prominent invasive neonatal pathogen, K1-antigenic Escherichia coli (K1EC), is also covered by a sialic acid capsule46. CD169+ macrophages have also been implicated in the pathogenesis of Listeria monocytogenes (LM)47. Reduced numbers of macrophages expressing CD169 could therefore account for impaired control of the three major neonatal invasive pathogens (GBS, K1EC and LM).
Previous studies on neonatal immunity depend heavily on human cord blood as a source of immune cells. An advance in our study is the ability to phenotype myeloid cells in neonatal mice, which required obtaining bone marrow from mice as young as three, and blood at six, days of age. Our findings in mice starkly contrast those using human cord blood neutrophils. In the latter, CD11b expression and function are shown to be reduced compared to blood neutrophils from later stages of life, and proposed as the basis for increased susceptibility to infections in neonatal life48-50. However, there are two caveats with cord blood studies. First, is their prenatal origin; human neonatal peripheral blood leukocytes are phenotypically distinct from cord blood leukocytes as early as one week into life, as demonstrated by a recent mass cytometry study2. We overcame this first limitation by direct assessment of peripheral blood neutrophils from human infants during the neonatal versus immediate post-neonatal period. Second, cord blood studies have relied primarily on purified microbial ligands or heat killed microbes51, which oversimplify the complexity of pathogens in the setting of infection. Our use of (i) bone marrow and blood directly from neonatal mice and (ii) live bacteria for in vivo infections or ex vivo assays revealed aspects of neonatal immunity that may have been overlooked in human cord blood experiments. Further, because dams in our experiments were never infected or vaccinated, we were able to investigate immune mechanisms without the confounding effects of maternal specific antibodies, which is difficult to accomplish with human studies. This allowed us to directly examine neonate-intrinsic molecular pathways, such as kinetics of myeloid development and consequences for microbial susceptibility.
We primarily focused on neutrophils, given their well-established role in immune control of gram-positive bacteria13. Neutrophils were historically thought to comprise a homogenous population due to their short lifespan and non-proliferative capacity, but there is increasing appreciation for their ability to undergo phenotypic diversification52. Few studies have investigated such diversity in the context of early life, and those that do have focused on myeloid-derived suppressor cells, or MDSCs53. We expanded on this by showing that neutrophils from another source of phenotypic diversification, ‘aged’ neutrophils, were prevalent in neonatal blood. Paralleling observations in adult mice, ‘aged’ neutrophils in neonates also possessed higher CD11b expression than ‘non-aged’ neutrophils, which further sustained the CD11b-driven protective advantage generated during neutrophil development in the bone marrow. Increased CD11b expression was also observed on Ly6C-expressing monocytes in neonatal mice, although (i) this increase was not observed on human neonatal monocytes in peripheral blood and (ii) Ccr2−/− and Ccr2+/+ neonatal mice had similar control of bacterial burden. These two observations suggested an unlikely role for Ly6C-expressing monocytes in mediating the protective effect against ‘non-neonatal’ pathogens, such as Spn. Because our model of invasive streptococcal infection spans only a few days, a direct contribution of neonatal adaptive immunity is unlikely. This is in line with existing evidence suggesting that lymphocytes are dispensable for systemic microbial immunity in early life54.
Molecular mechanisms driving increased CD11b surface expression on neonatal neutrophils as they develop in the bone marrow are likely multifaceted. Our data suggests downstream effectors actively inhibited by efferocytosis are involved in this process, as relief of their inhibition in early life would promote CD11b expression. Of note, one biological pathway inhibited by efferocytosis is the IL23–IL17–GCSF axis, which promotes granulopoeisis55. Efferocytosis of ‘aged’ neutrophils is a biological readout for sufficient neutrophil output in peripheries, and informs the hematopoietic compartment to reduce generation of neutrophils. A fundamental step in relaying this information is inhibition of GCSF production by bone marrow stromal cells, which is otherwise constitutive. Because neonates exhibit dampened efferocytosis, we speculate that differences in GCSF concentrations (and thus tonic sensing of GCSF by neutrophil progenitors) in the bone marrow microenvironment may drive differential CD11b expression. The potential effect of GCSF on priming CD11b-dependent neutrophil function could be decoupled from its effect on neutrophil numbers, as Itgam+/− neonates remained more susceptible to invasive Spn infection than Itgam+/+ neonates despite similar peripheral neutrophil counts.
Environmental factors can also influence host myeloid development and function. We and others have shown that the microbiota can functionally prime innate immunity under steady state conditions in both neonates and adults, promoting immunity against infectious insults at both mucosal and systemic tissues11,56-58. We saw no difference in CD11b expression on neutrophils from neonatal mice in our germ free versus conventional specific pathogen free facility, which excluded a role of the microbiota in priming CD11b expression and function. Other factors inherently different in early compared to later in life, such as diet, may also be at play. Neonates solely consume milk, which is high in dietary fat. Consumption of high fat diets in adults have been shown to dysregulate hematopoietic programs by promoting a myeloid over lymphoid shift in the bone marrow59,60, and such mice exhibit monocytosis and neutrophilia compared to adult mice fed regular chow61. It is possible that altered amounts of lipid metabolism byproducts in early life can prime CD11b expression and function. This correlates with the diminished CD11b surface expression in neutrophils from juvenile mice, which have begun consumption of solid foods as they wean off maternal milk.
Taken altogether, our results establish a mechanistic link between early life myeloid development, innate immune function and age-related patterns of infection susceptibility. We elucidate a previously unappreciated facet of neonatal immunity where an early life developmental defect (efferocytosis by macrophages) can prime enhanced bactericidal function (CD11b-dependent opsonophagocytosis by neutrophils) to resist invasive infection by two major pathogenic streptococci (Streptococcus pneumoniae & Streptococcus pyogenes). This is an immunological strategy to optimize the odds of survival as a newborn exits the womb into an environment filled with opportunistic microbes.
Limitations of Study
As there are no experimental models of invasive infection in humans, our study relied exclusively on murine models. Correlating mouse age with that of humans is inherently difficult without a more complete understanding of the relevant developmental and environmental factors affected by age. Many other questions remain. Multiple reports have documented that efferocytosis is impaired in the geriatric population, where defects in clearance of ‘aged’ neutrophils have also been observed62-64. The elderly are highly susceptible to invasive infection by ‘non-neonatal pathogens’, suggesting that phenotypic alterations to antimicrobial immunity that stem from impaired efferocytosis differ in old age compared to newborns. The effects of efferocytosis on infection susceptibility during old age were not addressed in our study. Finally, the stage of neutrophil development in early life where priming of CD11b surface expression occurs remains unknown.
STAR Methods
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jeffrey N. Weiser (Jeffrey.Weiser@nyulangone.org).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
This study did not generate new datasets or code.
Experimental Model and Subject Details
Ethics statement
This study was conducted according to guidelines outlined by the National Science Foundation Animal Welfare Requirements and the Public Health Service Policy on the Humane Care and Use of Laboratory Animals. The New York University Langone Health IACUC oversees the welfare, well-being, and proper care and use of all vertebrate animals.
Mice
Wild-type C57BL/6J mice obtained from Jackson Laboratories (Bar Harbor, ME), Itgam−/−, Itgam+/−, CD169-DTR, Cd169−/− mice, and Lyz2-cre Mertkfl/fl mice were bred and maintained in conventional animal facilities. For littermate breeders, Lyz2Cre/+ Mertkfl/fl mice were crossed with Mertkfl/fl mice65 to generate Cre+ and Cre− littermates. For non-littermate breeders, Lyz2Cre/Cre Mertkfl/fl mice were crossed with Mertkfl/fl mice to generate Lyz2Cre/+ Mertkfl/fl progeny, with sex- and age-matched Mertkfl/fl mice as controls. Littermates and non-littermates were used in experiments. Germ-free C57BL/6J mice were bred and maintained in flexible film isolators, and absence of microbes was confirmed via (1) aerobic culture in brain heart infusion, sabaraud and nutrient broth, and (2) qPCR for bacterial 16S and eukaryotic 18S ribosomal RNA through stool sampling on a monthly basis. Unless otherwise stated, neonatal mice used were between 6 – 9 days of life and housed with a dam throughout the duration of infection experiments. Juvenile mice were used between 3 – 4 weeks of life. Adult mice used were between 6 – 14 weeks of life. Both sexes of mice were used in experiments, except for GAS infection experiments, where only males were used66. Mice for control groups were sex- and age-matched to the experimental groups. For experiments involving CD169-DTR mice, both CD169-DTR and WT mice were administered 40ng/g body weight of diphtheria toxin (DT), with a pulse three days later at 4ng/g of diphtheria toxin followed by euthanasia or Spn infection another three days after (as described below).
Invasive streptococcal infection models
For Spn, P2406 (serotype 4, strain TIGR4; clinical isolate with streptomycin resistance) was used in infection experiments67, or P2453 (serotype 6, strain TIGR4; capsule switch mutant from parental strain P2406)68. Spn was grown statically in tryptic soy (TS) broth at 37°C to mid-log phase (optical density of 1.0 at 620nm). Quantification of Spn bacterial counts was done via serial plating on TS agar plates supplemented with 100μL catalase (30,000 U/mL, Worthington Biochemical) and 200μg/mL streptomycin, incubated overnight at 37°C and 5% CO2. For GAS, GAS5 (M-type 3, strain 950771; clinical isolate from a child with necrotizing fasciitis and sepsis) was used in infection experiments69. GAS was grown statically in Todd Hewitt broth supplemented with yeast extract (THY) at 37°C to an optical density of 1.0 at 620nm at a 1:10 subculture from an overnight 10mL culture inoculated with a single colony of GAS streaked on a TS agar plate supplemented with 5% sheep’s blood. Quantification of GAS bacterial counts was done via serial plating on TS agar plates supplemented with 5% sheep’s blood, incubated overnight at 37°C and 5% CO2. For GBS, GBS5 (serotype 3, COH-1; clinical isolate from newborn with sepsis) was used in infection experiments70. GBS strain was grown statically overnight in tryptic soy broth at 37°C. Quantification of GBS was done via serial plating on TS agar plates, incubated overnight at 37°C and 5% CO2.
For the intraperitoneal model of infection, neonatal, juvenile or adult mice were inoculated with 20uL, 40uL and 100uL of streptococci in Dulbecco’s phosphate buffered saline (PBS), respectively, at the following doses: Spn, 50-100 CFU/g body weight; GBS, 3 x 105 CFU/g body weight; GAS, 5-50 CFU/g body weight. Unless otherwise stated, dosing of Spn was normalized to body weight. For experiments involving depletions, either 150μg of neutrophil-depletion 1A8 antibody, 150μg of the isotype 2A3 control, or 25μg of cobra venom factor was administered IP 24 hours prior infection. At time points indicated following challenge, mice were euthanized via CO2 asphyxiation followed by cardiac puncture, and tissues harvested for appropriate analyses.
Method Details
Myeloid cell phenotyping & flow cytometry
For bone marrow cell phenotyping, bone marrow was collected from mice at postnatal day 3 (pooled tibia and femur from 3 - 4 mice per biological replicate) and day 6 (pooled tibia and femur from 2 −3 mice per biological replicate). Bones from mice at 9 days of age, juveniles and adults were representative of 1 mouse per biological replicate. Bone marrow was flushed out into 1x Hank’s Balanced Salt Solution (Corning). Bone marrow cell suspension was filtered using a 40μm strainer (Falcon), prior pelleting for other applications. For blood cell phenotyping, blood was obtained via cardiac puncture from mice at postnatal day 6 (pooled from 4 −7 mice per biological replicate), day 9 (pooled from 2 – 4 mice per biological replicate) and adult (1 mouse per biological replicate) mice.
Following lysis of red blood cells with ACK lysis buffer, single cell suspensions were pelleted. All incubations were performed at 4°C. Cells were re-suspended in LIVE/DEAD Fixable Aqua (ThermoFisher) at 1:1000 in PBS for 30 minutes for assessment of cell viability, followed by anti-CD16/CD32 at 1:200 in PBS + 1% bovine serum albumin (BSA) for 15 minutes for blocking of Fc receptors. Surface marker staining was performed for 30 minutes, with antibodies against anti-anti-CD45-APC/Cy7 (clone 30-F11, BD Biosciences), anti-CD11b-V450 (clone M1/70, BD Biosciences), anti-Ly6G-PerCP/Cy5.5 (clone 1A8, BD Biosciences) and (i) for bone marrow myeloid cells: anti-Ly6C-APC (clone HK1.4, Biolegend) or (ii) for blood neutrophils: anti-CXCR4-PE (clone L276F12, Biolegend) and anti-CD62L-APC (clone MEL-14, Biolegend) at 1:150 dilution in PBS + 1% BSA. Cells were then fixed in 4% paraformaldehyde in PBS for 15 minutes. All cytometry was performed on an LSRII flow cytometer (BD Biosciences) and data analysis was performed using FlowJo software (Tree Star). All median fluorescence intensity (MFI) data is normalized to values from adult mice for comparison across different ages, unless noted otherwise.
Complete blood count differentials
To enumerate white blood cells in peripheral blood, 20μL of blood was harvested through cardiac puncture of either neonatal (6 or 9 days of life), juvenile (3 – 4 weeks of life) or adult (6 – 12 weeks of life) mice. The blood was diluted in 80μL of cold PBS in an EDTA-coated microtainer tube (BD Biosystems), and samples were run on the Element HT5 Veterinary Hematology Analyzer (Heska) for data acquisition.
Neutrophil adoptive transfer
Bone marrow from either neonatal or adult mice were harvested and processed as described above. Resulting suspensions were then overlayed onto a Histopaque-1119 and Histopaque-1077 gradient and cells were spun at 2000rpm for 30 minutes at room temperature, with deceleration breaks turned off. Neutrophils were isolated from the interface of Histopaque-1119 and Histopaque-1077, washed in cold dPBS and resuspended to 5 x 107 cells/mL. Assessment of the purity in the granulocyte fraction from adult and neonatal mice, respectively, using flow cytometry yielded > 85%. At 4hpi, Spn-infected Itgam−/− recipient neonatal mice (infection as described above) were inoculated with 20uL of the suspension (~106 neutrophils) via the intraperitoneal route. Recipient mice were euthanized at 20 hpi to assess bacterial burden in the blood and spleen.
RNA isolation and quantitative RT-PCR
For spleen, lung and liver, tissues were harvested from neonatal and adult mice, then snap frozen in dry ice and stored at −80°C. Frozen tissues (~100mg to 200mg) were homogenized in 1mL of Trizol, and standard phenol-chloroform extraction was performed. For bone marrow and isolated neutrophils, cells were lysed in 600μL RLT lysis buffer (Qiagen) supplemented with 1% β-mercaptoethanol, and RNA was extracted using an isolation kit (RNeasy, Qiagen) as per manufacturer protocols. cDNA was generated using High Capacity cDNA Reverse Transcriptase Kit (Applied Biosystems, Thermo Fisher Scientific) as per manufacturer protocols. Reaction samples contained ~10ng cDNA and 0.5μM primers in Power SYBR Green PCR Master Mix (Applied Biosystems, Thermo Fisher) and tested in duplicates. qRT-PCR reactions were run in a 384 well plate (Bio-Rad) using CFX384™ Real-Time System (Bio-Rad). All genes were normalized to expression of Gapdh and fold changes in expression of relevant genes were quantified using the ΔΔCt method. Primer sequences are detailed in Table S1.
Tissue preparation for confocal microscopy
Tissues were processed as previously described47,71. Briefly, tissues were flash frozen in OCT media at −80°C. 20-um frozen tissue sections were sectioned using Leica CM3050S cryostat. Tissue were fixed with ice cold acetone for 15 minutes in −20°C followed by washing with 1x PBS. FcR blocked with anti-CD16/32 Fc block antibody (Clone 93, Biolegend) diluted in PBS containing 2% goat serum, and 2% fetal bovine serum (FBS) for 1 hour at room temperature. Sections were stained with the antibodies anti-CD169-ef660 (Clone Ser-4, Invitrogen), anti-MerTK-PE (clone DS5MMer, Biolegend), anti-F4/80-BV605 (clone BM8, Biolegend), anti-Ly6G-488 (clone 1A8, Biolegend), and anti-CD3-BV421 (clone 145-2C11, Biolegend) that were diluted in PBS containing 2% goat serum, 2% FBS, and 0.05% Fc block for 1 hour at room temperature. Images were acquired using a Zeiss LSM 880 confocal microscope (Carl Zeiss) with the Zen Black software. The imaging data were processed and analyzed using Imaris software version 9.0.1 (Bitplane; Oxford Instruments).
Ex vivo Spn opsonophagocytic and complement deposition assay
For the opsonophagocytosis assay, bone marrow cells were counted by trypan blue staining and adjusted to a density of 6 x 106 cells/mL in PBS. GFP-expressing Spn used in experiments, P2531, is a serotype 23, clinical isolate transformed with chromosomal DNA from GFP-expressing serotype 2 strain D39 (P2521 in our collection)72. Mid-log phase GFP-Spn was adjusted to a density of 1.5 x 108 cells/mL in PBS. 20μL of Spn was incubated with 20μL fresh mouse sera (as complement source) for 45 minutes at 180 rpm at 37°C. Following that, 20μL of bone marrow cells were added to the opsonized-bacteria, and incubated for 20 minutes at 180 rpm at 37°C, then immediately incubated at 4°C to stop the reaction. Cells were stained as described above, and neutrophil uptake is assessed as % of neutrophils with GFP-positive signal. Heat-inactivated sera (to degrade complement) was generated by incubating fresh adult mouse sera for 30 minutes at 56°C.
For the complement deposition assay, 2.5 x 106 Spn strain P112173 or P2406 in 25μL was incubated with 25μL mouse sera for 30 minutes at 37°C, followed by addition of 50μL of PBS in 20mM EDTA and immediate incubation at 4°C to stop the reaction. Spn was pelleted and then stained with 50μL of diluted FITC anti-mouse C3 antibody (MP Biomedicals) at 1:200 dilution for 30 minutes at 4°C. Spn was then fixed in 100μL of 4% paraformaldehyde in PBS for 20 minutes at room temperature, pelleted and resuspended for flow cytometry analysis.
Mass cytometry analysis of human peripheral blood neutrophils
Blood samples were drawn from newborns at the Karolinska University Hospital in Stockholm, Sweden. Samples were mixed with a stabilizer74 within 3 hours and cryopreserved. Mass cytometry experimental methods are detailed in Henrick et al. manuscript75.
All FCS-files were unrandomized using CyTOF software (version 6.0.626) and transferred with no further processing. Grid, an automated cell classification supervised algorithm, was used to first define reference subpopulations manually and then train a learning algorithm (XGBoost) to recognize the same subsets of cells in novel data76. Outputs from all mass cytometry experiments were merged, and batch differences removed using limma77 Immune cell data was reduced to include infant samples collected between 0 to 5 days and 31 to 81 days after birth, resulting in n = 75 and n = 31 samples respectively. The mean of each bin window was calculated and colored accordingly, and the package ggpubr was used to perform a Welsh T-test to compare both groups.
Quantification and Statistical Analysis
Differences in bacterial counts from infected mice were assessed using a non-parametric, Mann-Whitney test. Differences between two groups in median fluorescence intensity, percentages and absolute cell counts were analyzed using either an unpaired or paired Student’s t-test. Differences between three or more groups was analyzed using a one-way ANOVA test with Tukey’s multiple comparisons. Survival time course was compared between groups using the Kaplan-Meier’s log-rank test. All statistical analyses were performed on GraphPad Prism 8. All experiments were repeated at least twice. A p-value of < 0.05 is deemed statistically significant.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| APC/Cy7 Rat anti-Mouse CD45 (clone 30-F11) | BD Biosciences | Cat #: 557659; RRID: AB_396774 |
| V450 Rat anti-Mouse CD11b (clone M1/70) | BD Biosciences | Cat #: 560455; RRID: AB_1645266 |
| APC Rat anti-Mouse Ly6C (clone HK1.4) | Biolegend | Cat #: 128016; RRID: AB_1732076 |
| PerCP/Cy5.5 Rat anti-Mouse Ly6G (clone 1A8) | BD Biosciences | Cat #: 560602; RRID: AB_1727563 |
| APC Rat anti-Mouse CD62L (clone MEL-14) | Biolegend | Cat #: 104411; RRID: AB_313098 |
| PE Rat anti-Mouse CD184/CXCR4 (clone L276F12) | Biolegend | Cat #: 146505; RRID: AB_2562782 |
| Rat anti-Mouse CD16/32 (clone 93) | Biolegend | Cat #: 101302; RRID: AB_312801 |
| ef660 Rat anti-Mouse CD169 (clone SER-4) | Invitrogen | Cat #: 50-5755-82; AB_2574241 |
| PE Rat anti-Mouse MERTK (clone DS5MMER) | ThermoFisher Scientific | Cat #: 12-5751-82; RRID: AB_2572623 |
| BV605 Rat anti-Mouse F4/80 (clone BM8) | Biolegend | Cat #: 123133; RRID: AB_2562305 |
| AF488 Rat anti-Mouse Ly6G (clone 1A8) | Biolegend | Cat #: 127625; RRID: AB_2561340 |
| BV421 Hamster anti-Mouse CD3 (clone 145-2C11) | Biolegend | Cat #: 100335; RRID: AB_10898314 |
| PE Rat anti-mouse F4/80 (clone BM8) | Biolegend | Cat #: 123110 RRID: AB_893486 |
| PerCP-eFluor 710 Rat anti-Mouse MERTK (clone DS5MMER) | Invitrogen | Cat #:46-5751-82; RRID: AB_2688094 |
| PE Rat anti-Mouse CD169 (clone SER-4) | ThermoFisher Scientific | Cat #: 12-5755-82; RRID: AB_2572625 |
| FITC Goat anti-Mouse Complement C3 | MP Biomedicals, Inc | Cat #: 0855500; RRID: AB_2334931 |
| Anti-Ly6G (clone 1A8) | BioXCell | Cat #: BE0075-1; RRID: AB_1107721 |
| IgG2a control (clone 2A3) | BioXCell | Cat #: BE0089; RRID: AB_1107769 |
| Bacterial and virus strains | ||
| Streptococcus pneumoniae Serotype 4 (TIGR4) Strain P2406 | (Zafar et al., 2016) | N/A |
| Streptococcus pneumoniae GFP+ Serotype 23F Strain P2531 | This paper | N/A |
| Streptococcus pneumoniae Serotype 23F Strain P1121 | (McCool et al., 2002) | N/A |
| Streptococcus pneumoniae TIGR4 expressing Serotype 6A Capsule P2453 | (Zafar et al., 2017) | N/A |
| Streptococcus pyogenes GAS5 (M-type 3, strain 950771) | (Ashbaugh et al., 1998) | N/A |
| Streptococcus agalactiae GBS5 (serotype 3, WT COH1) | (Wilson and Weaver, 1985) | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Diphteria toxin | Sigma Aldrich | Cat #: D0564 |
| Catalase | Worthington Biochemical | Cat #: LS001896 |
| Cobra Venom Factor, Naja naja kaouthia | Millipore Sigma | Cat #: 233552 |
| Power SYBR Green PCR Master Mix | Applied Biosystems, ThermoFisher Scientific | Cat #: 4309155 |
| ACK Lysing Buffer | Gibco | Cat #: A10492-01 |
| LIVE/DEAD Fixable Aqua | ThermoFisher Scientific | Cat #: L34966 |
| BD Microtainer Tubes w/ K2E | BD Biosystems | Cat #: 365974 |
| Histopaque 1077 | Sigma | Cat #: 10771 |
| Histopaque 1119 | Sigma | Cat #: 11191 |
| QIAshredder | Qiagen | Cat #: 79656 |
| Critical commercial assays | ||
| RNeasy Mini Kit | Qiagen | Cat #: 74106 |
| High Capacity cDNA Reverse Transcriptase Kit | Applied Biosystems, ThermoFisher Scientific | Cat #: 4368813 |
| MiniElute PCR Purification Kit | Qiagen | Cat #: 28006 |
| DNA-free Kit, DNAse Treatment and Removal Reagents | Ambion, Life Technologies | Cat #: AM1906 |
| Experimental models: organisms/strains | ||
| Mouse: C57BL/6J | Jackson Laboratory | Stock #: 000664 |
| Mouse: Itgam−/− : B6.129S4-Itgamtm1Myd/J | Jackson Laboratory | Stock #: 003991 |
| Mouse: Itgam+/− | This paper | N/A |
| Mouse: CD169-DTR: B6;129-Siglec1<tm1(HBEGF)Mtka> | (Miyake et al., 2007) | RikenBRC: 04395 |
| Mouse: Cd169−/−: CD169-DTR KO | (Miyake et al., 2007) | RikenBRC: 04395 |
| Mouse: Mertkfl/fl | (Fourgeaud et al., 2016) | N/A |
| Mouse: Lyz2-Cre: B6.129P2-Lyz2tm1(cre)Ifo/J | Jackson Laboratory | Stock #: 004781 |
| Oligonucleotides | ||
| See Table S1 for qRT-PCR primer sequences | See Table S1 | N/A |
| Software and algorithms | ||
| GraphPad Prism | GraphPad | http://www.graphpad.com |
| FlowJo | FlowJo, LLC | http://www.flowjo.com |
| Imaris Software Version 9.0.1. | Bitplane; Oxford Instruments | http://www.bitplane.com |
Highlights.
Neonates resist Streptococcus pneumoniae invasive infection
Neonatal neutrophils have enhanced opsonin C3–integrin CD11b dependent phagocytosis
Dampened efferocytosis increases CD11bhi ‘aged’ neutrophils during early life
Dependence on CD11b opsonophagocytosis predicts age-related patterns of infection
Acknowledgements
We thank the laboratories of Drs. Iannis Aifantis and Ana Rodriguez for generously sharing their equipment, Dr. Adam Ratner for the COH1 Streptococcus agalactiae strain and assistance with GBS infection models, Dr. Victor Torres for a starter Itgam−/− mouse breeder pair, and Dr. Kirsten Kuipers for generating the P2531 strain. We acknowledge the NYULH Flow Cytometry & Cell Sorting Core and the NYULH Microscopy Laboratory (which are partially funded by NYU Cancer Center Support Grant NCI P30CA016087) for technical assistance and equipment support. We thank Dr. Christopher Hergott and Erica Ma for providing feedback on the manuscript. We thank Drs. Ken Cadwell, Shruti Naik, Christopher Park and members of the Weiser Laboratory for productive discussion. This study was supported by funding through the National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH) grants awarded to J.N.W. (R01 AI150893, R37 AI038446, R21 AI150867) and K.M.K. (R01 AI143861); the Knut and Alice Wallenberg Foundation (WAF2019.0181), ERC (StG15-677559) and Swedish Research council (2019-01495) awarded to P.B.; K. L. L-T. was supported by NIH grants F32AI143043 and T32AI007180-35. The graphical abstract was created with Biorender.com.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- 1.Klein JO, Baker CJ, Remington JS, and Wilson CB (2005). Infectious Diseases of the Fetus and the Newborn Infant (6th Edition) (Elsevier Saunders; ). https://doi-org.ezproxy.med.nyu.edu/10.1016/B0-72-160537-0/50003-7. [Google Scholar]
- 2.Olin A, Henckel E, Chen Y, Lakshmikanth T, Pou C, Mikes J, Gustafsson A, Bernhardsson AK, Zhang C, Bohlin K, and Brodin P (2018). Stereotypic Immune System Development in Newborn Children. Cell 174, 1277–1292 e1214. 10.1016/j.cell.2018.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaplan M, Rudensky B, and Beck A (1993). Perinatal infections with Streptococcus pneumoniae. Am J Perinatol 10, 1–4. 10.1055/s-2007-994687. [DOI] [PubMed] [Google Scholar]
- 4.Dawson KG, Emerson JC, and Burns JL (1999). Fifteen years of experience with bacterial meningitis. Pediatr Infect Dis J 18, 816–822. 10.1097/00006454-199909000-00014. [DOI] [PubMed] [Google Scholar]
- 5.Hoffman JA, Mason EO, Schutze GE, Tan TQ, Barson WJ, Givner LB, Wald ER, Bradley JS, Yogev R, and Kaplan SL (2003). Streptococcus pneumoniae infections in the neonate. Pediatrics 112, 1095–1102. 10.1542/peds.112.5.1095. [DOI] [PubMed] [Google Scholar]
- 6.Spaulding AB, Watson D, Dreyfus J, Heaton P, Grapentine S, Bendel-Stenzel E,and Kharbanda AB (2019). Epidemiology of Bloodstream Infections in Hospitalized Children in the United States, 2009-2016. Clin Infect Dis 69, 995–1002. 10.1093/cid/ciy1030. [DOI] [PubMed] [Google Scholar]
- 7.Germont Z, Bidet P, Plainvert C, Bonacorsi S, Poyart C, Biran V, Frerot A, Faye A, and Basmaci R (2020). Invasive Streptococcus pyogenes Infections in <3-Month-Old Infants in France: Clinical and Laboratory Features. Front Pediatr 8, 204. 10.3389/fped.2020.00204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bogaert D, De Groot R, and Hermans PW (2004). Streptococcus pneumoniae colonisation: the key to pneumococcal disease. Lancet Infect Dis 4, 144–154. 10.1016/S1473-3099(04)00938-7. [DOI] [PubMed] [Google Scholar]
- 9.Aniansson G, Alm B, Andersson B, Larsson P, Nylen O, Peterson H, Rigner P, Svanborg M, and Svanborg C (1992). Nasopharyngeal colonization during the first year of life. J Infect Dis 165 Suppl 1, S38–42. 10.1093/infdis/165-supplement_1-s38. [DOI] [PubMed] [Google Scholar]
- 10.Chaguza C, Senghore M, Bojang E, Gladstone RA, Lo SW, Tientcheu PE, Bancroft RE, Worwui A, Foster-Nyarko E, Ceesay F, et al. (2020). Within-host microevolution of Streptococcus pneumoniae is rapid and adaptive during natural colonisation. Nat Commun 11, 3442. 10.1038/s41467-020-17327-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clarke TB, Davis KM, Lysenko ES, Zhou AY, Yu Y, and Weiser JN (2010). Recognition of peptidoglycan from the microbiota by Nod1 enhances systemic innate immunity. Nat Med 16, 228–231. 10.1038/nm.2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kono M, Zafar MA, Zuniga M, Roche AM, Hamaguchi S, and Weiser JN (2016). Single Cell Bottlenecks in the Pathogenesis of Streptococcus pneumoniae. PLoS Pathog 12, e1005887. 10.1371/journal.ppat.1005887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Segal AW (2005). How neutrophils kill microbes. Annu Rev Immunol 23, 197–223. 10.1146/annurev.immunol.23.021704.115653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lewis ML, and Surewaard BGJ (2018). Neutrophil evasion strategies by Streptococcus pneumoniae and Staphylococcus aureus. Cell Tissue Res 371, 489–503. 10.1007/s00441-017-2737-2. [DOI] [PubMed] [Google Scholar]
- 15.Deniset JF, Surewaard BG, Lee WY, and Kubes P (2017). Splenic Ly6G(high) mature and Ly6G(int) immature neutrophils contribute to eradication of S. pneumoniae. J Exp Med 214, 1333–1350. 10.1084/jem.20161621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Paudel S, Baral P, Ghimire L, Bergeron S, Jin L, DeCorte JA, Le JT, Cai S, and Jeyaseelan S (2019). CXCL1 regulates neutrophil homeostasis in pneumonia-derived sepsis caused by Streptococcus pneumoniae serotype 3. Blood 133, 1335–1345. 10.1182/blood-2018-10-878082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsou CL., Peters W, Si Y, Slaymaker S, Aslanian AM, Weisberg SP, Mack M, and Charo IF (2007). Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J Clin Invest 117, 902–909. 10.1172/JCI29919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arnaout MA (1990). Structure and function of the leukocyte adhesion molecules CD11/CD18. Blood 75, 1037–1050. [PubMed] [Google Scholar]
- 19.Yuste J, Sen A, Truedsson L, Jonsson G, Tay LS, Hyams C, Baxendale HE, Goldblatt F, Botto M, and Brown JS (2008). Impaired opsonization with C3b and phagocytosis of Streptococcus pneumoniae in sera from subjects with defects in the classical complement pathway. Infect Immun 76, 3761–3770. 10.1128/IAI.00291-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gordon DL, Johnson GM, and Hostetter MK (1986). Ligand-receptor interactions in the phagocytosis of virulent Streptococcus pneumoniae by polymorphonuclear leukocytes. J Infect Dis 154, 619–626. 10.1093/infdis/154.4.619. [DOI] [PubMed] [Google Scholar]
- 21.Hyams C, Camberlein E, Cohen JM, Bax K, and Brown JS (2010). The Streptococcus pneumoniae capsule inhibits complement activity and neutrophil phagocytosis by multiple mechanisms. Infect Immun 78, 704–715. 10.1128/IAI.00881-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hostetter MK (1986). Serotypic variations among virulent pneumococci in deposition and degradation of covalently bound C3b: implications for phagocytosis and antibody production. J Infect Dis 153, 682–693. 10.1093/infdis/153.4.682. [DOI] [PubMed] [Google Scholar]
- 23.Abeyta M, Hardy GG, and Yother J (2003). Genetic alteration of capsule type but not PspA type affects accessibility of surface-bound complement and surface antigens of Streptococcus pneumoniae. Infect Immun 71, 218–225. 10.1128/IAI.71.1.218-225.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hyams C, Yuste J, Bax K, Camberlein E, Weiser JN, and Brown JS (2010). Streptococcus pneumoniae resistance to complement-mediated immunity is dependent on the capsular serotype. Infect Immun 78, 716–725. 10.1128/IAI.01056-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang D, Chen G, Manwani D, Mortha A, Xu C, Faith JJ, Burk RD, Kunisaki Y, Jang JE, Scheiermann C, et al. (2015). Neutrophil ageing is regulated by the microbiome. Nature 525, 528–532. 10.1038/nature15367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Uhl B, Vadlau Y, Zuchtriegel G, Nekolla K, Sharaf K, Gaertner F, Massberg S, Krombach F, and Reichel CA (2016). Aged neutrophils contribute to the first line of defense in the acute inflammatory response. Blood 128, 2327–2337. 10.1182/blood-2016-05-718999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Casanova-Acebes M, Pitaval C, Weiss LA, Nombela-Arrieta C, Chevre R, N. AG, Kunisaki Y, Zhang D, van Rooijen N, Silberstein LE, et al. (2013). Rhythmic modulation of the hematopoietic niche through neutrophil clearance. Cell 153, 1025–1035. 10.1016/j.cell.2013.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ioannou M, Hoving D, Aramburu IV, Temkin MI, De Vasconcelos NM, Tsourouktsoglou TD, Wang Q, Boeing S, Goldstone R, Vernardis S, et al. (2022).Microbe capture by splenic macrophages triggers sepsis via T cell-death-dependent neutrophil lifespan shortening. Nat Commun 13, 4658. 10.1038/s41467-022-32320-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adrover JM, Del Fresno C, Crainiciuc G, Cuartero MI, Casanova-Acebes M, Weiss LA, Huerga-Encabo H, Silvestre-Roig C, Rossaint J, Cossio I, et al. (2019). A Neutrophil Timer Coordinates Immune Defense and Vascular Protection. Immunity 51, 966–967. 10.1016/j.immuni.2019.11.001. [DOI] [PubMed] [Google Scholar]
- 30.Miyake Y, Asano K, Kaise H, Uemura M, Nakayama M, and Tanaka M (2007). Critical role of macrophages in the marginal zone in the suppression of immune responses to apoptotic cell-associated antigens. J Clin Invest 117, 2268–2278. 10.1172/JCI31990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.A-Gonzalez N, Guillen JA, Gallardo G, Diaz M, de la Rosa JV, Hernandez IH, Casanova-Acebes M, Lopez F, Tabraue C, Beceiro S, et al. (2013). The nuclear receptor LXRalpha controls the functional specialization of splenic macrophages. Nat Immunol 14, 831–839. 10.1038/ni.2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hong C, Kidani Y, N AG, Phung T, Ito A, Rong X, Ericson K, Mikkola H, Beaven SW, Miller LS, et al. (2012). Coordinate regulation of neutrophil homeostasis by liver X receptors in mice. J Clin Invest 122, 337–347. 10.1172/JCI58393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim JH, Podstawka J, Lou Y, Li L, Lee EKS, Divangahi M, Petri B, Jirik FR, Kelly MM, and Yipp BG (2018). Aged polymorphonuclear leukocytes cause fibrotic interstitial lung disease in the absence of regulation by B cells. Nat Immunol 19, 192–201. 10.1038/s41590-017-0030-x. [DOI] [PubMed] [Google Scholar]
- 34.Crocker PR, and Gordon S (1989). Mouse macrophage hemagglutinin (sheep erythrocyte receptor) with specificity for sialylated glycoconjugates characterized by a monoclonal antibody. J Exp Med 169, 1333–1346. 10.1084/jem.169.4.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.A-Gonzalez N, Bensinger SJ, Hong C, Beceiro S, Bradley MN, Zelcer N, Deniz J, Ramirez C, Diaz M, Gallardo G, et al. (2009). Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity 31, 245–258. 10.1016/j.immuni.2009.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scott RS, McMahon EJ, Pop SM, Reap EA, Caricchio R, Cohen PL, Earp HS, and Matsushima GK (2001). Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature 411, 207–211. 10.1038/35075603. [DOI] [PubMed] [Google Scholar]
- 37.Kiss RS, Elliott MR, Ma Z, Marcel YL, and Ravichandran KS (2006). Apoptotic cells induce a phosphatidylserine-dependent homeostatic response from phagocytes. Curr Biol 16, 2252–2258. 10.1016/j.cub.2006.09.043. [DOI] [PubMed] [Google Scholar]
- 38.Fond AM, Lee CS, Schulman IG, Kiss RS, and Ravichandran KS (2015). Apoptotic cells trigger a membrane-initiated pathway to increase ABCA1. J Clin Invest 125, 2748–2758. 10.1172/JCI80300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ishimoto Y, Ohashi K, Mizuno K, and Nakano T (2000). Promotion of the uptake of PS liposomes and apoptotic cells by a product of growth arrest-specific gene, gas6. J Biochem 127, 411–417. 10.1093/oxfordjournals.jbchem.a022622. [DOI] [PubMed] [Google Scholar]
- 40.Lysenko ES, Clarke TB, Shchepetov M, Ratner AJ, Roper DI, Dowson CG, and Weiser JN (2007). Nod1 signaling overcomes resistance of S. pneumoniae to opsonophagocytic killing. PLoS Pathog 3, e118. 10.1371/journal.ppat.0030118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Andre GO, Converso TR, Politano WR, Ferraz LF, Ribeiro ML, Leite LC, and Darrieux M (2017). Role of Streptococcus pneumoniae Proteins in Evasion of Complement-Mediated Immunity. Front Microbiol 8, 224. 10.3389/fmicb.2017.00224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Crocker PR, Kelm S, Dubois C, Martin B, McWilliam AS, Shotton DM, Paulson JC, and Gordon S (1991). Purification and properties of sialoadhesin, a sialic acid-binding receptor of murine tissue macrophages. EMBO J 10, 1661–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wessels MR, Rubens CE, Benedi VJ, and Kasper DL (1989). Definition of a bacterial virulence factor: sialylation of the group B streptococcal capsule. Proc Natl Acad Sci U S A 86, 8983–8987. 10.1073/pnas.86.22.8983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chang YC, Olson J, Louie A, Crocker PR, Varki A, and Nizet V (2014). Role of macrophage sialoadhesin in host defense against the sialylated pathogen group B Streptococcus. J Mol Med (Berl) 92, 951–959. 10.1007/s00109-014-1157-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lund SJ, Patras KA, Kimmey JM, Yamamura A, Butcher LD, Del Rosario PGB, Hernandez GE, McCoy AM, Lakhdari O, Nizet V, and Prince LS (2020). Developmental Immaturity of Siglec Receptor Expression on Neonatal Alveolar Macrophages Predisposes to Severe Group B Streptococcal Infection. iScience 23, 101207. 10.1016/j.isci.2020.101207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vimr E, and Lichtensteiger C (2002). To sialylate, or not to sialylate: that is the question. Trends Microbiol 10, 254–257. 10.1016/s0966-842x(02)02361-2. [DOI] [PubMed] [Google Scholar]
- 47.Perez OA, Yeung ST, Vera-Licona P, Romagnoli PA, Samji T, Ural BB, Maher L, Tanaka M, and Khanna KM (2017). CD169(+) macrophages orchestrate innate immune responses by regulating bacterial localization in the spleen. Sci Immunol 2. 10.1126/sciimmunol.aah5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abughali N, Berger M, and Tosi MF (1994). Deficient total cell content of CR3 (CD11b) in neonatal neutrophils. Blood 83, 1086–1092. [PubMed] [Google Scholar]
- 49.McEvoy LT, Zakem-Cloud H, and Tosi MF (1996). Total cell content of CR3 (CD11b/CD18) and LFA-1 (CD11a/CD18) in neonatal neutrophils: relationship to gestational age. Blood 87, 3929–3933. [PubMed] [Google Scholar]
- 50.Reddy RK, Xia Y, Hanikyrova M, and Ross GD (1998). A mixed population of immature and mature leucocytes in umbilical cord blood results in a reduced expression and function of CR3 (CD11b/CD18). Clin Exp Immunol 114, 462–467. 10.1046/j.1365-2249.1998.00743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang X, Zhivaki D, and Lo-Man R (2017). Unique aspects of the perinatal immune system. Nat Rev Immunol 17, 495–507. 10.1038/nri.2017.54. [DOI] [PubMed] [Google Scholar]
- 52.Silvestre-Roig C, Hidalgo A, and Soehnlein O (2016). Neutrophil heterogeneity: implications for homeostasis and pathogenesis. Blood 127, 2173–2181. 10.1182/blood-2016-01-688887. [DOI] [PubMed] [Google Scholar]
- 53.He YM, Li X, Perego M, Nefedova Y, Kossenkov AV, Jensen EA, Kagan V, Liu YF, Fu SY, Ye QJ, et al. (2018). Transitory presence of myeloid-derived suppressor cells in neonates is critical for control of inflammation. Nat Med 24, 224–231. 10.1038/nm.4467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wynn JL, Scumpia PO, Winfield RD, Delano MJ, Kelly-Scumpia K, Barker T, Ungaro R, Levy O, and Moldawer LL (2008). Defective innate immunity predisposes murine neonates to poor sepsis outcome but is reversed by TLR agonists. Blood 112, 1750–1758. 10.1182/blood-2008-01-130500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stark MA, Huo Y, Burcin TL, Morris MA, Olson TS, and Ley K (2005). Phagocytosis of apoptotic neutrophils regulates granulopoiesis via IL-23 and IL-17. Immunity 22, 285–294. 10.1016/j.immuni.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 56.Deshmukh HS, Liu Y, Menkiti OR, Mei J, Dai N, O'Leary CE, Oliver PM, Kolls JK, Weiser JN, and Worthen GS (2014). The microbiota regulates neutrophil homeostasis and host resistance to Escherichia coli K1 sepsis in neonatal mice. Nat Med 20, 524–530. 10.1038/nm.3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Khosravi A, Yanez A, Price JG, Chow A, Merad M, Goodridge HS, and Mazmanian SK (2014). Gut microbiota promote hematopoiesis to control bacterial infection. Cell Host Microbe 15, 374–381. 10.1016/j.chom.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hergott CB, Roche AM, Tamashiro E, Clarke TB, Bailey AG, Laughlin A, Bushman FD, and Weiser JN (2016). Peptidoglycan from the gut microbiota governs the lifespan of circulating phagocytes at homeostasis. Blood 127, 2460–2471. 10.1182/blood-2015-10-675173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Luo Y, Chen GL, Hannemann N, Ipseiz N, Kronke G, Bauerle T, Munos L, Wirtz S, Schett G, and Bozec A (2015). Microbiota from Obese Mice Regulate Hematopoietic Stem Cell Differentiation by Altering the Bone Niche. Cell Metab 22, 886–894. 10.1016/j.cmet.2015.08.020. [DOI] [PubMed] [Google Scholar]
- 60.Huang JY, Zhou QL, Huang CH, Song Y, Sharma AG, Liao Z, Zhu K, Massidda MW, Jamieson RR, Zhang JY, et al. (2017). Neutrophil Elastase Regulates Emergency Myelopoiesis Preceding Systemic Inflammation in Diet-induced Obesity. J Biol Chem 292, 4770–4776. 10.1074/jbc.C116.758748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nagareddy PR, Kraakman M, Masters SL, Stirzaker RA, Gorman DJ, Grant RW, Dragoljevic D, Hong ES, Abdel-Latif A, Smyth SS, et al. (2014). Adipose tissue macrophages promote myelopoiesis and monocytosis in obesity. Cell Metab 19, 821–835. 10.1016/j.cmet.2014.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Frisch BJ, Hoffman CM, Latchney SE, LaMere MW, Myers J, Ashton J, Li AJ, Saunders J 2nd, Palis J, Perkins AS, et al. (2019). Aged marrow macrophages expand platelet-biased hematopoietic stem cells via Interleukin1B. JCI Insight 5. 10.1172/jci.insight.124213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.De Maeyer RPH, van de Merwe RC, Louie R, Bracken OV, Devine OP, Goldstein DR, Uddin M, Akbar AN, and Gilroy DW (2020). Blocking elevated p38 MAPK restores efferocytosis and inflammatory resolution in the elderly. Nat Immunol 21, 615–625. 10.1038/s41590-020-0646-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lu RJ, Taylor S, Contrepois K, Kim M, Bravo JI, Ellenberger M, Sampathkumar NK, and Benayoun BA (2021). Multi-omic profiling of primary mouse neutrophils predicts a pattern of sex and age-related functional regulation. Nat Aging 1, 715–733. 10.1038/s43587-021-00086-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fourgeaud L, Traves PG, Tufail Y, Leal-Bailey H, Lew ED, Burrola PG, Callaway P, Zagorska A, Rothlin CV, Nimmerjahn A, and Lemke G (2016). TAM receptors regulate multiple features of microglial physiology. Nature 532, 240–244. 10.1038/nature17630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chella Krishnan K, Mukundan S, Alagarsamy J, Laturnus D, and Kotb M (2016). Host Genetic Variations and Sex Differences Potentiate Predisposition, Severity, and Outcomes of Group A Streptococcus-Mediated Necrotizing Soft Tissue Infections. Infect Immun 84, 416–424. 10.1128/IAI.01191-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zafar MA, Kono M, Wang Y, Zangari T, and Weiser JN (2016). Infant Mouse Model for the Study of Shedding and Transmission during Streptococcus pneumoniae Monoinfection. Infect Immun 84, 2714–2722. 10.1128/IAI.00416-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Abruzzo AR, Aggarwal SD, Sharp ME, Bee GCW, and Weiser JN (2022). Serotype-Dependent Effects on the Dynamics of Pneumococcal Colonization and Implications for Transmission. mBio, e0015822. 10.1128/mbio.00158-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ashbaugh CD, Warren HB, Carey VJ, and Wessels MR (1998). Molecular analysis of the role of the group A streptococcal cysteine protease, hyaluronic acid capsule, and M protein in a murine model of human invasive soft-tissue infection. J Clin Invest 102, 550560. 10.1172/JCI3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wilson CB, and Weaver WM (1985). Comparative susceptibility of group B streptococci and Staphylococcus aureus to killing by oxygen metabolites. J Infect Dis 152, 323–329. 10.1093/infdis/152.2.323. [DOI] [PubMed] [Google Scholar]
- 71.Ural BB, Yeung ST, Damani-Yokota P, Devlin JC, de Vries M, Vera-Licona P, Samji T, Sawai CM, Jang G, Perez OA, et al. (2020). Identification of a nerve-associated, lung-resident interstitial macrophage subset with distinct localization and immunoregulatory properties. Sci Immunol 5. 10.1126/sciimmunol.aax8756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kjos M, Aprianto R, Fernandes VE, Andrew PW, van Strijp JA, Nijland R, and Veening JW (2015). Bright fluorescent Streptococcus pneumoniae for live-cell imaging of host-pathogen interactions. J Bacteriol 197, 807–818. 10.1128/JB.02221-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McCool TL, Cate TR, Moy G, and Weiser JN (2002). The immune response to pneumococcal proteins during experimental human carriage. J Exp Med 195, 359–365. 10.1084/jem.20011576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Brodin P, Duffy D, and Quintana-Murci L (2019). A Call for Blood-In Human Immunology. Immunity 50, 1335–1336. 10.1016/j.immuni.2019.05.012. [DOI] [PubMed] [Google Scholar]
- 75.Henrick BM, Rodriguez L, Lakshmikanth T, Pou C, Henckel E, Arzoomand A, Olin A, Wang J, Mikes J, Tan Z, et al. (2021). Bifidobacteria-mediated immune system imprinting early in life. Cell 184, 3884–3898 e3811. 10.1016/j.cell.2021.05.030. [DOI] [PubMed] [Google Scholar]
- 76.Chen Y, Lakshmikanth T, Mikes J, and Brodin P (2020). Single-cell classification using learned cell phenotypes. bioRxiv. [Google Scholar]
- 77.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47. 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate new datasets or code.