

Abstract
Objective:
Congenital lymphatic anomalies (LAs) arise due to defects in lymphatic development and often present in utero as pleural effusion, chylothorax, nuchal and soft tissue edema, ascites, or hydrops. Many LAs are caused by single nucleotide variants, which are not detected on routine prenatal testing.
Methods:
Demographic data were compared between two subcohorts, those with clinically significant fetal edema (CSFE) and isolated fetal edema. A targeted variant analysis of LA genes was performed using American College of Medical Genetics criteria on whole exome sequencing (WES) data generated for 71 fetal edema cases who remained undiagnosed after standard workup.
Results:
CSFE cases had poor outcomes, including preterm delivery, demise, and maternal preeclampsia. Pathogenic and likely pathogenic variants were identified in 7% (5/71) of cases, including variants in RASopathy genes, RASA1, SOS1, PTPN11, and a novel PIEZO1 variant. Variants of uncertain significance (VOUS) were identified in 45% (32/71) of cases. In CSFEs, VOUS were found in CELSR1, EPHB4, TIE1, PIEZO1, ITGA9, RASopathy genes, SOS1, SOS2, and RAF1.
Conclusions:
WES identified pathogenic and likely pathogenic variants and VOUS in LA genes in 51% of fetal edema cases, supporting WES and expanded hydrops panels in cases of idiopathic fetal hydrops and fluid collections.
1 |. INTRODUCTION
Congenital lymphatic anomalies (LAs) often arise in the fetal period due to defects in lymphatic development. LAs can present as cystic lymphatic malformations (CLMs), primary lymphedema, and complex lymphatic anomaly, which include generalized lymphatic anomalies (GLAs), Kaposiform Lymphangiomatosis (KLA), Gorham-Stout Disease (GS), central conducting lymphatic anomaly (CCLA), and generalized lymphatic dysplasia (GLD).1,2 Increased nuchal translucency (NT) can occur secondary to aberrant lymphatic development, and many cystic hygromas (CHs) are fetal cervicofacial CLMs.3–6 Fetal CLMs also present in axillary and other soft tissues.7,8 Complex LAs are diffuse multifocal lymphatic hyperplasias that can involve the lungs, intestines, liver, spleen, soft tissues and bone.9,10 CCLAs are due to defects in the abdominal and thoracic lymphatic collecting ducts, resulting in backflow into the lymphatic capillary networks of the liver, intestines, and lungs.9–11 Complex LAs can cause fetal pleural/pericardial effusion, ascites, chylothorax, soft tissue edema, and nonimmune hydrops.6,9,12–16 Primary lymphedema is marked by the peripheral accumulation of the lymph in the soft tissues of extremities and genitalia.1,12,14,17,18 Overall, between 5% and 15% of cases of fetal nonimmune hydrops are suggested to develop due to lymphatic defects.12–14
LAs result from both germline autosomal dominant and recessive variants with varying degrees of penetrance as well as somatic variants.1 Lymphedema arises from inherited and de novo germline loss of function (LOF) variants in lymphangiogenic genes, ANGPT2, CCBE1, CELSR1, FLT4, FOXC2, GATA2, GJA1, GJC2, HGF, KIF11, IKBKG, SOX18, TIE1, PTPN14, and VEGFC.1 Variants in CALCRL, EPHB4, PIEZO1, and ITGA9 are associated with prenatal CCLA and GLD and precipitate nonimmune hydrops and chylothorax.19–22 LAs are often present as part of RASopathy syndromes with variants in the genes PTPN11, RAF1, NRAS, KRAS, HRAS, RASA1, RIT1, SOS1, and SOS2. LAs are also found as part of complex syndromes due to chromosomal abnormalities or variants in PTEN, TSC1, TSC2, and AKT1.1,2,12,17 Lastly, somatic variants in PIK3CA, NRAS, and KRAS have been described in CLMs and GLAs, which require genetic analysis of affected tissues.1,23–26
The ability to differentiate fetal LAs from other etiologies of hydrops and edema relies on genetic diagnosis, but the full scope of the genetic variants that contribute to LAs remains yet to be discovered. Thus, predicting outcomes of fetal edema remains challenging in the euploid, structurally normal fetus. LAs are often due to small nucleotide variants which are not detected by standard karyotyping and microarray. Diagnosis relies upon further testing via either targeted gene panels or whole exome sequencing (WES). Current genetic panels only query a subset of LA genes and the variants tested vary between clinical laboratories.27 Thus, genetic panels are not comprehensive. We queried for variants in LA genes using WES of parental-fetal trios from a cohort of idiopathic fetal fluid collection and hydrops, including increased NT, CH, cystic lesions, pleural effusion, chylothorax, ascites, and lymphedema. Our targeted analysis identified pathogenic and likely pathogenic variants and variants of uncertain significance (VOUS) in fetal edema cases. Many of these were not identified in a prior untargeted study.28 As predicting pregnancy outcomes in euploid fetal edema cases remains challenging,29,30 demographic data were compared between two subcohorts, clinically significant fetal edema (CSFE) versus isolated fetal edema (IFE). A better understanding of the LA variants which contribute to fetal hydrops and demographic factors associated with poor outcomes will aid in diagnosis and counseling in the prenatal period.
2 |. METHODS
2.1 |. Participants and study design
Fetal anomaly cases enrolled in a WES study at Columbia University (IRB:AAAO8009) between 2015 and 2019 were reviewed. Pathogenic WES findings for most fetal anomaly cases were previously published.28 In the current study, cases of pathologic fluid collection ascertained by prenatal ultrasound were identified, including increased NT (≥3.5 mm), CH, increased nuchal fold (≥6.0 mm), pleural effusion, chylothorax, ascites, skin and soft tissue edema, cystic structures consistent with CLM, and hydrops, defined as two or more fluid collections. Prenatal chylothorax was defined distinct from pleural effusion if prenatal thoracentesis was performed and ≥80% lymphocytes noted. To determine if there are any factors associated with the edema phenotype, fetal edema cases were subdivided into two subcohorts: cases with CSFE defined as requiring prenatal intervention, precipitating fetal outcome, or the effusion persisting postnatally and cases of IFE that resolved within the study period. Demographics and outcomes were compared between the two subcohorts. Outcomes included live birth, intrauterine fetal demise (IUFD), neonatal demise, spontaneous abortion (SAB), and iatrogenic abortion (IAB). Pregnancy end was defined as the end of pregnancy due to any of these outcomes. Known noncongenital lymphatic etiologies of fetal edema were excluded, including anemia preceding edema, immune hydrops, infection, skeletal-thoracic structural anomaly, twin cord/placental cord abnormality, fetal akinesia sequence, aneuploidy, and complex syndrome with unclear etiology of edema. Cases with concurrent gastrointestinal, genitourinary, and cardiac anomalies were not excluded as a number of complex syndromes copresent with LAs.1,17 Cases with diagnostic karyotype or microarray or limited data were excluded.
2.2 |. Clinical data, whole exome sequencing, and variant calling
Medical records and imaging were reviewed, and samples underwent WES at the Institute for Genomic Medicine with the NimblegenSeqCap EZ V2.0/3.0, SeqCap EZ HGSC VCRome, or xGenExome Research Panel v1.0 kits with the Illumina NovaSeq platform. Reads were aligned using human reference GRCh37 with Illumina DRAGEN Bio-IT Platform, and duplicates were marked with Picard (Broad Institute). Variants were called using Genome Analysis Toolkit (GATK) Best Practices v3.6 and annotated with ClinEff and Analysis Tool for Annotated Variants (ATAV) (in-house Analysis Tool for Annotated Variants).31 For LA variants with low alternative allele frequencies (≤0.4 for heterozygous and ≤0.9 for homozygous), Integrative Genomic Viewer (IGV), ATAV, and population data were reviewed by a multidisciplinary group, and variants likely to be artifacts were excluded. A modified version of our well-established diagnostic trio analysis framework was used (https://redmine.igm.cumc.columbia.edu/projects/atav/wiki/Diagnostic_Analysis_Workflow).28,32–34 Geographic ancestry was determined using a neural network pre-trained on samples with known ancestry, which generated probability estimates for each of the six groups (European, African, Latino, East Asian, South Asian, and Middle Eastern). An exome-based relatedness screen was performed employing KING to calculate pairwise kinship coefficients. Maternity/paternity was confirmed for de novo variants.
The primary endpoint was identification of pathogenic and likely pathogenic variants in LA genes and the identification of unique demographic factors between CSFE and IFE cases. The secondary endpoint was the identification of VOUS in LA genes. LA genes for targeted analysis were determined by review of the basic science and clinical literature and those documented in Online Mendelian Inheritance in Man (Table S1).1,17 All candidate genetic variants were classified by American College of Medical Genetics and Genomics (ACMG) and Association for Molecular Pathology criteria informed by ClinGen.35 Variants were reviewed by an interdisciplinary team including an expert in LAs and three clinical geneticists. ACMG VOUS among CSFE were determined to have potential clinical significance, given the strong gene–phenotype correlation of examined LA genes and absence of other diagnostic findings. CSFE variants were confirmed by Sanger sequencing except for Fetal0047F for which genomic DNA was not available. ACMG VOUS in the IFE subcohort are reported in Table S2.
2.3 |. Statistical analysis
Data were analyzed with Prism9. Primary and secondary outcomes are described as percentages. For demographic data, categorical variables are described as percentages and proportions and compared between groups with Fisher’s exact test and Chi-square test. Continuous variables are documented as mean values with interquartile ranges and compared between groups using an unpaired t-test or Welch’s t-test for data with standard deviation greater than twofold in difference. Normal distribution was examined by cross-checking t-test statistics with Mann–Whitney test statistics and histograms. A p < 0.05 was considered significant.
3 |. RESULTS
3.1 |. Study participants
Review of parental-fetal trios identified 111 fetal edema cases, 40 of which were excluded and 71 cases of abnormal fetal fluid collection were examined (Figure 1). Twenty-nine cases displayed CSFE defined as requiring prenatal intervention, precipitating fetal outcome, or the effusion persisting postnatally (Figure 2), while 42 cases were notable for IFE, which required no intervention and resolved within the study period. The CSFE subcohort was compared to the IFE subcohort to determine if there were characteristics unique to the two subcohorts (Table 1). In both subcohorts, over 70% of mothers had ancestry other than European. There was no difference in maternal/paternal age, nulliparity, history of anomaly, fetal sex, microarray finding, or maternal hypertension, diabetes, and BMI. CSFEs were diagnosed at a mean gestation age (GA) of 18 weeks, while IFEs were diagnosed at 11 weeks. The majority 69% (20/29) of CSFEs were hydropic, while 93% (39/42) IFEs presented with isolated nuchal edema. Pregnancy outcomes were significantly different between the two subcohorts. In the CSFEs, 31% (9/29) were live born, 41% (12/29) underwent IAB, 17% (5/29) experienced IUFD, 7% (2/29) neonatal demise, and 3% (1/29) underwent SAB. In the IFEs, 95% (40/42) were live born and 4% (2/42) underwent IAB. Live born CSFEs were delivered at a mean GA of 35 weeks versus 38 for IFEs. Mean GA at pregnancy end for CSFEs was 29 versus 37 weeks for IFEs. CSFEs trended toward having concurrent structural anomaly. Occurrence of preeclampsia with severe features or mirror syndrome occurred in 24% (5/21) of CSFEs versus 2.48% (2/41) of IFEs; 3.45% (1/28) of women with CSFEs used assisted reproductive technology, while 21% (9/42) did among IFEs.
FIGURE 1.
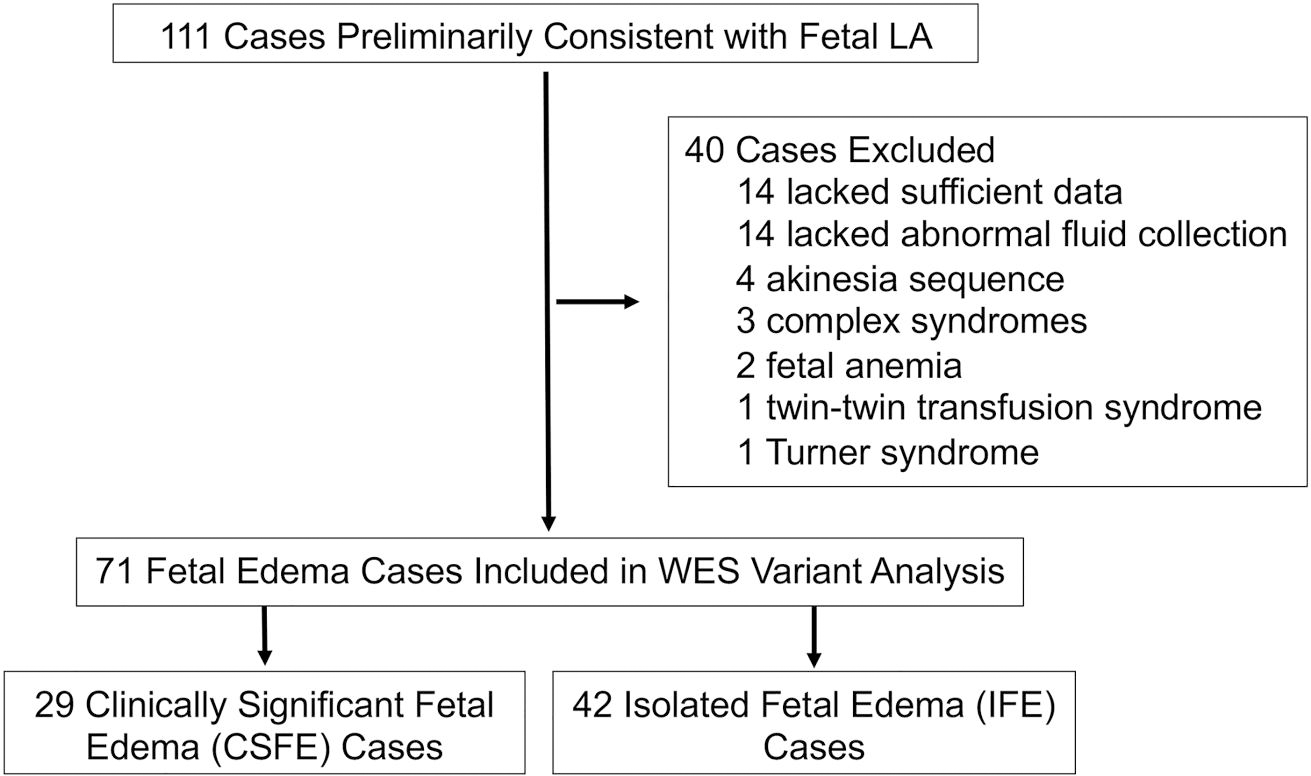
Flow chart of inclusion and exclusion criteria as applied to fetal edema cases.
FIGURE 2.
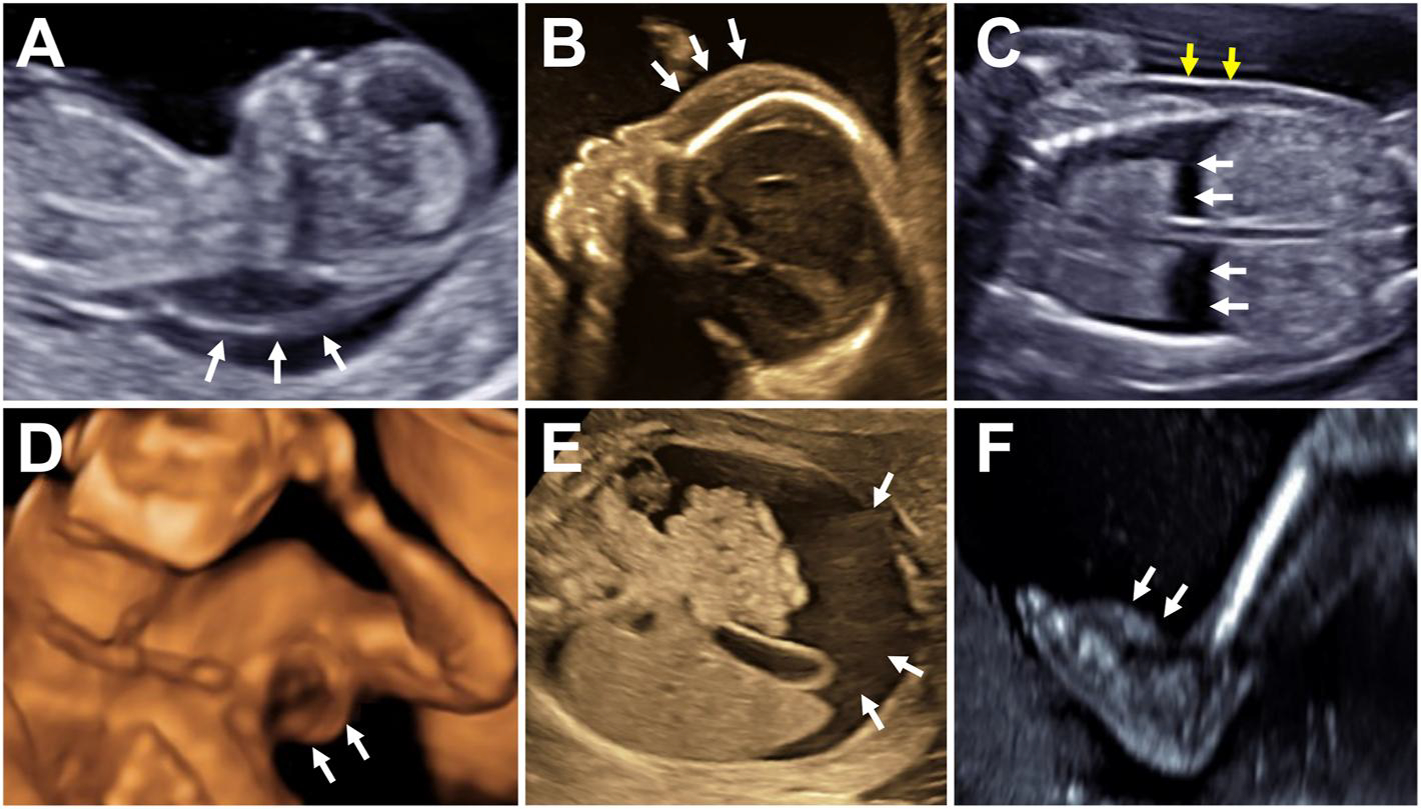
Spectrum of fetal LAs on prenatal ultrasound. (A) Cystic hygroma, 12w (white arrows). (B) Scalp soft tissue edema, 21w (white arrows). (C) Hydrops with bilateral pleural effusion (white arrows) and trunk soft tissue edema (yellow arrows), 19w. (D) 3D ultrasound of an axillary cystic lymphatic malformation, 20w (white arrows). (E) Ascites, 26w (white arrows). (F) Pedal lymphedema, 22w (white arrows). w, weeks’ gestation.
TABLE 1.
Characteristics and outcomes for CSFE versus IFE subcohorts.
| Characteristic | Clinically significant fetal edema | Isolated fetal edema | Test statistic |
|---|---|---|---|
| Mean maternal age—year (N, IQR, Std Dev)† | 31 (28, 27–35, 4.76) | 33 (42, 31–37, 5.24) | p = 0.2048 |
| Mean paternal age—year (N, IQR, Std Dev)† | 33 (28, 27–39, 8.40) | 36 (42, 31–39, 7.30) | p = 0.1618 |
| Nulliparous—no (N, %)° | 14 (28, 48.28) | 18 (42, 42.86) | p = 0.8087 |
| Use of ART—no (N, %)° | 1 (28, 3.45) | 9 (42, 21.43) | p = 0.0401 |
| Mean GA at diagnosis—week (N, IQR, Std Dev)‡ | 18 (23, 12–21, 5.77) | 11 (40, 11–12, 1.53) | p < 0.0001 |
| Microarray finding—no (N, %)° | 5 (29, 17.24) | 2 (41, 4.88) | p = 0.1175 |
| Prior pregnancy anomaly—no (N, %)° | 3 (29, 10.34) | 4 (42, 9.52) | p > 0.9999 |
| Parents′ consanguineous—no (N, %)° | 1 (29, 3.45) | 1 (42, 2.38) | p > 0.9999 |
| Prenatal phenotype—no (N, %)# | p < 0.0001 | ||
| Isolated nuchal edema | 6 (29, 20.69) | 39 (42, 92.86) | |
| Isolated non-nuchal fluid collection | 3 (29, 10.34) | 1 (42, 2.38) | |
| ≥2 abnormal fetal fluid collections | 20 (29, 68.97) | 2 (42, 4.76) | |
| Concurrent structural anomaly—no (N, %)° | 13 (28, 46.43) | 10 (42, 23.80) | p = 0.0693 |
| Fetal sex—no (N, %)° | p > 0.9999 | ||
| Female | 12 (29, 41.38) | 17 (42, 40.48) | |
| Male | 17 (29, 58.62) | 25 (42, 59.52) | |
| Maternal ancestry—no (N, %)° | p = 0.7783 | ||
| African | 2 (29, 6.90) | 5 (42, 11.90) | |
| Caucasian | 6 (29, 20.69) | 11 (42, 26.19) | |
| East Asian | 2 (29, 6.90) | 3 (42, 7.14) | |
| Hispanic | 8 (29, 27.59) | 15 (42, 35.71) | |
| Middle Eastern | 6 (29, 20.69) | 3 (42, 7.14) | |
| Mixed | 3 (29, 10.34) | 1 (42, 2.38) | |
| South Asian | 2 (29, 6.90) | 4 (42, 9.52) | |
| Maternal chr. hypertension—no (N, %)° | 1 (27, 3.70) | 3 (42, 7.14) | p > 0.9999 |
| Maternal diabetes—no (N, %)° | 0 (28, 0.00) | 3 (42, 7.14) | p = 0.2696 |
| Mean maternal BMI—(N, IQR, Std Dev)† | 25.14 (16, 22.08–26.08, 4.93) | 28.26 (40, 22.77–32.15, 6.99) | p = 0.1097 |
| Pregnancy outcome—no (N, %)° | p < 0.0001 | ||
| Live birth | 9 (29, 31.03) | 40 (42, 95.24) | |
| IAB | 12 (29, 41.38) | 2 (42, 4.76) | |
| IUFD | 5 (29, 17.24) | 0 (42, 0.00) | |
| Neonatal demise | 2 (29, 6.90) | 0 (42, 0.00) | |
| SAB | 1 (29, 3.45) | 0 (42, 0.00) | |
| Mean GA at live delivery—week (N, IQR, Std Dev)† | 35 (10, 33–38, 3.37) | 38 (40, 37–39, 1.97) | p = 0.0004 |
| Mean GA at pregnancy end—week (N, IQR, Std Dev)† | 29 (22, 23–36, 7.78) | 37 (42, 36–39, 4.94) | p < 0.0001 |
| Maternal preeclampsia spectrum—no (N, %)° | 5 (21, 23.81) | 2 (41, 2.38) | p = 0.0387 |
Note: Percentages may not total 100 due to rounding to the nearest two‐digit decimal place. Categorical variables are compared with Fisher's exact test ° and Chi‐square test#.
Continuous variables are compared between groups using an unpaired t-test† or Welch's t-test‡ for data with standard deviation greater than twofold in difference. Ancestry was as determined by genetic profiling.
Diabetes was type I or II, nongestational. Hypertension preceded pregnancy (chronic). Concurrent structural anomalies include those not related to abnormal fluid collection and diagnosed prenatally. Test statistics for fetal outcome and for ancestry are comparing live birth versus adverse outcome and Caucasian versus non-Caucasian, respectively. Pregnancy end includes all outcomes. All cases in this cohort were preeclampsia with severe features, or mirror syndrome though charts were reviewed for gestational hypertension, preeclampsia, preeclampsia with severe features, superimposed preeclampsia, eclampsia, and mirror syndrome. Significant statistical values are in bold.
Abbreviations: ART, assisted reproductive technology; CSFE, clinically significant fetal edema; GA, gestational age; IAB, iatrogenic abortion; IFE, isolated fetal edema; IQR, interquartile range; IUFD, intrauterine fetal demise (after 20w); SAB, spontaneous abortion (prior to 20w); Std Dev, standard deviation.
Further subanalysis of anomalies, which presented specifically in the first trimester in CSFEs (N = 11) and IFEs (N = 40), illustrated that most factors were similar between the two groups (Table S3). However, CSFEs which presented in the first trimester more often had multiple fluid collections; 82% (9/11) of these CSFEs had persistent/worsening edema or another structural anomaly on subsequent scans, while 18% (2/11) appeared structurally normal with resolution of fetal edema but had persistent nuchal edema and dysmorphic features after delivery. In contrast, only 23% (9/40) of IFEs were found to have a concurrent structural anomaly (CNS, renal, skeletal, and cardiac). Occurrence of preeclampsia was 22% (2/9) in the first-trimester CSFEs and 5% (2/40) in IFEs. Overall, pregnancy outcomes remained significantly different between the first-trimester CSFEs and IFEs.
3.2 |. Pathogenic and likely pathogenic variants in LA genes
Pathogenic and likely pathogenic variants were identified in 17% (5/29) of CSFEs (Table 2). Though not significant, diagnostic yield was lower for CSFE cases with isolated nuchal edema (0%; 0/9) than for cases with multiple fluid collections (25%; 5/20). Pathogenic and likely pathogenic variants were more common, though not significant, in cases with concurrent structural abnormality than without (23%; 3/13 vs. 13%; 2/15). No pathogenic or likely pathogenic variants were identified in IFEs.
TABLE 2.
Pathogenic and likely pathogenic variants in congenital lymphatic anomaly genes.
| Case | Prenatal phenotype | Postnatal phenotype | Concurrent structural anomalya | Fetal or neonatal interventionb/outcome | Gene/RefSeq ID | Genomic coordinate (GRCh37/hgl9)/nucleotide/protein alteration | Molecular consequence | OMIM genetic disorder/ MIM No. | Inheritance/ zygosity | Novel or PMID/ CLINVAR Ac. No | ACMG classifi cation |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Fetai0164F | Pleural effusion, ascites, hydrocele, skin and scalp edema, and polyhydramnios | NA | None | None/IUFD | RASA 1/NM_002890.3 | 5–86674312-AAG-Ac.2446_2447delGA p.Asp816Leufs*13 | Frameshift | Capillary malformation-arteriovenous malformation 1/608354 | Maternal/heterozygous | 30712878 | Likely pathogenic PVS1, PM2 |
| Feta10222F | Increased NT, scalp, skin and significant nuchal fold and shoulder edema, bilateral pleural effusion, ascites, and polyhydramnios | Bilateral pleural effusion, ascites, anasarca, respira tory di stress, dysmorphic features, webbed neck, pulmonic valve stenosis, and atrial septal defect | None | Neonatal ventilation, chest tu be placement and pulmonic valve repair complicated by prolonged postsurgical chylothorax/ live born | SOS V NM_005633.4 | 2–39251221-T-C c.H32A>G p.Thr378Ala | Missense | Noonan syndrome 4/610733 | Paternal/ heterozygous | 21387466 | Pathogenic PS1, PS4 (moderate), PM2, PM6 and, PP1 |
| Fetai0302F | Left > right pleural effusion, ascites, and polyhydramnios | Dysmorphic features, redundant nuchal skin folds, undescended testes, and respiratory distress | Bilateral ventricular hypertrophy, atypical or absent cavum septum pellucidum, and Blake's pouch cyst | Serial thoracentesis (2x) followed by thoracic shunt placement/ live born | PTPN11/NM_002834.5 | 12–112891083- G-C c.417G>C p.Glul39Asp | Missense | Noonan syndrome 1/163950 | Maternal/ heterozygous | 28363362 | Pathogenic PS1, PS2, PS3, PS4 (moderate), PM2, PP1, PP2, and PP3 |
| Feta10405F | Scalp, total body skin, nuchal fold edema, bilateral pleural effusion, ascites, and pericardial effusion | NA | Cardlomegaly, agenesis of the ductus venosus, placentomegaly, abnormal profile, and enlarged tongue, anemia | Percutaneous umbilical blood sampling with transfusion/ IUFD | PIEZ01/ NM_001142864.4 | 16–88805045- G- A c.565C>T p.Argl89* | Nonsense | Lymphatic malformation 6/616843 | Both parents/ homozygous | Novel | Likely pathogenic PVS1, PM2 |
| Fetal0485F | Bilateral pleural effusion, ascites, and unilateral shoulder edema | NA | Dilated left ventricle | None/IUFD | PTPN11/NM_002834.5 | 12–112891083- G-C c.417G>C p.Glul39Asp | Missense | Noonan syndrome 1/163950 | De Novo/ heterozygous | 28363362 | Pathogenic PS1, PS2, PS3, PS4 (moderate), PM2, PP1, PP2, and PP3 |
Abbreviations: CH, Cystic hygroma; IAB, iatrogenic abortion; IUFD, Intrauterine fetal demise; NT, Nuchal translucency; OMIM, Online Mendelian Inheritance in Man.
Concurrent structural anomalies are those that presented prenatally.
Intervention does not include iatrogenic delivery for non‐reassuring fetal monitoring or maternal indication.
Pathogenic and likely pathogenic variants included four genes linked to RASopathies, syndromes that often present with LAs.1 Three were heterozygous missense variants in Noonan syndrome genes, PTPN11 and SOS1. A c.417G>C variant in PTPN11 was identified in two CSFEs: one de novo and one inherited from a mother with Noonan syndrome. A SOS1 c.1132A>G variant was inherited from a father with Noonan syndrome. The fourth RASopathy variant was a c.2446_2447delGA variant in RASA1 predicted to cause LOF, which was inherited maternally, though no maternal phenotype was documented by the treating physician. We also identified a novel homozygous stop gained (LOF) variant c.565C>T in the GLD gene PIEZO1 inherited from consanguineous parents. These findings demonstrate that RASopathies can present as/with LAs and prenatal WES can aid in diagnosing GLD.
3.3 |. Variants of uncertain significance in LA genes
VOUS in LA genes were identified in 38% (11/29) of CSFEs with 6.9% (2/29) of these carrying VOUS in multiple LA genes (Tables 3 and 4). Similar to pathogenic and likely pathogenic variants, heterozygous VOUS were identified in RASopathy genes (SOS1, SOS2, and RAF1), inherited from unaffected parents. Novel VOUS were identified in two cases, a SOS2 c.1223A>G variant and a SOS1 c.421A>G variant. A previously reported RAF1 c.1895_1918delAGGATATCAATGCTTGCACGCTGA VOUS was identified that falls outside the commonly affected domains.36
TABLE 3.
VOUS in congenital lymphatic anomaly genes.
| Case | Prenatal phenotype | Postnatal phenotype | Concurrent structural anomalya | Fetal or neonatal interventionb/outcome | Gene/RefSeq ID | Genomic coordinate (GRCh37/hgl9)/nucleotide/protein alteration | Molecular consequence | OMIM genetic disorder/MIM No. | Inheritance/zygosity | Novel or PMID/CLINVAR Ac. No | ACMG classification |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Fetal0032F | CH, enlarged nuchal fold, total body anasarca with edema especially of the scalp and trunk with a small pleural effusion | NA | Cardiomegaly, small stomach, two vessel cords, skeletal growth delay, enlarged kidneys | None/IUFD | EPHB4/NM_004444.5 | 7–100421427-C-T c.250G>A p.Val84lle | Missense | Lymphatic malformation 7/617300 | Paternal/heterozygous | Novel | vous PM2 and BP4 |
| Fetal0038F | Right chylothorax, pericardial effusion | None | None | Serial thoracentesis (5x), maternal propranolol/liveborn | SOS2/NM_006939.4 | 14–50626778-T-Cc.l223A>G p.His408Arg | Missense | Noonan syndrome 9/616559 | Maternal/heterozygous | Novel | VOUS PM2, BP4 |
| Fetal0045F | CH | Dysmorphic features, right undescended testicle, hypertonia, spasticity, and redundant nuchal skin | None | None/liveborn |
EPHB4/NM_ 004444.5 |
7–100401090-T-C c.2957A>G p.Gln986Arg | Missense | Lymphatic malformation 7/617300 | Paternal/heterozygous | Novel | VOUS PM2, BP4, BP5 |
| Fetal0047F | Increased NT, bilateral pedal edema especially at the anterior and plantar surfaces | NA | None | None/IAB | TIE1/NM_001253357.2 | 1–43783709-C-T c.2888C>T p.Ala963Val | Missense | Lymphatic malformation 11/619401 | Maternal/heterozygous | Novel | VOUS PM2, BP4 |
| Fetal0052F | Left chylothorax | NA | None | Serial thoracentesis (2x), maternal propranolol/IAB | PIEZOl/NM_001142864.4 | 16–88808727-G-C c.264C>G p.Asp88Glu |
Missense | Dehydrated hereditary stomatocytosis with or without perinatal edema/194380 | Maternal/heterozygous | Novel | VOUS PM2, BP4 |
| Fetal0151F | Pericardial effusion, ascites, and hydrocele | Cardiomyopathy, dysplastic mitral value, respiratory distress, neonatal heart failure, dysmorphic features, and ascites | Cardiomyopathy and valvular dysfunction | Maternal digoxin, neonatal mechanical ventilation, and mitral valvuloplasty/neonatal demise | CELSR1/NM_014246.4 | 22–46792578-C-T c.5767G>A p.Vall923Met | Missense | Lymphatic malformation 9/619319 | Maternal/heterozygous | Novel | VOUS PM2, BP1 |
| Fetal0231F | CH | Dysmorphic features, redundant nuchal skin, fifth toe clinodactyly, and bilateral ventriculomegaly | Bilateral ventriculomegaly | None/Liveborn | PIEZ01/NM_001142864.4 | 16–88788059-C-G c.5290G>C p.Glul764Gln | Missense | Dehydrated hereditary stomatocytosis with or without perinatal edema/194380 | Maternal/heterozygous | VCV001049973.1 | vous PM2 |
| PIEZ01/NM_001142864.4 | 16–88800060-C-T c.2423G>A p.Arg808Gln | Missense | Dehydrated hereditary stomatocytosis with or without perinatal edema/194380 | Maternal/heterozygous | 30655378 | VOUS PP1, BS1 | |||||
| PIEZOl/NM_001142864.4 | 16–88800139-C-T c.2344G>A p.Gly782Ser | Missense | Dehydrated hereditary stomatocytosis with or without perinatal edema/194380 | Maternal/heterozygous | 30655378 | VOUSPP1, BS1 | |||||
| Fetal0310F | Increased nuchal fold, bilateral pleural effusion, total body skin edema, ascites, pericardial effusion, and polyhydramnios | Macrocephaly, anasarca, bilateral pleural effusion, hypospadias, and respiratory distress | None | Neonatal mechanical ventilation and bilateral chest tube placement/neonatal demise | PIEZOl/NM_001142864.4 | 16–88794058-G-A c.3208C>T p.Argl070Cys | Missense | Lymphatic malformation 6/616843 | Maternal uniparental disomy/homozygous | Novel | VOUS PM2, PP3 |
| Fetal0399F | Ascites, facial edema especially scalp and periorbital, lower extremity edema, and right pleural effusion | NA | None | None/IAB | SOSl/NM_005633.4 | 2–39283932-T-C c.421A>G p.llel41Val | Missense | Noonan syndrome 4/610733 | Paternal/heterozygous | Novel | VOUS PM2, BP4 |
| TIE1/NM_001253357.2 | 1-43773482-A-G c.929A>G p.His310Arg | Missense | Lymphatic malformation 11/619401 | Maternal/ heterozygous | Novel | VOUS PM2, BP4 | |||||
| Fetal0445F | Pleural effusion, other undescribed second abnormal fluid collection | NA | Unknown | Unknown/IAB | RAF1/NM_002880.4 | 3–12626041-GTCAGCGTGCAAGCATTGATATCCTGc.1895_1918delAGGATATCAATGCTTGCACGCTGA p.Glu632_Thr640delinsAla | Disruptive inframe deletion | Noonan syndrome 5/611553 | Paternal/heterozygous | VCV00092899 2.1 | VOUS PM2, PM4 |
| Feta10510F | Bilateral chylothorax, ascites | Pleural effusion, respiratory distress, and right lower quadra nt venous-lymphatic malformation | None | Serial thoracentesis (8x), chest tube placement, ventilation, propranolol, diuresis/live born | PIEZ01/NM_001142864.4 | 16–88798811-G-C c.2923C>G p.Leu975Val | Missense | Dehyd rated hereditary stomatocytosis with or without perinatal edema/194380 | Maternal/heterozygous | Novel | vous PM3, BP5 |
Abbreviations: CH, Cystic hygroma; IAB, iatrogenic abortion; IUFD, Intrauterine fetal demise; NT, Nuchal translucency; OMIM, Online Mendelian Inheritance in Man.
Concurrent structural anomalies are those that presented prenatally.
Intervention does not include iatrogenic delivery for non‐reassuring fetal monitoring or maternal indication.
TABLE 4.
Heterozygous VOUS in genes of uncertain inheritance.
| Case | Prenatal phenotype | Postnatal phenotype | Concurrent structural anomalya | Fetal or neonatal interventionb/outcome | Gene/RefSeq ID | Genomic coordinate (GRCh37/hgl9)/nucleotide/protein alteration | Molecular consequence | OMIM genetic disorder/MIM No. | Inheritance/zygosity | Novel or PMID/CLINVAR Ac. No | ACMG classification |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Fetal0399F | Ascites, facial edema especially scalp and periorbital, lower extremity edema, and right pleural effusion | NA | None | None/IAB | ITGA9/NM_002207.3 | 3–37493995-GC-AA c.l30_131delGCinsAA p.Ala44Asn | Missense | Congenital Chylothorax/NA | Paternal/heterozygous | Novel | VOUS PM1, PM2 |
| Fetal0403F | Bilateral pleural effusion, thickened nuchal fold, skin edema, ascites, and pericardial effusion | NA | None | None/IAB | ITGA9/NM_002207.3 | 3–37544736-C-T c.680C>T p.Thr227Met |
Missense | Congenital Chylothorax/NA | Paternal/heterozygous | Novel | VOUS PM2, PP3 |
| Fetal0523F | CH, skin edema of the chest, and pleural effusion | NA | None | None/SAB | ITGA9/NM_002207.3 | 3–37567549-G-A c.l360G>A p.Val454Met | Missense | Congenital Chylothorax/NA | Maternal/heterozygous | Novel | VOUS PM2, PP3 |
Abbreviations: CH, Cystic hygroma; IAB, iatrogenic abortion; OMIM, Online Mendelian Inheritance in Man; SAB, spontaneous abortion.
Concurrent structural anomalies are those that presented prenatally.
Intervention does not include iatrogenic delivery for non‐reassuring fetal monitoring or maternal indication.
VOUS in genes associated with GLA, primary lymphedema, and dehydrated hereditary stomatocytosis (DHS) with or without perinatal edema were identified in nine cases. Two novel heterozygous missense variants inherited from unaffected parents were found in EPHB4, c.250G>A, and c.2957A>G. One of these, Fetal0045F, is notable for carrying a pathogenic RERE LOF variant as previously reported.28,37 In two cases, novel heterozygous missense variants, inherited from an unaffected parent, c.2888C>T and c.929A>G, were identified in the lymphedema gene TIE1.38 A novel inherited heterozygous missense variant c.5767G>A was detected in the lymphedema gene, CELSR1.
For PIEZO1, a novel missense VOUS, c.3208C>T, was identified that was homozygous in the setting of maternal uniparental disomy, while five heterozygous missense variants were identified and inherited from unaffected mothers. One of these cases, Fetal0510F, also carried a pathogenic LOF GLMN variant, a gene associated with glomuvenous malformation, which was diagnosed postnatally. In one CSFE case, three PIEZO1 VOUS that have been previously described were identified in cis: c.5290G>C, c.2423G>A, and c.2344G>A.
In 17% (5/29) of CSFEs, a variant was detected for an autosomal recessive disorder consistent with the phenotype, but a second variant was not identified: ITGA9, CCBE1, and PTPN14. Unlike CCBE1 and PTPN14, the inheritance pattern for ITGA9 variants is not well established; thus, heterozygous VOUS may be of interest (Table 4). We identified novel missense ITGA9 VOUS inherited from unaffected parents, c.130_131delGCinsAA, c.680C>T, and c.1360G>A.
Heterozygous missense VOUS were identified in 45% (19/42) of IFEs (ANGPT2, CELSR1, FLT4, HGF, PIEZO1, PTEN, PTPN14, RIT1, TIE1, and TSC2) (Table S2). PIEZO1, CELSR1, and TIE1 VOUS in the IFE subcohort were distinct from the CSFE subcohort. Multiple variants were found in 9.5% (4/42) of IFEs.
4 |. DISCUSSION
Our targeted analysis of LA genes in 71 cases of idiopathic fetal edema identified pathogenic variants in 7% (5/71), and VOUS in 45% (32/71), for an overall yield of 51% (37/71). In CSFEs, pathogenic and likely pathogenic variants were identified in 17% (5/29) of cases. The CSFE subcohort had a slightly lower diagnostic yield than another publication with pathogenic and likely pathogenic variants identified in 29% of nonimmune hydrops cases due to all causes.30 The lower yield may be due to our targeted gene approach, as 7% (2/29) of CSFEs had diagnostic variants in non-LA genes (RERE and GLMN).28 Unlike CSFEs, pathogenic and likely pathogenic variants were not identified in IFEs. In CSFE, pathogenic and likely pathogenic variants were identified in fetal hydrops cases, but not those with isolated nuchal edema. A recent WES study had a similarly low yield in cases of isolated nuchal edema (1.8%) versus cases with structural abnormality or hydrops at presentation (22%) or later in pregnancy (32%).29 VOUS in LA genes were identified in 45% (13/29) of CSFEs and 45% (19/42) of IFEs; 38% (11/29) of CSFEs and 55% (23/43) of IFEs had no genetic findings, which may be due to several factors. Not all genes that contribute to LAs have been identified and were not included in our analyses. Secondly, somatic mutations often cause LAs, and germline WES will not detect these variants as it requires sequencing of affected tissues.9 Lastly, it is likely that some of these fetal edema cases were not secondary to LAs.
Four pathogenic and likely pathogenic variants were in RASopathy genes. RASopathies are heterogenous, overlapping disorders due to hyperactivating variants in the RAS/mitogen-activated protein kinase (MAPK) signaling cascade.36 RASopathy variants are a common etiology of hydrops and nuchal edema.29,30 In this study, CSFEs with RASopathy variants were all hydropic with pleural effusions and ascites and either with skin edema or polyhydramnios, similar to a recently described RASopathy cohort.39 CSFEs with VOUS in RASopathy genes also had hydrops and pleural effusions. Three pathogenic RASopathy variants (PTPN11, SOS1) were consistent with Noonan syndrome, an autosomal dominant syndrome of craniofacial and cardiac anomalies often accompanied by prenatal nuchal edema, chylothorax, and hydrops, as well as postnatal lymphedema, GLA, and chylothorax.1,17,36,40 The last likely pathogenic RASopathy variant was a frameshift truncation variant in RASA1. RASA1 LOF variants are associated with CM-AVM as a complex vascular anomaly that often concurrently presents with LAs.1,41,42 No features of CM-AVM were documented in this case though IUFD occurred and CM-AVM are rarely diagnosed prenatally.43 If CM-AVM was absent, this case may represent an expansion of the RASA1 genotype–phenotype association, which is supported by a RASA1 deletion variant recently identified in a case of fetal hydrops without CM-AVM.44
Analysis of CSFEs identified both likely pathogenic and VOUS variants in PIEZO1. Common features among this group included nuchal edema and pleural effusions that were similar to phenotypes observed in recently described PIEZO1 cases.39 Homozygous or compound heterozygous variants in PIEZO1 are associated with GLAs and marked fetal hydrops, while heterozygous variants are associated with DHS with or without perinatal edema.19,30,45–49 We identified a novel homozygous PIEZO1 missense VOUS in a fetal case of marked hydrops, where neonatal demise occurred on day of life one due to respiratory distress secondary to chylothorax. This variant is absent in large population databases, and there is a strong genotype-phenotype correlation. Additionally, heterozygous VOUS in PIEZO1 were identified in three cases. In one case of CH and dysmorphic features, three variants were identified in cis, two of which, c.2423G>A and c.2344G>A, had previously been identified in cis among several family members with DHS with perinatal lymphedema, and in one case, adult lymphedema.49,50 Though these variants are individually common in population databases, whether their occurrence in combination is rare enough to be associated with human disease remains unknown. While the other two heterozygous PIEZO1 VOUS are rare in the general population, PIEZO1 is a highly polymorphic gene, and functional studies and further clinical data are needed to classify their role in DHS and GLD.
The mechanism of DHS-related perinatal edema is unknown. Though DHS causes mild hemolytic anemia, significant fetal anemia has not been described in these cases and thus likely not the precipitant.30,48,49 DHS-associated perinatal edema may be lymphatic in etiology as evidenced by cases of CH, chylothorax, and adult onset lymphedema in the setting of heterozygous PIEZO1 variants.30,48,49 Conversely, homozygous LOF and compound heterozygous missense variants in PIEZO1 associated with GLD may have DHS like erythrocyte abnormalities though these are not well characterized.19,48,51 We identified a novel likely pathogenic homozygous LOF variant in PIEZO1 in a case of fetal hydrops that subsequently developed significant anemia of unknown etiology. Though transfusion improved anemia, the hydrops was unresolved and IUFD occurred. Given the timeline of hydrops preceding the onset of fetal anemia, anemia was not felt to be the precipitant. Whether this anemia was precipitated by PIEZO1 variant, which would be a novel presentation, remains unknown as neither fetal nor parental erythrocytes were examined.
Our WES analysis of CSFEs identified VOUS in several additional LA genes, EPHB4, TIE1, and CELSR1. Missense kinase inactivating variants in EPHB4 are associated with an autosomal dominant GLA that presents with hydrops and postnatal lymphedema.17,20 Though the EPHB4 VOUS we identified falls outside the kinase domain, a likely pathogenic frameshift truncating variant outside the kinase domain has been seen in another case of hydrops.20,29 Heterozygous TIE1 missense variants have been detected in individuals with lower extremity lymphedema, and we describe two novel TIE1 variants in fetuses with bilateral lower limb edema.38 In one fetus with isolated pedal edema, the variant falls within the commonly affected TIE1 kinase domain.38 We also identified a novel missense VOUS in CELSR1 in a fetus with ascites and hydrocele. While heterozygous CELSR1 LOF variants are also associated with lower extremity lymphedema, missense variants have been described but have yet to be established as disease causing.52–54
A heterozygous ITGA9 missense variant has been implicated in several cases of recurrent congenital chylothorax and fetal hydrops.21 The inheritance pattern for ITGA9 chylothorax is unknown; though an autosomal recessive mechanism has been proposed, the majority of cases are heterozygous inherited from unaffected parents.21,55 Given the reduced penetrance seen in other LAs, this may suggest autosomal dominant inheritance. We identified three heterozygous ITGA9 missense VOUS in CSFE with pleural effusions that fall outside the protein domain described to be associated with disease (Table 4). One case carried VOUS in ITGA9, SOS1, and TIE1 suggesting a multigenic cause of this fetal LA (Tables 3 and 4).
Small sample size limited our ability to identify predictive factors among first-trimester edema cases as to which would become clinically significant. Consistent with reported poor outcomes for hydropic fetuses, poor outcomes in the CSFE cases included preterm delivery, IUFD, neonatal demise, and maternal preeclampsia.16,30 These findings highlight the importance of early detection and management. Prompt clinical recognition of fetal edema followed by genetic diagnosis of LA could allow for intervention for both maternal and fetal benefit.16 Emerging therapies include maternal propranolol for congenital chylothorax, as well as the use of trametinib, a RAS/MAPK inhibitor, in RASopathy cases with significant lymphatic sequelae.56,57 Maternal propranolol is also an experimental therapy for fetal CLMs.58 Genetic diagnosis of LAs also aids in future pregnancy planning, as evidenced by one CSFE case, where the likely pathogenic PIEZO1 LOF variant was inherited in a homozygous manner in a subsequent pregnancy.
The major strength of this study is the targeted phenotype–genotype approach, which sheds light on the contribution of LAs to prenatal edema and hydrops and describes novel variants in LA genes. This study highlights a set of LA genes, though not exhaustive, which should be included in clinically available LA and hydrops gene panels to aid in diagnostic workup of idiopathic nonimmune hydrops and in suspected neonatal and pediatric LAs. This targeted gene approaches also allowed for the identification of novel VOUS in LA genes, which were otherwise overlooked in an undifferentiated WES analysis.28
Study limitations include the inclusion of non-LA cases in the fetal edema cohort. Although inclusion criteria were strict, fetal hydrops is often a nonspecific finding that may be precipitated by numerous nonlymphatic etiologies, including some structural anomalies that were not excluded due to their overlapping phenotypes with LA syndromes.16 This inclusion of non-LA cases in a targeted analysis of LA genes may have lowered the diagnostic yield. Our targeted gene approach also did not detect variants in genes outside our panel, which were found in 7% (2/29) of cases. It is possible that aborted cases could have resolved later and thus were included inappropriately among CSFEs. Conversely, LA phenotype in IFEs may have developed postnatally beyond the period of the study.t
5 |. CONCLUSION
Targeted analysis of LA genes identified variants in 51% (37/71) of fetal edema cases. This supports the use of next-generation sequencing and expanded LA panels for the prenatal diagnosis and further study of fetal LAs. We describe novel variants in several LA genes, including a likely pathogenic variant in PIEZO1, and VOUS in PIEZO1, ITGA9, CELSR1, EPHB4, and TIE1, as well as novel VOUS in RASopathy genes. Further work examining prenatal LAs and describing LA variants is needed.
Supplementary Material
Key points.
What’s known on this topic?
Congenital lymphatic anomalies (LAs) arise due to defects in lymphatic development.
Variants in several genes are known to cause LAs though many of variants remain unknown.
The full contribution of LA genes to fetal hydrops and nuchal edema remains unknown.
What does the study add?
The study identifies novel likely pathogenic variants and variants of uncertain significance via whole exome sequencing and analysis of genes associated with LAs.
Identified variants may improve diagnosis of fetal LAs among otherwise idiopathic fetal hydrops cases.
ACKNOWLEDGMENTS
We would like to thank all patients and families who participated in this study, the clinicians who enrolled and cared for them, Cynthia Masson and the Columbia University Department of Obstetrics and Gynecology for their support and for data access, Jimmy Duong for his statistical support, and June K. Wu for careful reading of this manuscript. Dr. Shawber received funding from the NIH/NICHD (R03HD092662) and the Department of Defense (W81XWH1910266). Dr. Wapner received funding from the Institute for Genomic Medicine Columbia University Irving Medical Center. Dr. Rogerson received funding from the CUIMC Dean’s Research Fellowship and NIH/NIDDK (T35DK093430).
Footnotes
CONFLICT OF INTEREST STATEMENT
None of the authors have conflicts of interest to disclose.
ETHICS STATEMENT
All studies were approved by Columbia University IRB (AAAO8009) and conformed to US Federal Policy for the Protection of Human Subjects standards.
SUPPORTING INFORMATION
Additional supporting information can be found online in the Supporting Information section at the end of this article.
DATA AVAILABILITY STATEMENT
The primary study submits all genotype and phenotype data to the database of Genotypes and Phenotypes (dbGaP) at the completion of the study as per agreement with the NIH. A copy of the data sharing certificate is available for documentation.
REFERENCES
- 1.Brouillard P, Boon L, Vikkula M. Genetics of lymphatic anomalies. J Clin Invest. 2014;124(3):898–904. 10.1172/JCI71614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anomalies ISftSoV. ISSVA Classification for Vascular Anomalies; 2014.
- 3.Bekker MN, van den Akker NM, de Mooij YM, Bartelings MM, van Vugt JM, Gittenberger-de Groot AC. Jugular lymphatic maldevelopment in Turner syndrome and trisomy 21: different anomalies leading to nuchal edema. Reprod Sci. 2008;15(3):295–304. 10.1177/1933719107314062 [DOI] [PubMed] [Google Scholar]
- 4.Bellini C, Rutigliani M, Boccardo FM, et al. Nuchal translucency and lymphatic system maldevelopment. J Perinat Med. 2009;37(6):673–676. 10.1515/JPM.2009.107 [DOI] [PubMed] [Google Scholar]
- 5.de Mooij YM, van den Akker NM, Bekker MN, Bartelings MM, van Vugt JM, Gittenberger-de Groot AC. Aberrant lymphatic development in euploid fetuses with increased nuchal translucency including Noonan syndrome. Prenat Diagn. 2011;31(2):159–166. 10.1002/pd.2666 [DOI] [PubMed] [Google Scholar]
- 6.Attar MA, Donn SM. Congenital chylothorax. Semin Fetal Neonatal Med. 2017;22(4):234–239. 10.1016/j.siny.2017.03.005 [DOI] [PubMed] [Google Scholar]
- 7.Farnaghi S, Kothari A. The value of early recognition of fetal lymphangioma. Australas J Ultrasound Med. 2013;16(3):147–152. 10.1002/j.2205-0140.2013.tb00103.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shamshirsaz AA, Stewart KA, Erfani H, et al. Cervical lymphatic malformations: prenatal characteristics and ex utero intrapartum treatment. Prenat Diagn. 2019;39(4):287–292. 10.1002/pd.5428 [DOI] [PubMed] [Google Scholar]
- 9.Makinen T, Boon LM, Vikkula M, Alitalo K. Lymphatic malformations: genetics, mechanisms and therapeutic strategies. Circ Res. 2021; 129(1):136–154. 10.1161/CIRCRESAHA.121.318142 [DOI] [PubMed] [Google Scholar]
- 10.Iacobas I, Adams DM, Pimpalwar S, et al. Multidisciplinary guidelines for initial evaluation of complicated lymphatic anomalies-expert opinion consensus. Pediatr Blood Cancer. 2020;67(1):e28036. 10.1002/pbc.28036 [DOI] [PubMed] [Google Scholar]
- 11.Ricci KW, Iacobas I. How we approach the diagnosis and management of complex lymphatic anomalies. Pediatr Blood Cancer. 2021; 69(S3):e28985. 10.1002/pbc.28985 [DOI] [PubMed] [Google Scholar]
- 12.Gordon K, Varney R, Keeley V, et al. Update and audit of the St George’s classification algorithm of primary lymphatic anomalies: a clinical and molecular approach to diagnosis. J Med Genet. 2020; 57(10):653–659. 10.1136/jmedgenet-2019-106084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sparks TN, Thao K, Lianoglou BR, et al. Nonimmune hydrops fetalis: identifying the underlying genetic etiology. Genet Med. 2019;21(6): 1339–1344. 10.1038/s41436-018-0352-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mardy AH, Chetty SP, Norton ME, Sparks TN. A system-based approach to the genetic etiologies of non-immune hydrops fetalis. Prenat Diagn. 2019;39(9):732–750. 10.1002/pd.5479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mastromoro G, Guadagnolo D, Giancotti A, et al. Recurrent prenatal PIEZO1-related lymphatic dysplasia: expanding molecular and ultrasound findings. Eur J Med Genet. 2021;64(1):104106. 10.1016/j.ejmg.2020.104106 [DOI] [PubMed] [Google Scholar]
- 16.Society for Maternal-Fetal M, Norton ME, Chauhan SP, Dashe JS. Society for maternal-fetal medicine (SMFM) clinical guideline #7: nonimmune hydrops fetalis. Am J Obstet Gynecol. 2015;212(2):127–139. 10.1016/j.ajog.2014.12.018 [DOI] [PubMed] [Google Scholar]
- 17.Martin-Almedina S, Mortimer PS, Ostergaard P. Development and physiological functions of the lymphatic system: insights from human genetic studies of primary lymphedema. Physiol Rev. 2021;101(4):1809–1871. 10.1152/physrev.00006.2020 [DOI] [PubMed] [Google Scholar]
- 18.Daniel-Spiegel E, Ghalamkarpour A, Spiegel R, et al. Hydrops fetalis: an unusual prenatal presentation of hereditary congenital lymphedema. Prenat Diagn. 2005;25(11):1015–1018. 10.1002/pd.1237 [DOI] [PubMed] [Google Scholar]
- 19.Fotiou E, Martin-Almedina S, Simpson MA, et al. Novel mutations in PIEZO1 cause an autosomal recessive generalized lymphatic dysplasia with non-immune hydrops fetalis. Nat Commun. 2015;6(1):8085. 10.1038/ncomms9085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martin-Almedina S, Martinez-Corral I, Holdhus R, et al. EPHB4 kinase-inactivating mutations cause autosomal dominant lymphatic-related hydrops fetalis. J Clin Invest. 2016;126(8):3080–3088. 10.1172/JCI85794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma GC, Liu CS, Chang SP, et al. A recurrent ITGA9 missense mutation in human fetuses with severe chylothorax: possible correlation with poor response to fetal therapy. Prenat Diagn. 2008;28(11): 1057–1063. 10.1002/pd.2130 [DOI] [PubMed] [Google Scholar]
- 22.Mackie DI, Al Mutairi F, Davis RB, et al. hCALCRL mutation causes autosomal recessive nonimmune hydrops fetalis with lymphatic dysplasia. J Exp Med. 2018;215(9):2339–2353. 10.1084/jem.20180528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blesinger H, Kaulfuss S, Aung T, et al. PIK3CA mutations are specifically localized to lymphatic endothelial cells of lymphatic malformations. PLoS One. 2018;13(7):e0200343. 10.1371/journal.pone.0200343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rodriguez-Laguna L, Agra N, Ibanez K, et al. Somatic activating mutations in PIK3CA cause generalized lymphatic anomaly. J Exp Med. 2019;216(2):407–418. 10.1084/jem.20181353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Manevitz-Mendelson E, Leichner GS, Barel O, et al. Somatic NRAS mutation in patient with generalized lymphatic anomaly. Angiogenesis. 2018;21(2):287–298. 10.1007/s10456-018-9595-8 [DOI] [PubMed] [Google Scholar]
- 26.Homayun-Sepehr N, McCarter AL, Helaers R, et al. KRAS-driven model of Gorham-Stout disease effectively treated with trametinib. JCI Insight. 2021;6(15). 10.1172/jci.insight.149831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Norton ME, Ziffle JV, Lianoglou BR, Hodoglugil U, Devine WP, Sparks TN. Exome sequencing vs targeted gene panels for the evaluation of nonimmune hydrops fetalis. Am J Obstet Gynecol. 2022; 226(1):128.e1–128.e11. 10.1016/j.ajog.2021.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Petrovski S, Aggarwal V, Giordano JL, et al. Whole-exome sequencing in the evaluation of fetal structural anomalies: a prospective cohort study. Lancet. 2019;393(10173):758–767. 10.1016/S0140-6736(18)32042-7 [DOI] [PubMed] [Google Scholar]
- 29.Mellis R, Eberhardt RY, Hamilton SJ, et al. Fetal exome sequencing for isolated increased nuchal translucency: should we be doing it? BJOG. 2022;129(1):52–61. 10.1111/1471-0528.16869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sparks TN, Lianoglou BR, Adami RR, et al. Exome sequencing for prenatal diagnosis in nonimmune hydrops fetalis. N Engl J Med. 2020;383(18):1746–1756. 10.1056/NEJMoa2023643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ren Z, Povysil G, Hostyk JA, Cui H, Bhardwaj N, Goldstein DB. ATAV: a comprehensive platform for population-scale genomic analyses. BMC Bioinf. 2021;22(1):149. 10.1186/s12859-021-04071-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhu X, Petrovski S, Xie P, et al. Whole-exome sequencing in undiagnosed genetic diseases: interpreting 119 trios. Genet Med. 2015;17(10):774–781. 10.1038/gim.2014.191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Need AC, Shashi V, Hitomi Y, et al. Clinical application of exome sequencing in undiagnosed genetic conditions. J Med Genet. 2012;49(6):353–361. 10.1136/jmedgenet-2012-100819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lippa N, Bier L, Revah-Politi A, et al. Diagnostic sequencing to support genetically stratified medicine in a tertiary care setting. Genet Med. 2022;24(4):862–869. 10.1016/j.gim.2021.12.010 [DOI] [PubMed] [Google Scholar]
- 35.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5): 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rauen KA. The RASopathies. Annu Rev Genom Hum Genet. 2013;14(1):355–369. 10.1146/annurev-genom-091212-153523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jordan VK, Fregeau B, Ge X, et al. Genotype-phenotype correlations in individuals with pathogenic RERE variants. Hum Mutat. 2018;39(5):666–675. 10.1002/humu.23400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Michelini S, Ricci M, Veselenyiova D, et al. TIE1 as a candidate gene for lymphatic malformations with or without lymphedema. Int J Mol Sci. 2020;21(18):6780. 10.3390/ijms21186780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu M, Smith CL, Biko DM, et al. Genetics etiologies and genotype phenotype correlations in a cohort of individuals with central conducting lymphatic anomaly. Eur J Hum Genet. 2022;30(9):1022–1028. 10.1038/s41431-022-01123-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bawle EV, Black V. Nonimmune hydrops fetalis in Noonan’s syndrome. Am J Dis Child. 1986;140(8):758–760. 10.1001/archpedi.1986.02140220040028 [DOI] [PubMed] [Google Scholar]
- 41.Burrows PE, Gonzalez-Garay ML, Rasmussen JC, et al. Lymphatic abnormalities are associated with RASA1 gene mutations in mouse and man. Proc Natl Acad Sci U S A. 2013;110(21):8621–8626. 10.1073/pnas.1222722110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Revencu N, Boon LM, Mendola A, et al. RASA1 mutations and associated phenotypes in 68 families with capillary malformation-arteriovenous malformation. Hum Mutat. 2013;34(12):1632–1641. 10.1002/humu.22431 [DOI] [PubMed] [Google Scholar]
- 43.Wooderchak-Donahue WL, Johnson P, McDonald J, et al. Expanding the clinical and molecular findings in RASA1 capillary malformation-arteriovenous malformation. Eur J Hum Genet. 2018;26(10):1521–1536. 10.1038/s41431-018-0196-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gallipoli A, MacLean G, Walia JS, Sehgal A. Congenital chylothorax and hydrops fetalis: a novel neonatal presentation of RASA1 mutation. Pediatrics. 2021;147(3):e2020011601. 10.1542/peds.2020-011601 [DOI] [PubMed] [Google Scholar]
- 45.Datkhaeva I, Arboleda VA, Senaratne TN, et al. Identification of novel PIEZO1 variants using prenatal exome sequencing and correlation to ultrasound and autopsy findings of recurrent hydrops fetalis. Am J Med Genet A. 2018;176(12):2829–2834. 10.1002/ajmg.a.40533 [DOI] [PubMed] [Google Scholar]
- 46.Liu N, Gao M, Yu Z. Dysfunction of dermal initial lymphatics of the arm and upper body quadrant causes congenital arm lymphedema. J Vasc Surg Venous Lymphat Disord. 2021;9(2):482–488. 10.1016/j.jvsv.2020.06.009 [DOI] [PubMed] [Google Scholar]
- 47.Chen Y, Jiang Y, Chen B, et al. Case report: whole exome sequencing revealed two novel mutations of PIEZO1 implicated in nonimmune hydrops fetalis. Front Genet. 2021;12:684555. 10.3389/fgene.2021.684555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Martin-Almedina S, Mansour S, Ostergaard P. Human phenotypes caused by PIEZO1 mutations; one gene, two overlapping phenotypes? J Physiol. 2018;596(6):985–992. 10.1113/JP275718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vignes S, Kaltenbach S, Garcon L, et al. PIEZO1-gene gain-of-function mutations with lower limb lymphedema onset in an adult: clinical, scintigraphic, and noncontrast magnetic resonance lymphography findings. Am J Med Genet A. 2022;188(1):243–248. 10.1002/ajmg.a.62476 [DOI] [PubMed] [Google Scholar]
- 50.Picard V, Guitton C, Thuret I, et al. Clinical and biological features in PIEZO1-hereditary xerocytosis and Gardos channelopathy: a retrospective series of 126 patients. Haematologica. 2019;104(8):1554–1564. 10.3324/haematol.2018.205328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Andolfo I, De Rosa G, Errichiello E, et al. PIEZO1 hypomorphic variants in congenital lymphatic dysplasia cause shape and hydration alterations of red blood cells. Front Physiol. 2019;10:258. 10.3389/fphys.2019.00258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Maltese PE, Michelini S, Ricci M, et al. Increasing evidence of hereditary lymphedema caused by CELSR1 loss-of-function variants. Am J Med Genet A. 2019;179(9):1718–1724. 10.1002/ajmg.a.61269 [DOI] [PubMed] [Google Scholar]
- 53.Gonzalez-Garay ML, Aldrich MB, Rasmussen JC, et al. A novel mutation in CELSR1 is associated with hereditary lymphedema. Vasc Cell. 2016;8:1. 10.1186/s13221-016-0035-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Erickson RP, Lai LW, Mustacich DJ, Bernas MJ, Kuo PH, Witte MH. Sex-limited penetrance of lymphedema to females with CELSR1 haploinsufficiency: a second family. Clin Genet. 2019;96(5):478–482. 10.1111/cge.13622 [DOI] [PubMed] [Google Scholar]
- 55.Choi I, Lee S, Kyoung Chung H, et al. 9-cis retinoic acid promotes lymphangiogenesis and enhances lymphatic vessel regeneration: therapeutic implications of 9-cis retinoic acid for secondary lymphedema. Circulation. 2012;125(7):872–882. 10.1161/CIRCULATIONAHA.111.030296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dori Y, Smith C, Pinto E, et al. Severe lymphatic disorder resolved with MEK inhibition in a patient with noonan syndrome and SOS1 mutation. Pediatrics. 2020;146(6). 10.1542/peds.2020-0167 [DOI] [PubMed] [Google Scholar]
- 57.Handal-Orefice R, Midura D, Wu JK, Parravicini E, Miller RS, Shawber CJ. Propranolol therapy for congenital chylothorax. Pediatrics. 2023;151(2):e2022058555. 10.1542/peds.2022-058555 [DOI] [PubMed] [Google Scholar]
- 58.Wu JK, Hooper ED, Laifer-Narin SL, Simpson LL, Kandel J, Shawber CJ. Initial experience with propranolol treatment of lymphatic anomalies: a case series. Pediatrics. 2016;138(3):e20154545. 10.1542/peds.2015-4545 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The primary study submits all genotype and phenotype data to the database of Genotypes and Phenotypes (dbGaP) at the completion of the study as per agreement with the NIH. A copy of the data sharing certificate is available for documentation.