


Abstract
Lymphoid Enhancer Factor-1 (LEF-1) is a member of a family of transcription factors that function as downstream mediators of the Wnt signal transduction pathway. In the absence of Wnt signals, specific LEF/TCF isoforms repress rather than activate gene targets through recruitment of the co-repressor CtBP. Characterization of the full-length human LEF-1 gene locus and its complete set of mRNA products shows that this family member exists as a unique set of alternatively spliced isoforms; none are homologous to TCF-1E/TCF-4E. Therefore LEF-1 is distinct from its TCF family members in that it cannot engage in activities specific to this isoform such as recruitment of the co-repressor CtBP. Expression of alternatively spliced LEF-1 isoforms are driven by a promoter that is highly active in lymphocyte cell lines. Transcription initiates within a TATA-less core promoter region that contains consensus binding sites for Sp1, an E box, an Initiator element and a LEF/TCF binding site, all juxtaposed to the start sites of transcription. The promoter is most active in a B lymphocyte cell line (Raji) in which the endogenous LEF-1 gene is silent, suggesting that the promoter region is actively repressed by a silencing mechanism.
INTRODUCTION
The Lymphoid Enhancer Factor (LEF)/TCF family of High Mobility Group (HMG) box transcription factors function in a developmental signaling pathway driven by Wnt proteins, secreted ligands that direct cell fate, polarity and growth changes in receptive cells. Wnt ligand recognition by transmembrane receptors at the cell surface initiates a signaling cascade that stabilizes the normally labile armadillo repeat protein β-catenin and re-directs it to the nucleus. In the nucleus, any of the four mammalian LEF/TCF proteins can bind tightly to Wnt-triggered nuclear β-catenin protein and through their single HMG DNA binding domains, tether the potent transcription activating domain in β-catenin to Wnt target genes. In the absence of Wnt signaling, some of the LEF/TCF factors have been shown to engage in active gene silencing through recruitment of co-repressors (1–4). Much of the current work is focused on the function of specific LEF/TCF proteins in Wnt signaling, but little is directed towards understanding how their full complement of alternative isoforms and their expression patterns influence signaling.
During embryogenesis Wnt signaling occurs at myriad sites of tissue differentiation and this is reflected in the pattern of expression of LEF/TCF proteins. Embryonic expression patterns for each of the four known mammalian LEF/TCF proteins (LEF-1, TCF-1, TCF-3 and TCF-4) show distinct but broad, overlapping distributions (5–8). Each of the LEF/TCF factors have been genetically inactivated in mice, and the phenotypes that result from these knock-outs are unique for each factor (8–11). Only a subset of the tissues that express an embryonic LEF/TCF are missing or damaged in the knock-out mice, suggesting a certain level of expression and functional redundancy among LEF/TCF proteins. Redundancy has been shown experimentally in LEF-1/TCF-1 double knock-out mice which die during embryogenesis with multiple, fatal deficiencies (12). However, LEF/TCF proteins are not entirely redundant. For example, both LEF-1 and TCF-1 are expressed to high levels in developing T lymphocytes in the thymus, but only TCF-1 knock-out mice have defects in T cell differentiation (10). Recently, transcription co-repressors have been shown to bind to TCF-1 and Xenopus TCF-3 and silence transcription in the absence of Wnt signaling (2–4). One of these co-repressors, CtBP, binds to the C-terminal tail of xTCF-3, a region that is encoded by alternative splicing and is most homologous to the C-terminus of TCF-1E and TCF-4E (although the CtBP binding site is present in TCF-4E and not TCF-1E) (2). We show in this manuscript that although this C-terminal ‘E’ region has been found to be a common gene product for all of the TCF genes, the LEF-1 gene is unique in that it cannot encode a similar sequence. Thus, the genetic locus for each family member may encode a unique subset of isoforms that are required at specific sites of differentiation.
Hours after birth, the broad patterns of LEF/TCF expression disappear and only very restricted patterns of LEF/TCF mRNA can be detected by northern analysis of mouse tissues (5). For example, both LEF-1 and TCF-1 mRNA are easily detected in the thymus, but very little mRNA can be detected in any other tissue (13–15). It is important to note that this type of northern data can be misleading; expression of LEF/TCFs has been detected by RT–PCR or in situ hybridization of tissues that appear negative by northern analysis. Such tissues include skin, hair follicles, intestine, colon and testes. These tissues are continually replenished by waves of differentiating cells that derive from a small population of mitotically active stem cells. With the exception of the thymus, which is loaded with mitotically active differentiating lymphocytes, stem cells comprise a small fraction of each tissue and are not easily detected by northern analysis. LEF/TCF expression has been detected in some of these small populations. The apparent unifying pattern of LEF-1 expression is that expression is silenced or dramatically down-regulated when cells reach a non-cycling, differentiated state. The best examples are from studies of differentiating B and T lymphocytes and cells at the base of hair follicles. In these tissues, LEF-1 expression can no longer be detected in mature B lymphocytes or in keratin secreting cells of the hair shaft, and only at very low levels in mature, immunocompetent T lymphocytes (7,14,16,17).
In contrast to these low levels of expression, moderate to high levels of LEF-1 are frequently detected in tumors, even in cancerous cells from tissues that are normally negative for LEF-1 expression (18; K.Hovanes and M.L.Waterman, unpublished observations). Either these cancers derive from a tiny fraction of cells that normally express LEF-1, or its expression is activated in the transformation process. Such is the case for LEF-1 expression in transformed cell lines derived from mature T and B lymphocytes, cells that normally do not express much LEF-1 mRNA. LEF-1 expression has also been detected frequently in melanomas and colon cancer (18,19). A frequent problem in colon cancer and melanomas involves ectopic and constitutive activation of the Wnt pathway due to genetic mutations that lead to a de-regulation of β-catenin. Thus, β-catenin becomes an abundant and constitutively available co-activator in the nucleus and if a LEF/TCF factor is present, inappropriate activation of gene targets and cell transformation is possible. Defining the mechanisms that drive this aberrant LEF-1 expression is important for understanding how the cancers initiate and how they progress to malignancy.
Here we describe the characterization of the LEF-1 gene, its multiple isoforms, its promoter and its preferential activity in lymphocytes. We find that the structure of the human LEF-1 gene shows remarkable conservation with the human TCF-1 gene, but that a few striking differences set it apart from its family members.
MATERIALS AND METHODS
Cell lines and culture conditions
Jurkat and Raji cells were grown in RPMI-1640 with 10% fetal bovine serum, 50 µM 2-mercaptoethanol and antibiotics. HeLa and Cos-7 cells were grown in Dulbecco’s modified Eagles medium (DMEM) with 10% fetal bovine serum and antibiotics. 293 cells were grown in MEM with 15% fetal bovine serum and antibiotics. PC12 cells were grown in DMEM with 10% heat-inactivated horse serum, 5% fetal bovine serum and antibiotics.
cDNA and genomic library screens
A LEF-1 open reading frame probe was used to screen 1 × 106 clones from a human fetal brain 5′-stretch plus cDNA library (Clontech, Palo Alto, CA). cDNA inserts were subcloned by insertion into the EcoRI site of Bluescript KS+ (Stratagene, La Jolla, CA). A probe derived from the 5′ untranslated region (5′-UTR) of the LEF-1 cDNA was used to screen a human testis 5′-stretch plus cDNA library; inserts were subcloned as above. PCR primers complementary to sequences in the first exon of LEF-1 were used to identify LEF-1 genomic sequences in an ordered array of pooled genomic PAC clones (20). One clone, PAC41F1, and smaller portions containing the 5′ half of the LEF-1 gene, were isolated by shotgun cloning of Spe1 fragments into the Spe1 site of Bluescript KS+. Clones containing the 5′-UTR were identified by colony hybridization using a probe derived from 5′-UTR of LEF-1 cDNA. A positive clone (5Ba) was identified and the 2881-bp insert was sequenced.
Reporter plasmid constructs and transient transfection assays
Reporter plasmids. To subclone portions of the genomic 5Ba clone for promoter analysis, fragments were excised with SacI (a site in the polylinker) and the following enzymes: with SacI at +305, Xho at +262, PpuMI at +78, StyI at –64 or AlwNI at –160. Fragments were subcloned as blunt-ended fragments in both orientations into pGL2-Basic and pGL2-Enhancer vector (Promega, Madison, WI) at the SmaI site. To create deletions, 5Ba was digested with AlwNI at –160, StyI at –64, PpuMI at +78 and XhoI at +262.
Transient transfection assays. Jurkat and Raji cells were transfected with 10 µg of the test plasmid and 0.5 µg of CMV-βgal plasmid using a BTX 600 electroporator set at 72 Ω, 2000 µF and 160 V (10 × 106 cells in 0.2 mm gap cuvettes, 200 µl serum free RPMI). COS cells were transfected using electroporator settings of 48 Ω, 1000 µF and 150 V (8 × 105 cells in 0.2 mm gap cuvettes, 400 µl of serum free DMEM). 293 cells were transfected using electroporator settings of 72 Ω, 800 µF and 190 V (5 × 106 cells in 0.2 mm gap cuvettes, 250 µl). HeLa cells (4 × 105 cells plated on 10 cm plates 24 h prior to transfection) were transfected using calcium phosphate, 20 µg of test plasmid and 2 µg of CMV-βgal. PC12 cells were transfected at 50% confluency with 7 µl of Lipofectin reagent (Gibco BRL, Gaithersburg, MD), 2 µg of reporter plasmid and 3 µg of TKβgal in 600 µl of OPTI-MEM I medium (pre-incubated at room temperature for 15 min). Luciferase activity was normalized with the β-galactosidase activity for each point.
RNase protection assay
Antisense probes A, B and C were prepared by T7 run-off transcription of human LEF-1 genomic clones containing the promoter and part of exon 1 (SpeI to EagI for A and B; SpeI to PpuMI for probe C) in Bluescript KS+ linearized with either EcoRI (probe A, probe C) or StyI (probe B). Run-off transcription was performed on 0.5 µg DNA, with 2 U of T7 RNA polymerase (Promega), [α-32P]UTP (4 µl, 800 Ci/mmol), 20 mM DTT, 1 mM rATP/rGTP/rCTP, 20 µM UTP/RNasin (2 U/µl, Promega). Reactions were stopped with RQ1 DNase [5 U (Promega); 37°C, 20 min] and phenol/chloroform extraction/ethanol precipitation. Labeled RNAs were resuspended in 100 µl of RNA hybridization solution: 1 M NaCl, 5 mM EDTA, 200 mM PIPES pH 6.4, 80% formamide. For each hybridization reaction (30 µl), 800 000 c.p.m. of labeled RNA was hybridized to 1 µg of polyA+ Jurkat or HeLa RNA at 85°C for 7 min and then 45°C for 14 h. Reactions were brought to 350 µl with 10 mM Tris pH 7.5, 300 mM NaCl, 5 mM EDTA and digested with RNaseA (25 µg/ml) and RNase T1 (0.01 U/µl) for 45 min at 30°C. Digestion was stopped with 10 µl 20% SDS and 10 µl 10 mg/ml Proteinase K at 37°C for 45 min followed by phenol/chloroform extraction and ethanol precipitation with 0.17 µg/ml glycogen. Samples were resuspended in 5 µl formamide loading buffer and analyzed on a 6% denaturing polyacrylamide gel. RNA markers were made by T7 run-off transcription of Bluescript KS+ linearized with XbaI (39 bp) or XhoI (104 bp). The 184-bp marker is undigested, full-length probe C.
DNase I footprint analysis
Crude whole cell extracts from Jurkat and HeLa cells or partially purified recombinant full-length LEF-1 protein were incubated with double-stranded DNA probes (5–15 fmol per reaction; single end-labeled on the 5′ end with [γ-32P]ATP) for 20 min on ice in a 50 µl reaction containing TM 0.05 M (50 mM Tris, pH 7.9, 12.5 mM MgCl2, 1 mM EDTA, 20% glycerol, 0.1% NP-40, 50 mM KCl). DNase I work-up procedures are as described (21). Preparations of crude whole cell extracts and recombinant LEF-1 protein have been previously described (13,22).
RESULTS
Genomic organization of the human LEF-1 gene
To identify the LEF-1 gene and define its complete set of alternatively spliced products, a total human genomic PAC library and three human cDNA libraries (testis, melanoma, human fetal brain) were screened using fragments of the human LEF-1 cDNA. The longest cDNA that had been previously isolated was 3.0 kb (13). However, northern analysis shows that the most abundant LEF-1 mRNA in cell lines is ∼3.6 kb. Using probes derived from the furthest 5′ portion of existing LEF-1 cDNAs to screen a human fetal brain cDNA library, an additional 600 nt stretch of 5′-UTR was identified. This 5′-UTR is highly GC-rich, difficult to sequence and therefore underrepresented in most cDNA libraries. The nucleotide sequence is not highly conserved between mouse and human LEF-1 because probes from this region do not detect mouse LEF-1 mRNA by northern analysis (K.Hovanes and M.L.Waterman, data not shown). As will be presented below, determining the start site of transcription established that the total length of the 5′-UTR is 1.2 kb, a length that is highly unusual for a eukaryotic message (see exon 1 in Fig. 1).
Figure 1.

Comparison of the genomic organization of the human LEF-1 gene with other LEF/TCF family members. A color diagram at the top summarizes the general domain structure of LEF/TCF proteins. For the genes, solid lines represent introns that are complete. Very large introns are indicated by double slashes, and introns that are not completely sequenced are shown by dashes. Alternative splicing patterns that generate all known human LEF-1, TCF-1, TCF-4 and Drosophila pangolin/dTCF isoforms are shown. Dashed splicing lines to exon 3a and 3b in the LEF-1 gene indicate that this intron and the exon structure are not completed. The most common alternatively spliced products are indicated by bold lines. Exon numbering for the LEF-1 and pop-1 gene are presented, numbering for human TCF-1 and pan are as previously established (24,28). The following genomic sequences were used: AC01095 (pangolin/dTCF); AF043703 (POP-1); AC011336 (TCF-1); AC008112 (TCF-4); AC000016 (LEF-1); AC21524 (LEF-1); AC004059 (LEF-1; no longer present in the database).
Using probes from the 5′ and 3′ regions of the larger, more complete cDNA, an ordered human genomic PAC library was screened. Three large PAC clones containing overlapping portions of the entire LEF-1 gene were identified. Southern analysis with probes from the 5′ and 3′ portions of the human cDNA was used to order the PACs. One PAC clone, PAC41F1, hybridized to 5′-UTR probes but not 3′-UTR probes and was chosen for mapping and sequence analysis as it was likely to contain the 5′ end and promoter of the LEF-1 gene. One Spe1 subclone was determined to contain the first exon and part of intron 1 (5Ba), and a second Spe1 subclone encoded the remainder of intron 1 through a portion of intron 3 (S2a). Recent genomic sequence information from human chromosome 4, which contained the LEF-1 locus starting within intron 3 was provided by the Stanford Human Genome Center (GenBank accession nos AC000016, AC21524). Combining this new sequence with our own sequence and mapping information of PAC41F1, a complete exon/intron structure of the LEF-1 gene was determined (Fig. 1). With the exception of introns 3 and 8, complete sequences of all exons and introns are known. The LEF-1 gene spans at least 52 kb and the positions of the exon/intron boundaries are shown in Figure 1. More recently, partial human genomic sequence from chromosome 17 covering the 3′ half of the human TCF-4 gene, and the complete genomic sequence of human TCF-1 on chromosome 5 became available (GenBank accession nos AC008112, AC011336 respectively). Likewise, the genomic organization of the nematode pop-1 gene (a LEF/TCF factor in Caenorhabditis elegans) was inferred by aligning its coding sequence with newly available genome sequence at the Sanger Web site (accession no. W10C8). The genomic organization for LEF-1, TCF-4 and pop-1 are presented in Figure 1 alongside the previously characterized genomic organization for human TCF-1 and the pan gene from Drosophila melanogaster. Also shown in Figure 1 is a schematic of the domain structure of LEF/TCF proteins from humans to nematodes. The most highly conserved domain is the HMG DNA binding/bending domain near the C-terminus. It is encoded by three exons in the human genes (orange and red exons). One interesting feature of the Drosophila pan gene is the fusion of the nuclear localization signal (NLS) to the 3′ half of the HMG box exon (Fig. 1, exon XIa) and the existence of a second, alternative version of this exon (exon XIb). These alternatively spliced products would produce isoforms that differ in the 3′ half of the HMG DNA binding domain, a change that is almost certain to alter DNA binding or bending characteristics (23). The intron sequences surrounding these exons are known for all the other LEF/TCFs shown in Figure 1. A search of these sequences for alternative HMG box coding sequences shows that neither the pop-1 gene nor any of the human LEF/TCF genes have the capacity to generate alternative DNA binding domains by splicing to another HMG exon. Thus this interesting feature is restricted so far to the pan gene only.
The next most highly conserved domain in LEF/TCF proteins is the β-catenin binding domain at the extreme N-terminus. All of the genes shown in Figure 1 encode this domain in a single exon, but for LEF-1, the β-catenin binding domain is encoded by a remarkably large exon that also includes the entire 1.2-kb 5′-UTR sequence (Fig. 1). Long 5′-UTRs are likely to preclude normal translation mechanisms that involve ribosome scanning. The entire 1.2 kb is highly GC-rich and difficult to sequence, further suggesting that scanning ribosomes would have trouble reaching the correct initiator methionine. We propose that the presence of a long, GC-rich UTR in LEF-1 must impose some form of post-transcriptional regulation to direct proper translation of LEF-1 coding sequences.
The last exon for each of the LEF/TCF genes is large and encodes some of the final residues in the alternative C-termini (purple exon, Fig. 1). Until now, only the TCF-1 and TCF-4 genes have been shown to generate alternative isoforms at the C-terminus (24,25). To generate these alternative C-termini, both TCF-1 and TCF-4 genes use alternative exons and different splice-acceptor sites in the common, final exon (designated XI.1–XI.4 for TCF-1 in Fig. 1). Each of these tails has been given a different alphabetic designation to distinguish them. At least for TCF-1 and TCF-4, the predominant forms contain a ‘B tail’ or an ‘E tail’ (indicated with bold lines for the splicing pattern, Fig. 1). Pan/dTCF, POP-1 and Xenopus, mouse and zebrafish TCF-3 so far are only known as proteins that contain an E tail (2,23,26,27). We have identified a LEF-1 cDNA from a human fetal brain library encoding a C-terminus with sequence similarity to the B tail. Exclusion of exon 11 is used to generate LEF-1B, but there is no example of alternative splice acceptor choice within the final exon 12 as there is for TCF-1 and TCF-4. Most cDNAs analyzed in this region encode a C-terminal domain designated LEF-1N, generated by including exon 11 and splicing directly to exon 12. We propose that the ‘B’ C-terminus is a conserved feature of the entire human LEF/TCF family. In contrast, the ‘E’ C-terminus, which is also common among most LEF/TCF proteins is not encoded by the LEF-1 gene (see below).
Additional LEF-1 cDNAs isolated from human testis and fetal brain libraries confirmed the previously identified alternative exon 6 and new alternative splices to exons in the third intron (Fig. 1, 3a and 3b). One isoform (3b) was identified as a cDNA clone from our human cDNA testis library, and the second, a matching EST clone from testis tissue (GenBank accession no. AI141511). Both cDNA clones contain exon 3 sequences followed by unique sequence. Intron 3 has not yet been entirely sequenced except for a region immediately following exon 3 and 8 kb preceding exon 4. Since exon 3a and 3b are not encoded in this partial intron sequence, the arrangment of these exons in the central portion of the intron is not known.
Conservation of exon/intron boundaries and sequences of alternatively spliced products
Positions of exon/intron boundaries are indicated within an amino acid alignment of human LEF-1, TCF-1, TCF-4 and D.melanogaster pangolin/dTCF (Fig. 2A) (28). The exon/intron boundaries surrounding the HMG DNA binding/bending domain (exons 8–10; double and triple underline, Fig. 2A) are precisely conserved as are the exon/intron boundaries for the β-catenin binding domain. The exons between the β-catenin binding domain and the HMG DNA binding domain exhibit much more variability, and this coincides with reduced amino acid sequence similarity. Amino acid alignments of C-terminal E tails are shown in Figure 2B. The first part of the E tail contains two regions enriched in basic and cysteine residues and is highly conserved from human to nematode (including Xenopus laevis TCF-3). A variant mouse TCF-4E protein (GenBank accession no. AF107299) carries a duplication of the first of these conserved motifs immediately preceding the beginning of the alignment shown in Figure 2B (25). The functional importance of this lys-, arg- and cys-rich motif is not yet known. Following this region, the C-terminal tails diverge in sequence and in length where the co-repressor CtBP is known to bind TCF-4 (QPLSLS, QPLSV) (2). A sequence similar to the CtBP binding motif (NPLSI) can be found in pangolin/dTCF. Since the intron sequences are known for the LEF-1 gene in this region, a search was undertaken to determine if an alternative exon was present to code for sequences that match the conserved regions in the E tail and the CtBP binding motif. No such coding sequences were identified in any of the introns; we conclude that another major difference, and so far a unique difference for LEF-1, is the absence of coding sequences for an E tail. Thus, expression of the LEF-1 locus does not produce a protein product capable of CtBP recruitment. In fact, whether any LEF-1 gene product can repress gene transcription has not been shown conclusively.
Figure 2.


Amino acid sequence and exon/intron boundary comparisons of conserved domains in human LEF-1, TCF-1, TCF-4 and Drosophila pan/dTCF protein. (A) Single, double and triple underlines mark the β-catenin binding domain, the HMG box and NLS in the HMG DNA binding domain respectively. Exon numbering corresponds to the LEF-1 gene, and intron positions are indicated by black triangles. The asterisk above the intron marker denotes alternative exons (shown below). (B) Amino acid alignment of the common E tails of LEF/TCF family members; the human LEF-1 gene does not encode an E tail. Three conserved motifs are boxed with the most highly conserved residues in bold and cysteines underlined. (C) Amino acid alignment of the known B tails. Conserved residues are highlighted in bold. (D) The most common C-terminal sequence of LEF-1 protein is designated as LEF-1N and an alignment with LEF-1 orthologs is shown. (E) The alternative exon(s) in intron 3 are shown with an alignment of translated sequence from a testis cDNA (this study, double underline) and an EST cDNA (dashed underline). The EST clone contains the unique sequence marked exon 3a, and both clones contain the sequences marked as exon 3b. Whether the LEF-1 testis cDNA is produced by exon skipping of 3a and splicing from exon 2 to 3b, or whether it is produced by using an internal splice acceptor is not yet known. The predicted amino acid sequences of the two clones are shown (EST sequence italicized); both would produce truncated LEF-1 protein products of 16 and 18 kDa, respectively.
Exclusion of exon 11 generates a LEF-1 isoform with a C-terminal sequence homologous to the B tails of TCF-1B and TCF-4B. An alignment of the B tails among the human LEF/TCFs is shown in Figure 2C. All three genes generate B tails by exon skipping and use of splice acceptor sequences in the final exon of the locus. So far, neither the Drosophila pan gene nor the C.elegans pop-1 gene has been shown to encode C-termini with this sequence; however, it may be that this isoform is as rare as it is for LEF-1. The most common C-terminal sequence for LEF-1 is shown in Figure 2D as LEF-1N. LEF-1 orthologs have been identified in mouse, X.laevis, Danio Reo and Gallus gallus; all of these orthologs encode a C-terminal region highly homologous to the ‘N’ tail of LEF-1.
Translation of the alternatively spliced sequences in the third intron (exons 3a, 3b) is also given (Fig. 2E). The unique sequence of our testis cDNA clone begins 30 nt into the EST AI141511 sequence (double-line and dashed lines, respectively), followed by a 142 nt sequence identical in both. The EST clone is a partial clone and ends within the stretch of unique sequence, while the testis cDNA clone continues for an additional 17 nt followed by exon 4 sequences to the 3′-UTR (Fig. 2E). None of the unique sequence matches the exon/intron junctions of exon 3 or exon 4. Therefore, they are not cDNA clones of partially processed LEF-1 mRNA. The 30 nt of unique sequence missing from the testis cDNA could either arise from use of a more 5′ splice acceptor sequence, or inclusion of an additional exon (referred to here as 3a). Although a function specific to these newly identified isoforms is not yet apparent, these alternative exons encode in-frame translation stop codons at different positions and would produce 16 and 18 kDa LEF-1 isoforms lacking the HMG DNA binding domain and NLS. As such, these proteins would be cytoplasmic and capable of interacting with β-catenin. Interestingly, the TCF-1 locus also encodes an alternative exon (Fig. 1, IIa) that provides an in-frame stop codon to produce a 16 kDa protein lacking DNA binding properties (24).
Identification of the LEF-1 promoter
The relative distribution and expression pattern of each of the alternatively spliced isoforms is not yet known and will require development of specific probes for detection. However each of them is likely to be encoded by similar sized mRNAs and to be initiated within an upstream promoter. To define the promoter, RNA probes derived from the genomic PAC41F1 clone were used in an RNase protection experiment. The assay identified four major protected fragments indicating multiple start sites of transcription (Fig. 3A). A primer extension assay confirmed these start sites, however extension products from the assay were at very low levels. Such difficulties of detection are most likely due to the inability of reverse transcriptase to extend through this region except under special conditions using high temperature and DMSO (K.Hovanes and M.L.Waterman, data not shown). The first start site falls within an Initiator-like consensus sequence (compare TCAGCG to TCAnYY) while the three tightly clustered start sites originate 19–25 nt downstream in a purine-rich region (Fig. 3B) (29). There are additional minor protected products that may represent weak start sites of transcription (Fig. 3A, probe C). However these minor products are not consistently seen with probes A and B and therefore they are not shown in Figure 3B. For simplicity, numbering will be based on the first, most 5′ start site. Thus the longest cDNA isolated begins at +27 and the first exon is 1401 nt in length with the first +1185 nt encoding a long 5′-UTR.
Figure 3.

Identification of transcription start sites of the human LEF-1 promoter. (A) RNase protection analysis of polyA+ RNA from LEF-1 expressing (Jurkat cells) and non-expressing (HeLa cells) were used to identify the 5′ end of endogenous LEF-1 mRNA. Uniformly labeled antisense probes A, B and C were generated by T7 transcription of a LEF-1 genomic clone (indicated schematically below) in the presence of [32P]UTP. Major products protected from RNase digestion after hybridization of the probes to the polyA+ RNA are indicated by black arrows. Size markers are [32P]UTP-labeled RNAs generated by T7 transcription of Bluescript KS+ (39, 104 nt) and undigested probe C (184 nt). (B) The genomic sequence surrounding the determined start sites designated as +1, +19, +21 and +25 (arrows). The longest cDNA identified from a human fetal brain cDNA library begins at the nucleotide shown with an open bar. Matches to binding sequences for transcription factor Sp1 and E box binding helix–loop–helix proteins are indicated in bold. Potential binding sites for GAGA factors are indicated in bold and boxed. The putative Initiator element surrounding the +1 start site is in bold. Regions of >20 nt with 90% purine or pyrimidine are underlined. Two regions protected in footprinting assays with Jurkat and HeLa whole cell extracts and recombinant full length human LEF-1 protein and Sp1 protein are designated by solid and dashed brackets, respectively (see Fig. 5).
The sequence surrounding the start site of transcription does not show any match to a TATA box or other basal element for RNA polymerase II transcription except for the Initiator-like sequence. Therefore to test whether bona fide start sites of transcription had been detected, fragments surrounding this region were tested by subcloning into a promoter-less reporter plasmid containing the luciferase gene and the SV40 enhancer (pGL2-Enhancer; Promega). These constructs were transiently transfected in Jurkat T lymphocytes (Fig. 4A). The largest fragment (–672, +305) exhibited an ∼15-fold increase in luciferase expression above the promoter-less vector, whereas the fragment inserted in the opposite orientation produced a 2-fold increase in luciferase activity above the empty vector control. These data suggest that a region within the 978-bp fragment is capable of directing transcription initiation in the proper orientation. To define the 3′ border of this fragment, several deletions were tested by removing sequences to +262, to +78 and then to –160 (Fig. 4A). Sequences between +262 and +78 have a positive effect on transcription but are not absolutely required. Removing the region containing the known start sites of transcription destroys promoter activity as there is no difference in luciferase expression between forward and reverse orientations of the inserted fragment. To define a minimal promoter region, the 5′ portion of the genomic fragment was truncated to –160 and then to –64 with varying amounts of sequence 3′ of the transcription start site (Fig. 4B). Sequences upstream of –160 have a slightly negative effect on transcription because transcription doubles to 37-fold activity above background in their absence (compare 15-fold expression for –672/+262 with 37-fold expression for –160/+262). Further deletion to –64 drops activity to 13-fold above background. However, if the promoter is truncated to +78, deletion from –160 to –64 has essentially no effect (compare –160/+78 and –64/+78, Fig. 4B). Likewise, a drop in transcription is observed when the promoter is truncated from +262 to +78, but only if sequences between –160 and –64 are present (compare –160/+262 and –160/+78 with –64/+262 and –64/+78; Fig. 4B). Perhaps there are co-operative interactions between factors binding upstream and downstream of the transcription start sites. Taken together, the data suggest that the minimal promoter region lies between –64 and +78. The sequence of this minimal region and additional upstream sequence is shown with the positions of the start sites and longest cDNA indicated (Fig. 3B; GenBank accession no. AF203908).
Figure 4.


Deletion analysis of the human LEF-1 promoter. (A) The 3′ end of the promoter was defined by analysis of each deletion fragment in the pGL2-enhancer luciferase reporter vector (Promega) in transiently transfected Jurkat T lymphocytes. Luciferase activity is shown as fold expression above empty, promoter-less pGL2-enhancer vector. Error bars reflect variation among duplicates; the entire experiment has been repeated four times. (B) The 5′ end of the minimal promoter was defined by deletion to the sites shown relative to the +1 start site, and analysis of the fragments in Jurkat cells.
A striking feature of the nucleotide sequence in the promoter is four long stretches of >90% purines or pyrimidines (underlined, Fig. 3B). A second interesting feature is the lack of a match to sequences encoding a TATA box but the presence of an initiator-like element surrounding the +1 start site. This sequence (TCAgCG) shows similarities to initiator sequences (TCAnYY) (29). There are also matches to three known DNA binding sites. The first to note is an E-box binding sequence (CAnnTG) 12 nt 5′ of +1. E box sequences are binding sites for helix–loop–helix proteins such as the E12/E47 proteins that are important for B lymphocyte differentiation (30,31). There is an Sp1 site 44 nt 5′ of the start site (bold, underlined, Fig. 3B). DNase I footprint analysis with purified Sp1 protein confirms that Sp1 is capable of recognizing this sequence (Fig. 5B). A similar pattern of protection over the Sp1 sequence can be observed with nuclear extracts from Jurkat and HeLa cells (Fig. 5A and B). Finally, there are at least four GAGAG sequences surrounding the transcription start site. GAGAG sequences serve as binding sites for the GAGA factor, a DNA binding protein well characterized in Drosophila (32,33). Drosophila GAGA factor interacts with target sequences that contain multiple GAGAG elements which increases DNA binding affinity and DNA bending. This multiply-bent DNA has been proposed to adopt a nucleosome-like structure, and GAGA bound regions are generally associated with open chromatin structures. However, as yet, a vertebrate ortholog of GAGA has not been identified.
Figure 5.
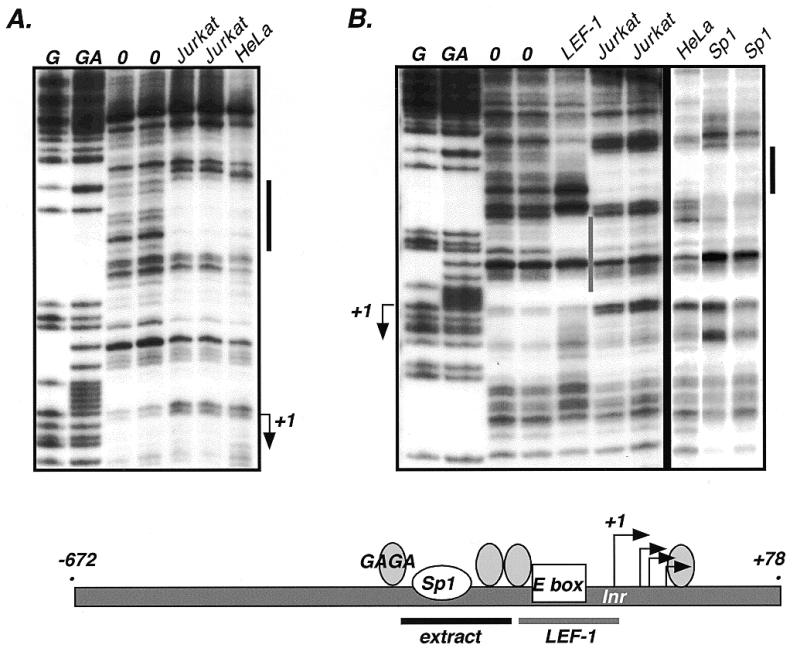
DNase I footprint analysis of the LEF-1 promoter. Whole cell extracts of Jurkat and HeLa cells and recombinant human LEF-1 protein and Sp1 protein were used in the analysis. The start sites of transcription are indicated by +1 and arrows. Maxam/Gilbert reactions (G, GA) were used to map the protected regions (indicated by solid bars).
LEF-1 binds to the minimal promoter
Human TCF-4 and β-catenin have recently been shown to activate TCF-1 expression through two LEF/TCF binding sites in the TCF-1 promoter (34). To survey the LEF-1 promoter region for LEF/TCF binding sites, a DNase I footprint assay was performed with recombinant LEF-1 protein and compared to footprint patterns with whole cell extracts from Jurkat and HeLa cells (Fig. 5A). All LEF/TCF family members recognize identical binding sites (YCTTTGWW) due to their highly conserved HMG DNA binding domains; therefore, use of recombinant LEF-1 should identify sites able to be recognized by any LEF/TCF family member (35,36). In addition, nuclear extracts from Jurkat cells (which express LEF-1) and HeLa cells (which do not express LEF-1) were used for comparison to determine whether cell-type differences in proteins binding to the promoter could be observed. As shown in Figure 5B, only one footprint, common to both the Jurkat and HeLa extracts and coinciding with the Sp1 footprint, was observed in the minimal promoter region. Interestingly, recombinant LEF-1 protein protected a sequence immediately 5′ to the +1 transcription start site (Fig. 5B). Within this footprint there is a weak match to the consensus LEF/TCF binding sequence. LEF-1 and TCF-1 are highly expressed in Jurkat cells and are capable of footprinting high affinity LEF/TCF sites from within a complex nuclear extract. If this site represented a high affinity LEF/TCF recognition site, a footprint over this region with the Jurkat extract should have been observed, and it was not. Co-transfection of a LEF-1 expression plasmid with or without a β-catenin expression plasmid did not alter activity of the core LEF-1 promoter in a reporter assay (–64, +78). While these observations suggest that this site may not be a functional LEF-1 site, we know that LEF-1 is a context-dependent transcription factor and now even LEF/β-catenin has been shown to be context dependent (37). In addition, known regulatory LEF/TCF binding sites include sequences that diverge significantly from the consensus binding sequence (38,39). It is possible that our transfections are not replicating the right context, either because additional required factors are missing in our cell lines or because the promoter needs a chromatin context, or the LEF-1 site is there to receive regulatory information from distal elements such as enhancers or silencers that are missing in our constructs. Experiments are currently underway to test these possibilities.
The LEF-1 promoter is preferentially active in lymphocyte cell lines
LEF-1 gene expression is best characterized in B and T lymphocyte cell lines which represent different stages of differentiation. Most immature and mature T lymphocyte cell lines exhibit high levels of LEF-1 gene expression, whereas in normal, non-transformed mature T lymphocytes there are only low levels of LEF-1 gene expression (10,13,14). In contrast, LEF-1 is highly expressed in immature stages of normal B lymphocytes and in B lymphocyte cell lines with an immature or pre-B cell phenotype, but LEF-1 mRNA is dramatically and rapidly down-regulated in both normal, mature B cells and transformed cells with a mature phenotype (14). The mechanism for this dramatic shut-off of expression is not known, but it occurs rapidly within hours, suggesting that an activator of LEF-1 gene expression is itself down-regulated, or a transcription silencing mechanism is activated. Even though no cell type-specific footprints (versus HeLa) could be observed within the promoter region, we wished to test whether the LEF-1 promoter was preferentially active in lymphocyte lines versus heterologous cell types. If so, we wished to test whether promoter activity was much lower in a mature B lymphocyte line (Raji) in which expression of endogenous LEF-1 gene expression was not detectable. A dramatic difference in promoter activity between Jurkat and Raji cells would suggest the loss of an activator. Using the largest LEF-1 promoter fragment (–672, +305) in a luciferase reporter plasmid that did not have any heterologous enhancer sequences (pGL2-Basic, Promega), we performed a survey of reporter gene expression activities in Jurkat and Raji cells as well as in two cell lines that express low levels of endogenous LEF-1 mRNA (293 human embryonic kidney and PC-12 human pheochromocytoma) and two cell lines that do not express endogenous LEF-1 mRNA (COS-7 monkey kidney and HeLa human cervical carcinoma) (Fig. 6). To enable comparisons across cell lines with varying transfection efficiencies, each promoter construct was tested in the forward and reverse orientations and data from each were normalized to the baseline activity of the promoterless vector. The results show dramatic differences in promoter activity in the different cell lines. Very high levels of promoter activity were observed in both Jurkat and Raji cells (82- and 133-fold over empty vector, respectively). Lower levels of promoter activity were observed in 293 and PC-12 cells, and minimal activity was observed in HeLa and COS-7 cells. For the most part, the activity of the isolated promoter fragment mimics the expression pattern of the endogenous LEF-1 gene with the striking exception of Raji cells. These results suggest that transcription factors exist in lymphocyte cell lines that enable high levels of transcription initiation from this promoter fragment and that other elements not present in the promoter fragment tested must direct the dramatic and rapid shut-down of LEF-1 gene expression in mature B lymphocytes.
Figure 6.

The LEF-1 promoter is preferentially active in lymphocyte cell lines. A LEF-1 promoter fragment (–672, +305), was subcloned into a pGL2-basic vector (no enhancer) in forward and reverse orientations. Reporter gene expression was assayed by transient transfection in Jurkat (human mature T lymphocyte), Raji (human mature B lymphocyte), HeLa (human cervical carcinoma), COS-7 (monkey kidney), 293 (human embryonic kidney) and PC-12 (human pheochromocytoma) cells. Reporter gene activity is given as fold expression relative to empty pGL2-basic vector.
DISCUSSION
Regulation of target gene expression by Wnt signals requires the presence of LEF/TCF factors in the nucleus. Their patterns of expression and the levels of alternative LEF/TCF isoforms may impart unique refinements to the potential for Wnt signal propagation in cells. In other words, different LEF/TCF isoforms may shape the level and time course of Wnt target gene expression. For example, the E tail of X.laevis xTCF-3 recruits the transcription co-repressor CtBP to down-regulate target genes. High levels of both CtBP and this isoform could dampen Wnt signaling. Many alternative isoforms have been described for human TCF-1, but only one alternative isoform has been described for human LEF-1 and two for human TCF-4. It is important to determine whether LEF-1 and the other LEF/TCF family members exist as a similar set of isoforms. Their conservation across family members and orthologs in other species would suggest that they contribute an important function. One of the new LEF-1 isoforms (LEF-1B) encodes a C-terminal amino acid sequence homologous to the prevalent TCF-1B and TCF-4B forms, suggesting an important role of this isoform in the actions of mammalian LEF/TCFs. On the other hand, the most universal of C-terminal tails observed in LEF/TCF proteins of all species is the E tail. It is thus striking that the LEF-1 gene does not appear to encode a similar isoform. A search of the complete nucleotide sequence of introns 9–11 of the LEF-1 gene for their capacity to encode one or more of the highly conserved amino acid motifs in the E tail (Fig. 2B) showed that no open reading frame exists that can code for a similar amino acid sequence. One function of the Xenopus TCF-3E tail and human TCF-4E tail is to recruit the transcription co-repressor CtBP but in addition, the motif rich in cysteines and basic residues, which is not involved in CtBP recruitment, must perform another important function. It appears unlikely that the LEF-1 gene produces an isoform that can act similarly.
The LEF-1N tail is not homologous to any known functional motif. This short tail is immediately juxtaposed to the HMG DNA binding domain and would be in close proximity to nucleic acid when LEF-1 binds and bends its cognate DNA sequence. However, deletion mutant forms of LEF-1 missing this tail are capable of high affinity DNA binding and bending indistinguishable from full-length LEF-1, thus this sequence does not contribute to these activities, at least in vitro (40). Likewise, a unique function or activity for the alternative LEF-1 isoform missing exon 6 is not known. Exon 6 encodes a part of the context-dependent transcription activation domain (CAD) of LEF-1, but the TCF-1 gene, which has not been shown to have a transcription activation domain, also expresses an isoform missing these sequences (Fig. 1, exon V). Therefore, a functional consequence of this alternative splice is likely to be independent of CAD activity and common to LEF/TCF proteins.
Equally important to proper Wnt signal propagation are the mechanisms that regulate LEF/TCF gene expression. Down-regulation of LEF/TCF expression would decrease the signaling capacity in the receptive cell, at least for changes in Wnt target gene activity. While it remains to be determined whether Wnt signaling plays a role in lymphocyte differentiation, these cells down-regulate LEF-1 expression during differentation. Thus, mature B lymphocytes do not express LEF-1, and mature T lymphocytes express only very low levels.
We have shown that the LEF-1 promoter is preferentially active in mature B and T lymphocyte cell lines. Whether these patterns of expression are due to aberrant gene activation or loss of the ability to down regulate LEF-1 expression will require a better understanding of the mechanisms that control transcription of this gene. Although the lymphocyte-specific activity was not reflected in differential DNase I protection patterns with Jurkat T lymphocyte extracts versus HeLa extracts, the transcription machinery driving expression may not be detectable on naked DNA templates with crude extracts. Several matches to known recognition elements are present including ones that match Sp1, E box and GAGA elements. Mutation of the E box element did not diminish promoter activity in Jurkat by >20% (K.Hovanes, unpublished observations), and co-transfection with an E47, an E box transcription factor prevalent in lymphocytes, did not affect promoter activity in Jurkat or COS-7 cells. Either this element functions only within the context of chromatin or within the entire LEF-1 locus, or we have not yet identified a cell line that expresses the cognate activator/repressor for this site.
The LEF-1 gene is rapidly down-regulated during differentiation, and our data suggest that this could occur by active silencing. The LEF-1 promoter is most active in Raji B lymphocytes even though the endogenous LEF-1 gene is not expressed in these cells. One interpretation of these data is that lymphocyte activators that can bind to the LEF-1 promoter are present in both Jurkat and Raji cells. These activators are prevented from working on the endogenous LEF-1 gene in Raji cells because the promoter is either actively silenced by distal cis-acting elements, and/or a silencing mechanism directs the formation of repressive chromatin structure over the promoter. Such distal cis-acting elements are not present in the promoter fragments we have studied and repressive chromatin structures are unlikely to form on promoter sequences present on transiently transfected plasmid DNA. To address both possibilities, we are currently examining the LEF-1 locus for distal regulatory elements and are examining the structure of the endogenous LEF-1 gene in Jurkat and Raji cells.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Jesus Munguia for technical assistance and Dr Katherine Jones for purified Sp1 protein. We also thank Dr Bert Semler for critical reading of the manuscript and advice on experimental approaches.
DDBJ/EMBL/GenBank accession no. AF203908
REFERENCES
- 1.Brown J. and Moon,R. (1998) Curr. Opin. Cell Biol., 10, 182–187. [DOI] [PubMed] [Google Scholar]
- 2.Brannon M., Brown,J.D., Bates,R., Kimelman,D. and Moon,R.T. (1999) Development, 126, 3159–3170. [DOI] [PubMed] [Google Scholar]
- 3.Roose J., Molenaar,M., Peterson,J., Hurenkamp,J., Brantjes,H., Moerer,P., van de Wetering,M., Destree,O. and Clevers,H. (1998) Nature, 395, 608–612. [DOI] [PubMed] [Google Scholar]
- 4.Cavallo R.A., Cox,R.T., Moline,M.M., Roose,J., Polevoy,G.A., Clevers,H., Peifer,M. and Bejsovec,A. (1998) Nature, 395, 604–608. [DOI] [PubMed] [Google Scholar]
- 5.Oosterwegel M., van de Wetering,M., Timmerman,J., Kruisbeek,A., Destree,O., Meijlink,F. and Clevers,H. (1993) Development, 118, 439–448. [DOI] [PubMed] [Google Scholar]
- 6.Korinek V., Barker,N., Willert,K., Molenaar,M., Roose,J., Wagenaar,G., Markman,M., Lamers,W., Destree,O. and Clevers,H. (1998) Mol. Cell. Biol., 18, 1248–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kratochwil K., Dull,M., Farinas,I., Galceran,J. and Grosschedl,R. (1996) Genes Dev., 10, 1382–1394. [DOI] [PubMed] [Google Scholar]
- 8.van Genderen C., Okamura,R., Farinas,I., Quo,R., Parslow,T., Bruhn,L. and Grosschedl,R. (1994) Genes Dev., 8, 2691–2703. [DOI] [PubMed] [Google Scholar]
- 9.Galceran J., Farinas,I., Depew,M.J., Clevers,H. and Grosschedl,R. (1999) Genes Dev., 13, 709–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Verbeek S., Izon,D., Hofhuis,F., Robanus-Maandag,E., te Riele,H., van de Wetering,M., Oosterwegel,M., Wilson,A., MacDonald,H.R. and Clevers,H. (1995) Nature, 374, 70–74. [DOI] [PubMed] [Google Scholar]
- 11.Korinek V., Barker,N., Moerer,P., van Donselaar,E., Huls,G., Peters,P.J. and Clevers,H. (1998) Nature Genet., 19, 379–383. [DOI] [PubMed] [Google Scholar]
- 12.Okamura R., Sigvardsson,M., Galceran,J., Verbeek,S., Clevers,H. and Grosschedl,R. (1998) Immunity, 8, 11–20. [DOI] [PubMed] [Google Scholar]
- 13.Waterman M.L., Fischer,W.H. and Jones,K.A. (1991) Genes Dev., 5, 656–669. [DOI] [PubMed] [Google Scholar]
- 14.Travis A., Amsterdam,A., Belanger,C. and Grosschedl,R. (1991) Genes Dev., 5, 880–894. [DOI] [PubMed] [Google Scholar]
- 15.van de Wetering M., Oosterwegel,M., Dooijes,D. and Clevers,H. (1991) EMBO J., 10, 123–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou P., Byrne,C., Jacobs,J. and Fuchs,E. (1995) Genes Dev., 9, 700–713. [DOI] [PubMed] [Google Scholar]
- 17.Oosterwegel M., van de Wetering,M. and Clevers,H. (1993) Thymus, 22, 67–81. [PubMed] [Google Scholar]
- 18.Porfiri E., Rubinfeld,B., Albert,I., Hovanes,K., Waterman,M. and Polakis,P. (1997) Oncogene, 15, 2833–2839. [DOI] [PubMed] [Google Scholar]
- 19.Rubinfeld B., Robbins,P., El-Gamil,M., Albert,I., Porfiri,E. and Polakis,P. (1997) Science, 275, 1790–1792. [DOI] [PubMed] [Google Scholar]
- 20.Church D.M., Yang,J., Bocian,M., Shiang,R. and Wasmuth,J.J. (1997) Genome Res., 7, 787–801. [DOI] [PubMed] [Google Scholar]
- 21.Jones K.A., Luciw,P.A. and Duchange,N. (1988) Genes Dev., 2, 1101–1114. [DOI] [PubMed] [Google Scholar]
- 22.Waterman M. and Jones,K. (1990) New Biologist, 2, 621–636. [PubMed] [Google Scholar]
- 23.van de Wetering M., Cavallo,R., Dooijes,D., van Beest,M., van Es,J., Loureiro,J., Ypma,A., Hursh,D., Jones,T., Bejsovec,A., Peifer,M., Mortin,M. and Clevers,H. (1997) Cell, 88, 789–799. [DOI] [PubMed] [Google Scholar]
- 24.Van de Wetering M., Castrop,J., Korinek,V. and Clevers,H. (1996) Mol. Cell. Biol., 16, 745–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee Y.J., Swencki,B., Shoichet,S. and Shivdasani,R.A. (1999) J. Biol. Chem., 274, 1566–1572. [DOI] [PubMed] [Google Scholar]
- 26.Molenaar M., van de Wetering,M., Oosterwegel,M., Peterson-Maduro,J., Godsave,S., Korinek,V., Roose,J., Destree,O. and Clevers,H. (1996) Cell, 86, 391–399. [DOI] [PubMed] [Google Scholar]
- 27.Lin R., Thompson,S. and Priess,J. (1995) Cell, 83, 599–609. [DOI] [PubMed] [Google Scholar]
- 28.Dooijes D., van Beest,M., van de Wetering,M., Boulanger,G., Jones,T., Clevers,H. and Mortin,M.A. (1998) Mol. Gen. Genet., 258, 45–52. [DOI] [PubMed] [Google Scholar]
- 29.Smale S.T., Jain,A., Kaufmann,J., Emami,K.H., Lo,K. and Garraway,I.P. (1998) Cold Spring Harb. Symp. Quant. Biol., 63, 21–31. [DOI] [PubMed] [Google Scholar]
- 30.Bain G., Robanus Maandag,E.C., te Riele,H.P., Feeney,A.J., Sheehy,A., Schlissel,M., Shinton,S.A., Hardy,R.R. and Murre,C. (1997) Immunity, 6, 145–154. [DOI] [PubMed] [Google Scholar]
- 31.Murre C., McCaw,P.S., Vaessin,H., Caudy,M., Jan,L.Y., Jan,Y.N., Cabrera,C.V., Buskin,J.N., Hauschka,S.D., Lassar,A.B. et al. (1989) Cell, 58, 537–544. [DOI] [PubMed] [Google Scholar]
- 32.Soeller W.C., Oh,C.E. and Kornberg,T.B. (1993) Mol. Cell. Biol., 13, 7961–7970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Farkas G., Gausz,J., Galloni,M., Reuter,G., Gyurkovics,H. and Karch,F. (1994) Nature, 371, 806–808. [DOI] [PubMed] [Google Scholar]
- 34.Roose J., Huls,G., van Beest,M., Moerer,P., van der Horn,K., Goldschmeding,R., Logtenberg,T. and Clevers,H. (1999) Science, 285, 1923–1926. [DOI] [PubMed] [Google Scholar]
- 35.Giese K., Amsterdam,A. and Grosschedl,R. (1991) Genes Dev., 5, 2567–2578. [DOI] [PubMed] [Google Scholar]
- 36.van de Wetering M. and Clevers,H. (1992) EMBO J., 11, 3039–3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nishita M., Hashimoto,M.K., Ogata,S., Laurent,M.N., Ueno,N., Shibuya,H. and Cho,K.W.Y. (2000) Nature, 403, 781–783. [DOI] [PubMed] [Google Scholar]
- 38.Sheridan P.L., Sheline,C.T., Cannon,K., Voz,M.L., Pazin,M.J., Kadonaga,J.T. and Jones,K.A. (1995) Genes Dev., 9, 2090–2104. [DOI] [PubMed] [Google Scholar]
- 39.Riese J., Yu,X., Munnerlyn,A., Eresh,S., Hsu,S.C., Grosschedl,R. and Bienz,M. (1997) Cell, 88, 777–787. [DOI] [PubMed] [Google Scholar]
- 40.Carlsson P., Waterman,M. and Jones,K. (1993) Genes Dev., 7, 2418–2430. [DOI] [PubMed] [Google Scholar]