

Abstract
Although KMT2D, also known as MLL2, is known to play an essential role in development, differentiation, and tumor suppression, its role in pancreatic cancer development is not well understood. Here, we discovered a novel signaling axis mediated by KMT2D, which links TGF-β to the activin A pathway. We found that TGF-β upregulates a microRNA, miR-147b, which in turn leads to post-transcriptional silencing of KMT2D. Loss of KMT2D induces the expression and secretion of activin A, which activates a non-canonical p38 MAPK-mediated pathway to modulate cancer cell plasticity, promote a mesenchymal phenotype, and enhance tumor invasion and metastasis in mice. We observed a decreased KMT2D expression in human primary and metastatic pancreatic cancer. Furthermore, inhibition or knockdown of activin A reversed the pro-tumoral role of KMT2D loss. These findings support a tumor-suppressive role of KMT2D in pancreatic cancer and identify miR-147b and activin A as novel therapeutic targets.
Keywords: MLL2, epithelial mesenchymal transition, metastasis, miR-147b, tumor suppressor
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is projected to become the second leading cause of cancer death in the United States by 2030, with an overall 5-year survival rate of less than 11%1,2. Early metastasis is one of the main reasons for poor survival for these patients. Epigenomic regulation and enhancer reprogramming are emerging as essential mechanisms for tumor progression and metastasis3,4. Whole genome sequencing recently revealed frequent mutations in epigenetic regulating genes in PDAC5,6. Some of these mutations are considered driver mutations that lead to altered chromatin structure, promoter accessibility, and gene transcription5,7. However, how epigenomic dysregulation promotes PDAC progression and metastasis is not well understood.
Whole genome sequencing recently discovered inactivating mutations of a histone modification enzyme, KMT2D (also known as MLL2), in up to 5% of PDAC cases, suggesting its tumor suppression potential8,9. KMT2D is also transcriptionally repressed in most PDACs through DNA methylation10. Additionally, KMT2D is mutated in 82% of neoadjuvant-treated PDACs in one study11, suggesting that KMT2D mutation may be associated with treatment resistance. KMT2D is one of the major histone methyltransferases for lysine 4 of histone 3 (H3K4) and mainly mono- and di-methylates H3K4 residue, which are predominant histone marks at distal promoters and enhancers12. Thus, KMT2D is known to play an essential role in establishing active promoter and enhancer landscapes, and cell-specific transcriptome. However, it remains poorly understood regarding how KMT2D contributes to PDAC progression and metastasis.
One of the critical regulators of metastasis and cancer cell plasticity is the transforming growth factor-β (TGF-β) pathway and the reversible epithelial to mesenchymal transition (EMT) process13. Alterations in TGF-β signaling, specifically SMAD4 mutation and inactivation, are found in more than 50% of PDAC cases. TGF-β serves as a tumor-suppressive signal in early PDAC but induces EMT and promotes tumor invasion and metastasis later in tumor progression14. The dual tumor suppressive and pro-tumorigenic role of TGF-β signaling requires a better understanding of this essential pathway during PDAC progression and metastasis.
Here, we uncovered a novel link between TGF-β signaling and KMT2D by miR-147b, and the subsequent upregulation of the EMT signaling by activin A, a member of the TGF-β superfamily. In addition, activin A promoted PDAC invasion through a non-canonical p38-dependent pathway. Furthermore, inhibition of activin A or p38 effectively reversed the pro-tumoral phenotype in KMT2D-knockout cells. These results not only revealed an essential novel molecular mechanism by which TGF-β promotes epigenetic reprograming, cancer cell plasticity, and PDAC progression and metastasis, but also discovered potential new therapeutic targets in PDAC patients with KMT2D inactivating mutations.
Methods
Cell culture and transfections
PDAC cell lines were obtained from the American Type Culture Collection and include: PANC-1 (RRID: CVCL_0480), BxPC-3 (RRID: CVCL_0186), MIA PaCa-2 (RRID: CVCL_0428), CFPAC-1 (RRID: CVCL_1119), HPAF-II (RRID: CVCL_0313), and AsPC-1 (RRID: CVCL_0152). UM28 was a patient derived primary PDAC cell line developed by Dr. Diane Simeone15. All human cell lines have been authenticated using short tandem repeat profiling within the last three years. All experiments were performed with mycoplasma-free cells. Cell lines were cultured in the appropriate medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin (GIBCO) at 37°C with 5% CO2. KMT2D siRNAs or scrambled siRNA (Dharmacon) (50 nM) were transfected into PDAC cells using Lipofectamine RNA iMAX reagent (Invitrogen) according to the manufacturer’s instructions. Synthesized guide RNAs (sgRNAs) and Cas9 protein (Dharmacon) were transfected into PANC-1 and BxPC-3 cell lines by RNA iMAX, followed by clonal cell isolation. Briefly, a single CRISPR/cas9-edited cell was plated in 96-well plate using limiting dilution method, and clonal cells were identified and expanded. The sequences of siRNAs and sgRNAs are listed in Supplementary Table 1.
Reagents and antibodies
All reagents and antibodies are listed in Supplementary Table 2.
Bru-seq and BruChase-seq
Bru-seq and BruChase-seq were performed as previously described16. log2FoldChange values from Bru-seq analysis were plotted for the two knockouts (vs. control) for the basal and classical gene sets as described by Moffitt et al.17 to create a heat map. The heat map was generated in R using the pheatmap package (v1.0.12). More detailed method was provided in the Supplementary Materials and Methods. The sequencing coverage and quality statistics for each sample are summarized in Supplementary Table 3.
Protein extraction and western blot analysis
For the preparation of cellular lysates, cultured cells were harvested with NP-40 based whole-cell lysis buffer as previously described18. Protein concentrations were measured using the Bradford assay (Bio-Rad). Protein samples were heated and separated on sodium dodecyl sulfate- PAGE or Tris-Acetate-PAGE gel-electrophoresis and transferred to PVDF membranes (Millipore). After blocking in PBS/Tween-20 containing 5% nonfat milk, the membranes were incubated with primary antibodies overnight at 4°C, followed by incubation with peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories) for 1 hour. Then the proteins were visualized using an ECL detection kit (Thermo Scientific).
RNA preparation and quantitative real-time PCR
Total RNAs and microRNAs were purified using the RNeasy and miRNeasy Mini Kit (QIAGEN). Reverse transcription of RNA was performed using the SuperScript ® III First-Strand Synthesis kit (Invitrogen) while the reverse transcription of microRNA was obtained using TaqMan™ MicroRNA Reverse Transcription Kit according to the manufacturer’s instructions. Quantitative real-time RT-PCR was performed using SYBR Green reagents and microRNA with TaqMan™ Universal Master Mix II (Applied Biosystems) in MicroAmp Optical 96-well reaction plates (Applied Biosystems). The 2−ΔΔCT method was used to measure the gene expression compared with the endogenous controls (U6 non-coding small nuclear RNA for miR-147b and GAPDH for mRNAs)19. Primers for miR-147b and U6 were purchased from Thermo Fisher Scientific, and other primers were designed using Primer-BLAST (https://www-ncbi-nlm-nih-gov.proxy.lib.umich.edu/tools/primer-blast/) and synthesized by Integrated DNA Technologies. The primer sequences are listed in Supplementary Table 4.
Dual luciferase reporter assay
Full length human KMT2D 3’ UTR sequence was cloned into pmirGLO Dual-Luciferase miRNA target expression vector (Promega). Cloned vector was transfected into 293FT cells with non-target miR or miR-147b mimic for 48 hours. Luminescence was measured using Dual-Glo™ luciferase assay (Promega), following the manufacturer’s instruction. Relative luciferase activity was calculated by normalizing reads from firefly luciferase activity to Renilla luciferase activity.
Patient pancreatic tissue samples, tissue microarrays, and clinicopathologic variables
Patients with pancreas resections for pancreatitis, cystic neoplasms, or PDAC from 2002 to 2015 at the University of Michigan Health System were included in the study. All Hematoxylin and Eosin (H&E) stained slides were reviewed, and diagnoses confirmed by a gastrointestinal pathologist (JS) and corresponding areas were selected. Duplicated 1 mm diameter tissue cores from a total of 311 patient tissue samples were punched/extracted and transferred to recipient tissue array blocks. Five TMAs were set up according to a standard protocol, as previously described20. H&E staining was performed using standard protocol.
Immunohistochemical analysis and scoring
IHC staining of the TMAs was completed using the KMT2D antibody (MilliporeSigma) as previously described20. The intensity of the staining was recorded as: 0 = negative; 1 = weak; 2 = moderate; 3 = strong. Final IHC staining score = staining intensity x positive cells%. The cases were divided into KMT2D high versus low groups with the median as the cutoff. The slides were scored by two independent pathologists blinded to the sample information. IHC staining of E-cadherin and Vimentin was quantified using ImageJ software. Five random fields were chosen for quantification. p-p38 staining was quantified by counting positive cells in 100 tumor cells in 5 randomly selected fields in the primary tumor. Each data point represents an individual field that was analyzed.
In vitro wound-healing and transwell invasion assays
Cell migratory ability was measured by wound healing assay, which was performed by seeding cells in complete media in 6-well plate for 24-48 hours until a confluent monolayer had formed as previously described21. Cell invasion assay was performed using a 24-well plate with 8-μm pore size chamber inserts (Corning). 0.5 - 1×105 cells in serum-free culture medium were seeded into the upper chamber per well with a Matrigel-coated membrane as previously described21.
Tumor sphere cultures
1 x 104 single cells were suspended in X-VIVO 10 Serum-free Hematopoietic Cell Medium (#04-380Q, Lonza) in six-well Ultra-Low Attachment Plates (Corning) for five days. For the KMT2D siRNA knockdown UM28 cell line, attached cells were transfected with the scrambled siRNA or KMT2D-targeted siRNA 24h before they were suspended and seeded. Pictures of the tumor sphere were taken using an inverted microscope (Olympus) and quantified manually.
ChIP-qPCR
ChIP was performed using the SimpleChIP Enzymatic Chromatin IP Kit (#9003 Cell Signaling) according to the manufacturer’s instructions with minor modifications. In brief, approximately 4 × 106 cells were formaldehyde-crosslinked, and chromatin was digested into 100-400 bp fragments by micrococcal nuclease. Then the digested chromatin was immunoprecipitated with H3 (#9003 Cell Signaling), IgG (#9003 Cell Signaling), H3K4me1(ab8895 Abcam), H3K4me2(#17677 EMD Millipore), H3K4me3(#9751 Cell Signaling), or H3K27ac (ab4729 Abcam) antibodies using the recommended dilutions. Washed and purified DNA were subjected to qPCR using SYBR Green reagents (Applied Biosystems) according to the manufacturer’s instructions. The primers directed against the INHBA promoter region were used in qPCR reactions. The enrichment of the PCR product in the target DNA fragment in each condition was compared to the % of DNA quantity in the input sample. The % input was calculated as follows: Percent Input = efficiency x 2(C[T] 2%Input Sample - C[T] IP Sample).
Establishment of orthotopic xenograft mouse model
Briefly, mice are anesthetized under isoflurane gas and placed on heated platform. Mice will receive carprofen (5 mg/kg) by s.c. or i.p. injection. The skin will be shaved by razor, disinfected with chlorhexidine or iodine scrub for three times, and washed for three times with sterile water or PBS. An incision was made at the left abdominal flank of 6-8 weeks old NSG mice, close to the splenic silhouette. The pancreas was pulled out of the peritoneal cavity. 1x106 BxPC-3 or PANC-1 cells were suspended in 40 μL mixed cell culture media and Matrigel (BD) at a 1:1 ratio and injected into the tail of the pancreas. The abdominal muscle and skin layers were sequentially closed with sutures. The animals were monitored every day for one week after surgery for any adverse effects, and once a week for four weeks to check for tumor growth. The study was terminated four weeks after tumor implantation surgery. Tumor tissue was harvested and processed for H&E and IHC staining.
Statistical analysis
ANOVA models were used to compare the expression of KMT2D in PDAC and its precursor lesions. Analysis was performed using GraphPad Prism software or SAS (version 9.4, SAS Institute) or Excel for all in vitro and in vivo studies. Results are presented as mean ± SD unless otherwise noted. Statistical significance among different groups was calculated as described in each figure. Significance is determined if p<0.05.
Results
TGF-β suppresses KMT2D expression by upregulating miR-147b
Although steady-state global transcriptional alterations induced by TGF-β have been studied, how TGF-β affects nascent RNA synthesis and RNA stability globally in PDAC has not been explored. To better understand TGF-β signaling in PDAC, we used Bru-seq and BruChase-seq technologies to capture the changes not only in de novo transcriptional landscape (Bru-seq), but also RNA stability (BruChase-seq), in TGF-β treated PDAC cells16. As expected, genes in the TGF-β and EMT pathways were transcriptionally upregulated with the treatment (Supplementary Fig. 1A). Other upregulated pathways included hypoxia, KRAS, TNFα, UV response, unfolded protein response, and glycolysis. We also observed that TGF-β treatment downregulated transcription of genes involved in interferon-alpha, interferon-gamma, E2F, oxidative phosphorylation, G2M checkpoint, fatty acid metabolism, estrogen response, reactive oxygen species, and DNA repair pathways (Supplementary Fig. 1A). Notably, Bru-seq analysis showed that the nascent RNA synthesis of KMT2D did not change (Fig. 1A, top panel). However, BruChase-seq revealed that the KMT2D mRNA level was decreased after 6 hours of uridine chase in cells treated with TGF-β compared to control (Fig. 1A, lower panel), indicating that TGF-β treatment resulted in degradation of KMT2D mRNA. To confirm this finding, we measured KMT2D mRNA and protein levels after TGF-β treatment. TGF-β treatment reduced KMT2D mRNA (Supplementary Fig. 1B) and protein levels (Fig. 1B) in multiple PDAC cell lines. Furthermore, selective TGF-β type I receptor inhibitor, SB505124, reversed TGF-β-induced KMT2D mRNA and protein reduction (Fig. 1C and Supplementary Fig. 1B, C), confirming that TGF-β signaling inhibits KMT2D expression.
Fig. 1.
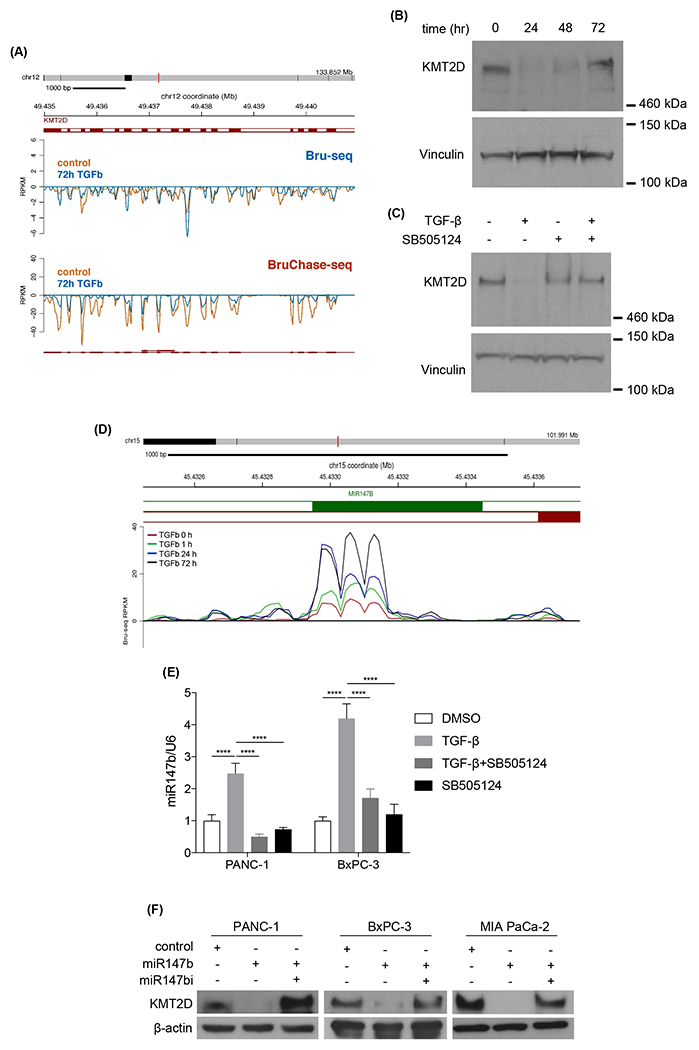
TGF-β decreases KMT2D expression through the upregulation of miR-147b. (A) Comparing KMT2D nascent mRNA synthesis (Bru-seq) and mRNA degradation (BruChase-seq) between vehicle control- and 72h TGF-β-treated PANC-1 cells. Exons were labeled as solid red bars and introns as white bars in between exons. (B) Western blot analysis of KMT2D protein in PANC-1 cells treated with TGF-β for 24, 48, and 72 hours compared to vehicle-treated cells. Vinculin was used as loading control. (C) Western blot analysis of KMT2D in BxPC-3 cells treated with TGF-β with or without a TGF-β inhibitor, SB505124, for 72 hours. Vinculin was used as loading control. (D) Bru-seq analysis of miR-147b transcription in PANC-1 cells treated with TGF-β for 1, 24, and 72 hours compared to vehicle-treated cells. (E) Quantitative real-time PCR of mature miR-147b in PANC-1 and BxPC-3 cells treated with DMSO or TGF-β for 48 hours with or without SB505124. U6 was used as the reference gene. (****p<0.0001, Two-way ANOVA with Dunnett’s multiple comparisons test, n=3) (F) Western Blot analysis of KMT2D in PANC-1, BxPC-3, and MIA PaCa-2 cells transfected with miR-147b mimic or miR-147b inhibitor (miR147i). β-actin was used as loading control.
The observation of decreased KMT2D mRNA after uridine chase without affecting nascent RNA synthesis in TGF-β- treated cells (Fig. 1A) led us to hypothesize that TGF-β increased KMT2D mRNA degradation. Since microRNA is one of the mechanisms of RNA degradation, we postulated that a microRNA might be upregulated by TGF-β treatment and subsequently led to KMT2D mRNA degradation. Using TargetScanHuman (www.targetscan.org), we identified microRNA-147b (miR-147b) as a candidate to target the 3’ UTR of KMT2D mRNA (Supplementary Fig. 1D). microRNA family conservation analysis showed that miR-147b is broadly conserved among mammals (Supplementary Fig. 1E), suggesting its vital function. Bru-seq data suggested that TGF-β induced miR-147b synthesis in a time-dependent manner (Fig. 1D). We hypothesized that TGF-β induces miR-147b expression, which in turn targets KMT2D mRNA for degradation. To confirm our hypothesis, we treated both PANC-1 and BxPC-3 cells with either TGF-β alone or together with TGF-β receptor inhibitor SB505124 and performed real-time PCR on steady-state RNA. TGF-β upregulated miR-147b in both cell lines, and SB505124 blocked this increase, confirming that TGF-β induces miR-147b expression (Fig. 1E). To confirm miR-147b reduces KMT2D expression, we transfected PDAC cell lines with either control miRNA, miR-147b mimic alone, or together with miR-147b inhibitor. miR-147b, but not control miRNA, decreased KMT2D protein expression in all cell lines, while miR-147b inhibitor reversed this effect (Fig. 1F), supporting that miR-147b suppresses KMT2D protein expression. To further confirm the direct regulation of KMT2D expression by miR-147b, we performed dual luciferase reporter functional assay. We observed a decrease in the luciferase activity in the presence of KMT2D 3’ UTR sequence in cells transfected with miR-147b mimic compared with control miRNA (Supplementary Fig. 1F), supporting the direct regulation of KMT2D expression by miR-147b. As a control, VHL, a previously reported miR-147b target22, also decreased when the cancer cells were transfected with miR-147b mimic and rescued by miR-147b inhibitor (miR-147bi) (Supplementary Fig. 1G). Moreover, we observed that inhibition of miR-147b in the presence of TGF-β restored KMT2D expression (Supplementary Fig. 1H). These results confirmed that TGF-β suppresses KMT2D expression via upregulating miR-147b.
Genetic and protein expression alterations of KMT2D in PDAC
To assess the genetic alterations of KMT2D in PDAC, we queried The Cancer Genome Atlas (TCGA), Queensland Centre for Medical Genomics (QCMG), and UT Southwestern (UTSW) sequencing databases. We found that 5-6% of PDAC genomes carried mutations, deletions, and alterations in the KMT2D gene (Fig. 2A). Query of RNAseq data in 1,457 human cancer cell lines from The Cancer Cell Line Encyclopedia revealed significant lower expression of KMT2D mRNA in PDAC cell lines compared to all human tumor-derived cell lines (Fig. 2B)23. Loss of function mutations and deletions of the KMT2D gene was observed in multiple PDAC cell lines (Fig. 2C). Furthermore, patients with altered KMT2D exhibited shorter disease-free survival compared to patients with wild-type KMT2D (Fig. 2D). These data suggest that KMT2D alterations are critical in PDAC progression. To investigate the relationship between genetic alterations of the KMT2D and the TGF-β pathway, we queried the TCGA and UTSW databases. KMT2D inactivating mutations and genetic alterations of critical genes in TGF-β signaling, including TGFB1, TGFBR2, and SMAD4, are mutually exclusive in PDAC patients (Fig. 2E). These data highly suggest that the function of KMT2D and the TGF-β pathway are likely epistatic, and alteration of one gene is sufficient to drive PDAC progression.
Fig. 2.
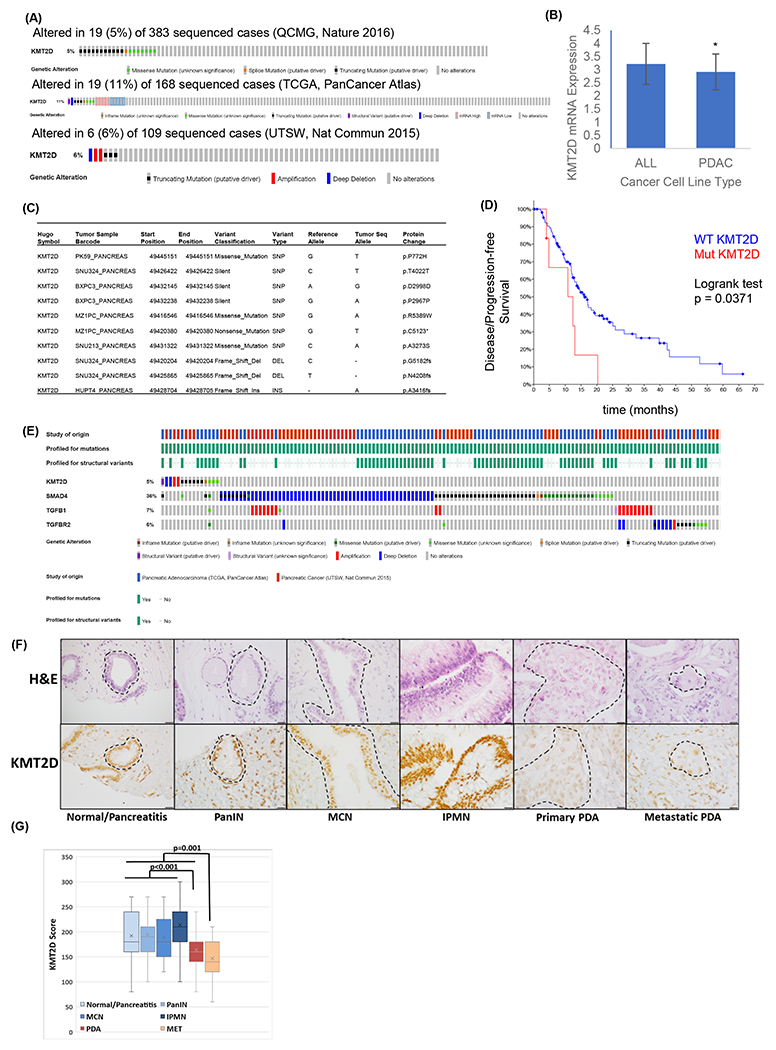
KMT2D mutation and expression profiles in PDAC patients. (A) Sequencing data of human PDACs (n=677) were queried from QCMG (top), TCGA (medium), and UTSW (bottom) databases and patients with KMT2D alterations were highlighted by various colored bars. (B) Comparison of KMT2D mRNA expression in 41 PDAC cell lines to all 1,457 human cancer cell lines based on the RNAseq data queried from the Cancer Cell Line Encyclopedia. * p<0.01. (C) Specific KMT2D mutations in representative PDAC cell lines. (D) Kaplan-Meier curve of disease-free survival in patients with either KMT2D gene mutation (Mut, n=8) or wild-type KMT2D (WT, n=141) queried from the TCGA database (n=149). (E) Patients with KMT2D, SMAD4, TGFB1, and TGFBR2 genetic alterations were queried from TCGA and UTSW databases. (F) Representative H&E and KMT2D immunohistochemical (IHC) staining in various stages of PDAC progression in TMA. Area inside the dashed lines mark the foci of interest. Scale bar = 20 μm. (G) IHC staining score of KMT2D in normal/pancreatitis (n=35), pancreatic intraepithelial neoplasm (PanIN, n=30), mucinous cystic neoplasm (MCN, n=13), intraductal papillary mucinous neoplasm (IPMN, n=38), primary (PDA, n=78) and metastatic (MET, n=19) PDAC from human TMA. One-way ANOVA was used to compare between groups.
To determine whether KMT2D protein expression is also altered in human PDAC and its precursor lesions, we used a tissue microarray (TMA) containing 213 duplicated human pancreatic tissue cores of benign pancreas (n=35), precursor lesions (pancreatic intraepithelial neoplasia [PanIN], intraductal papillary mucinous neoplasm [IPMN], and mucinous cystic neoplasm [MCN]) (n=81), and primary and metastatic PDAC (n=97) (Supplementary Table 5). Immunohistochemical (IHC) staining showed that KMT2D expression was lower in metastatic PDAC compared to all other lesions or normal pancreas/pancreatitis including primary PDAC (p=0.0004), and in primary PDAC compared to all normal/pancreatitis and precursor lesions (p=0.0000002) (Supplementary Table 5 and Fig. 2F, G). These results support that KMT2D may play an essential role in PDAC progression and metastasis.
Loss of KMT2D induces a basal-like and EMT gene signature
To investigate the biologic function of KMT2D in PDAC development, we used the CRISPR/Cas9 system to generate stable KMT2D knockout PDAC cells. We then performed Bru-seq analysis to investigate the global de novo transcriptional changes in KMT2D knockout cells. We identified 748 common differentially transcribed genes in both knockout clones (fold change >2 or <0.5); among which, 262 genes were upregulated, and 486 genes were downregulated (Fig. 3A). Gene Set Enrichment Analysis (GSEA) showed the top pathways that were upregulated included some of the most important oncogenic and metastasis-promoting pathways like MYC, EMT, E2F, and angiogenesis (Fig. 3B). The top downregulated pathways include estrogen response, P53, cholesterol homeostasis, KRAS, and bile acid metabolism (Fig. 3C). We next investigated whether KMT2D loss results in molecular subtype change as defined by recent report from Moffitt et al17. We found that KMT2D knockout cells took on a more basal-like gene signature compared to control cells (Fig. 3D), which is often associated with poor prognosis. Since EMT is one of the major processes associated with TGF-β signaling, cancer cell plasticity, and metastasis, we decided to focus our study on the link between KMT2D and EMT. There was a significant enrichment of EMT signaling in both KMT2D knockout clones (Fig. 3E). Our Bru-seq analysis showed that the epithelial marker, CDH1 (encoding E-cadherin), was markedly downregulated with KMT2D loss, a hallmark of EMT (Fig. 3F top). In contrast, a mesenchymal marker, VIM (encoding Vimentin), was markedly upregulated with KMT2D loss (Fig. 3F bottom).
Fig. 3.
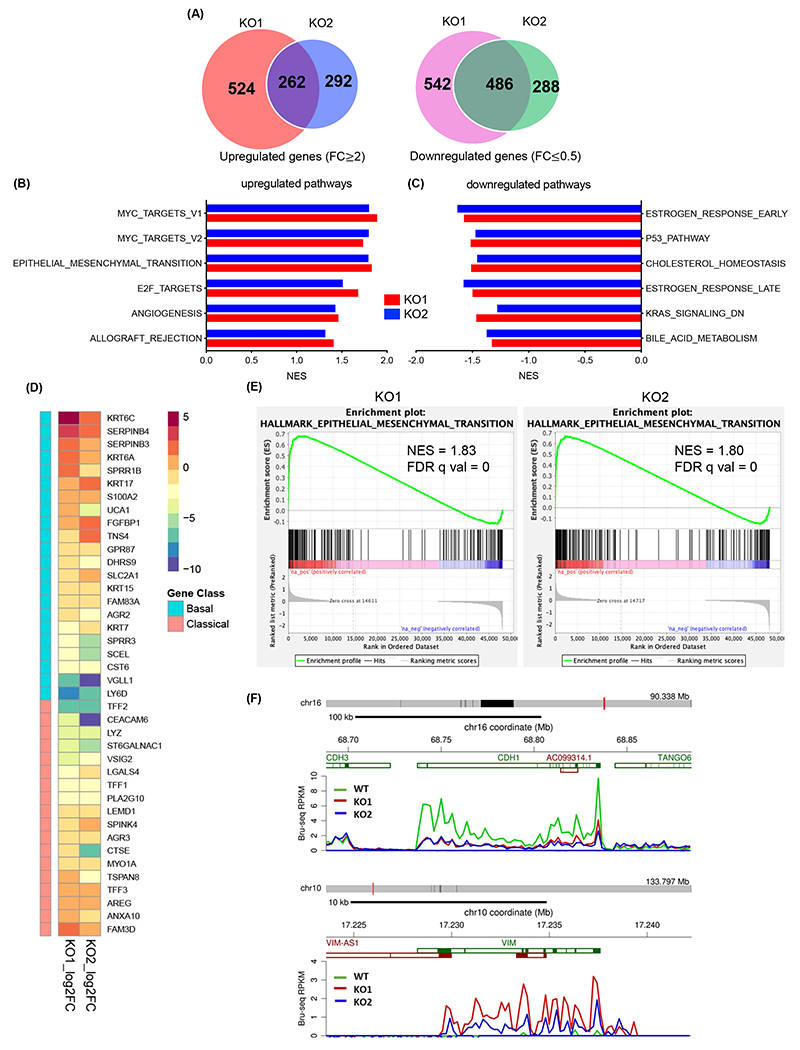
KMT2D knockout induced epigenetic reprograming. (A) Venn diagrams of differentially transcribed genes compared to control in 2 KMT2D knockout (KO) BxPC-3 clones using Bru-seq. (B, C) Top six hallmark pathways that are upregulated (B) or downregulated (C) in KMT2D KO BxPC-3 cells compared to control cells identified by Gene Set Enrichment Analysis (GSEA). (D) Heat map of altered gene signature in 2 KMT2D KO BxPC-3 clones according to the Moffitt gene sets defining the classical or basal-like subtypes of PDAC16. (E) EMT pathway signature enrichment plots in 2 KMT2D KO BxPC-3 clones. NES: normalized enrichment score, FDR q val: false discovery rate. (F) Bru-seq analysis showing de novo transcripts of CDH1 and VIM in wild-type (WT) and KMT2D KO BxPC-3 cells.
To further determine the impact of KMT2D loss on PDAC cell behavior, we analyzed the changes in cell morphology, migration, invasion, and tumorigenic features. KMT2D loss induced a spindled mesenchymal cell phenotype compared to the more epithelial cell morphology in control cells (Fig. 4A), similar to the cell morphology changes induced by TGF-β treatment which was reversed by TGF-β inhibitor SB505124 (Supplementary Fig. 2A). We observed a similar change in cell morphology in PANC-1 cells with CRISPR/Cas9 knockout of KMT2D and BxPC-3 cells with siRNA knockdown of KMT2D (Supplementary Fig. 2B–C). Additionally, the epithelial marker, E-cadherin, was downregulated, and mesenchymal markers, Vimentin and Snail, were upregulated in KMT2D-knockout cells compared to control cells (Fig. 4B, Supplementary Fig. 2D), indicative of EMT. The downregulation of E-cadherin and upregulation of mesenchymal markers resemble what was seen in cells treated with TGF-β and was reversible with TGF-β inhibitor SB505124 (Supplementary Fig. 2E). To confirm that these findings are due to the loss of KMT2D, we used two different siRNAs to knock-down KMT2D in 2 additional cell lines, including a patient-derived primary PDAC cell line, UM2815. Indeed, in both cell lines, E-cadherin expression was attenuated in KMT2D-knockdown cells (Fig. 4C), confirming the pro-mesenchymal effect of KMT2D-deficiency. Furthermore, KMT2D deficiency in normal human pancreatic ductal epithelial (HPDE) cells also showed decrease in CDH1 and increase in ZEB1 (Supplementary Fig. 2F), supporting that the concept of KMT2D loss inducing EMT can be generalized.
Fig. 4.
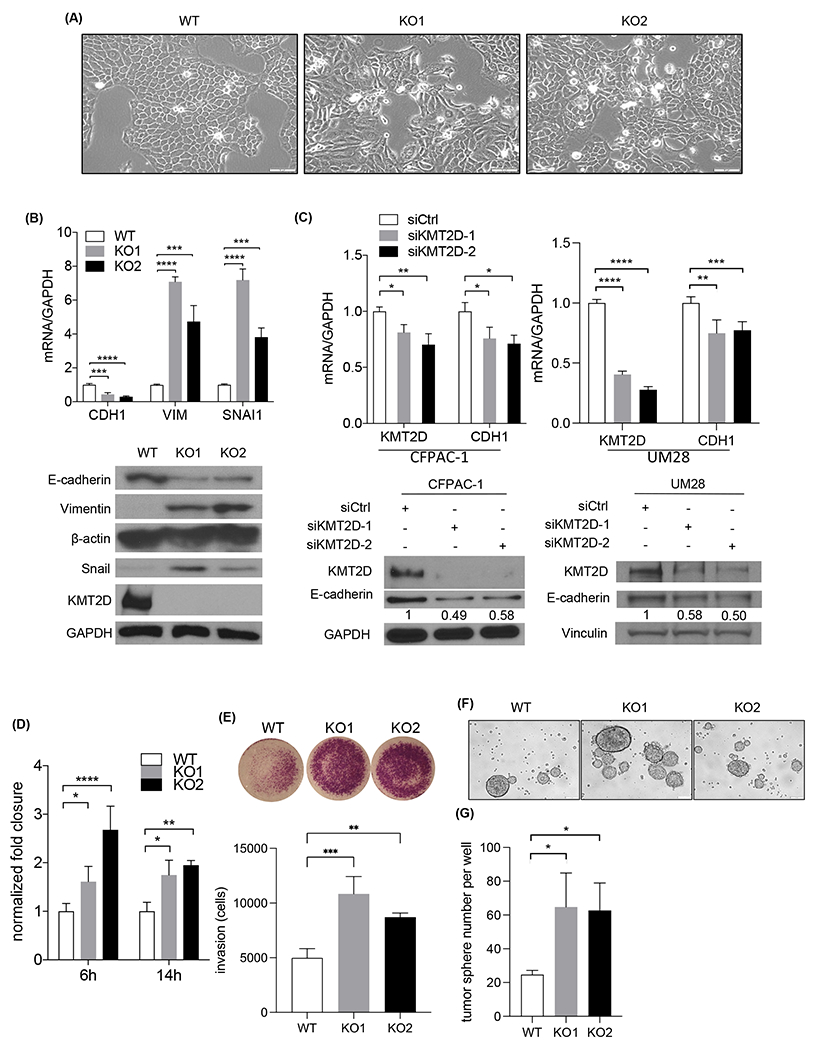
Loss of KMT2D promotes EMT, migration, invasion, and tumorigenicity. (A) Phase-contrast images of BxPC-3 wild-type (WT) and 2 KMT2D knockout (KO) clones in culture. Scale bar = 100 μm. (B) Western blot and quantitative real-time PCR of KMT2D, E-cadherin (CDH1), Vimentin (VIM), and Snail (SNAI1) expression in WT and 2 KMT2D KO BxPC-3 clones. Quantification of bands was shown relative to WT. GAPDH was used as control and reference. (***p<0.005, ****p<0.001, one-way ANOVA test with Dunnett’s multiple comparisons test, n=3) (C) Immunoblot and quantitative real-time PCR of KMT2D and E-cadherin (CDH1) in CFPAC1 and UM28 PDAC cells treated with scramble (siCtrl) or KMT2D siRNA (siKMT2D) for six days. GAPDH was used as control. Quantification of immunoblot bands were shown relative to siCtrl. (*p<0.05, **p<0.01, one-way ANOVA test with Dunnett’s multiple comparisons test (left) and unpaired student t-test (right), n=3) (D) Normalized fold wound closure in WT and KMT2D KO BxPC-3 cells at 6 and 14 hours after scratch. (*p<0.05, **p<0.01, ***p<0.005, ****p<0.001, two-way ANOVA test with Dunnett’s multiple comparisons test, n=3) (E) Invaded cells in KMT2D KO BxPC-3 cells normalized to WT at 48 hours after seeding by transwell invasion assay. (**p<0.01, ***p<0.005, one-way ANOVA test with Dunnett’s multiple comparisons test, n=3) (F) Representative pictures of WT and KMT2D KO BxPC-3 cell tumor spheres. (G) Quantification of tumor spheres in WT and KMT2D KO BxPC-3 cells. (*p<0.05, one-way ANOVA test with Dunnett’s multiple comparisons test, n=3)
Since KMT2D mainly acts as a mono- and di-methyltransferase of H3K4, we surveyed H3K4 methylation levels after KMT2D loss. As expected, we observed a global decrease in H3K4me1 and H3K4me2, but not H3K4me3, in both KMT2D knockout and siRNA knockdown cell lines (Supplementary Fig. 3A–B). Since EMT is associated with increased cell migration and invasion, we postulated that loss of KMT2D would promote cell migration and invasion. As predicted, both KMT2D knockout and knockdown cells exhibited enhanced ability of cell migration and invasion compared to control cells (Fig. 4D–E and Supplementary Fig. 3C–E). Since cancer cell plasticity is associated with enhanced tumor-forming ability, we hypothesize that loss of KMT2D promotes anchorage-independent cell growth potential. Indeed, KMT2D depletion led to more and larger tumor sphere formation compared to control cells (Fig. 4F–G, Supplementary Fig. 3F–G). These data indicated that loss of KMT2D promotes cancer cell plasticity, cell migration, invasion, and enhanced tumorigenic potential.
KMT2D deficiency activates activin signaling via a non-canonical pathway
To better understand how KMT2D depletion induces cancer cell plasticity and invasion, we turned our attention to the genes most consistently upregulated in KMT2D knockout cells according to our Bru-seq data (Supplementary Table 6). Among those genes, we found approximately 5-fold increase in nascent INHBA transcription in both KMT2D knockout clones in comparison to control cells (Fig. 5A). Because activin A (encoded by INHBA) is known to induce EMT in other cancers24–26, we decided to focus on INHBA. Quantitative real-time PCR on steady-state RNA confirmed a roughly 3-5-fold of increase in the INHBA mRNA level in both KMT2D knockout PDAC and HPDE cells (Fig. 5B and Supplementary Fig. 4A). Furthermore, enzyme-linked immunosorbent assay (ELISA) demonstrated an approximately higher than 9-fold increase in secreted activin A protein level in the media of KMT2D knockout cells (Fig. 5C). To exclude the off-target effect of the CRISPR/Cas9 system, we confirmed the upregulation of INHBA in 4 additional PDAC cell lines using KMT2D siRNA knockdown (Fig. 5D). To determine whether KMT2D deficiency induces INHBA expression by activating its promoter, we performed chromatin immunoprecipitation (ChIP)-qPCR to identify the changes of histone modification profile at the INHBA promoter region. Interestingly, despite a globally decreased H3K4me1 and H3K4me2 levels, enriched binding of H3K4me1, H3K4me2, and H3K27ac, but not H3K4me3, were observed at the INHBA promoter region in KMT2D knockout cells (Fig. 5E). Since H3K4me1 and H3K27ac are markers of active enhancers, our results suggest that KMT2D deficiency activates INHBA expression predominantly by activating its enhancer activity.
Fig. 5.
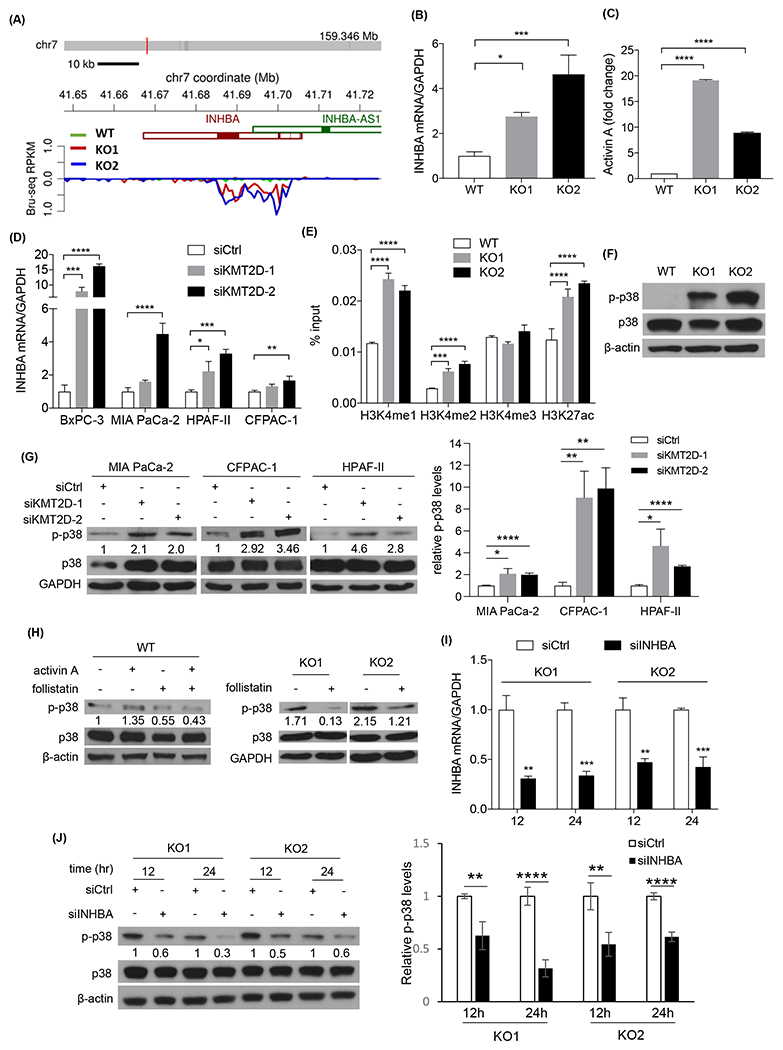
Depletion of KMT2D activates p38 MAPK by upregulating INHBA. (A) Bru-seq data of INHBA de novo transcription in wild-type (WT) and KMT2D knockout (KO) BxPC-3 cells. (B) Quantitative real-time PCR of INHBA in WT and KMT2D KO BxPC-3 cells. GADPH was used as the reference gene. (*p<0.05, ***p<0.005, one-way ANOVA test with Dunnett’s multiple comparisons test, n=3) (C) ELISA of secreted activin A protein in WT and KMT2D KO BxPC-3 cell media. (****p<0.001, two-way ANOVA test with Dunnett’s multiple comparisons test, n=2) (D) Quantitative real-time PCR of INHBA in 4 PDAC cell lines transfected with scramble (siCtrl) or 2 different KMT2D siRNAs (siKMT2D). GAPDH was used as the reference gene. (*p<0.05, **p<0.01, ***p<0.005, ****p<0.001, two-way ANOVA test with Dunnett’s multiple comparisons test, n=3) (E) Chromatin immunoprecipitation (ChIP) and real-time PCR assay of INHBA promoter region in WT and KMT2D KO BxPC-3 cells using H3K4me1, H3K4me2, H3K4me3, and H3K27ac antibodies. Results were normalized to inputs and expressed as % input. (***p<0.005, ****p<0.001, two-way ANOVA test with Dunnett’s multiple comparisons test, n=3) (F) Immunoblot of p38 and p-p38 in WT and KMT2D KO BxPC-3 cells. β-actin was used as loading control. (G) Immunoblot of p38 and p-p38 in three siCtrl or two different siKMT2D-transfected PDAC cell lines. GADPH was used as loading control. Average quantification of the p-p38 bands from three independent experiments was shown relative to siCtrl. (*p<0.05, **p<0.01, ****p<0.001, unpaired student t-test, n=3) (H) Western blot analysis of p38 and p-p38 in WT and KMT2D KO BxPC-3 cells treated with or without activin A or follistatin. Quantification of the p-p38 band was shown. (I) Real-time RT-PCR of INHBA in siCtrl or INHBA siRNA (siINHBA) transfected KMT2D KO BxPC-3 cells. GADPH was used as the reference gene. (**p<0.01, ***p<0.005, unpaired student t-test, n=3) (J) Immunoblot of p38 and p-p38 in KMT2D KO BxPC3 cells transfected with siINHBA or siCtrl for 12 or 24 hours. β-actin was used as loading control. Average quantification of the p-p38 bands from three independent experiments was shown relative to siCtrl. (**p<0.01, ****p<0.001, unpaired student t-test, n=3)
We queried the TCGA-PanCancer Atlas PDAC database and found that patients with altered INHBA have shorter disease-free survival compared to patients with wild-type INHBA (p=0.01, n=168, Supplementary Fig. 4B). Overall, 7% of PDAC patients have INHBA alterations, and most of these alterations are higher mRNA expression or amplification (Supplementary Fig. 4C). Additionally, INHBA and KMT2D alterations are mutually exclusive in PDAC based on TCGA-PanCancer Atlas PDAC database (Supplementary Fig. 4C), supporting that the function of KMT2D and INHBA are likely epistatic, and alteration of one gene is sufficient to drive PDAC progression. We then asked whether there was enrichment of classical or basal-like subtypes in PDAC tumors (TCGA) that were INHBA-altered. There was 7% more INHBA-altered PDACs have a basal-like gene signature compared to INHBA wild-type PDACs (Supplementary Fig. 4D), consistent with the trend we saw in KMT2D knockout cells (Fig. 3D). These data suggest that INHBA alterations are critical in PDAC progression.
There are two activin A signaling cascades: canonical and non-canonical. The canonical pathway signals through SMAD2/3/4 proteins27–29. The non-canonical pathway includes PI3K/Akt, ERK, JNK, and p38 mitogen-activated protein kinase (MAPK) signaling 30,31. To investigate which activin A pathway KMT2D deficiency induced, we surveyed both canonical and potential non-canonical pathways. There was no consistent change of p-SMAD2/3 in KMT2D knockout cells, suggesting that the canonical pathway is unlikely to be the mechanism (Supplementary Fig. 5A, B). Among non-canonical pathways, p38 MAPK was consistently activated in 4 different KMT2D knockout/knockdown cell lines (Fig. 5F–G). To determine if the expression of miR-147b recapitulates the signaling changes induced by KMT2D loss, we transfected miR-147b with or without miR-147b inhibitor into PDAC cells and asked whether EMT and p38 phosphorylation was induced. Indeed, miR-147b expression reduced E-cadherin expression and increased p38 phosphorylation, which was reversed by miR-147b inhibitor (Supplementary Fig. 5C), supporting the hypothesis that miR-147b signals through KMT2D. Additionally, activin A treatment increased p38 phosphorylation, while follistatin, an activin A inhibitor, prevented p38 phosphorylation in either activin-treated or untreated KMT2D wild type or knockout cells (Fig. 5H), strongly suggesting that activin A is the mediator between KMT2D loss and p38 MAPK activation. ERK, JNK, and Akt were largely unchanged or inconsistent upon loss of KMT2D (Supplementary Fig. 5B, D–F). These results suggested that KMT2D deficiency likely functions through the non-canonical activin A/p38 MAPK pathway. To confirm that activin A is specifically required for the activation of the p38 MAPK pathway upon KMT2D loss, we knocked down INHBA expression (siINHBA) in KMT2D knockout and wild type cells. INHBA knockdown attenuated p38 phosphorylation in KMT2D knockout cells, but not wild type cells (Fig. 5I–J, Supplementary Fig. 4E–F), confirming that loss of KMT2D activates the p38 MAPK pathway via activin A. Since TAK1 is known to mediate TGF-β signaling32,33 and is upstream of p38, we set out to determine if TAK1 mediates activin A-p38 signaling in KMT2D-deficient PDAC cells. We used a selective TAK1 inhibitor, Takinib, in our KMT2D knockout cells. The treatment decreased p38 phosphorylation while increased E-cadherin expression (Supplementary Fig. 5G). This finding suggests that the KMT2D-activin A-p38 signaling in PDAC cells is likely mediated by TAK1.
To determine whether activin A/p38 MAPK axis mediated the EMT and increased cell migration and invasion in KMT2D deficient cells, we knocked down INHBA in KMT2D deficient cells. The epithelial marker E-cadherin was upregulated (Fig. 6A), and tumor cell invasion was attenuated in INHBA knockdown cells (Fig. 6B). Furthermore, we treated wild type and KMT2D deficient cells with p38 MAPK inhibitor SB202190 and observed the upregulation of E-cadherin, especially in KMT2D deficient cells (Fig. 6C) and decreased tumor cell migration and invasion in a dose-dependent manner (Fig. 6D–E). As an evidence of treatment effectiveness, the phosphorylation of HSP27, a well-characterized downstream target of p38, was decreased upon SB202190 treatment (Fig. 6C). In addition, another p38 inhibitor SB203580 exhibited similar upregulation effect on E-cadherin (Supplementary Fig. 5H), supporting the specificity of SB202190. These results support that KMT2D depletion induced EMT, migration, and invasion are, at least in part, mediated by activin A/p38 MAPK pathway.
Fig. 6.
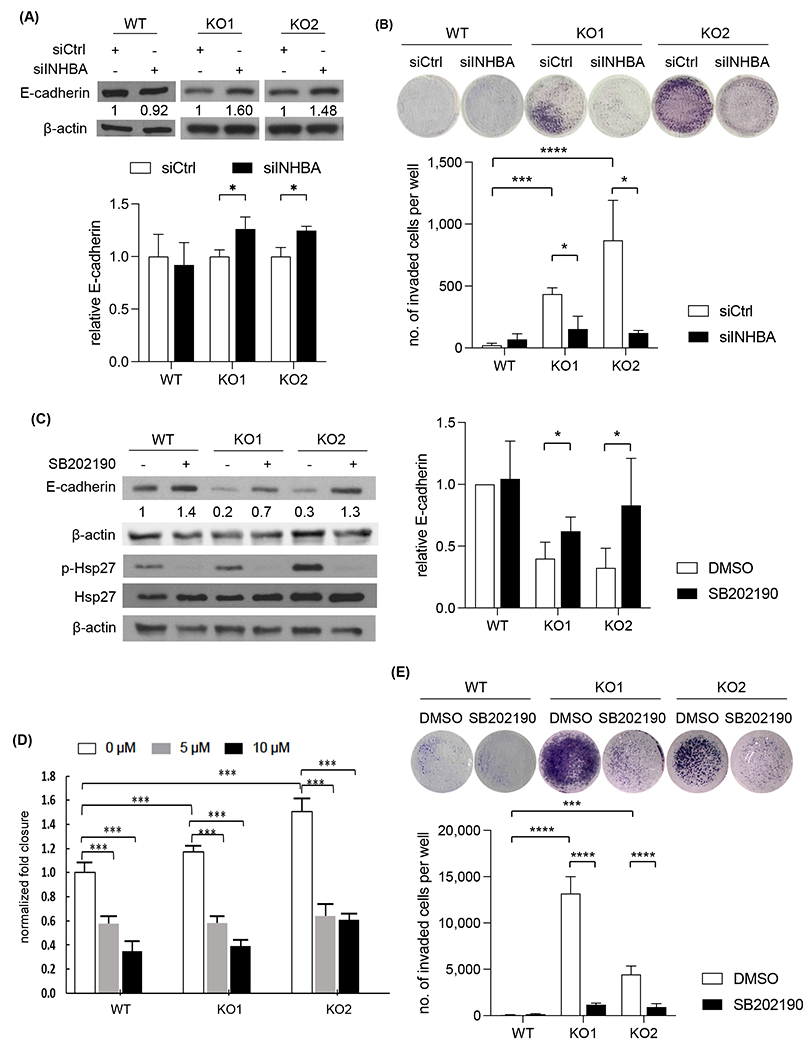
Depletion of INHBA or inhibition of p38 MAPK upregulated E-cadherin expression and attenuated PDAC cell migration and invasion. (A) Immunoblot of E-cadherin in KMT2D KO BxPC-3 cells transfected with either siCtrl or siINHBA. β-actin was used as loading control. Average quantification of the E-cadherin bands from three independent experiments was shown relative to siCtrl. (*p<0.05, unpaired student t-test, n=3) (B) Transwell invasion assay in KMT2D KO BxPC-3 cells transfected with siCtrl or siINHBA. (*p<0.05, unpaired student t-test, n=3) (C) Immunoblot of E-cadherin, Hsp27, and p-Hsp27 in KMT2D KO and WT BxPC-3 cells treated with 10 μM of SB202190 or DMSO control for 72 hours (E-cadherin) or 2 hours (Hsp27 and p-Hsp27). β-actin was used as loading control. Average quantification of the E-cadherin bands from three independent experiments was shown relative to control. (*p<0.05, unpaired student t-test, n=3) (D) Normalized fold closure in DMSO or 5 or 10 μM SB202190-treated KMT2D KO BxPC-3 cells by wound healing assay. (***p<0.005, ****p<0.001 Two-way ANOVA test with Dunnette’s multiple comparisons, n=3) (E) Transwell invasion assay of KMT2D KO BxPC-3 cells treated with either DMSO or 5 μM SB202190. (**p<0.01, ***p<0.005, unpaired student t-test, n=3)
Loss of KMT2D promotes tumor growth, metastasis, and EMT in vivo
To validate our findings in vivo, we injected KMT2D knockout or wild-type PDAC cells orthotopically into the pancreas of NOD-SCID IL2Rgammanull (NSG) mice. Mice implanted with KMT2D knockout cells developed bigger primary tumors (Fig. 7A–B) and metastases (Fig. 7C) compared to the control group. KMT2D knockout tumors had more mesenchymal or basal-like histology with decreased epithelial marker, E-cadherin, and increased mesenchymal marker, Vimentin, compared to control tumors (Fig. 7D, Supplementary Fig. 6A–B, reviewed by pathologists Jiaqi Shi, Yumei Gu, and Jing Yang), confirming the EMT process induced by KMT2D deficiency observed in vitro. Since BxPC3 cells are known to have squamous differentiation34,35, p63 IHC was performed. Consistent with previous reports, the wild-type tumor expressed p63 (Supplementary Fig. 6A–B). KMT2D knockout tumors maintained or had slightly increased p63 staining. Furthermore, we observed that p-p38 levels increased in tumors derived from KMT2D knockout cells compared to wild-type tumors (Fig. 7E). Additionally, there was a significant increase in activin A protein levels in KMT2D knockout tumors compared to wild type (Fig. 7F). In summary, our in vivo data supports our hypothesis that loss of KMT2D promotes metastasis and tumor progression by activating activin A/p38 MAPK pathway and EMT process.
Fig. 7.
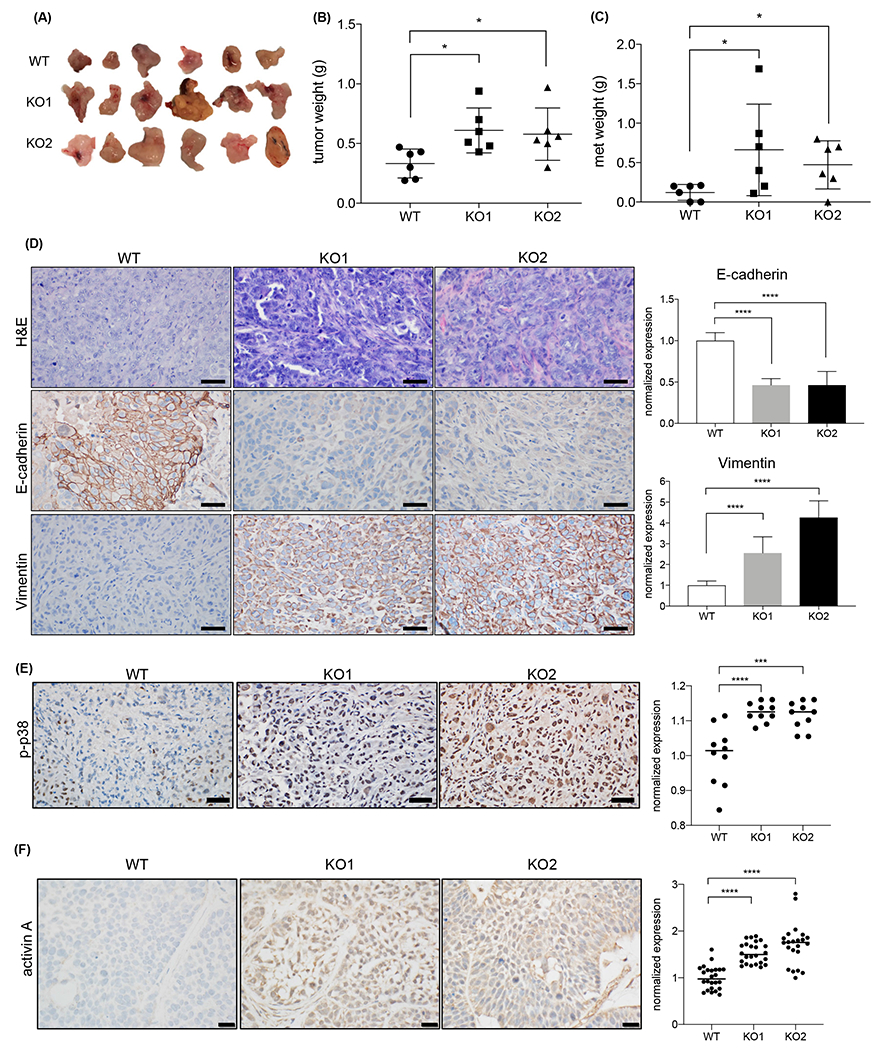
KMT2D depletion promotes both primary and metastatic PDAC growth and EMT in vivo. (A) Tumor harvested from WT and KMT2D KO BxPC-3 cell orthotopic xenograft four weeks after injection (n=6 each group). (B, C) Weight of primary (B) and metastatic (C) tumors harvested from WT and KMT2D KO orthotopic xenograft mouse model (*p<0.05, unpaired student t-test, n=6). (D) Representative H&E and IHC stains of E-cadherin and Vimentin in WT and KMT2D KO PANC-1 orthotopic xenograft tumors. (scale bar = 50 μm). Quantification of IHC stains of E-cadherin and Vimentin in WT and KMT2D KO tumors is represented as bar graphs. (**p<0.01, ***p<0.005, ****p<0.001, One-way ANOVA test with Tukey’s multiple comparisons, n=30) (E) Representative pictures of p-p38 IHC staining in PANC-1 xenograft tumor. Number of p-p38 positive cells in WT and KMT2D KO PANC-1 cell orthotopic xenograft tumors are plotted. Five random fields per animal were examined. Each data point represents one field. (***p<0.005, ****p<0.0001, One-way ANOVA test with Dunnett’s multiple comparisons, n=10). (F) Representative pictures and quantification of activin A IHC staining in BxPC-3 xenograft tumors. (****p<0.0001, One-way ANOVA test with Dunnett’s multiple comparisons, n=22-24)
Discussion
Treating pancreatic cancer patients is particularly challenging due to the early occurrence of metastasis. Multiple groups performed comparative genomic analysis between matching primary and metastatic tumors but failed to find metastasis-specific gene8,36. This observation led to the hypothesis that epigenetic alterations may regulate PDAC metastasis. Indeed, chromatin remodeling epigenetic regulators, including KMT2D, were identified as potential drivers for pancreatic cancer progression5. However, our understanding on how epigenetic dysregulation affects pancreatic cancer behavior is still elementary. Here, we discovered KMT2D as a new critical link between the TGF-β and non-canonical activin signaling to promote EMT and PDAC progression (Fig. 8).
Fig. 8.
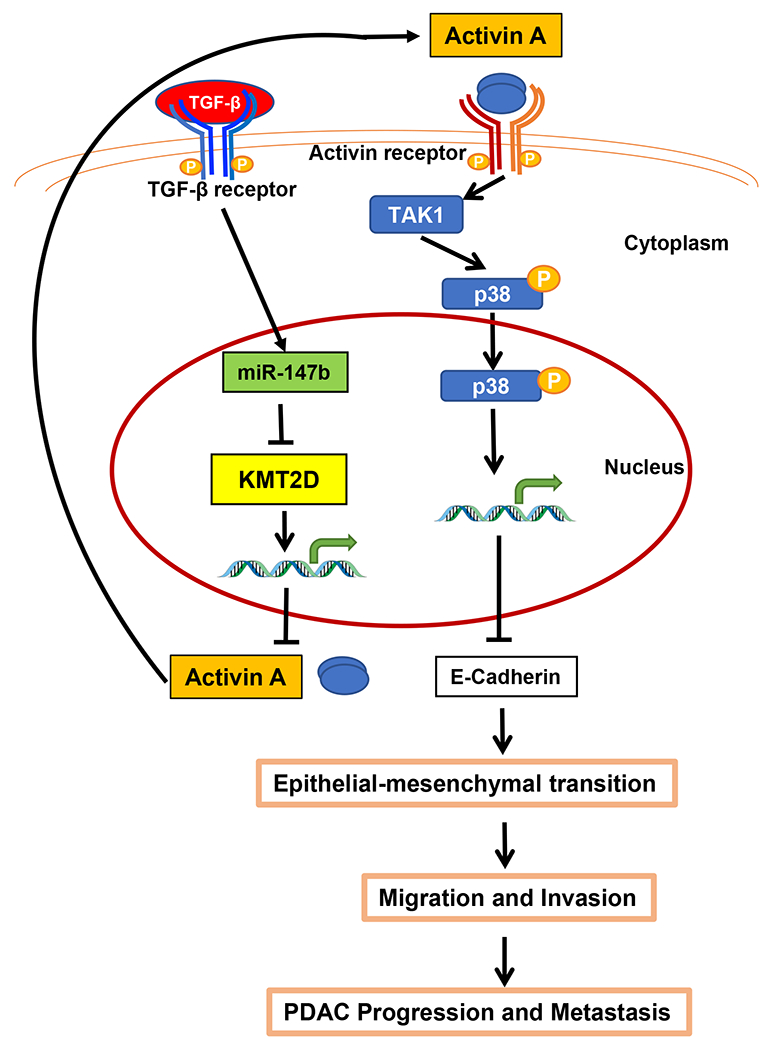
Overall hypothesis: KMT2D links TGF-β and activin A signaling. TGF-β induces KMT2D mRNA degradation by the upregulation of miR-147b. Decreased expression of KMT2D subsequently upregulates the transcription of INHBA/activin A, which then promotes EMT, cell migration and invasion, tumorigenicity, PDAC progression, and metastasis via a p38-mediated non-canonical pathway.
TGF-β is one of the most critical cytokines in pancreatic carcinogenesis. However, its highly context-dependent dual pro- and anti-tumor function is not fully understood and thus hindered the development of targeting strategies. Elevated TGF-β level is associated with poor survival in pancreatic cancer patients37. Tumor supportive role of TGF-β in pancreatic cancer sparked much interest in search for metastasis driving genes regulated by TGF-β. Global evaluation of the effect of TGF-β on PDAC cells has been performed on steady-state gene expression using either conventional high throughput RNA-Seq or gene microarray analysis38,39. However, the effect of TGF-β signaling on pancreatic cancer epigenetics is not well studied. Other groups have reported that TGF-β induces EMT through the upregulation of epigenetic regulators, KDM4B40 and KDM6B41. However, past studies did not identify how TGF-β regulated the expression of these epigenetic regulators. In this study, we used Bru-seq and BruChase-seq technologies to obtain a readout of RNA synthesis and stability in TGF-β treated cells. Bru-seq technology measures de novo RNA transcription independently from pre-existing RNAs (Bru-seq), as well as RNA stability (BruChase-seq)16. Using this unique technology, we discovered that TGF-β downregulates epigenetic regulatory protein, KMT2D, expression by post-transcriptional gene silencing.
Much work has been done to characterize distinct microRNAs expressed in pancreatic cancer compared to normal tissue42–44. We report here that miR-147b expression increases with TGF-β treatment, attenuates KMT2D expression, and subsequently allows the cancer cells to acquire a mesenchymal phenotype. Others have also reported the pro-tumor role of miR-147b37–39. For example, miR-147b increases the tumorigenicity in hepatocellular carcinoma by targeting ubiquitin-conjugating enzyme E2N45. In patients with hepatitis C-associated diffuse large B-cell lymphoma, high miR-147b level is correlated with poor prognosis46. Recent work in lung cancer showed that miR-147b endows enhanced drug resistance by altering tricarboxylic acid cycle22. These findings suggest the possibility of using miR-147b as a therapeutic target for multiple tumor types.
Alterations in the KMT2D gene have been reported for multiple types of cancers47. Inactivating KMT2D mutations have been associated with poor prognosis of some cancers such as non-small-cell lung cancer48, while high KMT2D expression predicts poor prognosis and promotes tumor progression in other cancers, including esophageal squamous cell carcinoma49. Dawkins and colleagues reported that reduced expression of KMT2D is associated with improved survival of PDAC patients50. The difference between the study by Dawkins et al. and our study is two-fold. First, they used ICGC dataset while we used TCGA dataset. Secondly, they compared KMT2D expression using ICGC gene expression profile data, while we compared between patients with KMT2D mutation and wild-type KMT2D. Therefore, the two studies represented two different datasets and KMT2D alterations. A more recent study by Koutsioumpa and colleagues showed that low KMT2D expression increased proliferation and tumorigenicity in PDAC by altering glucose and lipid metabolism10. Our study is in support of Koutsioumpa’s findings, showing that primary and metastatic PDACs had significantly lower expression of KMT2D and loss of KMT2D promoted PDAC invasion and migration, which supports a tumor-suppressive role of KMT2D in PDAC. Loss of KMT2D also induced a gene signature closely resembling the Moffitt basal-like subtype, which confers a significantly worse prognosis compared to the classical subtype17,51,52. These PDAC subtypes have distinct epigenetic landscapes that drive transcriptional alterations. Our study has suggested that KMT2D is a crucial epigenetic regulator that contributes to the basal-like subtype of PDAC. Furthermore, we discovered that activin A pathway was activated by KMT2D loss through non-canonical signaling via p38 MAPK.
Activin A belongs to the TGF-β superfamily and is best studied for its role in mesoderm cell fate determination during embryogenesis53,54. Activin A was recently shown to promote wound healing, angiogenesis, inflammation, immunity, and drive self-renewal and tumorigenicity of pancreatic cancer stem cells55–59. However, its role in pancreatic cancer development is not well studied. Like TGF-β, activin A post-natal expression is tightly regulated and highly tissue-specific. Normal pancreas expresses negligible activin A, according to the Human Protein Atlas. Activins are homo- or hetero-dimers of activin β subunits. Currently, there are three known bioactive activin dimers: activins A (βAβA), B (βBβB) and AB (βAβB)60,61. After secreted from the cell, activin A binds to ActRII/IIB, which preferentially recruits a type I activin receptor, ALK4 (activin receptor-like kinase 4), to form a heterodimer to recruit and phosphorylate SMAD2/3 in a similar mechanism to TGF-β27–29. In addition to the canonical pathway, non-canonical pathways, such as PI3K/Akt, ERK, JNK, and p38, have been associated with activin A function independent of SMAD activation30,31. In our study, we found that activin A and p38 MAPK are required for loss of KMT2D-induced EMT and enhanced tumor cell invasion since inhibition or knockdown of either activin or p38 MAPK attenuated EMT and tumor cell invasion induced by KMT2D loss. These findings revealed activin A and p38 MAPK as novel potential therapeutic targets for PDAC patients with low KMT2D.
In summary, we uncovered an essential crosstalk between TGF-β, KMT2D, and activin signaling to promote PDAC cell plasticity, basal-like gene signature, tumorigenicity, invasion, and metastasis. These findings shed light on the poorly understood functions of KMT2D and activin signaling in PDAC progression and filled in the gap of our knowledge on TGF-β function. More importantly, we identified miR-147b and activin A as novel potential therapeutic targets in PDAC.
Supplementary Material
Novelty and Impact:
The role of KMT2D in pancreatic cancer development remains elusive. We discovered that TGF-β upregulates miR-147b which in turn leads to the silencing of KMT2D. Loss of KMT2D activates a non-canonical p38 MAPK-mediated activin A pathway to modulate cancer cell plasticity and enhance tumor invasion and metastasis in mice. Furthermore, inhibition or knockdown of activin A reversed the pro-tumoral role of KMT2D loss. These findings identify miR-147b and activin A as novel therapeutic targets.
Acknowledgments
Dr. Jiaqi Shi is supported in part by the National Cancer Institute of the National Institutes of Health under award number K08CA234222 and R37CA262209. The authors would like to thank the lab members of Drs. Costas Lyssiotis, James Moon, and Timothy Frankel for sharing reagents, resources, and providing advice.
Abbreviations:
- ChIP
chromatin immunoprecipitation
- ELISA
enzyme-linked immunosorbent assay
- EMT
epithelial to mesenchymal transition
- GSEA
gene set enrichment analysis
- H3K4
lysine 4 of histone 3
- H&E
hematoxylin and eosin
- HPDE
human pancreatic ductal epithelial
- IHC
immunohistochemical
- IPMN
intraductal papillary mucinous neoplasm
- MAPK
mitogen-activated protein kinase
- MCN
mucinous cystic neoplasm
- NSG
NOD-SCID IL2Rgammanull
- PanIN
pancreatic intraepithelial neoplasia
- PBS
phosphate-buffered saline
- PDAC
pancreatic ductal adenocarcinoma
- sgRNAs
synthesized guide RNAs
- TCGA
the cancer genome atlas
- TGF-β
transforming growth factor-β
- TMA
tissue microarray
Footnotes
Conflict of Interest
The authors declare that they have no conflict of interest.
Ethics Statement
The use of patient tissue was approved by the Institutional Review Board at the University of Michigan (protocol number: HUM00098128). All animal studies were carried out under the protocol approved by the Institutional Animal Care and Use Committee at the University of Michigan (protocol ID: PRO00009389).
Data Availability Statement
All data needed for interpretation of the results are presented in this paper and its supplementary information files. The raw Bru-seq data generated in this study are available in GEO under accession number GSE199856 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi). Other data that support the findings of this study are available from the corresponding author upon request.
References:
- 1.Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM & Matrisian LM Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014; 74(11): 2913–2921. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Fuchs HE & Jemal A Cancer Statistics, 2021. CA Cancer J Clin. 2021; 71(1): 7–33. [DOI] [PubMed] [Google Scholar]
- 3.Rao RC & Dou Y Hijacked in cancer: the KMT2 (MLL) family of methyltransferases. Nat Rev Cancer. 2015; 15(6): 334–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roe JS, Hwang CI, Somerville TDD, Milazzo JP, Lee EJ, Da Silva B, Maiorino L, Tiriac H, Young CM, Miyabayashi K, Filippini D, Creighton B, Burkhart RA, Buscaglia JM, Kim EJ, Grem JL, Lazenby AJ, Grunkemeyer JA, Hollingsworth MA, Grandgenett PM, Egeblad M, Park Y, Tuveson DA & Vakoc CR Enhancer Reprogramming Promotes Pancreatic Cancer Metastasis. Cell. 2017; 170(5): 875–888 e820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, Johns AL, Miller D, Nones K, Quek K, Quinn MC, Robertson AJ, Fadlullah MZ, Bruxner TJ, Christ AN, Harliwong I, Idrisoglu S, Manning S, Nourse C, Nourbakhsh E, Wani S, Wilson PJ, Markham E, Cloonan N, Anderson MJ, Fink JL, Holmes O, Kazakoff SH, Leonard C, Newell F, Poudel B, Song S, Taylor D, Waddell N, Wood S, Xu Q, Wu J, Pinese M, Cowley MJ, Lee HC, Jones MD, Nagrial AM, Humphris J, Chantrill LA, Chin V, Steinmann AM, Mawson A, Humphrey ES, Colvin EK, Chou A, Scarlett CJ, Pinho AV, Giry-Laterriere M, Rooman I, Samra JS, Kench JG, Pettitt JA, Merrett ND, Toon C, Epari K, Nguyen NQ, Barbour A, Zeps N, Jamieson NB, Graham JS, Niclou SP, Bjerkvig R, Grutzmann R, Aust D, Hruban RH, Maitra A, Iacobuzio-Donahue CA, Wolfgang CL, Morgan RA, Lawlor RT, Corbo V, Bassi C, Falconi M, Zamboni G, Tortora G, Tempero MA, Gill AJ, Eshleman JR, Pilarsky C, Scarpa A, Musgrove EA, Pearson JV, Biankin AV & Grimmond SM Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015; 518(7540): 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, Miller DK, Christ AN, Bruxner TJ, Quinn MC, Nourse C, Murtaugh LC, Harliwong I, Idrisoglu S, Manning S, Nourbakhsh E, Wani S, Fink L, Holmes O, Chin V, Anderson MJ, Kazakoff S, Leonard C, Newell F, Waddell N, Wood S, Xu Q, Wilson PJ, Cloonan N, Kassahn KS, Taylor D, Quek K, Robertson A, Pantano L, Mincarelli L, Sanchez LN, Evers L, Wu J, Pinese M, Cowley MJ, Jones MD, Colvin EK, Nagrial AM, Humphrey ES, Chantrill LA, Mawson A, Humphris J, Chou A, Pajic M, Scarlett CJ, Pinho AV, Giry-Laterriere M, Rooman I, Samra JS, Kench JG, Lovell JA, Merrett ND, Toon CW, Epari K, Nguyen NQ, Barbour A, Zeps N, Moran-Jones K, Jamieson NB, Graham JS, Duthie F, Oien K, Hair J, Grutzmann R, Maitra A, Iacobuzio-Donahue CA, Wolfgang CL, Morgan RA, Lawlor RT, Corbo V, Bassi C, Rusev B, Capelli P, Salvia R, Tortora G, Mukhopadhyay D, Petersen GM, Munzy DM, Fisher WE, Karim SA, Eshleman JR, Hruban RH, Pilarsky C, Morton JP, Sansom OJ, Scarpa A, Musgrove EA, Bailey UM, Hofmann O, Sutherland RL, Wheeler DA, Gill AJ, Gibbs RA, Pearson JV, Waddell N, Biankin AV & Grimmond SM Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016; 531(7592): 47–52. [DOI] [PubMed] [Google Scholar]
- 7.Bird A DNA methylation patterns and epigenetic memory. Genes Dev. 2002; 16(1): 6–21. [DOI] [PubMed] [Google Scholar]
- 8.McDonald OG, Li X, Saunders T, Tryggvadottir R, Mentch SJ, Warmoes MO, Word AE, Carrer A, Salz TH, Natsume S, Stauffer KM, Makohon-Moore A, Zhong Y, Wu H, Wellen KE, Locasale JW, Iacobuzio-Donahue CA & Feinberg AP Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat Genet. 2017; 49(3): 367–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang J, Dominguez-Sola D, Hussein S, Lee JE, Holmes AB, Bansal M, Vlasevska S, Mo T, Tang H, Basso K, Ge K, Dalla-Favera R & Pasqualucci L Disruption of KMT2D perturbs germinal center B cell development and promotes lymphomagenesis. Nat Med. 2015; 21(10): 1190–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koutsioumpa M, Hatziapostolou M, Polytarchou C, Tolosa EJ, Almada LL, Mahurkar-Joshi S, Williams J, Tirado-Rodriguez AB, Huerta-Yepez S, Karavias D, Kourea H, Poultsides GA, Struhl K, Dawson DW, Donahue TR, Fernandez-Zapico ME & Iliopoulos D Lysine methyltransferase 2D regulates pancreatic carcinogenesis through metabolic reprogramming. Gut. 2019; 68(7): 1271–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yoon KA, Woo SM, Kim YH, Kong SY, Lee MK, Han SS, Kim TH, Lee WJ & Park SJ Comprehensive Cancer Panel Sequencing Defines Genetic Diversity and Changes in the Mutational Characteristics of Pancreatic Cancer Patients Receiving Neoadjuvant Treatment. Gut Liver. 2019; 13(6): 683–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yilmaz AS, Ozer HG, Gillespie JL, Allain DC, Bernhardt MN, Furlan KC, Castro LT, Peters SB, Nagarajan P, Kang SY, Iwenofu OH, Olencki T, Teknos TN & Toland AE Differential mutation frequencies in metastatic cutaneous squamous cell carcinomas versus primary tumors. Cancer. 2017; 123(7): 1184–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Massagué J TGFβ signalling in context. Nature Reviews Molecular Cell Biology. 2012; 13(616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heldin CH, Vanlandewijck M & Moustakas A Regulation of EMT by TGFbeta in cancer. FEBS Lett. 2012; 586(14): 1959–1970. [DOI] [PubMed] [Google Scholar]
- 15.Lefkofsky HB, Veloso A & Ljungman M Transcriptional and post-transcriptional regulation of nucleotide excision repair genes in human cells. Mutat Res. 2015; 776(9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Paulsen MT, Veloso A, Prasad J, Bedi K, Ljungman EA, Magnuson B, Wilson TE & Ljungman M Use of Bru-Seq and BruChase-Seq for genome-wide assessment of the synthesis and stability of RNA. Methods. 2014; 67(1): 45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moffitt RA, Marayati R, Flate EL, Volmar KE, Loeza SG, Hoadley KA, Rashid NU, Williams LA, Eaton SC, Chung AH, Smyla JK, Anderson JM, Kim HJ, Bentrem DJ, Talamonti MS, Iacobuzio-Donahue CA, Hollingsworth MA & Yeh JJ Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet. 2015; 47(10): 1168–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bereshchenko OR, Gu W & Dalla-Favera R Acetylation inactivates the transcriptional repressor BCL6. Nat Genet. 2002; 32(4): 606–613. [DOI] [PubMed] [Google Scholar]
- 19.Livak KJ & Schmittgen TD Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001; 25(4): 402–408. [DOI] [PubMed] [Google Scholar]
- 20.Nguyen N, Bellile E, Thomas D, McHugh J, Rozek L, Virani S, Peterson L, Carey TE, Walline H, Moyer J, Spector M, Perim D, Prince M, McLean S, Bradford CR, Taylor JM & Wolf GT Tumor infiltrating lymphocytes and survival in patients with head and neck squamous cell carcinoma. Head Neck. 2016; 38(7): 1074–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wen F, Shen A, Choi A, Gerner EW & Shi J Extracellular DNA in pancreatic cancer promotes cell invasion and metastasis. Cancer Res. 2013; 73(14): 4256–4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang WC, Wells JM, Chow KH, Huang H, Yuan M, Saxena T, Melnick MA, Politi K, Asara JM, Costa DB, Bult CJ & Slack FJ miR-147b-mediated TCA cycle dysfunction and pseudohypoxia initiate drug tolerance to EGFR inhibitors in lung adenocarcinoma. Nat Metab. 2019; 1(4): 460–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, Reddy A, Liu M, Murray L, Berger MF, Monahan JE, Morais P, Meltzer J, Korejwa A, Jane-Valbuena J, Mapa FA, Thibault J, Bric-Furlong E, Raman P, Shipway A, Engels IH, Cheng J, Yu GK, Yu J, Aspesi P Jr., de Silva M, Jagtap K, Jones MD, Wang L, Hatton C, Palescandolo E, Gupta S, Mahan S, Sougnez C, Onofrio RC, Liefeld T, MacConaill L, Winckler W, Reich M, Li N, Mesirov JP, Gabriel SB, Getz G, Ardlie K, Chan V, Myer VE, Weber BL, Porter J, Warmuth M, Finan P, Harris JL, Meyerson M, Golub TR, Morrissey MP, Sellers WR, Schlegel R & Garraway LA The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012; 483(7391): 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wamsley JJ, Kumar M, Allison DF, Clift SH, Holzknecht CM, Szymura SJ, Hoang SA, Xu X, Moskaluk CA, Jones DR, Bekiranov S & Mayo MW Activin upregulation by NF-κB is required to maintain mesenchymal features of cancer stem-like cells in non-small cell lung cancer. Cancer Res. 2015; 75(2): 426–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bashir M, Damineni S, Mukherjee G & Kondaiah P Activin-A signaling promotes epithelial-mesenchymal transition, invasion, and metastatic growth of breast cancer. NPJ Breast Cancer. 2015; 1(15007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bauer J, Ozden O, Akagi N, Carroll T, Principe DR, Staudacher JJ, Spehlmann ME, Eckmann L, Grippo PJ & Jung B Activin and TGFβ use diverging mitogenic signaling in advanced colon cancer. Mol Cancer. 2015; 14(182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gurdon JB, Harger P, Mitchell A & Lemaire P Activin signalling and response to a morphogen gradient. Nature. 1994; 371(6497): 487–492. [DOI] [PubMed] [Google Scholar]
- 28.Hashimoto O, Yamato K, Koseki T, Ohguchi M, Ishisaki A, Shoji H, Nakamura T, Hayashi Y, Sugino H & Nishihara T The role of activin type I receptors in activin A-induced growth arrest and apoptosis in mouse B-cell hybridoma cells. Cell Signal. 1998; 10(10): 743–749. [DOI] [PubMed] [Google Scholar]
- 29.Symes K, Yordan C & Mercola M Morphological differences in Xenopus embryonic mesodermal cells are specified as an early response to distinct threshold concentrations of activin. Development. 1994; 120(8): 2339–2346. [DOI] [PubMed] [Google Scholar]
- 30.McDowell N, Zorn AM, Crease DJ & Gurdon JB Activin has direct long-range signalling activity and can form a concentration gradient by diffusion. Curr Biol. 1997; 7(9): 671–681. [DOI] [PubMed] [Google Scholar]
- 31.Heldin CH, Landstrom M & Moustakas A Mechanism of TGF-beta signaling to growth arrest, apoptosis, and epithelial-mesenchymal transition. Curr Opin Cell Biol. 2009; 21(2): 166–176. [DOI] [PubMed] [Google Scholar]
- 32.Kim SI, Kwak JH, Na HJ, Kim JK, Ding Y & Choi ME Transforming growth factor-beta (TGF-beta1) activates TAK1 via TAB1-mediated autophosphorylation, independent of TGF-beta receptor kinase activity in mesangial cells. J Biol Chem. 2009; 284(33): 22285–22296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Safina A, Ren MQ, Vandette E & Bakin AV TAK1 is required for TGF-beta 1-mediated regulation of matrix metalloproteinase-9 and metastasis. Oncogene. 2008; 27(9): 1198–1207. [DOI] [PubMed] [Google Scholar]
- 34.Somerville TDD, Xu Y, Miyabayashi K, Tiriac H, Cleary CR, Maia-Silva D, Milazzo JP, Tuveson DA & Vakoc CR TP63-Mediated Enhancer Reprogramming Drives the Squamous Subtype of Pancreatic Ductal Adenocarcinoma. Cell Rep. 2018; 25(7): 1741–1755.e1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hamdan FH & Johnsen SA DeltaNp63-dependent super enhancers define molecular identity in pancreatic cancer by an interconnected transcription factor network. Proc Natl Acad Sci U S A. 2018; 115(52): E12343–e12352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Makohon-Moore AP, Zhang M, Reiter JG, Bozic I, Allen B, Kundu D, Chatterjee K, Wong F, Jiao Y, Kohutek ZA, Hong J, Attiyeh M, Javier B, Wood LD, Hruban RH, Nowak MA, Papadopoulos N, Kinzler KW, Vogelstein B & Iacobuzio-Donahue CA Limited heterogeneity of known driver gene mutations among the metastases of individual patients with pancreatic cancer. Nat Genet. 2017; 49(3): 358–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Friess H, Yamanaka Y, Buchler M, Ebert M, Beger HG, Gold LI & Korc M Enhanced expression of transforming growth factor beta isoforms in pancreatic cancer correlates with decreased survival. Gastroenterology. 1993; 105(6): 1846–1856. [DOI] [PubMed] [Google Scholar]
- 38.Maupin KA, Sinha A, Eugster E, Miller J, Ross J, Paulino V, Keshamouni VG, Tran N, Berens M, Webb C & Haab BB Glycogene expression alterations associated with pancreatic cancer epithelial-mesenchymal transition in complementary model systems. PLoS One. 2010; 5(9): e13002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.David CJ, Huang YH, Chen M, Su J, Zou Y, Bardeesy N, Iacobuzio-Donahue CA & Massague J TGF-beta Tumor Suppression through a Lethal EMT. Cell. 2016; 164(5): 1015–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li S, Wu L, Wang Q, Li Y & Wang X KDM4B promotes epithelial-mesenchymal transition through up-regulation of ZEB1 in pancreatic cancer. Acta Biochim Biophys Sin (Shanghai). 2015; 47(12): 997–1004. [DOI] [PubMed] [Google Scholar]
- 41.Ramadoss S, Chen X & Wang CY Histone demethylase KDM6B promotes epithelial-mesenchymal transition. J Biol Chem. 2012; 287(53): 44508–44517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M, Prueitt RL, Yanaihara N, Lanza G, Scarpa A, Vecchione A, Negrini M, Harris CC & Croce CM A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A. 2006; 103(7): 2257–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roldo C, Missiaglia E, Hagan JP, Falconi M, Capelli P, Bersani S, Calin GA, Volinia S, Liu CG, Scarpa A & Croce CM MicroRNA expression abnormalities in pancreatic endocrine and acinar tumors are associated with distinctive pathologic features and clinical behavior. J Clin Oncol. 2006; 24(29): 4677–4684. [DOI] [PubMed] [Google Scholar]
- 44.Bloomston M, Frankel WL, Petrocca F, Volinia S, Alder H, Hagan JP, Liu CG, Bhatt D, Taccioli C & Croce CM MicroRNA expression patterns to differentiate pancreatic adenocarcinoma from normal pancreas and chronic pancreatitis. Jama. 2007; 297(17): 1901–1908. [DOI] [PubMed] [Google Scholar]
- 45.Zhang E, Liu Q, Wang Y, Wang H, He L, Jin X & Li N MicroRNA miR-147b promotes tumor growth via targeting UBE2N in hepatocellular carcinoma. Oncotarget. 2017; 8(69): 114072–114080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Augello C, Gianelli U, Savi F, Moro A, Bonoldi E, Gambacorta M, Vaira V, Baldini L & Bosari S MicroRNA as potential biomarker in HCV-associated diffuse large B-cell lymphoma. J Clin Pathol. 2014; 67(8): 697–701. [DOI] [PubMed] [Google Scholar]
- 47.Ford DJ & Dingwall AK The cancer COMPASS: navigating the functions of MLL complexes in cancer. Cancer Genet. 2015; 208(5): 178–191. [DOI] [PubMed] [Google Scholar]
- 48.Ardeshir-Larijani F, Bhateja P, Lipka MB, Sharma N, Fu P & Dowlati A KMT2D Mutation Is Associated With Poor Prognosis in Non-Small-Cell Lung Cancer. Clin Lung Cancer. 2018; 19(4): e489–e501. [DOI] [PubMed] [Google Scholar]
- 49.Abudureheman A, Ainiwaer J, Hou Z, Niyaz M, Turghun A, Hasim A, Zhang H, Lu X & Sheyhidin I High MLL2 expression predicts poor prognosis and promotes tumor progression by inducing EMT in esophageal squamous cell carcinoma. J Cancer Res Clin Oncol. 2018; 144(6): 1025–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dawkins JB, Wang J, Maniati E, Heward JA, Koniali L, Kocher HM, Martin SA, Chelala C, Balkwill FR, Fitzgibbon J & Grose RP Reduced Expression of Histone Methyltransferases KMT2C and KMT2D Correlates with Improved Outcome in Pancreatic Ductal Adenocarcinoma. Cancer Res. 2016; 76(16): 4861–4871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Camolotto SA, Belova VK & Snyder EL The role of lineage specifiers in pancreatic ductal adenocarcinoma. J Gastrointest Oncol. 2018; 9(6): 1005–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Juiz NA, Iovanna J & Dusetti N Pancreatic Cancer Heterogeneity Can Be Explained Beyond the Genome. Front Oncol. 2019; 9(246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Smith JC, Price BM, Van Nimmen K & Huylebroeck D Identification of a potent Xenopus mesoderm-inducing factor as a homologue of activin A. Nature. 1990; 345(6277): 729–731. [DOI] [PubMed] [Google Scholar]
- 54.Nazareth EJP, Rahman N, Yin T & Zandstra PW A Multi-Lineage Screen Reveals mTORC1 Inhibition Enhances Human Pluripotent Stem Cell Mesendoderm and Blood Progenitor Production. Stem Cell Reports. 2016; 6(5): 679–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tao JJ, Cangemi NA, Makker V, Cadoo KA, Liu JF, Rasco DW, Navarro WH, Haqq CM & Hyman DM First-in-Human Phase I Study of the Activin A Inhibitor, STM 434, in Patients with Granulosa Cell Ovarian Cancer and Other Advanced Solid Tumors. Clin Cancer Res. 2019; 25(18): 5458–5465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Steller MD, Shaw TJ, Vanderhyden BC & Ethier JF Inhibin resistance is associated with aggressive tumorigenicity of ovarian cancer cells. Mol Cancer Res. 2005; 3(1): 50–61. [PubMed] [Google Scholar]
- 57.Lonardo E, Hermann PC, Mueller MT, Huber S, Balic A, Miranda-Lorenzo I, Zagorac S, Alcala S, Rodriguez-Arabaolaza I, Ramirez JC, Torres-Ruiz R, Garcia E, Hidalgo M, Cebrian DA, Heuchel R, Lohr M, Berger F, Bartenstein P, Aicher A & Heeschen C Nodal/Activin signaling drives self-renewal and tumorigenicity of pancreatic cancer stem cells and provides a target for combined drug therapy. Cell Stem Cell. 2011; 9(5): 433–446. [DOI] [PubMed] [Google Scholar]
- 58.Lonardo E, Frias-Aldeguer J, Hermann PC & Heeschen C Pancreatic stellate cells form a niche for cancer stem cells and promote their self-renewal and invasiveness. Cell Cycle. 2012; 11(7): 1282–1290. [DOI] [PubMed] [Google Scholar]
- 59.Antsiferova M & Werner S The bright and the dark sides of activin in wound healing and cancer. J Cell Sci. 2012; 125(Pt 17): 3929–3937. [DOI] [PubMed] [Google Scholar]
- 60.Green JB, New HV & Smith JC Responses of embryonic Xenopus cells to activin and FGF are separated by multiple dose thresholds and correspond to distinct axes of the mesoderm. Cell. 1992; 71(5): 731–739. [DOI] [PubMed] [Google Scholar]
- 61.Katz LH, Li Y, Chen JS, Munoz NM, Majumdar A, Chen J & Mishra L Targeting TGF-beta signaling in cancer. Expert Opin Ther Targets. 2013; 17(7): 743–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed for interpretation of the results are presented in this paper and its supplementary information files. The raw Bru-seq data generated in this study are available in GEO under accession number GSE199856 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi). Other data that support the findings of this study are available from the corresponding author upon request.