

Abstract
Purpose.
To overcome barriers to genomic testing for patients with rare cancers, we initiated a program to offer free clinical tumor genomic testing worldwide to patients with select rare cancer subtypes.
Patients and Methods.
Patients were recruited through social media outreach and engagement with disease-specific advocacy groups, with a focus on patients with histiocytosis, germ cell tumors, and pediatric cancers. Tumors were analyzed using the MSK-IMPACT next generation sequencing assay with the return of results to patients and their local physicians. Whole exome recapture was performed for female patients with germ cell tumors to define the genomic landscape of this rare cancer subtype.
Results.
333 patients were enrolled, and tumor tissue was received for 288 (86.4%), with 250 (86.8%) having tumor DNA of sufficient quality for MSK-IMPACT testing. Eighteen patients with histiocytosis have received genomically guided therapy to date, of whom 17 (94%) have had clinical benefit with a mean treatment duration of 21.7 months (range 6–40+). Whole exome sequencing of ovarian GCTs identified a subset with haploid genotypes, a phenotype rarely observed in other cancer types. Actionable genomic alterations were rare in ovarian GCT (28%), however, two patients with ovarian GCTs with squamous transformation had high tumor mutational burden, one of whom had a complete response to pembrolizumab.
Conclusion.
Direct-to-patient outreach can facilitate the assembly of cohorts of rare cancers of sufficient size to define their genomic landscape. By profiling tumors in a clinical laboratory, results could be reported to patients and their local physicians to guide treatment.
Introduction
Tumor genomic profiling is a standard component of the diagnostic evaluation of an increasing number of cancer subtypes. Genomic analysis of DNA derived from tumors and patient-matched normal DNA can confirm tumor diagnosis and subtyping, assess for heritable cancer risk, and identify actionable genomic alterations as a guide to therapy selection (1). While retrospective and more recently prospective clinical studies have defined the genomic landscape of common solid tumors, including cancers of the lung, colon, and prostate (2–7), the frequency of actionable genomic alterations remains poorly defined for many rare cancers. Additionally, despite recent tumor agnostic drug approvals, the paucity of data demonstrating the clinical benefit of tumor genomic profiling for patients with rare cancers limits insurance coverage and access to multi-gene next generation sequencing-based tumor genomic profiling with return of results at the point-of-care (8,9).
To overcome barriers to tumor genomic testing for patients with rare cancers, we initiated the Make-an-IMPACT program to offer free clinical tumor genomic testing worldwide to patients with select rare cancers. Patients were identified through social media outreach and additional crowdsourcing efforts such as partnerships with disease-specific advocacy groups. As opposed to prior discovery focused crowdsourcing initiatives (10), the Make-an-IMPACT program included clinical MSK-IMPACT tumor sequencing with return of results to patients to allow genomic findings to be used by local physicians to guide treatment selection.
As initial pilot cancer types, we focused on histiocytosis and female patients with germ cell tumors. Histiocytosis, which includes Langerhans Cell Histiocytosis and Erdheim-Chester Disease, was chosen given its rarity (4–10 cases per one million population), and the high likelihood that tumor genomic testing would alter clinical management. Somatic BRAF V600E mutations are present in approximately half of patients with Langerhans Cell Histiocytosis and Erdheim-Chester Disease, and robust and durable responses have been described with BRAF inhibitors for these entities, resulting in FDA approval of vemurafenib for BRAF V600E-mutated Erdheim-Chester Disease (11–15). MEK inhibitors have also been shown to confer therapeutic benefit for patients with histiocytosis whose tumors harbor mutations in several mitogen activated protein kinase (MAPK) pathway genes including RAS isoforms, ARAF, RAF1, and MAP2K1/2 (16). Female germ cell tumor was chosen given the lack of prior knowledge as to the molecular drivers of this rare cancer subtype (incidence of ~1/100,000 women/year) and the lack of treatment options for cisplatin-resistant disease (17). Patients with a diversity of pediatric and young adult tumors who were never offered tumor genomic profiling due to a lack of availability at their primary treatment site or because of gaps in insurance coverage were also eligible.
The primary goals were to 1) assess the feasibility of recruiting patients with rare cancers for tumor genomic profiling studies through direct-to-patient outreach via advocacy groups and social media platforms such as Facebook and Twitter, 2) determine whether tumor genomic profiling could identify actionable genomic alterations for patients with treatment refractory rare cancers, and 3) define the genomic landscape of rare tumor types such as ovarian germ cell tumors that are difficult to study at a single institution due to their low incidence.
METHODS AND MATERIALS
Accrual and Consent.
Beginning in 2016, patients were screened for eligibility for the Make-an-IMPACT program following a physician referral (49% of patients enrolled), referral by a disease specific advocacy group (27%) or following collection of contact information via the program website (24%). Following an initial screen to confirm eligibility, patients were sent an enrollment packet by mail, and patients who wished to proceed were then consented by telephone to an IRB approved protocol (NCT01775072), which allows for clinical tumor genomic profiling with return of results to patients as well as subsequent research analyses including whole exome sequencing. Diagnostic confirmation and clinical tumor genomic profiling were performed at no cost to the patient. Beginning in 2019, Memorial Sloan Kettering Cancer Center (MSK)’s electronic platform for virtual consenting, e-consent, became an option to facilitate these processes. Consent forms were available in a variety of languages and foreign language interpreters were available, if needed, for the consent discussion. Patient demographic and treatment data were collected directly from patients and/or their parent/guardian, if applicable, and through review of outside hospital medical records.
MSK-IMPACT testing.
Following consent, an FFPE tumor block or 20 unstained slides and at least one H&E slide were requested from the patient’s local physician. Patients were provided with a prepaid postage shipping container with an EDTA blood tube or a nail or saliva kit as a source of germline DNA. Centralized pathology review was performed with a pathology report transmitted to the local physician for all patients to confirm cancer subtype diagnosis and to ensure that sufficient tumor was present in the sample for next generation sequencing. Clinical tumor genomic profiling was performed using the Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT) assay (18,19). MSK-IMPACT is an FDA- authorized next generation sequencing assay that can detect mutations, copy number alterations and translocations in up to 505 cancer associated genes depending on the assay version. While analysis of germline DNA for pathogenic variants in genes associated with increased heritable cancer risk is now routinely performed for MSK patients undergoing MSK- IMPACT testing (20), clinical germline analysis was not performed on patients enrolled in the Make-an-IMPACT study. Select patients also had targeted RNA sequencing to confirm a suspected fusion identified by MSK-IMPACT or to assess for an occult fusion in samples with no identified mitogenic driver mutation (21).
Whole exome sequencing.
For select patients (n=59) with ovarian germ cell tumors, leftover sequencing libraries were used to perform whole exome sequencing. Briefly, whole exome recapture of the MSK-IMPACT tumor and normal sequencing libraries was performed using remaining barcoded library captured by hybridization using either the SureSelectXT Human All Exon V4 (Agilent 5190–4632) or xGen Exome Research Panel v1.0 (Integrated DNA Technologies) according to the manufacturer’s protocol. PCR amplification of the post-capture libraries was carried out for 8 cycles followed by sequencing as previously described (22). Indel realignment was performed using the Assembly Based ReAligner (ABRA) v.2.1240 and base quality recalibration was performed with GATK v.3.3–041. Somatic mutations were identified using MuTect v.1.1.442 and Vardict v.1.5.1 and recurrently mutated genes were identified using MutSig2CV (22,23). Cancer cell fraction (CCF) was calculated using ABSOLUTE based on variant allele frequency, purity, and local allelic copy number (24). Mutations with a CCF of at least 0.85 were deemed clonal. Samples with an estimated tumor purity of less than 0.10 by ABSOLUTE were excluded from subsequent analyses. Using final segmentation calls from ABSOLUTE, we defined copy number events as arm level if the event spanned at least 80% of the arm and affected at least one allele. Arm level amplifications were defined as arm level events with an absolute allelic copy number above 1.9 if the tumor sample was not whole-genome doubled or above 2.9 if the tumor sample was whole-genome doubled. We determined whether a tumor sample underwent whole genome doubling or had a haploid genomic profile by manually evaluating ABSOLUTE solutions (24). We were unable to confidently determine if any haploid tumors were whole genome doubled, and therefore WGD status for these tumors was not included in the co-mutation plot. Summary visualization of mutational profiles integrated with clinical variables was performed with CoMut (25). The mean fraction of amplified arms with a reciprocal arm level deletion (RLOH) was calculated as described previously (26).
Data Sharing.
All MSK-IMPACT results, including the MSK-IMPACT version used to analyze each individual tumor, and associated clinical data are available via the cBioPortal for Cancer Genomics (https://www.cbioportal.org/study/summary?id=makeanimpact_ccr_2023)(27) and as part of AACR Project GENIE (28). WES BAM files are deposited in dbGAP (Accession #phs001783: Exome Recapture and Sequencing of Prospectively Characterized Clinical Specimens From Cancer Patients)
RESULTS
Patient cohort/Demographic data.
Between March 8, 2016 and October 10, 2020, 359 cancer patients expressed interest in the Make-an-IMPACT program, and 333 met protocol eligibility and signed consent. The mean age was 31 years (range 1 – 89). Tumor tissue was received for 288 (86.4%) patients, of which 250 (86.8%) had DNA of sufficient quantity and quality for MSK-IMPACT testing. 63% of patients were enrolled from sites within the United States (24% from non-tertiary care facilities, Figure 1A). 124 patients (37%) from 17 countries were enrolled from sites outside the United States (Figure 1B). 27% of patients were referred by a disease advocacy group, 24% enrolled via the study website, and 49% were referred by a physician familiar with the Make-an-IMPACT program, most of whom initially were made aware of the program by an earlier patient enrolled via the study website or a diseases advocacy group. Among the patients who underwent successful tumor genomic profiling, the most common cancer types were histiocytosis (n = 84), female germ cell tumor (FGCT, n = 54), male germ cell tumor (n = 54) and adenoid cystic carcinoma (n = 19) (Figure 1C).
Figure 1. Country of Origin and CONSORT Diagram of the Make-an-IMPACT cohort.
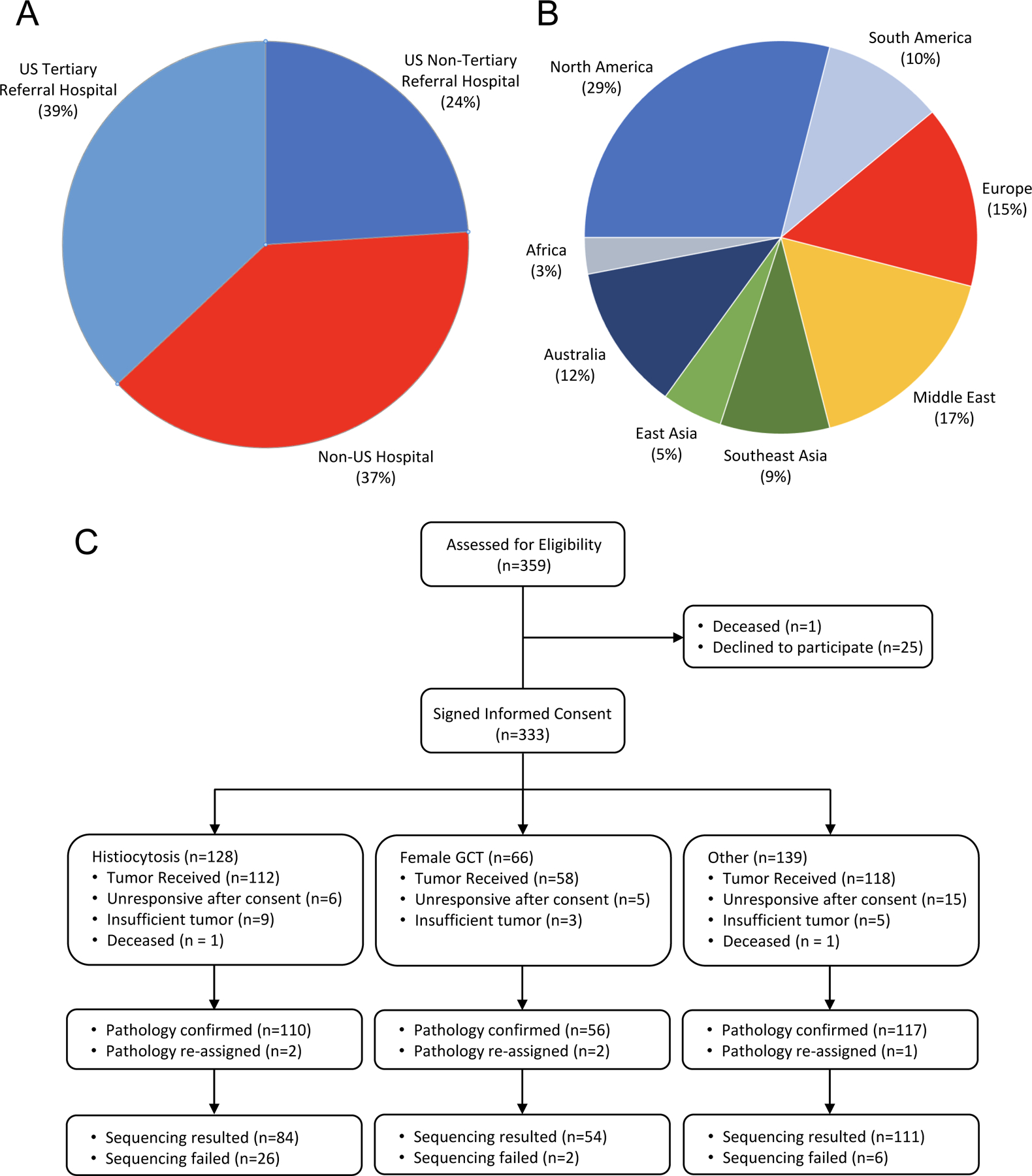
A. Fraction of patients recruited from sites outside the US, and from tertiary and non-tertiary centers in the United States.
B. International patients were enrolled from 17 countries worldwide including locations in North and South America, Europe, Asia, Australia, and Africa.
C. CONSORT diagram of the Make-an-IMPACT cohort.
Histiocytosis
128 patients with histiocytosis from 13 countries were consented for tumor genomic profiling, of whom 112 (87.5%) had sufficient tumor tissue for tumor genomic profiling. Patient demographic and treatment information for the histiocytosis cohort are summarized in Table 1. Central pathology review led to a change in diagnosis to inflammatory sclerosing fibrosis in one patient and to poorly differentiated cancer of unknown primary in a second. Of the remaining 110 patients, tumor genomic profiling was successful for 84 (76.3%). The higher-than-expected rate of technical failure (23.7%) for patients with histiocytosis likely reflects the high degree of stromal infiltration characteristic of histiocytic tumors. Potentially actionable genes most commonly mutated in the histiocytosis cohort were BRAF (33%), MAP2K1 (13%), KRAS (7%), and CSF-1R (2.4%)(Figure 2) (15). Actionable fusions were identified in four patients: 3 BRAF fusions (MS4A6A-BRAF, DOCK8-BRAF, HLA-A-BRAF) and one TFG-ALK fusion. A PRDX1-NTRK1 fusion was also subsequently detected by targeted RNA sequencing in a patient with histiocytosis in which no mutations were detected by DNA sequencing (Figure 2).
Table 1.
Patient demographic data for the Make-an-IMPACT Histiocytosis cohort (n=84)
| Age | Mean | 31.0 |
| Range | 1–77 | |
| Gender | Male | 44 |
| Female | 40 | |
| Race | White | 58 |
| Asian | 8 | |
| Black/African American | 5 | |
| Other/Unknown | 13 | |
| Ethnicity | Non-Hispanic | 74 |
| Hispanic | 7 | |
| Unknown | 3 | |
| Histology | Langerhans Cell Histiocytosis | 32 |
| Rosai-Dorman Disease | 16 | |
| Erdheim-Chester Disease | 14 | |
| Other Histiocytosis | 22 | |
| Primary Site | Lymph/Soft Tissue | 22 |
| Skin | 17 | |
| Bone | 17 | |
| Brain | 10 | |
| Other/Unknown | 18 | |
| Prior Lines of Therapy | 0 | 32 |
| 1 | 27 | |
| 2 | 13 | |
| 3 or more | 12 | |
| Treatments Received | Steroids | 10 |
| Cytotoxic Chemotherapy* | 31 | |
| Targeted Therapy** | 7 | |
| Radiation Therapy | 1 | |
| Interferon | 3 | |
| No prior systemic therapy | 32 | |
| Vital Status | AWD | 58 |
| NED | 16 | |
| DOD | 2 | |
| Unknown | 8 |
Cytotoxic Chemotherapy: Vinblastine/Vincristine (16), Cytarabine (6), Methotrexate (5), Clofarabine (2), Cladribine (1), Carboplatin/Etoposide/Ifosfamide (1)
Targeted Therapies: Trametinib (2), Cobimetinib (2), Vemurafenib (1), Imatinib (1), Sirolimus (1)
Figure 2: Genomic profile of patients with histiocytosis enrolled to the Make-an-IMPACT program and clinical benefit of genomically matched therapies.
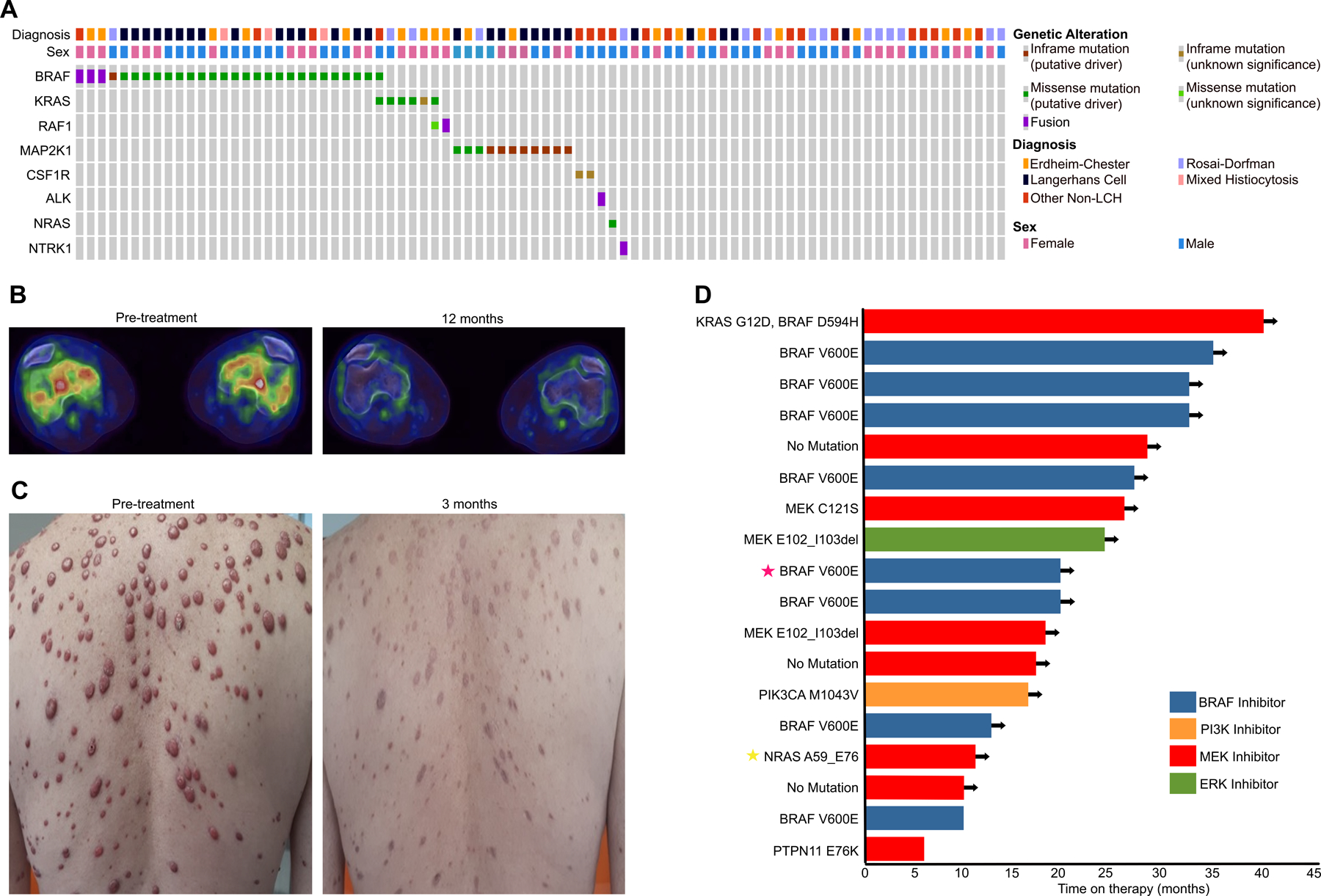
A. Oncoprint of potentially actionable genomic alterations in patients with histiocytosis.
B. Pre- and post-treatment fused axial FDG-PET/CT images of a patient with Erdheim- Chester Disease with a BRAF V600E mutant tumor. Pre-treatment PET (left) reveals symmetric, bilateral, intra-medullary FDG uptake involving the femoral condyles. Repeat PET imaging following 12 months of vemurafenib demonstrated a complete metabolic response.
C. Pre- and post-treatment images of a patient with Erdheim-Chester Disease with a NRAS A59_E79 in-frame deletion. Pre-treatment image (Left image) demonstrating extensive skin lesions. Marked flattening and regression of skin lesions following 3 months of cobimetinib (Right image).
D. Swimmers plot of patients with histiocytosis treated with targeted therapies selected based on their MSK-IMPACT results. Arrows designate ongoing treatment. Stars indicate the patients highlighted in B (red star) and C (yellow star).
Histiocytosis is managed with a variety of local and systemic therapies depending on the disease subtype and extent of organ involvement. Upon enrollment, 7 patients had received prior targeted therapies (trametinib (2), cobimetinib (2), vemurafenib (1), imatinib (1) and sirolimus (1), Table 1). To date, 18 patients have received targeted therapies based on the MSK-IMPACT results (Supplemental Table 1), including eight with BRAF V600E who were treated with a RAF inhibitor (vemurafenib or dabrafenib). Eight patients with BRAF V600E wildtype tumors were treated with a MEK inhibitors (trametinib, cobimetinib), including two with MAP2K1 mutations, one with co-occurrent KRAS G12D and BRAF D594H mutations, one with a NRAS A59_E76 mutation, one patient with a PTPN11 mutation and three with no mutations detected. An additional patient with a MAP2K1 mutation received an ERK inhibitor on a clinical trial, and one patient with histiocytosis with a PIK3CA mutation was treated with alpelisib.
Of the patients with histiocytosis who received targeted therapy, 17 of 18 exhibited clinical benefit based on local physician response assessment. Two examples of efficacious therapy implemented as a result of MSK-IMPACT tumor sequencing are highlighted in Figure 2B, 2C. Responses were durable with 16 patients still receiving genomically matched therapy with durations of 6 to 40 months (Figure 2D). In sum, the results highlight the feasibility and potential clinical benefit of recruiting patients with rare cancers such as histiocytosis who lack insurance coverage or local access to clinical tumor genomic profiling via direct-to-patient outreach.
Ovarian GCT
Ovarian GCT was chosen as a pilot to determine whether outreach via social media and disease advocacy groups could accelerate research for patients with rare cancers by facilitating the assembly of cohorts of sufficient size for genomic discovery. As GCTs can arise in extragonadal sites and as even less is known about the biology of such tumors, female patients with extragonadal primary GCT were also eligible (see Supplemental Table 2 for patient demographics). By combining the 54 female patients with GCT successfully sequenced via Make-an-IMPACT with female patients with GCT offered MSK-IMPACT testing through a prospective institution-wide tumor genomic profiling initiative at MSK, we were able to assemble a cohort of 83 female patients with GCT of whom 67 had ovarian primaries. Central pathology review was discordant with the local diagnosis in 2 cases; both had carcinomas with yolk sac differentiation (Figure 3A). Several patients also had transformation to secondary somatic malignancies (acute myelogenous leukemia, adenocarcinoma, or squamous carcinoma, Figure 3B) (29). Additional demographic and treatment information are summarized in Table 2.
Figure 3. Pathologic and genomic analysis of 83 female patients with germ cell tumors analyzed using the MSK-IMPACT assay.
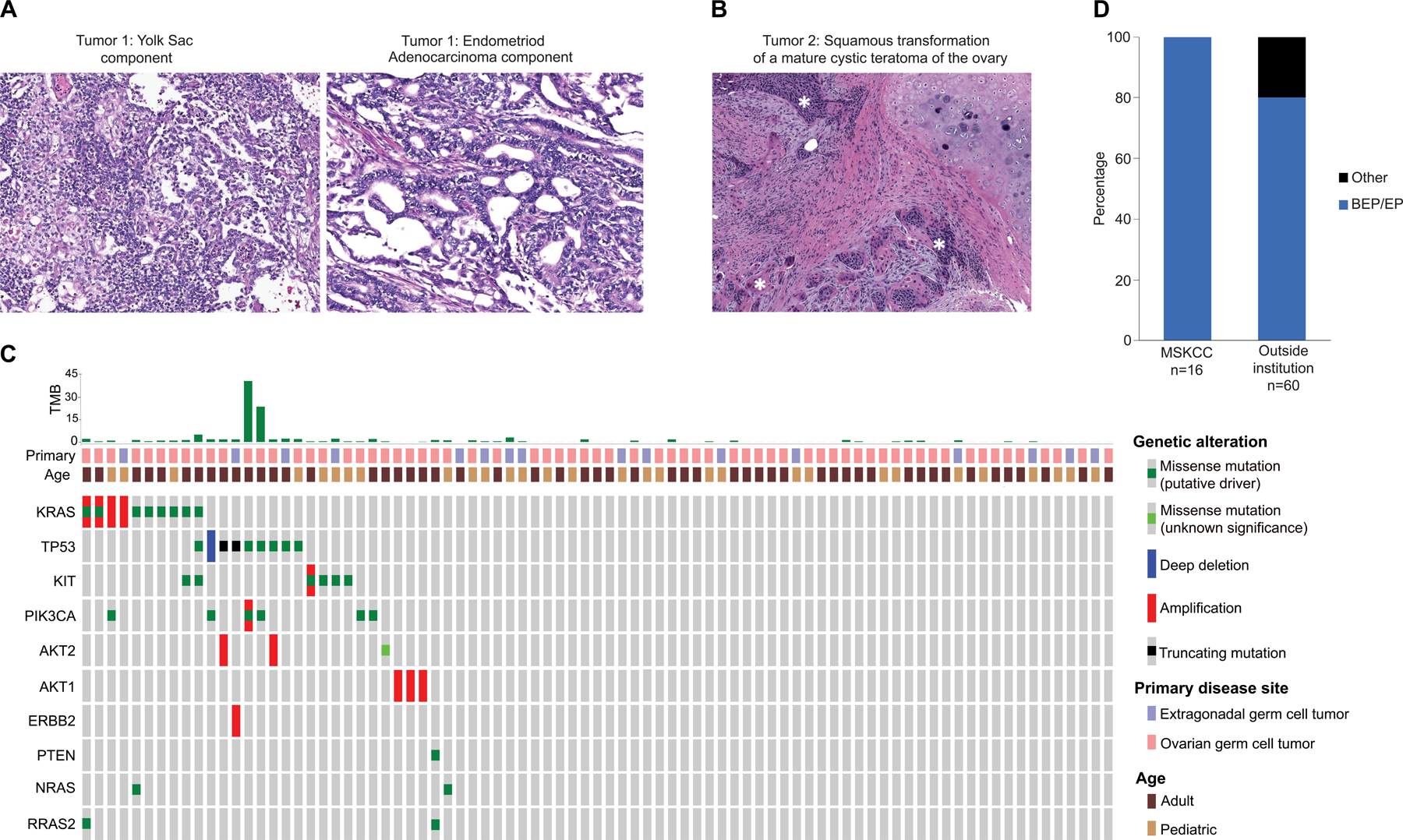
A. H&E Images of an endometrial cancer with yolk sac differentiation, misdiagnosed as a germ cell tumor. The tumor consists of solid, papillary and microcystic areas. Central pathology review identified areas of the tumor consistent with endometrioid adenocarcinoma, characterized by malignant glandular structures of confluent, cribriform glands. Immunohistochemical stains for glypican-3, SALL4, PAX8, CK7 and EMA supported the H&E impression of endometrial cancer with yolk sac differentiation.
B. H&E Image of an invasive keratinizing squamous cell carcinoma (highlighted in the image by white *) arising from a mature cystic teratoma of the ovary.
C. OncoPrint of select genomic alterations in germ cell tumors from female patients analyzed using the MSK-IMPACT targeted sequencing assay. The OncoPrint combines female patients with ovarian and extragonadal germ cell tumors enrolled via the Make-an-IMPACT program and Memorial Sloan Kettering Cancer Center (MSK)-treated patients analyzed as part of an institution-wide prospective sequencing initiative. Two patients, both ovarian GCTs with squamous transformation, had tumor mutational burden (TMB) high (TMB-H) tumors.
D. Fraction of patients in the MSK and non-MSK female GCT cohorts who received either bleomycin, etoposide, and cisplatin (BEP) or etoposide and cisplatin (or carboplatin in one patient) (EP) as first line chemotherapy. Chemotherapy was not recommended for 7 patients. The one Make-an-IMPACT patient treated with etoposide and carboplatin was not eligible for cisplatin due to renal insufficiency.
Table 2.
Patient demographic data for the combined (Make-an-IMPACT and MSK) ovarian GCT cohort (n=67)
| Age | Mean | 26.1 | |
| Range | 3–80 | ||
| Race | White | 54 | |
| Black/African American | 2 | ||
| Asian | 1 | ||
| Other/Unknown | 10 | ||
| Ethnicity | Non-Hispanic | 54 | |
| Hispanic | 10 | ||
| Unknown | 3 | ||
| Histology | Mixed Germ Cell Tumor | 20 | |
| Immature Teratoma | 17 | ||
| Yolk Sac tumor | 12 | ||
| Mature Teratoma | 12 | ||
| Dysgerminoma | 6 | ||
| Chemotherapy * | Bleomycin, Etoposide, Cisplatin | 45** | |
| Bleomycin, Etoposide, Cisplatin, Cyclophosphamide | 1 | ||
| Etoposide, Cisplatin | 4 | ||
| Etoposide, Carboplatin | 1 | ||
| Etoposide, Cisplatin, Ifosfamide | 1 | ||
| Carboplatin, Paclitaxel | 3 | ||
| Cisplatin, Paclitaxel | 3 | ||
| Carboplatin, Docetaxel | 1 | ||
| Cyclophosphamide, Etoposide | 1 | ||
| Chemotherapy not initially recommended*** | 7 | ||
| Mean Age**** | |||
| Surgery | unilateral salpingo-oopherectomy/oopherectomy | 46 | 22 |
| unilateral salpingo-oopherectomy + hysterectomy | 2 | 30.5 | |
| unilateral oopherectomy + partial salpingectomy | 1 | - | |
| bilateral salpingo-oopherectomy + hysterectomy | 12 | 45.6 | |
| bilateral salpingectomy + unilateral oopherctomy | 1 | - | |
| bilateral salpingo-oopherectomy | 1 | - | |
| bilateral ovarian cystectomy | 1 | - | |
| resection of pelvic tumor | 3 | 21.5 | |
| Vital Status | NED | 52 | |
| DOD | 11 | ||
| AWD | 4 |
First-line chemotherapy only
One patient was switched to bleomycin, etoposide, and carboplatin after Cycle 1 due to elevated creatinine.
1 of 7 patients received systemic chemotherapy after recurrence
Mean age was not calculated for categories with only one patient
Standard care for ovarian GCT includes staging surgery, and in patients with stage ≥1C disease, chemotherapy. Notably, the treatment received by patients with ovarian GCT differed between MSK and a subset of non-MSK patients. Unilateral salpingo-oophorectomy is recommended if feasible for children and young women with ovarian GCTs to avoid the long-term sequelae of early-onset estrogen deprivation. None of the MSK patients aged 30 or below underwent bilateral salpingo-oophorectomy whereas bilateral salpingo-oophorectomy was performed in 3 patients in this age group who received their initial surgery at an outside institution (Table 2). Similarly, bleomycin, etoposide and cisplatin (BEP) or etoposide and cisplatin (EP) were administered as first line chemotherapy for all female MSK patients with GCT (16/16), whereas BEP or EP (with carboplatin substituted for cisplatin due to impaired renal function in one patient) was the choice of first line chemotherapy in only 80% (48/60) of the female GCT patients who did not receive their initial chemotherapy at MSK, with several patients receiving treatment regimens that are standard care for high grade serous ovarian cancer such a platinum (cisplatin or carboplatin) and paclitaxel (Figure 3D and Table 2).
Similar to testicular GCT, oncogenic mutations in KIT, KRAS, and TP53 were observed in tumors from a minority of female GCT patients (Figure 3C) (30–32). The mean TMB was low at 2.86 (range 0–42.1, Figure 3C), however, two patients had high TMB (TMB-H) tumors (28.1 and 42.1 mut/MB), both of whom had ovarian germ cell tumors with malignant squamous transformation. 39 female patients with GCT developed disease recurrence after first-line chemotherapy of which 13 have died of disease, 7 are alive with active disease, and 63 have no evidence of disease (NED). To date, 4 female patients with GCT have received targeted therapy guided by the MSK-IMPACT sequencing results, including alpelisib for PIK3CA-mutant tumors and trastuzumab for a patient with ERBB2 amplification, none of whom had durable responses (Supplemental Table 1). One GCT with squamous transformation and a TMB-H tumor (42.1 mut/Mb) was treated with pembrolizumab and achieved a complete response, which is ongoing at 34 months.
As MSK-IMPACT failed to identify known or likely oncogenic mutations in 69% of the female GCTs, we performed whole exome recapture of 62 female GCT tumor/normal pairs from 59 patients using the tumor and germline sequencing libraries generated for clinical MSK-IMPACT testing. Mutational analysis of the whole exome data largely recapitulated findings from the MSK-IMPACT targeted sequencing results for genes covered by both (Figure 4A). Given the lack of treatment response noted in patients who received genomically matched therapy, we used the WES data to explore the clonality of known oncogenic alterations in KRAS, NRAS, KIT, PIK3CA, and TP53. Aside from alterations in NRAS, the majority of mutations detected in these genes were clonal (Figure 4B, Supplementary figure 1). In contrast to the sparsity of oncogenic mutations detected in female GCTs, large scale copy number events were nearly ubiquitous with over a fifth (n=13) of patients contributing at least one tumor sample demonstrating evidence of whole genome duplication. Notably, 17% (n=10) of female GCTs had a near-haploid genomic profile, a phenotype rarely observed in other cancer types, and this phenotype was mutually exclusive with 12p gain (Figure 4A, C) (33,34). Prior work by our group identified chromosome arm level amplifications with reciprocal deletions or reciprocal loss of heterozygosity (RLOH) events as a common feature of testicular GCT genomes (26,35). Similarly, we found that 51% of arm level deletions in female GCTs contained a compensatory reciprocal amplification after controlling for whole genome doubling (Figure 4D). By comparison, only 4% of arm level deletions have a compensatory amplification in ovarian serous cystadenocarcinoma (TCGA-OV).
Figure 4. Whole exome sequencing of female germ cell tumors.
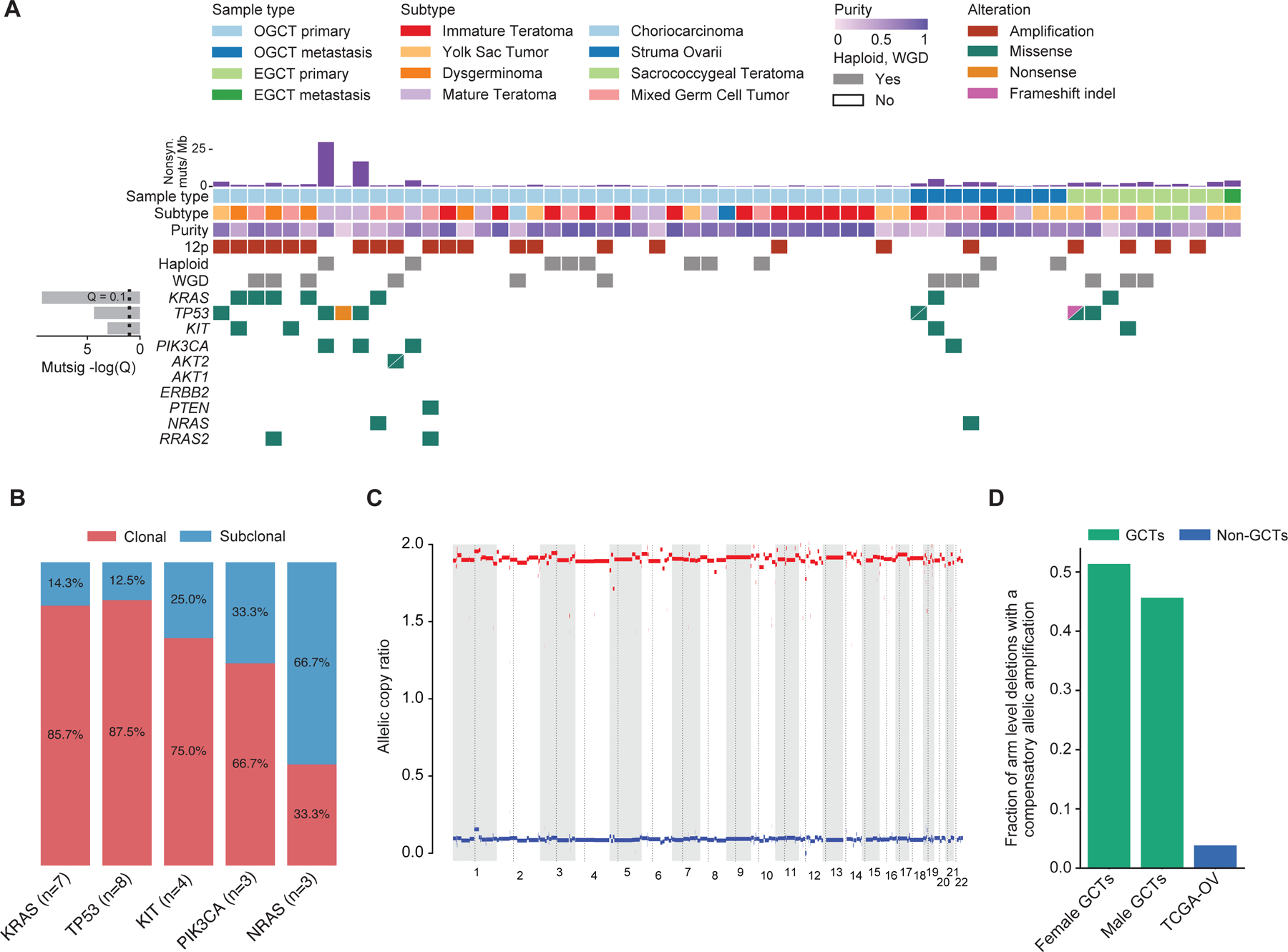
A. Genomic and clinical landscape of female germ cell tumors. Only one sequenced tumor sample from each patient was included. For patients with multiple samples sequenced, the sample with the highest inferred tumor purity as estimated by ABSOLUTE is shown (number displayed = 59). Each column represents a patient, and each row represents a genomic or clinical feature.
B. Percent of oncogenic mutations in KRAS, TP53, KIT, PIK3CA, and NRAS inferred as clonal or subclonal using estimated cancer cell fractions calculated by ABSOLUTE (Supplementary figure 1). “N” refers to the number of mutations in each gene represented.
C. Representative ABSOLUTE allelic copy ratio plot for a sample determined to have a haploid genomic profile (see Methods). Patient samples deemed haploid as indicated in panel A in the “haploid” row were mutually exclusive with tumors with 12p gain.
D. Fraction of arm level deletions with a compensatory amplification after controlling for whole- genome doubling in female GCTs, male GCTs, and the Cancer Genome Atlas ovarian serous cystadenocarcinoma cohort (TCGA-OV). The mean fractions of amplified arms with a reciprocal arm level deletion for men with GCTs and TCGA-OV was extracted from Taylor-Weiner et al., Nature, 2016. Only one sequenced tumor sample from each patient was included in the analysis. Samples with a haploid genomic profile were not included in the analysis.
Discussion
Tumor genomic profiling is increasingly used by oncologists to guide the selection of FDA-approved and investigational therapies in patients with advanced cancer. While few studies have explored the clinical utility of next generation sequencing in rare cancers, recent tumor agnostic approvals including pembrolizumab for MSI-H and TMB-H tumors and larotrectinib for tumors with NTRK fusions provide justification for clinical genomic profiling of all cancer patients for whom curative therapies are lacking. Access to tumor genomic testing for patients with rare cancers is often limited by a lack of insurance reimbursement or local testing expertise. Here, we sought to assess the feasibility of recruiting patients with rare cancer types for a tumor genomic profiling study via direct-to-patient outreach through patient advocacy groups and social media. As tumor genomic profiling was performed in a clinical laboratory, results were reported in real time to patients and their local physicians where they could be used to guide treatment selection.
As an initial pilot, we focused on histiocytosis, a rare cancer type in which actionable genomic alterations are common. 128 patients with histiocytosis residing in 13 countries were enrolled. 47/84 (56%) patients for whom sufficient tumor DNA for MSK-IMPACT could be obtained had a potentially actionable genomic alteration. Notably, the fraction of patients with histiocytosis for whom no mutations were detected by MSK-IMPACT was higher in the Make-an-IMPACT cohort than in our internal MSK cohort (18). This likely reflects selection bias, as a subset of patients with histiocytosis (8% of the histiocytosis cohort) were enrolled after more limited local molecular profiling was uninformative. Clinical benefit from matched therapy, as assessed by the local treating physician, was high with a mean time on therapy of 21.7 months, with 16/18 patients remaining on matched targeted therapy at last response assessment. Patients with histiocytosis often have slowly progressive disease and our expectation is that additional patients in the Make-an-IMPACT cohort will receive molecularly guided therapy upon progression to a symptomatic disease state. However, feedback from several international patients with histiocytosis and their local providers indicated that access to matched therapies has been either delayed or to date prevented by limited local insurance coverage.
Ovarian GCT was chosen as a pilot as limited prior knowledge was available as to the frequency of actionable genomic alterations in this rare cancer. By combining patients enrolled via the Make-an-IMPACT program with female patients with GCT offered MSK-IMPACT testing through a prospective institution-wide tumor genomic profiling initiative, we were able to assemble a cohort of 67 patients with ovarian GCT and 16 female patients with extragonadal GCT primaries for whom we have successfully generated MSK-IMPACT sequencing results. While potentially actionable genomic alterations were rare, two ovarian GCTs (both with squamous transformation) had TMB-H tumors, one of whom had a complete and durable response to pembrolizumab. Three additional patients received genomically matched therapy to date, two for hotspot oncogenic mutations in PIK3CA, neither of whom had a durable response. As the one patient with histiocytosis and an oncogenic PIK3CA mutation achieved a durable response to the PI3K inhibitor alpelisib, the lack of clinical benefit with alpelisib in patients with PIK3CA mutant GCT suggests that lineage specific differences may condition targeted therapy response in these two cancer types. As over half female patients with GCT had no oncogenic alterations identified by MSK-IMPACT, we also performed whole exome sequencing of 62 GCTs from 59 female patients to further characterize the genomic landscape of this rare cancer type. Similar to testicular GCTs, whole genome duplication and reciprocal loss of heterozygosity were common (26). A notable difference with testicular cancer was that 17% of the female GCTs had a fully haploid genomic profile, a rarity in common solid tumors, and a potential vulnerability that could be explored in future functional studies.
A formal histologic review was a component of the Make-an-IMPACT workflow to confirm the local tumor subtype classification and to select the optimal tumor region for DNA extraction. This central pathologic review revealed that several patients were misdiagnosed, including two patients diagnosed locally with GCT who in fact had carcinomas with yolk sac differentiation, entities that can be difficult to distinguish from GCTs with somatic malignant transformation. As neither of these patients responded to BEP, the lack of an accurate cancer subtype diagnosis may have negatively impacted their care. An additional pediatric patient diagnosed with rhabdomyosarcoma was reclassified on central review as an undifferentiated sarcoma. Genomic profiling identified an EWSR1-PATZ1 fusion, a fusion only recently described in cohorts of undifferentiated sarcomas and neuroepithelial tumors (36,37). A change in cancer subtype diagnosis led to a change in the recommended chemotherapy regimen for this child, highlighting the important role of tumor genomic profiling in sarcoma subtype classification and determination of the optimal therapeutic approach (38).
A notable observation from the female GCT cohort was that a substantial minority of patients (21%) did not receive the most commonly employed standard chemotherapy regimens for this disease but rather chemotherapy regimens commonly used for high grade serous ovarian cancer. Several pediatric and young adult patients with ovarian GCT also underwent bilateral salpingo-oophorectomy, a procedure generally avoided in patients with ovarian GCT given their often exquisite sensitivity to chemotherapy and the long-term adverse impact of early-onset estrogen deprivation. Taken together, our experience highlights that treatment at a specialized cancer center with expertise in rare cancers along with central pathologic review could improve outcomes by ensuring that patients receive the optimal standard care therapies for rare but potentially curative tumor types. While a centralized approach to the treatment of rare cancers has historically been viewed as logistically challenging, the rapid adoption of telehealth as a response to the COVID-19 pandemic suggests that centralized treatment of rare cancer patients by oncologists and pathologists with disease specific expertise is now feasible. Such efforts could be further facilitated by the adoption of digital pathology platforms that would facilitate central pathology review. In fact, the infrastructure created for the Make-an-IMPACT program including phone and eConsents was adopted broadly at Memorial Sloan Kettering Cancer Center as a response to the COVID-19 pandemic allowing our oncologists to continue to offer tumor genomic profiling to patients being evaluated and monitored largely via telehealth.
There were several limitations to the current study, many of which are limitations of real-world datasets more generally. For example, the timing of restaging studies was not prescribed and was thus variable and clinical benefit from matched therapy was quantified via local physician assessment and not by central radiology review. While all tumor sequencing was performed free of charge, obtaining often expensive matched targeted or immunotherapies was difficult and, in some cases, impossible, especially in countries with more limited health care resources. Despite these limitations, we found that social media outreach could facilitate the assembly of large cohorts of rare cancer patients which could then be used for discovery science, such as the finding of fully haploid genomes in a minority of female patients with GCT. Inclusion of clinical tumor profiling with return of results also allowed us to identify genomically guided therapies that proved effective in a minority of patients especially for patients in low resource settings. Future research initiatives linked to the Make-an-IMPACT cohort will seek to leverage social media and disease specific advocacy group outreach to explore survivorship questions such as fertility post-chemotherapy, which have been difficult to study for rare cancers such as ovarian GCTs.
Supplementary Material
Translational Relevance.
The utility to tumor genomic profiling remains poorly defined for many rare cancers, which are challenging to study due to their low incidence. In this study, we demonstrate that direct-to- patient outreach via patient advocacy groups and social media can facilitate studies of the genomic landscape of rare tumor subtypes and influence patient care with a focus on histiocytosis, ovarian germ cell tumors, and rare pediatric cancers. By profiling tumors in a clinical laboratory, results could be reported to patients and their local physicians where they could be used to guide treatment selection. For example, 17/18 (94%) patients with histiocytosis who received genomically guided therapy had clinical benefit with a mean treatment duration of 21.7 months. While actionable genomic alterations were rare in patients with ovarian germ cell tumors, high tumor mutational burden was identified in two patients with squamous transformation, one of whom had a complete and durable response to pembrolizumab.
Acknowledgement.
The authors thank Barbara Solit, Amy Heller and other members of the rare cancer advocacy community for assistance in patient recruitment. We also thank members of the Kravis Center for Molecular Oncology, the Integrated Genomics Organization and Diagnostic Molecular Pathology. This work was supported by Cycle for Survival, the Marie- Josée and Henry R. Kravis Center for Molecular Oncology, NIH R37-CA259260, NIH R37-CA222574, NIH R01-CA229624 and the NIH/NCI Cancer Center Support Grant, P30-CA008748.
Footnotes
Competing Interests: Y.L. has received research funding from AstraZeneca, GSK, and REPARE Therapeutics unrelated to this work. C.A. has received consulting fees from Eisai, Merk, Roche/Genentech, Abbvie, AstraZeneca and Repare Therapeutics and clinical trial funding from AstraZeneca. M.Y. has served as a consultant for Janssen Research and Development. O.A.-W. has served as a consultant for H3B Biomedicine, Foundation Medicine Inc, Merck, and Janssen, Loxo Oncology/Lilly and is on the Scientific Advisory Board of Envisagenics Inc and Harmonic Discovery Inc.; O.A.-W. has received prior research funding from H3B Biomedicine, Loxo Oncology/Lilly, and Nurix Therapeutics unrelated to the current manuscript. M.B. has served as a consultant for Eli Lilly, PetDx and received research funding from Grail. J.G.B has served as a consultant for Jazz Pharmaceuticals, was an uncompensated consultant on a DSMB for Springworks, Merck and Pfizer and served on pediatric advisory boards for BMS and Eisai. She receives institutional research support (but no salary support) for clinical trials from Roche, Merck, Amgen, Lilly, BMS, Eisai, Novartis, Loxo-oncology, Cellectar and Bayer. S.A.F. has received research support from AstraZeneca, Genentech/Roche, and Decibel Therapeutics is a consultant/advisory board member for Merck and BioNTech, and owns stock in Urogen, Allogene Therapeutics, Neogene Therapeutics, Kronos Bio, ByHeart, 76Bio, Vida Ventures, Inconovir, and Doximity. A.D. served as a consultant for Incyte, EUSA Pharma, Loxo Oncology and receives research support from Roche and Takeda. R.A.S. was paid annually for serving as assistant editor for one of the USCAP society journals. H.A-A. has served as a consultant for AstraZeneca and Paige.AI. D.R.F. has received research funding from Telix Pharmaceuticals, Decibel Therapeutics, Astellas, Royalties from Up-to-Date, and has served as a consultant for BioNTech. E.M.V. has served as an advisor/Consultant to Tango Therapeutics, Genome Medical, Genomic Life, Enara Bio, Manifold Bio, Monte Rosa, Novartis Institute for Biomedical Research, Riva Therapeutics, Serinus Bio, has received research support from Novartis, BMS, Sanofi, and has equity interests in Tango Therapeutics, Genome Medical, Genomic Life, Syapse, Enara Bio, Manifold Bio, Microsoft, Monte Rosa, Riva Therapeutics, Serinus Bio. E.L.D. discloses unpaid editorial support from Pfizer, Inc, and paid advisory board membership with Day One Biopharmaceuticals and Springworks Therapeutics. D.B.S. has served as a consultant for/received honorarium from Pfizer, Loxo Oncology/Lilly, Vividion Therapeutics, Scorpion Therapeutics, FORE Therapeutics, Fog Pharma, Elsie Biotechnologies, and BridgeBio. The remaining authors declare no potential conflicts of interest.
References
- 1.Chakravarty D, Solit DB. Clinical cancer genomic profiling. Nature reviews Genetics 2021;22(8):483–501 doi 10.1038/s41576-021-00338-8. [DOI] [PubMed] [Google Scholar]
- 2.Cancer Genome Atlas Research N. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014;511(7511):543–50 doi 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cancer Genome Atlas N Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012;487(7407):330–7 doi 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jordan EJ, Kim HR, Arcila ME, Barron D, Chakravarty D, Gao J, et al. Prospective Comprehensive Molecular Characterization of Lung Adenocarcinomas for Efficient Patient Matching to Approved and Emerging Therapies. Cancer Discov 2017;7(6):596– 609 doi 10.1158/2159-8290.CD-16-1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yaeger R, Chatila WK, Lipsyc MD, Hechtman JF, Cercek A, Sanchez-Vega F, et al. Clinical Sequencing Defines the Genomic Landscape of Metastatic Colorectal Cancer. Cancer cell 2018;33(1):125–36 e3 doi 10.1016/j.ccell.2017.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cancer Genome Atlas Research N. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015;163(4):1011–25 doi 10.1016/j.cell.2015.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abida W, Armenia J, Gopalan A, Brennan R, Walsh M, Barron D, et al. Prospective Genomic Profiling of Prostate Cancer Across Disease States Reveals Germline and Somatic Alterations That May Affect Clinical Decision Making. JCO Precis Oncol 2017;2017 doi 10.1200/PO.17.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lemery S, Keegan P, Pazdur R. First FDA Approval Agnostic of Cancer Site - When a Biomarker Defines the Indication. The New England journal of medicine 2017;377(15):1409–12 doi 10.1056/NEJMp1709968. [DOI] [PubMed] [Google Scholar]
- 9.Adashek JJ, Subbiah V, Kurzrock R. From Tissue-Agnostic to N-of-One Therapies: (R)Evolution of the Precision Paradigm. Trends Cancer 2021;7(1):15–28 doi 10.1016/j.trecan.2020.08.009. [DOI] [PubMed] [Google Scholar]
- 10.Painter CA, Jain E, Tomson BN, Dunphy M, Stoddard RE, Thomas BS, et al. The Angiosarcoma Project: enabling genomic and clinical discoveries in a rare cancer through patient-partnered research. Nat Med 2020;26(2):181–7 doi 10.1038/s41591-019-0749-z. [DOI] [PubMed] [Google Scholar]
- 11.Badalian-Very G, Vergilio JA, Degar BA, MacConaill LE, Brandner B, Calicchio ML, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood 2010;116(11):1919– 23 doi 10.1182/blood-2010-04-279083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hyman DM, Puzanov I, Subbiah V, Faris JE, Chau I, Blay JY, et al. Vemurafenib in Multiple Nonmelanoma Cancers with BRAF V600 Mutations. The New England journal of medicine 2015;373(8):726–36 doi 10.1056/NEJMoa1502309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diamond EL, Durham BH, Haroche J, Yao Z, Ma J, Parikh SA, et al. Diverse and Targetable Kinase Alterations Drive Histiocytic Neoplasms. Cancer Discov 2016;6(2):154–65 doi 10.1158/2159-8290.CD-15-0913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diamond EL, Subbiah V, Lockhart AC, Blay JY, Puzanov I, Chau I, et al. Vemurafenib for BRAF V600-Mutant Erdheim-Chester Disease and Langerhans Cell Histiocytosis: Analysis of Data From the Histology-Independent, Phase 2, Open-label VE-BASKET Study. JAMA oncology 2018;4(3):384–8 doi 10.1001/jamaoncol.2017.5029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Durham BH, Lopez Rodrigo E, Picarsic J, Abramson D, Rotemberg V, De Munck S, et al. Activating mutations in CSF1R and additional receptor tyrosine kinases in histiocytic neoplasms. Nat Med 2019;25(12):1839–42 doi 10.1038/s41591-019-0653-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Diamond EL, Durham BH, Ulaner GA, Drill E, Buthorn J, Ki M, et al. Efficacy of MEK inhibition in patients with histiocytic neoplasms. Nature 2019;567(7749):521–4 doi 10.1038/s41586-019-1012-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Veneris JT, Mahajan P, Frazier AL. Contemporary management of ovarian germ cell tumors and remaining controversies. Gynecol Oncol 2020;158(2):467–75 doi 10.1016/j.ygyno.2020.05.007. [DOI] [PubMed] [Google Scholar]
- 18.Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med 2017;23(6):703–13 doi 10.1038/nm.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. The Journal of molecular diagnostics : JMD 2015;17(3):251– 64 doi 10.1016/j.jmoldx.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mandelker D, Zhang L, Kemel Y, Stadler ZK, Joseph V, Zehir A, et al. Mutation Detection in Patients With Advanced Cancer by Universal Sequencing of Cancer- Related Genes in Tumor and Normal DNA vs Guideline-Based Germline Testing. Jama 2017;318(9):825–35 doi 10.1001/jama.2017.11137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Benayed R, Offin M, Mullaney K, Sukhadia P, Rios K, Desmeules P, et al. High Yield of RNA Sequencing for Targetable Kinase Fusions in Lung Adenocarcinomas with No Mitogenic Driver Alteration Detected by DNA Sequencing and Low Tumor Mutation Burden. Clin Cancer Res 2019;25(15):4712–22 doi 10.1158/1078-0432.CCR-19-0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jonsson P, Bandlamudi C, Cheng ML, Srinivasan P, Chavan SS, Friedman ND, et al. Tumour lineage shapes BRCA-mediated phenotypes. Nature 2019;571(7766):576–9 doi 10.1038/s41586-019-1382-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014;505(7484):495–501 doi 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, et al. Absolute quantification of somatic DNA alterations in human cancer. Nature biotechnology 2012;30(5):413–21 doi 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crowdis J, He MX, Reardon B, Van Allen EM. CoMut: visualizing integrated molecular information with comutation plots. Bioinformatics 2020;36(15):4348–9 doi 10.1093/bioinformatics/btaa554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taylor-Weiner A, Zack T, O’Donnell E, Guerriero JL, Bernard B, Reddy A, et al. Genomic evolution and chemoresistance in germ-cell tumours. Nature 2016;540(7631):114–8 doi 10.1038/nature20596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012;2(5):401–4 doi 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Consortium APG. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov 2017;7(8):818–31 doi 10.1158/2159-8290.CD-17-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taylor J, Donoghue MT, Ho C, Petrova-Drus K, Al-Ahmadie HA, Funt SA, et al. Germ cell tumors and associated hematologic malignancies evolve from a common shared precursor. The Journal of clinical investigation 2020;130(12):6668–76 doi 10.1172/JCI139682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen J, Li Y, Wu J, Liu Y, Kang S. Whole-exome sequencing reveals potential germline and somatic mutations in 60 malignant ovarian germ cell tumorsdagger. Biol Reprod 2021;105(1):164–78 doi 10.1093/biolre/ioab052. [DOI] [PubMed] [Google Scholar]
- 31.Van Nieuwenhuysen E, Busschaert P, Neven P, Han SN, Moerman P, Liontos M, et al. The genetic landscape of 87 ovarian germ cell tumors. Gynecol Oncol 2018;151(1):61–8 doi 10.1016/j.ygyno.2018.08.013. [DOI] [PubMed] [Google Scholar]
- 32.Heskett MB, Sanborn JZ, Boniface C, Goode B, Chapman J, Garg K, et al. Multiregion exome sequencing of ovarian immature teratomas reveals 2N near-diploid genomes, paucity of somatic mutations, and extensive allelic imbalances shared across mature, immature, and disseminated components. Mod Pathol 2020;33(6):1193–206 doi 10.1038/s41379-019-0446-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sukov WR, Ketterling RP, Wei S, Monaghan K, Blunden P, Mazzara P, et al. Nearly identical near-haploid karyotype in a peritoneal mesothelioma and a retroperitoneal malignant peripheral nerve sheath tumor. Cancer Genet Cytogenet 2010;202(2):123–8 doi 10.1016/j.cancergencyto.2010.07.120. [DOI] [PubMed] [Google Scholar]
- 34.Hmeljak J, Sanchez-Vega F, Hoadley KA, Shih J, Stewart C, Heiman D, et al. Integrative Molecular Characterization of Malignant Pleural Mesothelioma. Cancer Discov 2018;8(12):1548–65 doi 10.1158/2159-8290.CD-18-0804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cheng ML, Donoghue MTA, Audenet F, Wong NC, Pietzak EJ, Bielski CM, et al. Germ Cell Tumor Molecular Heterogeneity Revealed Through Analysis of Primary and Metastasis Pairs. JCO Precis Oncol 2020;4 doi 10.1200/PO.20.00166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Michal M, Rubin BP, Agaimy A, Kosemehmetoglu K, Rudzinski ER, Linos K, et al. EWSR1-PATZ1-rearranged sarcoma: a report of nine cases of spindle and round cell neoplasms with predilection for thoracoabdominal soft tissues and frequent expression of neural and skeletal muscle markers. Mod Pathol 2021;34(4):770–85 doi 10.1038/s41379-020-00684-8. [DOI] [PubMed] [Google Scholar]
- 37.Alhalabi KT, Stichel D, Sievers P, Peterziel H, Sommerkamp AC, Sturm D, et al. PATZ1 fusions define a novel molecularly distinct neuroepithelial tumor entity with a broad histological spectrum. Acta Neuropathol 2021;142(5):841–57 doi 10.1007/s00401-021-02354-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nacev BA, Sanchez-Vega F, Smith SA, Antonescu CR, Rosenbaum E, Shi H, et al. Clinical sequencing of soft tissue and bone sarcomas delineates diverse genomic landscapes and potential therapeutic targets. Nat Commun 2022;13(1):3405 doi 10.1038/s41467-022-30453-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.