

Abstract
Background:
To delineate the effects of integrin αv signaling in Marfan syndrome (MFS) and examine the potential efficacy of integrin αv blockade as a therapeutic strategy for MFS aneurysms.
Methods:
Induced pluripotent stem cells (iPSC) were differentiated into aortic smooth muscle cells (SMCs) of the second heart field (SHF) and neural crest (NC) lineages, enabling in vitro modeling of thoracic aortic aneurysm in MFS. Fbn1C1039G/+ MFS mice treated with integrin αv antagonist (GLPG0187) confirmed the pathologic role of integrin αv on aneurysm formation.
Results:
iPSC-derived MFS SHF SMCs overexpress integrin αv relative to MFS NC and healthy control SHF cells. Furthermore, downstream targets of integrin αv (FAK/AktThr308/mTORC1) were activated, especially in MFS SHF. Treatment GLPG0187 reduced p-FAK/p-AktThr308/mTORC1 activity in MFS SHF back to control SHF levels. Functionally, MFS SHF SMCs had increased proliferation and migration compared to MFS NC and control SMCs, which was then inhibited by GLPG0187 treatment. In the Fbn1C1039G/+ MFS mouse model, integrin αv, p-AktThr308, and downstream targets of mTORC1 proteins were elevated in the aortic root/ascending segment compared to littermate wild type control. Mice treated with GLPG0187 (age 6-14 weeks) resulted in reduced aneurysm growth, elastin fragmentation and normalization of the FAK/AktThr308/mTORC1 pathway. GLPG0187 treatment reduced the amount and severity of SMC modulation assessed by single cell RNA sequencing.
Conclusions
The integrin αv-FAK-AktThr308 signaling pathway is activated in iPSC SMCs from MFS patients, specifically from the SHF lineage. Mechanistically, this signaling pathway promotes SMC proliferation and migration in vitro. As biological proof of concept, GLPG0187 treatment slowed aneurysm growth and p-AktThr308 signaling in Fbn1C1039G/+ mice. Integrin αv blockade via GLPG0187 may be a promising therapeutic approach to inhibit MFS aneurysmal growth.
Graphical Abstract
Introduction
Marfan syndrome (MFS), a heritable connective tissue disorder resulting from fibrillin-1 (FBN1) mutation, affects 1 in 5,000 individuals.1,2 Premature death results from acute aortic events (dissection and rupture) secondary to aortic root aneurysm development.3,4 While beta-blocker and losartan regimens have therapeutic benefits, only surgical aortic root replacement significantly increases life expectancy in MFS patients.5 Although several studies suggest enhanced transforming growth factor-beta (TGF-β) signaling plays a key pathophysiological role in aneurysm development,6,7,8 uncertainty remains about the downstream molecular events, thus precluding development of preventative medical therapies.
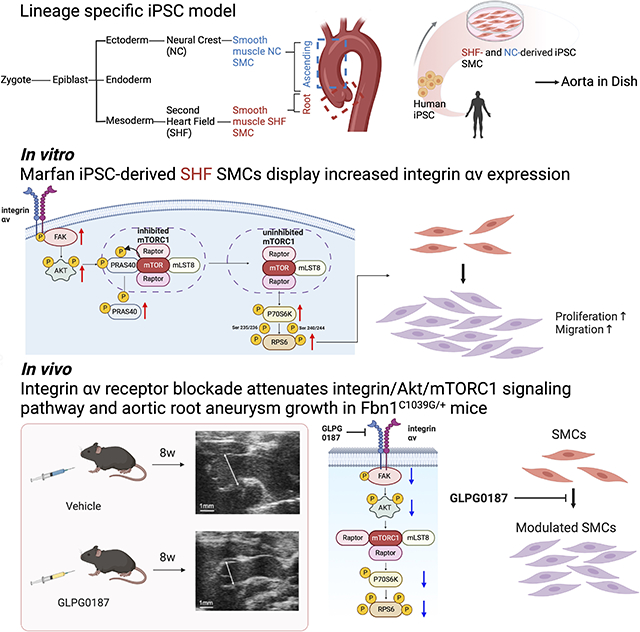
Vascular smooth muscle cells (SMC) populating the aortic root are derived from the second heart field (SHF) embryologic origin,9 while SMCs within the ascending aorta and arch originate from the neural crest (NC).10 Our group reported unique proteomic signatures in human MFS induced pluripotent stem cell (iPSC)-derived SMCs from both proximal thoracic aortic embryologic origins (SHF and NC).11 This analysis suggested FBN1 mutations induce lineage-specific proteomic signatures between MFS SHF SMCs and NC SMCs, theoretically explaining disease susceptibility to aortic root aneurysm formation. Among the proteins enriched in MFS SHF-derived SMCs, we identified integrin αv as a candidate disease marker which was further validated in human MFS aortic root aneurysm specimens.11 Furthermore, applying single-cell RNA sequencing (scRNAseq) to Fbn1C1039G/+ MFS mouse aortic aneurysm identified a signature of enhanced focal adhesion and cell-matrix interaction pathways in a subset of disease-specific modulated SMCs (modSMC).12 Collectively these data suggest an important role for integrin signaling, particularly via integrin αv-containing heterodimers within aortic SMC focal adhesions.
Integrins are transmembrane heterodimeric receptors that consists of 18 alpha (α) and 8 beta (β) subunits, forming 24 αβ heterodimers.13 Integrins can transduce both ECM-SMC biochemical and mechanical signals triggered by ligand binding or alteration in wall stress, tension or stiffness.14,15 Previous investigators have reported that integrin-induced signaling pathway activation potentially induces SMC proliferation, migration, survival, and phenotype modulation.16-18 Uniquely, SMCs are not terminally differentiated and maintain plasticity to shift from a normal contractile state towards a synthetic proliferative, migratory and proteolytic state, which contributes to aneurysm formation. Performing a proteomic screen during aneurysm formation in the MFS Fbn1C1039G/+ mouse model, Parker et al. reported enhanced integrin β3 protein expression, then illustrated in vitro overexpression in aortic SMCs potentiated non-canonical TGF-β signaling and SMC phenotype switching.19 In human MFS, the relationship between lineage-specific SMC integrin expression and the mechanistic role in aneurysm formation remains unknown. Moreover, the downstream events following MFS integrin-SMC mechanosignaling remain to be elucidated. In this study, we investigate the pathophysiology of enhanced integrin signaling utilizing a human MFS iPSC-derived SMC disease model and subsequently translate these mechanistic findings using the Fbn1C1039G/+ murine MFS aneurysm model. Herein, we report that: (1) integrin αv protein expression and downstream signaling is lineage-dependent; (2) integrin αv overexpression in iPSC-derived MFS SHF SMCs activates the FAK-Akt signaling cascade; (3) FAK-Akt-dependent mTORC1 activation enhances downstream proteins, P70S6K and RPS6, resulting in increased SMC proliferation and migration; and (4) systemic integrin blockade (GLPG0187) in vivo reduces FAK-Akt-mTORC1 activity, SMC modulation, and aneurysm formation in the Fbn1C1039G/+ Marfan mouse model.
Materials and Methods
Data Availability
All data and materials have been made publicly available at the Gene Expression Omnibus and can be accessed at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE223807.
Human
The Stanford Institutional Review Board (IRB) approved experiments involving human specimens. All patients included in this study gave informed consent for tissue banking and participation in human subject studies during elective cardiac surgery cases. As an age-matched control, PBMC-derived iPSC SMCs from a healthy male age 25 was utilized (donated by Dr. Joseph Wu, Stanford Cardiovascular Institute, Stanford University).
Cell cultures
iPSC culture: Human iPSC lines were produced at Stanford University Department of Genetics/Stem Cell Core Facilities. Peripheral blood was collected and centrifuged in heparinized cell preparation tubes (CPT, BD Biosciences, San Jose, CA). Plasma was discarded and the mononuclear cell layer was recovered, multiply washed, and cryopreserved. Briefly, cells were co-infected with Sendai virus vectors encoding KLF4, SOX2, OCT4, and MYC transcription factors. Cell lines were expanded on Geltrex coated culture dishes (Gibco/Thermo Fisher Scientific, Sunnyvale, CA), cultivated in mTeSR Plus medium (StemCell Technologies, Vancouver, Canada), and passaged using 0.5mM EDTA. The medium was changed daily starting within the initial 3 days of culture. Sendai virus clearance was evaluated between passages 15-20 by RT-qPCR with Sendai-specific primers. Pluripotent reprogramming efficiency was assessed through TRA1–60 and SSEA4 immunostaining.
iPSC differentiation
SHF and NC SMC differentiation was adapted from Cheng et al20. In brief, chemically defined medium was used as a vehicle for differentiation factors [CDM (500mL): 250mL of IMDM (Gibco/Thermo Fisher Scientific, Sunnyvale, CA) + 250mL F12 nutrient mix (Gibco/Thermo Fisher Scientific, Sunnyvale, CA) + 5mL chemically defined lipid concentrate (Sigma-Aldrich, Saint Louis, MO) + 250μL of transferrin (R&D Systems, Minneapolis, MN) + 350μL insulin (Roche, San Francisco, CA) + 20μL of monothioglycerol (Sigma-Aldrich), Saint Louis, MO]. For the NC lineage differentiation, cells were grown in a CDM plus SB431542 (10μM, Sigma-Aldrich, Saint Louis, MO) plus FGF2 (12ng/mL, R&D Systems, Minneapolis, MN) for 7 days. For the early mesoderm formation, cells were initially grown in CDM+FGF2 (20ng/mL, R&D Systems, Minneapolis, MN) plus LY294002 (10μM, Sigma-Aldrich, Saint Louis, MO) + BMP4 (10ng/mL, R&D Systems, Minneapolis, MN) for 36h. Further specification into SHF required CDM plus FGF2 (20ng/mL, R&D Systems, Minneapolis, MN) + BMP4 (50ng/mL, R&D Systems, Minneapolis, MN) for another 3.5 days. Effective differentiation into the SHF or NC progenitor intermediate population was confirmed with immunofluorescence staining against NKX2-5 and P75 respectively (Supplemental Figure 1). Upon obtaining the intermediate populations, cell monolayers were dissociated with TrypLE (Gibco/Thermo Fisher, Sunnyvale, CA) and cultured in SMC differentiation medium CDM plus PDGF-BB (10ng/mL, PeproTech, Rocky Hill, NJ) + TGF-β1 (2ng/mL, PeproTech, Rocky Hill, NJ) for 12 days. Following differentiation, newly formed SMCs were transitioned from Geltrex-coated plates to uncoated polystyrene flasks and media was changed to commercially available SMC medium (Medium 231 with SMC growth supplement, ThermoFisher Scientific, Sunnyvale, CA). Successful SMC differentiation was confirmed with immunofluorescence staining against mature SMC markers, ACTA2, CNN1, and TAGLN (Supplemental Figure 1).
Mice
All animal protocols were approved by the Administrative Panel on Laboratory Animal Care at Stanford University. Protocols followed the Institutional Animal Care and Use Committee. All experiments were performed with age-matched littermate control animals. Fbn1C1039G/+ mice (no. 012885) were purchased from the Jackson Laboratory (Bar Harbor, ME). Mice were mated exclusively within the Fbn1C1039G/+ colony. Marfan (Fbn1C1039G/+) and littermate control (Fbn1+/+) mice were utilized for all experiments. Animals were maintained in specific pathogen-free housing and fed standard laboratory diet (Envigo Teklad Global 18% Protein Rodent Diet).
Animal treatment groups
Fbn1C1039G/+ mice were treated intraperitoneally with either (1) GLPG0187 100mg/kg/d (MedChemExpress, Monmouth Junction, NJ); or (2) vehicle control (Dimethylsulfoxide (DMSO)) (ATCC, Manassas, Virginia) from age 6 to 14 weeks,21,22 Animals were euthanized at age 14 weeks (GLPG0187-treated Fbn1: males=6; females=7; vehicle control-treated Fbn1: males=6; females=7; GLPG0187-treated WT: males=6; females=7; vehicle control-treated WT: males=6; females=7). For scRNAseq experiments, either (1) GLPG0187 (100mg/kg/d) or (2) DMSO vehicle control treatment was administered from 6-16 weeks. N=2 male and n=2 female mice were pooled for single cell library preparation.
Echocardiography
Transthoracic echocardiography was performed at age 6, 8, 10, 12 and 14 weeks on mice sedated with 2% inhaled isoflurane (2-chloro-2-(difluoromethoxy)-1, 1, 1-trifluoroethane) (Baxter Healthcare Corporation, Deerfield, IL). Bodyweight, tibia length, and body length were recorded at each time point. The thorax was prepared with a depilatory agent. Images of the aortic root and mid ascending aorta (halfway between the sinotubular junction to the brachiocephalic artery) were acquired in parasternal long-axis with an MS700 70 MHz MicroScan transducer on the Vevo 2100 system (VisualSonics, Toronto, Canada). The most dilated portion of the aortic root sinuses and ascending aorta were measured during end-diastole. The aortic measurement was performed by two blinded investigators in triplicate.
Western blotting analysis
Proteins were extracted from iPSC-derived SMCs (MFS patients: n=3; donor control: n=1 biological replicates) or aortic root/ascending murine aortic tissue. To obtain enough protein, root and ascending aortic tissue were combined. Protein concentration was determined through bicinchoninic acid assay according to the manufacturer’s instructions (Thermo Scientific Pierce, Rockford, IL). Samples were denatured at 95°C for 5min in Laemmli’s sample buffer containing 2.5% 2-mercaptoethanol. An equal amount of protein samples was loaded on 4% to 15% polyacrylamide gels. Protein fractions separated onto the SDS-PAGE (4-20% Mini-PROTEAN TGX Gels, BIO-RAD, Hercules, CA) and transferred to nitrocellulose membranes (BIO-RAD, Hercules, CA)). The membranes were blocked with 3% w/v BSA (bovine serum albumin) for 30min and then probed with primary antibodies to detect: Integrin αv (1:5000) (Abcam, Cambridge, MA); p-FAK (1:1000) (Cell signaling Technology, Boston, MA); p-Aktthr308 (1:1000)(Cell signaling Technology, Boston, MA); Akt (1:2000) (Cell signaling Technology, Boston, MA); p-PRAS40 Thr246 (1:1000) (Cell signaling Technology, Boston, MA); p-RPS6Ser235/236 (1:2000) (Cell signaling Technology, Boston, MA); p-RPS6Ser240/244 (1:1000) (Cell signaling Technology, Boston, MA); RPS6 (1:1000) (Cell signaling Technology) and GAPDH (1:2000) (Cell signaling Technology) or β-Tubulin (1:1000) (Cell signaling Technology) as loading control/house-keeping marker. Since PRAS40 and RPS6 have MW of 40 and 31kDa, respectively, and are close to GAPDH (37kDa), - β-tubulin (55kDa) was used as a loading control instead. Secondary anti-rabbit-HRP (1:3000) (Cell signaling Technology, Boston, MA) antibodies were used to detect primary immune-complexes. Detection was performed using the ECL system (Thermo Fisher Scientific, Sunnyvale, CA). Band intensities were measured by densitometry scanning using ImageJ software (National Institute of Health, Bethesda, MD) and were normalized to GAPDH or β-Tubulin as a loading control.
Cell proliferation assay
Cells were cultured at a final cell density of 1.5 x 104 cells/cm2 on FN-coated (Corning BioCoat FN-coated plates, Corning, Glendale, AZ) or Col1-coated (Corning, Glendale, AZ) 96-well plates and cultured overnight in an incubator (37°C, 5% CO2). Media was changed to low serum SMC medium (Medium 231 with SMC growth supplement and 1% FBS) and incubated for 24h. Cells were then treated with TGF-β1 (2ng/ml) with or without GLPG0187 (10nM) or LY2584702 (1μM) for 24h. The GLPG0187 dose was determined from previously reported trials21 and also confirmed in dose titration study (1nM to 100nM). LY2584702 dose was referred from previous reports.23-25 BrdU solution (20μl) was added to each well and cell cultured for an additional 24h. Optical density (OD) was measured (450nm).
Cell Migration/Wound Closure Assay
Cells were plated on plastic 6-well plates (Corning, Glendale, AZ) at a density of 105 cells/cm2 in SMC medium (Medium 231 with SMC growth supplement). After 24h, media was changed to low serum SMC medium (Medium 231with SMC growth supplement and 1% FBS) and incubated for an additional 24h. A linear scratch was made along the surface of each well, denuding the SMCs and inducing an area free of the cell monolayer. GLPG0187 (10nM), LY2584702 (1μM) or vehicle control was added to wells and baseline images captured to define the boundaries of the scratch. After 12 and 24h, the scratch area was reimaged. Quantification of scratch area as a percent from t=0 was measured with ImageJ at 12 and 24h (n=3 per group).
Integrin-Mediated Cell Adhesion Assay
Surface integrin subunit identification on iPSC-derived SMCs was investigated using a fluorometric α/β integrin-mediated cell adhesion array combo kit (ECM535, MilliporeSigma, Burlington, MA). Assay detects integrins α1, α2, α3, α4, α5, αv, αvβ5, α5β1, β1, β3, and β6. MFS and Control iPSC-derived SMCs were treated with TGF-β1 (2ng/mL) in SMC medium for 24h then washed and resuspended in SMC culture medium. SMCs (1.0 x 105/well) were seeded and allowed to adhere for 2h at 37°C. After washing, the adherent SMCs were lysed and the assay performed using the fluorescence CyQuant™ GR dye according to manufacturer’s instructions.
iPSC-derived SHF SMC Integrin αv siRNA Transfection
Transient MFS SHF SMC integrin αv gene knock down was performed using either: (1) siRNA duplex against integrin αv (integrin αv Trilencer-27 human siRNA; Origene, Rockville, MD; Cat. No. SR302468) or (2) siRNA scrambled negative control (Origene; Rockville, MD; Cat. No. SR30004) following the manufacturer’s instructions. Briefly, SMCs (80% confluent) were plated in a 6-well dish and treated with siRNA (10nM) at 37°C, 5% CO2. After 72 h incubation period, immunocytochemistry and western blot analysis were performed.
Histology
Aortic root/ascending aortas were dissected, fixed in 4% paraformaldehyde, and embedded in Tissue-Tek OCT Compound Histomount (Sakura, Torrance, CA). Samples (7μm cross-sections) were stained with Accustain Elastin Verhoeff’s Van Gieson kit (Sigma Aldrich, Saint Louis, MO) according to the manufacturer’s instructions. Three aortic sections at 50μm increments from the most dilated portion of the aortic root were analyzed and imaged at x40 magnification using a Keyence BZ-X810 (Itasca, IL) microscope. For quantification, the average number of elastin breaks for elastic lamina using the whole circumference was measured by 2 investigators blinded to the genotype and treatment.
Murine liver and kidney were fixed in 4% paraformaldehyde solution for 24h at 4°C and stored 70% ethanol. The tissue was embedded in paraffin, sectioned (4μm), and stained with hematoxylin and eosin.
Enzyme-linked immunosorbent assay (ELISA)
Proteins were extracted from the aortic root and ascending aortic tissue of 14-week-old mice or iPSC-derived SMC lysates. For the aortic tissue of mice, we pooled root and ascending aortic tissue anatomic sections as done in our previous reports.26,27 Protein concentration was determined by a bicinchoninic acid assay. Human p-FAK ELISA kit (Cat#PEL-FAK-Y397-1, RayBiotech, Peachtree Corners, GA); Human p-AKTThr308 ELISA kit (Cat#67807, Cell signaling Technology); Human p-P70S6K (Thr421/Ser424) and Total P70S6K ELISA kit (Cat#PEL-P70S6K-T421-T, RayBiotech); Human p-RPS6Ser235/236 ELISA kit (Cat#7205C, Cell signaling Technology); and Human p-RPS6Ser240/244 ELISA kit (Cat#13911C, Cell signaling Technology); were used following manufacturer’s protocol. Twenty micrograms of total protein lysate were used per well in technical replicates. Results are expressed following colorimetric detection at 450nm using a multi-detection microplate reader (Synergy 2, Biotek, WA)
Human Akt pathway phosphorylation array
RayBio human and mouse Akt pathway phosphorylation array C series (RayBiotech, Norcross, GA) was used to assay Akt-dependent downstream phosphorylated proteins. Samples were collected in triplicates and 100μg of protein used. Arrays were processed according to the manufacturer’s instructions. Data were analyzed by densitometry scanning using ImageJ software and normalized to loading control. Reported signal intensity is the mean of the technical replicates.
Immunocytochemistry/Immunofluorescence (ICC/IF)
iPSC-derived SHF and NC differentiated SMCs were plated on glass coverslips (24h) then fixed for 10min in 4% PFA. Fixed cells were rehydrated with 1 x PBS, permeabilized with 0.2% Triton X-100, blocked in 1% BSA/0.3% Triton X-100/PBS for 60min. Samples were incubated in primary antibody against integrin αv (1:500) (Abcam, Cambridge, MA); p-FAK (1:100) (Cell signaling Technology, Boston, MA); Ki-67(1:250) (Abcam, ab16667); p-AktThr308 (1:500) (Cell signaling Technology) or isotype control (1:100) (Abcam, ab37415) 24h at 4°C. Secondary antibody was incubated for 1h at room temperature. Alexa Fluor-488 conjugated anti-rabbit secondary antibody (1:250; Thermo Fisher Scientific) and Texas Red-X phalloidin (1:400; Invitrogen) were used. Nuclei were stained with Hoechst reagent (1μg/mL) (bisBenzide H33258, Sigma-Aldrich, Saint Louis, MO) for 10min. Fluorescent images were captured at 20x magnification with a multichannel fluorescent microscope (Leica DM4000B, Buffalo Grove, IL). Reported signal intensity is the mean of the technical replicates.
Annexin V Apoptosis Assay
Fluorescence-Activated Cell Sorting (FACS) was performed at the Stanford Shared FACS Facility on a Sony SH800 instrument using a Dead Cell Apoptosis kit with Annexin V (V13241, Thermo Fisher Scientific). Single suspended cells were counted and diluted in annexin-binding buffer (1x106 cells/ml). FITC Annexin V (5μl) and PI (1μl) working solution were added to each 100μl of cell suspension and incubated for 15min at room temperature. After the incubation period, annexin buffer (400μl) was added and the stained cells were analyzed by flow cytometry. Cells were measured for fluorescence emission at 530nm and 575nm.
Mouse Aortic Dissociation and Single-Cell Suspension
Fbn1C1039G /+ mice and littermate controls were anesthetized and euthanized by cervical dislocation. For aortic root/ascending aorta samples, the aortic root was dissected completely and divided below the aortic valve. The ascending aorta was divided at the takeoff of the brachiocephalic artery. Aortas were transferred to a separate dish, further debrided, and rinsed in cold Hanks’ buffered saline solution (HBSS). Specimens were combined into groups with age and genotype-matched animals as biologic replicates, with equal male and female specimens. The aortas were then digested in enzymatic solution containing 2 U/mL Liberase TM (Roche no. 05401127001) and 2 U/mL elastase (Worthington no. LS002279) in HBSS for 1 hour. The cell suspension was filtered through a 70 μmol/L strainer, centrifuged, and resuspended in cold HBSS.
Fluorescence-Activated Cell Sorting (FACS)
FACS was performed at the Stanford Shared FACS Facility on a Sony SH800 instrument to sort cells and discard debris. Single cell suspensions were counted to ensure adequate cell density (>150 cells/μL) and immediately used for 10X Genomics single-cell capture.
Single Cell RNA Sequencing (scRNAseq)
All portions of the scRNAseq workflow (single-cell capture, library preparation, and quality control PCR) were performed at Stanford Genome Sequencing Service Center. Freshly sorted samples were diluted to target 5000 cells for capture. The cells were processed using a 10X Genomics microfluidics chip to generate barcoded Gel Bead-In Emulsions according to manufacturer’s protocols. Indexed single-cell libraries were then created according to 10X Genomics specifications. Samples were multiplexed and sequenced in pairs on an Illumina NovaSeq 6000 device by MedGenome Inc. (Foster City, CA).
scRNAseq Data Analysis
Raw base-call files from NovaSeq 6000 sequencer were demultiplexed and delivered as FASTQ files. The sequenced data were processed into expression matrices with the Cell Ranger Single-cell software suite 3.1.0 (https://www.10xgenomics.com/) on the Stanford Sherlock High Performance Computing Cluster. The reads were aligned to the mouse transcriptome (mm 10–3.0.0), cell barcodes and unique molecular identifiers were filtered and corrected using the cellranger aggr pipeline. The final output filtered expression matrices were imported into the Seurat package in R and built into Seurat objects using the CreateSeuratObject function. Filtering during this step included only genes detected in >3 cells and cells with >200 distinct genes. To exclude doublets/clumps and free RNA, thresholds for individual cell read counts (nCount_ RNA) and genes (nFeature_RNA) were determined for each data set by excluding indices (cells) with counts outside of normal distributions for these variables. Cells with >5% mitochondrial gene content were excluded.
Data normalization, scaling, and regression by mitochondrial content were then performed using the SCTransform command under default settings in Seurat. Principal component analysis and nonlinear dimensional reduction using uniform manifold approximation and projection. Cell clustering was then assessed across a range of predetermined resolution scales to ensure separation of known major aortic cell types without excessively subclustering. The FindAllMarkers function in Seurat was applied to perform parallel differential expression testing of all cells within a cluster versus all other cells in the data set via nonparametric Wilcoxon rank-sum test using default parameters.
16-week Fbn1C1039G/+SHF lineage-traced (Nkx2-5-cre knock-in combined with Cre-inducible tdTomato fluorescent reporter) publicly available dataset, from recently published work from our group, was reanalyzed (Gene Expression Omnibus repository under accession GSE186845).28
Statistical Analysis
All data are presented as means ± SD of 2 independent experiments unless otherwise stated. Statistical analysis in GraphPad Prism 9.5.0 (GraphPad Software, La Jolla, CA), except for the scRNAseq, which was performed using the Seurat package in R 4.1.0. Data were tested for normality using Kolmogorov-Smirnov or Shapiro-Wilk tests. For comparisons between two groups, a two-tailed t-test with Welch’s correction was used. When normality was not observed, non-parametric testing was performed with a Mann-Whitney test. For comparisons of three or more groups, normally distributed groups were compared with a one-way Brown-Forsythe and Welch ANOVA test with a Dunnett T3 correction or two-way ANOVA with a Bonferroni correction. Kruskal-Wallis with Dunn’s multiple comparison test was used for non-parametric multiple comparisons. A P-value of < 0.05 was considered statistically significant. Significance was assigned based on the P-value (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001). All experiments have been performed with technical replicates or repeats and three MFS biological replicates.
Results
Human MFS iPSC-derived SHF SMCs display increased integrin αv expression
MFS-iPSCs were generated from peripheral blood mononuclear cells from three Marfan patients. MFS1 is a 27-year-old female patient who required aortic root replacement for a 4.5cm aneurysm and concomitant severe mitral valve regurgitation. She harbors a missense mutation in exon 46 (NM_000138.5:c.5726T>C). MFS2 is a 20-year-old female patient who underwent elective aortic root replacement for a 5.0cm aneurysm. MFS2 has a two base pair deletion in exon 55 (NM_000138.5:c.6783_6784delinsGT) resulting in a frameshift mutation and premature stop codon. MFS3 is a 34-year-old male patient who required an aortic root replacement at age 12 and a second operation at age 34 for aortic insufficiency and a rapidly expanding aortic arch aneurysm. MFS3 has a missense mutation in exon 5 (NM_000138.5:c.493C>T). Control-iPSCs were generated from a 25-year-old healthy male patient. iPSCs were differentiated into either SHF or NC lineage-specific SMCs using a previously established protocol (Supplemental Figure I).11,20,29 Consistent with our previous reported work,11 immunoblot analysis confirmed the following: (1) integrin αv expression is enhanced in MFS SHF SMCs compared to control SHF SMCs (MFS1: 2.2; MFS2: 2.0; MFS3:2.2-fold change); (2) MFS and control NC SMCs express similar integrin αv expression (MFS1: 1.1; MFS2: 1.4; MFS3: 1.1-fold change); (3) MFS SHF SMC integrin αv expression is increased compared to MFS NC SMCs (MFS1: 2.1-fold change; MFS2: 1.5-fold change; MFS3: 2.0-fold change) (Figure 1A). Immunofluorescence staining confirmed enhanced integrin αv expression in MFS SHF compared to either MFS NC SMCs or control cell lines (Figure 1B). To determine if other integrin subtypes are upregulated on MFS SHF SMCs, we utilized a fluorometric α/β integrin-mediated cell adhesion assay. Integrins αv and α5 were the most differentially upregulated integrin α subunits and integrin β5 and β6 the most enhanced integrin β subunits (Supplemental Figure II). Both integrin αvβ5 and αvβ6 are RGD motif binding integrins.
Figure 1. Integrin αv, p-FAK, and p-AktThr308 protein levels in MFS iPSC SMCs.

A, Western blot analysis for integrin αv expression in iPSC SMCs illustrates increased expression in MFS SHF SMCs compared to control after treatment with TGF-β (2ng/ml) for 24h in low serum SMC medium (n=3 biological replicates). B, Immunofluorescence images of iPSC SMCs: integrin αv (green) and nuclei stained with Hoechst reagent (blue) (x20 magnification, scale bar = 50μm). C, p-FAK and E, p-AktThr308 western blot analysis of iPSC SMCs show increased expression in MFS SHF SMCs compared to control (n=3 biological replicates). D, p-FAK and F, p-AktThr308 (n=3 biological replicates) measured by ELISA. Groups tested by ELISA were compared by two-way ANOVA. Significance is assigned based on the P-value (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001).
Integrin αv activates FAK-Akt signaling pathway in Human MFS iPSC-derived SHF SMCs
While FAK/Akt signaling during aortic aneurysm formation has been reported,30-35 integrin αv-mediated FAK/Akt activation in human MFS lineage-specific SMCs has not been studied. Enhanced TGF-β activation is a recognized disease marker of uncertain mechanistic importance in both murine models and human MFS.8,36,37 To uniformly simulate this process in vitro, MFS and control SMCs were treated with 2ng/ml TGF-β1 for 24h for all subsequent experiments. Correlating with enhanced integrin αv expression, immunoblot analysis revealed enhanced p-FAK activation in MFS iPSC SHF SMCs compared to either MFS NC or the corresponding control lineage SMCs and confirmed by quantitative ELISA (p-FAK: MFS1 SHF: 1.4-fold; MFS2 SHF: 1.3-fold; MFS3 SHF: 1.2-fold) (Figure 1C-D). Similarly, MFS SHF SMC p-AktThr308 overexpression compared to control SHF SMC was also detected by immunoblotting and confirmed by quantitative ELISA (p-AktThr308: MFS1 SHF: 1.3-fold; MFS2 SHF: 1.4-fold; MFS3 SHF: 1.4-fold) (Figure 1E-F).
To further characterize integrin αv-dependent FAK and Akt activation in MFS iPSC-derived SHF and NC SMCs, loss- and gain-of-function experiments were performed. For loss-of-function, SMCs were treated with TGF-β1 (2ng/ml) and the RGD integrin blocker, GLPG0187 (inhibits integrins with integrin αv heterodimer subunit: αvβ1, αvβ3, αvβ5, αvβ6 and α5β1) for 24h.22,38-40 Confirming integrin αv participates in MFS SMC FAK/Akt activation, GLPG0187 significantly decreased p-FAK and p-AktThr308 signaling (p-FAK: MFS1 SHF: 0.6-fold; MFS2 SHF: 0.5-fold; MFS3 SHF: 0.5-fold; MFS1 NC: 0.8-fold; MFS2 NC: 0.8-fold; MFS3 NC: 0.8-fold (Figure 2A-B); p-AktThr308: MFS1 SHF: 0.6-fold; MFS2 SHF: 0.6-fold; MFS3 SHF: 0.5-fold; MFS1 NC: 0.7-fold; MFS2 NC: 0.7-fold; MFS3 NC: 0.6-fold (Figure 2C-D). Corroborating these results, immunofluorescence staining also illustrated that GLPG0187 reduced cytoplasmic expression of p-FAK and p-AktThr308 most substantially in the MFS SHF SMCs (Figure 3A-C). Next, gain-of-function experiments were performed by growing MFS iPSC-derived SHF SMCs on fibronectin (FN)-coated plates, which contain an RGD (arginine-glycine-aspartic acid) domain that binds and activates integrin αv.41-43 FN was selected among RGD-containing ECM substrates due to enhanced protein expression noted in both MFS and control SHF SMCs with multiplexed tandem mass spectrometry11 and enriched RNA expression in MFS-specific modulated SMCs.12 As a control, MFS SHF SMCs were plated on Collagen I (Col1)-coated plates which lack the RGD domain (Figure 3D). Both p-FAK and p-AktThr308 were increased specifically in MFS SHF SMCs cultured on FN-coated plates, however remained unchanged on Col1, thus confirming lineage-specific integrin-dependent activation of the FAK-Akt cascade (Figure 3E).
Figure 2. Effect of GLPG0187 on iPSC SMC FAK and AktThr308 phosphorylation.

MFS SHF and NC SMCs were treated with GLPG0187 (10nM) or vehicle control for 24h in low serum SMC medium. Protein expression of A, p-FAK, and C, p-AktThr308 was determined by western blot analysis (n=3 biological replicates). Representative blots are shown. B, D, Results were confirmed with quantitative ELISA (n=3 biological replicates). Two-way ANOVA was performed to examine the effect of GLPG0187 treatment. P-value (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001).
Figure 3. The p-FAK and p-Akt protein levels in iPSC SMCs following GLPG0187 treatment.

A, Immunofluorescence images of iPSC SHF and NC SMCs stained with Hoechst reagent (blue) and p-FAK antibody (green) (scale bar = 25μm) or B, p-AktThr308 antibody (green) (scale bar = 50μm) following GLPG0187 (10nM) or vehicle control. All images are representative of 4 independent experiments. C, Immunofluorescent staining was quantified as stained area/nuclei (n=4 technical replicates, at least 100 cells counted). D, Integrin αv activation, by growing cells on FN coated plates, increased cellular proliferation compared to Col1 coated plates. GLPG treatment reduced the level of proliferation. Left: Bright field (scale bar = 100 μm); Right: Fluorescent staining of iPSC SMCs: nuclei stained with Hoechst reagent (blue) and actin filaments with Phalloidin (red) (scale bar = 50μm) E, Representative western blot illustrates increased p-FAK and p-AktThr308 protein expression in MFS SHF vs. MFS NC SMCs when grown on FN-coated plates compared to Col1 coated plates (n=3 technical replicates per group).
Our laboratory previously reported dynamic SMC phenotype modulation during aneurysm progression in Marfan mice,12 although the initiating event remains unknown. ECM-mediated mechanical signaling via integrin mechanotransduction has been reported to potentially induce SMC phenotype modulation and increase SMC proliferation and migration.22 We hypothesized that enhanced integrin αv-mediated signaling plays an important initiating role during aneurysm formation by influencing MFS SMCs to become more proliferative and migratory. To test this hypothesis, functional proliferation and migration studies were performed to determine the role integrin αv plays in SMC phenotype modulation. MFS SHF SMCs treated with vehicle control showed a 1.5-fold increase in proliferation via BrdU cell proliferation assay compared to control cell lines, whereas GLPG0187 integrin αv blockade normalized cell proliferation to the level of MFS NC and control SMCs (Figure 4A). Similarly, Ki-67, a well-established marker for proliferating cells44,45 showed higher expression in MFS SHF-derived SMCs compared to MFS NC or control SHF. Consistently, GLPG0187 treatment suppressed Ki-67 expression (Figure 4B). To confirm that these findings are not a result of increased apoptosis from GLPG0187 treatment, flow cytometry for annexin V confirmed that GLPG0187 treatment actually decreased apoptosis compared to vehicle control (Supplemental Figure III). Cell migration (wound closure assay) illustrated that MFS SHF SMCs migrate faster at 24h compared to control SHF SMCs (p<0.001), whereas GLPG0187 integrin αv inhibition attenuated the migratory capacity (Figure 4C). Furthermore, MFS SHF SMCs on FN- vs. Col1-coated plates resulted in increased proliferation (1.5-fold) by Ki-67 and BrdU assay and returned to baseline with GLPG0187 blockade (Figure 4D-E).
Figure 4. MFS iPSC SMC proliferation and migration.

A, SHF and NC SMC proliferation determined with BrdU assay following GLPG0187 (10nM) or vehicle control treatment. Data expressed as mean ± SD (n=5-6 experimental replicates). Two-way ANOVA performed to examine the effect of GLPG0187 treatment. (**P < 0.01, ****P<0.0001). B, Immunofluorescence microscopy of iPSC SMCs (control (CTR) vs. MFS). DNA counterstain with Hoechst reagent (blue) and Ki-67 antibody (red) following vehicle control or GLPG0187 (10nM) treatment. Graph shows percent Ki-67 positive cells (n=4 technical replicates, scale bar = 100μm). Two-way ANOVA utilized. P-value (*P < 0.05, ****P < 0.0001). C, SMC migration measured by scratch assay (MFS SHF vs. CTR SHF SMCs). SMCs treated with vehicle control or GLPG0187 (10nM) for 24h (scale bar = 500 μm). Quantification reported as percentage cell-occupied area in scratch at 12 and 24h relative to the cell-occupied area at 0h (n=3 per group). D, MFS SHF iPSC SMCs plated on Col1- or FN-coated plates. Immunofluorescence microscopy of Ki-67 performed on SMCs treated with vehicle control or GLPG0187 (10nM) for 24h (scale bar = 100μm). E, SMC proliferation determined by BrdU assay after growing on Col1- or FN-coated plates. SMCs treated with vehicle control or GLPG0187. Data expressed as mean ± SD (n=3 biological each with 6-12 technical replicates). Groups were compared via one-way ANOVA. P-value (*P < 0.05, **P < 0.01, ****P < 0.0001).
MFS SHF SMC FAK/Akt overexpression activates mTOR pathway
After illustrating that higher MFS SHF SMC integrin αv density increases FAK/Akt pathway signaling, cellular proliferation, and SMC migration, we sought to reveal Akt downstream target proteins involved in spatially specific aneurysm formation. Eighteen phosphorylated proteins were screened with an Akt pathway phosphorylation array from MFS SHF SMCs grown on FN-coated plates either with or without GLPG0187 integrin αv inhibitor. Mechanistic target of rapamycin (phospho-mTOR-Ser2448), proline-rich Akt substrate of 40kDa (PRAS40 (P-Thr246)), Ribosomal protein S6 (RPS6 (P-Ser235/Ser236)), and P70-S6 Kinase (P70S6K (P-Thr421/Ser424)) were all increased when grown on FN and decreased following GLPG0187 treatment (Supplemental Figure IV). mTOR, PRAS40, Raptor, mLST8, and deptor comprise the mTOR protein complex 1 (mTORC1), which potentially increases protein synthesis and cellular growth via RPS6 and P70S6K activation.31,46,47,48 Pathway phosphorylation array results were validated with immunoblotting for PRAS40 (P-Thr246), RPS6 (P-Ser235/236), and RPS6 (P-Ser240/244) (Figure 5A), and by ELISA for PRAS40 (P-Thr246) (Figure 5B), p-P70S6K (Figure 5C), RPS6 (P-Ser235/236), and RPS6 (P-Ser240/244) (Figure 5D).
Figure 5. MFS SHF SMC FAK/Akt signaling activates mTORC1 pathway.

A, MFS SHF SMCs plated on Col1- or FN-coated plates and treated with vehicle control or GLPG0187 for 24h. Protein expression by western blot for p-PRAS40Thr246, p-RPS6Ser235/236, and p-RPS6Ser240/244 following RGD activation by FN. Representative western blot analysis (n=3 biological each with 6 technical replicates) with β-tubulin as internal control. B, p-PRAS40Thr246 (n=3 biological each with 5 technical replicates), C, p-P70S6K (n=3 biological each with 5 technical replicates), D, p-RPS6Ser235/236 (n=3 biological each with 5 technical replicates) and p-RPS6Ser240/244 (n=3 biological each with 5 technical replicates) quantified by ELISA for MFS SHF SMCs plated on Col1- or FN-coated plates treated with vehicle control or GLPG0187 for 24h. One-way ANOVA performed to examine effect of GLPG0187 treatment. P-value (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001).
Next, integrin αv-induced SMC proliferation and migration through the mTORC1 pathway was confirmed by blocking P70S6K with LY2584702 (Figure 6A). As expected, LY2584702 treatment of MFS SHF SMCs grown on FN significantly reduced proliferation by both Ki-67 and BrdU examination (Figure 6B). LY2584702 treatment also induced blunted MFS SHF SMC migration compared to non-treated cells in a wound closure assay (Figure 6C). p-RPS6, a downstream target of P70S6K in the mTORC1 pathway, had reduced phosphorylation in both Ser235/236 and Ser240/244 positions when treated with LY2584702 (Figure 6D). These results further validate that enhanced integrin αv activation signaling through Akt and mTORC1 modulates the SHF SMC towards a more proliferative and migratory cell, potentially exacerbating aneurysm formation.
Figure 6.

A, p-P70S6K ELISA for MFS SHF SMCs activated with FN-coated plates after treatment with vehicle control or LY2584702 (1 μM). B, Immunofluorescence microscopy of Ki-67-stained iPSC MFS SHF SMCs. DNA counterstain with Hoechst reagent (blue) and Ki-67 antibody (red) following treatment with either vehicle control or LY2584702 (1μM). Ki-67 and BrdU quantification shown in graph. Data represented as the mean ± SD (n=3 biological each with 12 technical replicates, scale bar = 50 μm). Welch’s t-test used for comparison between vehicle control vs. LY2584702 treated. P-value (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001). C, Migration assay performed on iPSC MFS SHF SMCs treated with either vehicle control or LY2584702 (1μM) (scale bar = 500 μm). Percentage of cell-occupied area in scratch at 12 and 24h was showed relative to the cell-occupied area at 0h. Graph was plotted with the average value of % closure (n=3 technical replicates). D, MFS SHF iPSC SMCs plated on Col1- or FN-coated cell culture plates and treated with LY2584702. p-RPS6Ser235/236 and p-RPS6Ser240/244 protein expression detected with western blot (n=3 biological each with 4 technical replicates) and ELISA (n=3 biological each with 5 technical replicates). Welch’s t-test was performed to examine the effect of LY2584702 treatment. P-value (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001) E, Loss-of-function studies performed using siRNA against scramble control siRNA, integrin αv siRNA, or untreated iPSC MFS SHF SMCs (n=2, biological replicates). Immunofluorescence staining for integrin αv (green) shows decreased expression in integrin αv siRNA-treated SMCs. Western blot reveals decreased p-FAK, p-PRAS40Thr246, p-RPS6Ser235/236, and p-RPS6Ser240/244.
Finally, because GLPG0187 blocks all RGD recognizing integrin heterodimers, integrin αv-specific loss of function experiments were performed to confirm that integrin αv mediates enhanced FAK/Akt/mTORC1 pathway signaling. MFS SHF SMCs were transfected with siRNA targeting either (1) integrin αv or (2) scrambled control, then western blotting performed for integrin αv and downstream FAK/mTORC1 signaling targets. Corroborating the GLPG0187 blockade experiments, decreased integrin αv expression was associated with a reduction in FAK/PRAS40/PRS6 signaling (Figure 6E)
Integrin αv receptor blockade attenuates aortic root aneurysm growth in Fbn1C1039G/+ mice
Our laboratory previously detected enhanced integrin αv expression in Fbn1C1039G/+ MFS aneurysmal aortic root and ascending aorta.11,19 Corroborating our findings from the iPSC-derived SHF SMCs, integrin αv, p-FAK and p-AktThr308 are increased in Fbn1C1039G/+ root/ascending aortic specimens compared to the corresponding WT littermate control aorta at ages 2, 6, 14, and 24 weeks (Figure 7A, Supplemental Figure VA-B). Immunofluorescence staining of aortic root and ascending aortic specimens revealed enhanced integrin αv expression in Fbn1C1039G/+ compared to WT mice at age 16 weeks. More specifically, integrin αv overexpression was predominately localized to the MFS aortic root (derived primarily from SHF) vs. ascending aorta (derived primarily from NC) (Figure 7B). To study the etiology of the noted lineage-specific integrin αv differential protein expression, scRNAseq was performed on 16-week (1) Fbn1C1039G/+SHF lineage-traced (Nkx2-5-cre knock-in combined with Cre-inducible tdTomato fluorescent reporter) and (2) WT littermate control mice. Aortic root/ascending aortic specimens were enzymatically digested and FACS performed to sort SHF- and nontraced (NC)-derived cells. Itgav gene expression was significantly increased in Fbn1C1039G/+SHF lineage-traced compared to WT control mice and detected predominately in the modulated SMC (modSMC) population (Figure 7C). ModSMCs are potentially implicated in MFS aneurysm formation.12,28,49 Because Fbn1C1039G/+ modSMCs from both lineages equally contribute to enhanced Itgav, we hypothesize that the noted SHF integrin αv protein expression differences are post-transcriptional.
Figure 7. Integrin αv upregulated in Fbn1C1039G/+ murine aortic root.

A, Western blot for integrin αv and p-AktThr308 in Fbn1C1039G/+ and wild type (WT) littermate control aortic root/ascending specimens at ages 6 (n=5, per group) and 14 (n=5, per group) weeks. B, Immunofluorescent staining for integrin αv in thoracic aorta (root and ascending) longitudinal sections. Integrin αv is enhanced in Fbn1C1039G/+ (right) compared to WT (left) aortic root specimens (scale bar = 500 μm). Magnification (20x) of the aortic root segments demonstrates Fbn1C1039G/+ integrin αv staining is localized within the medial layer (scale bar = 100 μm). C, Single cell RNAseq of 16-week SHF-lineage traced mice. Total dataset comprising all major aortic cell types (upper left). SMC (red) and modulated SMC (modSMC) (grey) clusters are similar between tdT-negative (NC) and SHF derived cells (lower left). Integrin αv (Itgav) gene expression is significantly increased in Fbn1C1039G/+ vs. WT SMCs (p=2.50x10−13). There is no difference in Itgav gene expression when comparing total SHF- vs. tdT neg (NC) lineage SMCs (p=1). When subtyped into SMC vs. modSMC, Itgav is enhanced in modSMCs in both tdT neg (NC) and SHF SMCs (p=7.36x10−26)
As biological proof of concept that integrin αv participates in MFS aneurysmal development, sex-matched Fbn1C1039G/+ and littermate WT mice were treated with either (1) GLPG0187 or (2) vehicle control from ages 6 to 14 weeks. At 14 weeks, Fbn1C1039G/+ mice had significantly increased aortic root diameter compared to WT mice (median aortic root diameter: 2.11mm [IQR: 1.79-2.25] vs. 1.52mm [IQR: 1.48-1.62]; n=24 and 19 respectively; P<0.0001) (Supplemental Figure VC). GLPG0187-treated Fbn1C1039G/+ mice had significantly reduced aortic root aneurysm diameter (1.85mm [IQR: 1.68-1.96] vs. 2.21mm [IQR: 1.99–2.26], p<0.001, n=13 per group, p<0.001) and growth rate (0.08mm/8wks [IQR: 0.01-0.19] vs. 0.38mm/8wks [IQR: 0.22-0.50], p=0.02) at 14 weeks compared to vehicle-treated Fbn1C1039G/+ mice. There was no significant difference in aortic root diameter in GLPG0187-treated WT mice compared to vehicle control WT mice (root: 1.41mm [IQR:1.34-1.55] vs. 1.50mm [IQR:1.37-1.65], p=0.3, n=13 per group) (Figure 8A). Of note, GLPG0187-treatment had no significant effect on ascending aortic diameter or growth rate (Supplemental Figure VD).
Figure 8. Integrin αv receptor blockade attenuates aortic root aneurysm growth in Fbn1C1039G/+ mice, in vivo.

A, Aortic root diameter (mm) and growth rate (mm/8wks)in WT and Fbn1C1039G/+ mice treated with either vehicle control or GLPG0187 (100mg/kg) measured with transthoracic echocardiography at ages 6 through 14 weeks (n=13, per group). Results presented as mean ± SD. * represents significant difference between MFS GLPG and MFS Vehicle. † represents significant difference between MFS Vehicle and WT Vehicle. B, Elastin lamina breaks was evaluated with elastin Van Gieson staining (EVG) in aortic root from Fbn1C1039G/+ mice treated with vehicle control or GLPG0187 (100mg/kg) or vehicle control at age 14 weeks (n=6, per group, scale bar = 50μm). C, Western blotting for p-AktThr308, p-RPS6Ser235/236 and p-RPS6Ser240/244 in aortic root/ascending aortic specimens from WT and Fbn1C1039G/+ mice treated with vehicle control or GLPG0187 (100mg/kg) at age 14 weeks. D, P-AktThr308 (n=6, per group), p-PRAS40 (n=3, per group), p-RPS6Ser235/236 (n=3, per group), and p-RPS6Ser240/244 (n=3, per group) expression were measured by ELISA. Welch’s t-test was performed to examine the effect of GLPG0187 treatment. P-value (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001).
Correlating with aneurysm reduction, histopathology showed decreased aortic root elastin fragmentation in GLPG0187-treated Fbn1C1039G/+ mice compared with vehicle control (0.84 [IQR: 0.75–1.01] vs. 1.09 [IQR: 0.99–1.18] breaks per lamina, p=0.015, n=6) (Figure 8B). No difference in ascending aorta elastin fragmentation was observed between MFS GLPG0187-treated Fbn1C1039G/+ mice compared with vehicle control (data not shown). Systemic GLPG0187 did not reveal any significant off target effects in either the liver or kidney (Supplemental Figure VIA). In addition, no effect was detected for growth in tibia length, body length or body weight (Supplemental Figure VIB). Finally, western blot and ELISA analysis on aortic specimens from GLPG0187-treated mice revealed a similar trend reported in the human MFS iPSC-derived SMC model system - significant reduction in p-AktThr308 (0.158 [0.130–0.178] vs. 0.195 [0.168–0.228], p=0.016, n=6), p-PRAS40 (0.186 [0.172–0.195] vs. 0.339 [0.291–0.409], p=0.046, n=3), p-RPS6Ser235/236 (0.154 [0.132–0.168] vs. 0.267 [0.244–0.28], p=0.002, n=3) and p-RPS6Ser240/244 (0.136 [0.112–0.164] vs. 0.258 [0.221–0.308], p=0.023, n=3) (Figure 8C-8D).
Utilizing scRNAseq, our laboratory previously reported that SMCs undergo phenotypic modulation (less contractile, more synthetic and proteolytic) during aneurysm formation in Fbn1C1039G/+ mice.12 To assess if GLPG0187-treatment inhibits SMC phenotypic modulation, Fbn1C1039G/+ mice were treated from 6-16 weeks with either (1) GLPG0187 or (2) vehicle control (n=2 male; n=2 female), then scRNAseq performed on isolated single cells from the aortic root/ascending aorta. Both libraries were pooled, sequenced on a NovaSeq 6000, and analyzed using the Seurat package in R. Cells (n=7357) from Fbn1C1039G/+ GLPG0187 and vehicle control treatment were jointly analyzed. A comparison between male and female mice, using Xist expression to identify female cells,50 showed similar clustering between genders with no significant difference in the expression of contractile genes or modSMC-specific genes (Supplementary Figure VIIA), therefore both genders were combined to increase power for the downstream analysis and no sex differences or interactions were investigated. All expected cell types were identified (SMCs, modSMCs, Fibroblasts, Valve interstitial cells, Endothelial cells, and Macrophages) in the Fbn1C1039G/+ aortic root/ascending aorta (Figure 9A). To identify changes within SMCs, we subsetted SMCs and modSMCs and showed a greater than 50% decrease in the percent of modSMCs when treated with GLPG0187 compared to vehicle-treated mice (10.4% vs. 24.3%). Among the 74 significantly differentially expressed genes (Supplementary Table 1), GLPG0187-treatment significantly increased the contractile genes, Cnn1 and Myh11. Mmp2 expression was reduced following GLPG0187 blockade, although this did not reach statistical significance (Figure 9B). Next, we used pseudotemporal trajectory to map the transcriptome of each SMC to demonstrate a gradual transition from a contractile SMC to modSMC. Tnfrsf11b and Vcam1, previously identified as strong late modSMC markers,12,51,52 were reduced following GLPG0187-treatment, therefore suggesting that integrin αv blockade both (1) decreases modulated SMC formation and (2) impedes the extent of SMC modulation (Figure 9C).
Figure 9. scRNAseq in GLPG-treated Fbn1C1039G/+ mice reveals reduced SMC phenotype modulation (modSMC).

A, Total dataset comprising all major aortic cell types in Fbn1C1039G/+ GLPG- and vehicle control-treated root/ascending aortic segments (left). Validated markers for major cell types (SMCs:Myh11; modSMCs:Tnfrsf11b; fibroblasts: Pi16; and valve interstitial cells: Tbx20) (right). B, SMC subsets consisting of SMCs and modulated SMCs (modSMCs). GLPG-treatment reduced the overall percentage of SMC phenotype modulation by over 50%. GLPG-treatment increased SMC contractility (Cnn1 and Myh11) and reduced proteolytic matrix metalloproteinase 2 (Mmp2) gene expression. C, Pseudotemporal scoring reflects SMC phenotype progression from quiescent SMCs to phenotype modulated SMCs. GLPG-treatment reduced modSMC markers including, Tnfrsf11b (osteoprotegerin) and Vcam1 (vascular cell adhesion molecule 1) at later pseudotime quantiles.
Discussion
The pathophysiology of aortic root aneurysm formation in MFS is a complex, multi-factorial process. In this study, we investigated integrin αv overexpression as a contributor to aortic root aneurysm formation by studying lineage-specific downstream mechanisms in human MFS iPSC-derived SMCs and translating these findings into the Fbn1C1039G/+ Marfan mouse model. Our overall hypothesis is that the enhanced SHF-specific integrin αv-FAK- Aktthr308 signaling modulates aortic SMCs into a synthetic, proliferative, and migratory cell contributing to localized aortic root aneurysm formation. To the best of our knowledge, this is the first study providing evidence that integrin αv activation contributes to enhanced aneurysm growth in vivo. Integrin αv and its downstream activated proteins may represent several novel targets for therapeutic drug development to slow aneurysm growth in MFS.
Discovering genetic mutations coding for proteins important in SMC contraction and adhesion as a cause of thoracic aortic aneurysm has suggested the importance of mechanosignaling in aortic wall homeostasis.15 SMC contraction requires attachment to the ECM via integrin-containing focal adhesions. While integrin plays an important role in cytoskeleton polymer assembly and mechanosignaling,41,53,54 there remains a gap in knowledge understanding the role it plays in focal dilation of the aortic root segment in MFS. We propose that a combination of lineage-specific integrin αv overexpression along with localized mechanical strain, ECM composition, and architecture contribute to regional SMC modulation in the aortic root. Supporting this hypothesis, Parker et al. investigated the ascending aortic proteome within the Fbn1C1039G/+ mouse model and noted an upregulation in integrin αvβ3 expression, a finding consistent with our previous experiment in iPSC-derived SMCs.11,19 They demonstrated that integrin overexpression enhanced non-canonical TGF-β activation of rapamycin-independent component of mammalian target of rapamycin (Rictor), leading to modified SMC proliferation, migration, and mitochondrial metabolism both in vivo and in vitro.
In this study, we sought to further characterize the molecular mechanism by which integrin αv influences SMC phenotype modulation, migration, and proliferation in MFS. We found that integrin αv RGD motif activation increased Akt-dependent mTORC1 activity in vitro, whereas RGD motif blockade with either GLPG0187 treatment or integrin αv siRNA ablation reduced mTORC1-induced SMC proliferation and migration. These findings were subsequently confirmed in vivo when integrin blockade reduced Akt activation, RPS6 phosphorylation (mTORC1 downstream target), SMC phenotype modulation and aneurysm growth in the MFS Fbn1C1039G/+ mouse model. Disinhibition of mTORC1 via Akt deactivation of PRAS40, an inhibitory component of the mTORC1 complex,55,56 fosters mTORC1 activation of RPS6, an essential component of the 40S ribosomal subunit, ultimately increasing protein synthesis and cell growth (Figure 10).57,58 Cellular proliferation diverts SMC ability for optimal contraction and induces ECM breakdown via increased MMP activity and phagocytosis. Loss of SMC contractile force further weaken ECM radial strength, ultimately leads to aneurysm formation.31,59 Corroborating our findings, using a conditional TGFBR2 SMC knockout mouse model, Li et al. showed aortic root aneurysm development was associated with increased mTOR phosphorylation. In contrast, mTOR inhibition with rapamycin prevented aortic degeneration, dilation, and dissection and was associated with decreased SMC proliferation and SMC medial expansion.60 Cao et al. similarly reported that rapamycin inhibited aortic aneurysm formation through mTOR-mediated downregulation of the proinflammatory mediators in a calcium chloride-induced rat thoracic aortic aneurysm model. They showed in vitro that rapamycin treatment prevented SMC modulation towards a synthetic proliferative state.61
Figure 10. Proposed pathologic mechanism of lineage-specific integrin αv-Akt-mTORC1-RPS6 signaling during MFS aortic root aneurysm formation.

Integrin αv signaling is enhanced in MFS aortic root SHF compared to adjacent NC SMCs. The mTOR protein complex 1 (mTORC1) is composed of mTOR, PRAS40, Raptor, mLST8, and deptor. Following integrin αv activation, Akt-dependent phosphorylation deactivates PRAS40, an inhibitory component of the mTORC1 complex. mTORC1 subsequently activates RPS6, an essential component of the 40S ribosomal subunit, ultimately increasing protein synthesis and cell growth. Enhanced SMC phenotype modulation, proliferation and migration participate in aneurysm development.
Vascular SMC phenotype modulation from a contractile quiescent to a synthetic proliferative state is a core marker of thoracic aortic aneurysm and may participate in aortic wall degeneration and ECM remodeling.12 Because lineage-specific MFS SHF SMC integrin αv is detected in immature iPSC-derived SMCs, we hypothesize integrin αv overexpression may play an important early role during aneurysm development.62 Perhaps during aortic wall development, increased integrin αv/Akt/mTORC1 pathway signaling triggers localized aortic root SMC modulation and resultant elastin degradation, further exacerbating SMC modulation towards a synthetic/proliferative/proteolytic state (positive feedback loop). Supporting this idea, utilizing a murine elastin insufficiency model, Li et al. revealed that elastin plays a regulatory role during arterial wall morphogenesis, illustrating that reduced elastin increases arterial wall SMC proliferation and migration.62 Noteworthy, although increased (1) SMC proliferation was detected in vitro and (2) aortic root SMC modulation in vivo, tunica media SMC density was similar between 16-week Fbn1C1039G/+ and WT aortic root specimens (Supplemental Figure VIII). Because aortic wall apoptosis is enhanced in this mouse model,63 increased Fbn1C1039G/+ SMC proliferation may be balanced by increased SMC apoptosis. In addition, because Fbn1C1039G/+ aortic diameter is larger than WT but with similar wall thickness, increased SMC proliferation may be masked by radial expansion of the aorta resulting in unchanged density of cells/mm2. Finally, histology was performed at a single time point and perhaps SMC proliferation differences were missed. However, we must acknowledge that SMC proliferation may not participate in MFS aneurysm pathology, but instead a marker for modulated SMCs.
We hypothesize that disease-causing FBN1 mutations differentially affect SMCs from distinct embryologic origins in the thoracic aorta, leading to focal molecular and biomechanical dysfunction in the aortic root segment. SHF-derived SMCs contribute to the aortic root where they intermingle with NC-derived SMCs. In contrast, in the ascending aorta, NC-SMCs are spatially more distinct. Because aneurysm formation predominantly affects the aortic root segment in MFS, we hypothesize that complex interactions between aortic root phenotypically modulated SHF- and NC-SMCs, each with uniquely perturbed biological functions and ECM-SMC mechanosignaling, participate in focal aneurysm formation. This hypothesis is supported by our recent scRNAseq MFS mouse model study illustrating that modulated SMCs are derived from both the SHF and NC and located focally in the root at the intersection of SHF and NC overlap. Modulated SMCs from each lineage display distinct transcriptomic signatures: SHF SMCs overexpressed collagen and leucine rich proteoglycans while NC SMCs expressed chondrogenic genes.28 Interestingly, Itgav gene expression is not differentially expressed between SMC embryologic origins in 16-week Fbn1C1039G/+ SHF lineage-traced mice, despite higher integrin αv protein expression in the aortic root vs. ascending (detected with immunofluorescence staining). Potential explanations include the following: (1) Itgav gene expression is not predictive of functional mature integrin αv protein expression due to post translational modifications and/or protein catabolism; and (2) integrin αv protein is enriched in the aortic root anatomically independent of embryological origin in vivo.
Several investigators have generated human iPSC-derived SMC subtypes from each embryologic origin to study mechanisms of vascular disease, including aneurysm formation. This model system has several benefits, including: (1) opportunity to study early pathologic mechanisms, opposed to primary SMC obtained from surgical specimens after aneurysms already developed; (2) ability to study an embryologic-specific SMC population, not contaminated by other cell types; and in theory (3) potential to study and predict future disease severity before aneurysm development (precision medicine). By discovering novel proteomic differences in MFS SHF vs. NC SMCs, then testing for related abnormal downstream molecular pathways in vitro, we highlight the role embryologic origin plays on varied biological responses within the root. Moreover, we validate the iPSC model system as a translatable system for MFS pathology and emphasize its potential as an “aorta in a dish” model to discover novel disease-causing signaling pathways and test potential therapeutic drug regimens (“clinical trial in a dish”). There are also challenges with using lineage-specific iPSC-derived SMC to recapitulate and study human disease. By definition, iPSC-derived SMCs are very immature and may not exactly represent the in vivo mature aorta. Moreover, it is difficult to model in vivo environmental conditions using a static 2D model. Nonetheless, in this study we confirm iPSC model system findings in the mouse model and identify the integrin-AKT-mTORC1-RPS6 pathway as a target for drug screening to reduce SMC phenotype modulation and potentially aneurysm growth in MFS.
Limitations
This study is limited by use of (1) age but only partially sex-matched iPSC donors and (2) non-isogenic control iPSCs. Genome wide demethylation during iPSC reprograming is more pronounced in female cells, however, sex-determination terminally differentiated SMC functionality is unknown. Demographics of the healthy donor control was limited to availability from collaborators. Future investigation will focus on generating CRISPR-corrected isogenic controls to circumvent erroneous findings linked to gender, age, and genetic background. Previous proteomic findings identified integrin αv as the most differentially expressed protein between MFS and the corresponding lineage-specific control cells, however the analysis was unable to establish the heterodimeric β subunit. Lastly, systemic use of integrin αv blocker potentially results in detrimental off-target effects, however, a recent phase I clinical trial for GLPG0187 in glioma demonstrated a favorable safety profile and dose tolerability.38
Supplementary Material
Highlights.
Marfan iPSC-derived SHF SMCs display increased integrin αv expression.
Integrin αv receptor blockade reduces Fbn1C1039G/+ aneurysm growth.
Integrin αv receptor blockade attenuates integrin/Akt/mTORC1 signaling pathway in Fbn1C1039G/+ mice.
Integrin αv receptor blockade limits tunica media SMC modulation.
Sources of Funding:
This work was supported by grants from National Institute of Health F32HL154681 (AJP), F32HL160058 (ARD), R01HL157949 (MPF)
Non-standard Abbreviations and Acronyms
- MFS
Marfan Syndrome
- iPSC
Induced pluripotent stem cell
- SMC
Smooth muscle cell
- SHF
Second heart field
- NC
Neural crest
- FAK
Focal adhesion Kinase
- FBN1
Fibrillin-1
- TGF-β
Transforming growth factor-beta
- ECM
Extracellular matrix
- FN
Fibronectin
- Col1
Collagen I
- mTORC1
Mechanistic target of rapamycin complex 1
- PRAS40
Proline-rich Akt substrate of 40kDa
- RPS6
Ribosomal protein S6
- P70S6K
P70-S6 Kinase
- scRNAseq
Single cell RNA sequencing
Footnotes
Disclosures: None
Data availability:
16-wk MFS GLPG treated dataset: Gene Expression Omnibus repository under accession GSE223807. 16-wks MFS SHF-lineage dataset: Gene Expression Omnibus repository under accession GSE186845
References:
- 1.Dietz HC, Cutting CR, Pyeritz RE, Maslen CL, Sakai LY, Corson GM, Puffenberger EG, Hamosh A, Nanthakumar EJ, Curristin SM, Stetten G, Meyers DA, Francomano CA. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352(6333):337–339. doi: 10.1038/352337a0 [DOI] [PubMed] [Google Scholar]
- 2.Judge DP, Dietz HC. Marfan’s syndrome. Lancet. 2005;366(9501):1965–1976. doi: 10.1016/S0140-6736(05)67789-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Saeyeldin A, Zafar MA, Velasquez CA, Ip K, Gryaznov A, Brownstein AJ, Li Y, Rizzo JA, Erben Y, Ziganshin BA, Elefteriades JA. Natural history of aortic root aneurysms in Marfan syndrome. Ann Cardiothorac Surg. 2017;6(6):625–632. doi: 10.21037/acs.2017.11.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pyeritz RE. Etiology and pathogenesis of the Marfan syndrome: current understanding. Ann Cardiothorac Surg. 2017;6(6):595–598. doi: 10.21037/acs.2017.10.04 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Milewicz DM, Dietz HC, Miller DC. Treatment of Aortic Disease in Patients With Marfan Syndrome. Circulation. 2005;111(11). doi: 10.1161/01.CIR.0000155243.70456.F4 [DOI] [PubMed] [Google Scholar]
- 6.Holm TM, Habashi JP, Doyle JJ, Bedja D, Chen Y, van Erp C, Lindsay ME, Kim D, Schoenhoff F, Cohn RD, Loeys BL, Thomas CJ, Patnaik S, Marugan JJ, Judge DP, Dietz HC. Noncanonical TGFβ signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science. 2011;332(6027):358–361. doi: 10.1126/science.1192149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dietz HC, Loeys B, Carta L, Ramirez F. Recent progress towards a molecular understanding of Marfan syndrome. Am J Med Genet. 2005;139C(1):4–9. doi: 10.1002/ajmg.c.30068 [DOI] [PubMed] [Google Scholar]
- 8.Matt P, Schoenhoff F, Habashi J, Holm T, Van Erp C, Loch D, Carlson OD, Griswold BF, Fu Q, De Backer J, Loeys B, Huso DL, McDonnell NB, Van Eyk JE, Dietz HC, the GenTAC Consortium. Circulating Transforming Growth Factor-β in Marfan Syndrome. Circulation. 2009;120(6):526–532. doi: 10.1161/CIRCULATIONAHA.108.841981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Waldo KL, Hutson MR, Ward CC, Zdanowicz M, Stadt HA, Kumiski D, Abu-Issa R, Kirby ML. Secondary heart field contributes myocardium and smooth muscle to the arterial pole of the developing heart. Dev Biol. 2005;281(1):78–90. doi: 10.1016/j.ydbio.2005.02.012 [DOI] [PubMed] [Google Scholar]
- 10.Jiang X, Rowitch DH, Soriano P, McMahon AP, Sucov HM. Fate of the mammalian cardiac neural crest. Development. 2000;127(8):1607–1616. [DOI] [PubMed] [Google Scholar]
- 11.Iosef C, Pedroza AJ, Cui JZ, Dalal AR, Arakawa M, Tashima Y, Koyano TK, Burdon G, Churovich SMP, Orrick JO, Pariani M, Fischbein MP. Quantitative proteomics reveal lineage-specific protein profiles in iPSC-derived Marfan syndrome smooth muscle cells. Sci Rep. 2020;10(1):20392. doi: 10.1038/s41598-020-77274-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pedroza AJ, Tashima Y, Shad R, Cheng P, Wirka R, Churovich S, Nakamura K, Yokoyama N, Cui JZ, Iosef C, Hiesinger W, Quertermous T, Fischbein MP. Single-Cell Transcriptomic Profiling of Vascular Smooth Muscle Cell Phenotype Modulation in Marfan Syndrome Aortic Aneurysm. Arterioscler Thromb Vasc Biol. 2020;40(9):2195–2211. doi: 10.1161/ATVBAHA.120.314670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takada Y, Ye X, Simon S. The integrins. Genome Biol. 2007;8(5):215. doi: 10.1186/gb-2007-8-5-215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Turner CJ, Badu-Nkansah K, Crowley D, van der Flier A, Hynes RO. α5 and αv integrins cooperate to regulate vascular smooth muscle and neural crest functions in vivo. Development. 2015;142(4):797–808. doi: 10.1242/dev.117572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Humphrey Jay D, Schwartz Martin A, Tellides George, Milewicz Dianna M. Role of Mechanotransduction in Vascular Biology. Circulation Research. 2015;116(8):1448–1461. doi: 10.1161/CIRCRESAHA.114.304936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giancotti FG. Integrin Signaling. Science. 1999;285(5430):1028–1033. doi: 10.1126/science.285.5430.1028 [DOI] [PubMed] [Google Scholar]
- 17.Jain M, Chauhan AK. Role of Integrins in Modulating Smooth Muscle Cell Plasticity and Vascular Remodeling: From Expression to Therapeutic Implications. Cells. 2022;11(4):646. doi: 10.3390/cells11040646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chakraborty R, Chatterjee P, Dave JM, Ostriker AC, Greif DM, Rzucidlo EM, Martin KA. Targeting smooth muscle cell phenotypic switching in vascular disease. JVS-Vascular Science. 2021;2:79–94. doi: 10.1016/j.jvssci.2021.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parker SJ, Stotland A, MacFarlane E, Wilson N, Orosco A, Venkatraman V, Madrid K, Gottlieb R, Dietz HC, Van Eyk JE. Proteomics reveals Rictor as a noncanonical TGF-β signaling target during aneurysm progression in Marfan mice. Am J Physiol Heart Circ Physiol. 2018;315(5):H1112–H1126. doi: 10.1152/ajpheart.00089.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheung C, Bernardo AS, Pedersen RA, Sinha S. Directed differentiation of embryonic origin-specific vascular smooth muscle subtypes from human pluripotent stem cells. Nat Protoc. 2014;9(4):929–938. doi: 10.1038/nprot.2014.059 [DOI] [PubMed] [Google Scholar]
- 21.Reeves KJ, Hurrell JE, Cecchini M, van der Pluijm G, Down JM, Eaton CL, Hamdy F, Clement-Lacroix P, Brown NJ. Prostate cancer cells home to bone using a novel in vivo model: Modulation by the integrin antagonist GLPG0187: Model for tracking early prostate cancer homing to bone. Int J Cancer. 2015;136(7):1731–1740. doi: 10.1002/ijc.29165 [DOI] [PubMed] [Google Scholar]
- 22.van der Horst G, van den Hoogen C, Buijs JT, Cheung H, Bloys H, Pelger RCM, Lorenzon G, Heckmann B, Feyen J, Pujuguet P, Blanque R, Clément-Lacroix P, van der Pluijm G. Targeting of αv-Integrins in Stem/Progenitor Cells and Supportive Microenvironment Impairs Bone Metastasis in Human Prostate Cancer. Neoplasia. 2011;13(6):516–IN9. doi: 10.1593/neo.11122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Erdem C, Lee AV, Taylor DL, Lezon TR. Inhibition of RPS6K reveals context-dependent Akt activity in luminal breast cancer cells. Hall B, ed. PLoS Comput Biol. 2021;17(6):e1009125. doi: 10.1371/journal.pcbi.1009125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hipolito VEB, Diaz JA, Tandoc KV, Oertlin C, Ristau J, Chauhan N, Saric A, Mclaughlan S, Larsson O, Topisirovic I, Botelho RJ. Enhanced translation expands the endo-lysosome size and promotes antigen presentation during phagocyte activation. Cadwell K, ed. PLoS Biol. 2019;17(12):e3000535. doi: 10.1371/journal.pbio.3000535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen B, Yang L, Zhang R, Gan Y, Zhang W, Liu D, Chen H, Tang H. Hyperphosphorylation of RPS6KB1, rather than overexpression, predicts worse prognosis in non-small cell lung cancer patients. Coleman WB, ed. PLoS ONE. 2017;12(8):e0182891. doi: 10.1371/journal.pone.0182891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Merk DR, Chin JT, Dake BA, Maegdefessel L, Miller MO, Kimura N, Tsao PS, Iosef C, Berry GJ, Mohr FW, Spin JM, Alvira CM, Robbins RC, Fischbein MP. miR-29b participates in early aneurysm development in Marfan syndrome. Circ Res. 2012;110(2):312–324. doi: 10.1161/CIRCRESAHA.111.253740 [DOI] [PubMed] [Google Scholar]
- 27.Tashima Y, He H, Cui JZ, Pedroza AJ, Nakamura K, Yokoyama N, Iosef C, Burdon G, Koyano T, Yamaguchi A, Fischbein MP. Androgens Accentuate TGF-β Dependent Erk/Smad Activation During Thoracic Aortic Aneurysm Formation in Marfan Syndrome Male Mice. J Am Heart Assoc. 2020;9(20):e015773. doi: 10.1161/JAHA.119.015773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pedroza AJ, Dalal AR, Shad R, Yokoyama N, Nakamura K, Cheng P, Wirka RC, Mitchel O, Baiocchi M, Hiesinger W, Quertermous T, Fischbein MP. Embryologic Origin Influences Smooth Muscle Cell Phenotypic Modulation Signatures in Murine Marfan Syndrome Aortic Aneurysm. Arteriosclerosis, Thrombosis, and Vascular Biology. 2022;42(9):1154–1168. doi: 10.1161/ATVBAHA.122.317381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Granata A, Serrano F, Bernard WG, McNamara M, Low L, Sastry P, Sinha S. An iPSC-derived vascular model of Marfan syndrome identifies key mediators of smooth muscle cell death. Nat Genet. 2017;49(1):97–109. doi: 10.1038/ng.3723 [DOI] [PubMed] [Google Scholar]
- 30.Harada T, Yoshimura K, Yamashita O, Ueda K, Morikage N, Sawada Y, Hamano K. Focal Adhesion Kinase Promotes the Progression of Aortic Aneurysm by Modulating Macrophage Behavior. ATVB. 2017;37(1):156–165. doi: 10.1161/ATVBAHA.116.308542 [DOI] [PubMed] [Google Scholar]
- 31.Li G, Wang M, Caulk AW, Cilfone NA, Gujja S, Qin L, Chen PY, Chen Z, Yousef S, Jiao Y, He C, Jiang B, Korneva A, Bersi MR, Wang G, Liu X, Mehta S, Geirsson A, Gulcher JR, Chittenden TW, Simons M, Humphrey JD, Tellides G. Chronic mTOR activation induces a degradative smooth muscle cell phenotype. Journal of Clinical Investigation. 2020;130(3):1233–1251. doi: 10.1172/JCI131048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ghosh A, Lu G, Su G, McEvoy B, Sadiq O, DiMusto PD, Laser A, Futchko JS, Henke PK, Eliason JL, Upchurch GR. Phosphorylation of AKT and Abdominal Aortic Aneurysm Formation. The American Journal of Pathology. 2014;184(1):148–158. doi: 10.1016/j.ajpath.2013.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yamashita O, Yoshimura K, Nagasawa A, Ueda K, Morikage N, Ikeda Y, Hamano K. Periostin Links Mechanical Strain to Inflammation in Abdominal Aortic Aneurysm. Aikawa E, ed. PLoS ONE. 2013;8(11):e79753. doi: 10.1371/journal.pone.0079753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu M, Chen C, Cao Y, Qi R. Inhibitory effects of doxycycline on the onset and progression of abdominal aortic aneurysm and its related mechanisms. European Journal of Pharmacology. 2017;811:101–109. doi: 10.1016/j.ejphar.2017.05.041 [DOI] [PubMed] [Google Scholar]
- 35.Huang C, Fang X, Xie X, Liu Y, Xu D, Meng X, Long J. Effect of miR-126 on the Proliferation and Migration of Vascular Smooth Muscle Cells in Aortic Aneurysm Mice Under PI3K/AKT/mTOR Signaling Pathway. Mol Biotechnol. 2021;63(7):631–637. doi: 10.1007/s12033-021-00327-6 [DOI] [PubMed] [Google Scholar]
- 36.Kim KL, Yang JH, Song SH, Kim JY, Jang SY, Kim JM, Kim JA, Sung KI, Kim YW, Suh YL, Suh W, Kim DK. Positive Correlation Between the Dysregulation of Transforming Growth Factor-β1 and Aneurysmal Pathological Changes in Patients With Marfan Syndrome. Circ J. 2013;77(4):952–958. doi: 10.1253/circj.CJ-12-0874 [DOI] [PubMed] [Google Scholar]
- 37.Nataatmadja M, West J, West M. Overexpression of Transforming Growth Factor-β Is Associated With Increased Hyaluronan Content and Impairment of Repair in Marfan Syndrome Aortic Aneurysm. Circulation. 2006;114(1_supplement). doi: 10.1161/CIRCULATIONAHA.105.000927 [DOI] [PubMed] [Google Scholar]
- 38.Cirkel GA, Kerklaan BM, Vanhoutte F, der Aa AV, Lorenzon G, Namour F, Pujuguet P, Darquenne S, de Vos FYF, Snijders TJ, Voest EE, Schellens JHM, Lolkema MP. A dose escalating phase I study of GLPG0187, a broad spectrum integrin receptor antagonist, in adult patients with progressive high-grade glioma and other advanced solid malignancies. Invest New Drugs. 2016;34(2):184–192. doi: 10.1007/s10637-015-0320-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li Y, Drabsch Y, Pujuguet P, Ren J, van Laar T, Zhang L, van Dam H, Clément-Lacroix P, ten Dijke P. Genetic depletion and pharmacological targeting of αv integrin in breast cancer cells impairs metastasis in zebrafish and mouse xenograft models. Breast Cancer Res. 2015;17(1):28. doi: 10.1186/s13058-015-0537-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mao R, Doyon G, Gordon IO, Li J, Lin S, Wang J, Le THN, Elias M, Kurada S, Southern B, Olman M, Chen M, Zhao S, Dejanovic D, Chandra J, Mukherjee PK, West G, Van Wagoner DR, Fiocchi C, Rieder F. Activated intestinal muscle cells promote preadipocyte migration: a novel mechanism for creeping fat formation in Crohn’s disease. Gut. 2022;71(1):55–67. doi: 10.1136/gutjnl-2020-323719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Humphries JD, Byron A, Humphries MJ. INTEGRIN LIGANDS. J Cell Sci. 2006;119(Pt 19):3901–3903. doi: 10.1242/jcs.03098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xiao T, Takagi J, Coller BS, Wang JH, Springer TA. Structural basis for allostery in integrins and binding to fibrinogen-mimetic therapeutics. Nature. 2004;432(7013):59–67. doi: 10.1038/nature02976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xiong JP, Stehle T, Zhang R, Joachimiak A, Frech M, Goodman SL, Arnaout MA. Crystal Structure of the Extracellular Segment of Integrin αVβ3 in Complex with an Arg-Gly-Asp Ligand. Science. 2002;296(5565):151–155. doi: 10.1126/science.1069040 [DOI] [PubMed] [Google Scholar]
- 44.Endl E, Hollmann C, Gerdes J. Chapter 18 Antibodies against the Ki-67 protein: Assessment of the growth fraction and tools for cell cycle analysis. In: Methods in Cell Biology. Vol 63. Elsevier; 2001:399–418. doi: 10.1016/S0091-679X(01)63022-X [DOI] [PubMed] [Google Scholar]
- 45.Schwarting R, Gerdes J, Niehus J, Jaeschke L, Stein H. Determination of the growth fraction in cell suspensions by flow cytometry using the monoclonal antibody Ki-67. Journal of Immunological Methods. 1986;90(1):65–70. doi: 10.1016/0022-1759(86)90384-4 [DOI] [PubMed] [Google Scholar]
- 46.Copp J, Manning G, Hunter T. TORC-specific phosphorylation of mammalian target of rapamycin (mTOR): phospho-Ser2481 is a marker for intact mTOR signaling complex 2. Cancer Res. 2009;69(5):1821–1827. doi: 10.1158/0008-5472.CAN-08-3014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jhanwar-Uniyal M, Wainwright JV, Mohan AL, Tobias ME, Murali R, Gandhi CD, Schmidt MH. Diverse signaling mechanisms of mTOR complexes: mTORC1 and mTORC2 in forming a formidable relationship. Adv Biol Regul. 2019;72:51–62. doi: 10.1016/j.jbior.2019.03.003 [DOI] [PubMed] [Google Scholar]
- 48.Harter PN, Jennewein L, Baumgarten P, Ilina E, Burger MC, Thiepold AL, Tichy J, Zörnig M, Senft C, Steinbach JP, Mittelbronn M, Ronellenfitsch MW. Immunohistochemical Assessment of Phosphorylated mTORC1-Pathway Proteins in Human Brain Tumors. Alonso MM, ed. PLoS ONE. 2015;10(5):e0127123. doi: 10.1371/journal.pone.0127123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pedroza AJ, Shad R, Dalal AR, Yokoyama N, Nakamura K, Hiesinger W, Fischbein MP. Acute Induced Pressure Overload Rapidly Incites Thoracic Aortic Aneurysmal Smooth Muscle Cell Phenotype. Hypertension. 2022;79(4):e86–e89. doi: 10.1161/HYPERTENSIONAHA.121.18640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Saichi M, Ladjemi MZ, Korniotis S, Rousseau C, Ait Hamou Z, Massenet-Regad L, Amblard E, Noel F, Marie Y, Bouteiller D, Medvedovic J, Pène F, Soumelis V. Single-cell RNA sequencing of blood antigen-presenting cells in severe COVID-19 reveals multi-process defects in antiviral immunity. Nat Cell Biol. 2021;23(5):538–551. doi: 10.1038/s41556-021-00681-2 [DOI] [PubMed] [Google Scholar]
- 51.Cheng P, Wirka RC, Kim JB, Kim HJ, Nguyen T, Kundu R, Zhao Q, Sharma D, Pedroza A, Nagao M, Iyer D, Fischbein MP, Quertermous T. Smad3 regulates smooth muscle cell fate and mediates adverse remodeling and calcification of the atherosclerotic plaque. Nat Cardiovasc Res. 2022;1(4):322–333. doi: 10.1038/s44161-022-00042-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wirka RC, Wagh D, Paik DT, Pjanic M, Nguyen T, Miller CL, Kundu R, Nagao M, Coller J, Koyano TK, Fong R, Woo YJ, Liu B, Montgomery SB, Wu JC, Zhu K, Chang R, Alamprese M, Tallquist MD, Kim JB, Quertermous T. Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat Med. 2019;25(8):1280–1289. doi: 10.1038/s41591-019-0512-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bax DanielV Bernard SE, Lomas A, Morgan A, Humphries J, Shuttleworth CA, Humphries MJ, Kielty CM. Cell Adhesion to Fibrillin-1 Molecules and Microfibrils Is Mediated by α5β1 and αvβ3 Integrins. Journal of Biological Chemistry. 2003;278(36):34605–34616. doi: 10.1074/jbc.M303159200 [DOI] [PubMed] [Google Scholar]
- 54.Taylor-Weiner H, Ravi N, Engler AJ. Traction forces mediated by integrin signaling are necessary for definitive endoderm specification. J Cell Sci. 2015;128(10):1961–1968. doi: 10.1242/jcs.166157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wiza C, Nascimento EBM, Ouwens DM. Role of PRAS40 in Akt and mTOR signaling in health and disease. American Journal of Physiology-Endocrinology and Metabolism. 2012;302(12):E1453–E1460. doi: 10.1152/ajpendo.00660.2011 [DOI] [PubMed] [Google Scholar]
- 56.Völkers M, Sussman M. mTOR/PRAS40 interaction: Hypertrophy or proliferation. Cell Cycle. 2013;12(23):3579–3580. doi: 10.4161/cc.26822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Martin-Perez J, Thomas G. Ordered phosphorylation of 40S ribosomal protein S6 after serum stimulation of quiescent 3T3 cells. Proceedings of the National Academy of Sciences. 1983;80(4):926–930. doi: 10.1073/pnas.80.4.926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Krieg J, Hofsteenge J, Thomas G. Identification of the 40 S ribosomal protein S6 phosphorylation sites induced by cycloheximide. J Biol Chem. 1988;263(23):11473–11477. [PubMed] [Google Scholar]
- 59.Estrada AC, Irons L, Rego BV, Li G, Tellides G, Humphrey JD. Roles of mTOR in thoracic aortopathy understood by complex intracellular signaling interactions. PLOS Computational Biology. 2021;17(12):e1009683. doi: 10.1371/journal.pcbi.1009683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li W, Li Q, Jiao Y, Qin L, Ali R, Zhou J, Ferruzzi J, Kim RW, Geirsson A, Dietz HC, Offermanns S, Humphrey JD, Tellides G. Tgfbr2 disruption in postnatal smooth muscle impairs aortic wall homeostasis. J Clin Invest. 2014;124(2):755–767. doi: 10.1172/JCI69942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cao J, Wu Q, Geng L, Chen X, Shen W, Wu F, Chen Y. Rapamycin inhibits CaCl2-induced thoracic aortic aneurysm formation in rats through mTOR-mediated suppression of proinflammatory mediators. Molecular Medicine Reports. 2017;16(2):1911–1919. doi: 10.3892/mmr.2017.6844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li DY, Brooke B, Davis EC, Mecham RP, Sorensen LK, Boak BB, Eichwald E, Keating MT. Elastin is an essential determinant of arterial morphogenesis. Nature. 1998;393(6682):276–280. doi: 10.1038/30522 [DOI] [PubMed] [Google Scholar]
- 63.Emrich FC, Okamura H, Dalal AR, Penov K, Merk DR, Raaz U, Hennigs JK, Chin JT, Miller MO, Pedroza AJ, Craig JK, Koyano TK, Blankenberg FG, Connolly AJ, Mohr FW, Alvira CM, Rabinovitch M, Fischbein MP. Enhanced caspase activity contributes to aortic wall remodeling and early aneurysm development in a murine model of Marfan syndrome. Arterioscler Thromb Vasc Biol. 2015;35(1):146–154. doi: 10.1161/ATVBAHA.114.304364 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data and materials have been made publicly available at the Gene Expression Omnibus and can be accessed at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE223807.
16-wk MFS GLPG treated dataset: Gene Expression Omnibus repository under accession GSE223807. 16-wks MFS SHF-lineage dataset: Gene Expression Omnibus repository under accession GSE186845