

Abstract
Objectives:
Hepatocytic CEACAM1 plays a critical role in NASH pathogenesis, as bolstered by the development of insulin resistance, visceral obesity, steatohepatitis and fibrosis in mice with global Ceacam1 (Cc1) deletion. In contrast, VECadCre+Cc1fl/fl mice with endothelial loss of Cc1 manifested insulin sensitivity with no visceral obesity despite elevated NF-κB signaling and increased systemic inflammation. We herein investigated whether VECadCre+Cc1fl/fl male mice develop hepatic fibrosis and whether this is mediated by increased production of endothelin1 (ET1), a transcriptional NF-κB target.
Methods:
VECadCre+Et1.Cc1fl/fl mice with combined endothelial loss of Cc1/Et1 genes were generated. Histological and immunohistochemical analyses were conducted on their livers and on liver tissue biopsies from adult patients undergoing bariatric surgery or from patients with NASH diagnosis receiving liver transplant.
Results:
Hepatic fibrosis and inflammatory infiltration developed in VECadCre+Cc1fl/fl liver parenchyma. This was preceded by increased ET1 production and reversed with combined endothelial loss of Et1. Conditioned media from VECadCre+Cc1fl/fl, but not VECadCre+Et1.Cc1fl/fl primary liver endothelial cells activated wild-type hepatic stellate cells; a process inhibited by bosentan, an ETAR/ETBR dual antagonist. Consistently, immunohistochemical analysis of liver biopsies from patients with NASH showed a decline in endothelial CEACAM1 in parallel with increased plasma endothelin1 levels and progression of hepatic fibrosis stage.
Conclusions:
The data demonstrated that endothelial CEACAM1 plays a key role in preventing hepatic fibrogenesis by reducing autocrine endothelin1 production.
Keywords: Fibrosis, Endothelin1, Endothelial cells, Hepatic stellate cells, Inflammation
Graphical abstract
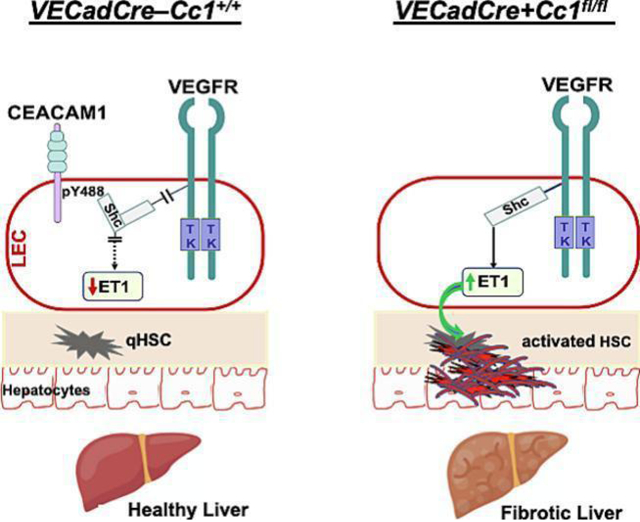
1. Introduction
Hepatic fibrosis is a critical pathology in non-alcoholic steatohepatitis (NASH), a leading cause of liver-related death worldwide [1]. It is characterized by stellate cells activation involving extracellular matrix accumulation [2]. To date, there is no FDA-approved specific therapeutic modality against fibrotic liver disease, owing to our limited knowledge of key underlying mechanisms and to disease heterogeneity [3–6]. Moreover, most of the animal models fail to replicate faithfully the human disease [2]. Whereas metabolic abnormalities are implicated in NAFLD and related drugs are repurposed against fibrosis [7], the role of insulin resistance in hepatic fibrosis has not been fully resolved. Accordingly, the need for therapies directed primarily against hepatic fibrosis remains largely unmet [4].
Carcinoembryonic Antigen-related Cell Adhesion Molecule1 (CEACAM1) is a transmembrane glycoprotein that is expressed in all liver cells, including hepatocytes and endothelial cells. Obese subjects with insulin resistance and NAFLD manifest reduction in hepatic CEACAM1 levels [8] and CEACAM1 level constitute a key determinant of liver donor quality [9]. Consistent with its role in promoting insulin clearance in hepatocytes, mice with global (Cc1−/−), or with liver-specific deletion/inactivation of CEACAM1 display impaired insulin clearance with resultant hyperinsulinemia-driven insulin resistance and NASH features, including chicken-wire bridging fibrosis [10, 11]. Hepatic fibrosis develops in parallel with hyperinsulinemia-driven induction of plasma endothelin1 (ET1) with a reciprocal suppression of nitric oxide (NO) production [11]. This could tip the vasomotor balance towards contractility, which would activate hepatic stellate cells (HSC) to cause fibrosis [2]. Rescuing CEACAM1 in hepatocytes reversed hyperinsulinemia and endothelial dysfunction in parallel to ET1 levels and hepatic fibrosis in Cc1−/− nulls [11, 12]. Collectively, this assigns a protective role for hepatocytic CEACAM1 against hepatic fibrosis.
Hepatic fibrosis also implicates a cascade of biochemical changes in endothelial cells, including disruption of their integrity and mesenchymal transformation, loss of fenestration in the sinusoid, recruitment of neutrophils and defective autophagy [13, 14]. Consistent with the role of endothelial cell CEACAM1 in promoting cell differentiation [15] and regulating barrier function [16], Cc1−/− mice develop endothelial and vascular dysfunction [12, 16]. Similarly, mice with endothelial loss of Ceacam1 (VECadCre+Cc1fl/fl) exhibit disturbed endothelial cell junctions and endothelial dysfunction (characterized by reduced eNOS-dependent NO production) together with increased circulating cytokine levels driven by hyperactivated NF-κB pathways in endothelial cells [17].
Despite their heightened pro-inflammatory state, VECadCre+Cc1fl/fl mice did not exhibit insulin resistance or hepatic steatosis. Thus, we sought to examine whether they develop hepatic fibrosis and to delineate the underlying mechanisms in the absence of hyperinsulinemia.
2. Materials and methods
2.1. Generation of mice
As previously detailed [17], VECadCre+Cc1fl/fl (hereafter VECad+Cc1fl/fl) were generated by crossing Ceacam1(Cc1)loxp/loxp with mice expressing Cre under the transcriptional control of VECadherin promoter (Jackson Laboratories, Bar Harbor, ME). Mice were backcrossed with C57BL/6J.Endothelin1loxp/loxp(Et1loxp/loxp) mice, generated by Prof. M.Yanagisawa (University of Tsukuba, Japan) [18] and generously provided by Dr. D.E.Kohan (University of Utah). Mice were then bred to homozygosity and analyzed by qRT-PCR using gene-specific primers (Table S1) to identify VECad+Et1.Cc1fl/fl double mutants and their double-Flox littermates (VECad–Et1.Cc1fl/fl) (Table S2).
Mice were housed in a 12h-light/dark cycle and fed ad libitum a standard chow (Harlan Teklad 2016; Haslett, MI). Only male mice were examined per approved protocols (16-11-011 and 16-H-025) at Ohio University.
2.2. Isolation of primary cells and media transfer
Primary liver endothelial cells (LEC) were isolated from ketamine/xylazine-anesthetized 2-to-3–month-old mice [12] and hepatic stellate cells (HSC) from mice of ≥8 months of age [19]. Cells were maintained at 37°C-5%CO2 in DMEM (Gibco Lab, Gaithersburg, MD) supplemented with 10% fetal bovine serum, 1% gentamycin, and 1% antibiotic/antimycotic (Gibco).
HSC from VECad−Cc1+/+ controls were incubated in 6-well plates in DMEM (Gibco) at 50–60% confluence. Media were collected and cells washed with PBS (Gibco) before re-incubation for 24h in the collected control media or in media from single or double LEC mutants or their controls. Cells were incubated with nilotinib (1μM) (Sigma-Aldrich, St Louis MO) [20] to inhibit platelet-derived growth factor-B (PDGF-B) or bosentan (10μM) (Sigma-Aldrich), an ETA/ETB receptor dual antagonist [21].
Liver sinusoidal endothelial cells (LSECs) were isolated as detailed in supplemental data [22]. Briefly, cells were collected from perfused livers, purified on a Percoll density gradient, and incubated in 24-well collagen-coated plates for 3 days before being subjected to analysis of gene expression in cells and endothelin1 levels in media.
2.3. Evaluation of clinical human liver biopsies
Routine standard of care human liver tissue biopsies were obtained from adult male patients undergoing bariatric surgery or from patients with a NASH diagnosis receiving an orthotopic liver transplant. All research was conducted in accordance with both the Declarations of Helsinki and Istanbul and was approved by IRB-protocols (201700650 and 201900298) at the University of Florida. All subjects were recruited and consented between August 2019 and December 2021 (Table S3). Patients or the public were not involved in the design, or conduct, or reporting, or dissemination plans of the research. Blood and tissue samples were deidentified after collection and prior to analysis. Biopsies were immediately stored in 10% buffered formalin for 24h, paraffin embedded (FFPE), sectioned and then processed for regular histological evaluation by a blinded clinical pathologist. Fibrosis was graded per Brunt Criteria [23].
As detailed in Supplemental data, FFPE sections were analyzed by immunohistochemical staining with mouse anti-CD68 and rabbit anti-MPO, and by triple immunofluorescence staining with rabbit anti-CEACAM1 and mouse anti-CD31, followed by Alexa Fluor 488 anti-rabbit IgG (H+L) and Alexa Fluor 555 anti-mouse IgG (H+L).
3. Results
3.1. Hepatic inflammation
Like Cc1−/− nulls [11], H&E-staining of liver sections revealed no parenchymal ballooning or altered hepatocellular architecture in 8-month-old VECad+Cc1fl/fl mice (Fig. 1A, panel d). Unlike Cc1−/− [11]; however, their liver sections displayed no lipid deposition (Fig. 1A, panel d), and their hepatic and plasma triacylglycerol levels were normal (Table 1). Consistent with normal plasma insulin and NEFA [17], their livers manifested unchanged mRNA levels of lipogenic genes [Srebp-1c and fatty acid synthase (Fasn)] and of Cd36 fatty acid translocase (Table S4).
Fig. 1. Histological and immunohistochemical analysis.
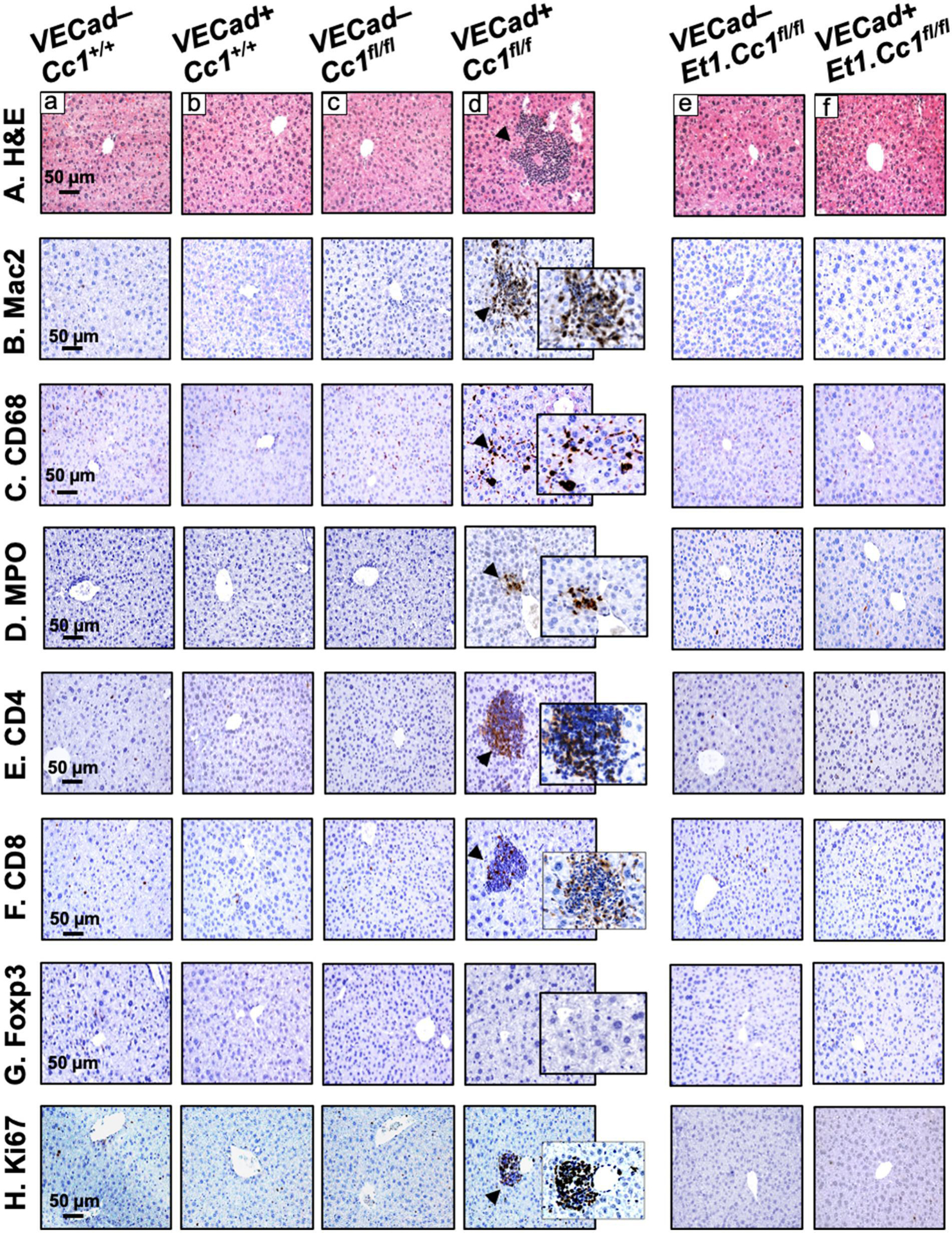
Livers were removed from 8-month-old VECad+Cc1fl/fl male mice, their 3 littermate controls and 9-month-old VECad+Et1.Cc1fl/fl male mice and their VECadCre–Et1.Cc1fl/fl double littermate Flox controls (n=4–5/genotype). (A) H&E staining of liver sections to identify foci of inflammatory cell infiltrates (arrowheads) in single mutants (panel d) but not in their littermate controls (panel a-c) or double mutants (panel f) and their double Flox controls (panel e). (B-H) liver sections were subjected to immunohistochemical analysis with: (B) Mac-2 to assess macrophage recruitment and (C) CD68 macrophage activation, (D) MPO to determine neutrophil accumulation, (E-F) CD4 and CD8 to immunostain T cells, (G) FoxP3 to evaluate anti-inflammatory Treg pool, and (H) Ki67 to examine proliferation. Representative images taken at 50μm magnification are shown with insets at 20μm.
Table 1:
Plasma and tissue biochemistry in male VECadCre+Cc1fl/fl mice and their control littermates at 8 months of age
| VECadCre−Cc1+/+ | VECadCre+Cc1+/+ | VECadCre−Cc1fl/fl | VECadCre+Cc1fl/fl | |
|---|---|---|---|---|
| Hepatic TG (μg/mg protein) | 54.3 ± 2.9 | 59.9 ± 3.8 | 60.4 ± 4.8 | 54.4 ± 5.1 |
| Plasma TG (mg/dl) | 69.0 ± 3.9 | 70.2 ± 4.2 | 72.8 ± 4.5 | 69.8 ± 4.5 |
| Plasma IL-6 (pg/ml) | 3.7 ± 0.4 | 4.1 ± 0.3 | 7.2 ± 1.0 | 39.1 ± 4.8*†‡ |
| Plasma TNFα (pg/ml) | 12.5 ± 2.4 | 15.2 ± 2.9 | 10.6 ± 2.9 | 25.9 ± 3.1*†‡ |
| Hepatic NAD/NADH | 1.1 ± 0.1 | 1.2 ± 0.1 | 1.1 ± 0.1 | 3.3 ± 0.2*†‡ |
| Plasma 8-Isoprostane (pg/ml) | 5.1 ± 0.4 | 4.8 ± 0.3 | 5.6 ± 0.4 | 14.0 ± 0.6*†‡ |
| Plasma ALT (mU/ml) | 39.6 ± 2.4 | 31.8 ± 2.0 | 37.6 ± 5.0 | 66.9 ± 5.9*†‡ |
| Plasma AST (mU/ml) | 135.8 ± 10.9 | 112.2 ± 12.0 | 130.5 ± 15.2 | 186.9 ± 16.8*†‡ |
| Plasma MMP2 (ng/ml) | 836.1 ± 30.1 | 753.3 ± 22.1 | 774.3 ± 52.8 | 1279. ± 160.*†‡ |
| Plasma MMP13 (pg/ml) | 324.7 ± 69.2 | 263.1 ± 86.9 | 272.2 ± 83.1 | 295.2 ± 58.6 |
| Plasma TIMP1 (pg/ml) | 1.8 ± 0.2 | 1.5 ± 0.1 | 1.5 ± 0.2 | 3.3 ± 0.6*†‡ |
| Plasma endothelin1 (pg/ml) | 4.8 ± 1.2 | 4.5 ± 1.2 | 4.2 ± 1.3 | 8.8 ± 0.3*†‡ |
| Plasma SDF-1α (ng/ml) | 47.5 ± 2.4 | 45.4 ± 1.8 | 44.9 ± 3.6 | 82.8 ± 5.3*†‡ |
| Plasma PDGF-B (ng/ml) | 5.0 ± 0.3 | 4.7 ± 0.3 | 5.2 ± 0.3 | 12.8 ± 0.7*†‡ |
Male mice (8 months of age, n=5/genotype) were fasted from 5:00 p.m. until 11:00 a.m. the next morning before blood was drawn and tissues were excised. Values are expressed as mean ± SEM.
P<0.05 vs VECadCre–Cc1+/+,
P<0.05 vs VECadCre+Cc1+/+,
P<0.05 vs VECadCre–Cc1fl/fl.
Mutant livers displayed mononuclear inflammatory foci formation in the perivascular areas at 8 (Fig. 1A, panel d vs a-c), but not at 3–6 months of age (Figs. S2A and S3A). This was likely facilitated by increased endothelial cell permeability, marked by lower hepatic VE-Cadherin protein level in their liver endothelial cells (LEC), owing to a rise in ADAM10 metalloprotease [17]. Accordingly, Western analysis showed lower VE-Cadherin with a reciprocally higher ADAM10 levels in VECad+Cc1fl/fl liver lysates relative to their Flox (VECad−Cc1fl/fl), Cre (VECad+Cc1+/+) and wild-type (VECad−Cc1+/+) controls starting at 8 months of age (Figs. 2A.a and S4A vs S3B).
Fig. 2. Analysis of fibrosis.
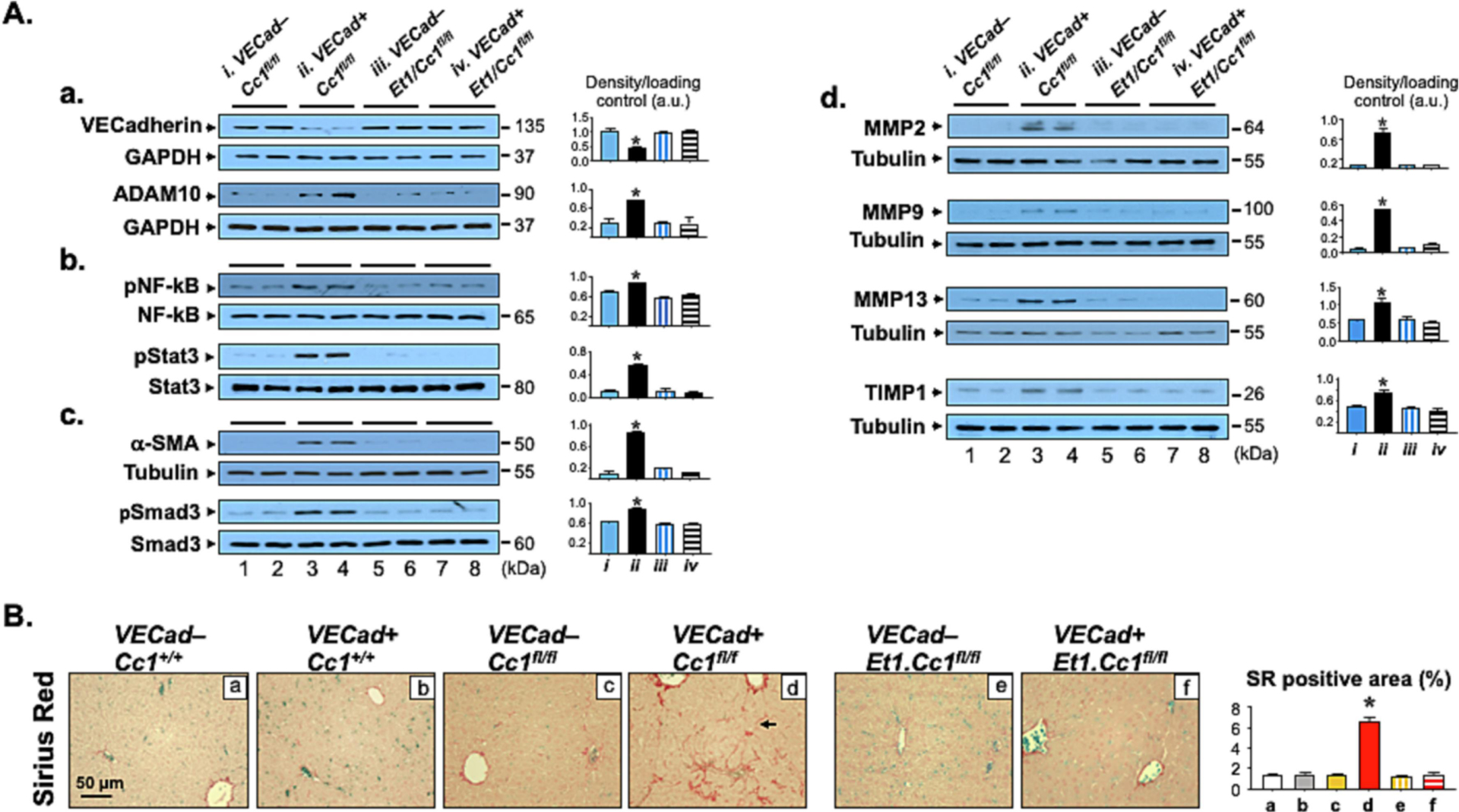
Livers from mice described in Fig. 1 were excised to carry out: (A) Western Blot analysis on liver lysates from VECad+Cc1fl/fl male mice (lane 3–4) and their VECad−Cc1fl/fl littermate Flox controls (lane 1–2), and from VECad+Et1.Cc1fl/fl double mutants (lane 7–8) and their VECad–Et1.Cc1fl/fl double Floxed controls (lane 5–6). Representative gels indicate the protein levels of VE-Cadherin and ADAM10, normalized against GAPDH applied on parallel gels (panel a), the activation (phosphorylation) of NF-κB and Stat3 inflammatory pathways using phospho-antibodies normalized against total loaded NF-κB and Stat3 proteins analyzed on parallel gels (panel b), α-SMA protein levels, normalized against tubulin, and the amount of phosphorylated Smad3 normalized against total protein loaded to assess TGFβ activation (panel c), and MMP-2, -9, -13 and TIMP-1 protein content, normalized against tubulin (panel d). Gels represent two different mice/genotype. The apparent molecular mass (kDa) is indicated at the right hand-side of each gel. Each group represent 2 different mice/genotype. Gels were scanned using image J (v1.53t) and the density of the test band was divided by that of its corresponding loading control and represented in arbitrary units (a.u) in the accompanying graph. (B) Sirius Red staining was performed to detect interstitial and bridging (arrow) chicken-wire deposition of collagen fibers in VECad+Cc1fl/fl (panel d). Representative images are shown. 8–10 randomly selected high-power fields (20x) per sample were imaged and quantified with ImageJ (v1.53t) to determine the percentage of Sirius red (SR) stain area within the whole area of imaged hepatic tissue. Briefly, each image was RGB-stacked, subjected to standardized thresholding of individual channel to isolated Sirius Red stain and then quantified as %area. Image quantifications were averaged and the mean within each experimental group was plotted in the accompanying graph.
8-month-old VECad+Cc1fl/fl livers exhibited an elevation in macrophage pool [increased F4/80 mRNA levels (Table S4)], activation and recruitment [elevated immunostained Mac2 and CD68 respectively (Fig. 1B–C, panels d vs a-c)]. Immunohistochemical (IHC) analysis of MPO demonstrated neutrophil accumulation in perivascular areas in VECad+Cc1fl/fl, but not in control livers (Fig. 1D, panel d vs a-c). Accordingly, the expression of hepatic neutrophil markers (MPO, Elastase, S100A8 and PR3) was higher in mutant livers (Table S4). Consistent with increased T-cell lymphocytes recruitment to injured liver [24], mRNA (Table S4) and protein levels of pro-inflammatory CD4+T and CD8+T cells were higher in VECad+Cc1fl/fl livers (Fig. 1E–F, panels d vs a-c). In contrast, there was no increase in the anti-inflammatory Treg immunostain (Foxp3) (Fig. 1G, panel d vs a-c), or in hepatic Foxp3 and IL-10 expression (Table S4). Together with elevated hepatic IFNγ and unaltered IL-4 and IL-13 mRNA levels (Table S4), this points to CD4+Th1 response in mutants.
NF-κB was basally activated (phosphorylated) in VECad+Cc1fl/fl LEC (Fig. S5A) and [17]. This resulted from reduced Shc sequestration by CEACAM1 and the reciprocal increase in its coupling to the vascular endothelial growth factor receptor [25]. Consistently, NF-κB was more activated in 8-month-old VECad+Cc1fl/fl livers (Figs. 2A.b and S4B) to contribute to their higher levels of hepatic (Table S4) and plasma pro-inflammatory IL-6 and TNFα cytokines at this age (Table 1 vs Table S5). IL-6 could activate Stat3-mediated pro-inflammatory pathways in LEC (Fig. S5A) and in 8-month-old mutant livers (Figs. 2A.b and S4B). Combined with NF-κB activation by TNFα, this could induce Tlr2/4 and Mcp-1/Ccl2 transcription (Tables S4 and S6) while repressing that of Irf-8 to induce Cd11b+ macrophage pool [26].
3.2. Hepatic fibrosis
Consistent with the role of endothelial CEACAM1 in promoting cell differentiation [15], the expression of endothelial cell markers (Cd31 and Tie2) was repressed in mutant LEC reciprocally to the increase in mesenchymal genes expression (α-Sma, Fsp-1, Snail and Slug) (Table S6). Consistently, α-SMA protein level was higher in mutant LEC (Fig. S5B) and livers (Fig. 2A.c) starting at 6 months (Fig. S3B), concomitantly with the activation of the canonical TGFβ–Smad2/3 profibrogenic pathway (Fig. 2A.c, Smad2 not shown). This was supported by reduced expression of TGFβ–Smad2/3 inhibitor, Smad7 (Tables S4 and S6), and by increased protein content of its downstream target, SNAIL, in mutant LEC (Fig. S5B) [27]. Mutant LEC manifested increased proliferation [marked by elevated PCNA protein levels (Fig. S5C)] to contribute to elevated Ki67 immunostaining in the perivascular region of mutant livers (Fig. 1H, panel d vs a-c)]. This points to increased myofibroblastic features of mutant LEC.
Endothelial loss of Ceacam1 caused a progressive reduction in NO bioavailability by deactivating Akt/eNOS pathway in LEC [17]. Consistently, NADPH oxidase-1 and -4 (Nox1, Nox4) expression was ~two-to-threefold higher in mutant livers (Table S4) and LEC (Table S6), and NAD/NADH level was higher in VECad+Cc1fl/fl livers (Table 1). Together with increased plasma 8-isoprostane levels at 6–8 months of age (Tables 1 and S5), this points to increased oxidative stress and lipid peroxidation in VECad+Cc1fl/fl mice.
Because oxidative stress synergizes with TGFβ signaling to induce liver injury [13], we examined whether VECad+Cc1fl/fl mice developed hepatic fibrosis. Sirius Red stain showed an extensive periportal (arrowhead) and interstitial (arrow) chicken-wire pattern of collagen deposition in VECad+Cc1fl/f livers starting at 8 months (Fig. 2B vs S3A, panels d vs a-c) with a brunt score of 3 vs 0–1 in controls. Consistently, the expression of pro-fibrogenic genes (α-Sma, Col6α3, procollagen 1, Tgfβ) was higher in mutant livers (Tables S4 and S8) and LEC (Table S6). This could drive liver injury [increased Txn, Nqo, Nrf1 and Hgf mRNA levels (Table S4)] and dysfunction [progressive increase in plasma alanine transaminase (ALT) and aspartate aminotransferase (AST) in VECad+Cc1fl/fl mice at 6–8 months of age (Tables 1 and S5)].
Activation of MAP kinase, NF-κB and TGFβ-Smad pathways contribute to the regulation of the expression of the matrix metalloproteinases (MMPs) and the Tissue Inhibitor of Metalloproteinases (TIMPs) [28] that are involved not only in the inflammatory response to injury and myofibroblast activation to produce collagen, but also in the resolution of excess collagen and other extracellular matrix components during tissue remodeling [29]. Consistently, VECad+Cc1fl/fl mice displayed elevated hepatic protein levels of MMP2, MMP9, MMP13 and TIMP1 (Fig. 2A.d) and plasma levels of MMP2 and TIMP1, but not MMP13 (Table 1) relative to their Flox controls. Whereas MMP9 and TIMP1 promote fibrosis (with TIMP1 stimulating myofibroblast activation and inhibiting excess matrix resolution), MMP2 antagonizes it by increasing collagen clearance [30]. In contrast, MMP13 promotes both myofibroblast activation and collagen clearance [29, 30]. Thus, it is possible that unaltered MMP13 plasma levels in VECad+Cc1fl/fl mice reflects the tipping of its activity towards its pro-fibrogenic action. Regardless, together the data suggest that endothelial loss of CEACAM1 causes hepatic fibrosis by increasing collagen production and regulating a net effect of MMPs and TIMPs that favors fibrosis.
3.3. Endothelin1 release from VECad+Cc1fl/fl endothelial cells
ET1, predominantly produced in endothelial cells, promotes fibrosis via multiple mechanisms [31]. Thus, we investigated the effect of endothelial Ceacam1 deletion on autocrine ET1 production. Depleting Ceacam1 activated NF-κB in LEC [17] and subsequently, induced the expression of its targets, ET1 and its receptor A (ETAR), by four-to-sevenfold (Tables S4 and S6) [32]. NF-κB could also induce the transcription of hypoxia inducible factor-1α (HIF-1α) [33] in LEC (by ~8-fold) (Table S6) to elevate its protein levels (Fig. 3A.a). HIF-1α could provide a positive feedback mechanism on ET1 transcription [34] to contribute to its higher content in LEC media (Fig. 3A.b) and in plasma of VECad+Cc1fl/fl relative to controls starting at 6 months of age (Tables 1 and S5).
Fig. 3.
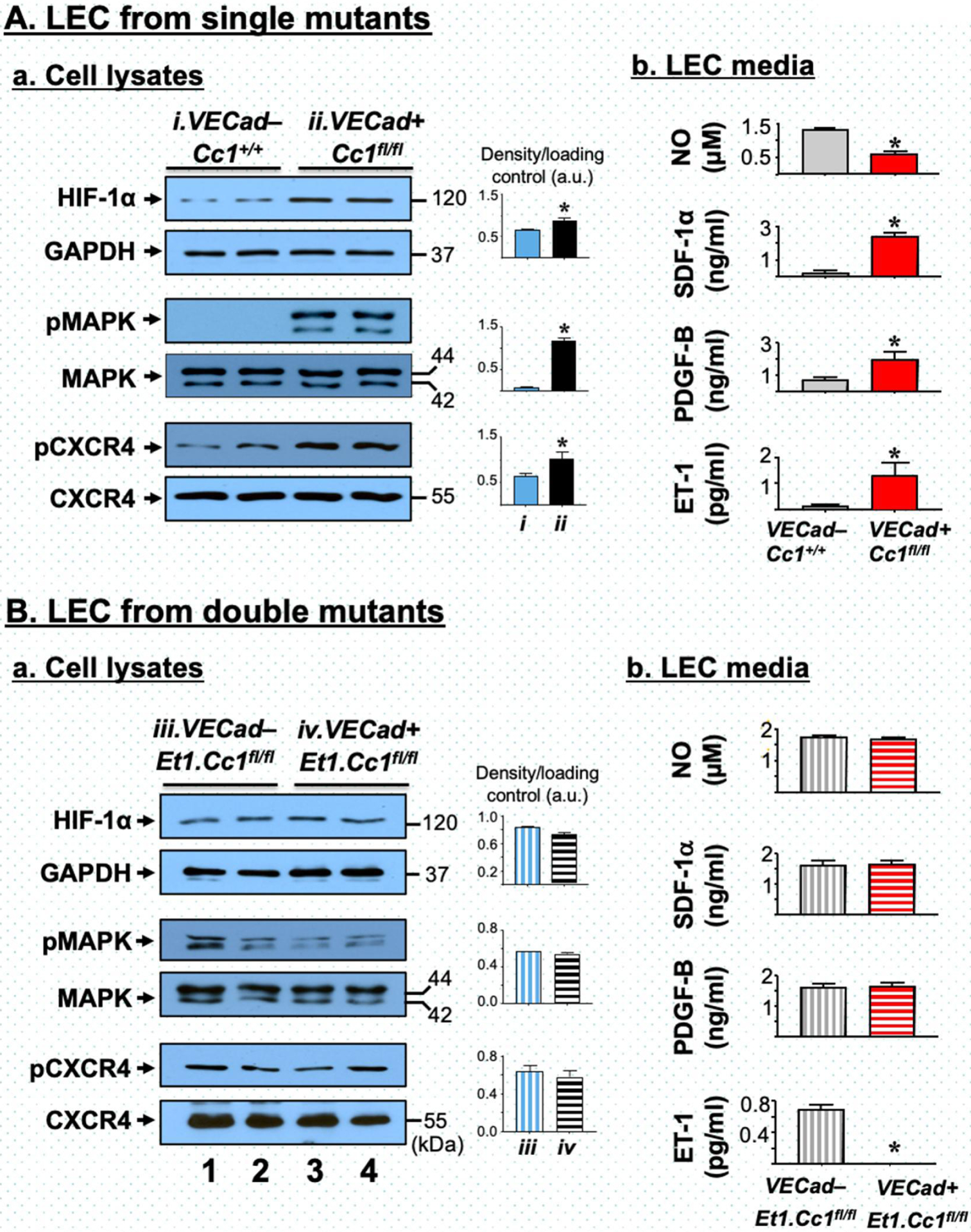
Analysis of primary liver endothelial cells. Liver endothelial cells (LEC) were prepared from 2-month-old VECad+Cc1fl/fl single mutants (A) and VECad+Et1.Cc1fl/fl double mutants (B) with their respective Flox controls. In panels a, Western blot analysis was performed on LEC lysates, as described in the legend to Fig. 2 with lane 1–2 depicting 2 different LEC preparations from control mice and lane 3–4 from mutant mice. Band densities were calculated as in the legend to Fig. 2 and represented in the accompanying graphs. In panels b, levels of NO, SDF-1α, PDGF-B and ET1 in LEC media were determined in triplicate. Values were expressed as mean ± SEM. *P<0.05 vs controls.
Similarly, plasma levels of the pro-fibrogenic platelet-derived growth factor-B (PDGF-B) were elevated in VECad+Cc1fl/fl mice relative to their control littermates starting at 6 months (Tables 1 and S5). Activated NF-κB could stimulate Pdgf-B transcription directly [35] or indirectly via activating HIF-1α [33] to yield an ~7-fold increase in its expression (Table S6) and release into mutant LEC media (Fig. 3A.b). Reciprocally, HIF-1α could induce Tlr3 and Tlr4 transcription [36] to activate NF-κB and subsequently, stimulate Tlr2 and Tlr9 expression [32] in mutant LEC (Table S6) and livers (Table S4). Combined activation of TLR3/TGFβ and TLR9/NF-κB pathways with repressed expression of Smad7 by IFN-β (Tables S4 and S6) could induce Pdgf-B transcription [37].
As in [17], Ceacam1 deletion caused an increase in Ras-MAPK activation in LEC (Fig. 3A.a). This could induce HIF-1α levels (Fig. 3A.a) and activation to stimulate the transcription of stromal cell-derived factor (SDF-1α) and PDGF-B [34] (Table S6), and elevate their levels in VECad+Cc1fl/fl LEC media (Fig. 3A.b) and plasma starting at 6 months (Tables 1 and S5). Moreover, CXCR4 transcription (~10-fold) (Table S6) and phosphorylation by SDF-1α binding (Fig. 3A.a) were induced in mutant LEC. With CXCR7 expression being reciprocally repressed (Table S6), this points to a preferential activation of SDF-1α/CXCR4 signaling to contribute to the high PDGF-B production in mutant LEC [38].
In addition to PDGF-B, mutant LEC manifested an ~7-fold increase in PDGFR-β expression versus a 2-to-4-fold increase in the expression of PDGF-A,-C,-D, and PDGFR-α (Table S6). Thus, Ceacam1 deletion caused a more stimulatory effect on PDGF-B/PDGFR-β than other family members.
3.4. Conditioned media from VECad+Cc1fl/fl LEC activate wild-type hepatic stellate cells
Because ET1 and PDGF-B induce fibrogenesis [20, 31], we then examined whether media transferred from VECad+Cc1fl/fl LEC (KO-Med) could activate wild-type (WT) HSC. As Fig. 4A–B shows, incubating WT-HSC with KO-Med repressed peroxisome proliferator-activated receptor-γ (PPARγ) expression, indicating higher HSC activation, relative to incubating HSC in media from wild-type LEC (WT-Med) and in regular culture media (Reg-Med) (Fig. 4A–B, – bars of KO-Med vs Reg-Med and WT-Med). Consistently, KO-Med induced the mRNA levels of PDGF-B and its receptor, α-SMA, FSP-1 and Desmin by two-to-fourfold. Supplementing the media with nilotinib (nilo) (Fig. 4A), an inhibitor of PDGFR-β tyrosine kinase [20] or with bosentan (BOS) (Fig. 4B), a dual antagonist of ETAR/ETBR [21], reversed HSC activation by KO-Med (Figs. 4A and 4B, respectively, + vs – bars).
Fig. 4.
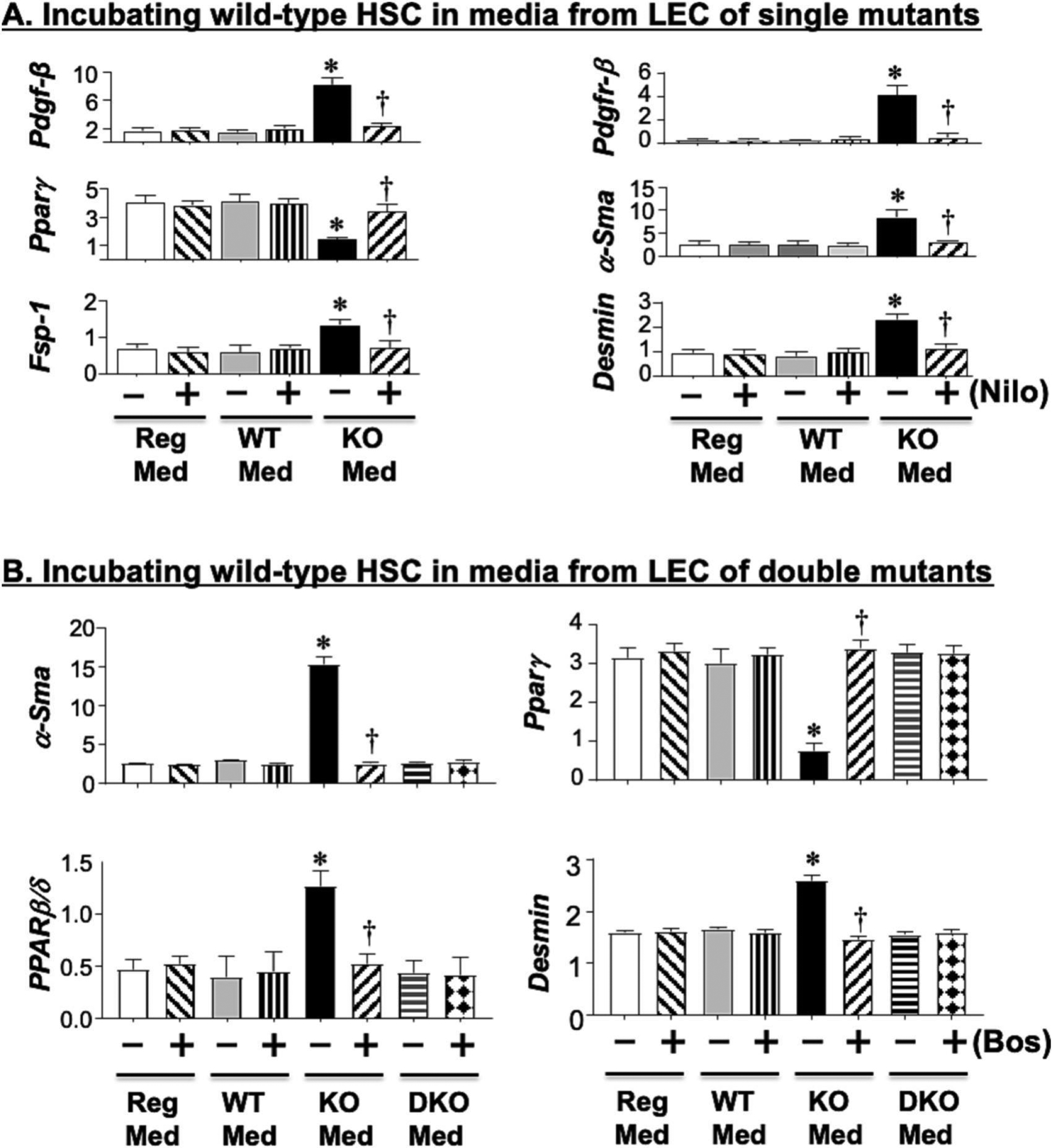
Activation of hepatic stellate cells by media transferred from liver endothelial cells. (A) Primary hepatic stellate cells (HSC) from wild-type (WT) mice were incubated with either regular media (Reg-Med), media transferred from LEC of WT controls (WT-Med) or media transferred from LEC of VECad+Cc1fl/fl mice (KO-Med) in the presence (+) or absence (−) of Nilotinib (Nilo), an inhibitor of PDGFR-B kinase. HSC activation was determined by qRT-PCR analysis of mRNA levels of genes relative to Gapdh in triplicate. Values were expressed as mean ± SEM. *P< 0.05 vs Reg-Med and WT-Med without Nilo, †P< 0.05 vs KO-Med without Nilo. (B) same as A, but in addition to KO-Med, media was also transferred from LEC of VECad+Et1.Cc1fl/fl mice (DKO-Med) in the presence (+) or absence (−) of Bosentan (Bos), a dual antagonist of endothelin1 receptor-A and -B.
3.5. Combined loss of endothelin1 reverses hepatic fibrosis in VECad+Cc1fl/fl mice
Given the pro-fibrogenic and the pro-inflammatory functions of ET1, we then investigated whether its rise causes hepatic fibrosis in VECad+Cc1fl/fl mice. To this end, we examined whether its combined deletion from endothelial cells (Fig. S1 and Table S2) would reverse the histopathological abnormalities caused by loss of endothelial Ceacam1. As expected, ET1 levels were negligible in LEC from VECad+Et1.Cc1fl/fl double mutants compared to their double-Flox controls (VECad–Et1.Cc1fl/fl) (Table S7) and in their media (Fig. 3B.b). This translated into negligible plasma ET1 levels in 9-month-old mutants relative to their age-matched double-Flox and double-Cre (VECad+Et1.Cc1+/+) controls (Table 2).
Table 2:
Plasma and tissue biochemistry of male VECadCre+Et1.Cc1fl/fl double mutant mice and their double Flox and double Cre control littermates at 9 months of age
| VECadCre+Et1.Cc1+/+ | VECadCre-Et1.Cc1fi/fi | VECadCre+Et1.Cc1fi/fl | |
|---|---|---|---|
| Body weight (BW) (g) | 31.4 ± 0.3 | 30.1 ± 0.1 | 29.6 ± 0.5 |
| % WAT/BW | 2.3 ± 0.3 | 2.1 ± 0.4 | 2.2 ± 0.3 |
| Hepatic TG (μg/mg protein) | 57.2 ± 3.1 | 59.4 ± 3.6 | 56.9 ± 2.8 |
| Plasma TG (mg/dl) | 71.0 ± 4.7 | 69.9 ± 4.1 | 70.2 ± 3.5 |
| Plasma endothelin1 (pg/ml) | 6.1 ± 0.2 | 5.5 ± 1.2 | Negl |
| Plasma MMP2 (ng/ml) | 826.9 ± 45.6 | 843.8 ± 59.9 | 757.8 ± 23.3 |
| Plasma MMP13 (pg/ml) | 234.4 ± 89.1 | 246.6 ± 41.7 | 305.9 ± 64.4 |
| Plasma TIMP1 (pg/ml) | 1.4 ± 0.2 | 1.7 ± 0.7 | 1.4 ± 0.1 |
| Plasma ALT (mU/ml) | 39.7 ± 7.8 | 39.9 ± 5.2 | 35.9 ± 3.5 |
| Plasma AST (mU/ml) | 140.3 ± 11.9 | 136.1 ± 13.5 | 144.1 ± 10.4 |
| Plasma PDGF-B (ng/ml) | 6.0 ± 0.5 | 5.6 ± 0.4 | 5.4 ± 0.3 |
| Plasma NO (μM) | 35.1 ± 1.5 | 34.5 ± 1.0 | 35.4 ± 0.9 |
| Hepatic NAD/NADH | 1.3 ± 0.3 | 1.1 ± 0.2 | 1.4 ± 0.4 |
| Plasma 8-Isoprostane (pg/ml) | 5.5 ± 0.4 | 5.2 ± 0.5 | 4.9 ± 0.3 |
| Plasma IL-6 (pg/ml) | 3.7 ± 0.9 | 5.5 ± 1.9 | 6.1 ± 1.8 |
| Plasma TNFα (pg/ml) | 9.6 ± 2.7 | 10.8 ± 3.3 | 9.9 ± 2.9 |
Male mice (9 months of age, n=5/genotype) were fasted from 5:00 p.m. until 11:00 a.m. the next morning before blood was drawn and tissues were excised and plasma was processed. White adipose tissue (WAT) was collected and calculated relative to body weight (BW) to determine visceral adiposity. Values are expressed as mean ± SEM.
As Fig. 2B reveals, there was no significant collagen deposition in the liver sections of 9-month-old double VECad+Et1.Cc1fl/fl mutants (Panel f) and their double-Flox controls (Panel e) (Brunt fibrosis score of 0–1), as it did in VECad+Cc1fl/fl single mutants (Panel d, fibrosis score of 3). Consistently, α-Sma mRNA (Table S7) and protein levels (Fig. 2A.c) were normalized in parallel to TGFβ–Smad2/3 inactivation in VECad+Et1.Cc1fl/fl relative to VECad+Cc1fl/fl livers. Similarly, the expression of other pro-fibrogenic genes were normalized in LEC (Table S7) and liver lysates (Table S8) of double versus single mutants. Consistently, combined loss of endothelial Et1 and Cc1 reversed the increase in plasma (Table 2) and hepatic levels of MMPs and TIMP1 (Fig. 2A.d) and hepatic injury (Table S8), and restored liver function [normal ALT and AST levels (Table 2)] caused by loss of endothelial Ceacam1.
In contrast to single mutants, conditioned media from VECad+Et1.Cc1fl/fl failed to induce the mRNA levels of α-Sma, Fsp-1, desmin and PPARβ/δ, and to repress PPARγ mRNA levels in WT-HSC (Fig. 4B). This ET1–free conditioned media exhibited normal PDGF-B levels (Fig. 3B.b), as expected from normal HIF-1α levels, and restored MAPK and SDF-1α/CXCR4 signaling [34, 38] in LEC from VECad+Et1.Cc1fl/fl mice (Fig. 3B.a). This translated into normal plasma PDGF-B levels in double mutants (Table 2). Together, this demonstrated that the rise in ET1 played a key role in hepatic fibrosis in mice with endothelial loss of Ceacam1, and that this was partly mediated by increased PDGF-B production.
Elevated ET1 levels inactivate eNOS to lower NO bioavailability [39]. Consistently, its deletion restored NO production, as reflected by normal NO levels in the LEC media (Fig. 3B.b) and plasma (Table 2) of VECad+Et1.Cc1fl/fl relative to single VECad+Cc1fl/fl mutants. It also normalized hepatic NAD/NADH and plasma 8-isoprostane levels (Table 2). Together with normalization of Nox-1/4 mRNA levels in LEC (Table S7) and livers (Table S8), the data demonstrated that the double mutation reversed the redox imbalance caused by the loss of endothelial Ceacam1 alone.
Consistent with NF-κB activation by ET1 [40], Et1 co-deletion blunted hepatic NF-κB activation (Fig. 2A.b). This repressed the transcription (Tables S7 and S8) and plasma levels (Table 2) of IL-6 and TNFα pro-inflammatory cytokines. Accordingly, the expression of pro-inflammatory genes were normalized in LEC (Table S7) and livers (Table S8) of double mutants, and H&E staining did not detect inflammatory infiltration in their liver sections (Fig. 1A, panel f vs d). As expected from inactivation of TGFβ and NF-κB [41], the combined endothelial loss of Et1 with Ceacam1 restored the expression of endothelial markers [VE-Cadherin (Fig. 2A.a) and Cd31 and Tie2 (Table S7)] with a reciprocal reduction in α-SMA protein level (Fig. 2A.c), and the mRNA of other mesenchymal genes [Fsp-1, Snail and Slug (Table S7)]. This limited LEC proliferation as reflected by the absence of Ki67 immunostaining in the double mutant livers (Fig. 1H). By recovering VE-Cadherin protein levels (Fig. 2A.a), endothelial Et1 deletion restored LEC membrane integrity to impede macrophage and neutrophil recruitment and infiltration, as supported by the loss of Mac2, CD68, and MPO immunostaining in the livers of double mutants (Figs. 1B–D, panels f vs d). Additionally, the double mutants exhibited a loss of immunostained CD4+T and CD8+T cells (Fig. 1E–F, panels d vs a-c). Together, this underscores the critical role of elevated ET1 production in the pathogenesis of hepatic fibrosis caused by endothelial CEACAM1 loss.
3.6. Reduced endothelial CEACAM1 expression in patients with progressive fibrosis
We next examined whether hepatic CEACAM1 expression is reduced in patients with fibrosis. Patients were predominantly non-Hispanic whites (24:4:8 non-Hispanic whites:Hispanics:Non-Hispanic Blacks) (Table S3). IHC analysis detected a progressive increase in MPO and CD68 immunostaining as fibrosis stage increased from 0 to 4 (Fig. 5A), with most FS4 patients having cirrhosis driven by NASH.
Fig. 5.
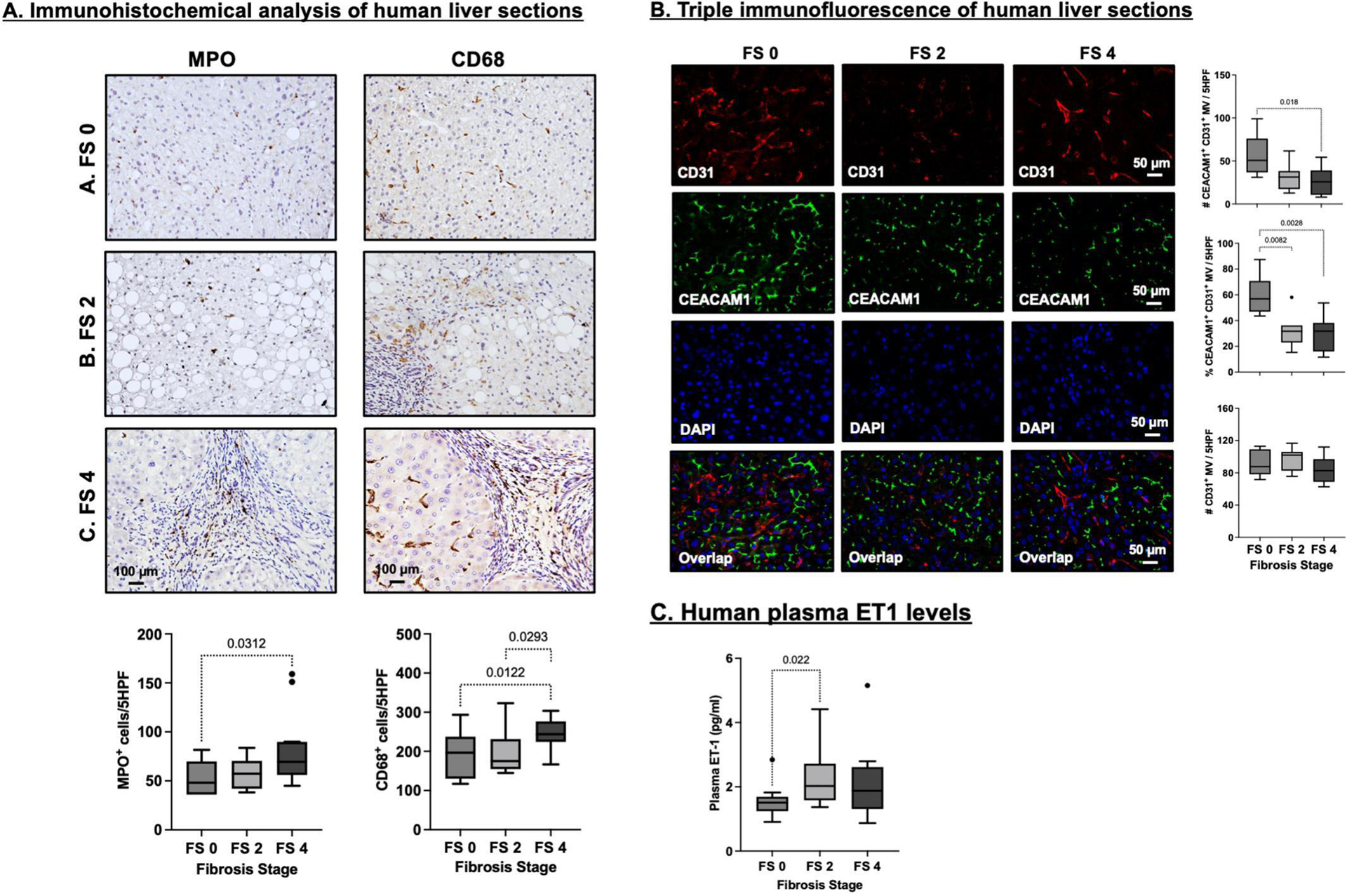
Analysis of biopsies of liver sections and plasma from patients at different stages of fibrosis. (A) Representative images of immunohistochemical analysis of myeloperoxidase+ (MPO+) and CD68+ cells in human liver biopsies graded by a blinded pathologist with fibrosis stage 0 (FS 0; n=7), fibrosis stage 2 (FS 2; n=6) and fibrosis stage 4 (FS 4; n=12). The number of positive cells per 5 randomly selected high-power fields were blindly quantified in quadruplicate assessments, averaged and plotted for each sample. Data are presented as Tukey box-and-whisker plots: whiskers are inner fences at 1.5 times the interquartile range. Boxes represent the interquartile ranges, dots outlying values and lines the median values for each study group. P<0.05 are indicated. (B) Triple immunofluorescence analysis and quantification of CEACAM1 in CD31+ micro-vessels of human liver biopsies graded by a blinded pathologist with FS 0 (n=7), FS 2 (n=7) and FS 4 (n=9). CD31 is shown in red (Alexa Fluor 555), CEACAM1 in green (Alexa Fluor 488) and Dapi nuclear staining in blue. The number of double positive CEACAM-1/CD31 micro vessels, the number of single CD31 positive micro-vessels and the percentage of double positive CEACAM1/CD31 micro vessels per 5 high power fields were quantified. (C) ELISA in duplicate of ET1 levels in plasma of patients with FS 0 (n=15), FS 2 (n=7) and FS 4 (n=14). Data were analyzed and presented as in (A) above.
Reciprocally, immunofluorescence analysis (Fig. 5B) showed a progressive decline in CEACAM1 expression in CD31+ endothelial cells (top and middle graph) with no change in CD31+ cell number (lower graph) as fibrosis advanced (lower graph). This inversely correlated with an increase in plasma ET1 (Fig. 5C).
To further examine the CEACAM1/ET1 axis in humans, we re-analyzed a scRNA sequencing data set consisting of healthy liver samples (n=5) and liver cells isolated from explants of patients with advanced fibrosis-cirrhosis (n=4) driven by NASH and alcoholic steatohepatitis without viral hepatitis [42]. The patient with primary biliary cholangitis was not included because this data set did not contain many LSECs. We used the annotated data [42, 43] to subset and recluster the mesenchymal, endothelial and small hepatocyte populations (Fig. 6A) using cell-type specific markers (Fig. 6B). The regular LEC and LSEC populations express VECadherin (Cdh5) at similar levels and cells percentages. CEACAM1 was detected predominantly in LSECs (as opposed to 11% of LEC pool) and to a lesser extent in small hepatocytes of healthy human livers while being virtually absent in LSECs (and small hepatocytes) from cirrhotic livers (Fig. 6C). Reciprocally, ET1 was almost undetected in LSECs from healthy individuals but was remarkably high in LSECs from cirrhotic patients (Fig. 6C). Interestingly, ET1 levels in the regular LEC pool did not change with the disease state (Fig. 6C). Finally, only a small portion of CEACAM1+LSECs expressed ET1 in healthy and diseased livers (Fig. 6D, 21.7% and 37.5%, respectively).
Fig. 6.
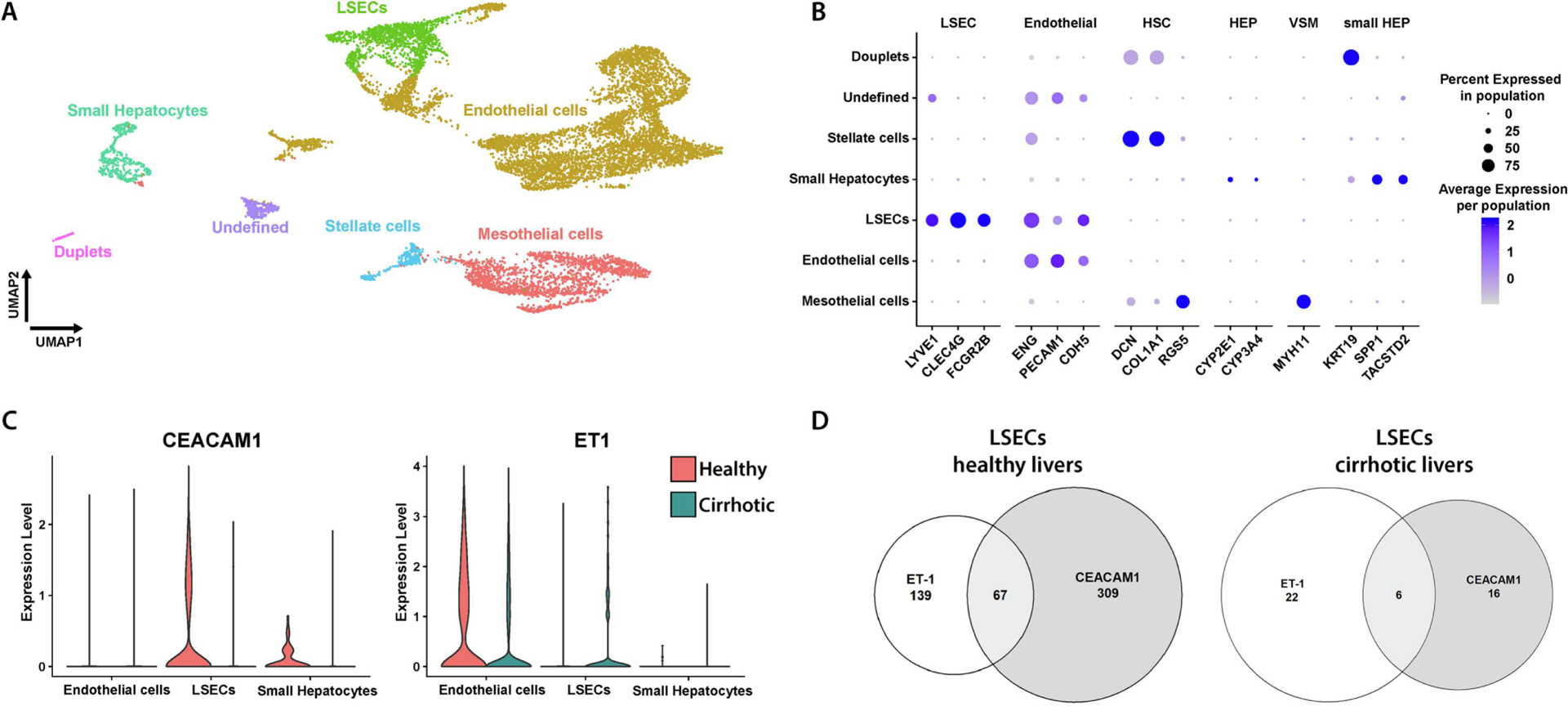
Reclustering of different cell populations in the liver explants. (A) Uniformed Manifold Approximation and Projection (UMAP) visualization of annotated clusters in the healthy and cirrhotic livers. (B) Dotplot of expression of cell-type specific markers in each annotated cluster. (C) Violin plot representing CEACAM1 and ET1 mRNA levels in endothelial cells, LSECs and hepatocytes from scRNA-sequencing data from human livers. The expression levels in red represent healthy liver (n=5) explants and those in teal represent cirrhotic liver (n=4) explants. (D) Venn diagram of gene expression of CEACAM1 (grey section) and ET1 (white section) expressed in healthy and cirrhotic LSECs. The overlapping region (light grey) represents the cells positive for both CEACAM1 and ET1.
3.7. Increased Endothelin1 production in liver sinusoidal endothelial cells from VECad+Cc1fl/fl mice
Since CEACAM1 was detected predominantly in human LSECs by comparison to the regular LEC pool, we then examined the effect of deleting Ceacam1 on ET1 production and cell damage from murine LSECs. Depleting Ceacam1 (Table S2) caused an ~2-fold increase in Et1 levels in the LSEC media (Fig. 7A) and mRNA content (Fig. 7B). In contrast to ETBR, ETAR (Etar) mRNA was significantly increased in VECad+Cc1fl/fl LSECs (Fig. 7B). This effect of Ceacam1 deletion was absent in LSECs from VECad+Et.Cc1fl/fl double mutants, consistent with a key role for the amplification of ET1 pro-fibrogenic signaling in LSECs of single mutant mice. In contrast to their Flox LSEC controls, the mRNA levels of cell damage marker genes in chronic liver disease (Fabp4, vwf and vwfa1) [43] were higher in LSECs from VECad+Cc1fl/fl, but not VECad+Et.Cc1fl/fl mice (Fig. 7C).
Fig. 7. Analysis of liver sinusoidal endothelial cells (LSECs).
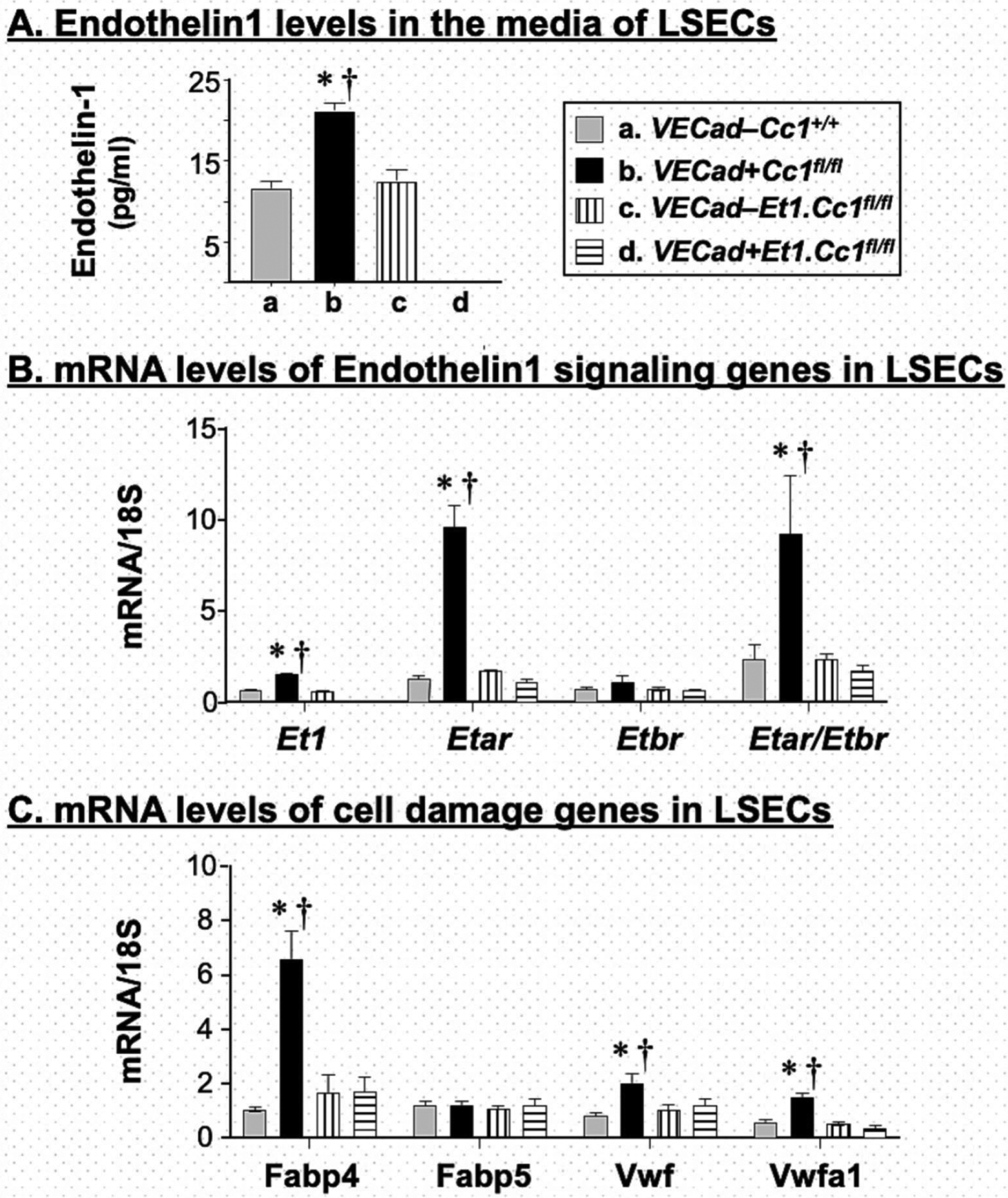
LSECs were isolated from mice at 2–3 months of mice, allowed to grow for 3 days in 3 separate wells of 24-well plates before (A) the media was collected to assess ET1 levels and cells were lysed for qRT-PCR analysis of mRNA levels of genes involved in (B) ET1 signaling and (C) cell damage. Measurements were done in duplicate. Values were expressed as mean ± SEM. *P<0.05 vs control/each genotype and †P<0.05 VECad+Cc1fl/fl.ET1fl/fl vs VECad+Cc1fl/fl.
4. Discussion
The current studies demonstrated that loss of endothelial CEACAM1 caused hepatic fibrosis. Fibrosis in VECad+Cc1fl/fl mice with endothelial loss of Ceacam1 was marked by a net effect of changes in MMPs and TIMPs that tips the balance towards increased deposition of collagen fibers during extracellular remodeling. Fibrosis was mediated by increased endothelin1 production, as supported by reversed phenotype with combined loss of endothelin1 and Ceacam1 genes in endothelial cells. Increased endothelin1 production in VECad+Cc1fl/fl mice was not only from the general LEC pool, but also from their LSECs in parallel to increased expression of signature LSEC cell damage genes in chronic liver disease [43]. These findings in mice were further supported by scRNA-sequencing analysis showing lower CEACAM1 mRNA levels in LSECs of patients with advanced fibrosis/cirrhosis in parallel to increased endothelin1 production. Moreover, endothelial CEACAM1 gradually decreased with advancement of fibrosis in inverse correlation with plasma endothelin1 levels in patients regardless of their ethnicity.
VECad+Cc1fl/fl mice displayed endothelial dysfunction, increased plasma cytokine levels (TNFα and IL-6) levels while maintaining insulin sensitivity [17]. The current studies showed that these mice also developed spontaneous hepatic bridging fibrosis and perivascular inflammatory infiltration without hepatic steatosis. As in Cc1−/− nulls [11] and NASH patients [24, 26], inflammation in VECad+Cc1fl/fl livers was marked by a CD4+Th1 response. This agrees with the well-characterized loss of the anti-inflammatory function of LSECs during HSC activation [44], and with the reported reduction of Ceacam1 expression in LSECs of experimentally cirrhotic mice [22].
Endothelial loss of Ceacam1 also disturbed VE-Cadherin/β-catenin complex formation to cause barrier malfunction and facilitate trans-endothelial leukocyte migration [16] which could contribute to HSC activation and hepatic fibrosis [2]. Consistent with HIF-1α repressing VE-Cadherin to promote endothelial cell permeability in part by inducing endothelin1 and its A receptors [45], the combined loss of endothelin1 restored VE-Cadherin levels in endothelial cells and subsequently, restricted macrophage and neutrophil infiltration to VECad+Cc1fl/fl liver parenchyma.
Exclusive reconstitution of CEACAM1 to hepatocytes reversed hyperinsulinemia-driven endothelin1 production in parallel to hepatic fibrosis in Cc1−/− nulls [11, 12]. Reversed hepatic fibrosis in VECad+Cc1fl/fl livers upon concomitant endothelin1 deletion demonstrated a distinct anti-fibrotic role for endothelial CEACAM1 mediated by reducing autocrine endothelin1 production independent of its anti-hyperinsulinemic role in hepatocytes.
Mechanistically, loss of endothelial CEACAM1 reduced Shc sequestration and caused a reciprocal increase in its coupling to vascular endothelial growth factor [25] and insulin receptors [17] followed by activation of its downstream NF-κB pathways, which could induce endothelin1 and PDGF-B directly [35] or indirectly via HIF-1α–mediated pathways [33, 34]. Endothelin1, in turn, can provide a positive feedback mechanism to activate HIF-1α, MAPK and SDF-1α/CXCR4 signaling pathways leading to PDGF-B production. Elevated PDGF-B/PDGFR-β signaling synergizes with MAPK and NF-κB [32] to cause proliferation of mesenchymal cells as well as resident HSC and their activation. The role of PDGF-B in this process was supported by activation of wild-type HSC by conditioned media of VECad+Cc1fl/fl endothelial cells in the absence of nilotinib, a PDGFR-β tyrosine kinase inhibitor. Given that nilotinib also inhibits TGFβ, it is possible that this small molecule inhibited PDGF-B synthesis via TGFβ [37] in addition to inhibiting its signaling via PDGFR-β receptors.
Moreover, the expression of MMPs and TIMPs is regulated at least partly by the coordinated effort of MAP kinase, NF-κB and TGFβ-Smad pathways [28] that are activated in CEACAM1-depleted endothelial cells, as shown in the current and previous reports [17]. Increased levels of circulating MMPs and TIMP1 in parallel to endothelin1 have been found in patients with metabolic syndrome and cardiovascular disease [46, 47]. Whether the loss of endothelial Ceacam1 is implicated in this association is not clear and warrants further investigation.
5. Strengths and Weaknesses
The current studies provide an in vivo demonstration of a novel mechanistic link between a distinct CEACAM1/endothelin1 signaling module in endothelial cells and hepatic fibrosis, a critical modality of NASH. This was supported by the gradual decrease in endothelial CEACAM1 levels as the fibrosis stage progressed in patients with NASH. Given that these cells could be more easily targeted than hepatocytes [44], endothelial CEACAM1 can become a marker of disease progression and a basis for pharmacogenomic therapy with limited side effects. Despite this progress, NASH is a heterogenous disease with a progressive clinical course and our patient collection, albeit extensive, may fail to capture such complexity. In addition, our data do not allow for conclusions based on gender, race, ethnicity or socioeconomic factors that affect the course of the disease in humans. Further analysis is required to translate our observations to human NASH diagnosis, fibrosis progression, and treatment.
6. Conclusions
The current findings assign a key role for endothelial cell CEACAM1 in preventing hepatic injury and fibrosis by reducing autocrine endothelin1 production independent of its metabolic role in hepatocytes.
Supplementary Material
Highlights.
Mice with endothelial loss of CEACAM1 causes hepatic fibrosis
Hepatic fibrosis is mediated by endothelin1 production in endothelial cells
Endothelial CEACAM1 is reduced with progression of fibrosis in NASH patients
Plasma endothelin1 is increased with progression of fibrosis in NASH patients
Acknowledgments
We thank Prof. D.Accili and Dr. S.Kumarasamy for their critical reading of the manuscript.
Financial support
This work was supported by NIH grants: R01-HL112248, R01-DK054254, R01-DK124126 and R01-MD012579 to S.M.N. and S.D., K08-DK113244 to AZ. H.M. is partially supported by DK111378, S.V. by FWO 1243121N, L.A.vG. by FWO G071922N and B.B.S. by Deutsche Forschungsgemeinschaft Grant SI-1558/6-1. S.M.N. is partially supported by OHF J.J.Kopchick Eminent Research Chair.
Abbreviations
- CEACAM1
Carcinoembryonic Antigen-related Cell Adhesion Molecule1
- Cc1 −/−
Global Ceacam1 knockout mice
- VECad+(Et1).Cc1fl/fl
VECadherin-Cre transgenics with homozygous null Cc1 without or with endothelin1 (Et1) alleles
- VECad–(Et1).Cc1fl/fl
Flox littermate controls
- VECad–(Et1).Cc1 +/+
Wild-type littermate controls
- VECad+(Et1).Cc1+/+
VECadherin-Cre littermate controls
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Credit authorship Contribution statement
H.T.M., H.E.G., R.A., H.L.S., J.K.L., G.D.B., M.S.J., A.D.L., B.B.S., I.A-P., T.S.S., S.V., and S.D. researched data. H.T.M., planned and organized experiments, collected and analyzed data, and drafted the manuscript. H.E.G., S.V. and S.D. contributed to the preparation of the illustrations. B.B.S., H.M., L.A.vG, A.Z. and S.D. discussed data and edited the manuscript. S.M.N. oversaw the work, including its conception and study design, analyzed data, led scientific discussions and reviewed/edited the manuscript.
Declaration of competing interest
None declared.
Data availability statement
All data relevant to the study are included in the article/online supplemental information
REFERENCES
- [1].Younossi ZM. Non-alcoholic fatty liver disease - A global public health perspective. J Hepatol. 2019;70:531–44. [DOI] [PubMed] [Google Scholar]
- [2].Friedman SL, Pinzani M. Hepatic fibrosis 2022: Unmet needs and a blueprint for the future. Hepatology. 2022;75:473–88. [DOI] [PubMed] [Google Scholar]
- [3].Eslam M, Valenti L, Romeo S. Genetics and epigenetics of NAFLD and NASH: Clinical impact. J Hepatol. 2018;68:268–79. [DOI] [PubMed] [Google Scholar]
- [4].Arrese M, Arab JP, Barrera F, Kaufmann B, Valenti L, Feldstein AE. Insights into Nonalcoholic Fatty-Liver Disease Heterogeneity. Semin Liver Dis. 2021;41:421–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Masoodi M, Gastaldelli A, Hyotylainen T, Arretxe E, Alonso C, Gaggini M, et al. Metabolomics and lipidomics in NAFLD: biomarkers and non-invasive diagnostic tests. Nat Rev Gastroenterol Hepatol. 2021;18:835–56. [DOI] [PubMed] [Google Scholar]
- [6].Tilg H, Adolph TE, Dudek M, Knolle P. Non-alcoholic fatty liver disease: the interplay between metabolism, microbes and immunity. Nat Metab. 2021;3:1596–607. [DOI] [PubMed] [Google Scholar]
- [7].Dufour JF, Anstee QM, Bugianesi E, Harrison S, Loomba R, Paradis V, et al. Current therapies and new developments in NASH. Gut. 2022;71:2123–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lee W The CEACAM1 expression is decreased in the liver of severely obese patients with or without diabetes. Diagn Pathol. 2011;6:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Nakamura K, Kageyama S, Kaldas FM, Hirao H, Ito T, Kadono K, et al. Hepatic CEACAM1 expression indicates donor liver quality and prevents early transplantation injury. The Journal of clinical investigation. 2020;130:2689–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Lee SJ, Heinrich G, Fedorova L, Al-Share QY, Ledford KJ, Fernstrom MA, et al. Development of nonalcoholic steatohepatitis in insulin-resistant liver-specific S503A carcinoembryonic antigen-related cell adhesion molecule 1 mutant mice. Gastroenterology. 2008;135:2084–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Helal RA, Russo L, Ghadieh HE, Muturi HT, Asalla S, Lee AD, et al. Regulation of hepatic fibrosis by carcinoembryonic antigen-related cell adhesion molecule 1. Metabolism. 2021;121:154801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Russo L, Muturi HT, Ghadieh HE, Wisniewski AM, Morgan EE, Quadri SS, et al. Liver-specific rescuing of CEACAM1 reverses endothelial and cardiovascular abnormalities in male mice with null deletion of Ceacam1 gene. Mol Metab. 2018;9:98–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Sun X, Harris EN. New aspects of hepatic endothelial cells in physiology and nonalcoholic fatty liver disease. Am J Physiol Cell Physiol. 2020;318:C1200–C13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hammoutene A, Rautou PE. Role of liver sinusoidal endothelial cells in non-alcoholic fatty liver disease. J Hepatol. 2019;70:1278–91. [DOI] [PubMed] [Google Scholar]
- [15].Rueckschloss U, Kuerten S, Ergun S. The role of CEA-related cell adhesion molecule-1 (CEACAM1) in vascular homeostasis. Histochem Cell Biol. 2016;146:657–71. [DOI] [PubMed] [Google Scholar]
- [16].Ghavampour S, Kleefeldt F, Bommel H, Volland J, Paus A, Horst A, et al. Endothelial barrier function is differentially regulated by CEACAM1-mediated signaling. FASEB J. 2018;32:5612–25. [DOI] [PubMed] [Google Scholar]
- [17].Muturi HT, Khuder SS, Ghadieh HE, Esakov EL, Noh H, Kang H, et al. Insulin Sensitivity Is Retained in Mice with Endothelial Loss of Carcinoembryonic Antigen Cell Adhesion Molecule 1. Cells. 2021;10:2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kisanuki YY, Emoto N, Ohuchi T, Widyantoro B, Yagi K, Nakayama K, et al. Low blood pressure in endothelial cell-specific endothelin 1 knockout mice. Hypertension. 2010;56:121–8. [DOI] [PubMed] [Google Scholar]
- [19].Mederacke I, Dapito DH, Affo S, Uchinami H, Schwabe RF. High-yield and high-purity isolation of hepatic stellate cells from normal and fibrotic mouse livers. Nat Protoc. 2015;10:305–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Liu Y, Wang Z, Kwong SQ, Lui ELH, Friedman SL, Li FR, et al. Inhibition of PDGF, TGF-beta, and Abl signaling and reduction of liver fibrosis by the small molecule Bcr-Abl tyrosine kinase antagonist Nilotinib. J Hepatol. 2011;55:612–25. [DOI] [PubMed] [Google Scholar]
- [21].Davenport AP, Hyndman KA, Dhaun N, Southan C, Kohan DE, Pollock JS, et al. Endothelin. Pharmacol Rev. 2016;68:357–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Su T, Yang Y, Lai S, Jeong J, Jung Y, McConnell M, et al. Single-Cell Transcriptomics Reveals Zone-Specific Alterations of Liver Sinusoidal Endothelial Cells in Cirrhosis. Cell Mol Gastroenterol Hepatol. 2021;11:1139–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Brunt EM, Janney CG, Di Bisceglie AM, Neuschwander-Tetri BA, Bacon BR. Nonalcoholic steatohepatitis: a proposal for grading and staging the histological lesions. Am J Gastroenterol. 1999;94:2467–74. [DOI] [PubMed] [Google Scholar]
- [24].Seki E, Schwabe RF. Hepatic inflammation and fibrosis: functional links and key pathways. Hepatology. 2015;61:1066–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Abu Helal R, Muturi HT, Lee AD, Li W, Ghadieh HE, Najjar SM. Aortic Fibrosis in Insulin-Sensitive Mice with Endothelial Cell-Specific Deletion of Ceacam1 Gene. Int J Mol Sci. 2022;23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Tacke F, Zimmermann HW. Macrophage heterogeneity in liver injury and fibrosis. J Hepatol. 2014;60:1090–6. [DOI] [PubMed] [Google Scholar]
- [27].Su J, Morgani SM, David CJ, Wang Q, Er EE, Huang YH, et al. TGF-beta orchestrates fibrogenic and developmental EMTs via the RAS effector RREB1. Nature. 2020;577:566–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Vincenti MP, Brinckerhoff CE. Signal transduction and cell-type specific regulation of matrix metalloproteinase gene expression: can MMPs be good for you? J Cell Physiol. 2007;213:355–64. [DOI] [PubMed] [Google Scholar]
- [29].Duarte S, Baber J, Fujii T, Coito AJ. Matrix metalloproteinases in liver injury, repair and fibrosis. Matrix Biol. 2015;44–46:147–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Giannandrea M, Parks WC. Diverse functions of matrix metalloproteinases during fibrosis. Dis Model Mech. 2014;7:193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Barton M, Yanagisawa M. Endothelin: 30 Years From Discovery to Therapy. Hypertension. 2019;74:1232–65. [DOI] [PubMed] [Google Scholar]
- [32].Suryavanshi SV, Kulkarni YA. NF-kappabeta: A Potential Target in the Management of Vascular Complications of Diabetes. Front Pharmacol. 2017;8:798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].van Uden P, Kenneth NS, Rocha S. Regulation of hypoxia-inducible factor-1alpha by NF-kappaB. Biochem J. 2008;412:477–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Krock BL, Skuli N, Simon MC. Hypoxia-induced angiogenesis: good and evil. Genes Cancer. 2011;2:1117–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Au PY, Martin N, Chau H, Moemeni B, Chia M, Liu FF, et al. The oncogene PDGF-B provides a key switch from cell death to survival induced by TNF. Oncogene. 2005;24:3196–205. [DOI] [PubMed] [Google Scholar]
- [36].Han S, Xu W, Wang Z, Qi X, Wang Y, Ni Y, et al. Crosstalk between the HIF-1 and Toll-like receptor/nuclear factor-kappaB pathways in the oral squamous cell carcinoma microenvironment. Oncotarget. 2016;7:37773–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Chow EK, O’Connell RM, Schilling S, Wang XF, Fu XY, Cheng G. TLR agonists regulate PDGF-B production and cell proliferation through TGF-beta/type I IFN crosstalk. EMBO J. 2005;24:4071–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Hamdan R, Zhou Z, Kleinerman ES. SDF-1alpha induces PDGF-B expression and the differentiation of bone marrow cells into pericytes. Mol Cancer Res. 2011;9:1462–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ramzy D, Rao V, Tumiati LC, Xu N, Sheshgiri R, Miriuka S, et al. Elevated endothelin-1 levels impair nitric oxide homeostasis through a PKC-dependent pathway. Circulation. 2006;114:I319–26. [DOI] [PubMed] [Google Scholar]
- [40].Rosano L, Spinella F, Bagnato A. Endothelin 1 in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2013;13:637–51. [DOI] [PubMed] [Google Scholar]
- [41].Piera-Velazquez S, Jimenez SA. Endothelial to Mesenchymal Transition: Role in Physiology and in the Pathogenesis of Human Diseases. Physiol Rev. 2019;99:1281–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ramachandran P, Dobie R, Wilson-Kanamori JR, Dora EF, Henderson BEP, Luu NT, et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature. 2019;575:512–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Verhulst S, van Os EA, De Smet V, Eysackers N, Mannaerts I, van Grunsven LA. Gene Signatures Detect Damaged Liver Sinusoidal Endothelial Cells in Chronic Liver Diseases. Front Med (Lausanne). 2021;8:750044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Lafoz E, Ruart M, Anton A, Oncins A, Hernandez-Gea V. The Endothelium as a Driver of Liver Fibrosis and Regeneration. Cells. 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Jiang W, Sun Y, Wang H, Hu Z, Song J, Meng C, et al. HIF-1alpha Enhances Vascular Endothelial Cell Permeability Through Degradation and Translocation of Vascular Endothelial Cadherin and Claudin-5 in Rats With Burn Injury. J Burn Care Res. 2021;42:258–68. [DOI] [PubMed] [Google Scholar]
- [46].Yu AP, Tam BT, Yau WY, Chan KS, Yu SS, Chung TL, et al. Association of endothelin-1 and matrix metallopeptidase-9 with metabolic syndrome in middle-aged and older adults. Diabetol Metab Syndr. 2015;7:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].De Ciuceis C, Rizzoni D, Palatini P. Microcirculation and Physical Exercise In Hypertension. Hypertension. 2023;80:730–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data relevant to the study are included in the article/online supplemental information