

Abstract
SIX5 (previously known as myotonic dystrophy associated homeodomain protein-DMAHP) is a member of the SIX [sine oculis homeobox (Drosophila) homologue] gene family which encodes proteins containing a SIX domain adjacent to a homeodomain. To investigate the DNA binding specificities of these two domains in SIX5, they were expressed as GST fusion proteins, both separately and together. Affinity purified recombinant proteins and cell lysates from bacteria expressing the recombinant proteins were used in gel retardation assays with double stranded oligonucleotides representing putative DNA binding sites. The putative sites included two in the promoter region of DMPK (dystrophia myotonica protein kinase) and the previously characterised murine Six4 DNA binding site in the Na+/K+ ATPase α1 subunit gene (ATP1A1) regulatory element (ARE). None of the recombinant proteins showed any affinity for the two putative sites in DMPK. However, the two recombinant proteins containing the homeodomain both formed at least one specific complex with the ARE. The recombinant protein containing both domains formed a second specific complex with the ARE, assumed to be a dimer complex. Finally, a whole genome PCR-based screen was used to identify genomic DNA sequences to which SIX5 binds, as an initial stage in the identification of genes regulated by SIX5.
INTRODUCTION
Six mammalian members of the SIX [sine oculis homeobox (Drosophila) homogue] family have been identified (Six1–6) and they all encode at least two functional domains (1–4). In addition to the 60 amino acid homeodomain, the SIX domain (approximately 116 amino acids) shows a high degree of homology between family members and lies immediately N-terminal to the predicted homeodomain (5). Homologues have also been identified in Drosophila, Caenorhabditis elegans, Xenopus laevis, chick, newt, killifish/medaka and zebrafish (4,6–12).
SIX5 shows greater homology to SIX4 (also known as AREC3) than to any other SIX protein (amino acid sequence similarity is 70.3% in the SIX domain and homeodomain). Murine Six4 was originally identified as one of the cell type-specific factors that bind to the ATP1A1 regulatory element (ARE). DNase I footprinting and methylation experiments identified the binding site of murine Six4 as GGTGTCAGGTTGC (conserved in human, mouse, rat and horse) and a possible minimum sequence for binding as GGNGNCNGGTTGC (13). A full-length mouse Six4 cDNA was used to synthesise GST–Six4 recombinant fusion proteins containing different regions of Six4 protein (2). The SIX domain and the homeodomain were both required for specific binding to GGTGTCAGGTTGC, although the homeodomain alone bound specifically to some other, unidentified, region of the ARE. This is a similar situation to that found in the Paired and POU classes of homeodomain proteins, in which the presence of two domains is required for specific DNA binding (14–16).
Murine Six2 and Six5, but not Six3, also bind specifically to the Six4 binding site in the ARE (3). The sequence of this binding site differs from all previously reported homeodomain binding sites, the majority of which contain a core tetranucleotide ATTA (17). It is known that arginine at position five of the homeodomain, which is conserved in 95% of known homeodomains, interacts with the core ATTA sequence (17). SIX5 and SIX4 have a valine at this position and other members of the SIX family have serine or threonine. Therefore, it is possible that SIX proteins will not bind to a target sequence with a typical ATTA core. The amino acid at position 50 of the homeodomain normally recognises the two bases immediately 5′ to the core sequence. A lysine at position 50 (as present in all members of the SIX family) is known to specify a target sequence of GGATTA. Therefore, if SIX5 does in fact bind to an ATTA core sequence this would be the predicted recognition site.
SIX5 is situated downstream of the DMPK (dystrophia myotonica protein kinase) gene and the unstable (CTG)n repeat associated with myotonic dystrophy type 1 (DM1) (1,18–25). DM1 is the most prevalent form of adult onset muscular dystrophy, with an estimated incidence of 1 in 8000, and is inherited as an autosomal dominant disorder. It is characterised by muscle weakness and atrophy although symptoms are highly variable and multisystemic (26).
An enhancer element that controls the expression of SIX5 was identified within a DNase I hypersensitive site adjacent to the (CTG)n repeat and alleles with large expansions were shown to lose hypersensitivity (24,25). A 2–4-fold reduction in the steady-state transcript levels of SIX5 from the expanded allele, compared to the normal allele of DM1 patients, was seen in fibroblasts and skeletal muscle cells. This implies that expansion of the (CTG)n repeat alters local chromatin structure in such a way as to reduce expression of SIX5, which in turn is implicated in the pathogenesis of DM1 (25). Other groups have also reported a reduction in the levels of SIX5 mRNA synthesis from the DM1 allele in primary myoblast cultures and post-mortem muscle, heart, brain and liver tissues from DM1 patients (27–29). In contrast, other reports indicated that levels of SIX5 expression were unchanged in fibroblast cell lines and adult skeletal muscle from DM1 patients, compared to normal controls (30,31). However, allele-specific PCR has recently shown that expression of SIX5 from the expanded allele is reduced in the DM1 fibroblast cell lines (32).
SIX5 could potentially regulate DMPK, as both genes have been shown to be expressed in a similar range of tissues in both human and mouse and levels of expression from both genes have been shown to be altered in DM1 patients (1,25,27,33–39). We identified a site similar to that bound by Six4, in the promoter region of human DMPK, along with a GGATTA consensus site (which is conserved in both mouse and human).
In this study GST–SIX5 fusion proteins were used in gel retardation assays with short double stranded DNA fragments representing putative DNA binding sites, to investigate DNA binding targets of SIX5 and the functions of its two conserved domains in DNA binding specificity. A whole human genome PCR-based screen was also performed with recombinant protein to identify other potential targets of SIX5. We hypothesised that: (i) SIX5 protein regulates DMPK expression by binding to one of the two putative binding sites within its promoter; (ii) SIX5 is partially responsible for the regulation of ATP1A1 by binding to a sequence within the ARE; (iii) the SIX domain and the homeodomain of SIX5 are both required for sequence-specific binding. We also hypothesised that as SIX5 is expressed in a wide range of tissues affected by the multisystemic disorder DM1, that SIX5 binds to and regulates a number of genes that are involved in the manifestation of the phenotype.
MATERIALS AND METHODS
Expression and purification of GST–SIX5 recombinant proteins
Recombinant proteins for use in gel retardation assays and the whole genome PCR-based screen were generated using the pGEX bacterial expression system (Amersham Pharmacia Biotech, Bucks, UK). The gene fragments to be expressed were subcloned into the MCS of pGEX4T3. From a 500 ml culture, 20 ml of bacterial cell lysate were obtained and, where required, purified by affinity chromatography (40). GST-fusion proteins were eluted in 6 ml of elution buffer [50 mM Tris–HCl (pH 8.0), 120 mM NaCl, 10 mM DTT, 1 mM PMSF, 20 mM reduced glutathione]. For GST–SIX, a BamHI–AccI fragment of a genomic SIX5 clone (nucleotides 1310–1731, accession number X84813) was subcloned into pGEX4T3. The predicted fusion protein contains SIX5 amino acids G65–Y206 (assuming the translation start site is at position 1118–1120 of accession number X84813). For GST–-HD, the homeobox (180 bp) plus 131 bp of flanking sequence was amplified from a 1.15 kb RT–PCR product subclone (1) by PCR [primers: KJDMF and SIX5-R.SEH, previously described by Winchester et al. (39)] and subcloned into pGEX4T3 (SIX5 amino acids V183–E285). These two overlapping clones were used to generate GST–SIX+HD by inserting an AccI fragment of the GST–HD construct into the AccI site of the GST–SIX construct (SIX5 amino acids G65–E285) maintaining the correct reading frame (Fig. 1).
Figure 1.

GST–SIX5 recombinant proteins. A schematic diagram of SIX5 cDNA indicating the regions expressed as GST-fusion proteins (GST–SIX, GST–HD and GST–SIX+HD). PCR primers, KJDMF and SIX5-R.SEH are indicated. Amino acid positions assume that the translation start site is at 1118–1120 of accession number X84813.
Generation of putative DNA binding sites
Forty base-pair double stranded oligonucleotides representing putative DNA binding sites and flanking regions were created by annealing two single stranded oligonucleotides to give a sequence with a BamHI compatible site at one end and an EcoRI compatible site at the other end. The oligonucleotide pairs are shown below with the putative binding sites in bold and the restriction endonuclease sites in lower case.
ARE1 and ARE2 represent –96 to –62 of ATP1A1 (13) and include the 13 bp Six4 binding site and 21 bp of flanking sequence within the ARE (this entire sequence is conserved between human, rat and mouse). A mutant ARE sequence that differs by a single nucleotide and has been shown not to bind Six4 was also produced (oligonucleotides, AREmut1 and AREmut2) (2). The mutated base in each strand is underlined. ARE-like1 and ARE-like2 (nucleotides 726–760, accession number L08835) represent the ARE-like sequence in the DMPK promoter. GGATTA1 and GGATTA2 (nucleotides 1311–1346, accession number L08835) represent the GGATTA consensus site present in the promoter region of DMPK. The double stranded oligonucleotides were subcloned into the BamHI–EcoRI sites of pBluescript“ SK(+) phagemid (Stratagene, La Jolla, CA).
Gel retardation assays
Aliquots of each plasmid DNA (1–2 µg), containing the cloned putative DNA binding sites, were digested with XbaI and HindIII [for pBluescript SK(+) plasmids] or NotI (for pGEM plasmids, used in whole genome PCR). Vector and insert were end-radiolabelled with [α-32P]dCTP using the Klenow fragment of DNA polymerase I.
Ten microliters of affinity purified recombinant protein (~6 pmol) or a 1/10 dilution of unpurified cell lysate diluted in 1.5× DNA–protein binding buffer [50 mM Tris–HCl (pH 8.0), 120 mM NaCl, 10 mM DTT, 1 mM PMSF], 7% (v/v) glycerol, 0.01 µg/µl poly[dI/dC]·poly[dI/dC], 3.33 × 10–3% (w/v) bromophenol blue, 100 cps radiolabelled probe (~3 fmol) and H2O to bring the total volume to 15 µl were mixed on ice. They were incubated for 20 min at room temperature. Binding reactions were analysed by electrophoresis in non-denaturing 8 or 4% (w/v) polyacrylamide gels [50 mM Tris–glycine (pH 9.4), 0.1 mM EDTA, 8 or 4% (w/v) acrylamide:bis (37.5:1), 0.12% (w/v) APS, 0.06% (v/v) TEMED] for 3 h at 200 V.
Whole genome PCR
The whole genome PCR screen was based on a modification of published methods (41,42). In summary, 1 µg aliquots of high molecular weight human lymphocyte DNA were digested to completion with either Sau3AI or Tsp509I. The Sau3AI digest was then ligated to linkers consisting of 5 µg of complementary oligonucleotides V-SU (5′-GATCGGACTTGCTACGGTA-ATCAG-3′) and V-NL (3′-CCTGAACGATGCCATTAGTC-5′) annealed together. The Tsp509I digest was ligated to linkers consisting of 5 µg of complementary oligonucleotides Tsp (5′-AATTGGACTTGCTACGGTAATCAG-3′) and V-NL annealed together. The DNA was heated to 95°C, precipitated using 1.3 vol of propan-2-ol and 0.3 vol of 10 M ammonium acetate, air dried and resuspended in 10 µl H2O. This created two independent linker libraries.
Linkered genomic DNA (300 ng; either Sau3AI cleaved or Tsp509I cleaved) was mixed with 200 µl of a 50% (v/v) slurry of GST–SIX+HD bound to glutathione coated Sepharose“ 4B beads (~4 µg of protein) in an Eppendorf tube. The sample was rotated at room temperature for 1 h before centrifugation at 10 000 g for 5 min and the supernatant removed. The beads were washed in 1 ml of 1× DNA–protein binding buffer (as used in the gel retardation assay). Bound DNA was then eluted by the addition of 100 µl 1× DNA–protein binding buffer plus 1 M NaCl. The sample was rotated at room temperature for 30 min before centrifugation at 10 000 g for 5 min and the eluate removed to a fresh tube. The DNA was precipitated with propan-2-ol, air dried and resuspended in 10 µl H2O, before being used as a PCR template.
PCR was carried out using Perkin Elmer AmpliTaq Gold‘, with the following conditions: 0.5 µM of primer V-NL, 0.2 mM each dNTP, 1.25 U Taq polymerase, 5% (v/v) DMSO and 1× Gene Amp PCR buffer in a 25 µl total volume. Thirty cycles of PCR were performed, consisting of 1 min at 94°C, 1 min at 59°C and 1 min at 72°C, followed by 7 min at 72°C. The templates used were 5 µl of eluted, bound and precipitated DNA, 5 µl of 1/10 (all rounds) 1/100 (rounds 3–5) and 1/1000 (round 5) dilutions of the DNA. PCR product (100–400 ng) was used in another round of selection.
Rounds of binding and PCR were repeated five times until discrete bands could be seen after electrophoresis in a 1% (w/v) agarose gel. The discrete bands were subcloned into pGEM“-T Easy vector (Promega, Madison, WI) following the manufacturer’s instructions. Clones were sequenced using T7 and SP6 sequencing primers. Each individual sequence was tested for its ability to bind GST–SIX+HD in a gel retardation assay with 100-fold molar excess of unlabelled ARE as a competitor.
DNA sequencing
DNA sequencing was performed under standard conditions on either an ABI 373A sequencer or an ABI 377 sequencer (Perkin Elmer, Beaconsfield, UK), by the Molecular Biology Support Unit at the University of Glasgow.
RESULTS
Characterisation of the DNA binding properties of GST–SIX5 recombinant proteins
The SIX domain of SIX5 (GST–SIX), the homeodomain (GST–HD) and the SIX and homeodomain together (GST–SIX+HD), were expressed as recombinant GST-fusion proteins in a bacterial expression system (Fig. 1). Affinity purified GST-fusion proteins and cell lysates from bacteria expressing these recombinant proteins were tested for their ability to bind to the putative DNA binding sites (ARE, AREmut, ARE-like and GGATTA, see Materials and Methods) using gel retardation assays (Fig. 2).
Figure 2.
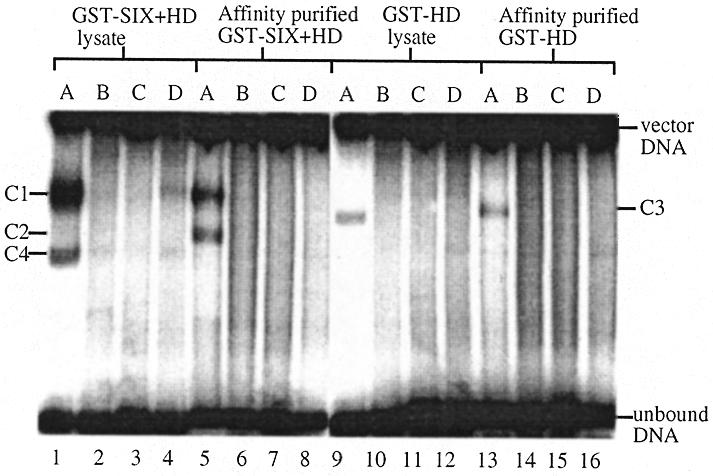
Sequence selective binding by GST–SIX5 recombinant proteins. The binding activities of 10 µl of 1/10 dilution of cell lysates from bacterial cultures expressing GST–SIX+HD or GST–HD were compared to the binding activities of 6 pmol of the respective affinity purified recombinant proteins. ARE (A), ARE-like (B), GGATTA (C) and AREmut (D) (3 fmol) were used as probes. C1–4 indicate the retarded complexes formed.
Binding reactions using affinity purified fusion proteins gave the following results. (i) Two retarded complexes (C1 and C2) formed when GST–SIX+HD bound to the ARE fragment (lane 5). No complexes were seen when it was mixed with the mutant ARE, which differs by a single nucleotide (lane 8), or either of the putative sites in DMPK (lanes 6 and 7), indicating that GST–SIX+HD does not bind to these targets. (ii) A single complex (C3) was formed, intermediate in mobility between the two complexes of GST–SIX+HD, when GST–HD bound to the ARE fragment (lane 13). It did not form complexes with the mutant ARE (lane 16) or either of the putative sites in DMPK (lanes 14 and 15). (iii) GST–SIX and the GST-tag, showed no binding activity to any of the oligonucleotides (results not shown).
Binding reactions were also performed using bacterial cell lysates containing over-expressed recombinant GST–SIX+HD (lanes 1–4), GST–HD (lanes 9–12) or GST-tag protein (results not shown). GST–SIX could not be assayed in this manner due to its insolubility in the cell lysate. The intensity of the upper complex (C1), but not the lower complex (C2/C4), formed by the binding of GST–SIX+HD to the ARE was greatly enhanced in the presence of the cell lysate (lane 1). No binding to any of the other oligonucleotides was visible (lanes 2–4). The cell lysate did not increase the intensity of the complex formed by the binding of GST–HD to the ARE (lane 9). Lysate from bacteria expressing the GST-tag protein did not bind to any of the oligonucleotides (results not shown). These experiments indicated that either a cofactor was present in the cell lysate that enhanced the binding activity of GST–SIX+HD or that the activity or concentration of the recombinant protein itself was reduced by affinity purification. It was not possible to estimate the concentrations of the over-expressed recombinant GST-fusion proteins present in the bacterial cell lysates by protein assay or SDS–PAGE analysis, due to the abundance of other bacterial proteins. Therefore, cell lysate from bacteria expressing GST-tag protein was added to a binding reaction consisting of 5.4 pmol of affinity purified GST–SIX+HD and the ARE. About 80% (w/w) of the ARE formed a single retarded complex (C1) (Fig. 3). This experiment indicates the presence of a cofactor in the cell lysate that enhances the binding of SIX5 to the ARE.
Figure 3.
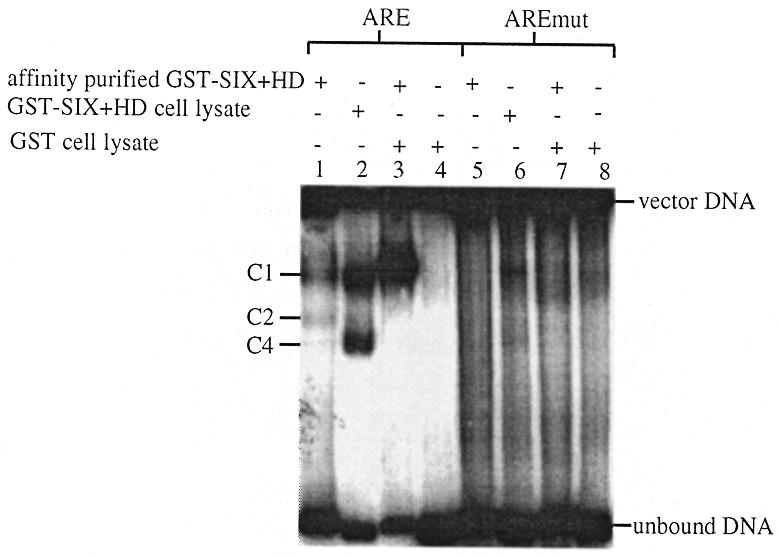
Gel retardation analysis of GST–SIX+HD ± cell lysate. The binding activity of affinity purified GST–SIX+HD combined with cell lysate from bacteria expressing recombinant GST was investigated by gel retardation analysis with 3 fmol of ARE and AREmut. Lanes 1 and 5 contain 10 µl of a 1/10 dilution of GST–SIX+HD cell lysate, lanes 3 and 7 contain 9 µl (5.4 pmol) of affinity purified GST–SIX+HD and 1 µl of undiluted GST cell lysate and lanes 4 and 8 contain 10 µl of a 1/10 dilution of GST cell lysate. C1–4 indicate the retarded complexes formed.
Characterisation of the binding enhancement cofactor present in the bacterial cell lysate
To characterise the cofactor present in the bacterial cell lysate that enhanced the binding of GST–SIX+HD to the ARE, aliquots of lysate from bacteria expressing the GST-tag protein were modified by a number of techniques. These were (i) size fractionation using a Centricon 10 column (Millipore, Watford, UK) to give one fraction containing proteins >10 kDa and one fraction containing proteins with a molecular weight <10 kDa; (ii) digestion with 50 µg/ml proteinase K at 55°C overnight (the proteinase K was inactivated by heating to 95°C for 10 min), (iii) boiling for 45 min, (iv) dialysis. The modified lysates were added to binding reactions consisting of 5.4 pmol of affinity purified GST–SIX+HD and the ARE as in the previous experiment. The enhancement activity was lost from the cell lysates that contained only proteins with a molecular weight <10 kDa or had been treated with proteinase K (Fig. 4). This experiment indicated that the cofactor is a heat-stable protein with a molecular weight >10 kDa.
Figure 4.

Characterisation of the binding enhancement cofactor present in the bacterial cell lysate. The enhancement activity of cell lysate from bacteria expressing recombinant GST that had been (i) size fractionated, (ii) proteinase K treated, (iii) boiled, (iv) dialysed, was compared to that of whole untreated lysate. Assays contained 9 µl (5.4 pmol) of affinity purified GST–SIX+HD and 1 µl of appropriate GST cell lysate. Controls included an assay containing no bacterial cell lysate and one containing no affinity purified GST–SIX+HD.
Two known heat-stable bacterial DNA bending proteins (HU and FIS from Escherichia coli) that modulate the activity of a variety of DNA binding proteins (43) were tested to see if they could enhance the binding of GST–SIX+HD to the ARE. Concentrations of these proteins ranging from 0.005 to 3 ng/µl were tested. No enhancement activity was detected (results not shown).
Whole genome PCR
In order to identify gene promoters regulated by SIX5 a whole genome PCR-based screening method was used to identify human genomic DNA sequences bound by GST–SIX+HD protein.
GST–SIX+HD was bound to glutathione-coated Sepharose“ 4B beads. The immobilised protein was incubated with two human genomic DNA linker libraries (Sau3AI and Tsp509I). Bound DNA was eluted and amplified by PCR. This selection process was repeated five times, at which point discrete products could be visualised by agarose gel electrophoresis (Fig. 5). The final selected products were subcloned into pGEM“-T Easy vector and sequenced. Seven independent fragments were identified from each of the two libraries; the average length of which was 420 bp (range 336–524 bp). None of the selected fragments contains the known Six4 binding site (within the ARE). Each fragment was tested by gel retardation assays with cell lysate from bacteria expressing either GST–SIX+HD or GST-tag protein. All 14 fragments formed specific complexes with GST–SIX+HD; five of these (clones SauD, SauE, TspA, TspB and TspC) also formed other retarded complexes as indicated by a gel shift in the absence of GST–SIX+HD. No complexes were formed in the absence of bacterial cell lysate (Fig. 6). The binding of GST–SIX+HD to five of the fragments (clones SauD, TspA, TspC, TspD and TspE) was not competed by the presence of 100-fold molar excess of unlabelled ARE.
Figure 5.
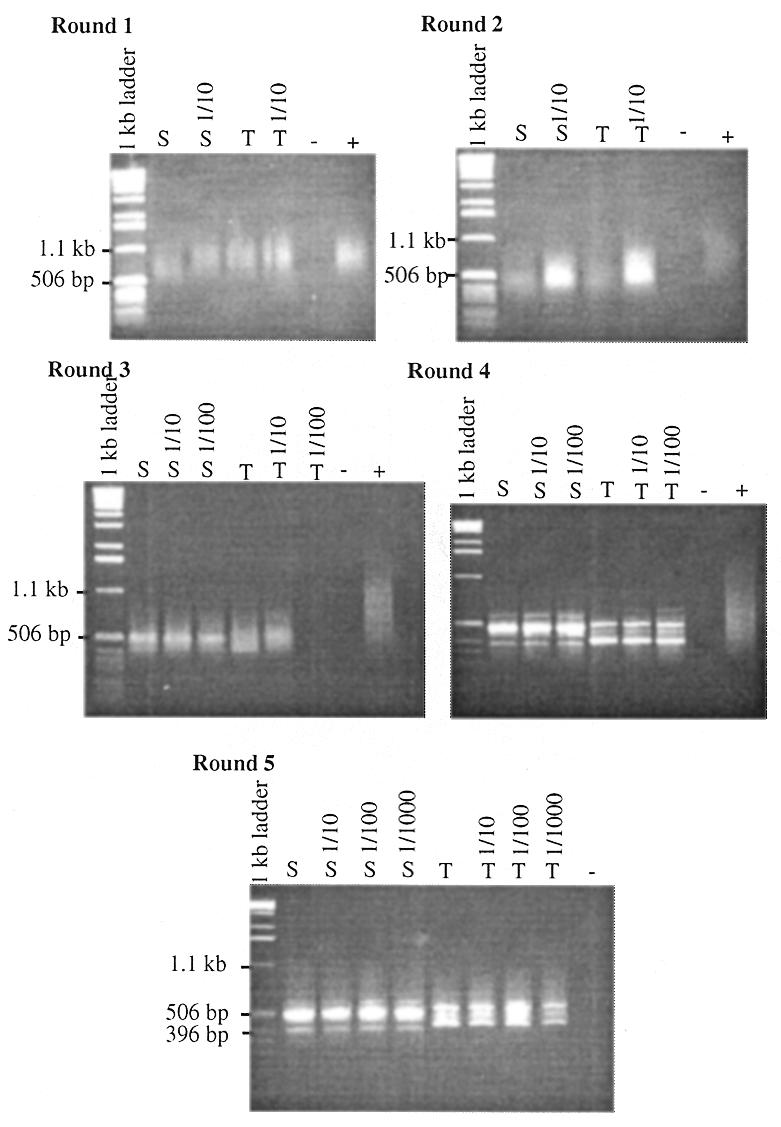
Whole genome PCR. Five rounds of binding and PCR were performed using two linker libraries: Sau3AI (S) and Tsp509I (T) (Materials and Methods). After each round of binding, 5 µl of eluate and of 1/10–1/1000 dilutions of eluate were amplified by PCR using primer V-NL. Fifty percent (v/v) of each reaction was electrophoresed on a 1% (w/v) agarose gel. Linkered genomic DNA was used as a positive control for each round of PCR (+) and H2O as a negative control (–).
Figure 6.
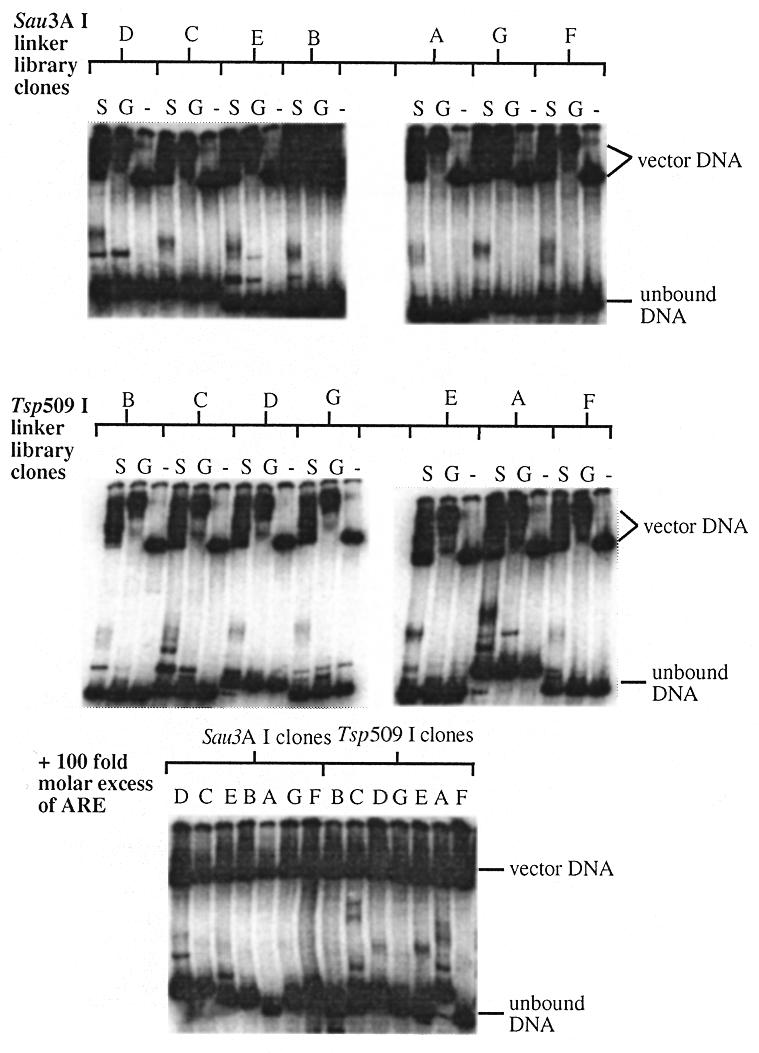
Gel retardation analysis of sequences selected by whole genome PCR. The selected sequences from each linker library were tested for binding to recombinant SIX5 by gel retardation assays with cell lysates from bacteria expressing GST–SIX+HD (S) or GST (G). Negative control assays (containing no protein) were also performed (–). Gel retardation assays were then repeated using cell lysate from bacteria expressing GST–SIX+HD and a 100-fold molar excess of unlabelled ARE fragment to determine specific binding.
The sequences of all 14 fragments were used in BLAST searches of the GenBank, EMBL, DDBJ and PDB databases. TspE shows 92% identity, over its entire length of 388 bp, to a portion of the 5′ flanking and promoter region of the human dopamine D5 receptor gene (DRD5) This sequence maps from –965 to –578 of DRD5 (accession number U21164) and starts 1161 bp 3′ of the initiation site (44). However, TspE shows 97% identity to the homologous region of DRD5 transcribed pseudogene 1 (ϕDRD5-1). A second DRD5 transcribed pseudogene (ϕDRD5-2) also exists which has been reported to have ~95% identity with DRD5 and ϕDRD5-1 for 1.9 kb upstream of the coding region of DRD5 (45–47). Both pseudogenes contain differences in their coding regions that make them incabable of encoding functional receptors. Unfortunately the 5′UTR sequence of ϕDRD5-2 is not present in any of the databases. However, all three genes have been mapped to separate chromosomes; DRD5 to chromosome 4, ϕDRD5-1 to chromosome 2 and ϕDRD5-2 to chromosome 1 (48,49). Using a somatic cell hybrid DNA panel (HGMP) we have mapped TspE to chromosome 1, indicating that it is likely to be from the 5′UTR of ϕDRD5-2.
TspC contains non-alphoid repetitive sequence identical to such sequences in clones from the cat eye syndrome region of chromosome 22q11.2 (13421–13803, accession number AP000540). No potential genes or promoters were identified in this region using NIX analysis (HGMP). NIX is a WWW tool for the running and viewing of the results of many DNA analysis programs on a DNA sequence. These programs include GRAIL, Fex, Hexon, MZEF, Genemark, Genefinder, FGene, BLAST (against many databases) Polyah, RepeatMasker and tRNAscan. SauD, TspA and TspD show no significant matches to any sequences in the databases.
Of the sequences that were shown to be competed by excess ARE, SauB contains sequence from the genomic region containing the BLU gene (1659–17405, accession number AC002481 ). SauG is 99% identical to genomic DNA from chromosome 21q22.2, Down syndrome region (73900–74312, accession number AP000162). No potential genes or promoters were identified in this region using NIX analysis. TspF contains a repetitive element present on chromosome 22q13.3 (55710–56114, accession number AL022327). No potential genes or promoters were identified in this region using NIX analysis. The other six sequences show no significant matches to any sequences in the databases. MEME (multiple EM for motif elicitation) analysis (50) was performed separately on the five sequences not competed by the ARE and the nine sequences competed by the ARE. This program identifies highly conserved regions within a group of related DNAs. No conserved motifs were identified. Table 1 summarises details of all the clones tested by gel retardation assay with GST–SIX+HD. The 14 sequences identified by the whole genome PCR-based screen have been submitted to GenBank and their accession numbers are shown.
Table 1. Summary of GST–SIX+HD–DNA binding results.
| Clone | Sequence length (bp) | Gel shift with GST–SIX+HD | Sequence identity | Competed by ARE | Contains MEF3 site | GenBank accession no. | |
|---|---|---|---|---|---|---|---|
|
In vitro |
ARE |
40 |
Yes |
ATP1A1 |
N/D |
Yes |
N/A |
| |
AREmut |
40 |
No |
N/A |
N/D |
No |
N/A |
| |
ARE-like |
40 |
No |
DMPK |
N/D |
Yes |
N/A |
| |
GGATTA |
40 |
No |
DMPK |
N/D |
No |
N/A |
| Genomic screen |
SauA |
336 |
Yes |
N/K |
Yes |
No |
AF242566 |
| |
SauB |
448 |
Yes |
BLU |
Yes |
No |
AF242567 |
| |
SauC |
524 |
Yes |
N/K |
Yes |
No |
AF242568 |
| |
SauD |
424 |
Yes |
N/K |
No |
No |
AF242569 |
| |
SauE |
437 |
Yes |
N/K |
Yes |
Yes |
AF242570 |
| |
SauF |
464 |
Yes |
N/K |
Yes |
No |
AF242571 |
| |
SauG |
414 |
Yes |
Genomic seq on Chr 21 |
Yes |
No |
AF242572 |
| |
TspA |
469 |
Yes |
N/K |
No |
Yes |
AF242573 |
| |
TspB |
359 |
Yes |
N/K |
Yes |
No |
AF242574 |
| |
TspC |
383 |
Yes |
Genomic seq on Chr 22 |
No |
No |
AF242575 |
| |
TspD |
434 |
Yes |
N/K |
No |
No |
AF242576 |
| |
TspE |
388 |
Yes |
øDRD5-2 |
No |
No |
AF242577 |
| |
TspF |
406 |
Yes |
Genomic seq on Chr 22 |
Yes |
No |
AF242578 |
| TspG | 391 | Yes | N/K | Yes | No | AF242579 |
N/A, not applicable; N/K, not known; N/D, not done.
DISCUSSION
Gel retardation assays with GST–SIX5 recombinant proteins indicated that SIX5 does not bind to a GGATTA consensus site present in the promoter of DMPK. Although GGATTA is recognised by homeodomain proteins that possess a lysine at position 50 of the homeodomain it is an arginine at position 5 of the homeodomain that contacts the ATTA core sequence. SIX5 (and other members of the SIX family) contain a lysine at position 50 but lack an arginine at position five and therefore this is not an unexpected result. It also appears that SIX5 does not bind to the ARE-like sequence in the promoter of DMPK and therefore if SIX5 is regulating DMPK expression, it is unlikely to be doing so directly by binding to either of these putative binding sites.
Both GST–SIX+HD and GST–HD bound to the ARE but not to the mutant ARE (which differed by only 1 bp), which indicates that this binding is highly sequence-specific. This is highly suggestive that SIX5 is involved in the regulation of ATP1A1. This is of particular interest as Na+/K+ ATPase activity has been shown to be reduced in DM1 patients (51,52). Although we obtained no evidence that the SIX domain on its own binds to the ARE, when present with the homeodomain (as in the endogenous protein) it affects the binding affinity. It is possible that the higher molecular weight complex observed in the gels is formed by a protein dimer and the lower molecular weight complex by a protein monomer binding to the DNA and that the SIX domain is required for dimerisation to occur. Although homeodomain proteins can bind as monomers in vitro, evidence is now emerging that many also bind as dimers. The paired family of homeodomain proteins have been shown to bind as both heterodimers and homodimers to palindromic DNA sequences. The binding of one homeodomain molecule was reported to increase the affinity of a second molecule 300-fold (53).
The presence of the bacterial cell lysate greatly enhanced the formation of the putative dimer complex which suggests that it contains a cofactor that alters the structure of the DNA in such a way as to increase the likelihood of dimer formation. There are many examples of prokaryotic and eukaryotic factors that can bend DNA to enhance the interaction of two or more transcription factors (54). It appears that a cofactor is present in the bacterial crude cell lysate, that is sufficiently similar to a natural cofactor present in human cells, to alter the structure of the DNA enough to enhance dimerisation. We have shown that this cofactor is a heat-stable protein with a molecular weight >10 kDa.
The SIX domain has been shown in Drosophila to be important for protein–protein interactions. Drosophila sine oculis (so), the founding member of the SIX family, and eyes absent (eya), also an eye development protein in Drosophila, interact in yeast and in vitro through the SIX domain of so and an evolutionarily conserved domain in eya (55). Recently murine Six and Eya proteins have also been shown to specifically interact through their homologous conserved domains (56,57). The experiments described in this paper provide further evidence that the SIX domain is involved in protein–protein interactions as it was shown to be necessary for the putative homodimer complex to form.
The whole genome PCR-based screen identified 14 independent sequences, five of which were bound by GST–SIX+HD even in the presence of 100× molar excess of the ARE fragment. This indicates that DNA sequence or sequences exist for which SIX5 has a greater affinity than it does for the ARE and may also partly explain why the screen did not identify the ARE sequence. However, it should also be noted that a screen of this kind will only identify binding sites that are in a restriction fragment of a length that can easily be amplified by PCR (in this case ~500 bp was optimal). DNase I footprinting is currently being performed to identify the exact protein–DNA binding site within each of the selected sequences. From this a consensus binding site for SIX5 may be determined. Although MEME analysis failed to identify a consensus sequence it should be noted that this program does not allow gaps within the motifs and it is known that not all the positions of the 13 bp sequence within the ARE are involved in Six4 binding (13). Recently, murine Six1, Six2, Six4 and Six5 were shown to bind to a MEF3 (myogenic enhancing factor 3) consensus sequence (TCAGGTT) within the myogenin promoter, thus implicating them in the myogenesis pathway (57,58). This could explain how down-regulation of SIX5 expression leads to the delay in muscle maturation associated with congenital DM1 and the muscle atrophy in classical DM1 patients. MEF3 sites are also present in the promoter regions of aldolase A and cardiac troponin C, which may also be involved in DM1 muscle and heart symptoms (59–62). Interestingly, a MEF3 site is present in the ARE (to which we have shown SIX5 binds) and absent in AREmut (to which SIX5 does not bind; the first C is changed to a G). However, the ARE-like sequence within the promoter of DMPK, to which this study has shown that SIX5 does not bind, also contains a MEF3 site indicating that the sequence flanking the MEF3 site may be important in specifying the SIX5 target DNA site. Of the 14 sequences identified by the whole genome PCR screen only TspA and SauE contain a MEF3 site. Together these data suggest that there are several (possibly related) potential in vitro sequence-specific binding sites for SIX5 (and other members of the SIX family) of which all or a subset may be functionally active. It is likely that in vivo the affinities of members of the SIX family for the various DNA binding sites are specified by particular SIX–EYA or SIX–SIX complexes. The down-regulation of SIX5 could disrupt pathways in which other SIX family members (but not SIX5) are involved if they bind to DNA and or protein sequences unoccupied by SIX5 complexes.
We concluded that the sequence of clone TspE is from ϕDRD5-2. If the DRD5 gene 5′ flanking and promoter region is also shown to contain a SIX5 binding site this could indicate an interesting mechanism for the regulation of this gene and its transcriptionally active pseudogenes, as the homologous region within DRD5 is situated between the transcription and translation start sites. SIX5 may therefore interfere with transcription of DRD5. Alternatively, SIX5 may bind to the RNA at this position and thus act as a translational repressor. A Drosophila homeodomain protein, bicoid, which binds DNA and transcriptionally activates different target genes has also been shown to repress the translation of caudal by binding to the 3′UTR of the mRNA (63). It has been reported that the dopamine receptors of pituitary prolactin cells might be impaired in DM1 patients (64). DM1 patients suffer from psychological problems (26) and one can speculate that this may be due to defects in their dopamine receptors. Therefore, DRD5 is a candidate for involvement in the development of the DM1 phenotype. Further studies into DRD5 mRNA and protein levels in DM1 patients and normal controls are being initiated to address this question.
The sequence of SauB is from the human BLU gene within the lung cancer region on chromosome 3p21.3 and contains both exonic and intronic sequence. DNase I footprinting will tell us if the binding site is exonic or intronic giving us an insight into a possible regulatory mechanism for this gene.
The other 12 sequences identified by the whole genome PCR screen show no significant matches to any genes in the databases although SauG, TspC and TspF have been localised to particular genomic loci. All 12 sequences are being characterised further to identify any genes to which they are linked.
In conclusion we have shown that in vitro the homeodomain of human SIX5 acts as a sequence-specific DNA binding domain and that the SIX domain is necessary for the formation of a stable putative homodimer complex that enhances this binding activity. We have also identified five DNA sequences to which SIX5 binds with greater affinity than it does to the ARE and have identified two genes potentially regulated by SIX5.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Drs Mark Bailey, Martin Boocock, Graham Brock and Richard Wilson for helpful discussions and critical reading of the manuscript. We also thank Sue Macauley of the MBSU, for sequencing the clones identified by the whole genome PCR-based screen and Graham Hamilton for assistance with computing. The HU and FIS proteins were gifts from Dr Martin Boocock. This work was funded by a MDG prize studentship (S.E.H.) and MDG grant number RA3/434 (C.L.W. and K.J.J.).
REFERENCES
- 1.Boucher C.A., King,S.K, Carey,N., Krahe,R. Winchester,C.L., Rahman,S., Creavin,T., Meghji,P., Bailey,M.E.S., Chartier,F.L. et al. (1995) Hum. Mol. Genet., 4, 1919–1925. [DOI] [PubMed] [Google Scholar]
- 2.Kawakami K., Ohto,H., Ikeda,K. and Roeder,R.G. (1996) Nucleic Acids Res., 24, 303–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kawakami K., Ohto,H., Takizawa,T. and Saito,T. (1996) FEBS Lett., 393, 259–263. [DOI] [PubMed] [Google Scholar]
- 4.Toy J., Yang,J.M., Leppert,G.S. and Sundin,O.H. (1998) Proc. Natl Acad. Sci. USA, 95, 10643–10648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oliver G., Mailhos,A., Wehr,R., Copeland,N.G., Jenkins,N.A. and Gruss,P. (1995) Development, 121, 4045–4055. [DOI] [PubMed] [Google Scholar]
- 6.Bovolenta P., Mallamaci,A. and Boncinelli,E. (1996) Int. J. Dev. Biol., 1, 73–74. [PubMed] [Google Scholar]
- 7.Loosli F., Koster,R. and Wittbrodt,J. (1997) Dev. Biol., 186, B24. [Google Scholar]
- 8.Znoiko I.Y., Znoiko,S.L., Zinoveva,R.D. and Mitashov,V.I. (1997) Izvestiya Akademii Nauk Seriya Biologicheskaya, 654–659. [PubMed] [Google Scholar]
- 9.Zuber M.E., Perron,M., Bang,A., Holt,C.E. and Harris,W.A. (1997) Dev. Biol., 186, B20. [Google Scholar]
- 10.Bovolenta P., Mallamaci,A., Puelles,L. and Boncinelli,E. (1998) Mech. Dev., 70, 201–203. [DOI] [PubMed] [Google Scholar]
- 11.Kobayashi M., Toyama,R., Takeda,H., Dawid,I.B. and Kawakami,K. (1998) Development, 125, 2973–2982. [DOI] [PubMed] [Google Scholar]
- 12.Seo H.C., Drivenes,O., Ellingsen,S. and Fjose,A. (1998) Mech. Dev., 73, 45–57. [DOI] [PubMed] [Google Scholar]
- 13.Suzuki-Yagawa Y., Kawakami,K. and Nagano,K. (1992) Mol. Cell. Biol., 12, 4046–4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ingraham H.A., Flynn,S.E., Voss,J.W., Albert,V.R., Kapiloff,M.S., Wilson,L. and Rosenfeld,M.G. (1990) Cell, 61, 1021–1033. [DOI] [PubMed] [Google Scholar]
- 15.Treisman J., Harris,E. and Desplan,C. (1991) Genes Dev., 5, 594–604. [DOI] [PubMed] [Google Scholar]
- 16.Verrijzer C.P., Alkema,M.J., van Weperen,W.W., Van Leeuwen,H.C., Strating,M.J.J. and van der Vliet,P.C. (1992) EMBO J., 11, 4993–5003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gehring W.J., Affolter,M. and Burglin,T. (1994) Annu. Rev. Biochem., 63, 487–526. [DOI] [PubMed] [Google Scholar]
- 18.Aslanidis C., Jansen,G., Amemiya,C., Shutler,G., Mahadevan,M., Tsilfidis,C., Chen,C., Alleman,J., Wormskamp,N.G., Vooijs,M. et al. (1992) Nature, 355, 548–551. [DOI] [PubMed] [Google Scholar]
- 19.Brook J.D., McCurrach,M.E., Harley,H.G., Buckler,A.J., Church,D., Aburatani,H., Hunter,K., Stanton,V.P., Thirion,J.P., Hudson,T. et al. (1992) Cell, 68, 799–808. [DOI] [PubMed] [Google Scholar]
- 20.Buxton J., Shelbourne,P., Davies,J., Jones,C., Van Tongeren,T., Aslandis,C., de Jong,P., Jansen,G., Anvret,M., Riley,B. et al. (1992) Nature, 355, 547–548. [DOI] [PubMed] [Google Scholar]
- 21.Fu Y.H., Pizzuti,A., Fenwick,R.G.,Jr, King,J., Rajnarayan,S., Dunne,P.W., Dubel,J., Nasser,G.A., Ashizawa,T., de Jong,P. et al. (1992) Science, 255, 1256–1258. [DOI] [PubMed] [Google Scholar]
- 22.Harley H.G., Rundle,S.A., Reardon,W., Myring,J., Crow,S., Brook,J.D., Harper,P.S. and Shaw,D.J. (1992) Lancet, 339, 1125–1128. [DOI] [PubMed] [Google Scholar]
- 23.Mahadevan M., Tsilfildis,C., Sobourin,L., Shutler,G., Amemiya,C., Jansen,G., Neville,C., Narang,M., Barcelo,J., O’Hoy,K. et al. (1992) Science, 255, 1253–1255. [DOI] [PubMed] [Google Scholar]
- 24.Otten A.D. and Tapscott,S.J. (1995) Proc. Natl Acad. Sci. USA, 92, 5465–5469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klesert T.R., Otten,A.D., Bird,T.D. and Tapscott,S.J. (1997) Nature Genet., 16, 402–406. [DOI] [PubMed] [Google Scholar]
- 26.Harper P.S. (1989) Myotonic Dystrophy. 2nd Edn. WB Saunders Company, London.
- 27.Thornton C.A., Wymer,J.P., Simmons,Z., McClain,C. and Moxley,R.T. (1997) Nature Genet., 16, 407–409. [DOI] [PubMed] [Google Scholar]
- 28.Gennarelli M., Pavoni,M., Amicucci,P., Angelini,C., Menegazzo,E., Zelano,G., Novelli,G. and Dallapiccola,B. (1999) Neuromuscul. Disord., 9, 215–219. [DOI] [PubMed] [Google Scholar]
- 29.Korade-Mirnics Z., Tarleton,J., Servidei,S., Casey,R.R., Gennarelli,M., Pegoraro,E., Angelini,C. and Hoffman,E.P. (1999) Hum. Mol. Genet., 8, 1017–1023. [DOI] [PubMed] [Google Scholar]
- 30.Hamshere M.G., Newman,E.E., Alwazzan,M., Athwal,B.S. and Brook,J.D. (1997) Proc. Natl Acad. Sci. USA, 94, 7394–7399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eriksson M., Ansved,T., Edstrom,L., Anvret,M. and Carey,N. (1999) Hum. Mol. Genet., 8, 1053–1060. [DOI] [PubMed] [Google Scholar]
- 32.Alwazzan M., Newman,E., Hamshere,M.G. and Brook,J.D. (1999) Hum. Mol. Genet., 8, 1491–1497. [DOI] [PubMed] [Google Scholar]
- 33.Carango P., Noble,J.E., Marks,H.G. and Funanage,V.L. (1993) Genomics, 18, 340–348. [DOI] [PubMed] [Google Scholar]
- 34.Fu Y.H., Friedman,D.L., Richards,S., Pearlman,J.A., Gibbs,R.A., Pizzuti,A., Ashizawa,T., Perryman,M.B., Scarlato,G., Fenwick,R.G.,Jr et al. (1993) Science, 260, 235–238. [DOI] [PubMed] [Google Scholar]
- 35.Sabourin L.A., Mahadevan,M.S., Narang,M., Lee,D.S.C., Surh,L.C. and Korneluk,R.G. (1993) Nature Genet., 4, 233–238. [DOI] [PubMed] [Google Scholar]
- 36.Krahe R., Ashizawa,T., Abbruzzese,C., Roeder,E., Carango,P., Giacanelli,M., Funanage,V.L. and Siciliano,M.J. (1995) Genomics, 28, 1–14. [DOI] [PubMed] [Google Scholar]
- 37.Wang J., Pegoraro,E., Menegazzo,E., Gennarelli,M., Hoop,R.C., Angelini,C. and Hoffman,E.P. (1995) Hum. Mol. Genet., 4, 599–606. [DOI] [PubMed] [Google Scholar]
- 38.Heath S.K., Carne,S., Hoyle,C., Johnson,K.J. and Wells,D.J. (1997) Hum. Mol. Genet., 6, 651–657. [DOI] [PubMed] [Google Scholar]
- 39.Winchester C.L., Ferrier,R.K., Sermoni,A., Clark,B.J. and Johnson,K.J. (1999) Hum. Mol. Genet., 8, 481–492. [DOI] [PubMed] [Google Scholar]
- 40.Smith D.B. and Johnson,K.S. (1988) Gene, 67, 31–40. [DOI] [PubMed] [Google Scholar]
- 41.Kinzler K.W. and Voglestein,B. (1989) Nucleic Acids Res., 17, 3645–3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fainsod A., Margalit,Y., Haffner,R. and Gruenbaum,Y. (1991) Nucleic Acids Res., 19, 4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Finkel S.E. and Johnson,R.C. (1992) Mol. Microbiol., 6, 3257–3265. [DOI] [PubMed] [Google Scholar]
- 44.Beischlag T.V., Marchese,A., Meador-Woodruff,J.H., Damask,S.P., O’Dowd,B.F., Tyndale,R.F., Van Tol,H.H.M., Seeman,P. and Niznik,H.B. (1995) Biochemistry, 34, 5960–5970. [DOI] [PubMed] [Google Scholar]
- 45.Grandy D.K., Zhang,Y.A., Bouvier,C., Zhou,Q.Y., Johnson,R.A., Allen,L., Buck,K., Bunzow,J.R., Salon,J., Civelli,O. et al. (1991) Proc. Natl Acad. Sci. USA, 88, 9175–9179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nguyen T., Bard,J., Jin,H., Tarusico,D., Ward,D.C., Kennedy,J.L., Weinshank,R., Seeman,P. and O’Dowd,B.F. (1991) Gene, 109, 211–218. [DOI] [PubMed] [Google Scholar]
- 47.Marchese A., Beischlag,T.V., Nguyen,T., Niznik,H.B., Weinshank,R.L., George,S.R. and O’Dowd,B.F. (1995) Gene, 154, 153–158. [DOI] [PubMed] [Google Scholar]
- 48.Eubanks J.H., Altherr,M., Wagner-McPherson,C., McPherson,J.D., Wasmuth,J.J. and Evans,G.A. (1992) Genomics, 12, 510–516. [DOI] [PubMed] [Google Scholar]
- 49.Grandy D.K., Allen,L.J., Zhang,Y., Mageis,R.E. and Civelli,O. (1992) Genomics, 13, 968–973. [DOI] [PubMed] [Google Scholar]
- 50.Bailey T.L. and Elkan,C. (1994) Proceedings of the Second International Conference on Intelligent Systems for Molecular Biology. AAAI Press, Menlo Park, CA.
- 51.Benders A.A.G.M., Timmermans,J.A.H., Oosterhof,A., Ter Laak,H.J., van Kuppevelt,T.H.M.S.M., Wevers,R.A. and Veerkamp,J.H. (1993) Biochem. J., 293, 269–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Benders A., Wevers,R.A. and Veerkamp,J.H. (1996) Acta Physiol. Scand., 156, 355–367. [DOI] [PubMed] [Google Scholar]
- 53.Wilson D., Sheng,G., Lecuit,T., Dostatni,N. and Desplan,C. (1993) Genes Dev., 7, 2120–2134. [DOI] [PubMed] [Google Scholar]
- 54.Werner M.H. and Burley,S.K. (1997) Cell, 88, 733–736. [DOI] [PubMed] [Google Scholar]
- 55.Pignoni F., Hu,B.R., Zavitz,K.H., Xiao,J.A., Garrity,P.A. and Zipursky,S.L. (1997) Cell, 91, 881–891. [DOI] [PubMed] [Google Scholar]
- 56.Heanue T.A., Reshef,R., Davis,R.J., Mardon,G., Oliver,G., Tomarev,S., Lassar,A.B. and Tabin,C.J. (1999) Genes Dev., 13, 3231–3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ohto H., Kamada,S., Tago,K., Tominaga,S.-I., Ozaki,H., Sato,S. and Kawakami,K. (1999) Mol. Cell. Biol., 19, 6815–6824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Spitz F., Demignon,J., Porteu,A., Kahn,J.-P., Daegelen,D. and Maire,P. (1998) Proc. Natl Acad. Sci. USA, 95, 14220–14225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hidaka K., Yamamoto,I., Arai,Y. and Mukai,T. (1993) Mol. Cell. Biol., 13, 6469–6478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Parmacek M.S., Ip,H.S., Jung,F., Shen,T., Martin,J.F., Vora,A.J., Olson,E.N. and Leiden,J.M. (1994) Mol. Cell. Biol., 14, 1870–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Salminen M., Lopez,S., Maire,P., Kahn,A. and Daegelen,D. (1996) Mol. Cell. Biol., 16, 76–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Spitz F., Salminen,M., Demignon,J., Kahn,A., Daegelen,D. and Maire,P. (1997) Mol. Cell. Biol., 17, 656–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dubnau J. and Struhl,G. (1996) Nature, 379, 694–699. [DOI] [PubMed] [Google Scholar]
- 64.Sakuma H., Takase,S., Mizuno,Y., Teramura,K. and Hanew,K. (1988) Tohoku J. Exp. Med., 156, 291–298. [DOI] [PubMed] [Google Scholar]