

Abstract
The expression of MHC-II and CD86 on the surface of antigen presenting cells (APCs) must be tightly regulated to foster antigen-specific CD4 T cell activation and to prevent autoimmunity. Surface expression of these proteins is regulated by their dynamic ubiquitination by the E3 ubiquitin ligase March-I. March-I promotes turnover of peptide-MHC-II complexes on resting APCs and termination of March-I expression promotes MHC-II and CD86 surface stability. In this review we will highlight recent studies examining March-I function in both normal and pathological conditions.
The regulated expression of MHC-II and CD80/CD86 are essential for the generation of an effective CD4 T cell response. In APCs such as dendritic cells (DCs), MHC-II expression is regulated both transcriptionally and post-transcriptionally. Resting DCs constitutively express peptide-MHC-II (pMHC-II) complexes by a process in which newly-synthesized MHC-II bind “self” peptides in endosomal antigen processing compartments (reviewed in [1]). These complexes are continually expressed on the DC surface and their turnover is regulated by ubiquitin-dependent lysosomal degradation catalyzed by the E3 ubiquitin ligase March-I (reviewed in [2]). March-I is constitutively expressed in professional hematopoietic APCs including DCs, B cells, and monocytes/macrophages where it regulates surface expression of MHC-II and CD86 [3–5]. Whereas March-I is not expressed in epithelial cells, the closely-related March-I homolog March-8 regulates MHC-II expression in cortical epithelial cells in the thymus [6] [7]. A recent study revealed dysregulated expression of MHC-II on the surface of neutrophils and eosinophils in March-I-deficient mice [8], demonstrating that March-I is also expressed on these cells that can function as “atypical” APCs under certain conditions [9]. Activation of professional APCs by engagement of TLR abrogates MHC-II transcription and also terminates expression of March-I, thereby protecting newly-generated pMHC-II from lysosomal degradation and stabilizing their expression on the plasma membrane [3] [4,10,11]. CD86 is also a well-characterized target of March-I in APCs [12] and it is generally assumed that the only physiological March-I substrates are MHC-II and CD86 [8]. As will be discussed below, given recent studies in a variety of experimental systems, this assumption should perhaps be modified. In this review we will summarize recent studies examining March-I structure, substrate specificity, and function in both normal and pathophysiological conditions.
What Does March-I Do and How Does It Work?
MARCH-I is an E3 ubiquitin ligase which, together with its unidentified E2 ubiquitin ligase partner(s), ubiquitinates MHC-II and CD86 in hematopoietic APCs [8]. It is our opinion that the mechanism of action of March-I on its substrates is relatively straightforward, as summarized in Figure 1 for MHC-II. MHC-II constitutively recycles following clathrin-independent endocytosis in APCs [13,14], and ubiquitination by March-I, either at the cell surface or in early endosomes [15], re-routes internalized MHC-II to a pathway of lysosomal degradation [16]. The same mechanism likely holds for CD86 as well, however CD86 internalization and recycling have not been extensively investigated. Essentially identical results are obtained in APCs in which the MHC-II and/or CD86 ubiquitination sites have been mutated, revealing a direct role for MHC-II and CD86 ubiquitination (or lack thereof) in their intracellular trafficking [16,17]. So the consequences of MHC-II and CD86 ubiquitination by March-I are really quite simple; in immature APCs (which express March-I) surface expressed MHC-II and CD86 are internalized, ubiquitinated, and degraded whereas in mature APCs (or March-I-deficient APCs) the termination of March-I expression results in the return of internalized pMHC-II and CD86 to the cell surface, thereby protecting newly-formed pMHC-II complexes from degradation in activated/mature APCs.
Figure 1.
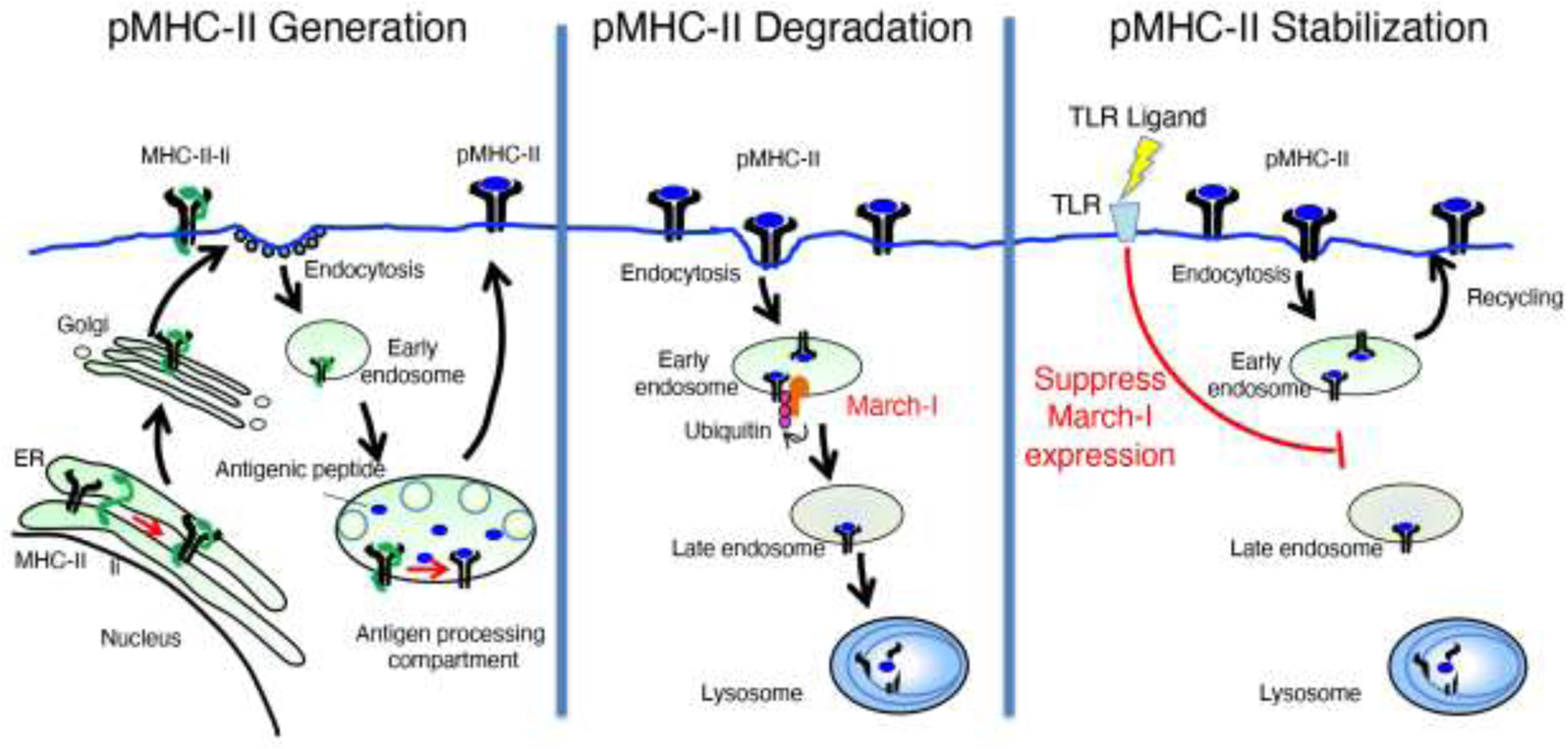
LEFT PANEL: MHC-II αβ dimers associate with Invariant chain (Ii) in the endoplasmic reticulum, traffic through the Golgi apparatus, and are delivered to the plasma membrane. MHC-II-Ii complexes are internalized by clathrin-mediated endocytosis and traffic to multivesicular antigen processing compartments. Some of these complexes sort onto the intralumenal vesicles of multivesicular antigen processing compartments, where Ii is degraded until only a small fragment of Ii remains bound to MHC-II. Following removal of residual Ii-derived peptides from the peptide binding groove of MHC-II and high affinity peptide binding, pMHC-II complexes traffic to the plasma membrane.
MIDDLE PANEL: After arrival at the cell surface, pMHC-II internalizes to early endosomes by clathrin-independent endocytosis. In resting DCs and B cells, pMHC-II is ubiquitinated by the E3 ubiquitin ligase March-I either at the plasma membrane or in early endosomes and is targeted for lysosomal degradation.
RIGHT PANEL: Activation of APCs (for example, by engagement of TLR ligands) results in rapid down-regulation of March-I expression. Internalized pMHC-II is therefore not ubiquitinated and returns to the plasma membrane by Rab11a-dependent recycling, thereby prolonging pMHC-II half-life and stabilizing pMHC-II complexes on the plasma membrane.
While we know that APC activation terminates March-I transcription [11] [10,18], little is known about how March-I protein is degraded in APCs. March-I itself is the target of ubiquitination (although not likely by itself) and March-I degradation is blocked by lysosomal proteinase inhibitors [19–21]. These data argue that March-I protein turnover is regulated by ubiquitination-dependent lysosomal sorting and degradation, and while we have recently identified Ube2D1 as an E2 ubiquitin ligase that controls March-I ubiquitination [19], the E3 ligase that ubiquitinates March-I remains unknown. Being a RING-domain containing E3 ubiquitin ligase, March-I function depends on E2 ligase activity, although this too remains largely unexplored. Recent studies have revealed that UBL3 is a “co-factor” required for March-I function in APCs [22]. UBL3 is not an E2 ligase and it is not clear how it works, however the myriad of defects observed in March-I knock-out APCs (described below) are also observed in UBL3 knock-out mice, highlighting the role that UBL3 plays in regulating March-I function.
March-I Substrates
March-I is generally thought to be expressed exclusively in hematopoietic APCs. Although a wide-range of substrates can be ubiquitinated by March-I when overexpressed (including CD44, CD98, FAS, STING, and transferrin receptor) [23,24], a recent comprehensive analysis of protein expression in wild-type, March-I knock-out, and March-8 knock-out hematopoietic APCs, non-hematopoietic APCs, and granulocyes/monocytes revealed dysregulated expression of only MHC-II and CD86 but not the above-mentioned proteins [8], strongly suggesting that MHC-II and CD86 are the sole substrates for March-I and March-8 in APCs. The type I-IFN signaling mediator STING is elevated in March-I knock-out spleen cells and the protection of March-I-deficient mice from death following malaria infection is dependent on type I-IFN and T cells [24]. While these data suggest that STING is a March-I substrate, given the plethora of defects present in March-I-deficient mice (described below), it is unclear at present if regulation of STING expression is a direct consequence of March-I-mediated ubiquitination or an indirect consequence of immune cell dysfunction.
Whereas MHC-II and CD86 are the only clearly recognized substrates of March-I in APCs, a recent study has shown that March-I protein expression is stimulated by HCMV-infection of human fibroblasts and this is important to reduce the expression of the transferrin receptor on infected cells [25]. HCMV does not encode any E3 ligases and seems to have coopted March-I to reduce transferrin receptor expression on infected cells, thereby reducing the potentially toxic effects of elevated intracellular iron levels. The transferrin receptor was found to be a March-I substrate when overexpressed in HeLa cells [23], however this is the first report (to our knowledge) of transferrin receptor being an in vivo substrate for March-I. In addition to this unexpected role of endogenous March-I in regulating transferrin receptor expression, Ablack et al. have shown that CD98 expression is increased in CD8 T cells isolated from March-I-deficient mice [26]. The sensitivity of these CD8 T cells to anti-CD98 mAb-dependent suppression of T cell proliferation is March-I-dependent, leading the authors to conclude that March-I is expressed in activated CD8 T cells [26] [27]. Thus while in APCs the only March-I substrates appear to be MHC-II and CD86, induced expression of March-I in non-APCs can lead to ubiquitination and downregulation of other substrates that are important for immune function. How it is that March-I does NOT dysregulate expression of transferrin receptor or CD98 in APCs remains unknown and seems to be as topic worthy of investigation.
Function of March-I-Deficient APCs
Studies of immune function in March-I knock-out or MHC-II K225R ubiquitination mutant mice generally assume that the APCs are “normal” and that any observed defect can be attributed solely to dysregulation of MHC-II and/or CD86 expression. However, recent data has shown that this is not the case. Two studies examining DCs isolated from either March-I knock-out or MHC-II K225R mice revealed that these cells have a profoundly altered phenotype of surface protein expression and gene transcription as compared to their wild-type counterparts [28,29]. Although MHC-II K225R DCs and B cells process and present large amounts of specific pMHC-II on their surface [16,30,31] and these complexes remain on the cell surface for an extended period of time [11] [16] [4], these APCs are very poor stimulators of naïve CD4 T cells [12] [32,33] [28] [29]. Curiously, these DCs are also poor stimulators of CD8 T cells by MHC-I, which is not a March-I substrate [34]. They also secrete IL-12 and TNF-α very poorly when directly stimulated with LPS in vitro [12,28], a parameter that is unrelated to MHC-II or CD86 expression. The expression of many surface proteins, including CD18, CD24, CD11c, CD8, and DEC205, is dysregulated on the surface of MHC-II ubiquitination-mutant DCs [29,32–34] and these cells have also been reported to have reduced expression of lipid rafts and tetraspanin web microdomain proteins [33]. For reasons that are not fully understood, March-I-deficient or MHC-K225R mutant mice also have significantly reduced numbers of spleen DCs [29,35] and regulatory T cells [30,36], further confounding interpreting data of immune function in these mice.
Recent studies have shown that complement component 3 (C3) binds to MHC-II on the surface of DCs, and enhanced surface expression of MHC-II in MHC-II-ubiquitination-deficient DCs leads to dramatically enhanced trogocytosis of DC membrane proteins by marginal zone B cells that is complement receptor 2 dependent [35]. In addition, it has been reported that March-I-deficiency leads to extremely high expression of MHC-II on neutrophils, eosinophils, and monocytes in the spleen and lung [8]. The extent to which these cells could now play an unexpected role in antigen presentation, especially in models of pulmonary diseases, makes simple interpretation of data from such studies nearly impossible. Precisely how dysregulated expression of MHC-II alters DC function so profoundly is unknown (these defects are not observed in mice possessing ubiquitination-defective CD86 molecules [32]), however simply attributing altered immune function in MHC-II mutant mice to “enhanced pMHC-II stability or surface expression” is unfounded.
March-I as an Antiviral Factor
While trying to make lentivirus expressing the March-I homolog March-8, Tada et al. found their March-8-containing lentiviral particles were far less infectious than control particles [37]. In an attempt to understand this phenomenon, they went on to show that March-8 also inhibits HIV infectivity by downregulating HIV gp120 from the cell surface [37]. This has now been confirmed by other groups [38–40] and more recently both March-I and March-2 have also been shown to downregulate HIV gp120 surface expression and viral infectivity [38]. Curiously, unlike the effects of March-I on MHC-II, March-8-mediated downregulation of gp120 surface expression is not a direct consequence of gp120 endocytosis and degradation [39,40]. Instead, March-8 leads to retention of gp160 in the trans-Golgi network due to impaired carbohydrate processing that prevents cleavage of gp160 by the proteinase furin [40]. This activity of March-8 was blocked when a cytosolic tyrosine in March-8 was mutated [39]. The identity of the March-8 substrate, how this tyrosine regulates March-8 activity, and whether March-I and March-2 downregulate HIV gp120 by the same mechanism remains to be determined. This model of March-dependent intracellular retention of HIV gp160 has been challenged, however, as Umthong et al. have recently demonstrated that March-I and March-8 directly target gp160 for degradation in lysosomes and do not lead to its intracellular accumulation [41].
Like HIV gp120, the envelope proteins of vesicular stomatitis virus (VSV-G), LCMV, Lassa virus, SARS-COV-2, and the influenza A virus M2 protein are directly ubiquitinated by March-I and/or March-8 and are then degraded by lysosomal proteolysis [39,42] [41]. The tyrosine mutant of March-8 that lost the ability to downregulate HIV gp120 expression was still able to ubiquitinate and downregulate VSV-G [39], highlighting two distinct mechanisms of suppressing viral replication by March-8.
March-I as a Tumor Biomarker
The relationship between March-I expression and prognosis for cancer recovery has recently been examined in two studies [43,44]. Unfortunately, these studies do not agree with each other, with one study showing high March-I expression in lung adenocarcinoma (and other tumors) predicting a favorable clinical outcome [43] while another showed that high March-I expression in oral squamous cell carcinoma correlates with poor prognosis [44]. In the case of oral squamous cell carcinoma it was postulated that high levels of March-I lead to the degradation of the phosphatase PHLPP2, thereby leading to enhanced levels of phosphor-AKT and cell proliferation [44]. While it is an intriguing possibility that during tumorigenesis March-I expression is induced, March-I protein expression in these tumor cells must be confirmed since normal epithelial cells do not express March-I.
Role of March-I in Diabetes
The role of March-I in insulin-dependent diabetes has also recently been examined. March-I-deficient mice have been reported to have increased insulin sensitivity, suggesting that March-I is a negative regulator of insulin receptor signaling [45]. In this study the authors showed that March-I can ubiquitinate and degrade insulin receptor β-chain in HeLa and HepG2 cells, however expression of endogenous insulin receptors in March-I-deficient mice was not examined. These data are consistent with the hypothesis that March-I constitutively regulates expression of insulin receptors in vivo. By contrast, using these same March-I-deficient mice Majdoubi et al. reported that March-I-deficiency leads to enhanced obesity-induced insulin resistance [46]. Adoptive-transfer of spleen CD8 T cells from March-I knock-out mice exacerbated insulin resistance in their model and these T cells were skewed toward an effector memory or resident memory phenotype and had reduced capacity to generate proinflammatory cytokines [46]. The same results were not obtained in CD8 T cells isolated from spleens of MHC-II K225R ubiquitination-deficient mice however, showing that these effects are not due to dysregulation of MHC-II expression in March-I knock-out mice. Why CD8 T cells in March-I-deficient mice are detrimental and whether this is a direct consequence of March-I ubiquitination of an unknown substrate in CD8 T cells or is indirectly due to environmental factors present in March-I knock-out spleens remains to be determined.
Role of March-I in Asthma
Allergic asthma is an inflammatory disease that is exacerbated by Th2 cytokines and recent data suggests that March-I plays a role in regulating T cell skewing in this disease. March-I-deficient mice have reduced eosinophilia, IgE, and Th2 cells in a house dust mite-induced model of asthma [47] and Th2 cells isolated from lungs of March-I-deficient asthmatic mice display an exhausted phenotype [48]. Another study examining ovalbumin-induced allergic asthma confirmed the finding that Th2 cells and Th2 cytokine-production were reduced in March-I-deficient mice, however other parameters of disease found in the studies of Castellanos et al. were not observed in this model [49]. It was proposed that reduced Th2 cell development was due to sustained T cell signaling by enhanced MHC-II expression in March-I-deficient lymph node-resident DCs [47], however given data showing that March-I-deficiency leads to reduced cytokine production and poor antigen presentation [12,32] [28], this hypothesis requires further study.
Conclusion
Although March-I was first described by Bartee et al, nearly 20 years ago [23], only recently have scientists begun to examine immune function in mice lacking March-I or its homolog March-8. The inability to ubiquitinate MHC-II leads to a variety of APC defects that could well explain altered immune function in MHC-II ubiquitination-deficient mice observed in cancer, diabetes, and asthma. Given the recently described ability of March-I to degrade viral envelope proteins, it will be important to investigate whether pharmacological regulation of March-I function could serve as an antiviral therapy. Additional studies in March-I-deficient mice, keeping in mind the confounding effects of altered function in March-I-deficient APCs, will be important to clarify further the role of March-I in immunity.
Acknowledgements
This work was supported by the Intramural Research Program of the National Institutes of Health (P.A.R.).
Footnotes
Declaration of interest: none
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Roche PA, Furuta K: The ins and outs of MHC class II-mediated antigen processing and presentation. Nat Rev Immunol 2015, 15:203–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu H, Mintern JD, Villadangos JA: MARCH ligases in immunity. Curr Opin Immunol 2019, 58:38–43. [DOI] [PubMed] [Google Scholar]
- 3.Matsuki Y, Ohmura-Hoshino M, Goto E, Aoki M, Mito-Yoshida M, Uematsu M, Hasegawa T, Koseki H, Ohara O, Nakayama M, et al. : Novel regulation of MHC class II function in B cells. Embo J 2007, 26:846–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cho KJ, Walseng E, Ishido S, Roche PA: Ubiquitination by March-I prevents MHC class II recycling and promotes MHC class II turnover in antigen-presenting cells. Proc Natl Acad Sci U S A 2015, 112:10449–10454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mittal SK, Cho KJ, Ishido S, Roche PA: Interleukin 10 (IL-10)-mediated Immunosuppression: MARCH-I INDUCTION REGULATES ANTIGEN PRESENTATION BY MACROPHAGES BUT NOT DENDRITIC CELLS. J Biol Chem 2015, 290:27158–27167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu H, Jain R, Guan J, Vuong V, Ishido S, La Gruta NL, Gray DH, Villadangos JA, Mintern JD: Ubiquitin ligase MARCH 8 cooperates with CD83 to control surface MHC II expression in thymic epithelium and CD4 T cell selection. J Exp Med 2016, 213:1695–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.von Rohrscheidt J, Petrozziello E, Nedjic J, Federle C, Krzyzak L, Ploegh HL, Ishido S, Steinkasserer A, Klein L: Thymic CD4 T cell selection requires attenuation of March8-mediated MHCII turnover in cortical epithelial cells through CD83. J Exp Med 2016, 213:1685–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **8.Schriek P, Liu H, Ching AC, Huang P, Gupta N, Wilson KR, Tsai M, Yan Y, Macri CF, Dagley LF, et al. : Physiological substrates and ontogeny-specific expression of the ubiquitin ligases MARCH1 and MARCH8. Curr Res Immunol 2021, 2:218–228. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study is a comprehensive analysis of surface protein expression on a wide-variety of professional and non-professional APCs isolated from March-I and March-8-deficient mice. The data strongly argue that the only physiological substrates for March-I and March-8 are MHC-II and CD86.
- 9.Kambayashi T, Laufer TM: Atypical MHC class II-expressing antigen-presenting cells: can anything replace a dendritic cell? Nat Rev Immunol 2014, 14:719–730. [DOI] [PubMed] [Google Scholar]
- 10.Young LJ, Wilson NS, Schnorrer P, Proietto A, ten Broeke T, Matsuki Y, Mount AM, Belz GT, O’Keeffe M, Ohmura-Hoshino M, et al. : Differential MHC class II synthesis and ubiquitination confers distinct antigen-presenting properties on conventional and plasmacytoid dendritic cells. Nat Immunol 2008, 9:1244–1252. [DOI] [PubMed] [Google Scholar]
- 11.De Gassart A, Camosseto V, Thibodeau J, Ceppi M, Catalan N, Pierre P, Gatti E: MHC class II stabilization at the surface of human dendritic cells is the result of maturation-dependent MARCH I down-regulation. Proc Natl Acad Sci U S A 2008, 105:3491–3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ohmura-Hoshino M, Matsuki Y, Mito-Yoshida M, Goto E, Aoki-Kawasumi M, Nakayama M, Ohara O, Ishido S: Cutting edge: requirement of MARCH-I-mediated MHC II ubiquitination for the maintenance of conventional dendritic cells. J Immunol 2009, 183:6893–6897. [DOI] [PubMed] [Google Scholar]
- 13.McCormick PJ, Martina JA, Bonifacino JS: Involvement of clathrin and AP-2 in the trafficking of MHC class II molecules to antigen-processing compartments. Proc Natl Acad Sci U S A 2005, 102:7910–7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walseng E, Bakke O, Roche PA: Major histocompatibility complex class II-peptide complexes internalize using a clathrin- and dynamin-independent endocytosis pathway. J Biol Chem 2008, 283:14717–14727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Furuta K, Walseng E, Roche PA: Internalizing MHC class II-peptide complexes are ubiquitinated in early endosomes and targeted for lysosomal degradation. Proc Natl Acad Sci U S A 2013, 110:20188–20193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walseng E, Furuta K, Bosch B, Weih KA, Matsuki Y, Bakke O, Ishido S, Roche PA: Ubiquitination regulates MHC class II-peptide complex retention and degradation in dendritic cells. Proc Natl Acad Sci U S A 2010, 107:20465–20470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baravalle G, Park H, McSweeney M, Ohmura-Hoshino M, Matsuki Y, Ishido S, Shin JS: Ubiquitination of CD86 is a key mechanism in regulating antigen presentation by dendritic cells. J Immunol 2011, 187:2966–2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walseng E, Furuta K, Goldszmid RS, Weih KA, Sher A, Roche PA: Dendritic cell activation prevents MHC class II ubiquitination and promotes MHC class II survival regardless of the activation stimulus. J Biol Chem 2010, 285:41749–41754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lei L, Bandola-Simon J, Roche PA: Ubiquitin-conjugating enzyme E2 D1 (Ube2D1) mediates lysine-independent ubiquitination of the E3 ubiquitin ligase March-I. J Biol Chem 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jabbour M, Campbell EM, Fares H, Lybarger L: Discrete domains of MARCH1 mediate its localization, functional interactions, and posttranscriptional control of expression. J Immunol 2009, 183:6500–6512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bourgeois-Daigneault MC, Thibodeau J: Autoregulation of MARCH1 expression by dimerization and autoubiquitination. J Immunol 2012, 188:4959–4970. [DOI] [PubMed] [Google Scholar]
- *22.Liu H, Wilson KR, Firth AM, Macri C, Schriek P, Blum AB, Villar J, Wormald S, Shambrook M, Xu B, et al. : Ubiquitin-like protein 3 (UBL3) is required for MARCH ubiquitination of major histocompatibility complex class II and CD86. Nat Commun 2022, 13:1934. [DOI] [PMC free article] [PubMed] [Google Scholar]; Using a genome-wide CRISPR knock-out screen UBL3 was found to be critical for March-I function in DCs and macrophages. UBL3 deficiency phenocopies the functional defects observed in March-I-deficient mice, highlighting its role in March-I function.
- 23.Bartee E, Mansouri M, Hovey Nerenberg BT, Gouveia K, Fruh K: Downregulation of major histocompatibility complex class I by human ubiquitin ligases related to viral immune evasion proteins. J Virol 2004, 78:1109–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu J, Xia L, Yao X, Yu X, Tumas KC, Sun W, Cheng Y, He X, Peng YC, Singh BK, et al. : The E3 ubiquitin ligase MARCH1 regulates antimalaria immunity through interferon signaling and T cell activation. Proc Natl Acad Sci U S A 2020, 117:16567–16578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martin M, Sandhu P, Kumar R, Buchkovich NJ: The Immune-Specific E3 Ubiquitin Ligase MARCH1 Is Upregulated during Human Cytomegalovirus Infection to Regulate Iron Levels. J Virol 2022, 96:e0180621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ablack JN, Metz PJ, Chang JT, Cantor JM, Ginsberg MH: Ubiquitylation of CD98 limits cell proliferation and clonal expansion. J Cell Sci 2015, 128:4273–4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ablack JN, Ortiz J, Bajaj J, Trinh K, Lagarrigue F, Cantor JM, Reya T, Ginsberg MH: MARCH Proteins Mediate Responses to Antitumor Antibodies. J Immunol 2020, 205:2883–2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *28.Kim HJ, Bandola-Simon J, Ishido S, Wong NW, Koparde VN, Cam M, Roche PA: Ubiquitination of MHC Class II by March-I Regulates Dendritic Cell Fitness. J Immunol 2021, 206:494–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *29.Wilson KR, Jenika D, Blum AB, Macri C, Xu B, Liu H, Schriek P, Schienstock D, Francis L, Makota FV, et al. : MHC Class II Ubiquitination Regulates Dendritic Cell Function and Immunity. J Immunol 2021, 207:2255–2264. [DOI] [PubMed] [Google Scholar]; This paper, together with that of Kim et al. above, examine the function of APCs in March-I-deficient and MHC-II K225R ubiquitination-defective DCs. The studies reveal altered spleen DC numbers and phenotype as well as dramatically reduced MHC-I and MHC-II antigen presentation in vivo and in vitro by DCs in MHC-II ubiquitination-deficient mice.
- 30.Oh J, Wu N, Baravalle G, Cohn B, Ma J, Lo B, Mellman I, Ishido S, Anderson M, Shin JS: MARCH1-mediated MHCII ubiquitination promotes dendritic cell selection of natural regulatory T cells. J Exp Med 2013, 210:1069–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bannard O, McGowan SJ, Ersching J, Ishido S, Victora GD, Shin JS, Cyster JG: Ubiquitin-mediated fluctuations in MHC class II facilitate efficient germinal center B cell responses. J Exp Med 2016, 213:993–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ishikawa R, Kajikawa M, Ishido S: Loss of MHC II ubiquitination inhibits the activation and differentiation of CD4 T cells. Int Immunol 2014, 26:283–289. [DOI] [PubMed] [Google Scholar]
- 33.Oh J, Perry JSA, Pua H, Irgens-Moller N, Ishido S, Hsieh CS, Shin JS: MARCH1 protects the lipid raft and tetraspanin web from MHCII proteotoxicity in dendritic cells. J Cell Biol 2018, 217:1395–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wilson KR, Liu H, Healey G, Vuong V, Ishido S, Herold MJ, Villadangos JA, Mintern JD: MARCH1-mediated ubiquitination of MHC II impacts the MHC I antigen presentation pathway. PLoS One 2018, 13:e0200540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *35.Schriek P, Ching AC, Moily NS, Moffat J, Beattie L, Steiner TM, Hosking LM, Thurman JM, Holers VM, Ishido S, et al. : Marginal zone B cells acquire dendritic cell functions by trogocytosis. Science 2022, 375:eabf7470. [DOI] [PubMed] [Google Scholar]; This study found that elevated complement component 3 on the surface of MHC-II-ubiquitination mutant DCs allow marginal zone B cells to capture DC surface proteins by the process of trogocytosis. It is postulated that one function of March-I is to limit surface expression of MHC-II (and complement component 3) on the surface of DCs, thereby limiting trogocytosis of DC membrane proteins by marginal zone B cells.
- 36.Liu H, Wilson KR, Schriek P, Macri C, Blum AB, Francis L, Heinlein M, Nataraja C, Harris J, Jones SA, et al. : Ubiquitination of MHC Class II Is Required for Development of Regulatory but Not Conventional CD4(+) T Cells. J Immunol 2020, 205:1207–1216. [DOI] [PubMed] [Google Scholar]
- 37.Tada T, Zhang Y, Koyama T, Tobiume M, Tsunetsugu-Yokota Y, Yamaoka S, Fujita H, Tokunaga K: MARCH8 inhibits HIV-1 infection by reducing virion incorporation of envelope glycoproteins. Nat Med 2015, 21:1502–1507. [DOI] [PubMed] [Google Scholar]
- *38.Zhang Y, Tada T, Ozono S, Yao W, Tanaka M, Yamaoka S, Kishigami S, Fujita H, Tokunaga K: Membrane-associated RING-CH (MARCH) 1 and 2 are MARCH family members that inhibit HIV-1 infection. J Biol Chem 2019, 294:3397–3405. [DOI] [PMC free article] [PubMed] [Google Scholar]; Expression of March-I, March-2, or March-8 inhibits HIV infection by downregulating HIV Env from the surface of virus-producing cells, thereby reducing incorporation of HIV Env into virions and limiting viral infectivity.
- 39.Zhang Y, Tada T, Ozono S, Kishigami S, Fujita H, Tokunaga K: MARCH8 inhibits viral infection by two different mechanisms. Elife 2020, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu C, Li S, Zhang X, Khan I, Ahmad I, Zhou Y, Li S, Shi J, Wang Y, Zheng YH: MARCH8 Inhibits Ebola Virus Glycoprotein, Human Immunodeficiency Virus Type 1 Envelope Glycoprotein, and Avian Influenza Virus H5N1 Hemagglutinin Maturation. mBio 2020, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Umthong S, Lynch B, Timilsina U, Waxman B, Ivey EB, Stavrou S: Elucidating the Antiviral Mechanism of Different MARCH Factors. mBio 2021, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu X, Xu F, Ren L, Zhao F, Huang Y, Wei L, Wang Y, Wang C, Fan Z, Mei S, et al. : MARCH8 inhibits influenza A virus infection by targeting viral M2 protein for ubiquitination-dependent degradation in lysosomes. Nat Commun 2021, 12:4427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu Z, Liu J, Liu Z, Zhang H: MARCH1 as a novel immune-related prognostic biomarker that shapes an inflamed tumor microenvironment in lung adenocarcinoma. Front Oncol 2022, 12:1008753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu L, Guo B, Han Y, Xu S, Liu S: MARCH1 silencing suppresses growth of oral squamous cell carcinoma through regulation of PHLPP2. Clin Transl Oncol 2022, 24:1311–1321. [DOI] [PubMed] [Google Scholar]
- 45.Nagarajan A, Petersen MC, Nasiri AR, Butrico G, Fung A, Ruan HB, Kursawe R, Caprio S, Thibodeau J, Bourgeois-Daigneault MC, et al. : MARCH1 regulates insulin sensitivity by controlling cell surface insulin receptor levels. Nat Commun 2016, 7:12639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Majdoubi A, Lee JS, Kishta OA, Balood M, Moulefera MA, Ishido S, Talbot S, Cheong C, Alquier T, Thibodeau J: Lack of the E3 Ubiquitin Ligase March1 Affects CD8 T Cell Fate and Exacerbates Insulin Resistance in Obese Mice. Front Immunol 2020, 11:1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *47.Castellanos CA, Ren X, Gonzalez SL, Li HK, Schroeder AW, Liang HE, Laidlaw BJ, Hu D, Mak ACY, Eng C, et al. : Lymph node-resident dendritic cells drive T(H)2 cell development involving MARCH1. Sci Immunol 2021, 6:eabh0707. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that March-I-mediated turnover of MHC-II and CD86 is essential for the development of TH2 T cell responses and, for a first time, links the genetic variants of March-I with asthma risk in humans.
- 48.Castellanos CA, Hiam-Galvez KJ, Ishido S, Satpathy AT, Shin JS: MARCH1 Controls an Exhaustion-like Program of Effector CD4+ T Cells Promoting Allergic Airway Inflammation. Immunohorizons 2022, 6:684–692. [DOI] [PubMed] [Google Scholar]
- 49.Kishta OA, Sabourin A, Simon L, McGovern T, Raymond M, Galbas T, Majdoubi A, Ishido S, Martin JG, Thibodeau J: March1 E3 Ubiquitin Ligase Modulates Features of Allergic Asthma in an Ovalbumin-Induced Mouse Model of Lung Inflammation. J Immunol Res 2018, 2018:3823910. [DOI] [PMC free article] [PubMed] [Google Scholar]