

Abstract
Purpose:
Jansen-deVries syndrome is a neurodevelopmental condition attributed to pathogenic variants in exons 5–6 of PPM1D. As the full phenotypic spectrum and natural history remain to be defined, we describe a large cohort of children and adults with Jansen-deVries syndrome.
Methods:
Retrospective cohort study of 37 individuals from 34 families with disease-causing variants in PPM1D leading to Jansen-deVries syndrome. Clinical data were provided by treating physicians and/or families.
Results:
Of the 37 individuals, 27 were male and 10 female, with median age 8.75 years (range 8 months - 62 years). Four families document autosomal dominant transmission, and 32/34 probands were diagnosed via exome sequencing. The facial gestalt, including a broad forehead and broad mouth with a thin and tented upper lip, was most recognizable between 18–48 months of age. Common manifestations included global developmental delay (35/36, 97%), hypotonia (25/34, 74%), short stature (14/33, 42%), constipation (22/31, 71%), and cyclic vomiting (6/35, 17%). Distinctive personality traits include a hyper-social affect (21/31, 68%) and moderate-to-severe anxiety (18/28, 64%).
Conclusions:
Jansen-deVries is a clinically-recognizable neurodevelopmental syndrome with a characteristic personality and distinctive facial features. The association of pathogenic variants in PPM1D with cyclic vomiting bears not only medical attention but also further pathogenic and mechanistic evaluation.
Keywords: Jansen-deVries syndrome, PPM1D, cyclic vomiting, hypersocial personality, developmental delay
Introduction:
Jansen-deVries syndrome (JdVS) was first delineated as a syndromic form of intellectual disability in 2017 in a cohort of 14 individuals with truncating variants in PPM1D 1. All disease-causing variants in this initial cohort were found in the penultimate or last exons (5 and 6) of this gene and all were de novo 1. Prior to this report, PPM1D had been identified as a candidate gene for intellectual disability due to its enrichment for de novo variants in two large cohorts of individuals with intellectual disability 2, 3. In their report, Jansen et al describe in further detail the seven individuals reported in these larger meta-analyses2, 3 in addition to seven new individuals. They highlight several features shared among those with pathogenic variants in PPM1D 1. Intellectual disability was seen in all but one individual (though that person had learning difficulties) and other notable features included short stature, recurrent craniofacial features such as a broad forehead, a broad mouth with thin upper lip, small hands and feet, and gastrointestinal symptoms such as poor feeding, constipation, and cyclic vomiting. A particular behavioral phenotype was also noted, with an outgoing hyper-social personality, increased sensitivity to sound, nearly universal anxiety, and a high pain threshold commonly reported. Indeed, the condition was initially referred to as “intellectual developmental disorder with gastrointestinal difficulties and high pain threshold” (IDDGIP), but later renamed Jansen-De Vries syndrome (OMIM#617450). The molecular mechanism is attributed to a truncated protein product that escapes nonsense mediated decay but has lost the nuclear localization signal while maintaining the PPM-type phosphatase domain in exon 5 1; thus, disease-causing PPM1D variants may exert their effects in a dominant negative or gain-of-function mechanism.
Since this publication in 2017, 10 additional individuals have been published 4–10 that both reiterate and expand upon this phenotype with novel features including cleft lip and palate (one case) 8, bladder exstrophy (one case) 5, syndactyly 9, and unilateral renal dysplasia with the diagnosis found on fetal exome 6. Six of these 10 have been diagnosed by exome sequencing (ES) 4–6, 9, one by genome sequencing (GS) 10 and three via a ~5000 gene panel (Illumina TruSight One) 7, 8. In order to further elucidate the spectrum of manifestations of this rare neurodevelopmental condition, we now report an additional 34 families with Jansen-deVries syndrome, highlighting the unique developmental and behavioral phenotype, facial gestalt, and gastrointestinal symptoms through physician reports and family interviews.
Materials and Methods:
Editorial Policies and Ethical Considerations:
Participants 8, 23, and 30 were enrolled in the Manton Center for Orphan Disease Research’s IRB-approved protocol at Boston Children’s Hospital. For all other cases, informed consent to participate, including publication of photographs where relevant, was obtained at the local institution via their own institutional guidelines. De-identified data for all patients was collected and analyzed under an IRB protocol at Boston Children’s Hospital.
Individuals were identified via referral from clinicians, identification through GeneMatcher 11, and via connections made through the Jansen-deVries family support group. Phenotypic data were collected via parent interview or information provided by the treating geneticists. Twenty three parent interviews (CJC) were conducted via phone call, video call, or in person visits (five families). All parents provided additional information via email.
Results:
Demographics
We ascertained 37 individuals from 34 families, including the first report of familial cases, with two families involving an affected mother and child, a third involving an affected son, mother and grandmother, and a fourth involving an affected father and daughter. Across the cohort, there were 27 male and 10 female individuals. Median age at ascertainment was 8.75 years (IQR 5–12.25 years), with a range from 8 months to 62 years (though many families reported age at diagnosis of two to three years). Families were from North America, Europe, and Australia.
Molecular Diagnosis
Diagnosis was made via ES in all but two families, where the diagnosis was made by gene panel. All variants fell in exons 5 or 6 in PPM1D and were either frameshift variants or missense variants leading to premature termination. Two variants were recurrent, with the c.1210C>T, p.(Gln404Ter) variant observed in four separate families and the c.1280G>A, p.(Trp427Ter) variant also observed in four families. The variants in these 34 families are represented in Figure 1. Variants were de novo in 30/34 families, with the remaining four families documenting autosomal dominant inheritance.
Figure 1.
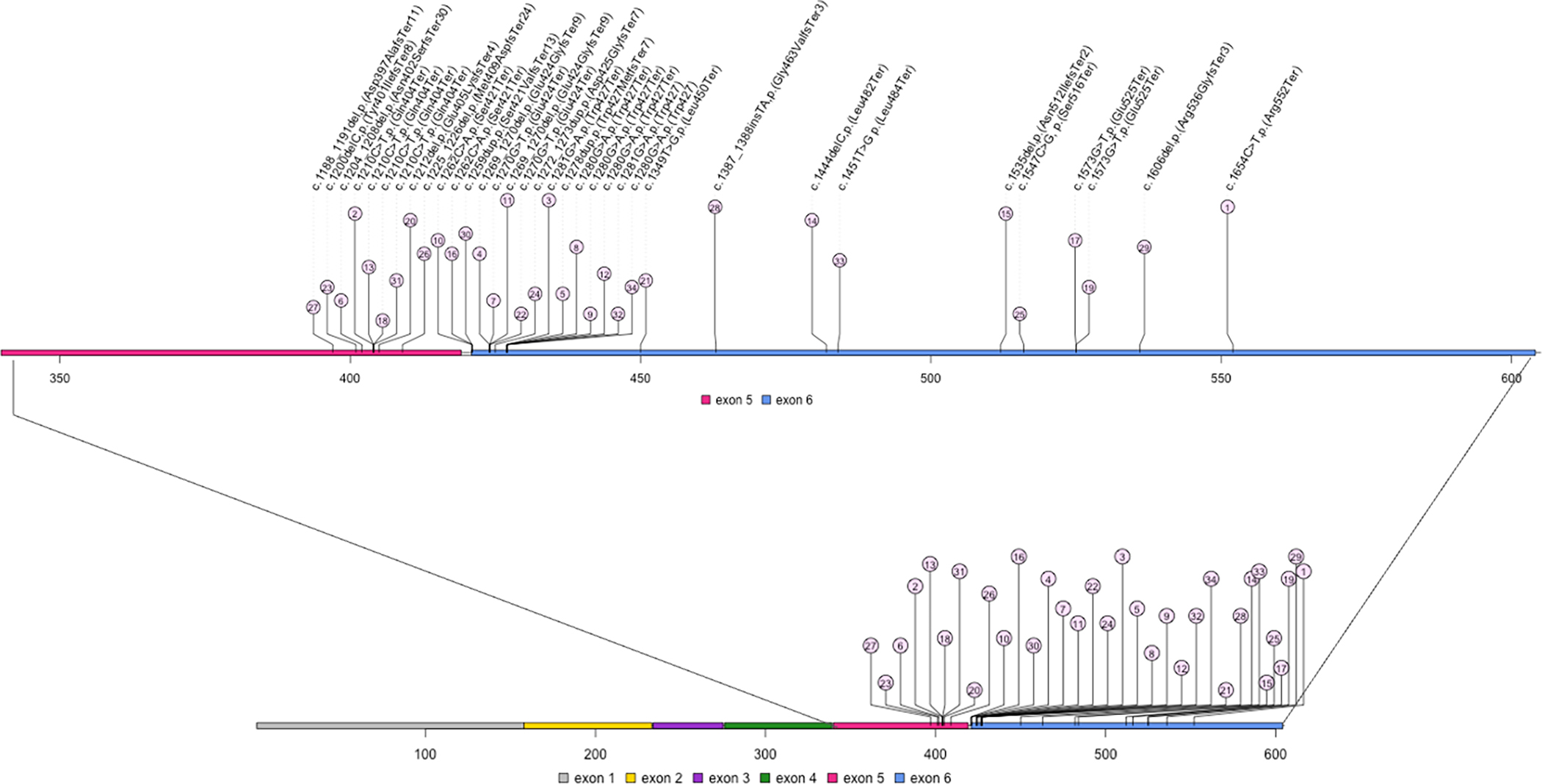
Spectrum of variants identified in PPM1D. Variants are identified by position in the amino acid sequence. The number within the circle indicates the case number from this series. All variants fall within exons 5 and 6 of PPM1D, which comprises six exons total.
Perinatal History
Polyhydramnios was noted in 6/34 (18%) pregnancies and gestational diabetes in 6/34 (18%) with no other salient prenatal features. Most individuals were born full term (three preterm) and anthropometric measurements at birth were generally within the normal range. Six individuals (24%) had neonatal complications including need for resuscitation at birth (five), tachypnea (two), apneic spells (one), and hypoglycemia (one). Common features noted in early infancy were hypotonia (25/34, 74%), poor feeding (two with dysphagia/choking in infancy), and constipation (22/31, 71%) (Supplemental Table 1).
Growth
Growth parameters were notable for short stature (Z-score of ≤ −2) in 14/33 (42%), with only three individuals having a Z-score for height of ≤ −3 and a median Z-score for height of −1.3. Low weight (Z-score of ≤ −2) was noted in 10/35 individuals (29%) and two were overweight with the rest at normal weight for age and a median Z-score for weight of −0.5. Head circumference was normal in the majority with available measurements (20), with seven (35%) having a Z-score ≤ −2 (Figure 2).
Figure 2.
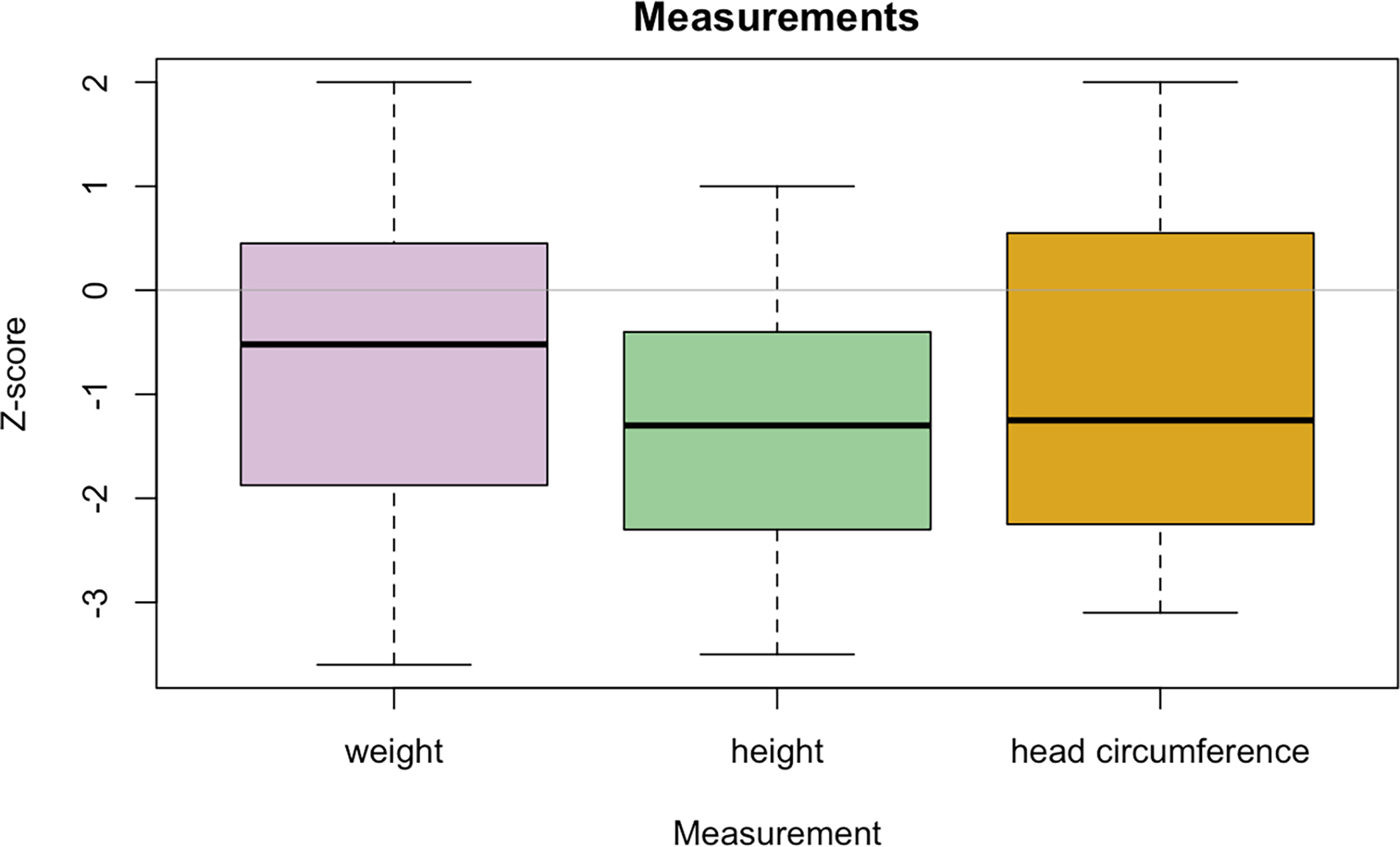
Box and whisker plot depicting the Z-score for anthropomorphic measurements at last examination. The line within the box represents the median, with the box boundaries representing the interquartile range and whiskers represent range. Data on height were available for 33 participants, weight for 35, and head circumference for 20.
Development
Developmental milestones are described in Supplemental Table 2. Motor skills were generally delayed, with median age at independent walking of 25 months (IQR 22–28.5 months). Gait was described as abnormal in several cases, with four described as walking with a wide-based gait and some experiencing frequent falls. Most individuals were able to run, some ride a bicycle, and one played ice hockey. Mild hypotonia appeared to improve with age but was often documented in older children as well. Language skills were also delayed, with a wide spectrum of ages at first words: from 12 months to 15 years, with a median of 20 months (IQR 16 – 33 months). Two individuals were non-verbal and one lost language at 16 years of age.
Intellectual skills varied; those who had intelligence quotient (IQ) testing (8/34, 24%) had median IQ of 70 (IQR 58.25 – 72.75). One person had severe intellectual disability. Eight of 29 individuals (28%) were noted to have features of autism spectrum disorder (with possible autism in one additional child), 17/29 (58%) had been diagnosed with attention deficit hyperactivity disorder (ADHD) with possible ADHD in two. Eighteen of 28 (64%) have a formal diagnosis of anxiety and 21/31 individuals (68%) are described as hypersocial. In at least five cases, there was clinical suspicion for Williams Syndrome due to the similar personality, echolalia, perseveration and overlapping physical exam findings such as nail hypoplasia. Other behavioral issues include aggression (8/22, 36%) and issues with impulsivity, with several children described as having “no sense of danger”. Hypersexuality was a concern in some older patients. Of the affected parents or grandparents within the familial cases in our cohort, these adults had some limited work experience and at least one had completed high school in regular classes. Two of the affected women ran their households, were able to drive, shop, handle money and had other unaffected children. At least in the families reported here, fertility appears to have been normal.
Physical examination
A common facial gestalt was noted, particularly the broad mouth with a thin and tented upper lip though these features were less noticeable after school age (Figure 3A–D). The hands were noted to be small due to short fingers (palm size normal) in 22/34 (65%) and feet were also noted to be small in 22/35 (63%) (Figure 3E). Few actual measurements were available. Nails were often hypoplastic (15/34, 44%). Common facial features include a broad forehead (27/37, 73%), thin upper lip (33/37, 89%), broad mouth (35/37, 95%), and downturned corners of the mouth (26/37, 70%) (Supplemental Table 1). Other occasional facial features seen included a smooth philtrum (3, 8%), midface hypoplasia (2, 5%), upturned nose (10, 27%) small/widely-spaced teeth (4, 11%), hypertelorism (1), low posterior hairline and bushy eyebrows (1), everted/thick lower lip (1), deep set eyes (1), metopic prominence (2, 5%), notched and hypoplastic alae nasi (1), underfolded pinnae with hypoplastic superior crus of the antihelix (1), high anterior hairline, high arched palate (2, 5%), broad nasal tip (1), midline nasal groove (1), small ears (2, 5%), and dental crowding (1). Face2Gene® (FDNA, Inc) research analysis also supports a specific craniofacial phenotype (Figure 4).
Figure 3.



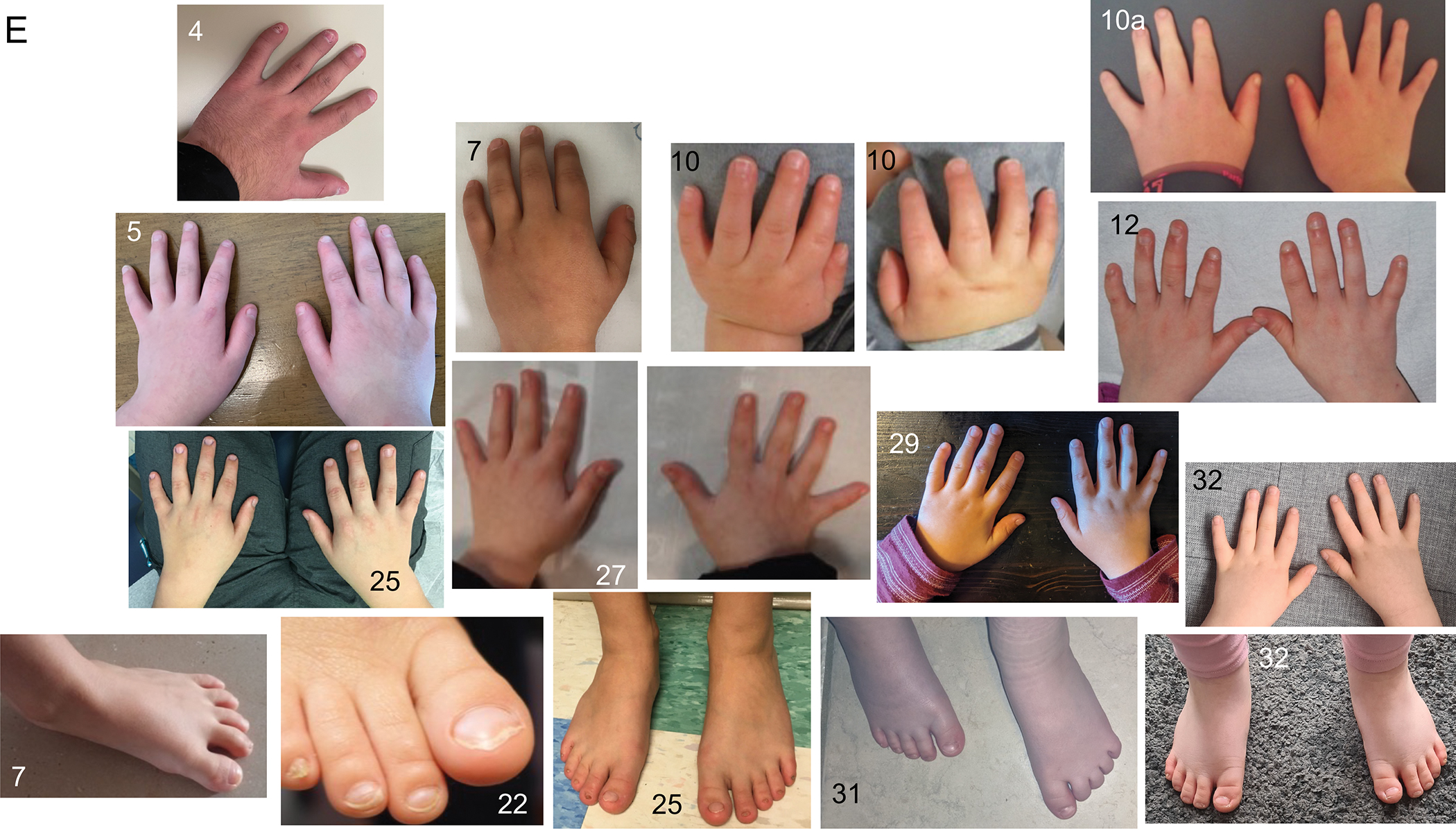
(A) In infancy and early childhood, the common facial gestalt is more easily recognizable, particularly the broad forehead and wide mouth with thin upper lip. (B) School age. (C) Adulthood. (D) Familial cases. (E) Hands and feet of affected individuals, illustrating the small size and hypoplastic nails in some.
Figure 4.

Face2Gene facial composite from a cohort of children with Jansen-deVries Syndrome (Cohort 1) compared to controls.
Brain imaging
Brain imaging via MRI was performed in 17 individuals, with seven having a normal brain MRI and the remainder having a variety of abnormalities, including parenchymal gliosis in 2/17 (12%), and the following abnormalities each seen once: small pituitary, mega cisterna magna and mild ventriculomegaly, hippocampal rotation, shortening of the corpus callosum and inferior vermian hypoplasia, “underdeveloped” cerebellum, Chiari I malformation, corpus callosum hypoplasia, and arachnoid cyst.
Gastrointestinal complaints
Gastroesophageal reflux disease (GERD) was noted in 16 individuals with two undergoing fundoplication and three requiring gastrostomy tube feeding for weeks to months. Six patients were noted to have issues with gagging or choking on liquids and 20 had unexplained vomiting episodes with six being diagnosed with cyclic vomiting syndrome (CVS). The vomiting episodes were often associated with hypoglycemia leading to frequent emergency department visits and/or hospital admission. Extensive evaluation for metabolic/mitochondrial dysfunction in at least four of these children has been negative. Constipation was reported in 22 individuals, though this may be more prevalent than reported, and constipation tended to improve with age.
Other health concerns
Well-controlled seizures of inconsistent types developed in two children and one had a single febrile seizure. Four individuals had recurrent migraine with vomiting, often developing after years of recurrent vomiting episodes. Four were diagnosed with growth hormone deficiency, though most were not tested. Fourteen were noted to have recurrent otitis media (including both individuals with high-arched palates) and another three had recurrent infections. Immunologic evaluations, when done, were non-diagnostic. Visual problems were noted in 17/23 individuals (74%), including hypermetropia (4/23, 17%), myopia (4/23, 17%) and strabismus (7/23, 30%). Other major anomalies were rare – one person had Tetralogy of Fallot but no other cardiac anomalies were reported in this cohort.
More recently, concerns for a possible neuroinflammatory complication of this condition have emerged, with three people having unexplained developmental regression. One individual was diagnosed with Pediatric Autoimmune Neuropsychiatric Disorder Associated with Streptococcal infections (PANDAS) at age six12, one experienced loss of language at age 16, and one had recurrent episodes of inability to walk and regression of other skills at age 6–7. A unifying etiology for these abnormalities has not been uncovered. As somatic PPM1D variants have been implicated in various malignancies13, 14, the potential for increased cancer risk in people with Jansen-de Vries syndrome has also been raised. At this time there have been no malignancies noted in this cohort, although we are aware of one child with neuroblastoma who has a pathogenic variant in PPM1D (S. Jansen, personal communication).
Literature Review
In order to describe the frequency of commonly-noted manifestations of this condition we reviewed our 37 cases, the 10 additional individuals reported in the literature9 and the original 14 cases described in 20171 for a total of 61 individuals (Figure 5). Full phenotypic details were not available for all cases, particularly regarding neurodevelopmental outcomes, and certain cases were identified in the context of a broader phenotype-driven cohort such as autism and developmental delays 15 or specific birth defects (bladder exstrophy)5.
Figure 5.
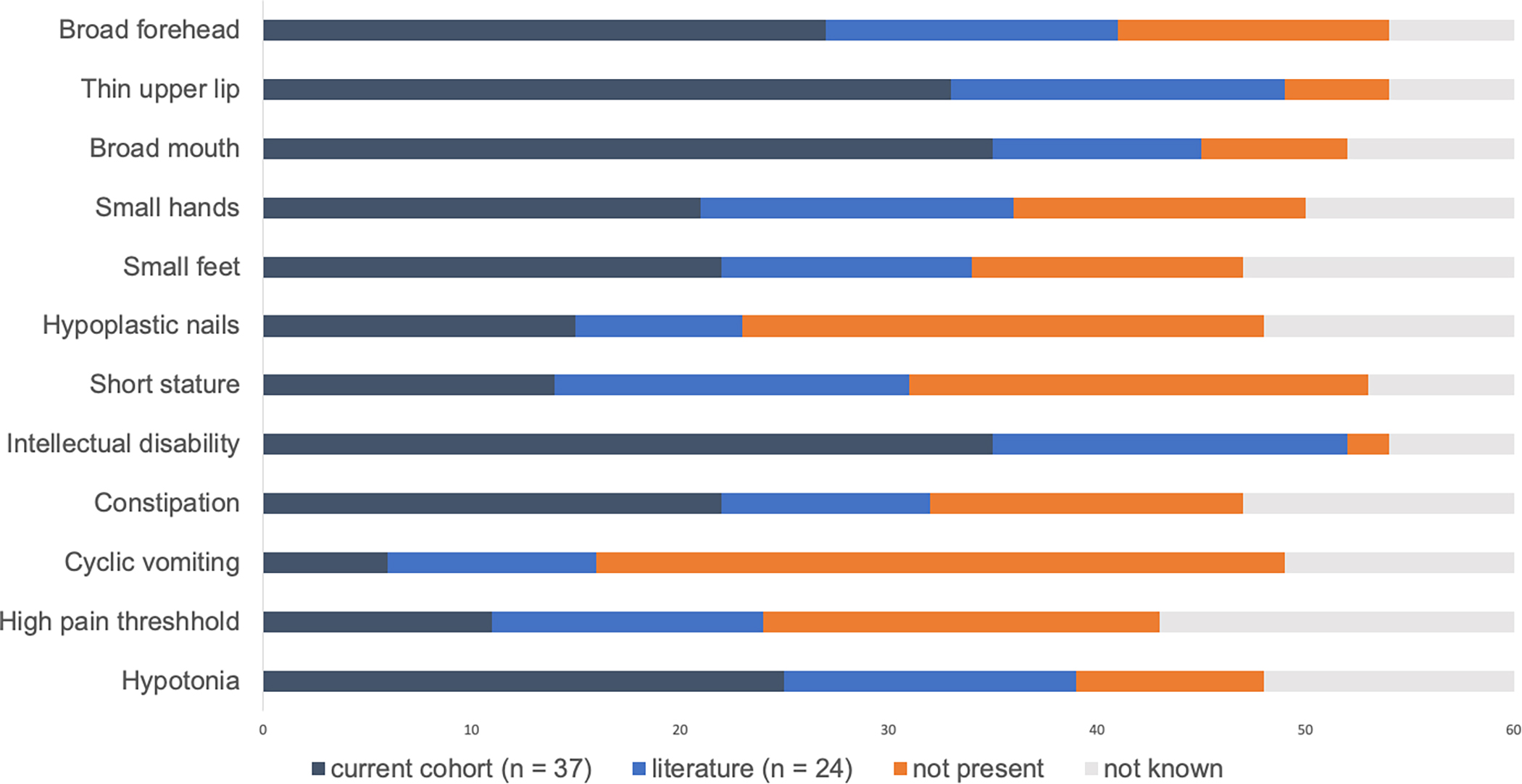
Frequency of salient features within our cohort and published literature cases. Dark blue bars represent the number cases within our cohort, light blue bars represent cases published in the literature, orange bars represent the number without the feature, and grey bars indicate the number for whom the feature is not reported.
Discussion:
We describe a cohort of 37 individuals from 34 families with disease-causing variants in PPM1D and review the existing literature in order to further elaborate on distinctive and recurrent features of Jansen-de Vries syndrome and refine the phenotype and genotypic spectrum. These results highlight the unique neurodevelopmental phenotype of this condition, a particular facial gestalt, and common medical comorbidities, with all affected individuals harboring heterozygous truncating variants in either the last or penultimate exon of PPM1D. The documentation of vertical transmission of Jansen de Vries syndrome in four families is consistent with autosomal dominant inheritance. As in other neurodevelopmental and chromosomal microdeletion or duplication syndromes, the ascertained probands were in general significantly more severely affected than their affected parent.
Regarding the neurodevelopmental phenotype of Jansen-deVries syndrome, a distinct personality has been described including hyper-social behavior and hyperacusis in addition to anxiety, somewhat similar to that described in Williams-Beuren syndrome 16 - though many individuals in our cohort may be too young to clearly detect either hyper-sociality or anxiety. Global developmental delay was nearly universal, though speech delay was more prominent than motor delay. All individuals did ultimately ambulate independently with a broad range of motor skills achieved, and most were able to communicate verbally, with a range of cognitive functioning described in those who have had formal testing. Some features, such as the aggression and hypersexuality, appear to emerge in some adolescents and older patients 10. Additionally, many individuals in our cohort have been diagnosed with ADHD, and about 25% with autism-like features, as was also seen in a large neurodevelopmental cohort where one of eight described with damaging variants in PPM1D had autism, while the others were diagnosed with developmental delay or intellectual disability15.
The physical features as described in our cohort support a unique facial gestalt for Jansen-de Vries syndrome, and, indeed, a computational facial recognition model was able to discern a distinct pattern for individuals with pathogenic variants PPM1D compared to controls with intellectual disability 17. Short stature was seen in 42% of our cohort and 74% of literature cases, and small hands or feet have also been seen frequently along with hypoplastic nails. As many other features seen in Jansen-deVries syndrome such as hypotonia and feeding difficulties (particularly in infancy) are also commonly seen in other genetic neurodevelopmental disorders, additional features unique to Jansen-deVries syndrome that have been seen in multiple families include the gastrointestinal issues, particularly CVS. While CVS was only formally diagnosed in six individuals in our cohort and another 10 in the literature, this is a relatively rare and distinct condition and it is therefore unusual to see so many cases within this group; vomiting not formally described as cyclic was reported in another thirteen individuals in our cohort. While also rare in our cohort, the developmental regression possibly suggestive of neuroinflammatory dysregulation and seen in three individuals in this cohort certainly bears further study.
In terms of the management following a diagnosis of Jansen-deVries syndrome, typically identified via molecular genetic testing and supported by the above clinical features, commonly-addressed medical issues include: anxiety, growth and short stature, vomiting, and behavioral issues such as aggression. No firm management guidelines can be universally endorsed as most reports of successes and failures are anecdotal and rely on parent reports. However, we offer the below guidance based upon our clinical experience:
Anxiety: This symptom seems almost universal in well-described patients and is early in onset, often by 18 months of age. It seems to intensify in school age and persists into adulthood. Helpful strategies include adherence to routine, calming techniques, learned coping strategies, meditation and psychotherapy. Medications including selective serotonin reuptake inhibitors (SSRIs) have been used with variable success and many families have explored using cannabidiol products with some suggestions of improvement. Further studies are indicated.
Growth: Short stature is frequent in Jansen-deVries syndrome and many parents have asked about growth hormone (GH) treatment. Given the positive experiences with GH in Prader-Willi syndrome18, parents have extrapolated and wondered about improving hypotonia and development with early GH. Four children in our cohort have been found to be growth hormone deficient and placed on treatment, although in at least one child no improvement in growth has been noted. No demonstrable improvement in cognition or development have been documented in the treated children. At this point there is no evidence for empiric treatment with GH in the absence of documented GH deficiency.
Vomiting: Prolonged, unexpected episodes of vomiting with nausea, dehydration and hypoglycemia have been seen in a number of children and adults with Jansen-de Vries syndrome – both those with and without an official diagnosis of CVS. The etiology appears obscure, as is often the case with CVS, even in individuals without an underlying genetic disorder. Triggers have included intercurrent illness, excitement, sleep deprivation, changes in schedule, and travel. Extensive evaluations for mitochondrial or genetic metabolic abnormalities in several people with Jansen-deVries syndrome have been unrevealing. There are no universally recommended treatments although patients and physicians have used ondansetron, cyproheptadine, amitriptyline, and Coenzyme Q with variable outcomes. Early recognition and treatment with medication and, if needed, intravenous fluids are recommended for acute episodes. The CVS support group may be of assistance to families (cvsaonline.org). Results of further studies are anticipated 19.
Behavior: Most children with Jansen-de Vries Syndrome are outgoing, delightful and happy children who are affectionate and much enjoyed by their families and care providers. Autistic features, when present, were usually mild and these diagnoses were based frequently on the child’s lack of boundaries, and unusual social interactions with lack of the normal response to social cueing. Behavioral abnormalities ranging from meltdowns and tantrums in younger children to aggression and anger in older individuals with Jansen-deVries syndrome are common. As in other syndromes and typical children, the treatment is supportive and must be individualized. Strategies for managing hypersexuality in some older patients may be challenging. These issues are quite similar to those seen in Williams syndrome where the outgoing personality, lack of boundaries and lack of fear or appreciation of consequences contribute to risk. Approaches such as long-term protection against pregnancy and monitoring the environment along with supportive counseling are essential. For many families affected by Jansen-deVries syndrome, enrollment in the support group, Facebook group and WhatsApp chat have proven valuable. (https://jansen-devries.org)
The molecular mechanisms giving rise to this constellation of findings remain to be fully elucidated. In a cohort of over 10,000 people with neurodevelopmental phenotypes (ID, DD, autism), enrichment for de novo variants in PPM1D was observed 15. PPM1D is noted to be missense constrained but not loss-of-function constrained in the gnomAD database, which fits with the finding that the mechanism of disease in for Jansen-deVries syndrome entails escape from nonsense-mediated decay 20. PPM1D is known to be a negative regulator of p53 and other tumor suppressors, and somatic variants are found in multiple malignancies14, 21. There are several individuals with stop gain variants designated as ClinVar pathogenic/likely pathogenic in gnomAD, though at least one is present in an older individual; these may represent somatic variants acquired in the setting of clonal hematopoiesis of indeterminate potential, as has been reported with this gene, particularly in association with myelodysplastic syndromes 22, 23 and has been previously seen in other highly-penetrant, autosomal dominant conditions 24. To our knowledge, there are no reports of individuals with Jansen-de Vries syndrome and concurrent malignancies.
Overall, our findings support a distinct phenotype associated with for Jansen-de Vries syndrome, with hallmark features including the facial gestalt, small hands and feet often with hypoplastic nails, the distinctive personality pattern, short stature, cyclic vomiting, moderate to severe anxiety and a high pain threshold. Disease-causing variants in our cohort fell exclusively within exons 5 and 6 of PPM1D as has been previously described. As ES is now recommended as a first-line test for those with intellectual disability25, it is likely that additional novel variants in other exons of this gene will be identified, and the clinical features seen in our cohort differentiated from the features seen with variants in other exons. To date the features seen in those with pathogenic variants in other exons seem generally dissimilar to those seen with variants in exons 5 and 6 and should not be confused with Jansen-deVries Syndrome12. Further research into the manifestations of damaging germline variants in exons 1–4 of PPM1D is warranted. Other areas in need of future research related to this condition include tailored treatments of common manifestations such as anxiety, short stature, and cyclic vomiting, and further investigation into the incidence and etiology of developmental regression and possible neuroinflammatory features.
Supplementary Material
Acknowledgments:
The authors would like to thank the devoted parents who spent hours in interviews and in the collection of medical records, as well as the dedicated founders of the Jansen-deVries support group.
Funding:
H.M.L. is supported by the NIH/NIMH (R21 MH131740) and the NIH/NICHD (P30 HD071593) to the Albert Einstein College of Medicine’s Rose F. Kennedy Intellectual and Developmental Disabilities Research Center. M.H.W. is supported by NIH/NICHD K23 HD102589. S.S. is supported by NIH/NINDS K23 NS119666. The content is solely the responsibility of the authors and does not necessarily represent the official view of the National Institutes of Health. B.B.A.d.V. is supported by the Dutch Organization for Health Research and Development: ZON-MW grant 912-12-109. B.C. is a senior clinical investigator of the Research Foundation- Flanders.
Footnotes
Conflict of interest disclosures: The authors have no relevant conflicts of interest to report.
Ethics approval and patient consent statement: Three participants were enrolled in the Manton Center for Orphan Disease Research’s IRB-approved protocol at Boston Children’s Hospital. For all other cases, informed consent to participate, including publication of photographs where relevant, was obtained at the local institution via their own institutional guidelines. De-identified data for all patients was collected and analyzed under an IRB protocol at Boston Children’s Hospital.
Data availability statement:
No new sequencing data were generated for the purpose of this study. Data supporting the results reported in this manuscript are provided in the supplemental tables. Additional de-identified data may be available upon request and contingent upon a data use agreement. Please contact the corresponding author to initiate such a request.
References
- 1.Jansen S, Geuer S, Pfundt R et al. De Novo Truncating Mutations in the Last and Penultimate Exons of PPM1D Cause an Intellectual Disability Syndrome. Am J Hum Genet. 2017;100(4):650–8. doi: 10.1016/j.ajhg.2017.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lelieveld SH, Reijnders MR, Pfundt R et al. Meta-analysis of 2,104 trios provides support for 10 new genes for intellectual disability. Nat Neurosci. 2016;19(9):1194–6. doi: 10.1038/nn.4352. [DOI] [PubMed] [Google Scholar]
- 3.Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature. 2017;542(7642):433–8. doi: 10.1038/nature21062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li Z, Du C, Zhang C et al. Novel truncating variant of PPM1D penultimate exon in a Chinese patient with Jansen-de Vries syndrome. Mol Genet Genomic Med. 2020;8(3):e1120. doi: 10.1002/mgg3.1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pitsava G, Feldkamp ML, Pankratz N et al. Exome sequencing of child-parent trios with bladder exstrophy: Findings in 26 children. Am J Med Genet A. 2021;185(10):3028–41. doi: 10.1002/ajmg.a.62439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou X, Wang Y, Shao B et al. Molecular diagnostic in fetuses with isolated congenital anomalies of the kidney and urinary tract by whole-exome sequencing. J Clin Lab Anal. 2020;34(11):e23480. doi: 10.1002/jcla.23480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuroda Y, Murakami H, Yokoi T et al. Two unrelated girls with intellectual disability associated with a truncating mutation in the PPM1D penultimate exon. Brain Dev. 2019;41(6):538–41. doi: 10.1016/j.braindev.2019.02.007. [DOI] [PubMed] [Google Scholar]
- 8.Porrmann J, Rump A, Hackmann K et al. Novel truncating PPM1D mutation in a patient with intellectual disability. Eur J Med Genet. 2019;62(1):70–2. 10.1016/j.ejmg.2018.05.006. [DOI] [PubMed] [Google Scholar]
- 9.Tsai MM, Lee NC, Chien YH, Hwu WL, Tung YC. Short stature leads to a diagnosis of Jansen-de Vries syndrome in two unrelated Taiwanese girls: A case report and literature review. J Formos Med Assoc. 2022;121(4):856–60. doi: 10.1016/j.jfma.2021.12.022. [DOI] [PubMed] [Google Scholar]
- 10.Martin Fernandez-Mayoralas D, Fernandez-Perrone AL, Jimenez de Domingo A, Alba Menendez A, Fernandez-Jaen A. [Jansen-de Vries syndrome. First case diagnosed in Spain]. Neurologia (Engl Ed). 2021;36(4):330–2. doi: 10.1016/j.nrl.2020.06.006. [DOI] [PubMed] [Google Scholar]
- 11.Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Human mutation. 2015;36(10):928–30. doi: 10.1002/humu.22844 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trifiletti R, Lachman HM, Manusama O et al. Identification of ultra-rare genetic variants in pediatric acute onset neuropsychiatric syndrome (PANS) by exome and whole genome sequencing. Sci Rep. 2022;12(1):11106. doi: 10.1038/s41598-022-15279-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Machiela MJ, Myers TA, Lyons CJ et al. Detectible mosaic truncating PPM1D mutations, age and breast cancer risk. J Hum Genet. 2019;64(6):545–50. doi: 10.1038/s10038-019-0589-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khadka P, Reitman ZJ, Lu S et al. PPM1D mutations are oncogenic drivers of de novo diffuse midline glioma formation. Nat Commun. 2022;13(1):604. doi: 10.1038/s41467-022-28198-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coe BP, Stessman HAF, Sulovari A et al. Neurodevelopmental disease genes implicated by de novo mutation and copy number variation morbidity. Nat Genet. 2019;51(1):106–16. doi: 10.1038/s41588-018-0288-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martens MA, Wilson SJ, Reutens DC. Research Review: Williams syndrome: a critical review of the cognitive, behavioral, and neuroanatomical phenotype. J Child Psychol Psychiatry. 2008;49(6):576–608. doi: 10.1111/j.1469-7610.2008.01887.x. [DOI] [PubMed] [Google Scholar]
- 17.van der Donk R, Jansen S, Schuurs-Hoeijmakers JHM et al. Next-generation phenotyping using computer vision algorithms in rare genomic neurodevelopmental disorders. Genet Med. 2019;21(8):1719–25. doi: 10.1038/s41436-018-0404-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Drabik M, Lewinski A, Stawerska R. Management of Prader-Labhart-Willi syndrome in children and in adults, with particular emphasis on the treatment with recombinant human growth hormone. Pediatr Endocrinol Diabetes Metab. 2022;28(1):64–74. doi: 10.5114/pedm.2022.112861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pizzol AmAdams KA, Curry C, Calvo PL. Is cyclic vomiting syndrome part of Jansen deVries syndrome? Abstract presented at: European Society for Pediatric Gastroenterology, Hepatology, and Nutrition 54th Annual Meeting; June 22–25, 2022. [Google Scholar]
- 20.Coban-Akdemir Z, White JJ, Song X et al. Identifying Genes Whose Mutant Transcripts Cause Dominant Disease Traits by Potential Gain-of-Function Alleles. Am J Hum Genet. 2018;103(2):171–87. doi: 10.1016/j.ajhg.2018.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Graf JF, Mikicic I, Ping X. Substrate spectrum of PPM1D in the cellular response to DNA double-strand breaks. iScience. 2022;25(9):104892. doi: 10.1016/j.isci.2022.104892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Panagiota V, Meggendorfer M, Kubasch A et al. Impact of PPM1D mutations in patients with myelodysplastic syndrome and deletion of chromosome 5q. Am J Hematol. 2021;96(6):E207–E10. doi: 10.1002/ajh.26162. [DOI] [PubMed] [Google Scholar]
- 23.Zink F, Stacey SN, Norddahl GL et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood. 2017;130(6):742–52. doi: 10.1182/blood-2017-02-769869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carlston CM, O’Donnell-Luria AH, Underhill HR et al. Pathogenic ASXL1 somatic variants in reference databases complicate germline variant interpretation for Bohring-Opitz Syndrome. Hum Mutat. 2017;38(5):517–23. doi: 10.1002/humu.23203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Manickam K, McClain MR, Demmer LA et al. Exome and genome sequencing for pediatric patients with congenital anomalies or intellectual disability: an evidence-based clinical guideline of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2021;23(11):2029–37. doi: 10.1038/s41436-021-01242-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No new sequencing data were generated for the purpose of this study. Data supporting the results reported in this manuscript are provided in the supplemental tables. Additional de-identified data may be available upon request and contingent upon a data use agreement. Please contact the corresponding author to initiate such a request.