

Abstract
Atrial fibrillation (AF) is the most common sustained cardiac arrhythmia and a major cause of stroke and morbidity. The strongest genetic risk factors for AF in humans are variants on chromosome 4q25, near the paired-like homeobox transcription factor 2 gene PITX2. Although mice deficient in Pitx2 (Pitx2+/−) have increased AF susceptibility, the mechanism remains controversial. Recent evidence has implicated hyperactivation of the cardiac ryanodine receptor (RyR2) in Pitx2 deficiency, which may be associated with AF susceptibility. We investigated pacing-induced AF susceptibility and spontaneous Ca2+ release events in Pitx2 haploinsufficient (+/−) mice and isolated atrial myocytes to test the hypothesis that hyperactivity of RyR2 increases susceptibility to AF, which can be prevented by a potent and selective RyR2 channel inhibitor, ent-verticilide. Compared with littermate wild-type Pitx2+/+, the frequency of Ca2+ sparks and spontaneous Ca2+ release events increased in permeabilized and intact atrial myocytes from Pitx2+/− mice. Atrial burst pacing consistently increased the incidence and duration of AF in Pitx2+/− mice. The RyR2 inhibitor ent-verticilide significantly reduced the frequency of spontaneous Ca2+ release in intact atrial myocytes and attenuated AF susceptibility with reduced AF incidence and duration. Our data demonstrate that RyR2 hyperactivity enhances SR Ca2+ leak and AF inducibility in Pitx2+/− mice via abnormal Ca2+ handling. Therapeutic targeting of hyperactive RyR2 in AF using ent-verticilide may be a viable mechanism-based approach to treat atrial arrhythmias caused by Pitx2 deficiency.
Keywords: Pitx2, cardiac ryanodine receptor (RyR2), calcium, atrial fibrillation, ent-verticilide
Graphical abstract
1. Introduction
Atrial fibrillation (AF) is the most common cardiac arrhythmia in the general population, with increasing incidence and prevalence worldwide, and associated with increased risk of death, stroke, heart failure, and dementia [1]. Although knowledge of the pathophysiology of AF has expanded greatly during the last decade, effective antiarrhythmic drug therapy to prevent AF remains elusive, likely because the mechanisms leading to AF are multifactorial: electrophysiological, molecular, and structural alterations [2–6].
Although still somewhat controversial, one cellular mechanism proposed to lead to AF initiation and maintenance is abnormal intracellular calcium (Ca2+) handling caused by cardiac ryanodine receptor (RyR2) hyperactivity, which is thought to cause rapid focal ectopic activity. RyR2 channels release Ca2+ from the sarcoplasmic reticulum (SR) to coordinate cardiac excitation-contraction coupling. However, dysfunctional RyR2-mediated diastolic SR Ca2+ release in atrial myocytes is associated with substantially increased AF risk in catecholaminergic polymorphic ventricular tachycardia (CPVT) animal models [7,8]. Additionally, RyR2-mediated triggered activity that contribute to the pathophysiology of AF can occur through enhanced RyR2 Ca2+ sensitivity by phosphorylation or oxidation [9–12].
Large-scale genome-wide association studies (GWAS) have identified chromosome 4q25 variants near the paired-like homeodomain transcription factor 2 (PITX2) gene as the strongest genetic risk factor for AF in humans [13,14]. Pitx2 is an atrial-selective transcription factor (Pitx2c isoform) that helps establish the left-right asymmetry of the atria during fetal development and is also an upstream transcription regulator of atrial electric function [15]. In mice, heterozygous deletion of Pitx2 (Pitx2+/−) causes increased AF susceptibility when challenged by programmed stimulation [16,17]. Pitx2 haploinsufficiency causes a developmental patterning defect associated with expanded pacemaker gene expression, altered ion channel expression, Ca2+ handling, and cell-cell coupling, which have all been associated with AF risk [18–20]. A recent study showed that RyR2 is upregulated and hyperphosphorylated in mutant Pitx2 human and mouse hearts, which suggests that excessive diastolic Ca2+ release and the resulting spontaneous electrical activity in atrial cardiomyocytes could be the cause of AF susceptibility in vivo [21]. Mice with heterozygous deletion of the Pitx2 gene are viable without obvious fibrosis or structural defects [17]. Hence, Pitx2+/− mice provide a good model to study the AF biology associated with Pitx2 loss of function, and test therapeutic efficacy of drugs in a genetically-defined form of AF.
Here, we investigated the AF susceptibility and Ca2+ handling in Pitx2+/− mice and isolated atrial myocytes to test the hypothesis that hyperactivity of RyR2 causes spontaneous Ca2+ release in atrial myocytes and increase susceptibility to AF by promoting DADs and triggered activity. We further hypothesized that Ca2+-triggered AF in Pitx2+/− mice can be prevented by a mechanism-based approach of suppressing spontaneous Ca2+ release with ent-verticilide, which is a potent and selective RyR2 Ca2+ release channel inhibitor [22].
2. Materials and Methods
2.1. Animal use and isolation of atrial myocytes
The use of animals was approved by the Animal Care and Use Committee of Vanderbilt University Medical Center (animal protocol No. M1900057–00) and performed in accordance with National Institutes of Health guidelines. Male and female paired-like homeodomain 2 transcription factor deficient (Pitx2+/−) and their littermate wild type (WT, Pitx2+/+) mice at the age between 16 and 30 weeks old were used for all experiments. To measure AF susceptibility, 16–19 week old mice underwent a transesophageal atrial pacing protocol. Mice were euthanized ~2–3 weeks after the in vivo measurements for myocyte isolation and tissue procurement. Atrial myocytes from 20- to 30-week-old Pitx2+/− or littermate WT mice were isolated from the heart by collagenase and protease digestion as previously described [7]. Cells were then washed twice by gravity sedimentation for 20 minutes at room temperature in standard Tyrode solution with 0.2 mM CaCl2. The final suspension contained 0.6 mM Ca2+ and the cells were immediately used for intact intracellular Ca2+ measurements and Ca2+ sparks.
2.2. Ca2+ spark measurements in permeabilized atrial myocytes
Ca2+ spark measurements from permeabilized atrial cardiomyocytes were carried at as previously reported [23]. Briefly, cells were plated on laminin-coated glass slides and allowed to attach for >10 minutes. Cells were then washed once and permeabilized for one minute in an internal solution containing saponin, to selectively permeabilize the outer membrane. The internal solution contained (in mM) L-aspartic acid potassium salt (120), KCl (15), potassium phosphate (5), HEPES (10), MgCl2 (0.6), EGTA (0.5), dextran (4%), and CaCl2 to give a free Ca2+ concentration of 54 nM. Free Ca2+ was calculated using MaxChelator (https://somapp.ucdmc.ucdavis.edu/pharmacology/bers/maxchelator/). After cell permeabilization, cells were incubated in internal solution containing Fluo-4 to detect Ca2+ sparks. After 10 minutes, Sparks were recorded using an Olympus inverted confocal microscope equipped with a Yokogawa spinning disk with 50 μm pinhole and 40X silicone objective (1.25 NA). Excitation was achieved via a 100-mW solid state diode laser (488 nm) and the emission was collected using a Hamamatsu CMOS camera. Spark detection analysis was performed using the SparkMaster plugin for ImageJ [24] and statistical analysis was carried out in R using the hierarchical clustering script provided by Sikkel et al [25]. Data were log2-transformed prior to analysis (as necessary for parametric tests) and reported as normalized to the WT genotype for that day.
2.3. Intracellular Ca2+ measurements in intact atrial myocytes
Atrial myocytes were pre-incubated for 2 hours with DMSO or ent-verticilide (0.5 μM). Myocytes were then loaded with Fura-2 acetoxymethyl ester (Fura-2, AM; Invitrogen) as described previously [22]. Briefly, isolated single atrial myocytes were incubated with 2 μM Fura-2, AM for 7 minutes, washed 2 × 10 minutes with normal Tyrode (NT) solution containing 250 μM probenecid. The composition of NT used for Fura-2 loading and washing was (in mM): 134 NaCl, 5.4 KCl, 1.2 CaCl2, 1 MgCl2, 10 Glucose, and 10 HEPES (pH adjusted to 7.4 with NaOH). After Fura-2 loading, experiments were conducted in NT solution containing 1 μM isoproterenol and 2 mM CaCl2 (all solutions retained DMSO or ent-verticilide). Atrial myocytes were electrically paced at 3 Hz field stimulation for 20 seconds, followed by no electrical stimulation for 40 seconds for quantification of spontaneous Ca2+ release events. Myocytes were then immediately exposed to 10 mM caffeine in NT solution for 5 seconds to estimate total SR Ca2+ content. Intracellular Ca2+ transients were recorded using a dual-beam excitation fluorescence photometry setup (IonOptix Corp.). All experiments were conducted at room temperature. 3 Hz paced Ca2+ transients were analyzed for Ca2+ parameters using IonWizard7 data analysis software (IonOptix Corp. Milton, MA).
2.4. Isolation of sarcoplasmic reticulum vesicles
Cardiac SR vesicles were isolated from left-ventricular tissue of fresh porcine hearts as previously described [26]. SR vesicles were flash-frozen and stored at −80 °C.
2.5. Ca-ATPase activity assay
SERCA2a activity was measured in porcine cardiac SR vesicles using an enzyme-linked NADH-coupled ATPase assay. To each well of a 96-well microplate, we applied DMSO or test compound (ent-verticilide, thapsigargin), cardiac SR to 10 μg/mL, and assay mix containing 50 mM MOPS (pH 7.0), 100 mM KCl, 1 mM EGTA, 0.2 mM NADH, 1 mM phosphoenolpyruvate, 10 IU/mL of pyruvate kinase, 10 IU/mL of lactate dehydrogenase, and 7 μM of the Ca2+ ionophore A23187 (Sigma). CaCl2 was added to result in free Ca2+ of the indicated concentrations. After 20 min of compound contact at room temperature, the assay was started upon addition of MgATP for a final concentration of 5 mM, and absorbance was read at 340 nm in a SpectraMax Plus384 microplate spectrophotometer (Molecular Devices, Sunnyvale, CA).
2.6. Transesophageal electrical pacing with surface electrocardiogram (ECG)
Mice were anesthetized with inhaled isoflurane (3% for induction, 1.5–2% for maintenance) while breathing spontaneously and placed in the supine position on a heating pad. Surface electrocardiograms (ECG) were obtained by subcutaneous placement of 27-gauge needles in each limb, and recorded continuously using amplifiers (AD Instruments) and LabChart 8 software. An octopolar 2F electrode catheter (CIB’ER MOUSE™; NuMED, Inc) was placed through the mouth in the esophagus, and situated properly for atrial capturing using a programmable stimulator, followed by measurement of the atrial diastolic capture threshold. Bipolar atrial pacing was performed using a stimulus amplitude twice threshold with a pulse width of 2 ms, as described previously [27]. To test for susceptibility of AF, burst atrial pacing was performed at six different cycle lengths (50, 40, 30, 25, 20, and 15 ms) for 15 seconds, each as described previously [7]. Between all pacing trains, there was a recovery time of 30 seconds. AF episodes were defined as rapid atrial activity and irregular ventricular rate lasting more than 1 second. The duration of AF was measured from the end of the pacing train to the first P wave with regular atrioventricular conduction after termination of AF. The number of induced episodes and total duration of AF were calculated for each animal. If an AF episode lasted for 10 min, the procedure was terminated. DMSO (vehicle group) or ent-verticilide (10 mg/kg, intraperitoneal injection) was administered to mice 15 minutes prior to the study. Isoproterenol (1.5 mg/kg, intraperitoneal (i.p.) injection) was administered before the burst atrial pacing. At the end of the procedure, the esophageal catheter and subcutaneous needles were removed, and isoflurane discontinued. The animal was placed in a cage with food and water and observed until recovery.
2.7. Statistical analysis
Statistical analyses were performed using Prism v6.0.1 (GraphPad Software, Inc.). The specific statistical tests used are reported in the figure and table legends. For all data, mean and standard deviation are provided. Results were considered statistically significant if the probability (p) value was less than 0.05 as the threshold to reject the null hypothesis. Ca-ATPase data were fit in Prism using the equation: .
3. Results
3.1. Hyperactive RyR2-mediated Ca2+ release in permeabilized atrial myocytes from Pitx2+/− mice
Ca2+ sparks are elementary sarcoplasmic reticulum (SR) Ca2+ release events generated by spontaneous opening of intracellular RyR2 Ca2+ release channels [28]. Hence, to test the hypothesis that hyperactivity of RyR2 causes spontaneous Ca2+ release in atrial myocytes isolated from the Pitx2 deficient mouse model, we first examined RyR2 activity by measuring Ca2+ sparks in permeabilized atrial myocytes with saponin.
Compared to wild-type Pitx2+/+ littermates, atrial myocytes from Pitx2+/− mice exhibited a significant increase in Ca2+ spark frequency (+/+ veh 1.0, 95% CI 0.96–1.04 vs +/− veh 1.15, 95% CI 1.1–1.2; Fig. 1A) and spark-mediated SR Ca2+ leak (+/+ veh 1.0, 95% CI 0.97–1.03 vs +/− veh 1.09, 95% CI 1.05–1.13; Fig. 1D). In addition, the amount of Ca2+ released during each Ca2+ spark (measured as spark amplitude and spark mass) was also significantly increased in the Pitx2+/− group (amplitude: +/+ veh 1.0, 95% CI 0.98-1.02 vs +/− veh 1.07, 95% CI 1.02–1.11, mass: +/+ veh 1.0, 95% CI 0.97–1.03 vs +/− veh 1.08, 95% CI 1.02-1.13; Fig. 1B & C). We next investigated the effect of the RyR2 selective inhibitor, ent-verticilide [22]. Application of 3 μM ent-verticilide in Pitx2+/− myocytes significantly reduced the frequency of Ca2+ sparks, spark amplitude, and spark mass (Fig. 1A – C), without depletion of SR Ca2+ stores (Fig. 1E). Consequently, spark-mediated SR Ca2+ leak was drastically reduced by ent-verticilide (Fig. 1D), indicating that Pitx2 deficiency leads to increased RyR2 activity, which is reduced by selective RyR2 inhibition. To determine whether ent-verticilide inhibits Ca2+ sparks via effects on the cardiac SR Ca2+ ATPase (SERCA2a), we next measured the Ca2+-dependent ATP hydrolysis in SR microsomes from porcine hearts, in the presence of ent-verticilide at concentrations ranging 0.03 – 30 μM. Thapsigargin was used as a positive control for inhibition of SERCA2a. Within the tested [ent-verticilide] range, we observed no effect on the Ca2+-ATPase activity (Figure 2A). In concurrent control measurements, thapsigargin induced the typical complete inhibition, with low-nanomolar IC50 (Figure 2B).
Figure 1. Ca2+ spark properties in permeabilized atrial cardiomyocytes.
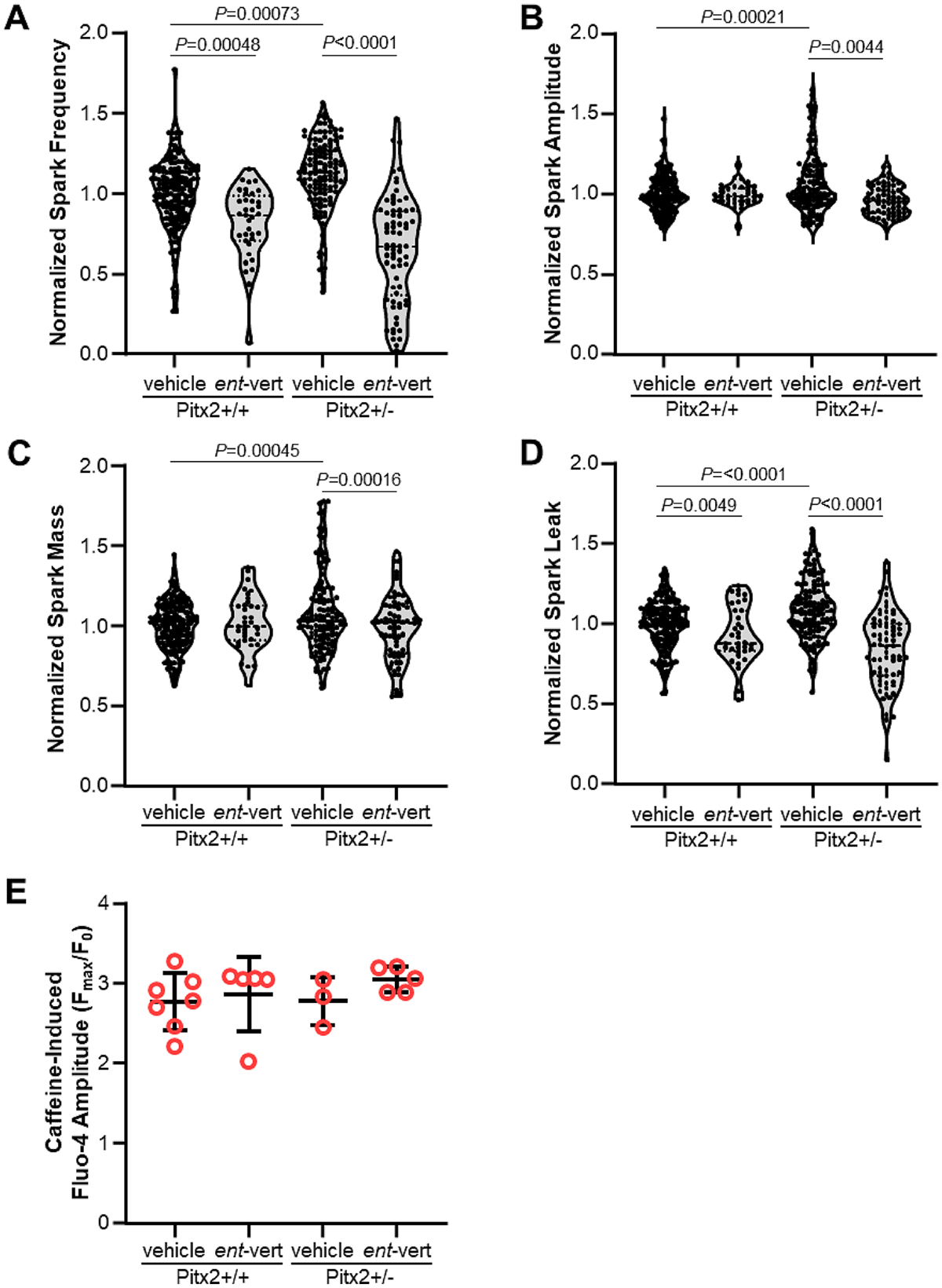
(A) Ca2+ spark frequency, (B) amplitude, (C) mass, and (D) leak. Figure panels shown with full range of values and a box bounding the 25th and 75th percentiles with median. N: Pitx2+/+ (vehicle) = 168 cells from 6 mice; Pitx2+/+ (ent-vert) = 36 cells from 2 mice; Pitx2+/− (vehicle) = 127 cells from 6 mice; Pitx2+/− (ent-vert) = 70 cells from 4 mice normalized to values from +/+ mice by day of experiment. Bonferroni-adjusted P values calculated using hierarchical clustering. (E) SR load measured by caffeine-induced Ca2+ transient amplitude. Individual values with mean ± SD.
Figure 2. ent-Verticilide did not inhibit cardiac sarco(endo)plasmic reticulum Ca2+ ATPase (SERCA2a) function.
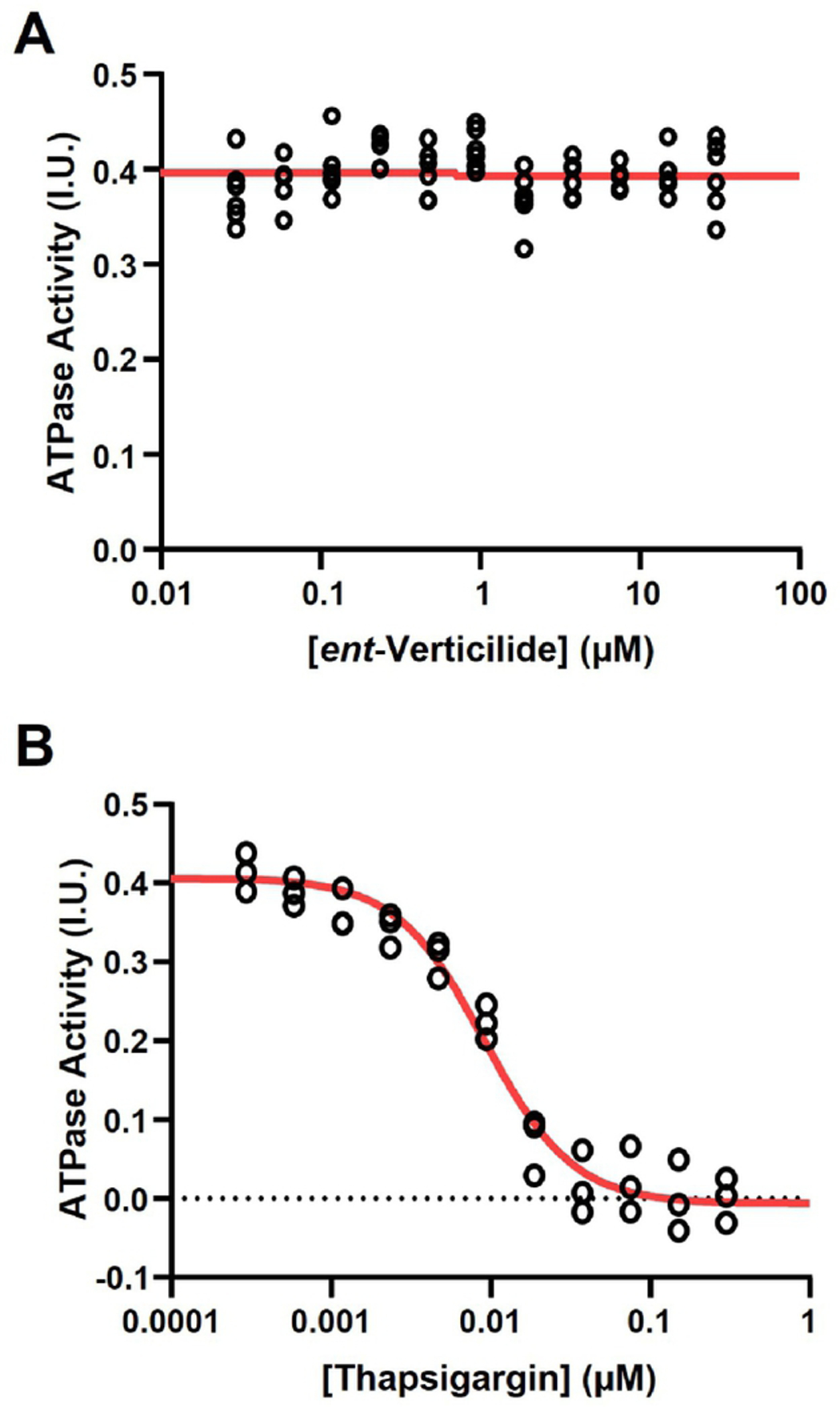
(A) SERCA2a ATPase activity (calculated as VpCa5.4-VpCa8) in the presence of ent-verticilide at the indicated concentrations. N = 6 replicates per concentration tested. (B) Positive control: SERCA ATPase activity in the presence of thapsigargin (SERCA2a inhibitor) at the indicated concentrations. N = 3 replicates at each concentration tested. Red lines show the result of fitting the equation: .
3.2. RyR2 inhibition with ent-verticilide suppresses spontaneous SR Ca2+ release in intact atrial myocytes
We next investigated intracellular Ca2+ handling in intact atrial myocytes. Spontaneous Ca2+ release (SCR) events are the cellular mechanism responsible for delayed afterdepolarizations that can trigger premature beats and evoke atrial ectopy and arrhythmogenesis [7]. Intracellular Ca2+ was monitored in Fura-2, AM-loaded intact atrial myocytes treated with the beta-adrenergic agonist isoproterenol to simulate catecholaminergic stress. After a rapid pacing train with 3 Hz pacing, we quantified spontaneous diastolic Ca2+ release events in isolated myocytes from Pitx2+/− and littermate wild-type (+/+) mice (Fig 3A). Consistent with the increased rate of RyR2-mediated Ca2+ sparks (Fig. 1), spontaneous Ca2+ release events were significantly increased in Pitx2+/− compared to Pitx2+/+ myocytes, consistent with abnormal Ca2+ handling caused by hyperactive RyR2 in Pitx2+/− mice. Inhibition of RyR2 with ent-verticilide (0.5 μM) had no effect on Ca2+ handling in wild-type Pitx2+/+ myocytes, whereas in Pitx2+/− cells, ent-verticilide significantly reduced the frequency of SCR events compared to vehicle (veh 6.97 ± 5.62 vs. ent-vert 2.36 ± 2.55, p<0.0001; Fig 3B). At the same time, ent-verticilide normalized the increased Ca2+ transient amplitude (-veh 0.79 ± 0.19 vs. ent-vert 0.67 ± 0.14, p=0.0090; Fig 3C) and increased fractional SR Ca2+ release (veh 0.87 ± 0.15 vs. ent-vert 0.78 ± 0.13, p=0.0293; Fig 3D) observed in Pitx2+/− to values similar to those observed in wild-type atrial myocytes. There were no statistically significant differences in other Ca2+ handling parameters such as diastolic Ca2+ levels in the cytoplasm, time to peak, decay rate of the Ca2+ transient (a measure of sarcoplasmic reticulum Ca2+-ATPase [29]), SR Ca2+ content or caffeine-induced Ca2+ decay kinetics (a measure of Na+-Ca2+ exchanger function [30]) (Table 1). Ca2+ transient alternans – a beat-to-beat change in the Ca2+ transient amplitude especially at fast pacing rates – has been associated with increased risk for arrhythmias and sudden cardiac death [31,32]. However, we did not observe Ca2+ alternans in any group during 3 Hz pacing, the fastest rate that allowed continuous capture in atrial myocytes. Taken together, Pitx2+/− atrial myocytes exhibit RyR2 hyperactivity resulting in increased spontaneous RyR2-mediated Ca2+ release, which can be prevented by RyR2 inhibition with ent-verticilide.
Figure 3. ent-Verticilide reduced spontaneous Ca2+ release (SCR) in Pitx2+/− atrial myocytes.
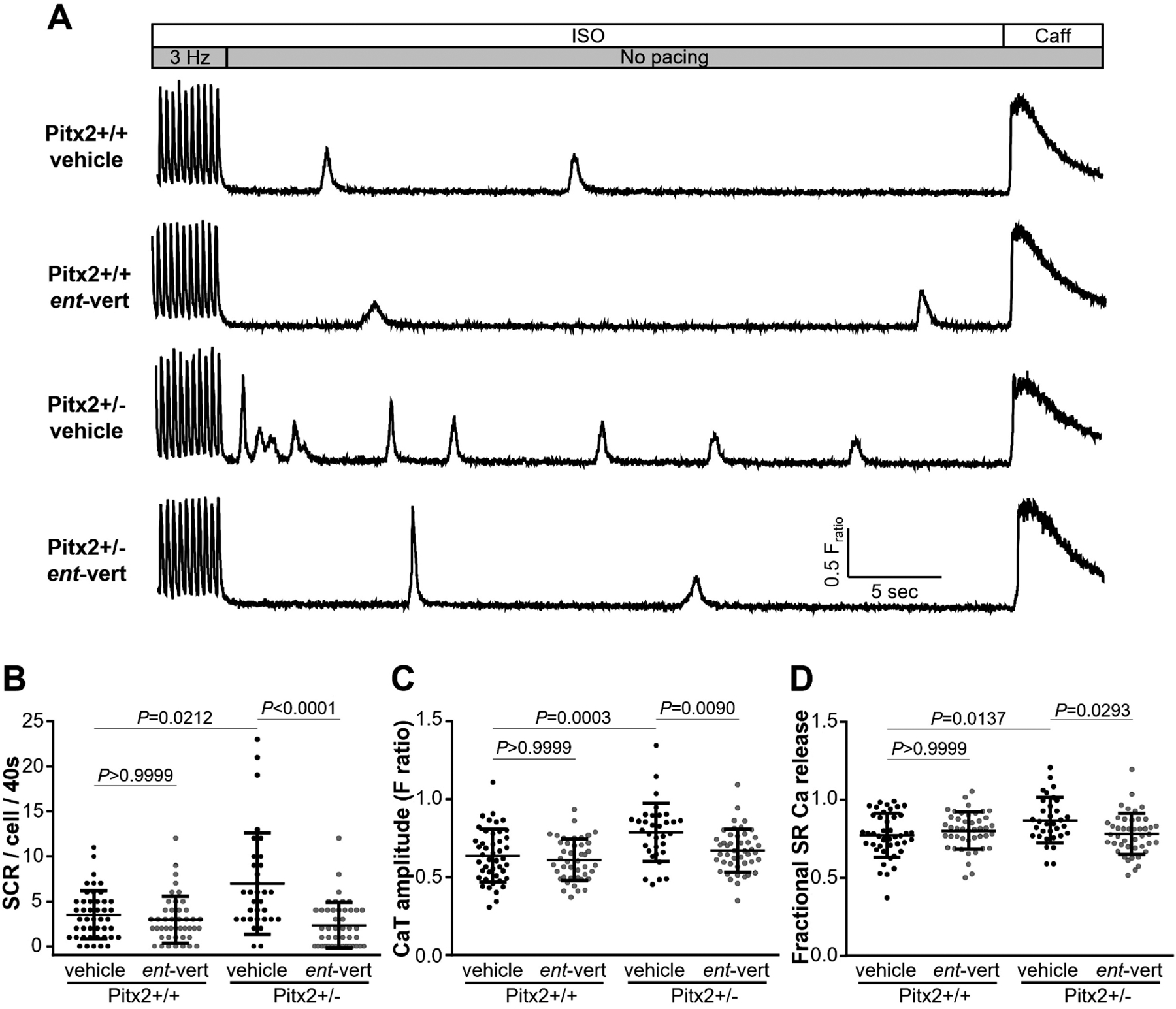
Isolated intact atrial myocytes were pre-treated with vehicle or ent-verticilide for 2.5 hours and loaded with Fura-2 acetoxymethyl to measure spontaneous Ca2+ release (SCR) events. (A) Representative Ca2+ transient from intact atrial myocytes. Atrial myocytes were field-stimulated at 3 Hz for 20 s followed by 40 s recording of SCR events in 1 μM isoproterenol (ISO) containing Tyrode solution. Application of 10 mM caffeine (Caff) was used to measure SR Ca2+ content. Summary data of SCR events (B), Ca2+ transient amplitude (C), and fractional SR Ca2+ release (D). Data are mean ± SD. n=45, 44, 33 and 45 cells from 4–6 mice in each group. P values calculated using ANOVA multiple comparisons with Dunn’s or Bonferroni’s test.
Table 1. Intracellular Ca2+ measurements in intact atrial myocytes.
Ca2+ handling parameters of Fura-2 acetoxymethyl-ester loaded atrial myocytes paced at 3 Hz followed by caffeine (10 mM) application. n=45 cells for +/+ vehicle, 44 cells for +/+ ent-verticilide, 33 cells for +/− vehicle, and 45 cells for +/− ent-verticilide. All data displayed as mean ± SD and analyzed using ANOVA multiple comparisons with Bonferroni-adjusted P values. CaT – pacing-induced Ca2+ transient, Caff.T – caffeine-induced Ca2+ transient
| (A) Pitx2+/+ vehicle (n=45) | (B) Pitx2+/+ ent-vert (n=44) | (C) Pitx2+/− vehicle (n=33) | (D) Pitx2+/− ent-vert (n=45) | P values | |||
|---|---|---|---|---|---|---|---|
| (A) vs (B) | (A) vs (C) | (C) vs (D) | |||||
| Diastolic Ca2+ (FRatio) | 0.60 ± 0.07 | 0.56 ± 0.06 | 0.60 ± 0.09 | 0.59 ± 0.05 | 0.10 | >0.99 | >0.99 |
| Time to peak (ms) | 30.79 ± 5.04 | 29.45 ± 3.07 | 29.17 ± 3.03 | 29.36 ± 4.12 | 0.78 | 0.54 | >0.99 |
| CaT decay rate constant (ms) | 114.2 ± 23.0 | 123.6 ± 24.6 | 114.1 ± 14.7 | 114.9 ± 18.8 | 0.24 | >0.99 | >0.99 |
| SR Ca2+ content (FRatio) | 0.83 ± 0.17 | 0.78 ± 0.16 | 0.91 ± 0.15 | 0.86 ± 0.09 | 0.89 | 0.10 | >0.99 |
| Caff.T decay rate constant (s) | 2.95 ± 0.27 | 2.85 ± 0.38 | 2.85 ± 0.32 | 2.99 ± 0.32 | >0.99 | >0.99 | 0.53 |
3.3. Adrenergic stimulation increases AF susceptibility in Pitx2 deficient mice
Next, we determined in vivo AF susceptibility of Pitx2+/− mice using a transesophageal (TE) atrial burst pacing protocol. The TE atrial stimulation protocol is widely used to initiate pacing-induced AF [7,33–35] and is a survival procedure that allows for repeated measurement in the same mouse. However, TE atrial pacing can cause vagal stimulation (evidenced by atrioventricular (AV) block), which causes AF induction even in normal mice. Hence, pacing episodes with AV block were excluded to increases the specificity of the TE burst pacing protocol [27]. Atrial burst pacing resulted in a significantly higher AF incidence (27.3 % vs 75.0 %, P=0.039, Fischer exact test, n=11 and 12 mice) and total AF burden (7.6 ± 17.2 sec vs 27.6 ± 33.2 sec, P=0.027, Mann-Whitney test) in Pitx2+/− mice compared with wild-type Pitx2+/+ littermates. Addition of a catecholamine challenge with isoproterenol (iso) before burst pacing stimulation increased AF burden further in Pitx2+/− mice, and sustained AF was also observed (persistent AF over 300 sec). Fig 4A shows representative ECG traces of pacing-induced AF. Atrial pacing-induced AF was observed in 96 % of Pitx2+/− mice (21 of 22 mice), whereas wild-type littermates had only a 50 % AF incidence (7 of 14 mice; Fig 4B). Moreover, sustained AF – defined as persistent AF lasting more than 300 seconds – was significantly increased in Pitx2+/− mice (+/+ 21.4 % vs. +/− 81.8 %, P=0.0005 Fischer Exact Test; Fig 4B). Total AF burden was also increased in Pitx2+/− compared to Pitx2+/+ mice (Fig 4C).
Figure 4. Pixt2+/− mice exhibited increased susceptibility to atrial fibrillation (AF) by transesophageal atrial burst pacing.
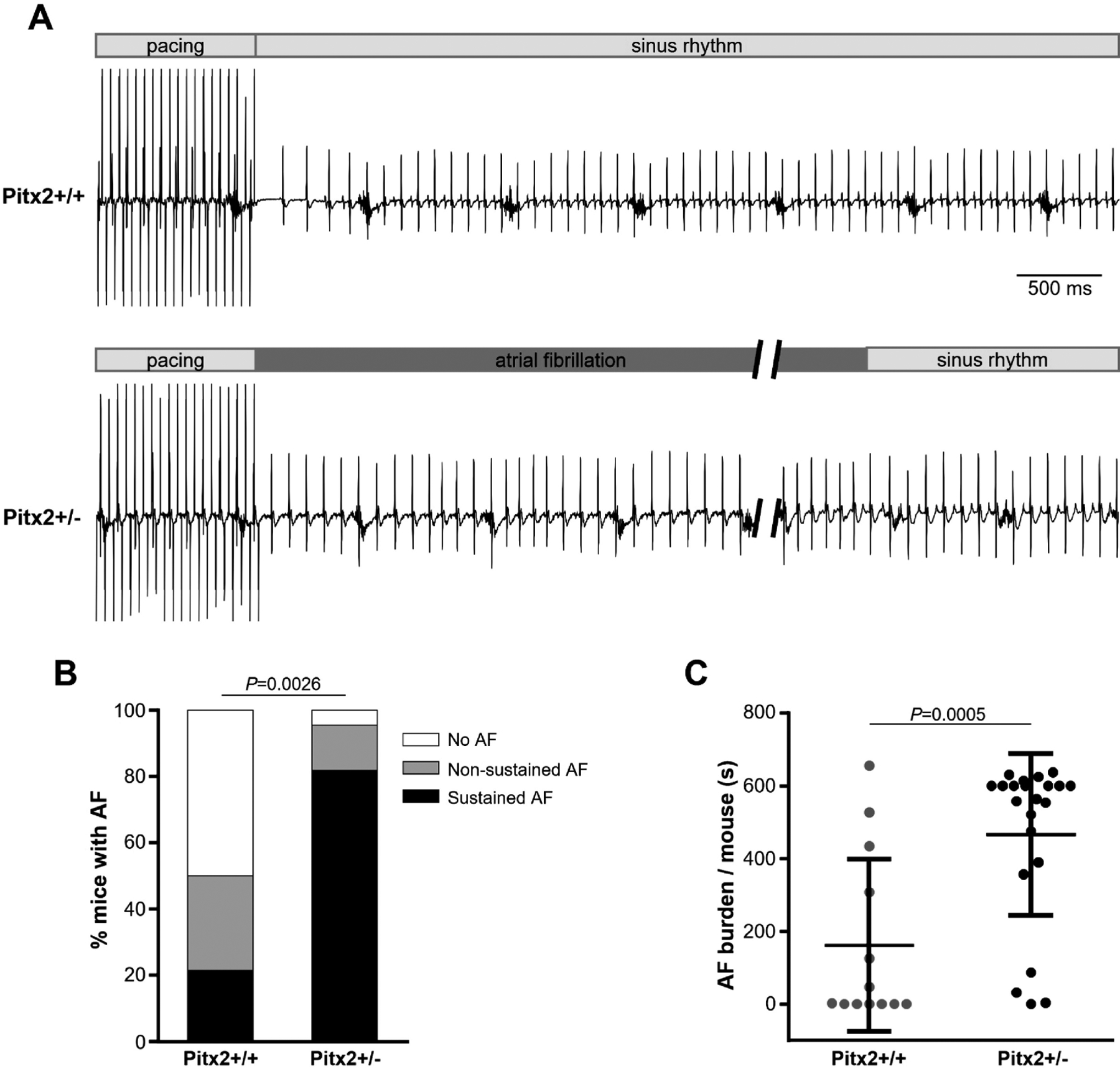
(A) Representative ECG traces of littermate WT (upper) and Pitx2+/− (lower) mice after an episode of burst pacing protocol in the presence of isoproterenol (1.5 mg/kg, i.p. injection). Pitx2+/− mice exhibit inducible AF followed by spontaneous conversion to sinus rhythm. (B) Incidence of inducible AF (+/+ 50.0 % vs. +/− 95.5 %; non-sustained AF + sustained AF). Sustained AF was defined as continuous AF over 300 sec induced by any single pacing train (+/+ 21.4 % vs. +/− 81.8 %, P=0.0005). P values were obtained using Fisher’s exact test. (C) Summary of total AF burden in WT (N=14 mice) and Pitx2 deficiency (N=22 mice). Data are mean ± SD. P values were obtained using the Mann-Whitney test.
On the other hand, ECG parameters such as heart rate, RR, PR, QRS and QT intervals were not statistically different between Pitx2+/− mice and wild-type littermates. TE pacing programmed stimulation was used to determine Wenckebach cycle length (WCL) and atrioventricular effective refractory period (AVERP) at a basic cycle length of 100 msec. There were no significant differences between Pitx2+/− and wild-type littermates (Table 2). Our data corroborate the previously reported AF susceptibility and ECG parameters for these mice [16].
Table 2. Baseline cardiac electrophysiological parameters in Pitx+/+ and Pitx2+/− mice.
ECG, electrocardiogram; RR, time interval between two consecutive R wave peaks; PR, interval from the onset of P wave to the start of QRS complex; QRS, time interval from start of Q wave to end of S wave; QT, interval from start of Q wave to end of T wave; AVERP, atrioventricular nodal effective refractory period; WCL, Wenckebach cycle length. All data displayed as mean ± SD and analyzed using unpaired t-test.
| ECG intervals (ms) | Pitx2+/+ (n=14 mice) | Pitx2+/− (n=22 mice) | P values |
|---|---|---|---|
| Heart rate | 564.2 ± 27.1 | 563.1 ± 33.4 | 0.914 |
| RR | 106.6 ± 5.3 | 106.9 ± 6.3 | 0.869 |
| PR | 35.6 ± 1.8 | 34.9 ± 2.1 | 0.293 |
| QRS | 10.4 ± 0.5 | 10.5 ± 0.8 | 0.755 |
| QT | 46.9 ± 1.9 | 47.8 ± 2.4 | 0.270 |
| AVERP | 44.6 ± 3.6 | 45.8 ± 3.7 | 0.324 |
| WCL | 71.7 ± 2.9 | 70.5 ± 3.3 | 0.290 |
3.4. ent-Verticilide prevents pacing-induced AF in Pitx2 deficient mice
To test the hypothesis that RyR2 hyperactivity is the underlying molecular mechanism responsible for pacing induced AF in Pitx2+/− mice, we used the RyR2 inhibitor ent-verticilide, which does not block membrane ion channels and has no effect on the cardiac action potential [22]. Both Pitx2+/+ and Pitx2+/− mice underwent a TE atrial burst pacing protocol after vehicle injection, followed by the same pacing protocol in the presence of ent-verticilide one week later. Intraperitoneal injection of ent-verticilide 15 min prior to the TE pacing significantly reduced AF inducibility from 100 % to 65 % in Pitx2+/− mice, whereas there was no significant effect in wild-type Pitx2+/+ (veh 54.6% vs. ent-vert 45.5%; Fig 5A, B). ent-Verticilide significantly reduced the incidence of sustained AF (veh 90 % vs. ent-vert 25 %, P<0.0001 Fischer Exact Test; Fig 5B) to values similar to wild-type Pitx2+/+ mice, but had no effect on sustained AF in wild-type Pitx2+/+ mice (veh 27.3% vs. ent-vert 27.3%; Fig 5A). ent-Verticilide also significantly reduced the total AF duration in Pitx2+/− mice (veh 508.0 ± 183.8 sec vs. ent-vert 169.1 ± 233.5 sec; Fig 5D), but had no significant effect in Pitx2+/+ mice (veh 148.0 ± 241.1 sec vs. ent-vert 154.7 ± 265.4 sec; Fig 5C). These data demonstrate that ent-verticilide treatment suppresses both incidence and total duration of AF in Pitx2+/− mice. Taken together, our results indicate that in Pitx2 deficient mice, RyR2 hyperactivity causes AF susceptibility via spontaneous Ca2+ release, which can be prevented in vivo by the selective RyR2 inhibitor, ent-verticilide.
Figure 5. Anti-arrhythmic efficacy of ent-verticilide on pacing-induced AF in both Pitx2+/+ and Pitx2+/− mice.
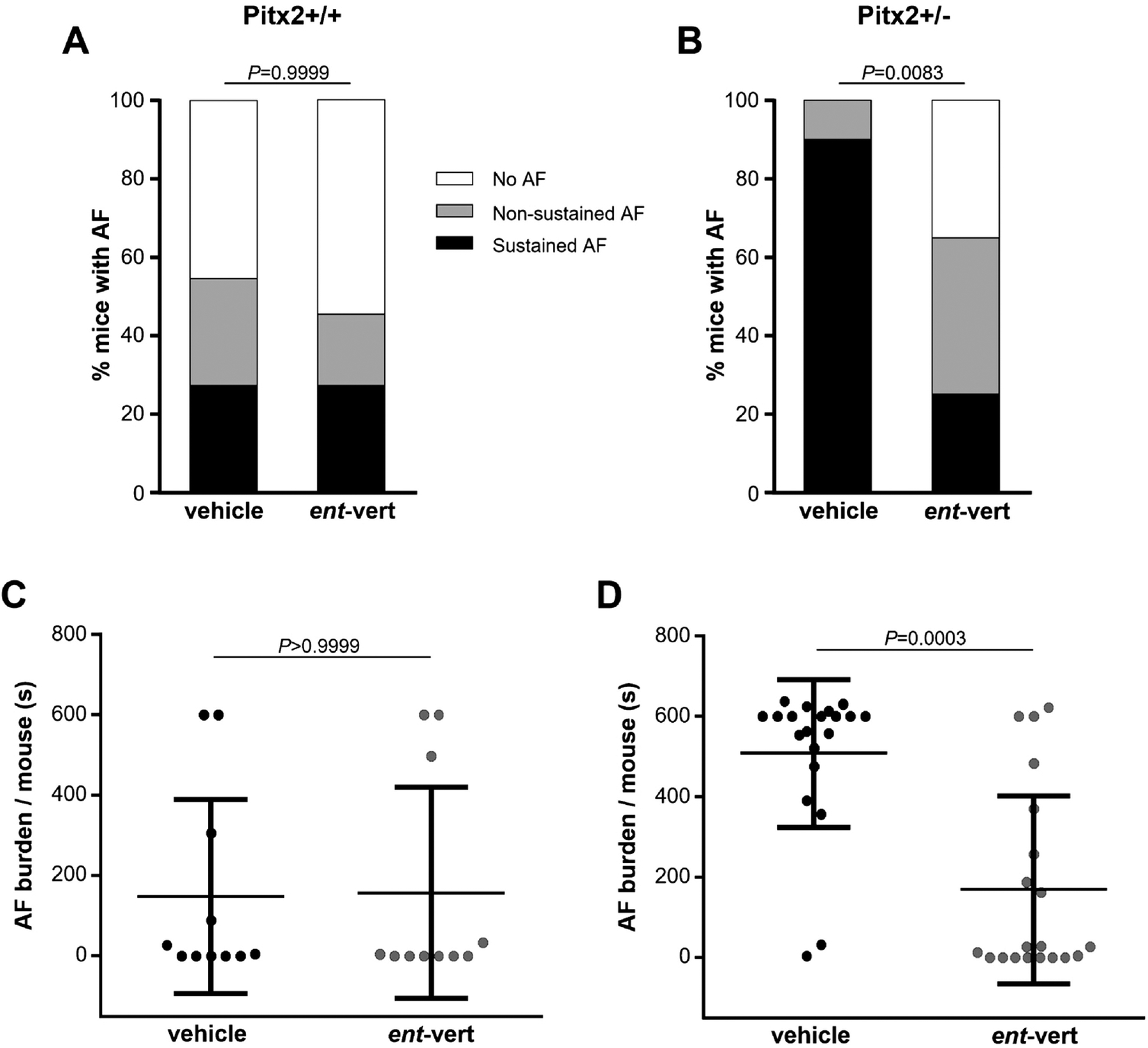
(A, B) Incidence of inducible AF (Pitx2+/+: vehicle 54.6 % vs. ent-vert 45.5 %, Pitx2+/−: vehicle 100.0 % vs. ent-vert 65.0 %; non-sustained AF + sustained AF) by burst pacing stimulation in Pitx2+/+ and Pitx2+/− mice. Sustained AF was defined as continuous AF over 300 sec induced by any single pacing train. ent-Verticilide significantly reduced sustained AF compared to vehicle group in Pitx2+/− mice (vehicle 90.0 % vs. ent-vert 25.0 %, P<0.0001). (C, D) Summary of total AF burden in Pitx2+/+ and Pitx2+/− mice (N=11 and 20 mice, respectively). Data are mean ± SD. P values were obtained using Fisher’s exact test and Wilcoxon matched-pairs signed rank test.
4. Discussion
In this study, we report that Pitx2 deficiency in mice causes RyR2 hyperactivity and spontaneous SR Ca2+ release in atrial cardiomyocytes, and AF susceptibility in vivo. Given the efficacy of a selective inhibitor of RyR2 both in vitro and in vivo, our data demonstrate mechanistic causality from enhanced Ca2+ wave frequency in single atrial myocytes to AF inducibility in mice. Moreover, our data provide proof-of-concept for therapeutic efficacy of RyR2 inhibition with ent-verticilide in Pitx2-linked AF risk.
4.1. Dysfunction of RyR2-mediated Ca2+ handling for atrial arrhythmogenesis in Pitx2 deficiency
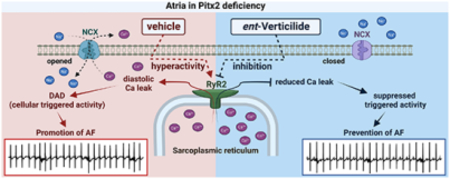
Human GWAS studies have identified sequence variation within 4q25 as conferring increased susceptibility to AF [13,14,36,37]. The Pitx2 gene is in this region and considered a prime candidate for the development of atrial arrhythmias. Consistent with the hypothesis that loss of function in Pitx2 causes AF risk, heterozygous deletion of Pitx2 in mice causes an increased tendency of atrial flutter and tachycardia, with reduced atrial action potential duration postulated as a mechanism of AF induction [16,17]. Interestingly, we find increased AF risk in Pitx2 mice without a reduction in action potential duration in vivo (as evidenced by the normal effective atrial refractory period, Table 2). As such, other mechanisms are probably important. It is known that Pitx2 plays a key role in left-right asymmetry and has regulatory effects on gene networks including microRNAs, and membrane effector genes [38]. Especially, Pitx2 modulates microRNAs and thereby regulates distinct ion channel, gap junction cell-cell communication and beta-adrenergic signaling, resulting in electrical remodeling and calcium abnormalities in humans with 4q25 variants and in mutant mouse models [20,39]. Moreover, Pitx2 has been linked to defective Ca2+ homeostasis such as regulating Ca2+-handling gene expression via Wnt signaling, increased SERCA2a expression, SR Ca2+ load, and RyR2 phosphorylation [18,21], which is pointing to a pivotal role of Pitx2 for AF associated with excessive spontaneous Ca2+ release-induced electrical activity. Therefore, we focused in our study on the role of RyR2 for AF inducibility in this mouse model. Our results show that the frequency of Ca2+ sparks and RyR2-mediated Ca2+ leak is significantly increased in isolated atrial myocytes from Pitx2+/− mice (Fig. 1), which is the first direct evidence of RyR2 hyperactivity in the Pitx2+/− mouse model. In addition, we find that the abnormal RyR2 activity increased diastolic Ca2+ release events in intact Pitx2+/− atrial myocytes (Fig. 3). Although we did not directly examine the molecular mechanism for RyR2 hyperactivity here, it is well established that diastolic SR Ca2+ leak can be mediated by enhanced RyR2 phosphorylation [21,40,41], increased RyR2 expression [42–44], or SR Ca2+ overload [44], which have all been documented in the Pitx2 mouse model [18,20], and which are key contributors to atrial arrhythmogenesis in humans with different forms of AF. Spontaneous SR Ca2+ release via RyR2 activates the electrogenic Na+-Ca2+ exchanger (NCX), resulting in a depolarizing transient inward current that is referred to as delayed afterdepolarization (DAD). DADs can trigger premature action potentials leading to proarrhythmic ectopic activity, and hence AF [45]. Generally, the amplitude of Ca2+ transient is dynamically regulated and depends on both the RyR2 open probability and the SR Ca2+ content. Our data show that the SR Ca2+ content was not changed, whereas the Ca2+ transient amplitude was increased (Fig. 3), indicating that the increased spontaneous Ca2+ release events are caused by increased RyR2 open probability rather than SR Ca2+ overload [46].
Our transesophageal burst pacing protocol shows a significant increase on AF inducibility in Pitx2 deficient mice. In an AF mouse model that has increased SR Ca2+ leak caused by RyR2 mutations, the prevalence of pacing-induced AF was not suppressed by beta-blocker, which was interpreted as evidence that catecholamines did not contribute importantly in triggering AF [8]. In contrast, our data show more frequent and persistent AF on catecholaminergic challenge with isoproterenol in the Pitx2+/− mouse model. This result supports the hypothesis that Pitx2 plays a more complex role beyond RyR2 regulation, which is consistent with Pitx2’s well-established regulatory roles in heart development, as well as in a highly complex gene regulatory network including beta-adrenergic signaling [17,18,39]. Nevertheless, our results demonstrate that targeting RyR2 hyperactivity is an effective approach for reducing AF susceptibility in Pitx2 deficiency.
4.2. Effect of RyR2 inhibition with ent-verticilide on AF susceptibility
RyR2 dysfunction due to its hyperactivity, mutations, and abnormal regulation by its binding partners, typically manifest as increased SR Ca2+ leak and spontaneous Ca2+ release events, which ultimately increase AF susceptibility [37,40–42]. This establishes a critical role of RyR2 in atrial arrhythmia risk in humans and mice. Consequently, stabilizing RyR2 and preventing diastolic Ca2+ leak could attenuate AF inducibility. Our experimental study here provides proof of concept for this approach in a genetic model of AF susceptibility due to Pitx2 haploinsufficiency.
AF is the most prevalent cardiac arrhythmia requiring antiarrhythmic drug therapy. Rhythm-control therapy has a beneficial effect when the treatment begins early. Current FDA-approved anti-arrhythmic drugs targeting RyR2 have short-term efficacy, but carry substantial risks when used long-term, particularly drug-induced pro-arrhythmia. For instance, an antiarrhythmic drug currently in clinical use, flecainide, effectively suppresses RyR2-mediated spontaneous Ca2+ release, but it also blocks a Na channel [47,48]. In addition, the dual RyR2 and Na channel inhibitor R-propafenone prevents AF induction through reduction of spontaneous Ca2+ elevation in mouse model compared to S-propafenone (no RyR2 inhibition) [7]. Unfortunately, when tested in humans, both R- and S-propafenone increase the rate of inducible atrial flutter due to Na channel blockade [49]. We previously reported that ent-verticilide is effective against ventricular arrhythmias triggered by RyR2-medicated Ca2+ release and is more potent as an inhibitor of RyR2 compared with the benchmark compounds dantrolene, tetracaine, and flecainide [22]. Hence, we here tested ent-verticilide for efficacy against arrhythmogenic spontaneous Ca2+ release and pacing-induced AF. Consistent with our previous study in ventricular cardiomyocytes, we observed the reduction of both spontaneous Ca2+ release events and Ca2+ transient amplitude in the presence of ent-verticilide in intact Pitx2+/− atrial myocytes, without changing the other Ca2+ parameters (Figs. 2, 3 and Table 1), demonstrating that ent-verticilide selectively inhibits RyR2 hyperactivity without altering SERCA2a function and SR Ca2+ content. ent-Verticilide inhibition of the spontaneous Ca2+ release in vitro translated into activity against pacing-induced AF in vivo (Fig 5), indicating that ent-verticilide effectively attenuates the susceptibility of atrial, as well as ventricular arrhythmia, by selective inhibition of RyR2-mediated Ca2+ release. Furthermore, since RyR2 inhibition with ent-verticilide saturates at 25% [22] and does not reduce skeletal muscle function, it has a large therapeutic window in vivo [50]. Our experimental data demonstrates that pathophysiological RyR2-mediated Ca2+ release triggers pacing-induced AF, and ent-verticilide prevents the initiation of AF by selective targeting the RyR2 channel in mouse model. However, research to support the ent-verticilide pharmacological relevance in AF patients is still needed.
In summary, the reduction in AF burden and incidence by the RyR2 specific inhibitor ent-verticilide supports the hypothesis that diastolic Ca2+ leak by resting RyR2 hyperactivity is responsible for AF susceptibility in the setting of Pitx2 deficiency. These results suggest a potential therapeutic role for using a selective RyR2 inhibitor such as ent-verticilide to prevent AF.
Highlights.
Pitx2 deficiency causes RyR2 hyperactivity and increased Ca2+ spark leak in permeabilized atrial myocytes and atrial fibrillation susceptible in vivo
Increased spontaneous Ca2+ release events in Pitx2 deficient atrial cardiomyocytes were prevented by RyR2 inhibition with ent-verticilide
Adrenergic stimulation aggravated atrial fibrillation susceptibility in Pitx2 deficient mice
RyR2 inhibition with ent-Verticilide effectively prevented pacing-induced atrial fibrillation
Acknowledgment
This research was supported in part by the National Institutes of Health National Heart, Lung, and Blood Institute [R35 HL144980 (to B.C.K.), R01 HL151223 (to J.N.J., B.C.K., R.L.C.), R01 HL139065 and HL138539 (to R.L.C); the PhRMA Foundation Postdoctoral Fellowship (to D.J.B.); the American Heart Association Arrhythmia and Sudden Death Strategically Focused Research Network grant [19SFRN34830019 (to B.C.K.)]; and the Leducq Foundation grant [18CVD05 (to B.C.K.)].
Non-standard Abbreviations and Acronyms
- RyR2
Cardiac ryanodine receptor
- Pitx2
Paired-like homeodomain transcription factor 2
- AF
Atrial fibrillation
- SR
Sarcoplasmic reticulum
- Ca2+
Calcium
- WT
Wild type
- CPVT
Catecholaminergic polymorphic ventricular tachycardia
- GWAS
Genome-wide association studies
- DAD
Delayed afterdepolarization
- NT
Normal Tyrode
- SERCA2a
Sarcoplasmic reticulum Ca2+-ATPase
- ECG
Electrocardiogram
- SCR
Spontaneous Ca2+ release
- TE
Transesophageal
- AV
Atrioventricular
- WCL
Wenckebach cycle length
- AVERP
Atrioventricular effective refractory period
- NCX
Sodium-Calcium exchanger
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
R.L.C. holds equity in and serves as executive officer for Photonic Pharma LLC. This relationship has been reviewed and managed by the University of Minnesota. Photonic Pharma had no role in this study. The other authors have no actual or perceived conflicts of interest with the contents of this article.
References
- [1].Freeman J v, Wang Y, Akar J, Desai N, Krumholz H. National trends in atrial fibrillation hospitalization, readmission, and mortality for Medicare beneficiaries, 1999–2013. Circulation. 2017;135(13):1227–39. [DOI] [PubMed] [Google Scholar]
- [2].Darbar D, Roden DM. Genetic mechanisms of atrial fibrillation: impact on response to treatment. Nat Rev Cardiol. 2013;10(6):317–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Kirchhof P, Bax J, Blomstrom-Lundquist C, Calkins H, Camm AJ, Cappato R, et al. Early and comprehensive management of atrial fibrillation: proceedings from the 2nd AFNET/EHRA consensus conference atrial fibrillation entitled ‘research perspectives in atrial fibrillation’. Europace. 2009;11(7):860–85. [DOI] [PubMed] [Google Scholar]
- [4].Benjamin EJ, Chen PS, Bild DE, Mascette AM, Albert CM, Alonso A, et al. Prevention of atrial fibrillation: report from a national heart, lung, and blood institute workshop. Circulation. 2009;119(4):606–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Wijffels MCEF, Kirchhof CJHJ, Dorland R, Allessie MA. Atrial fibrillation begets atrial fibrillation: a study in awake chronically instrumented goats. Circulation. 1995;92(7):1954–68. [DOI] [PubMed] [Google Scholar]
- [6].Wakili R, Voigt N, Kääb S, Dobrev D, Nattel S, others. Recent advances in the molecular pathophysiology of atrial fibrillation. J Clin Invest. 2011;121(8):2955–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Faggioni M, Savio-Galimberti E, Venkataraman R, Hwang HS, Kannankeril PJ, Darbar D, et al. Suppression of spontaneous ca elevations prevents atrial fibrillation in calsequestrin 2-null hearts. Circ Arrhythm Electrophysiol. 2014;7(2):313–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Shan J, Xie W, Betzenhauser M, Reiken S, Chen BX, Wronska A, et al. Calcium leak through ryanodine receptors leads to atrial fibrillation in 3 mouse models of catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2012;111(6):708–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Dobrev D, Wehrens XHT. Calcium-mediated cellular triggered activity in atrial fibrillation. J Physiol. 2017;595(12):4001–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Nattel S, Dobrev D. The multidimensional role of calcium in atrial fibrillation pathophysiology: mechanistic insights and therapeutic opportunities. Eur Heart J. 2012;33(15):1870–7. [DOI] [PubMed] [Google Scholar]
- [11].Delfiner MS, Nofi C, Li Y, Gerdes AM, Zhang Y. Failing hearts are more vulnerable to sympathetic, but not vagal stimulation--induced, atrial fibrillation-ameliorated with dantrolene treatment. J Card Fail. 2018;24(7):460–9. [DOI] [PubMed] [Google Scholar]
- [12].Chelu MG, Sarma S, Sood S, Wang S, van Oort RJ, Skapura DG, et al. Calmodulin kinase II--mediated sarcoplasmic reticulum Ca 2+ leak promotes atrial fibrillation in mice. J Clin Invest. 2009;119(7):1940–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gudbjartsson DF, Arnar DO, Helgadottir A, Gretarsdottir S, Holm H, Sigurdsson A, et al. Variants conferring risk of atrial fibrillation on chromosome 4q25. Nature. 2007;448(7151):353–7. [DOI] [PubMed] [Google Scholar]
- [14].Ellinor PT, Lunetta KL, Albert CM, Glazer NL, Ritchie MD, Smith A v, et al. Meta-analysis identifies six new susceptibility loci for atrial fibrillation. Nat Genet. 2012;44(6):670–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Franco D, Sedmera D, Lozano-Velasco E. Multiple roles of Pitx2 in cardiac development and disease. J Cardiovasc Dev Dis. 2017;4(4):16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wang J, Klysik E, Sood S, Johnson RL, Wehrens XHT, Martin JF. Pitx2 prevents susceptibility to atrial arrhythmias by inhibiting left-sided pacemaker specification. Proceedings of the National Academy of Sciences. 2010;107(21):9753–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kirchhof P, Kahr PC, Kaese S, Piccini I, Vokshi I, Scheld HH, et al. PITX2c is expressed in the adult left atrium, and reducing Pitx2c expression promotes atrial fibrillation inducibility and complex changes in gene expression. Circ Cardiovasc Genet. 2011;4(2):123–33. [DOI] [PubMed] [Google Scholar]
- [18].Lozano-Velasco E, Hernandez-Torres F, Daimi H, Serra SA, Herraiz A, Hove-Madsen L, et al. Pitx2 impairs calcium handling in a dose-dependent manner by modulating Wnt signalling. Cardiovasc Res. 2016;109(1):55–66. [DOI] [PubMed] [Google Scholar]
- [19].Tao G, Kahr PC, Morikawa Y, Zhang M, Rahmani M, Heallen TR, et al. Pitx2 promotes heart repair by activating the antioxidant response after cardiac injury. Nature. 2016;534(7605):119–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Tao Y, Zhang M, Li L, Bai Y, Zhou Y, Moon AM, et al. Pitx2, an atrial fibrillation predisposition gene, directly regulates ion transport and intercalated disc genes. Circ Cardiovasc Genet. 2014;7(1):23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Herraiz-Mart\’\inez A, Llach A, Tarifa C, Gand\’\ia J, Jiménez-Sabado V, Lozano-Velasco E, et al. The 4q25 variant rs13143308T links risk of atrial fibrillation to defective calcium homoeostasis. Cardiovasc Res. 2019;115(3):578–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Batiste SM, Blackwell DJ, Kim K, Kryshtal DO, Gomez-Hurtado N, Rebbeck RT, et al. Unnatural verticilide enantiomer inhibits type 2 ryanodine receptor-mediated calcium leak and is antiarrhythmic. Proceedings of the National Academy of Sciences. 2019;116(11):4810–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wleklinski MJ, Kryshtal DO, Kim K, Parikh SS, Blackwell DJ, Marty I, et al. Impaired Dynamic Sarcoplasmic Reticulum Ca Buffering in Autosomal Dominant CPVT2. Circ Res. 2022;131(8):673–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Picht E, Zima A v, Blatter LA, Bers DM. SparkMaster: automated calcium spark analysis with ImageJ. American Journal of Physiology-Cell Physiology. 2007;293(3):C1073--C1081. [DOI] [PubMed] [Google Scholar]
- [25].Sikkel MB, Francis DP, Howard J, Gordon F, Rowlands C, Peters NS, et al. Hierarchical statistical techniques are necessary to draw reliable conclusions from analysis of isolated cardiomyocyte studies. Cardiovasc Res. 2017;113(14):1743–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Gruber SJ, Cornea RL, Li J. Peterson KC, Schaaf TM, Gillispie GD, et al. Discovery of enzyme modulators via high-throughput time-resolved FRET in living cells. J Biomol Screen. 2014;19(2):215–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Murphy MB, Kim K, Kannankeril PJ, Subati T, van Amburg JC, Barnett J. v, Murray KT, Optimizing transesophageal atrial pacing in mice to detect atrial fibrillation, American Journal of Physiology-Heart and Circulatory Physiology. 322 (2022) H36--H43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Cheng H, Lederer WJ, Cannell MB, Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle, Science (1979). 262 (1993) 740–744. [DOI] [PubMed] [Google Scholar]
- [29].Diaz ME, Trafford AW, O’neill SC, Eisner DA, Measurement of sarcoplasmic reticulum Ca2+ content and sarcolemmal Ca2+ fluxes in isolated rat ventricular myocytes during spontaneous Ca2+ release, J Physiol. 501 (1997) 3–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Bers DM, Calcium fluxes involved in control of cardiac myocyte contraction, Circ Res. 87 (2000) 275–281. [DOI] [PubMed] [Google Scholar]
- [31].Edwards JN, Blatter LA. Cardiac alternans and intracellular calcium cycling. Clin Exp Pharmacol Physiol. 2014;41(7):524–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Short B Understanding Ca2+ alternans. J Gen Physiol. 2021;153(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Fukui A, Takahashi N, Nakada C, Masaki T, Kume O, Shinohara T, Teshima Y, Hara M, Saikawa T, Role of leptin signaling in the pathogenesis of angiotensin II--mediated atrial fibrosis and fibrillation, Circ Arrhythm Electrophysiol. 6 (2013) 402–409. [DOI] [PubMed] [Google Scholar]
- [34].Schrickel JW, Bielik H, Yang A, Schimpf R, Shlevkov N, Burkhardt D, Meyer R, Grohé C, Fink K, Tiemann K, others, Induction of atrial fibrillation in mice by rapid transesophageal atrial pacing, Basic Res Cardiol. 97 (2002) 452–460. [DOI] [PubMed] [Google Scholar]
- [35].Verheule S, Sato T, Everett IV T, Engle SK, Otten D, der Lohe M, Nakajima HO, Nakajima H, Field LJ, Olgin JE, Increased vulnerability to atrial fibrillation in transgenic mice with selective atrial fibrosis caused by overexpression of TGF-$β$1, Circ Res. 94 (2004) 1458–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Schott J-J, Charpentier F, Peltier S, Foley P, Drouin E, Bouhour J-B, Donnelly P, Vergnaud G, Bachner L, Moisan J-P, others, Mapping of a gene for long QT syndrome to chromosome 4q25–27, Am J Hum Genet. 57 (1995) 1114. [PMC free article] [PubMed] [Google Scholar]
- [37].Kääb S, Darbar D, van Noord C, Dupuis J, Pfeufer A, Newton-Cheh C, Schnabel R, Makino S, Sinner MF, Kannankeril PJ, others, Large scale replication and meta-analysis of variants on chromosome 4q25 associated with atrial fibrillation, Eur Heart J. 30 (2009) 813–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Hernandez-Torres F, Rodr\’\iguez-Outeiriño L, Franco D, Aranega AE, Pitx2 in embryonic and adult myogenesis, Front Cell Dev Biol. 5 (2017) 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Bai J, Lu Y, Zhu Y, Wang H, Yin D, Zhang H, Franco D, Zhao J, Understanding PITX2-Dependent Atrial Fibrillation Mechanisms through Computational Models, Int J Mol Sci. 22 (2021) 7681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Voigt N, Li N, Wang Q, Wang W, Trafford AW, Abu-Taha I, Sun Q, Wieland T, Ravens U, Nattel S, others, Enhanced sarcoplasmic reticulum Ca2+ leak and increased Na+-Ca2+ exchanger function underlie delayed afterdepolarizations in patients with chronic atrial fibrillation, Circulation. 125 (2012) 2059–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Heijman J, Muna AP, Veleva T, Molina CE, Sutanto H, Tekook M, Wang Q, Abu-Taha IH, Gorka M, Künzel S, others, Atrial myocyte NLRP3/CaMKII nexus forms a substrate for postoperative atrial fibrillation, Circ Res. 127 (2020) 1036–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Chiang DY, Kongchan N, Beavers DL, Alsina KM, Voigt N, Neilson JR, Jakob H, Martin JF, Dobrev D, Wehrens XHT, others, Loss of microRNA-106b-25 cluster promotes atrial fibrillation by enhancing ryanodine receptor type-2 expression and calcium release, Circ Arrhythm Electrophysiol. 7 (2014) 1214–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Beavers DL, Wang W, Ather S, Voigt N, Garbino A, Dixit SS, Landstrom AP, Li N, Wang Q, Olivotto I, others, Mutation E169K in junctophilin-2 causes atrial fibrillation due to impaired RyR2 stabilization, J Am Coll Cardiol. 62 (2013) 2010–2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Voigt N, Heijman J, Wang Q, Chiang DY, Li N, Karck M, Wehrens XHT, Nattel S, Dobrev D, Cellular and molecular mechanisms of atrial arrhythmogenesis in patients with paroxysmal atrial fibrillation, Circulation. 129 (2014) 145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Bers DM, Cardiac excitation--contraction coupling, Nature. 415 (2002) 198–205. [DOI] [PubMed] [Google Scholar]
- [46].Dobrev D, Wehrens XHT, Calmodulin kinase II, sarcoplasmic reticulum Ca2+ leak, and atrial fibrillation, Trends Cardiovasc Med. 20 (2010) 30–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Galimberti ES, Knollmann BC, Efficacy and potency of class I antiarrhythmic drugs for suppression of Ca2+ waves in permeabilized myocytes lacking calsequestrin, J Mol Cell Cardiol. 51 (2011) 760–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Watanabe H, Chopra N, Laver D, Hwang HS, Davies SS, Roach DE, Duff HJ, Roden DM, Wilde AAM, Knollmann BC, Flecainide prevents catecholaminergic polymorphic ventricular tachycardia in mice and humans, Nat Med. 15 (2009) 380–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Shoemaker MB, Yoneda ZT, Crawford DM, Akers WS, Richardson T, Montgomery JA, Phillips S, Shyr Y, Saavedra P, Estrada JC, others, A mechanistic clinical trial using (R)-versus (S)-propafenone to test RyR2 (ryanodine receptor) inhibition for the prevention of atrial fibrillation induction, Circ Arrhythm Electrophysiol. 15 (2022) e010713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Blackwell DJ, Smith AN. Do TQ, Gochman A, Schmeckpeper J, Hopkins CR, Akers WS, Johnston JN, Knollmann BC. In vivo pharmacokinetic and pharmacodynamic properties of the antiarrhythmic molecule ent-verticilide. J Pharmacol Exp Ther. (2023). Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]