

Abstract
Background:
The current treatment paradigm of imatinib-resistant metastatic gastrointestinal stromal tumor (GIST) does not incorporate KIT/PDGFRA genotypes in therapeutic drug sequencing, except for PDGFRA exon 18-mutant GIST that are indicated for avapritinib treatment. Here, ctDNA sequencing was used to analyze plasma samples prospectively collected in the phase III VOYAGER trial to understand how the KIT/PDGFRA mutational landscape contributes to tyrosine-kinase inhibitor (TKI) resistance and to determine its clinical validity and utility.
Patients and Methods:
VOYAGER (N=476) compared avapritinib with regorafenib in patients with KIT/PDGFRA-mutant GIST previously treated with imatinib and 1 or 2 additional TKIs (NCT03465722). KIT/PDGFRA ctDNA mutation profiling of plasma samples at baseline and end-of-treatment was assessed with 74-gene Guardant360® CDx. Molecular subgroups were determined and correlated with outcomes.
Results:
386/476 patients with KIT/PDGFRA-mutant tumors underwent baseline (pre-trial treatment) ctDNA analysis; 196 received avapritinib, and 190 received regorafenib. KIT and PDGFRA mutations were detected in 75.1% and 5.4%, respectively. KIT resistance mutations were found in the activation loop (A-loop; 80.4%) and ATP-binding pocket (ATP-BP; 40.8%); 23.4% had both. An average of 2.6 KIT mutations were detected per patient; 17.2% showed 414 different KIT resistance mutations. Of all pathogenic KIT variants, 28.0% were novel, including alterations in exons/codons previously unreported. PDGFRA mutations showed similar patterns. ctDNA-detected KIT ATP-BP mutations negatively prognosticated avapritinib activity, with a median progression-free survival (mPFS) of 1.9 versus 5.6 months for regorafenib. mPFS for regorafenib did not vary regardless of the presence or absence of ATP-BP/A-loop mutants and was greater than mPFS with avapritinib in this population. Secondary KIT ATP-BP pocket mutation variants, particularly V654A, were enriched upon disease progression with avapritinib.
Conclusions:
ctDNA sequencing efficiently detects KIT/PDGFRA mutations and prognosticates outcomes in patients with TKI-resistant GIST treated with avapritinib. ctDNA analysis can be used to monitor disease progression and provide more personalized treatment.
Keywords: Avapritinib, ctDNA, GIST, KIT, PDGFRA, regorafenib
INTRODUCTION
Detection and monitoring of cancer-related mutations in circulating tumor DNA (ctDNA) theoretically allows capture of the spatial and temporal evolution of tumors across therapeutic interventions.1 Although mutations are ubiquitous in all cancers, neoplasms driven by targetable genomic alterations may benefit the most from implementation of ctDNA assessments to determine the mutational status of the underlying disease at the time of treatment. In this sense, gastrointestinal stromal tumor (GIST), the most common sarcoma histological subtype, constitutes a paradigm to address the clinical impact of ctDNA determination.2–4
The majority of GISTs are driven by gain-of-function mutations in genes encoding KIT (~70%) or PDGFRA (~15%) receptor tyrosine kinases.5–7 Accordingly, targeted inhibition of these receptors with first-line imatinib achieves major responses and durable clinical benefit.7–9 KIT oncogenic signaling remains the primary oncogenic driver after imatinib failure, as emergence of secondary mutations in KIT is the primary cause of imatinib resistance in ~90% of KIT-mutant GISTs.4,10 Previous tumor tissue-based series showed these mutations cluster in the ATP-binding pocket (ATP-BP; encoded by exons 13 and 14) and the activation loop (A-loop; encoded by exons 17 and 18).10–13 Approved tyrosine kinase inhibitors (TKIs) for the treatment of imatinib-resistant GIST are sunitinib (second line), regorafenib (third line), and ripretinib (fourth line), each displaying an inhibitory spectrum against certain secondary KIT mutations.14–16 Of note, several studies support that KIT primary and secondary genotype correlates with the activity of some or all of these TKIs.10,11,13,17,18 Similarly, avapritinib, a type I TKI approved for the treatment of PDGFRA D842V-mutant GIST, specifically targets the kinase A-loop, and resistant subpopulations emerge through secondary mutations in the ATP-BP.19,20 This, in turn, confirms the relevance of KIT and PDGFRA signaling throughout GIST progression and reinforces the variable activity of anti-GIST TKIs between GIST-specific molecular subsets.
Despite these data, the current treatment paradigm of imatinib-resistant metastatic GIST does not consider the presence of specific KIT/PDGFRA mutations to drive therapeutic choices due to limited clinical data validating these approaches, and patients continue to be treated sequentially following the regulatory approval order of the different available TKIs. Several studies have attempted to provide evidence to support use of ctDNA determination to guide therapeutic decisions.3 A smaller, retrospective study of 243 patients with GIST developed a receiver-operating characteristic sensitivity curve for ctDNA testing and found patient outcomes superior to historical controls when management incorporates ctDNA testing.4 However, the rarity of this disease together with the heterogeneity of technologies used in previous studies have limited the understanding of GIST progression and the development of ctDNA analysis technology as a clinical tool. Herein, we evaluated prospectively collected ctDNA molecular data from the phase III VOYAGER trial21 to understand the evolving landscape of TKI progression and expand on the potential clinical utility of ctDNA determination in patients with advanced GIST who experienced progression after 2 or 3 lines of treatment.
PATIENTS AND METHODS
Patients
Main inclusion and exclusion criteria were as follows: eligible patients were aged 18 years or older, had histologically confirmed unresectable/metastatic GIST, and were previously treated with imatinib and up to 2 additional TKIs. Patients were not eligible if previously treated with either avapritinib, regorafenib, or 4 or more different TKIs. Complete inclusion/exclusion criteria can be found in Kang et al.21
Study design
The open-label, randomized (1:1), multicenter phase III VOYAGER trial compared oral avapritinib (300 mg QD in continuous 28-day treatment cycles) versus regorafenib (160 mg QD for 21 days every 28 days [3 weeks on, 1 week off]) in patients with GIST previously treated with imatinib and a maximum of 2 other additional TKIs (NCT03465722). Random assignment was stratified by third- and fourth-line TKI treatment, geographic region, and PDGFRA D842V status measured by ctDNA. Crossover from regorafenib to avapritinib was allowed for patients with centrally confirmed radiologic disease progression. Further design and main clinical outcome details were published previously.21
The study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines. The Protocol was approved by the institutional review board or independent ethics committee at each study center. All patients provided written informed consent for data collection supporting these analyses.
Sample collection
Plasma samples were obtained from patients with advanced GIST enrolled in VOYAGER at baseline prior to initiation of either avapritinib or regorafenib. Additionally, end-of-treatment plasma samples were collected from patients progressing on avapritinib.
Sequencing and data analysis
Tumor mutation profiling of plasma at baseline and end-of-treatment was assessed with 74-gene Guardant360® CDx sequencing technology. Following the recommendations of the manufacturer, only 2 samples failed quality control evaluations and were consequently discarded from downstream analyses. KIT/PDGFRA-detected variants were annotated using Ensembl’s Variant Effect Predictor tool, restricting the prioritization to RefSeq canonical transcripts (KIT: NM_000222.3; PDGFRA: NM_006206.6). Nonsynonymous variants were considered pathogenic when meeting at least 3 of the following criteria in order of priority: (1) ClinVar significance is available and (likely) pathogenic, (2) at least 3 predictors (SIFT, PolyPhen-2) or meta-predictors (BayesDel, ClinPred, REVEL) report the variant as pathogenic/damaging, (3) any domain of clinical-biological relevance is affected, (4) the variant is not frequent in the general population (minor allele frequency <1%), and (5) the locus is conserved in mammals.
Statistical analysis
The Kaplan-Meier method was used to estimate progression-free survival (PFS) probabilities. The Cox regression model was used to assess hazard ratios and 95% confidence intervals (CI). To assess statistical associations, Chi-square (discrete variables) and Wilcoxon rank-sum tests (continuous variables) were used for univariate statistical analyses. Logistic regressions were used for multivariate analyses. Descriptive statistics were used to summarize cohort data. The cutoff date for these analyses was March 9, 2020.
RESULTS
Baseline characteristics and ctDNA detection
Baseline ctDNA analysis was performed in 386/476 patients (81.1%) enrolled in the trial, 196 in the avapritinib arm and 190 in the regorafenib arm. A total of 327 (84.7%) patients received avapritinib or regorafenib in the third line, and 59 (15.3%) in the fourth line. Demographics and baseline characteristics did not differ between treatment arms in the subgroup analyzed with ctDNA compared to the overall population (Supplementary Table S1).
All patients had KIT- or PDGFRA-mutant GIST by inclusion criteria. ctDNA mutations in any gene were found in 333/386 patients (86.3%) and showed similar distribution across treatment arms (Figure 1). Of the patients with any ctDNA mutations identified, at least 1 KIT or PDGFRA pathogenic variant was detected in the plasma of 250/333 (75.1%) and 18/333 (5.4%) patients, respectively. Median ctDNA variant allele frequency was 0.55% (range, 0.02–86.47%) for all ctDNA mutations, and 1.08% (range, 0.03–86.47%) and 1.38% (range, 0.11–20.32%) for KIT and PDGFRA mutations, respectively. Clinical variables correlating with the presence of KIT or PDGFRA mutations in plasma in the multivariate analysis were colorectal and gastric primary tumor locations, presence of liver metastases, and the sum of diameters from target lesions (Supplementary Table S2).
Figure 1.
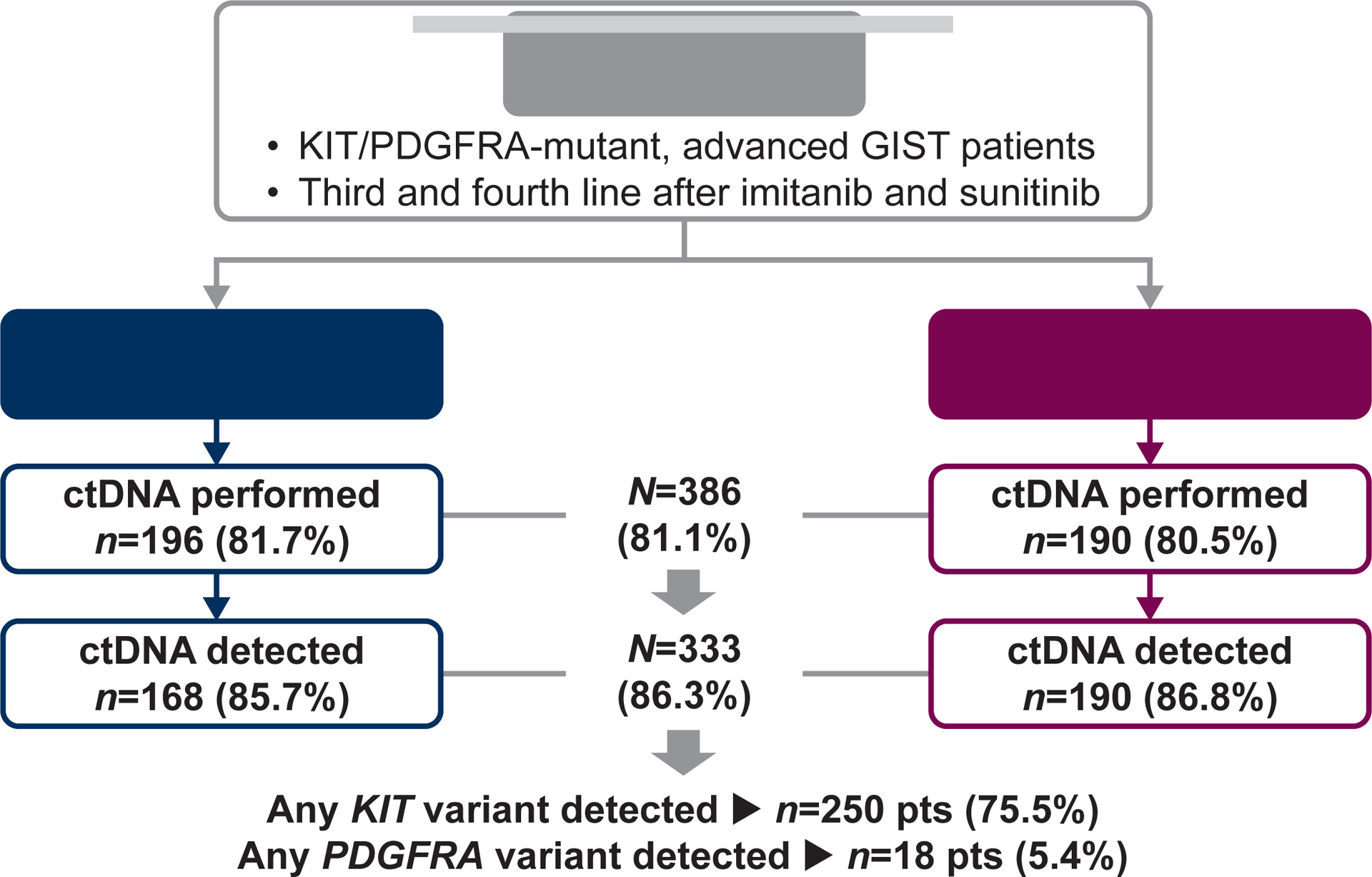
Flow chart of ctDNA analyses performed and mutational findings.
KIT mutational landscape in imatinib-resistant GIST
A total of 641 KIT pathogenic variants, 179 of which were unique, were detected in the subset of 250 patients with at least 1 KIT mutation detected by ctDNA testing prior to treatment on the VOYAGER study. In this population, KIT primary and secondary mutations were found in 94.0% and 73.6% of patients, respectively. Both KIT primary and secondary mutations were found simultaneously in 68.0% of patients, while 26.4% had only KIT primary variants in the absence of any KIT resistance mutation, and 6.0% had at least 1 KIT resistance mutation without detecting the primary. The majority of these genetic alterations were missense (45.3%), followed by in-frame deletions (39.1%), complex indels (7.8%), and insertions (5.0%). Overall, the distribution of these proportions was similar between treatment arms (Supplementary Table S3).
Primary and secondary KIT mutations are distributed across hotspot regions of the kinase, although these data largely originate from scattered tumor tissue series prior to the advent of modern next-generation sequencing.10,11,13,17,18 The ctDNA profile of KIT primary mutations largely paralleled that from the early series, with 78.8% of alterations in KIT exon 11, 19.9% in exon 9, and 1.3% in exon 13 (Supplementary Table S3). Notably, the frequency of codons affected across KIT exon 11 follows the same distribution of historical tissue-based analysis, which supports the reliability of ctDNA sequencing in GIST (Figure 2). KIT secondary mutations in the A-loop—encoded by exons 17 and 18—were more frequent (80.4%) than in the ATP-BP (40.8%)—encoded by exons 13 and 14. Additionally, 23.4% of patients with KIT secondary mutations had both ATP-BP and A-loop mutations. Although some specific codons were more commonly found (i.e. D820, N822, V654, Y823) (Figure 2), ctDNA analysis demonstrated for the first time a widespread distribution of secondary mutations across multiple KIT codons and exons (Figure 2; Supplementary Figure S1A). The frequency of all amino acid changes can be seen in Supplementary Table S3.
Figure 2. KIT affected exons and codons.
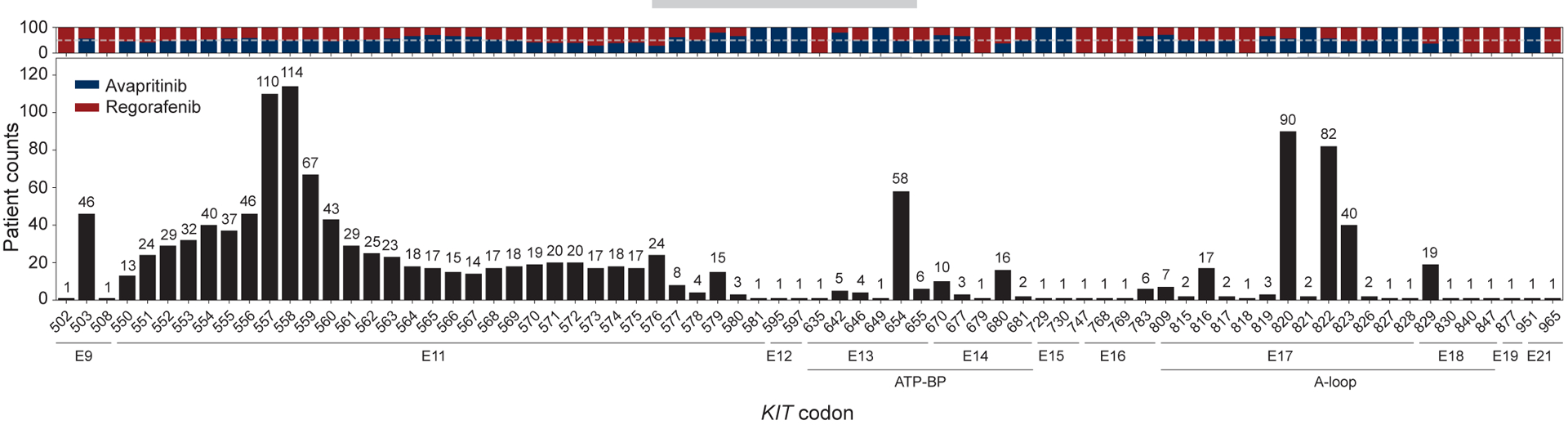
The distribution of codons affected by any KIT variant was similar between treatment arms, which is better observed in those with higher frequency.
Notably, the determination of ctDNA in this large cohort of patients with GIST in the third- or fourth-line setting after disease progression on imatinib and sunitinib allowed identification of 50 novel pathogenic KIT variants, 28.0% of the total identified. Although half of them were deletions, insertions, or indels affecting KIT exon 11, a total of 14 KIT variants were missense substitutions affecting a wide range of exons. Interestingly, secondary in-frame deletions (N=6) and in-frame insertions (N=2) were identified for the first time in KIT exon 17 (Supplementary Table S4).
PDGFRA mutational landscape in imatinib-resistant GIST
Similar mutational patterns were observed across the 18 PDGFRA-mutant GISTs as determined by ctDNA. Half of the patients had PDGFRA mutations in the A-loop, with the most frequent being the D842V substitution (27.8%), which agrees with known frequencies (Supplementary Figure S1B; Supplementary Figure S2; Supplementary Table S5). However, ctDNA analysis also detected pathogenic PDGFRA mutations in several other exons beyond the expected 12, 14, and 18. Furthermore, ctDNA analyses provided insight on resistance mutations, which remains largely unexplored. Only 3 patients showed secondary mutations in PDGFRA (Supplementary Figure S2; Supplementary Table S5). As occurred with KIT, 7 novel pathogenic PDGFRA variants were found by ctDNA determination (Supplementary Table S4).
Heterogeneity of KIT and PDGFRA resistance mutations
The spectrum of secondary resistance mutations in GIST that can be identified by ctDNA is currently unclear, as it has been only investigated in few studies with small numbers of patients using a wide range of technologies with varying sensitivities. Herein, among KIT-mutant tumors, multiple KIT mutations were commonly detected with an average of 2.6 mutations per patient. The majority of patients had 1–3 KIT variants detected, although 17.2% of the patients had substantial heterogeneity, with 4–14 different KIT mutations detected in plasma (Figure 3). There were no statistically significant differences between patient distributions in the avapritinib and regorafenib arms, based on the number of KIT mutations. Only 3/18 PDGFRA-mutant GISTs had more than 1 variant detected in plasma, 2 patients with 2 each, and 1 patient with 5 (Supplementary Figure S2B). The extent of KIT mutational heterogeneity did not correlate with any of the baseline clinicopathological features (Supplementary Table S2).
Figure 3. KIT mutational heterogeneity.
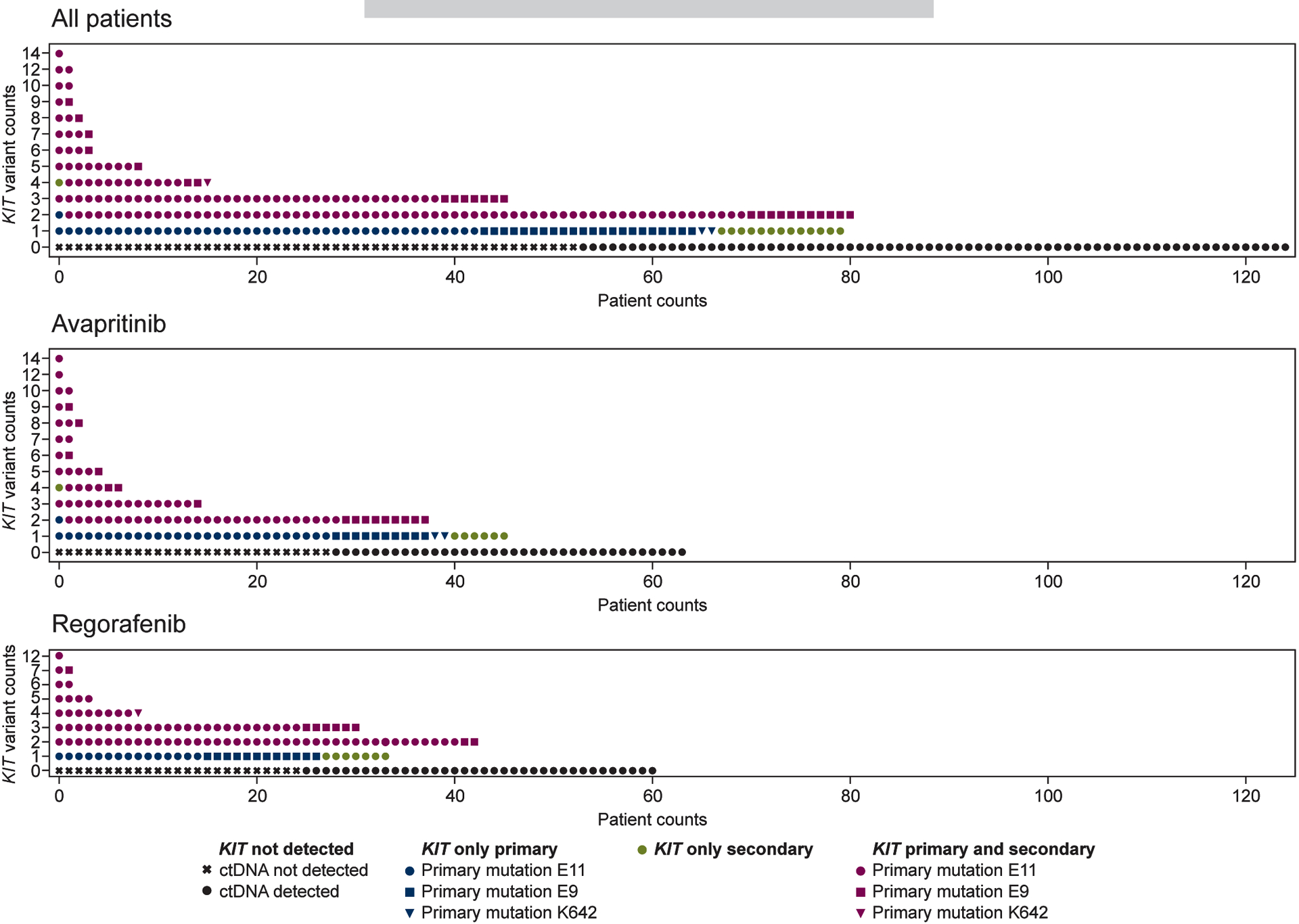
Multiple KIT mutations were commonly detected among KIT-mutant tumors.
KIT ctDNA determination correlates with clinical efficacy in avapritinib- and regorafenib-treated patients with GIST
Current treatment of metastatic KIT-mutant GIST relies on sequential administration of TKIs solely based on their regulatory approval order, without considering the evolving GIST mutational landscape. Therefore, we investigated whether ctDNA analysis has a prognostic role in this setting. Of the 386 patients with a baseline ctDNA test performed, 352 had baseline measurable target lesions. In this subset of patients, median PFS (mPFS) for avapritinib and regorafenib was 3.8 and 5.6 months, respectively, which did not differ from the total population (Supplementary Figure S3).
The detection of KIT secondary mutations in the ATP-BP was a negative prognostic factor of avapritinib clinical efficacy, with a mPFS of 1.9 months (Figure 4A). Indeed, the absence of ATP-BP mutations in plasma correlated with avapritinib activity in patients, with a mPFS of 5.6 months (Supplementary Figure S4A). In comparison, regorafenib efficacy in patients with GIST harboring ATP-BP mutations detected in the ctDNA was significantly better than that of avapritinib, with a mPFS of 5.7 months (P<0.001; Figure 4A). The antitumor activity of regorafenib did not differ regardless of the presence or absence of KIT ATP-BP resistance mutations in plasma (5.7 vs 5.6 months, P=0.829; Supplementary Figure S4B). Also, any dimensional shrinkage per RECIST in patients with ATP-BP mutations was more frequently observed in patients treated with regorafenib (65.7% vs 25.8%, P=0.002).
Figure 4. KIT ATP-BP and A-loop mutations in ctDNA are prognostic of TKI activity in GIST.
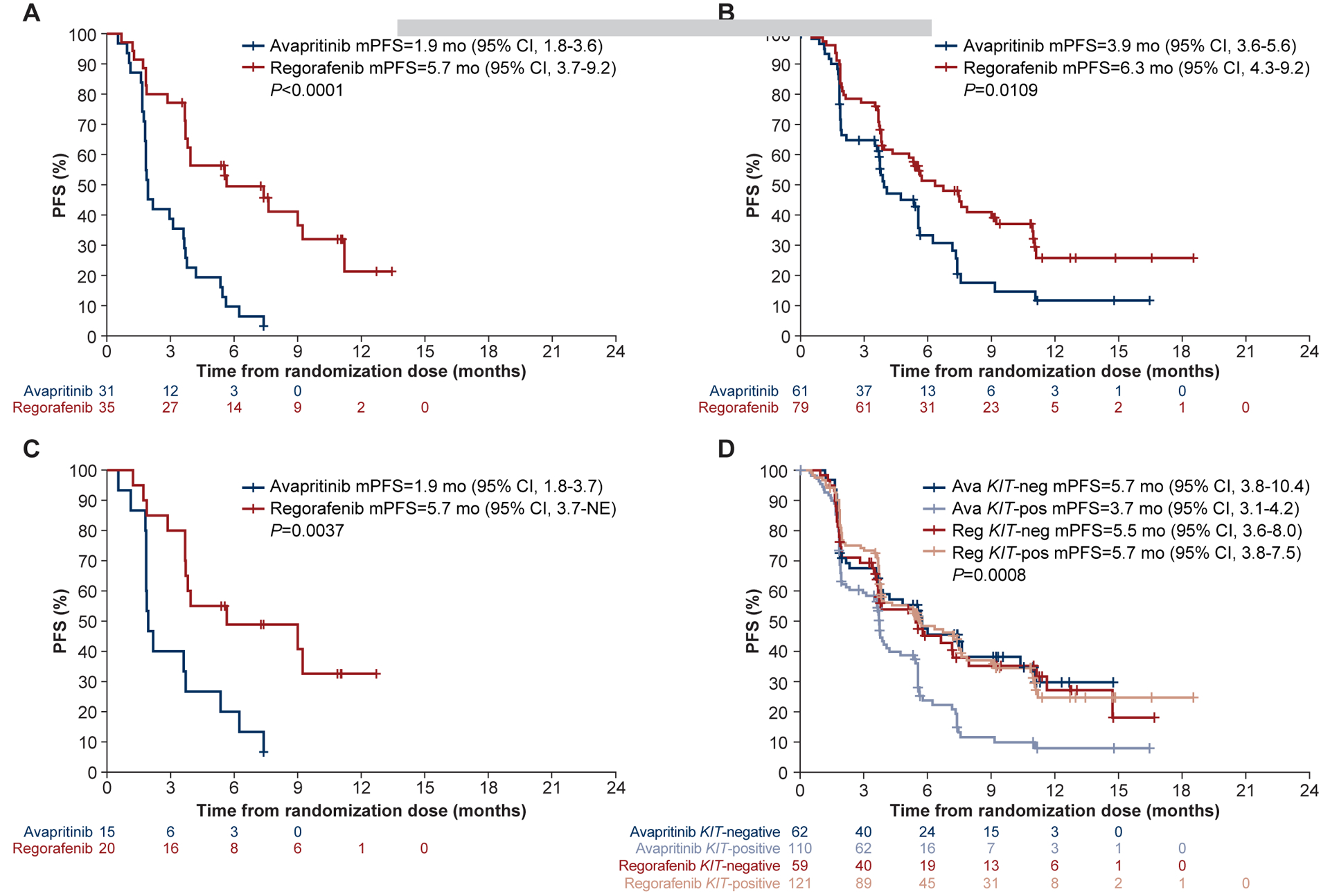
Regorafenib had greater efficacy than avapritinib in patients with an ATP-BP mutation (A), a KIT A-loop mutation (B), and those with both ATP-BP and A-loop mutations (C). (D) Avapritinib improved PFS in the absence of a KIT mutation, while regorafenib was relatively unaffected. Ava, avapritinib; CI, confidence interval; mPFS, median progression-free survival; NE, not estimable; neg, negative; pos, positive; Reg, regorafenib.
The detection of KIT A-loop mutants in plasma was prognostic of better mPFS with regorafenib compared to avapritinib (6.3 vs 3.9 months, P=0.011; Figure 4B). However, in the absence of ATP-BP mutations, the presence of a KIT A-loop mutation alone in ctDNA was neither prognostic of clinical efficacy with avapritinib nor regorafenib, likely indicating better avapritinib activity in the absence of ATP-BP mutants (Supplementary Figure S4C–D). Finally, the simultaneous presence of ATP-BP and A-loop mutations in plasma was also a negative prognostic factor of avapritinib versus regorafenib efficacy (1.9 vs 5.7 months, P=0.004; Figure 4C). Accordingly, PFS curves in this subset of patients tended to overlap with outcomes from patients with GIST harboring only ATP-BP mutations, confirming their relevance to determine avapritinib/regorafenib treatment outcomes in patients with GIST (Supplementary Figure S4E–F).
The number and heterogeneity of mutations found in plasma can be associated with outcomes. We first studied KIT-positive versus KIT-negative ctDNA populations, since KIT is the clonal driver mutation, and all patients had KIT- or PDGFRA-mutant GIST by inclusion criteria. From the 386 patients who underwent ctDNA testing, KIT mutations were not detected in 136 (avapritinib, N=72; regorafenib, N=64). The 18 patients with PDGFRA-mutant GIST were excluded from this analysis. Surprisingly, avapritinib and regorafenib displayed different behavior (P=0.0008, Figure 4D): while patients with KIT-positive ctDNA had a shorter mPFS with avapritinib (3.7 vs 5.7 months), regorafenib activity was not affected by the detection of KIT variants in plasma (5.5 vs 5.7 months). The heterogeneity of KIT mutations detected in plasma was further evaluated. However, increased heterogeneity (>3 mutations) did not impact mPFS in either treatment arm (Supplementary Figure S4G–H). Together, the detection of KIT mutations in ctDNA correlates with outcomes in patients with metastatic GIST. Particularly, the determination of specific regions mutated across the KIT gene is relevant to determine the sensitivity or resistance of specific TKIs before treatment initiation.
Individual ctDNA mutations in KIT and PDGFRA determine TKI sensitivity and resistance in metastatic GIST
Preclinical studies suggested that individual primary and secondary mutations in KIT and PDGFRA can predict the efficacy of TKIs either approved or having shown activity in GIST. However, little supportive correlative science is available so far beyond the first-line setting.10,11,13,17,18 Here, individual KIT secondary ctDNA mutations were not associated overall withTKI response/progression. This can be explained by the heterogeneity of KIT secondary mutations with different sensitivity profiles and the predominance of disease stabilization as best response outcomes. However, the presence of baseline KIT exon 13 V654A mutations in plasma, together with other less common substitutions in the ATP-BP (T670, N680), tended to correlate against avapritinib activity, with only 1 patient showing a RECIST partial response compared to the 4 partial responses achieved by regorafenib (Figure 5A). The proportion of partial responses with avapritinib or regorafenib did not differ substantially regardless of the individual secondary mutation in the most common codons across the A-loop (exons 17 and 18), although alterations found in the last part of the A-loop (Y823, A829) were arguably better surrogate markers of activity for regorafenib compared with avapritinib (Figure 5A).
Figure 5. KIT/PDGFRA primary and secondary mutations determined in ctDNA in GIST are prognostic of TKI activity.
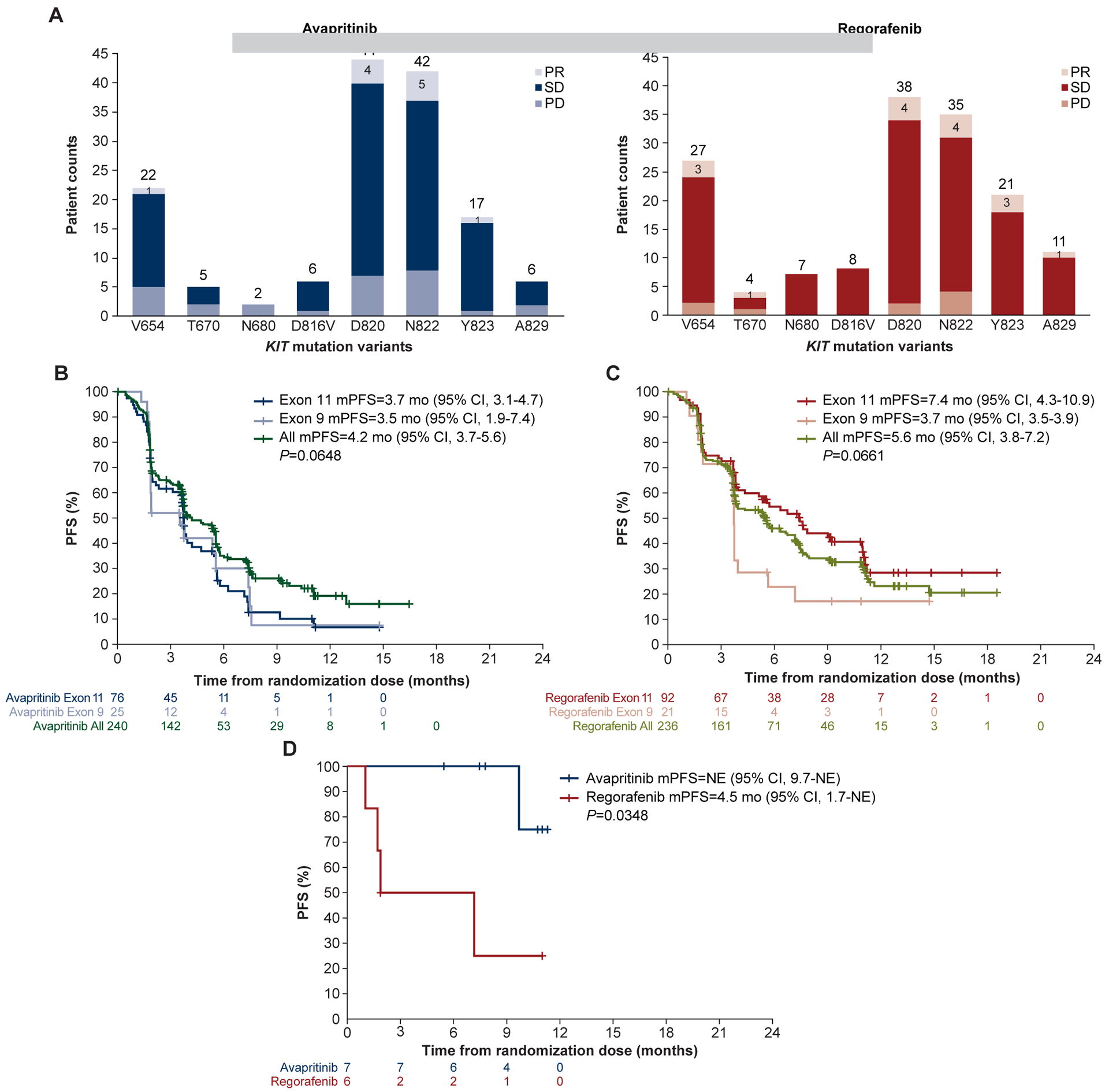
(A) KIT secondary mutations did not significantly affect the number of partial responses between treatments arms. Avapritinib efficacy was similar between primary KIT exon 9 and 11 mutations (B), whereas regorafenib trended to be more effective in patients with a primary exon 11 mutation (C). (D) Avapritinib had greater efficacy in patients with a primary PDGFRA exon 18 D842V mutation than regorafenib. CI, confidence interval; mPFS, median progression-free survival; NE, not estimable; PD, progressive disease; PR, partial response; SD, stable disease.
The specific exonic or amino acid involvement of primary mutations in KIT and PDGFRA can predict the clinical efficacy of first- and second-line imatinib and sunitinib regardless of the status of the secondary mutations. Herein, avapritinib appeared to have similar efficacy regardless of the location of primary mutations in KIT, displaying a mPFS of 3.7 and 3.5 months, respectively, against KIT exon 11 and exon 9 mutants. However, the detection of primary KIT exon 11 mutations in the plasma of patients treated with regorafenib showed a nonsignificant trend (P=0.067) towards an improved mPFS in comparison with those patients harboring a primary KIT exon 9 mutation (7.9 vs 3.7 months, Figure 5B and 5C). Likewise, ctDNA detection of a primary PDGFRA exon 18 D842V mutation demonstrated improved clinical efficacy with avapritinib, as the mPFS was not reached relative to 4.5 months with regorafenib (P=0.035; Figure 5D). This, in turn, agrees with prior data confirming the high activity of avapritinib in this molecular subgroup of GIST as well as the little clinical efficacy of other TKIs approved for treatment of metastatic GIST.19,22
Serial ctDNA determinations detect the emergence of resistant subpopulations
To further understand the sensitivity/resistance profile of avapritinib, plasma samples from 41 patients with GIST were collected at the time of disease progression, and 40 could be analyzed. Compared to baseline, there was a significant increase in the detection of KIT secondary mutations (P=0.022) at the time of disease progression. Although this increase was largely polyclonal, KIT ATP-BP pocket variants were found highly enriched (P=0.001; Figure 6A), particularly V654A. Subclonal dynamics of a representative case shows that, in the molecular context of KIT mutational heterogeneity, there is an allele frequency increase in a resistant subpopulation harboring the KIT exon 13 V654A substitution that parallels the primary clonal KIT exon 9 mutation. Conversely, subpopulations with secondary mutations in the A-loop remained inhibited at the time of avapritinib progression (Figure 6B). Together, ctDNA analyses at the time of disease progression are useful to determine the resistant subclones responsible for TKI resistance, confirm the accuracy of baseline ctDNA determination in prognosticating treatment outcomes, are helpful in selecting next-line therapy, and are overall consistent with the known mechanism of action for avapritinib specifically targeting the A-loop of KIT and PDGFRA.
Figure 6. Serial KIT ctDNA determinations detect resistance.
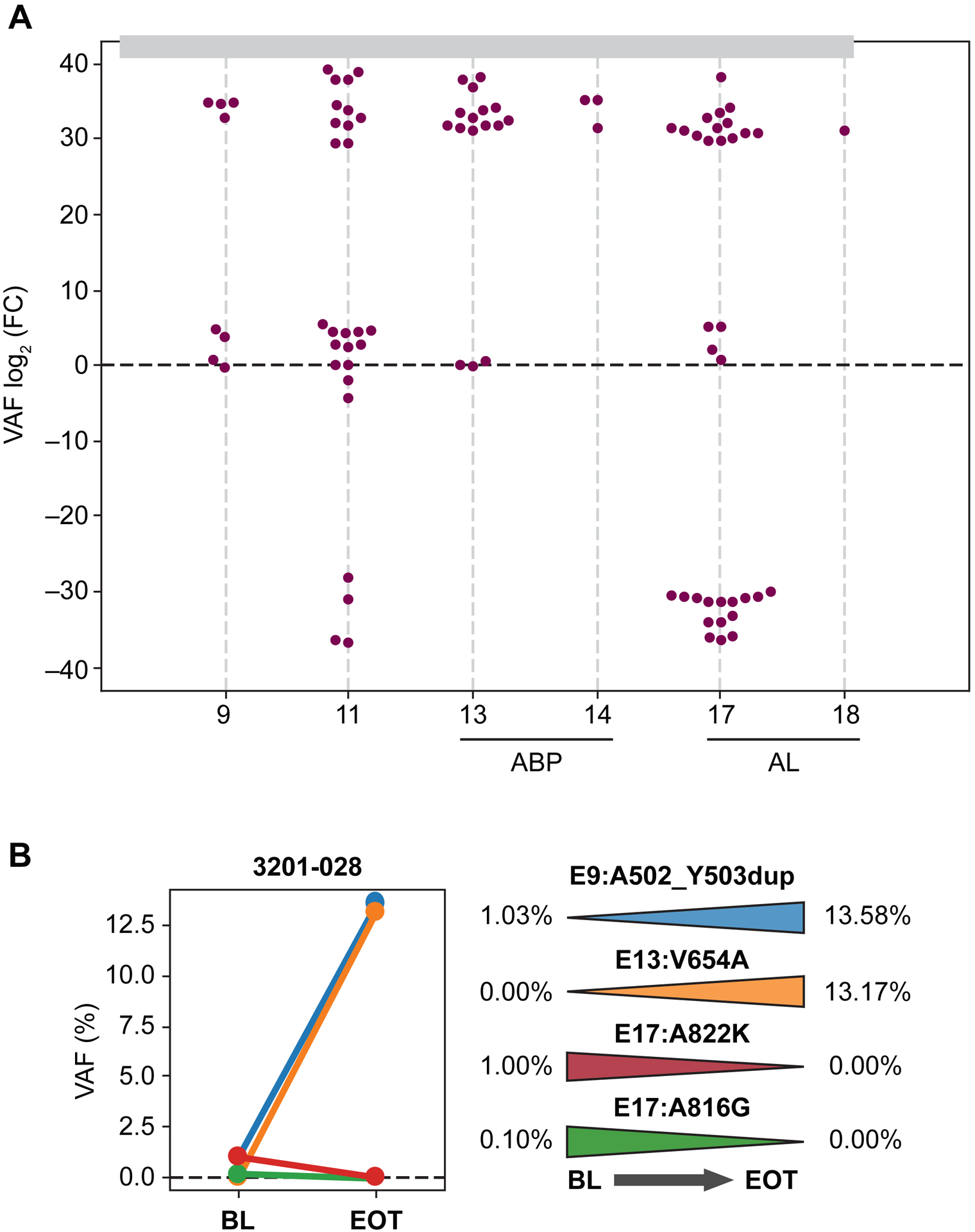
(A) KIT secondary mutation detection significantly increased upon disease progression. (B) Representative subclonal evolution showing increased allele frequency in subpopulations with a KIT exon 13 V654A mutation, in parallel to primary clonal KIT exon 11 mutations. ABP, ATP-BP; AL, activation loop; BL, baseline; EOT, end-of-treatment; FC, fold change; VAF, variable allele frequency.
DISCUSSION
GIST constitutes an excellent model to evaluate implementation of ctDNA as a routine clinical tool to guide cancer treatment decisions. Supporting this view is the fact that oncogenic activation of KIT or PDGFRA is the essential driver for GIST development and maintenance. Unlike most mutant KIT/PDGFRA-driven tumors, the main mechanism of resistance in GIST is the polyclonal emergence of cis-allele, secondary mutations in the primary oncogenic driver, either KIT (~90% of patients) or PDGFRA, and preclinical studies consistently show that TKIs used in GIST treatment display drug-specific activity against subsets of the KIT/PDGFRA mutational spectrum.10,11,13,17,18,20 However, only a limited number of studies have analyzed mechanisms of tumor progression after first-line imatinib, and they did not establish the clinical role of ctDNA determination in therapeutic decision-making.3 Thus, this study evaluates, for the first time, the clinical validity and potential clinical utility of ctDNA determination in a large cohort of patients with metastatic GIST with plasma samples collected prospectively in the VOYAGER trial.
The detection rate of KIT and PDGFRA mutations in ctDNA was 69%, with an average allele frequency of ~1%. Although these third- or fourth-line patients typically have bulky disease, ctDNA shedding in GIST is comparatively lower than other cancer types,4 which appears common for many sarcomas.23–25 Regardless of these tumor-related limitations, primary mutations in known exonic regions of KIT and PDGFRA were found at similar proportions as described previously. This also relates to the profile of codons affected within KIT exon 11, which often includes complex insertions, deletions, or indels that could be detected in plasma.7 Furthermore, baseline ctDNA analysis from the VOYAGER trial uncovered a shift in resistance patterns after 2–3 lines of treatment. Namely, KIT secondary mutations in the A-loop were predominant at the expense of a decrease in clones harboring ATP-BP mutations—known to be the most common at the onset of imatinib failure13—which is consistent with the selective pressure exerted by second-line sunitinib, which has potency against ATP-BP mutant clones. Another relevant aspect was an arguably low level of complex heterogeneity, with 82% of the patients displaying only 1–3 KIT variants. Although it is likely that some mutations exist below our threshold of detection, accurate prognosis of outcomes may increae the possibility that undetected clones are not clinically meaningful, even when present. Finally, this ctDNA analysis also determined a greater spectrum of KIT pathogenic variants compared with previous studies, including exons and codons previously not known to be affected. Together, this analysis of the mutational spectrum of acquired resistance as demonstrated by ctDNA reinforces the critical role of KIT oncogenic signaling and provides the basis for future drug development in GIST after progression to various lines of treatment.
This study also demonstrates that ctDNA assessment might predict TKI activity in metastatic GIST in the third- and fourth-line setting. Thus, although the VOYAGER trial did not yield significant mPFS differences between avapritinib and regorafenib, ctDNA determination revealed the detection of ATP-BP mutations in plasma was a strong negative prognostic marker of avapritinib activity. Conversely, patients with GIST harboring A-loop mutations in the absence of ATP-BP achieved equal benefit from avapritinib and regorafenib. This, in turn, is consistent with the known mechanism of action of avapritinib, its superior efficacy in patients with tumors harboring the PDGFRA D842V mutation, and the enrichment of KIT exon 13 V654A subpopulations observed at the time of progression. Surprisingly, regorafenib appeared to have similar activity irrespective of the type of KIT secondary mutation, while prior preclinical studies set V654A as a potential liability.13 It is conceivable the multikinase nature of regorafenib allows blockage of several other signaling intermediates downstream of KIT relevant for its oncogenic function and mediation of GIST cell growth, thus compensating for incomplete upstream KIT inhibition.
In summary, we have shown ctDNA sequencing reliably detects KIT/PDGFRA mutations and correlates with outcomes in patients with metastatic, TKI-resistant GIST treated with avapritinib or regorafenib. Therefore, consideration of ctDNA to monitor disease progression and provide more personalized treatment options in patients with GIST should continue to be explored.
Supplementary Material
Figure S1. Distribution of affected KIT (A) and PDGFRA (B) exons in GIST ctDNA.
Figure S2. Distribution of affected PDGFRA codons (A) and mutational heterogeneity (B).
Figure S3. KIT ctDNA determination prognosticates avapritinib (A) and regorafenib (B) efficacy vs the total population. CI, confidence interval; mPFS, median progression-free survival.
Figure S4. ctDNA determination and avapritinib treatment outcomes in GIST. Absence of KIT ATP-BP mutation prognosticated improved avapritinib activity (A), whereas regorafenib antitumor activity did not differ regardless of ATP-BP mutation status (B). The presence of a KIT A-loop mutation alone was not prognostic of efficacy with avapritinib (C) or regorafenib (D). Presence of both an ATP-BP and A-loop mutation was prognostic of avapritinib (E) and regorafenib (F) outcomes in patients with GIST. (G), (H) Increased KIT mutational heterogeneity (>3 mutations) did not impact PFS in either treatment arm. ABP, ATP-BP; AL, activation loop; CI, confidence interval; mPFS, median progression-free survival; NE, not estimable.
Table S1. Demographics and baseline characteristics.
ctDNA, circulating tumor DNA; ECOG, Eastern Cooperative Oncology Group.
Table S2. Association between clinical variables and detection of KIT or PDGFRA ctDNA.
CAT, computed axial tomography; CI, confidence interval; coef, coefficient; ctDNA, circulating tumor DNA; OR, odds ratio; std err, standard error; TKI, tyrosine kinase inhibitor.
Table S3. KIT mutations and distribution.
CDS, coding sequence; HGNC, HUGO Gene Nomenclature Committee; HGVSc, Human Genome Variation Society coding sequence; HGVSp, Human Genome Variation Society protein sequence.
Table S4. KIT and PDGFRA novel mutations found through ctDNA sequencing.
ctDNA, circulating tumor DNA; CDS, coding sequence; HGNC, HUGO Gene Nomenclature Committee; HGVSc, Human Genome Variation Society coding sequence; HGVSp, Human Genome Variation Society protein sequence.
Table S5. PDGFRA mutations and distribution.
CDS, coding sequence; HGNC, HUGO Gene Nomenclature Committee; HGVSc, Human Genome Variation Society coding sequence; HGVSp, Human Genome Variation Society protein sequence.
HIGHLIGHTS.
This study comprehensively documents the landscape of KIT and PDGFRA mutations in metastatic, imatinib-resistant GIST
The selective pressure exerted with prior lines promotes a shift toward increased resistant subpopulations in the KIT A-loop
Individual mutations in KIT/PDGFRA determine TKI sensitivity and resistance in metastatic GIST
ctDNA-detected KIT/PDGFRA mutations in imatinib-resistant GIST prognosticate third- or fourth-line TKI treatment outcomes
ACKNOWLEDGEMENTS
The authors would like to thank the patients, their families, all investigators, clinical research staff, sites, and Blueprint employees involved in this study. Editorial support was provided Miranda Bader-Goodman, PhD, of the Healthcare Consultancy Group, supported by Blueprint Medicines Corporation according to Good Publication Practice guidelines (https://doi.org/10.7326/M22-1460). The sponsor was involved in the study design, collection, analysis, and interpretation of data, as well as data checking of information provided in the manuscript. However, ultimate responsibility for opinions, conclusions, data interpretation, and manuscript writing lies with the authors.
FUNDING
This project was funded in part by the Fero Foundation, ISCIII PI19/01271 and PI22_00720, and the Asociación Española Contra el Cáncer (AECC CLSEN20004SERR), all to CS; ISCIII FI20/00275 to DG-P. The trial was funded by Blueprint Medicines Corporation. The general work of MCH has been supported by grants from the Department of Veterans Affairs (1 I01 BX005358-01A1) and the NIH National Cancer Institute (1 R21 CA263400-01) and by philanthropic donations from the GIST Cancer Research Fund and the Jonathan David Foundation. JCT is supported by a NIH/NCI Cancer Center Support Grant P30CA240139 and The Childhood Cancer Research Project.
CONFLICTS OF INTEREST
CS has received research funding (institution) from IDRX, Blueprint, Karyopharm, Pfizer, Deciphera, and Bayer; consulting fees (advisory role) from IDRX, CogentBio, Immunicum AB, Deciphera and Blueprint; payment for lectures from Deciphera, PharmaMar, Pfizer, Bayer and Blueprint; and travel grants from PharmaMar, Gilead, Pfizer, and Bayer.
SBa has received honoraria from Novartis, Pfizer, Bayer, Pharmamar, and GlaxoSmithKline; reports consulting or advisory role for Blueprint Medicines Corporation, Bayer, Lilly, Deciphera, Nanobiotix, Daiichi Sankyo, Exelixis, Janssen-Cilag, ADC Therapeutics, Mundipharma, and GlaxoSmithKline; research funding from Blueprint Medicines Corporation, Novartis, and Incyte; travel and accommodation expenses from Pharmamar.
DG-P declares no conflicts of interest.
Y-K K reports consulting or advisory role for DAEHWA Pharmaceutical, Bristol Myers Squibb, Zymeworks, ALX Oncology, Amgen, Novartis, Macrogenics, Surface Oncology, and Blueprint Medicines Corporation.
RLJ reports consulting or advisory role for Lilly, Immune Design, Merck Serono, Adaptimmune, Daiichi Sankyo, Eisai, Morphotek, TRACON Pharma, Immodulon Therapeutics, Deciphera, PharmaMar, Blueprint Medicines Corporation, Clinigen Group, Epizyme, Boehringer Ingelheim, Bayer, Karma Oncology, and UpToDate; research funding from GlaxoSmithKline; travel and accommodation, expenses from PharmaMar.
PR has received honoraria from Bristol Myers Squibb, MSD, Novartis, Roche, Lilly, Pfizer, Pierre Fabre, Sanofi, and Merck; reports consulting or advisory role for Novartis, Blueprint Medicines Corporation, and Bristol Myers Squibb.
OM is an employee and shareholder of Amgen, Inc.
MCH reports stock and other ownership interests in MolecularMD; honoraria from Novartis; consulting or advisory role for MolecularMD, Novartis, Blueprint Medicines Corporation, Deciphera, C Stone Pharmaceuticals, Zai Labs, and Theseus Pharmaceuticals; patents, royalties, and other intellectual property regarding patent on treatment of GIST-licensed to Novartis; expert testimony for Novartis.
WDT has a leadership role in Certis Oncology Solutions, Atropos, and Innova Therapeutics; reports owning stock and other ownership interests in Certis Oncology Solutions, and Atropos; consulting or advisory role for EMD Serono, Lilly, Daiichi Sankyo, Blueprint Medicines Corporation, Agios, NanoCarrier, Deciphera, C4 Therapeutics, Mundipharma, Adcendo, Ayala Pharmaceuticals, Kowa Pharmaceutical, Servier, and AbMaxBio; research funding from Novartis, Lilly, Plexxikon, Daiichi Sankyo, TRACON Pharma, Blueprint Medicines Corporation, Immune Design, BioAtla, and Deciphera; patents, royalties, and other intellectual property regarding Companion Diagnostic for CDK4 inhibitors—14/854,329, Enigma and CDH18 as companion Diagnostics for CDK4 inhibition—SKI2016-021-03
KN is an employee and equity holder of Blueprint Medicines Corporation.
AG is an employee and equity holder of Blueprint Medicines Corporation.
HS is an employee and equity holder of Blueprint Medicines Corporation.
SBi reports no conflicts of interest.
PS has received honoraria from Deciphera, Blueprint Medicines Corporation, Boehringer and Ingelheim; consulting or advisory role for Blueprint Medicines Corporation, Ellipses Pharma, Adaptimmune, Intellisphere, Transgene, Deciphera, Exelixis, Boehringer Ingelheim, Medscape, Guided Clarity, Ysios Capital, and Studiecentrum voor Kernenergie;research funding from CoBioRes NV, Eisai, G1 Therapeutics, Novartis, and PharmaMar; travel and accommodations expenses from MSD, Ipsen, and Boehringer Ingelheim.
MAP has received research funding (institution) from Novartis; consulting fees (advisory role) from Roche, PharmaMar; lecture fee from Pfizer, Eli Lilly.
MvM reports consulting or advisory role for Deciphera, and Exelixis; research funding from ArQule, Novartis, Blueprint Medicines Corporation, Deciphera, Gradalis, Springworks Therapeutics, Lilly, Arog, Genmab, and ASCO; travel and accommodations expenses from Deciphera Pharmaceuticals, and NCCN.
JCT reports consulting or advisory role for Novartis, Lilly, Janssen, and Blueprint Medicines Corporation, Deciphera, Daiichi Sankyo, Epizyme, Agios, C4 Therapeutics, AADI, and Bayer.
SG reports owning stock and other ownership interests in Abbott Laboratories; consulting or advisory role for Blueprint Medicines Corporation, Deciphera, Bayer, Lilly, UpToDate, Research to Practice, MORE Health, Daiichi, and Kayothera; research funding from Blueprint Medicines Corporation, Deciphera, Daiichi Sankyo RD Novare, Merck, Eisai, SpringWorks Therapeutics, Theseus, and IDRX; patents, royalties, and other intellectual property reported for UptoDateExpert; testimony for Bayer; other relationship reported for Research to Practice, and WCG.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
These data were presented in part at the 2022 ASCO Annual Meeting: César Serrano, Sebastian Bauer, David Gómez-Peregrina, Yoon-Koo Kang, Robin L. Jones, Piotr Rutkowski, Olivier Mir, Michael C. Heinrich, William D. Tap, Kate Newberry, Alexandra Grassian, Stephen G. Miller, Hongliang Shi, Patrick Schöffski, Maria A. Pantaleo, Margaret von Mehren, Jonathan C. Trent, Suzanne George. Circulating tumor DNA (ctDNA) analyses of the phase III VOYAGER trial: KIT mutational landscape and outcomes in patients with advanced gastrointestinal stromal tumor (GIST). J Clin Oncol. 2022;40(16 suppl): Abstract 101.
REFERENCES
- 1.Heitzer E, Haque IS, Roberts CES, et al. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat Rev Genet. 2019;20(2):71–88. [DOI] [PubMed] [Google Scholar]
- 2.Serrano C, George S. Gastrointestinal stromal tumor: challenges and opportunities for a new decade. Clin Cancer Res 2020;26(19):5078–5085. [DOI] [PubMed] [Google Scholar]
- 3.Gómez-Peregrina D, García-Valverde A, Pilco-Janeta D, et al. Liquid biopsy in gastrointestinal stromal tumors: ready for prime time? Curr Treat Options Oncol. 2021;22(4):32. [DOI] [PubMed] [Google Scholar]
- 4.Arshad J, Roberts A, Ahmed J, et al. Utility of circulating tumor DNA in the management of patients with GI stromal tumor: analysis of 243 patients. JCO Precis Oncol. 2020;4:66–73. [DOI] [PubMed] [Google Scholar]
- 5.Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-KIT in human gastrointestinal stromal tumors. Science. 1998;279(5350):577–580. [DOI] [PubMed] [Google Scholar]
- 6.Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299(5607):708–710. [DOI] [PubMed] [Google Scholar]
- 7.Blay JY, Kang YK, Nishida T, et al. Gastrointestinal stromal tumours. Nat Rev Dis Primers. 2021;7(1):22. [DOI] [PubMed] [Google Scholar]
- 8.Casali PG, Zalcberg J, Le Cesne A, et al. Ten-year progression-free and overall survival in patients with unresectable or metastatic GI stromal tumors: Long-term analysis of the European Organisation for Research and Treatment of Cancer, Italian Sarcoma Group, and Australasian Gastrointestinal Trials Group Intergroup phase III randomized trial on imatinib at two dose levels. J Clin Oncol. 2017;35(15):1713–1720. [DOI] [PubMed] [Google Scholar]
- 9.Demetri GD, von Mehren M, Blanke CD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347(7):472–480. [DOI] [PubMed] [Google Scholar]
- 10.Liegl B, Kepten I, Le C, et al. Heterogeneity of kinase inhibitor resistance mechanisms in GIST. J Pathol. 2008;216(1):64–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heinrich MC, Corless CL, Blanke CD, et al. Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J Clin Oncol. 2006;24(29):4764–4774. [DOI] [PubMed] [Google Scholar]
- 12.Wardelmann E, Merkelbach-Bruse S, Pauls K, et al. Polyclonal evolution of multiple secondary KIT mutations in gastrointestinal stromal tumors under treatment with imatinib mesylate. Clin Cancer Res. 2006;12(6):1743–1749. [DOI] [PubMed] [Google Scholar]
- 13.Serrano C, Marino-Enriquez A, Tao DL, et al. Complementary activity of tyrosine kinase inhibitors against secondary KIT mutations in imatinib-resistant gastrointestinal stromal tumours. Br J Cancer. 2019;120(6):612–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Demetri GD, van Oosterom AT, Garrett CR, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368(9544):1329–1338. [DOI] [PubMed] [Google Scholar]
- 15.Demetri GD, Reichardt P, Kang YK, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381(9863):295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blay JY, Serrano C, Heinrich MC, et al. Ripretinib in patients with advanced gastrointestinal stromal tumours (INVICTUS): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2020;21(7):923–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heinrich MC, Corless CL, Demetri GD, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21(23):4342–4349. [DOI] [PubMed] [Google Scholar]
- 18.Heinrich MC, Maki RG, Corless CL, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol. 2008;26(33):5352–5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heinrich MC, Jones RL, von Mehren M, et al. Avapritinib in advanced PDGFRA D842V-mutant gastrointestinal stromal tumour (NAVIGATOR): a multicentre, open-label, phase 1 trial. Lancet Oncol. 2020;21(7):935–946. [DOI] [PubMed] [Google Scholar]
- 20.Grunewald S, Klug LR, Muhlenberg T, et al. Resistance to avapritinib in PDGFRA-driven GIST is caused by secondary mutations in the PDGFRA kinase domain. Cancer Discov. 2021;11(1):108–125. [DOI] [PubMed] [Google Scholar]
- 21.Kang YK, George S, Jones RL, et al. Avapritinib versus regorafenib in locally advanced unresectable or metastatic GI stromal tumor: a randomized, open-label phase III study. J Clin Oncol. 2021;39(28):3128–3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Evans EK, Gardino AK, Kim JL, et al. A precision therapy against cancers driven by KIT/PDGFRA mutations. Sci Transl Med. 2017;9(414):eaao1690. [DOI] [PubMed] [Google Scholar]
- 23.Shu Y, Wu X, Tong X, et al. Circulating tumor DNA mutation profiling by targeted next generation sequencing provides guidance for personalized treatments in multiple cancer types. Sci Rep. 2017;7(1):583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Serrano C, Vivancos A, Lopez-Pousa A, et al. Clinical value of next generation sequencing of plasma cell-free DNA in gastrointestinal stromal tumors. BMC Cancer. 2020;20(1):99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arshad J, Barreto-Coelho P, Jonczak E, et al. Identification of genetic alterations by circulating tumor DNA in leiomyosarcoma: a molecular analysis of 73 patients. J Immunother Precis Oncol. 2020;3(2):64–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Distribution of affected KIT (A) and PDGFRA (B) exons in GIST ctDNA.
Figure S2. Distribution of affected PDGFRA codons (A) and mutational heterogeneity (B).
Figure S3. KIT ctDNA determination prognosticates avapritinib (A) and regorafenib (B) efficacy vs the total population. CI, confidence interval; mPFS, median progression-free survival.
Figure S4. ctDNA determination and avapritinib treatment outcomes in GIST. Absence of KIT ATP-BP mutation prognosticated improved avapritinib activity (A), whereas regorafenib antitumor activity did not differ regardless of ATP-BP mutation status (B). The presence of a KIT A-loop mutation alone was not prognostic of efficacy with avapritinib (C) or regorafenib (D). Presence of both an ATP-BP and A-loop mutation was prognostic of avapritinib (E) and regorafenib (F) outcomes in patients with GIST. (G), (H) Increased KIT mutational heterogeneity (>3 mutations) did not impact PFS in either treatment arm. ABP, ATP-BP; AL, activation loop; CI, confidence interval; mPFS, median progression-free survival; NE, not estimable.
Table S1. Demographics and baseline characteristics.
ctDNA, circulating tumor DNA; ECOG, Eastern Cooperative Oncology Group.
Table S2. Association between clinical variables and detection of KIT or PDGFRA ctDNA.
CAT, computed axial tomography; CI, confidence interval; coef, coefficient; ctDNA, circulating tumor DNA; OR, odds ratio; std err, standard error; TKI, tyrosine kinase inhibitor.
Table S3. KIT mutations and distribution.
CDS, coding sequence; HGNC, HUGO Gene Nomenclature Committee; HGVSc, Human Genome Variation Society coding sequence; HGVSp, Human Genome Variation Society protein sequence.
Table S4. KIT and PDGFRA novel mutations found through ctDNA sequencing.
ctDNA, circulating tumor DNA; CDS, coding sequence; HGNC, HUGO Gene Nomenclature Committee; HGVSc, Human Genome Variation Society coding sequence; HGVSp, Human Genome Variation Society protein sequence.
Table S5. PDGFRA mutations and distribution.
CDS, coding sequence; HGNC, HUGO Gene Nomenclature Committee; HGVSc, Human Genome Variation Society coding sequence; HGVSp, Human Genome Variation Society protein sequence.