

Abstract
There is growing interest in identifying antibodies that protect against infectious diseases, especially pathogens for which no vaccine is yet available and those for which natural exposure does not confer protection. Many of these challenging pathogens express complex arrays of virulence-associated proteins that aid in immune avoidance to allow for growth as opportunistic or latent infections. Some organisms have developed strategies to selectively destroy pathogen-directed antibodies, while others create decoy epitopes that trick the host immune system into generating antibodies that are at best non-protective and, at worst, enhance pathogenesis. Design of pathogen-resistant antibodies can enhance protection and guide development of vaccine immunogens against these complex pathogens. In this review, we will discuss general strategies for design of antibodies resistant to specific immune defense mechanisms.
Keywords: Passive immunization, vaccines, immune evasion, Fc engineering, bispecific antibody, antibody-drug conjugate
1. Introduction
The immune system provides critical defenses against invading pathogens which are increasingly important in the face of rising resistance against small molecule drugs and a shrinking pipeline of new antimicrobials. Protective antibodies are key components of any immune response The es they can neutralize secreted toxins, block interactions with host cells and recruit immune system components. These antibodies can be elicited by natural infection, vaccination or be administered passively as purified proteins. Indeed, monoclonal antibodies are now available to treat five infectious diseases: respiratory syncytial virus (RSV), anthrax, recurrent Clostridium difficile, COVID-19 and Ebola.
The earliest efforts used the entire pathogen with its complex set of antigens in attenuated or inactivated vaccines. However, subsequent efforts aimed to identify a pathogen’s Achilles’ heel: a single target that cripples the pathogen when bound by an antibody. This approach supported the design of successful vaccines against tetanus, diphtheria and other diseases and antibodies against RSV, anthrax and C. difficile. Nevertheless, it has proven challenging to develop effective vaccines and antibody therapeutics against more complex pathogens for which natural immunity is often insufficient to prevent re-infection. These pathogens encode diverse proteins whose expression is orchestrated by complex regulatory pathways, with few antigens expressed across different disease states. Expression of functionally redundant virulence factors and strain variation can further complicate target selection. In addition, many pathogens have evolved diverse immune evasion strategies and, in some cases, can avoid elimination after phagocytosis. Even pathogens with relatively few genes, such as the influenza and SARS-CoV-2 viruses, have managed to rapidly evade immune responses against the few proteins targeted, emphasizing the need for more sophisticated antibodies.
This review will focus on general strategies used by bacterial and viral pathogens to evade capture by antibodies and protein engineering efforts to overcome them. These strategies include identification of antibodies that target conserved but poorly accessible or rare epitopes. Antibodies can be engineered to undermine pathogen efforts to degrade, capture or block antibodies that would otherwise trigger potent protective immune responses. Finally, antibodies can disrupt pathogen schemes to suppress complement activation or shelter inside cells. These approaches are expected to contribute to development of successful therapeutics for otherwise challenging pathogens.
2. Targeting pathogens that shield vulnerable epitopes
A key requirement for antibody targeting is facile recognition of pathogen-associated molecules. The best targets are expressed by most if not all strains in different tissues and during multiple stages of infection, are readily accessible, and either perform critical functions for disease progression or recruit opsonins that mediate pathogen destruction. Unsurprisingly, this provides selective pressures for pathogens to conceal these vulnerable epitopes. Structure-function studies of viral glycoproteins proteins in complex with neutralizing and non-neutralizing antibodies have revealed common mechanisms of antibody escape, including antigenic drift, epitope shielding and immune redirection to dominant but non-protective epitopes. These insights, in conjunction with new epitope-specific and target-agnostic antibody discovery tools (1, 2), support design of more resilient antibody therapeutics.
2.1. Antibodies binding conserved epitopes that resist antigenic drift
Considerable effort has been invested in identifying antibodies that target conserved epitopes on enveloped viruses, including RSV, influenza, HIV and coronaviruses, in order to develop broadly reactive antibodies and vaccines that protect against many strains. These viruses employ a fusogenic glycoprotein to invade host cells that first attaches to a host cell receptor and then undergoes dramatic conformational changes to bring the viral and host membranes together and mediate membrane fusion. Efforts to elicit neutralizing antibodies have focused on the receptor binding domain to block this initial interaction. Indeed, this region is often immunodominant and antibodies binding key epitopes can be potently neutralizing. Seminal work with the RSV F fusion protein demonstrated that the pre-fusion conformation contains many more neutralizing epitopes than the post-fusion conformation and that these can be lost during conformational change (3). Antibody D25 binds the pre-fusion site Ø to block receptor binding and neutralize RSV 10–50-fold more potently than the antibody in current clinical use (palivizumab) (4). Because of these promising results, D25 is progressing through clinical trials as nirsevimab with an extended half-life Fc (5).
Identification of such potent antibodies has been challenging for other viruses, due in part to high sequence variation in the receptor binding domain which often limits antibody neutralization to a few strains. In the case of SARS-CoV-2, multiple antibodies blocking interactions between the spike protein and ACE2 receptor received emergency use authorization only to be rendered ineffective due to mutations in circulating variants (6, 7) (Figure 1). Since these viruses continue to bind the ACE2 receptor, efforts now focus on identifying antibodies that engage more conserved but less exposed residues responsible for ACE2 binding that are expected to be less susceptible to antigenic drift (8, 9). Nanobodies are particularly useful in this situation, as their small size and long CDR3 loops can access epitopes that are not available to IgG antibodies and are therefore less susceptible to antigenic drift resulting from human immune system pressures. One effort with immunized camels identified hundreds of CDR3 nanobody families targeting five new neutralizing epitopes on spike (10, 11). Among these were ultra-potent nanobodies that bind with picomolar affinities and neutralize multiple SARS-CoV-1 and −2 strains in animal models, while their high stabilities support nasal administration to achieve high concentrations at the site of infection.
Figure 1. Key domains and antibody-targeted vulnerable sites in viral fusogen proteins from SARS-CoV-2, HIV-1, and influenza.
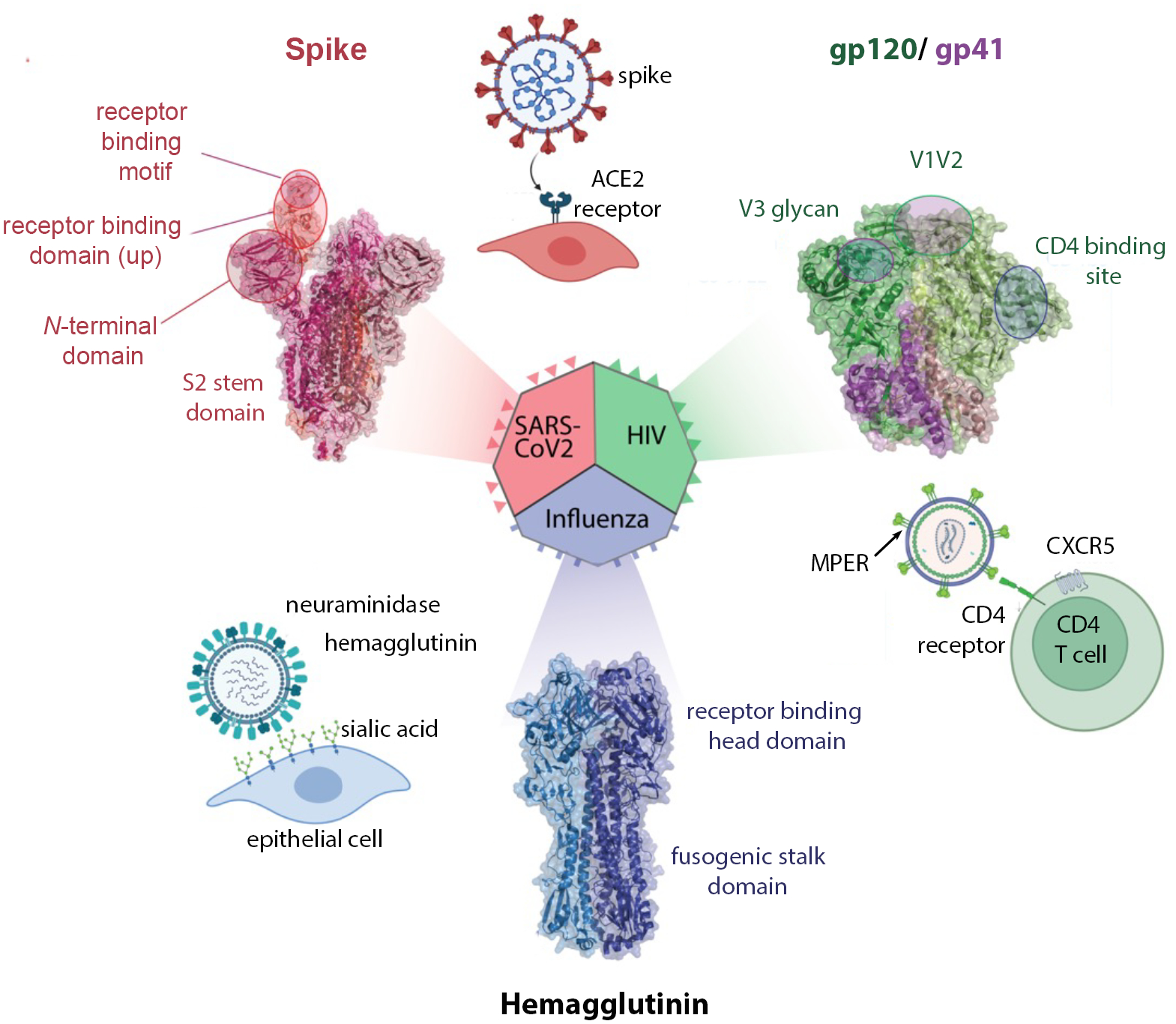
For each virus, the cellular receptors are shown as well as structure of the fusogen with key domains and epitopes identified. Structures for influenza hemagglutinin H3 from PDB 4FNK, for HIV-1 envelope PDB 5CJX, for SARS-CoV-2 spike from PDB 7SXT.
An alternate approach to identification of antibodies binding rare, conserved neutralizing epitopes is the identification of those binding functionally constrained epitopes. On fusogenic glycoproteins, this includes the highly conserved region that undergoes conformational change. In influenza, serum antibodies binding this region correlated with protection in an infection study (12) and it appears less susceptible to viral evasion (13). Whereas most neutralizing antibodies bind strain-specific epitopes in the receptor binding “head” domain of hemagglutinin, those binding the fusogenic “stalk” domain can neutralize multiple virus groups by inhibiting the conformational rearrangements necessary for membrane fusion in the low pH endosomes and by recruiting Fc effector functions (14) (Figure 1). Guthmiller, et al identified a highly conserved hemagglutinin anchor epitope that is common in the human B memory repertoire (15). Antibodies binding this epitope stabilize hemagglutinin near the viral membrane to neutralize a broad range of H1 strains as well as potential pandemic H2 and H5 strains and the common A388V stalk escape variant. Similarly, the 3A3 antibody (16) and 7A3 nanobody (17) both target the highly conserved beta-coronavirus hinge to bind a wide range of strains, with 7A3 shown to protect human ACE2-expressing mice from the Delta SARS-CoV-2 infection.
These successes have stimulated interest in identification of highly conserved epitopes on other target proteins, including those expressed by complex bacterial pathogens. Protective antibodies binding the bacterial surface can directly mediate phagocytosis by Fc receptors or activate complement, leading to C3b opsonization and phagocytosis or direct lysis. A key constraint is that the antigen surface density must be sufficiently high to support the antibody and receptor clustering necessary to trigger phagocytosis and complement; in the case of Neisseria meninigitidis, this appears to be >757 proteins per cell (18). The target protein should also be essential, since bactericidal antibodies can drive antigen loss, as has been observed for the pertactin protein expressed by Bordetella pertussis (19). To this end, antibodies binding the conserved and protective Streptococcal pneumonia antigen pneumococcal histidine triad protein (PhtD) were discovered by screening human donor cells (20). These bind antigen on encapsulated bacteria from several serotypes and protected mice against primary and secondary challenges (21) to offer a potential alternative to individually targeting the >100 distinct capsule serotypes. Similarly, conserved regions of the Lyme disease pathogen Borrelia burgdorferi outer surface protein VlsE (variable major protein-like sequence, expressed) have been identified, although their therapeutic potential remains to be defined (22). These reports are exciting because they suggest that antibodies binding bacterial surface proteins can be used to complement those binding bacterial carbohydrates.
2.2. Antibodies accessing concealed epitopes
Solvent-exposed epitopes are accessible to antibodies which exerts pressure for antigenic drift to evade antibody binding. Accordingly, pathogens can shade conserved and functionally important epitopes. One strategy is presentation of highly immunogenic but non-protective decoy epitopes to limit antibody recognition of adjacent vulnerable epitopes. For example, antibody IgG22 binds an epitope conserved on MERS-CoV, SARS-CoV-1 and −2 stem domains and prophylactically protected mice from MERS-CoV and SARS-CoV-2 lethal challenges (23). Antibodies binding this highly conserved epitope are rare since it is adjacent to an immunodominant and hyper-variable loop. Deleting this loop from the immunogen elicited high levels of IgG22-like antibodies that protected mice against different β-human coronaviruses. Similarly, the human cytomegalovirus gB fusogen contains a highly conserved “site 1” spanning residues 69–78 in antigenic domain 2 that elicits potently protective antibodies. These are effectively masked by antibodies binding the adjacent “site 2” spanning residues 50–54 and the immunodominant antigenic domain 1 which both elicit primarily non-neutralizing antibodies (24). Extrapolating from the IgG22 report, future immunogen design efforts may be able to focus immune responses on gB site 1 and sub-dominant but protective epitopes in other antigens.
A second strategy employed by pathogens is the use of glycosylation sites to shield vulnerable epitopes. The HIV-1 envelope is decorated with >25 N-linked glycosylation sites that shield broadly neutralizing epitopes including the V1V2 site, comprising the first and second variable regions on gp120 (Figure 1). Antibodies binding V1V2 correlated with protection in the Phase III RV144 Thai vaccine trial by mediating Fc effector functions (antibody-dependent cellular phagocytosis and toxicity as well as complement activation), rather than broad neutralization (25). This region elicits cross-reactive antibodies in many donors and but requires extensive somatic hypermutation and long CDRH3 loops to access the epitope (26). The CAP256-VR26 antibody lineage binds the V1V2 region and elicits potent Fc-mediated protection (27) while tolerating the loss of spike glycans N160 and N156 unlike prior V1V2 binding antibodies, such as PG9 (26). Immunization with cocktails of V1V2-scaffold immunogens that varied in glycosylation and multimerization elicited modestly neutralizing but functionally active Fc responses in macaques (28). Unlike other broadly HIV neutralizing antibodies which require years of maturation and extensive somatic hypermutation, CAP256-VRC26.08 contained far fewer mutations which could simplify strategies to elicit similar antibodies by vaccination.
A third strategy is structural blockade of vulnerable epitopes. For example, receptor binding epitopes may be transiently exposed only when needed to engage a receptor. This was reported for the SARS-CoV-1/ 2 cross-reactive antibody CR3022 which binds a cryptic epitope which is concealed when the receptor binding domain is in the “down” state, but exposed when this domain converts to the “up” state to engage receptor (29). Similarly, the broadly neutralizing anti-HIV antibodies 3BNC117 and 10–1074 bind only when CD4 engages the HIV-1 envelope to transiently expose vulnerable CD4 binding site (30, 31). Co-administration of soluble CD4 or CD4 mimics exposes the epitope for antibody binding; accordingly, non-human primates vaccinated with HIV-1 gp120 variants showed improved antibody-dependent cellular cytotoxicity against HIV-1 infected T cells only in the presence of a small molecule CD4 mimic (32). The membrane-proximal external region of the gp41 envelope protein elicits the broadest neutralizing monoclonal antibodies (e.g., 4E10, LN01, VRC42) (33), but these are rare as this epitope is only transiently exposed during the pre- to post-fusion transition (34, 35). These reports suggest that co-administration of two antibodies may be required to access some epitopes: one to stabilize the target protein in an open state (e.g., a receptor mimic) and a second antibody to bind the sensitive epitope.
2.3. Antibodies binding targets despite antigenic variability
The examples described above highlight the challenges of targeting vulnerable epitopes on viruses that require just a few proteins to mediate receptor binding and membrane fusion. The application of these strategies to bacterial pathogens expressing many more surface antigens presents additional challenges since, in many cases, the key molecule to target is not readily apparent. For example, influenza has just 8 gene segments whereas the opportunistic pathogen Pseudomonas aeruginosa has 321 core essential genes shared by all strains with a pangenome of 5,316 genes distributed across different isolates (36). Further complicating matters, many of the surface proteins are shielded from immune access by a capsule or expressed at low levels incompatible with triggering Fc functions.
Given their abundance, accessibility and immunogenicity, bacterial carbohydrates (lipopolysaccharides and capsular polysaccharides), have been primary targets for antibodies and vaccination. Indeed, vaccination with glycoconjugates of the N. meningitidis serogroup A capsule nearly eliminated clinical cases of this serogroup. Unfortunately, these molecules are also highly variable: while vaccines targeting P. aeruginosa lipopolysaccharide elicit protective immunity in animal models (37) there are >20 O-antigen serotypes, many with multiple subtypes (38), which would necessitate development of serotype-specific therapeutics. To explore this option, antibody KBPA101 was developed to target the O-11 serotype common in clinical isolates with a 600 nM functional affinity. It potently promotes opsonophagocytic killing due to its formulation as a pentavalent IgM isotype and entered clinical trials (39). As a more general approach, antibody F598 was developed to target the surface polysaccharide poly-N-acetyl-d-glucosamine expressed by a range of pathogenic bacteria. This antibody induces complement-dependent opsonic and bactericidal effects against multiple organisms including Listeria monocytogenes, Streptococci pyogenes and Burkholderia species (40). Structural analyses of antibody-polysaccharide complexes have revealed the importance of acetyl groups in recognition that anticipates development of additional potent yet specific anti-polysaccharide antibodies (41–44).
Given the diversity of bacterial carbohydrates and challenges in generating antibodies against these flexible molecules, conserved surface proteins may be better suited as antibody targets. An antibody binding the essential Escherichia coli outer membrane export protein BamA was identified which binds a single extracellular loop to disrupt BamA function. Antibody binding induced bacterial stress responses, disrupted outer membrane integrity and killed bacteria at sub-nanomolar concentrations (45). However, since outer membrane proteins are buried beneath the surface carbohydrates, this antibody was only effective against strains with a minimal lipo-polysaccharide structure. The same group identified antibodies against a different E. coli outer membrane protein, LptD, only to find that no antibody-accessible loop impacts protein function (46). Together, these reports highlight the challenges of selecting appropriate bacterial targets a priori. To instead isolate antibodies using a target-agnostic approach, DiGiandomenico et al (47) identified antibodies from infected and healthy humans that bound intact P. aeruginosa cells and screened these for opsonophagocytic killing. A panel of single gene knock-out P. aeruginosa isolates revealed that all selected antibodies bound the surface-exposed polysaccharide Psl, which is conserved across multiple strains and expressed across multiple disease states. Cam-003 binds 85% of 173 P. aeruginosa clinical isolates and, despite an affinity of just 144 nM, provided strong protection in a mouse acute lethal pneumonia model. It recently completed Phase II clinical trials as part of a bispecific antibody (48).
Pathogen adaptability necessitates creativity in antibody targeting approaches. This can include antibody discovery strategies using antigenic “baits” from multiple strains to identify cross-reactive antibodies against a known target, an approach that has identified a range of antibodies binding different viral epitopes. To expand these approaches to more complex bacterial pathogens, target-agnostic approaches followed by extensive screening for the desired function has shown promise. While not discussed here, prediction of immune escape pathways via machine learning, computation (49), as well as in vivo (50) and in vitro pathogen passaging (51–53) is also expected to support development of resilient antibody therapeutics. These reports highlight the promise of identifying therapeutics antibodies for challenging diseases such as tuberculosis (54) and malaria (55).
3. Targeting pathogens that undermine antibody Fc functions
While many efforts have focused on the development of neutralizing or blocking antibodies, the contributions of Fc effector functions to protection are being increasingly recognized. These can be mediated by human IgG1 and IgG3 isotypes, which bind host Fc receptors to mediate antibody-dependent cellular cytotoxicity (ADCC) by natural killer cells, antibody-dependent cellular phagocytosis by neutrophils and macrophages, and complement-dependent cytotoxicity. These responses rely upon Fc interactions with activating Fc receptors such as the complement component C1q and classical Fc receptors CD16A, CD32A and CD64 that are highly dependent on antigen density and epitope accessibility on the target cell surface (reviewed in (56)). Although Fc interactions can lead to antibody dependent enhanced disease, most notably with dengue infection (57, 58), they typically represent a powerful arm of the adaptive immune response that can complement neutralizing antibodies. Unfortunately, this has also provided selective pressures for pathogens to evade these functions by degrading or sequestering antibodies. Antibodies engineered to resist cleavage, block protease activities or resist antibody sequestration present opportunities to defuse these immune evasion strategies.
3.1. Antibodies that resist pathogenic protease activities
Enzymes such as papain (from the tropical fruit papaya, Carica papaya) and pepsin (an enzyme in the mammalian digestive tract) are commonly used to generate Fab and F(ab’)2 antibody fragments by cleaving sequences in the upper and lower hinge regions, respectively (59). Several human matrix metalloproteinases (MMPs) associated with tumor invasion and inflammation, such as MMP-3 and MMP-7, can also cleave immunoglobulins (60), suggesting these features are common across phylogenies. As part of an immune evasion repertoire, bacteria can secrete enzymes with homologous functions. For example, the cysteine protease Streptococcal pyrogenic exotoxin B (SpeB), Immunoglobulin G-degrading enzyme (IdeS) and endoglycosidase (EndoS) are secreted by Streptococcus pyogenes and cleave antibodies. While SpeB has broad specificity for immunoglobulins (61), IdeS is specific for the IgG hinge (62) and EndoS cleaves the conserved sugar. Similarly, Staphylococcus aureus produces glutamyl endopeptidase V8 (GluV8) that cleaves immunoglobulins in the upper hinge (63) while P. aeruginosa secretes abundant quantities of elastase B (LasB) that also cleaves immunoglobulins, possibly in the hinge region (64).
While useful for biotechnology applications, antibody cleavage can be detrimental to a protective immune response. Indeed, the addition of protease inhibitors decreased antibody cleavage while increasing C3b complement deposition and neutrophil phagocytosis of S. aureus bacteria (65). Most IgG-specific proteases employ a two-step process: one heavy chain hinge is cleaved, generating an intermediate, singly-cut product (60, 63), followed by slower cleavage of the second heavy chain to generate Fc and Fab or F(ab’)2 fragments. Singly-cleaved IgG retains antigen binding activity but is no longer able to promote effector functions (66). Auto-antibodies that bind cleaved IgG1 are observed in many individuals, showing that this intermediate product is immunogenic and physiologically relevant (67, 68). This accumulation of cleaved antibodies on the bacterial surface inhibits access of intact antibodies to antigens, thereby reducing recruitment of Fc effector functions. Hence, even incomplete antibody cleavage can effectively evade immune responses (69).
To restore Fc effector functions lost by antigen-bound and cleaved antibodies, the use of antibodies that recognize the new epitopes created by cleavage has been explored (Figure 2A). Intact antibodies can reconstitute the severed link between the target cell and Fc receptors on phagocytic cells. This approach was evaluated by Jordan et al. (67) using antibodies binding the S. aureus surface that are rapidly cleaved by GluV8 in a rabbit model of infection. The authors showed that immunization with peptides similar to antibody hinge sequences elicited a strong antibody response that specifically recognized GluV8-cleaved antibodies. Rabbits immunized with hinge-like peptides had reduced S. aureus colonization levels and sera from these animals recovered complement dependent cytotoxicity in an in vitro assay, versus an unimmunized group.
Figure 2. Antibodies that resist pathogenic protease activities to sustain recognition by host immune proteins, such as the C1q component of complement and CD32a Fc receptor found on phagocytes.
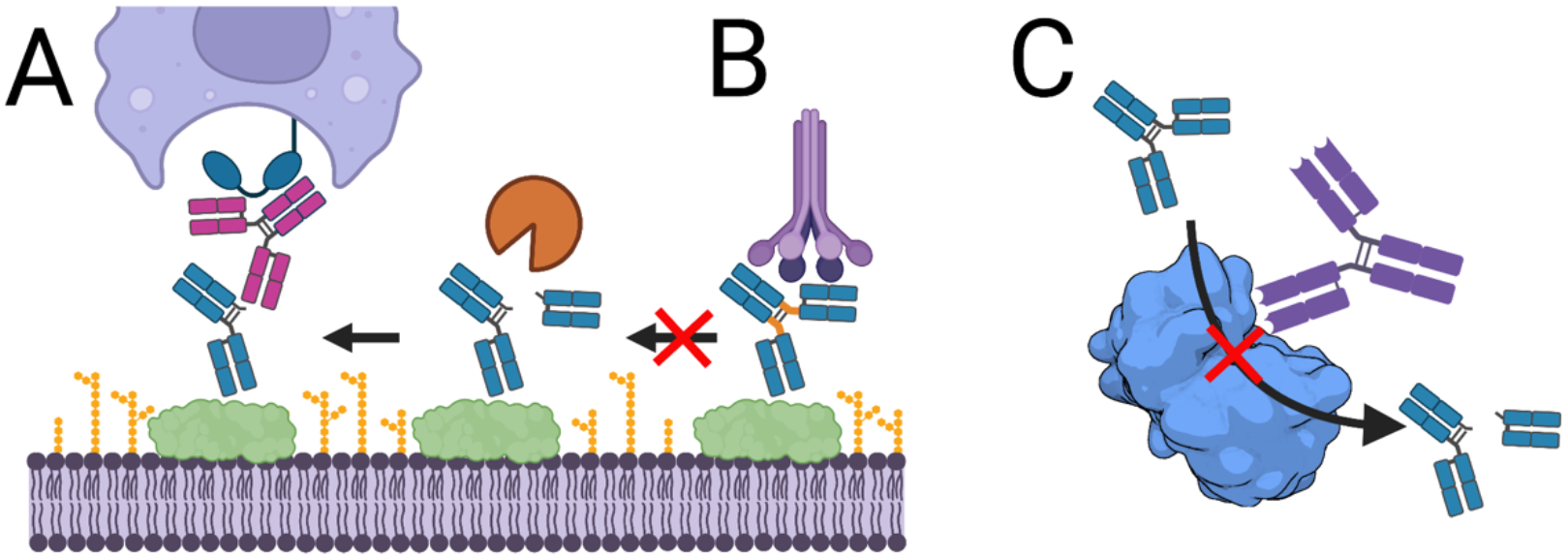
A, Antibodies recognizing hinge epitopes exposed after cleavage restore Fc functions to cleaved antibodies. B, Antibodies with engineered hinge regions no longer serve as suitable substrates for pathogenic proteases. C, Antibodies that block the activity of pathogenic proteases by directly blocking access to the active site (shown) or non-competitive (allosteric) mechanisms that bind an alternate enzyme epitope protect antibody functions. Shown is the structure of LasB (PDB 3DBK).
Engineering antibodies to resist cleavage is another strategy to counter proteolytic degradation. While IgG1 is the most abundant isotype in the human body and the most common isotype for therapeutic antibodies, IgG2 is generally more resistant to proteolytic cleavage (70). However, IgG2 binds more weakly to CD16a and C1q than IgG1, and accordingly mediates reduced Fc-dependent killing and complement deposition (71). Efforts to combine the resilience of IgG2 with the function of IgG1 resulted in chimeras in which the IgG2 lower hinge and CH2 sequences replaced those of IgG1. Unfortunately, antibodies also bind classical Fc receptors and C1q via interactions on the conserved lower hinge-CH2 interface, particularly residues E233/L234/L235/G236/G237/P238 (EU numbering) (72), an accessible region that serves as a substrate for many proteases. Accordingly, the introduction of mutations to increase antibody resistance to cleavage also negatively impacted Fc receptor binding and recruitment of immune effector functions.
To address the need for antibody protease-resistance while preserving effector functions, the Fc was engineered to recover the effector functions lost in an IgG1/IgG2 hybrid antibody. Kinder et al. (73) first introduced the lower hinge of an IgG2 domain in an IgG1 by introducing the substitutions E233P/L234V/L235A and deleting Gly236. As expected, this construct lost its ability to promote complement killing and phagocytosis in in vitro assays. Mutations previously established to selectively activate either complement killing or opsonophagocytosis were then introduced into this chimera. Variant 2h-DE (S239D/I332E) restored FcγR binding while 2h-AA (K326A/E333A) restored complement killing. Combining these mutations to make 2h-DAA (239D/K326A/E333A) and 2h-AEA (K326A/I332E/E333A) restored both complement killing and opsonophagocytic activities while retaining resistance to cleavage by multiple proteases (S. pyogenes IdeS, S. aureus GluV8, MMP-3 and MMP-12)(Figure 2B). Incorporation of protease-resistant Fc domains may support development of antibody therapeutics to treat bacterial infections.
Directly targeting proteases with neutralizing antibodies is an alternative approach to block proteolytic activity and has the advantages of blocking cleavage of all protease targets. This approach was initially explored for tumor-associated MMPs, resulting in identification of antibodies that act as competitive inhibitors with Ki values ~5 nM (74). It was extended to the P. aeruginosa pseudolysin (LasB) protease which supports early infection by cleaving elastin, collagen, IgA and IgG and complement proteins (for review, see (75)). Small molecule inhibitors suffer from poor specificity or activity, although recent molecules were reported to have Ki values of ~120–160 nM (76). As an alternative approach, Santajit et al. identified antibodies blocking LasB activity from the lymphocytes of healthy volunteers (77). Two clones were shown to bind LasB with modest EC50 values of ~100 nM and demonstrated concentration-dependent inhibition enzyme activity (Figure 2C). Given that this protease is conserved and abundantly expressed by P. aeruginosa, LasB neutralization may improve the efficacy of vaccine-elicited or therapeutic antibodies against this pathogen in the early stages of lung infection (78).
Antibody therapeutics that are resistant to proteolytic cleavage may improve their protective capacity and can be achieved by identification of Fc substitutions that resist degradation. An attractive feature of Fc engineering is that once a suitably engineered Fc is developed, it can be combined with Fab arms binding any antigen. Alternatively, an antibody blocking enzyme activity could be developed for each protease of interest. If supported by studies with monoclonal antibodies, appropriately engineered protease immunogens could enhance protection conferred by naturally and vaccine-elicited polyclonal antibodies.
3.2. Antibodies that block bacterial Fc binding proteins
Various bacterial pathogens express virulence factors that bind conserved antibody sequences. These captured antibodies can then shield the pathogen, blocking immune access to other surface antigens, antagonizing Fc effector functions and even altering immune cell signaling. While multiple organisms secrete antibody-binding proteins, including the S. aureus second immunoglobulin-binding protein (Sbi), S. pyogenes proteins G and M and Peptostreptococcus magnus protein L (79), the S. aureus protein A is the most studied (Figure 3). Protein A is expressed by all colonizing isolates and is comprised of five homologous domains each able to bind the Fc of all human IgG isotypes except IgG3 and all mouse IgG isotypes except IgG1 with high-affinity (Kd values of 2.6 to 14 nM) (80, 81). When attached to the bacterial cell wall via a C-terminal anchor, it prevents antibody-dependent complement activation by interfering with IgG hexamer formation (82) and prevents opsonophagocytic killing by directly blocking FcRn binding to reduce circulating half-life (83) and sterically blocking Fc interactions with the low affinity Fc receptors (84). Additionally, some protein A is released from the bacterial surface and to bind the framework of VH3-type B-cell receptors. Cross-linking of these receptors activates B cells, leading to a clonal expansion and apoptotic collapse that limits anti-Staphylococcus responses (85).
Figure 3. Antibodies that resist capture by Fc binding protein.
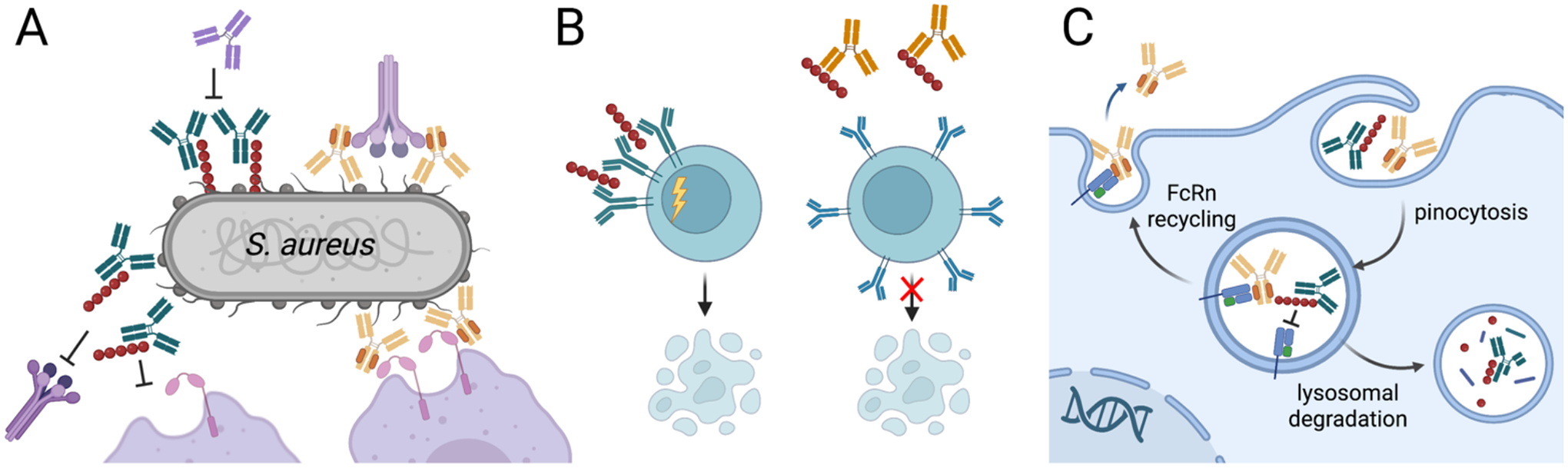
A. S. aureus protein A disrupts antibody responses in multiple ways, which can be restored by Fc domains with reduced protein A affinity (most IgG3 allotypes or engineered IgG1 domains) or antibodies that bind protein A to block Fc capture. A, Membrane-bound protein A can block antibody Fc binding to the low affinity Fc receptors CD16b and CD32a, block the Fc hexamerization required for efficient recruitment of C1q and activation of complement and shield the bacterial surface from recognition by antibody Fab domains. Secreted protein A can B, crosslink- VH3 domains to trigger B cell receptor activation and apoptotic collapse and C, block antibody recycling by Fc/FcRn binding to reduce antibody half-life. Unmodified antibodies shown in green, Fc engineered antibodies shown in yellow, anti-protein A antibodies shown in orange.
Directly targeting protein A with the antigen-binding paratopes of monoclonal antibodies has been explored to block its Fc binding activities and support bacterial opsonophagocytosis. Antibody 514G3 binds an epitope that is accessible on the bacterial surface in the presence of serum antibodies. When produced as a human IgG3, the only human isotype not captured by protein A due to an H435R substitution present in 24 of 29 allotypes (86), 514G3 mediated enhanced S. aureus killing versus an isotype control in an opsonophagocytic assay (87). In a mouse model, it demonstrated protection when administered prior to lethal challenge with methicillin-resistant S. aureus, while a lower antibody dose exhibited synergy with vancomycin antibiotic treatment. Antibody 514G3 was subsequently evaluated in a Phase I/II study in patients hospitalized with S. aureus bacteremia (NCT02357966), although results have yet to be reported. Antibody 3F6 binds protein A and the related protein Sbi to block Fc capture and B cell cross-linking effects. Passive immunization with 3F6 as a mouse IgG2a protected neonatal mice against bloodstream infection and allowed for higher serum IgG titers against other S. aureus antigens (88, 89).
Design of antigens that elicit strong 512G3 and 3F6-like protein A responses has been pursued, with the goal of reducing protein A affinity for the Fc. An initial detoxified variant SpAKKAA with lysine substitutions in residues 9 and 10 and alanine at residues 36 and 37 in each protein A domain exhibited negligible Fc and 100-fold reduced VH3 affinity. Unlike wild-type protein A, immunization with SpAKKAA elicited high-titer antibodies that promoted opsonophagocytic killing and protected mice against bloodstream S. aureus infection. However, SpAKKAA continued to cause animal distress, suggesting it retains some B cell superantigen activities. Accordingly, a second rational design effort combined the Q9E/Q10E changes and also altered residue S33 to E or T (SpAQQE and SpAQQT) to further reduced Vh3 cross-linking capacity (90). These variants exhibit negligible affinity for Fc and Fab while showing reduced mast cell degranulation and retaining the immunogenic and protective qualities of SpAKKAA. Design of immunogens that lose affinity for immune components and thereby focus the resulting antibody repertoire on disrupting immune protein capture has promise for this class of antigen.
3.3. Antibodies engineered to resist capture by Fc binding proteins
To support development of monoclonal antibodies for therapeutic use, the antibody Fc domain can be engineered to reduce affinity for pathogenic Fc binding proteins. Prior work demonstrated that antibodies with higher affinity for activating CD16a and CD32a host receptors due to afucosylation or amino acid sequence changes have enhanced effector functions that are relevant for cancer (91–93), but the potential impacts on infectious disease responses are less well understood. The combination of a human IgG1 Fc with two residue changes that reduce protein A binding (H435R and Y436F) (94) with variable regions binding the abundant surface glycopolymer wall teichoic acid on S. aureus mediated efficient bacterial phagocytosis regardless of whether the bacteria expressed a fully functional protein A (84). Subsequent rational Fc engineering identified variant R-QQV with four amino acid changes in human IgG1 (H435R, T307Q, Q311V, A387V) that reduced protein A but not FcRn binding. When combined with variable regions from the protein A binding antibody 3F6, 3F6-hIgG1R-QVV demonstrated reduced kidney colonization and abscess formation in mice with protection appearing to correlate with enhanced activation of C1q at the bacterial surface (95). Complementary approaches could develop IgG3 therapeutics which have multiple advantages in addition to protein A resistance (96) or Hexabodies whose enhanced Fc-Fc interactions may limit protein A effects.
Analogous Fc binding proteins are expressed by herpes viruses, including the gE/gI of alpha-herpes viruses and gp68 and RL11 family of the beta-herpes virus cytomegalovirus. These bind all human IgG isotypes, with gE/gI and gp68 binding near the CH2/CH3 elbow to competitively inhibit FcRn binding (97, 98), while gp34 binds near the hinge to inhibit binding of classical Fcγ receptors such as CD16a. When these viral Fc receptors are expressed on an infected cell surface, they can capture the Fc domain of antibodies binding adjacent viral glycoproteins. This antagonism inhibits Fc binding to and signaling through host Fc receptors on an immune cell (99, 100) and can result in antibody internalization (99, 101) for lysosomal degradation or repackaging to cloak the virion with antibodies (102, 103). These proteins appear to limit antibody efficacy; for instance, HSV gE/gI complex inhibits antibody mediated elimination of HSV infected cells in vitro and in vivo (104). Similarly, antibodies binding cytomegalovirus proteins more potently bound human Fc receptors and induced ADCC when virus strains lacking the Fc receptors were used to infect fibroblasts (105).
This suggests that Fc domains could be engineered to enhance the potency of non-neutralizing anti-herpes virus responses such as ADCC. An initial report modified the antibody Fc to include the S239D and I332E residues which increase Fc-CD16A affinity by 100-fold (91). When combined with variable regions binding the viral glycoprotein gB which is expressed on the infected cell surface and used in in vitro NK cell degranulation assays, a ~3-fold increase was observed for modified versus wild-type IgG1 when multiple antibodies binding the same antigen were pooled (106). Reasoning that Fc modifications to instead reduce viral Fc receptor binding while retaining affinity for host receptors would provide a wider therapeutic window, we used directed evolution and yeast display to engineer the human IgG1 Fc domain for 70-fold reduced gp68 affinity and 45-fold reduced gp34 affinity but unaltered binding to FcRn and CD16A. When combined with Fab arms binding the major viral glycoprotein gB, modified Fc G2 eliminated antibody internalization while restoring CD16a signaling and ADCC activities when incubated with CMV-infected fibroblasts and human NK cells in vitro (107).
Preventing pathogen sequestration of immune molecules can allow the host immune system to respond effectively to infection. In the S. aureus example, antibody 514G3 blocks protein A binding to any antibody Fc while use of the IgG3 Fc prevents protein A capture of the therapeutic. By understanding antibody-escape pathways elicited by immune evasion genes, we can design better therapeutics. By analogy, Fc binding proteins may undermine the efficacy of multiple antibodies, including those targeting other bacterial pathogens and herpes viruses. Advances in Fc engineering technologies (56) provides opportunities to develop designer Fc domains that mediate potent effector functions. These reports provide proof-of-concept for antibodies targeting protein A that may be extended to other Fc binding proteins.
4. Targeting pathogens that undermine host immunity
Antibodies binding conserved and accessible targets often require host immune responses to prevent or resolve an infection. In these situations, opsonized pathogens can be destroyed by a variety of mechanisms, including complement lysis, macrophage/ neutrophil phagocytosis or natural killer-mediated killing of infected cells. For example, simply tagging a pathogen with an antibody to mediate phagocytosis via Fc receptors instead of the natural invasion pathway can result in delivery of the pathogen to the lysosome for destruction (108). In response, bacterial and viral pathogens have evolved strategies to evade these immune functions, including proteins that disrupt the complement cascade or impair the ability of host immune cells to capture and destroy internalized pathogens. Antibodies blocking immune-evasive proteins can restore immune function while engineered antibodies can target bacteria in their intracellular hiding places.
4.1. Antibodies that preserve complement activities
The classical complement pathway is activated by abundant surface antigens that allow for binding and hexamerization of IgG antibodies or, more effectively, binding by the naturally pentameric or hexameric IgM molecule (Figure 4). These antibodies can then engage the six globular heads of the C1q protein to activate a proteolytic cascade (109). The resulting C3b deposition on the pathogen surface mediates phagocytosis by engaging complement receptors on leukocytes, particularly complement receptor 3 (CR3). If C3b densities are high, the lytic membrane attack complex can form to lyse gram-negative bacteria and virions. Since not all antigens are spatially presented to support efficient formation of IgG hexamers, residue changes (E430G and E345K or R) were identified that independently increase the Fc-Fc interactions supporting hexamer formation, leading to increased C1q binding and 5–7-fold enhanced complement activation (110). When combined with an antibody targeting a conserved N. gonorrhoeae lipo-oligosaccharide epitope, the Hexabody Fc mediated enhanced bacterial clearance from mice as compared to an unmodified Fc using mechanisms that required complement activation only (111).
Figure 4. Microbial disruption of the classical complement cascade by recruiting inhibitors or degrading complement proteins.
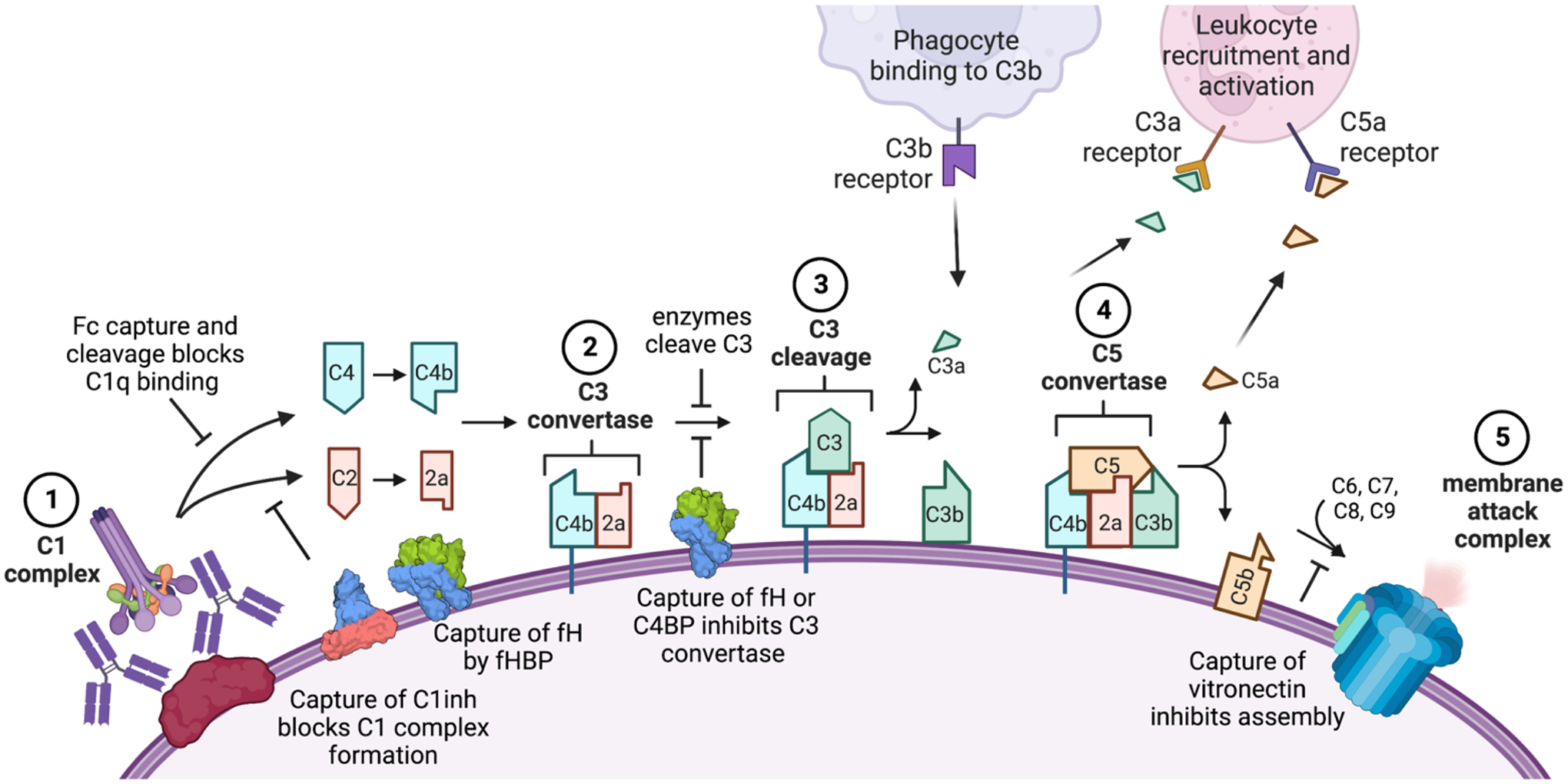
Key steps of the classical pathway of antibody activation shown: (1) the complement proteins C1q and then C1r and C1s bind the microbial surface or IgG/ IgM to form the C1 complex; (2) this cleaves C2 and C4 to produce C4b and C2a which form the C3 convertase; (3) this cleaves C3 to release C3a and deposit C3b covalently on the cell surface; (4) when C3b levels are high, it joins the C3 convertase to form the C5 convertase and deposit C5b on the surface; (5) components C6, C7, C8 and C9 join C5b to form the membrane attack complex and lyse the target cell. The lectin pathway follows a similar cascade but is initiated by the mannose binding lectin complex which recruits C1q, while the alternate pathway results from spontaneous C3 cleavage and C3b deposition to join the cascade at the C3 convertase step using an alternate C3b/Bb complex. Many of these steps can be inhibited by pathogen components, including proteins that bind or cleave the antibody Fc to inhibit C1q recruitment (e.g., protein A, staphylokinase); proteins that recruit host complement regulators (e.g., the Neisseria factor H binding protein [fHBP] which recruits factor H [fH] and B. pertussis Vag8 which recruits C1 inhibitor [C1inh]) and enzymes that degrade complement components (e.g., staphylokinase depletion of C3 away from the microbial surface). Engineering efforts to overcome these strategies include use of Hexabodies whose altered Fc domains favor hexamerization and C1q binding, antibodies altered to resist capture by Fc binding proteins or cleavage by bacterial proteases and antibodies that target microbial evasion proteins to block their functions.
Complement evasion strategies are common in bacterial pathogens, including the production of capsules to shield antibody access to antigens, surface antigens that recruit complement inhibitors and proteases that cleave complement proteins (Figure 4). Antibodies targeting these antigens could simultaneously block inhibitor binding and recruit C1q to the pathogen surface, thereby contributing to bacterial killing (112). This has been demonstrated most convincingly with antibodies binding N. meningitidis, an important cause of bacterial meningitis and sepsis for which the classical complement pathway dominates bactericidal killing (111). When three antibodies binding different epitopes on the N. meningitidis factor H binding protein (fHBP) were compared, only the antibody blocking recruitment of the factor H (fH) complement inhibitor showed human complement-mediated bacterial lysis (113). The high antibody/fH affinity was likely critical for the antibody to successfully inhibit the ~1000-fold weaker affinity fH/fHBP interaction. Factor H binding protein is included in two licensed meningococcal serogroup B vaccines and induces high titers of bactericidal antibodies (114). However, fHBP is speculated to form complexes with factor H in serum which would limit development of protective antibodies that competitively inhibit the fH/fHBP interaction. To address this, an engineered fH immunogen was developed that includes two amino acid changes (R41S and H248L) that reduce fH affinity by >250-fold (115). After immunization of Rhesus macaques, this modified fHBP elicited three-fold higher serum IgG titers and 150-fold higher serum bactericidal titers than the native fHBP which correlated with increased deposition of the complement component C4b on live bacteria.
These data suggest that targeting complement-evading antigens expressed by other organisms may support vaccine development efforts. For example, a goal for future B. pertussis vaccines is to better limit human colonization, yet the organism is quite resistant to complement-mediated lysis. Accordingly, its four proteins involved in complement inhibition (the C4b-binding protein Fha, the C1 esterase inhibitor binding Vag8, as well as BapC and the Bordetella resistant to killing A protein BrkA) are attracting interest as future vaccine antigens and may benefit from engineering as did fHBP (116).
4.2. Antibodies that protect leukocyte functions
Many bacteria produce leukotoxins, which impair the abilities of immune cells to capture and destroy pathogens. Staphylococcus aureus produces a suite of pore-forming leukocidins with redundant and complementary functions, including alpha-hemolysin (Hla) and the five bicomponent cytotoxins HlgAB, HlgCB, LukSF, LukED and LukGH (117). Antibodies binding Hla have been evaluated in the clinic, with Suvratoxumab appearing promising but not meeting endpoints in Phase 2 clinical trials (118), while Tosatoxumab (119) is currently in Phase 3 clinical trials to evaluate prevention of ventilator-associated pneumonia caused by S. aureus (NCT03816956). To simultaneously neutralize multiple toxins and provide superior protection, a monoclonal antibody was engineered to bind a single epitope conserved among Hla and four additional leukocidins (120). A high affinity anti-Hla antibody was initially isolated from a human yeast display library, followed by randomization and selection to select for antibodies binding a highly conserved microdomain involved in pore formation. This effort yielded a single antibody that binds Hla, HlgB, LukF and LukD with Kd values <2.2 nM for each antigen and protected mice in lethal pneumonia and bacteremia models (121). This was combined with a second antibody specifically targeting the leucocidin LukGH (122). A cocktail of these two antibodies neutralized six different S. aureus toxins met Phase 1 endpoints (123) and is currently being evaluated in phase II clinical trials (NCT02940626) under the name ASN100.
Even if opsonizing antibodies bind conserved epitopes on the pathogen surface, secretion of leukotoxins may limit bacterial killing after phagocytosis. In this situation, antibodies blocking leukotoxin activities are expected to complement opsonizing antibodies. This is the case for Bordetella species, including B. pertussis, which secrete the 177-kDa adenylate cyclase toxin. The repeat-in-toxin (RTX) domain binds leukocytes via the αMβ2 integrin receptor, after which the N-terminal catalytic domain is translocated to the cytosol, where it binds calmodulin and rapidly converts available ATP to cAMP (124). This activity dysregulates cell signaling and reduces opsonophagocytosis and bacterial clearance during the early stages of infection (125, 126). We showed that neutralizing antibodies competitively inhibiting RTX-receptor binding (127, 128) and synergize with opsonizing anti-pertactin antibodies in a mouse pneumonia model (129). The same concept was illustrated by Tkaczyk et al, who showed that an opsonizing antibody binding ClfA synergized with an antibody neutralizing Hla in lethal pneumonia and bacteriemia models of S. aureus infection (130).
4.3. Antibody-antibiotic conjugates targeting internalized bacteria
While intracellular bacteria are traditionally viewed as having obligate or facultative intracellular lifestyles, there is an increasing appreciation that phagocytosed bacteria can serve as a reservoir of viable organisms to seed infection (131). For example, S. aureus is readily phagocytosed by macrophages and neutrophils (131). While most internalized bacteria are killed, a fraction resist killing and use the phagocytes to mediate dissemination to other sites. For S. aureus specifically, intracellular survival helps the bacteria evade innate immune defenses and destroy neutrophils by programmed necrosis (132) with leukocidins mediating bacterial survival/escape through induction of programmed necrosis (133). Moreover, many viruses perform membrane fusion events required for entry into host cells in the low pH endosome (e.g., influenza and Ebola). In both cases, the intracellular pathogen is largely inaccessible to antibodies and antibiotics. Clever engineering strategies can deliver multi-functional antibodies to these intracellular organisms to support pathogen destruction by natural immune cell functions or co-delivered antibiotic.
Opsonizing antibodies have been engineered to co-localize antibiotics or other anti-virulence molecules with intracellular pathogens. Multi-functional antibody-antibiotic conjugates rely on the antibody component binding an abundant bacterial surface antigen to tag it for phagocytosis into an endosome, which may harbor previously internalized bacteria. Local proteases then release the antibiotic to kill all bacteria within that vesicle. This strategy was developed by Lehar et al. (134) to target S. aureus using an antibody binding the conserved β-O-linked N-acetylglucosamine sugar on cell wall teichoic acid. This was expressed as a ThioMab with six unpaired cysteines to allow conjugation to a peptide linker and antibiotic. In the neutrophil phagosome, cathepsin B cleaved the linker to release the antibiotic, a rifamycin derivative which retains potent antibacterial activity in low pH environments (134, 135). In vitro, the antibody-antibiotic conjugate killed bacteria internalized by macrophages, endothelial and epithelial cells. In mice, prophylactic administration prevented kidney colonization better than vancomycin, while neither the antibody alone nor pooled sera were able to prevent infection spread. The promise of this strategy is supported by favorable Phase I clinical trial results of the Staph-targeting DSTA4637S (NCT03162250) (136).
This strategy depletes poorly accessible bacterial reservoirs that would otherwise present a source of recurrent infection. While bactericidal activity is a key requirement for new antibiotics, these often possess undesirable host toxicities. Conjugation to an antibody extends the circulating half-life and localizes the antibiotic to the infection site before release. This strategy has the potential to diminish systemic toxicities while achieving high local antibiotic concentrations within the phagosome necessary to kill the bacteria. Accordingly, it has been extended to P. aeruginosa, using an antibody binding the abundant lipo-polysaccharide O antigen to enhance antibiotic potency ~100-fold (from single digit micromolar minimum inhibitory concentrations), indicating that antibody-antibiotic conjugates could revitalize antibiotics with modest potencies (137). It may also be appropriate for obligate pathogens such as Burkholderia species (138).
4.4. Antibodies that access internalized pathogens
Bispecific antibodies use an analogous approach in which one binding site tags the pathogen for phagocytosis, while the other performs a complementary function, such as blockade of virulence factors that render the intracellular site hospitable to the pathogen. The most advanced therapeutic in this is class is MEDI3902, which recently completed Phase 2 clinical trials (NCT02696902) (139). One arm of MEDI3902 uses the Cam-003 binding site to bind the abundant Psl exopolysaccharide on the P. aeruginosa surface, activate complement and mediate neutrophil phagocytosis. The other arm binds PcrV to block type III secretion of toxins such as ExoS into the host cell cytosol. This blockade supports phagosome acidification and enhanced bacterial killing in preclinical experiments but did not meet endpoints in Phase II trials, perhaps due to small sample size (140, 141). A similar approach was used to neutralize filoviruses including Ebola, whose cryptic glycoprotein receptor binding site is exposed by proteases present in late endosomes, a site generally inaccessible to antibodies. Bispecific antibodies were generated with one arm blocking the receptor binding site at endosomal pH while the other arm mediated phagocytosis and endosomal delivery by binding a conserved non-neutralizing epitope on the virion (142). This concept could be extended to block other proteins that support pathogen escape or maintain the endosome as a hospital environment, such as the S. aureus leukotoxins (133).
The above strategies require the antibody and pathogen to engage prior to internalization which may present challenges for treating an established infection or eliminating all intracellular pathogens. As an alternative, antibodies can be delivered separately to join pathogens already present in an endosome or lysosome. This can occur by natural pinocytosis and FcRn-mediated antibody transport across epithelial cells; during this process, antibody-containing endosomes can fuse with those containing influenza virions to neutralize hemagglutinin and prevent fusion of the influenza virus and host cell membranes (143). If antibody-bound virions escape endosomes as part of the natural infective cycle, the cytosolic E3 ubiquitin ligase and Fc receptor TRIM21 can direct proteasomal-destruction. This appears to be the mechanism by which VP6-specific antibodies protect against rotavirus (144). Alternatively, antibodies can be directed to the lysosome by receptor-mediated endocytosis when designed to target the mannose-6-phophate receptor used to deliver enzyme replacement therapies (145). This approach was used to develop a second-generation bispecific Ebola endosomal neutralizing antibody that is resistant to viral escape (146).
The strategies described here target intracellular pathogens using complementary approaches: blockade of cytotoxic mechanisms that prevent host cell killing of internalized bacteria and targeted delivery of an antibiotic with the bacterium into the phagosome to mediate intracellular bacterial killing. In both cases, identification of a bacterial surface antigen as an antibody target is essential for success.
5. Conclusions
Opportunities for pathogens to cause disease have increased with increasing population densities, travel and immunocompromised individuals. Antibody engineering tools help define mechanisms of pathogen immune evasion and counter them by identifying functionally relevant antibody targets, engineering antibodies that render antibody evasion tactics useless and hunting pathogens hidden inside host cells. These efforts are performed with the knowledge that any new therapeutic will exert selective pressures and may drive emergence of organisms with altered traits. However, the persistence of many pathogens coupled, with the emergence of new pathogens underscores the need for innovative approaches to prevent and treat infections.
Figure 5. Antibodies that target intracellular pathogens.
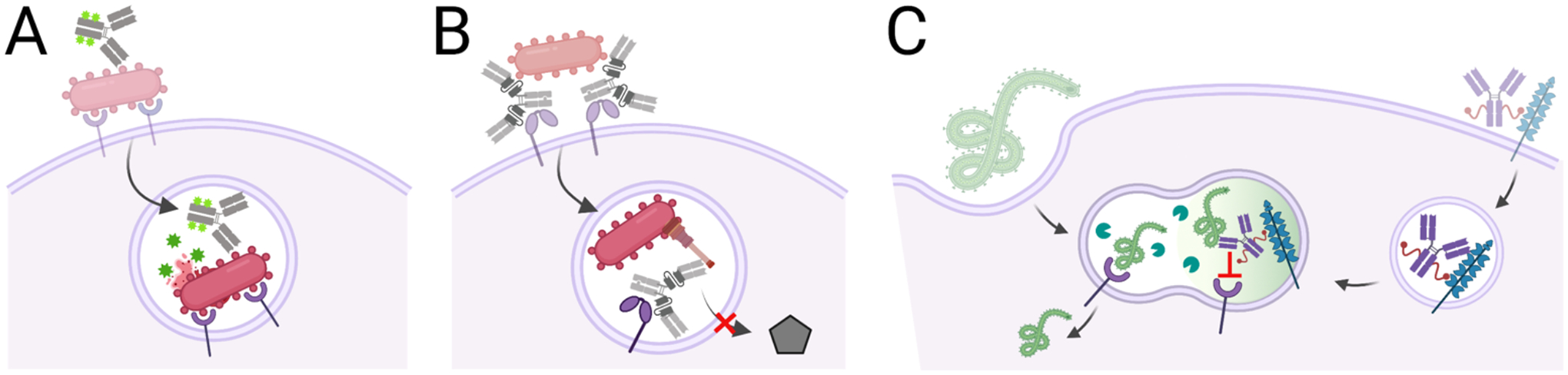
A, Antibody-antibiotic conjugates bind bacterial surface antigens and are internalized with the bacteria by bacterial or immune-mediated mechanisms into endosomal compartments. In this environment, the antibiotic is released to kill co-localized bacteria. B, Bi-specific antibody uses one binding site to bind the Psl antigen on the P. aeruginosa surface and mediate phagocytosis. Once in the endosome, that other antibody binding site blocks Type III secretion to support endosome acidification and bacterial killing. C, Antibodies block Ebola-receptor interactions in the endo-lysosome by hijacking the mannose-6-phosphate receptor to mediate antibody transfer to an endo-lysosome which may already contain Ebola virions. Once co-localized, the antibody blocks glycoprotein-receptor interactions and viral escape into the cytosol.
Summary Points.
Complex pathogens excel at concealing key epitopes from antibody detection and immune evasion. Effective antibody therapeutics can access vulnerable sites using strategies such as ultra-long CDR3 loops.
While non-neutralizing antibodies that trigger complement and Fc receptor activation are increasingly recognized as key components of successful immune responses, their contributions to protection are not fully understood and merit additional study.
Multiple bacterial and viral pathogens evade antibody effector functions by binding or cleaving antibody Fc domains. Antibodies engineered to resist capture or cleavage may confer enhanced protection.
Many pathogens enter the host cell cytosol from the protected space of an endosome or phagolysosome. Antibodies engineered to access these intracellular spaces and block pathogens at the site of entry may help eliminate these protected reservoirs.
Many pathogens recruit complement regulatory proteins to their surface to avoid triggering this arm of the immune system. Antibodies binding these surface proteins to block inhibitor recruitment may support complement activation while also serving as an opsonin.
Acknowledgements
This work was supported by the National Institutes of Health (AI155453 to J.A.M.), the Welch Foundation (#F-1767 to J.A.M.) and an NSF GRFP to A.N.Q. All figures made with BioRender. The authors thank Annalee Nguyen and Kelli Hager for helpful comments on the manuscript. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funders.
Footnotes
Disclosure statement JAM and ANQ are listed as inventors on patents describing antibody Fc domains that resist capture by CMV.
Literature cited
- 1.Setliff I, Shiakolas AR, Pilewski KA, Murji AA, Mapengo RE, et al. 2019. High-Throughput Mapping of B Cell Receptor Sequences to Antigen Specificity. Cell 179: 1636–46 e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pedrioli A, Oxenius A. 2021. Single B cell technologies for monoclonal antibody discovery. Trends Immunol 42: 1143–58 [DOI] [PubMed] [Google Scholar]
- 3.Ngwuta JO, Chen M, Modjarrad K, Joyce MG, Kanekiyo M, et al. 2015. Prefusion F-specific antibodies determine the magnitude of RSV neutralizing activity in human sera. Sci Transl Med 7: 309ra162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhu Q, McLellan JS, Kallewaard NL, Ulbrandt ND, Palaszynski S, et al. 2017. A highly potent extended half-life antibody as a potential RSV vaccine surrogate for all infants. Sci Transl Med 9: eaaj1928 [DOI] [PubMed] [Google Scholar]
- 5.Bergeron HC, Tripp RA. 2022. Breakthrough therapy designation of nirsevimab for the prevention of lower respiratory tract illness caused by respiratory syncytial virus infections (RSV). Expert Opin Investig Drugs 31: 23–29 [DOI] [PubMed] [Google Scholar]
- 6.Cao Y, Wang J, Jian F, Xiao T, Song W, et al. 2022. Omicron escapes the majority of existing SARS-CoV-2 neutralizing antibodies. Nature 602: 657–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.VanBlargan LA, Errico JM, Halfmann PJ, Zost SJ, Crowe JE Jr., et al. 2022. An infectious SARS-CoV-2 B.1.1.529 Omicron virus escapes neutralization by therapeutic monoclonal antibodies. Nat Med [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jette CA, Cohen AA, Gnanapragasam PNP, Muecksch F, Lee YE, et al. 2021. Broad cross-reactivity across sarbecoviruses exhibited by a subset of COVID-19 donor-derived neutralizing antibodies. Cell Rep 36: 109760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Starr TN, Czudnochowski N, Liu Z, Zatta F, Park YJ, et al. 2021. SARS-CoV-2 RBD antibodies that maximize breadth and resistance to escape. Nature 597: 97–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xiang Y, Huang W, Liu H, Sang Z, Nambulli S, et al. 2022. Superimmunity by pan-sarbecovirus nanobodies. Cell Rep 39: 111004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xiang Y, Nambulli S, Xiao Z, Liu H, Sang Z, et al. 2020. Versatile and multivalent nanobodies efficiently neutralize SARS-CoV-2. Science 370: 1479–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ng S, Nachbagauer R, Balmaseda A, Stadlbauer D, Ojeda S, et al. 2019. Novel correlates of protection against pandemic H1N1 influenza A virus infection. Nature Medicine 25: 962–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doud MB, Lee JM, Bloom JD. 2018. How single mutations affect viral escape from broad and narrow antibodies to H1 influenza hemagglutinin. Nature Communications 9: 1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DiLillo DJ, Tan GS, Palese P, Ravetch JV. 2014. Broadly neutralizing hemagglutinin stalk-specific antibodies require FcgammaR interactions for protection against influenza virus in vivo. Nat Med 20: 143–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guthmiller JJ, Han J, Utset HA, Li L, Lan LY, et al. 2022. Broadly neutralizing antibodies target a haemagglutinin anchor epitope. Nature 602: 314–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang Y, Nguyen AW, Hsieh C-L, Silva R, Olaluwoye OS, et al. 2021. Identification of a conserved neutralizing epitope present on spike proteins from all highly pathogenic coronaviruses. bioRxiv [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hong J, Kwon HJ, Cachau R, Chen CZ, Butay KJ, et al. 2021. Camel nanobodies broadly neutralize SARS-CoV-2 variants. bioRxiv : the preprint server for biology: 2021.10.27.465996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Biagini M, Spinsanti M, De Angelis G, Tomei S, Ferlenghi I, et al. 2016. Expression of factor H binding protein in meningococcal strains can vary at least 15-fold and is genetically determined. Proc Natl Acad Sci U S A 113: 2714–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martin SW, Pawloski L, Williams M, Weening K, DeBolt C, et al. 2015. Pertactin-Negative Bordetella pertussis Strains: Evidence for a Possible Selective Advantage. Clin Infect Dis 60: 223–7 [DOI] [PubMed] [Google Scholar]
- 20.Huang J, Gingerich AD, Royer F, Paschall AV, Pena-Briseno A, et al. 2021. Broadly Reactive Human Monoclonal Antibodies Targeting the Pneumococcal Histidine Triad Protein Protect against Fatal Pneumococcal Infection. Infect Immun 89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gingerich AD, Royer F, McCormick AL, Scasny A, Vidal JE, Mousa JJ. 2022. Synergistic Protection against Secondary Pneumococcal Infection by Human Monoclonal Antibodies Targeting Distinct Epitopes. J Immunol [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li L, Di L, Akther S, Zeglis BM, Qiu W. 2022. Evolution of the vls Antigenic Variability Locus of the Lyme Disease Pathogen and Development of Recombinant Monoclonal Antibodies Targeting Conserved VlsE Epitopes. Microbiol Spectr 10: e0174322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hsieh CL, Werner AP, Leist SR, Stevens LJ, Falconer E, et al. 2021. Stabilized coronavirus spike stem elicits a broadly protective antibody. Cell Rep 37: 109929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schrader JW, McLean GR. 2007. Location, location, timing: analysis of cytomegalovirus epitopes for neutralizing antibodies. Immunol Lett 112: 58–60 [DOI] [PubMed] [Google Scholar]
- 25.Kim JH, Excler JL, Michael NL. 2015. Lessons from the RV144 Thai phase III HIV-1 vaccine trial and the search for correlates of protection. Annu Rev Med 66: 423–37 [DOI] [PubMed] [Google Scholar]
- 26.Doria-Rose NA, Schramm CA, Gorman J, Moore PL, Bhiman JN, et al. 2014. Developmental pathway for potent V1V2-directed HIV-neutralizing antibodies. Nature 509: 55–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Richardson SI, Lambson BE, Crowley AR, Bashirova A, Scheepers C, et al. 2019. IgG3 enhances neutralization potency and Fc effector function of an HIV V2-specific broadly neutralizing antibody. PLOS Pathogens 15: e1008064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hessell AJ, Powell R, Jiang X, Luo C, Weiss S, et al. 2019. Multimeric Epitope-Scaffold HIV Vaccines Target V1V2 and Differentially Tune Polyfunctional Antibody Responses. Cell Rep 28: 877–95.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yuan M, Wu NC, Zhu X, Lee CD, So RTY, et al. 2020. A highly conserved cryptic epitope in the receptor-binding domains of SARS-CoV-2 and SARS-CoV. Science [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bournazos S, Klein F, Pietzsch J, Seaman MS, Nussenzweig MC, Ravetch JV. 2014. Broadly neutralizing anti-HIV-1 antibodies require Fc effector functions for in vivo activity. Cell 158: 1243–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Veillette M, Coutu M, Richard J, Batraville L-A, Dagher O, et al. 2015. The HIV-1 gp120 CD4-Bound Conformation Is Preferentially Targeted by Antibody-Dependent Cellular Cytotoxicity-Mediating Antibodies in Sera from HIV-1-Infected Individuals. Journal of Virology 89: 545–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Madani N, Princiotto AM, Mach L, Ding S, Prevost J, et al. 2018. A CD4-mimetic compound enhances vaccine efficacy against stringent immunodeficiency virus challenge. Nature Communications 9: 2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kwong PD, Wilson IA. 2009. HIV-1 and influenza antibodies: seeing antigens in new ways. Nat Immunol 10: 573–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pinto D, Fenwick C, Caillat C, Silacci C, Guseva S, et al. 2019. Structural Basis for Broad HIV-1 Neutralization by the MPER-Specific Human Broadly Neutralizing Antibody LN01. Cell Host Microbe 26: 623–37.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krebs SJ, Kwon YD, Schramm CA, Law WH, Donofrio G, et al. 2019. Longitudinal Analysis Reveals Early Development of Three MPER-Directed Neutralizing Antibody Lineages from an HIV-1-Infected Individual. Immunity 50: 677–91.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Poulsen BE, Yang R, Clatworthy AE, White T, Osmulski SJ, et al. 2019. Defining the core essential genome of Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 116: 10072–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fisher MW, Devlin HB, Gnabasik FJ. 1969. New immunotype schema for Pseudomonas aeruginosa based on protective antigens. J Bacteriol 98: 835–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Knirel YA. 1990. Polysaccharide antigens of Pseudomonas aeruginosa. Crit Rev Microbiol 17: 273–304 [DOI] [PubMed] [Google Scholar]
- 39.Horn MP, Zuercher AW, Imboden MA, Rudolf MP, Lazar H, et al. 2010. Preclinical in vitro and in vivo characterization of the fully human monoclonal IgM antibody KBPA101 specific for Pseudomonas aeruginosa serotype IATS-O11. Antimicrob Agents Chemother 54: 2338–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cywes-Bentley C, Skurnik D, Zaidi T, Roux D, Deoliveira RB, et al. 2013. Antibody to a conserved antigenic target is protective against diverse prokaryotic and eukaryotic pathogens. Proc Natl Acad Sci U S A 110: E2209–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Henriques P, Dello Iacono L, Gimeno A, Biolchi A, Romano MR, et al. 2020. Structure of a protective epitope reveals the importance of acetylation of Neisseria meningitidis serogroup A capsular polysaccharide. Proc Natl Acad Sci U S A 117: 29795–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Soliman C, Walduck AK, Yuriev E, Richards JS, Cywes-Bentley C, et al. 2018. Structural basis for antibody targeting of the broadly expressed microbial polysaccharide poly-N-acetylglucosamine. J Biol Chem 293: 5079–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fong R, Kajihara K, Chen M, Hotzel I, Mariathasan S, et al. 2018. Structural investigation of human S. aureus-targeting antibodies that bind wall teichoic acid. MAbs 10: 979–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ozdilek A, Huang J, Babb R, Paschall AV, Middleton DR, et al. 2021. A Structural Model for the Ligand Binding of Pneumococcal Serotype 3 Capsular Polysaccharide-Specific Protective Antibodies. mBio 12: e0080021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Storek KM, Auerbach MR, Shi H, Garcia NK, Sun D, et al. 2018. Monoclonal antibody targeting the beta-barrel assembly machine of Escherichia coli is bactericidal. Proc Natl Acad Sci U S A 115: 3692–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Storek KM, Chan J, Vij R, Chiang N, Lin Z, et al. 2019. Massive antibody discovery used to probe structure-function relationships of the essential outer membrane protein LptD. Elife 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.DiGiandomenico A, Warrener P, Hamilton M, Guillard S, Ravn P, et al. 2012. Identification of broadly protective human antibodies to Pseudomonas aeruginosa exopolysaccharide Psl by phenotypic screening. J Exp Med 209: 1273–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ali SO, Yu XQ, Robbie GJ, Wu Y, Shoemaker K, et al. 2019. Phase 1 study of MEDI3902, an investigational anti-Pseudomonas aeruginosa PcrV and Psl bispecific human monoclonal antibody, in healthy adults. Clin Microbiol Infect 25: 629 e1–29 e6 [DOI] [PubMed] [Google Scholar]
- 49.Verkhivker GM, Agajanian S, Oztas DY, Gupta G. 2021. Atomistic Simulations and In Silico Mutational Profiling of Protein Stability and Binding in the SARS-CoV-2 Spike Protein Complexes with Nanobodies: Molecular Determinants of Mutational Escape Mechanisms. ACS Omega 6: 26354–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bashor L, Gagne RB, Bosco-Lauth AM, Bowen RA, Stenglein M, VandeWoude S. 2021. SARS-CoV-2 evolution in animals suggests mechanisms for rapid variant selection. Proceedings of the National Academy of Sciences 118: e2105253118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meijers M, Vanshylla K, Gruell H, Klein F, Lässig M. 2021. Predicting in vivo escape dynamics of HIV-1 from a broadly neutralizing antibody. Proceedings of the National Academy of Sciences 118: e2104651118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Starr TN, Greaney AJ, Addetia A, Hannon WW, Choudhary MC, et al. 2021. Prospective mapping of viral mutations that escape antibodies used to treat COVID-19. Science 371: 850–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee JM, Huddleston J, Doud MB, Hooper KA, Wu NC, et al. 2018. Deep mutational scanning of hemagglutinin helps predict evolutionary fates of human H3N2 influenza variants. Proceedings of the National Academy of Sciences 115: E8276–E85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Watson A, Li H, Ma B, Weiss R, Bendayan D, et al. 2021. Human antibodies targeting a Mycobacterium transporter protein mediate protection against tuberculosis. Nat Commun 12: 602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gonzales SJ, Clarke KN, Batugedara G, Garza R, Braddom AE, et al. 2022. A Molecular Analysis of Memory B Cell and Antibody Responses Against Plasmodium falciparum Merozoite Surface Protein 1 in Children and Adults From Uganda. Front Immunol 13: 809264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Delidakis G, Kim JE, George K, Georgiou G. 2022. Improving Antibody Therapeutics by Manipulating the Fc Domain: Immunological and Structural Considerations. Annu Rev Biomed Eng 24: 249–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shukla R, Ramasamy V, Shanmugam RK, Ahuja R, Khanna N. 2020. Antibody-Dependent Enhancement: A Challenge for Developing a Safe Dengue Vaccine. Frontiers in Cellular and Infection Microbiology 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang TT, Sewatanon J, Memoli MJ, Wrammert J, Bournazos S, et al. 2017. IgG antibodies to dengue enhanced for FcγRIIIA binding determine disease severity. Science 355: 395–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Parke JA, Avis PJ. 1964. The Effect of Digestion with Papain and Pepsin Upon the Antitoxic Activity of Rabbit Antibody. Immunology 7: 248–60 [PMC free article] [PubMed] [Google Scholar]
- 60.Gearing AJ, Thorpe SJ, Miller K, Mangan M, Varley PG, et al. 2002. Selective cleavage of human IgG by the matrix metalloproteinases, matrilysin and stromelysin. Immunol Lett 81: 41–8 [DOI] [PubMed] [Google Scholar]
- 61.Collin M, Olsen A. 2001. Effect of SpeB and EndoS from Streptococcus pyogenes on human immunoglobulins. Infect Immun 69: 7187–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.von Pawel-Rammingen U, Johansson BP, Bjorck L. 2002. IdeS, a novel streptococcal cysteine proteinase with unique specificity for immunoglobulin G. EMBO J 21: 1607–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ryan MH, Petrone D, Nemeth JF, Barnathan E, Bjorck L, Jordan RE. 2008. Proteolysis of purified IgGs by human and bacterial enzymes in vitro and the detection of specific proteolytic fragments of endogenous IgG in rheumatoid synovial fluid. Mol Immunol 45: 1837–46 [DOI] [PubMed] [Google Scholar]
- 64.Fick RB Jr., Baltimore RS, Squier SU, Reynolds HY. 1985. IgG proteolytic activity of Pseudomonas aeruginosa in cystic fibrosis. J Infect Dis 151: 589–98 [DOI] [PubMed] [Google Scholar]
- 65.Fernandez Falcon MF, Echague CG, Hair PS, Nyalwidhe JO, Cunnion KM. 2011. Protease inhibitors decrease IgG shedding from Staphylococcus aureus, increasing complement activation and phagocytosis efficiency. J Med Microbiol 60: 1415–22 [DOI] [PubMed] [Google Scholar]
- 66.Brezski RJ, Vafa O, Petrone D, Tam SH, Powers G, et al. 2009. Tumor-associated and microbial proteases compromise host IgG effector functions by a single cleavage proximal to the hinge. Proc Natl Acad Sci U S A 106: 17864–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jordan RE, Fernandez J, Brezski RJ, Greenplate AR, Knight DM, et al. 2016. A peptide immunization approach to counteract a Staphylococcus aureus protease defense against host immunity. Immunol Lett 172: 29–39 [DOI] [PubMed] [Google Scholar]
- 68.Brezski RJ, Luongo JL, Petrone D, Ryan MH, Zhong D, et al. 2008. Human anti-IgG1 hinge autoantibodies reconstitute the effector functions of proteolytically inactivated IgGs. J Immunol 181: 3183–92 [DOI] [PubMed] [Google Scholar]
- 69.Brezski RJ, Jordan RE. 2010. Cleavage of IgGs by proteases associated with invasive diseases: an evasion tactic against host immunity? MAbs 2: 212–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Brezski RJ, Oberholtzer A, Strake B, Jordan RE. 2011. The in vitro resistance of IgG2 to proteolytic attack concurs with a comparative paucity of autoantibodies against peptide analogs of the IgG2 hinge. MAbs 3: 558–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vidarsson G, Dekkers G, Rispens T. 2014. IgG subclasses and allotypes: from structure to effector functions. Front Immunol 5: 520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Duncan AR, Winter G. 1988. The binding site for C1q on IgG. Nature 332: 738–40 [DOI] [PubMed] [Google Scholar]
- 73.Kinder M, Greenplate AR, Grugan KD, Soring KL, Heeringa KA, et al. 2013. Engineered protease-resistant antibodies with selectable cell-killing functions. J Biol Chem 288: 30843–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nam DH, Lee KB, Kruchowy E, Pham H, Ge X. 2020. Protease Inhibition Mechanism of Camelid-like Synthetic Human Antibodies. Biochemistry 59: 3802–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Everett MJ, Davies DT. 2021. Pseudomonas aeruginosa elastase (LasB) as a therapeutic target. Drug Discov Today 26: 2108–23 [DOI] [PubMed] [Google Scholar]
- 76.Leiris S, Davies DT, Sprynski N, Castandet J, Beyria L, et al. 2021. Virtual Screening Approach to Identifying a Novel and Tractable Series of Pseudomonas aeruginosa Elastase Inhibitors. ACS Med Chem Lett 12: 217–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Santajit S, Kong-Ngoen T, Chongsa-Nguan M, Boonyuen U, Pumirat P, et al. 2021. Human Single-Chain Antibodies That Neutralize Elastolytic Activity of Pseudomonas aeruginosa LasB. Pathogens 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wolska K, Szweda P. 2009. Genetic features of clinical Pseudomonas aeruginosa strains. Pol J Microbiol 58: 255–60 [PubMed] [Google Scholar]
- 79.Smith EJ, Visai L, Kerrigan SW, Speziale P, Foster TJ. 2011. The Sbi protein is a multifunctional immune evasion factor of Staphylococcus aureus. Infect Immun 79: 3801–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Van Loghem E, Frangione B, Recht B, Franklin EC. 1982. Staphylococcal protein A and human IgG subclasses and allotypes. Scand J Immunol 15: 275–8 [DOI] [PubMed] [Google Scholar]
- 81.Choe W, Durgannavar TA, Chung SJ. 2016. Fc-Binding Ligands of Immunoglobulin G: An Overview of High Affinity Proteins and Peptides. Materials (Basel) 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cruz AR, Boer MAD, Strasser J, Zwarthoff SA, Beurskens FJ, et al. 2021. Staphylococcal protein A inhibits complement activation by interfering with IgG hexamer formation. Proc Natl Acad Sci U S A 118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Blumberg LJ, Humphries JE, Jones SD, Pearce LB, Holgate R, et al. 2019. Blocking FcRn in humans reduces circulating IgG levels and inhibits IgG immune complex-mediated immune responses. Sci Adv 5: eaax9586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cruz AR, Bentlage AEH, Blonk R, de Haas CJC, Aerts PC, et al. 2022. Toward Understanding How Staphylococcal Protein A Inhibits IgG-Mediated Phagocytosis. J Immunol 209: 1146–55 [DOI] [PubMed] [Google Scholar]
- 85.Roben PW, Salem AN, Silverman GJ. 1995. VH3 family antibodies bind domain D of staphylococcal protein A. J Immunol 154: 6437–45 [PubMed] [Google Scholar]
- 86.Boero E, Cruz AR, Pansegrau W, Giovani C, Rooijakkers SHM, et al. 2022. Natural Human Immunity Against Staphylococcal Protein A Relies on Effector Functions Triggered by IgG3. Front Immunol 13: 834711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Varshney AK, Kuzmicheva GA, Lin J, Sunley KM, Bowling RA Jr., et al. 2018. A natural human monoclonal antibody targeting Staphylococcus Protein A protects against Staphylococcus aureus bacteremia. PLoS One 13: e0190537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Thammavongsa V, Rauch S, Kim HK, Missiakas DM, Schneewind O. 2015. Protein A-neutralizing monoclonal antibody protects neonatal mice against Staphylococcus aureus. Vaccine 33: 523–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kim HK, Emolo C, DeDent AC, Falugi F, Missiakas DM, Schneewind O. 2012. Protein A-specific monoclonal antibodies and prevention of Staphylococcus aureus disease in mice. Infect Immun 80: 3460–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Shi M, Chen X, Sun Y, Kim HK, Schneewind O, Missiakas D. 2021. A protein A based Staphylococcus aureus vaccine with improved safety. Vaccine 39: 3907–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lazar GA, Dang W, Karki S, Vafa O, Peng JS, et al. 2006. Engineered antibody Fc variants with enhanced effector function. Proc Natl Acad Sci U S A 103: 4005–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Forero-Torres A, de Vos S, Pohlman BL, Pashkevich M, Cronier DM, et al. 2012. Results of a phase 1 study of AME-133v (LY2469298), an Fc-engineered humanized monoclonal anti-CD20 antibody, in FcgammaRIIIa-genotyped patients with previously treated follicular lymphoma. Clin Cancer Res 18: 1395–403 [DOI] [PubMed] [Google Scholar]
- 93.Rugo HS, Im SA, Cardoso F, Cortes J, Curigliano G, et al. 2021. Efficacy of Margetuximab vs Trastuzumab in Patients With Pretreated ERBB2-Positive Advanced Breast Cancer: A Phase 3 Randomized Clinical Trial. JAMA Oncol 7: 573–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Jendeberg L, Nilsson P, Larsson A, Denker P, Uhlen M, et al. 1997. Engineering of Fc(1) and Fc(3) from human immunoglobulin G to analyse subclass specificity for staphylococcal protein A. J Immunol Methods 201: 25–34 [DOI] [PubMed] [Google Scholar]
- 95.Chen X, Schneewind O, Missiakas D. 2022. Engineered human antibodies for the opsonization and killing of Staphylococcus aureus. Proc Natl Acad Sci U S A 119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chu TH, Patz EF Jr., Ackerman ME. 2021. Coming together at the hinges: Therapeutic prospects of IgG3. MAbs 13: 1882028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sprague ER, Wang C, Baker D, Bjorkman PJ. 2006. Crystal structure of the HSV-1 Fc receptor bound to Fc reveals a mechanism for antibody bipolar bridging. PLoS Biol 4: e148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sprague ER, Reinhard H, Cheung EJ, Farley AH, Trujillo RD, et al. 2008. The human cytomegalovirus Fc receptor gp68 binds the Fc CH2-CH3 interface of immunoglobulin G. J Virol 82: 3490–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ndjamen B, Joshi DS, Fraser SE, Bjorkman PJ. 2016. Characterization of Antibody Bipolar Bridging Mediated by the Human Cytomegalovirus Fc Receptor gp68. J Virol 90: 3262–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jenks JA, Goodwin ML, Permar SR. 2019. The Roles of Host and Viral Antibody Fc Receptors in Herpes Simplex Virus (HSV) and Human Cytomegalovirus (HCMV) Infections and Immunity. Front Immunol 10: 2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Frank I, Friedman HM. 1989. A novel function of the herpes simplex virus type 1 Fc receptor: participation in bipolar bridging of antiviral immunoglobulin G. J Virol 63: 4479–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Manley K, Anderson J, Yang F, Szustakowski J, Oakeley EJ, et al. 2011. Human cytomegalovirus escapes a naturally occurring neutralizing antibody by incorporating it into assembling virions. Cell Host Microbe 10: 197–209 [DOI] [PubMed] [Google Scholar]
- 103.Kolb P, Hoffmann K, Sievert A, Reinhard H, Merce-Maldonado E, et al. 2021. Human cytomegalovirus antagonizes activation of Fcgamma receptors by distinct and synergizing modes of IgG manipulation. Elife 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lubinski JM, Lazear HM, Awasthi S, Wang F, Friedman HM. 2011. The Herpes Simplex Virus 1 IgG Fc Receptor Blocks Antibody-Mediated Complement Activation and Antibody-Dependent Cellular Cytotoxicity In Vivo. Journal of Virology 85: 3239–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Corrales-Aguilar E, Trilling M, Hunold K, Fiedler M, Le VT, et al. 2014. Human cytomegalovirus Fcgamma binding proteins gp34 and gp68 antagonize Fcgamma receptors I, II and III. PLoS Pathog 10: e1004131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Vlahava V-M, Murrell I, Zhuang L, Aicheler RJ, Lim E, et al. 2021. Monoclonal antibodies targeting nonstructural viral antigens can activate ADCC against human cytomegalovirus. The Journal of Clinical Investigation 131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Qerqez AN HK, Lee AG, Mishra AK, Delidakis G, Chowdhury A, Nguyen AW, Georgiou G, McLellan JS, Hengel H, and Maynard JA. 2023. Uncoupling antibody binding to host and viral Fc receptors restores antibodymediated NK cell activation against HCMV-infected cells. submitted [Google Scholar]
- 108.Joller N, Weber SS, Muller AJ, Sporri R, Selchow P, et al. 2010. Antibodies protect against intracellular bacteria by Fc receptor-mediated lysosomal targeting. Proc Natl Acad Sci U S A 107: 20441–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Diebolder CA, Beurskens FJ, de Jong RN, Koning RI, Strumane K, et al. 2014. Complement is activated by IgG hexamers assembled at the cell surface. Science 343: 1260–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.van Kampen MD, Kuipers-De Wilt L, van Egmond ML, Reinders-Blankert P, van den Bremer ETJ, et al. 2022. Biophysical Characterization and Stability of Modified IgG1 Antibodies with Different Hexamerization Propensities. J Pharm Sci 111: 1587–98 [DOI] [PubMed] [Google Scholar]
- 111.Gulati S, Beurskens FJ, de Kreuk BJ, Roza M, Zheng B, et al. 2019. Complement alone drives efficacy of a chimeric antigonococcal monoclonal antibody. PLoS Biol 17: e3000323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Meri S, Jordens M, Jarva H. 2008. Microbial complement inhibitors as vaccines. Vaccine 26 Suppl 8: I113–7 [DOI] [PubMed] [Google Scholar]
- 113.Giuntini S, Reason DC, Granoff DM. 2011. Complement-mediated bactericidal activity of anti-factor H binding protein monoclonal antibodies against the meningococcus relies upon blocking factor H binding. Infect Immun 79: 3751–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Biolchi A, Tomei S, Santini L, La Gaetana R, Mori E, et al. 2021. Four-component Meningococcal Serogroup B Vaccine Induces Antibodies With Bactericidal Activity Against Diverse Outbreak Strains in Adolescents. Pediatr Infect Dis J 40: e66–e71 [DOI] [PubMed] [Google Scholar]
- 115.Granoff DM, Giuntini S, Gowans FA, Lujan E, Sharkey K, Beernink PT. 2016. Enhanced protective antibody to a mutant meningococcal factor H-binding protein with low-factor H binding. JCI Insight 1: e88907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Thiriard A, Raze D, Locht C. 2018. Diversion of complement-mediated killing by Bordetella. Microbes Infect 20: 512–20 [DOI] [PubMed] [Google Scholar]
- 117.Badarau A, Trstenjak N, Nagy E. 2017. Structure and Function of the Two-Component Cytotoxins of Staphylococcus aureus - Learnings for Designing Novel Therapeutics. Adv Exp Med Biol 966: 15–35 [DOI] [PubMed] [Google Scholar]
- 118.Francois B, Jafri HS, Chastre J, Sanchez-Garcia M, Eggimann P, et al. 2021. Efficacy and safety of suvratoxumab for prevention of Staphylococcus aureus ventilator-associated pneumonia (SAATELLITE): a multicentre, randomised, double-blind, placebo-controlled, parallel-group, phase 2 pilot trial. Lancet Infect Dis 21: 1313–23 [DOI] [PubMed] [Google Scholar]
- 119.Francois B, Mercier E, Gonzalez C, Asehnoune K, Nseir S, et al. 2018. Safety and tolerability of a single administration of AR-301, a human monoclonal antibody, in ICU patients with severe pneumonia caused by Staphylococcus aureus: first-in-human trial. Intensive Care Med 44: 1787–96 [DOI] [PubMed] [Google Scholar]
- 120.Rouha H, Badarau A, Visram ZC, Battles MB, Prinz B, et al. 2015. Five birds, one stone: neutralization of alpha-hemolysin and 4 bi-component leukocidins of Staphylococcus aureus with a single human monoclonal antibody. MAbs 7: 243–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Szijarto V, Guachalla LM, Hartl K, Varga C, Badarau A, et al. 2017. Endotoxin neutralization by an O-antigen specific monoclonal antibody: A potential novel therapeutic approach against Klebsiella pneumoniae ST258. Virulence 8: 1203–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Rouha H, Weber S, Janesch P, Maierhofer B, Gross K, et al. 2018. Disarming Staphylococcus aureus from destroying human cells by simultaneously neutralizing six cytotoxins with two human monoclonal antibodies. Virulence 9: 231–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Magyarics Z, Leslie F, Bartko J, Rouha H, Luperchio S, et al. 2019. Randomized, Double-Blind, Placebo-Controlled, Single-Ascending-Dose Study of the Penetration of a Monoclonal Antibody Combination (ASN100) Targeting Staphylococcus aureus Cytotoxins in the Lung Epithelial Lining Fluid of Healthy Volunteers. Antimicrob Agents Chemother 63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Stein RL. 2022. Kinetic Studies of the Activation of Bordetella pertussis Adenylate Cyclase by Calmodulin. Biochemistry [DOI] [PubMed] [Google Scholar]
- 125.Gray MC, Hewlett EL. 2011. Cell cycle arrest induced by the bacterial adenylate cyclase toxins from Bacillus anthracis and Bordetella pertussis. Cell Microbiol 13: 123–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Fedele G, Schiavoni I, Adkins I, Klimova N, Sebo P. 2017. Invasion of Dendritic Cells, Macrophages and Neutrophils by the Bordetella Adenylate Cyclase Toxin: A Subversive Move to Fool Host Immunity. Toxins (Basel) 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Goldsmith JA, DiVenere AM, Maynard JA, McLellan JS. 2021. Structural basis for antibody binding to adenylate cyclase toxin reveals RTX linkers as neutralization-sensitive epitopes. PLoS Pathog 17: e1009920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Goldsmith J, DiVenere A, Maynard J, McLellan J. 2022. Structural Basis for Non-canonical Integrin Engagement by Bordetella Adenylate Cyclase Toxin. Cell Reports, in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.DiVenere AM, Amengor D, Silva RP, Goldsmith JA, McLellan JS, Maynard JA. 2022. Blockade of the Adenylate Cyclase Toxin Synergizes with Opsonizing Antibodies to Protect Mice against Bordetella pertussis. mBio 13: e0152722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Tkaczyk C, Kasturirangan S, Minola A, Jones-Nelson O, Gunter V, et al. 2017. Multimechanistic Monoclonal Antibodies (MAbs) Targeting Staphylococcus aureus Alpha-Toxin and Clumping Factor A: Activity and Efficacy Comparisons of a MAb Combination and an Engineered Bispecific Antibody Approach. Antimicrob Agents Chemother 61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Thwaites GE, Gant V. 2011. Are bloodstream leukocytes Trojan Horses for the metastasis of Staphylococcus aureus? Nat Rev Microbiol 9: 215–22 [DOI] [PubMed] [Google Scholar]
- 132.Rungelrath V, Porter AR, Malachowa N, Freedman BA, Leung JM, et al. 2021. Further Insight into the Mechanism of Human PMN Lysis following Phagocytosis of Staphylococcus aureus. Microbiol Spectr 9: e0088821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.DuMont AL, Yoong P, Surewaard BG, Benson MA, Nijland R, et al. 2013. Staphylococcus aureus elaborates leukocidin AB to mediate escape from within human neutrophils. Infect Immun 81: 1830–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lehar SM, Pillow T, Xu M, Staben L, Kajihara KK, et al. 2015. Novel antibody-antibiotic conjugate eliminates intracellular S. aureus. Nature 527: 323–8 [DOI] [PubMed] [Google Scholar]
- 135.Staben LR, Koenig SG, Lehar SM, Vandlen R, Zhang D, et al. 2016. Targeted drug delivery through the traceless release of tertiary and heteroaryl amines from antibody-drug conjugates. Nat Chem 8: 1112–19 [DOI] [PubMed] [Google Scholar]
- 136.Zhou C, Lehar S, Gutierrez J, Rosenberger CM, Ljumanovic N, et al. 2016. Pharmacokinetics and pharmacodynamics of DSTA4637A: A novel THIOMAB antibody antibiotic conjugate against Staphylococcus aureus in mice. MAbs 8: 1612–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kajihara KK, Pantua H, Hernandez-Barry H, Hazen M, Deshmukh K, et al. 2021. Potent Killing of Pseudomonas aeruginosa by an Antibody-Antibiotic Conjugate. mBio 12: e0020221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Taylor A, Jenner D, Rowland C, Laws T, Norville I, Prior J. 2021. Monoclonal Antibodies Opsonize Burkholderia spp. and Reduce Intracellular Actin Tail Formation in a Macrophage Infection Assay. J Bacteriol 203: e0024421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Chastre J, Francois B, Bourgeois M, Komnos A, Ferrer R, et al. 2022. Safety, efficacy, and pharmacokinetics of gremubamab (MEDI3902), an anti-Pseudomonas aeruginosa bispecific human monoclonal antibody, in P. aeruginosa-colonised, mechanically ventilated intensive care unit patients: a randomised controlled trial. Crit Care 26: 355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Heimer SR, Evans DJ, Stern ME, Barbieri JT, Yahr T, Fleiszig SM. 2013. Pseudomonas aeruginosa utilizes the type III secreted toxin ExoS to avoid acidified compartments within epithelial cells. PLoS One 8: e73111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Thanabalasuriar A, Surewaard BG, Willson ME, Neupane AS, Stover CK, et al. 2017. Bispecific antibody targets multiple Pseudomonas aeruginosa evasion mechanisms in the lung vasculature. J Clin Invest 127: 2249–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wec AZ, Nyakatura EK, Herbert AS, Howell KA, Holtsberg FW, et al. 2016. A “Trojan horse” bispecific-antibody strategy for broad protection against ebolaviruses. Science 354: 350–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Bai Y, Ye L, Tesar DB, Song H, Zhao D, et al. 2011. Intracellular neutralization of viral infection in polarized epithelial cells by neonatal Fc receptor (FcRn)-mediated IgG transport. Proc Natl Acad Sci U S A 108: 18406–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Caddy SL, Vaysburd M, Wing M, Foss S, Andersen JT, et al. 2020. Intracellular neutralisation of rotavirus by VP6-specific IgG. PLoS Pathog 16: e1008732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.LeBowitz JH, Grubb JH, Maga JA, Schmiel DH, Vogler C, Sly WS. 2004. Glycosylation-independent targeting enhances enzyme delivery to lysosomes and decreases storage in mucopolysaccharidosis type VII mice. Proc Natl Acad Sci U S A 101: 3083–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wirchnianski AS, Wec AZ, Nyakatura EK, Herbert AS, Slough MM, et al. 2021. Two Distinct Lysosomal Targeting Strategies Afford Trojan Horse Antibodies With Pan-Filovirus Activity. Front Immunol 12: 729851. [DOI] [PMC free article] [PubMed] [Google Scholar]