

Abstract
Adrenocortical carcinoma (ACC) is a rare cancer in which tissue-specific differentiation is paradoxically associated with dismal outcomes. The differentiated ACC subtype CIMP-high is prevalent, incurable, and routinely fatal. CIMP-high ACC possess abnormal DNA methylation and frequent β-catenin activating mutations. Here, we demonstrated that ACC differentiation is maintained by a balance between nuclear, tissue-specific β-catenin-containing complexes and the epigenome. On chromatin, β-catenin bound master adrenal transcription factor SF1 and hijacked the adrenocortical super-enhancer landscape to maintain differentiation in CIMP-high ACC; off chromatin, β-catenin bound histone methyltransferase EZH2. SF1/β-catenin and EZH2/β-catenin complexes present in normal adrenals persisted through all phases of ACC evolution. Pharmacologic EZH2 inhibition in CIMP-high ACC expelled SF1/β-catenin from chromatin and favored EZH2/β-catenin assembly, erasing differentiation and restraining cancer growth in vitro and in vivo. These studies illustrate how tissue-specific programs shape oncogene selection, surreptitiously encoding targetable therapeutic vulnerabilities.
Keywords: adrenocortical carcinoma, Polycomb, EZH2, PRC2, CIMP, DNA methylation, adrenal, epigenetics, cancer, Wnt, β-catenin, NR5A1, steroidogenic factor 1, ACC
INTRODUCTION
Deranged epigenetic patterning is a hallmark of cancer (1,2). CIMP-high is a recurrent epigenetic signature defined by abnormal DNA methylation in promoter CpG islands (CpGi). These CpGi are normally repressed by histone H3 lysine 27 trimethylation (H3K27me3), written by the histone methyltransferase EZH2 as part of the Polycomb repressive complex 2 (PRC2) (3). PRC2 is required for embryonic and stem cell pluripotency, restricting lineage specification and differentiation (4). EZH2/PRC2 actions are crucial for many cancers (5). Together, these observations have seeded a model that CIMP-high maintains cancer cell stemness, possibly through redundant or cooperative DNA methylation-dependent silencing of PRC2 targets (6,7).
Though prevalent across cancer types, CIMP-high has variable prognostic significance. In the rare endocrine cancer adrenocortical carcinoma (ACC), the 30–40% of patients with CIMP-high disease experience rapid metastatic recurrence and death (8,9). Genetically, CIMP-high ACC are distinguished by recurrent somatic alterations leading to constitutive activation of the cell cycle and the Wnt/β-catenin pathway (8–10). Specifically, 80% of CIMP-high ACC possess driver alterations in cell cycle genes TP53, CDK4, CCNE1, or CDKN2A; and >50% possess driver alterations in Wnt/β-catenin pathway genes ZNRF3, CTNNB1 (encoding β-catenin), and APC (8,9). Classically, Wnt pathway activation culminates in β-catenin co-activation of TCF/LEF-driven transcription and expression of programs that facilitate stem cell and tissue maintenance (11). This is also true in the adrenal cortex, the tissue of origin of ACC, where cell type specification is established by integration of paracrine and endocrine cues (12). All adrenocortical cells express master transcription factor SF1 (NR5A1). Wnt/β-catenin prevents differentiation of SF1+ cells into a population termed the zona fasciculata (zF). This antagonizes pituitary hormone ACTH, which expands and differentiates the zF to produce glucocorticoids (Figure 1A).
Figure 1: Differentiated, Wnt-active ACC subtype CIMP-high possesses aberrant PRC2 target hypermethylation with high EZH2 coupled to H3K27me3.
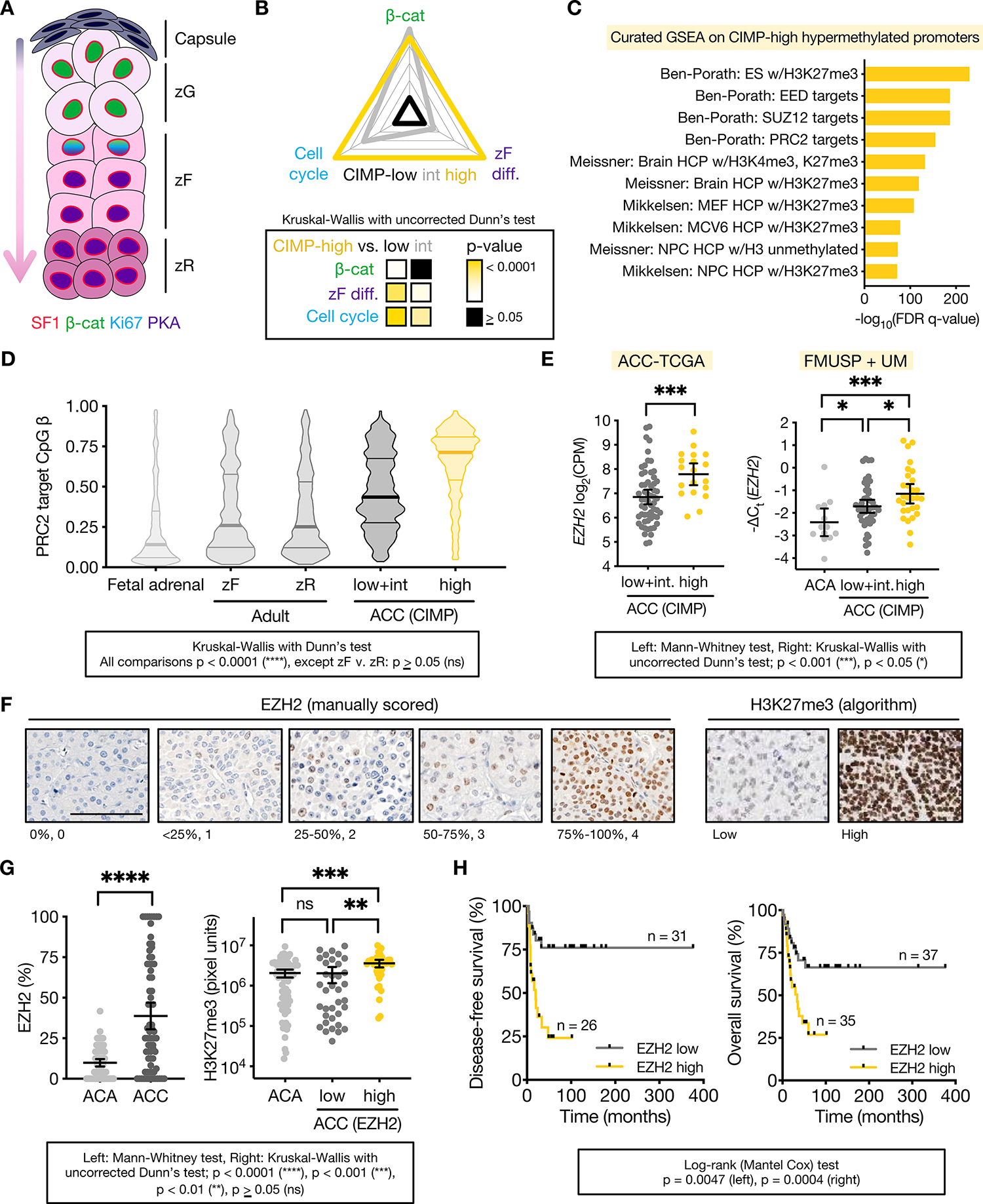
A. Corticocapsular unit of adrenocortical homeostasis depicting peripheral mesenchymal cells (capsule) and human cortical populations zona glomerulosa (zG), zona fasciculata (zF), and zona reticularis (zR), that produce mineralocorticoids, glucocorticoids, and androgens, respectively. Differentiation in the cortex is centripetal, and zG, zF, and zR cells are supplied by peripheral capsular and cortical progenitors (arrow). The entire cortex is SF1 positive. ZG cells possess active Wnt/β-catenin signaling and lower zF/zR cells possess active ACTH signaling through protein kinase A (PKA). Wnt/β-catenin signaling fades in the upper zF, and these cells respond to ACTH/PKA with proliferation (Ki67). Mice do not have a zR.
B. GSVA was used on ACC-TCGA RNA-seq to calculate the expression score of genes that define zF differentiation or are regulated in a cell-cycle- or Wnt-dependent manner across ACC-TCGA. Score validation detailed in Methods and Supp Fig 1. Radar plot depicts average score for each ACC CIMP class, with values mapped onto an arbitrary scale along each axis. Heatmap below depicts p-value for comparisons.
C. 10 most significant gene sets from curated GSEA on genes with promoters targeted for hypermethylation in CIMP-high ACC.
D. Violin plot of PRC2 target CpGi methylation measured by Illumina 850k or 450k methylation array in fetal (n=3) or adult adrenal (zF, zR; n=4 each) and ACC from ACC-TCGA (n=79). Lines at median and quartiles.
E. EZH2 expression in ACC-TCGA by RNA-seq (n=78), left, and independent cohort by qPCR (n=102, FMUSP+UM classified by CIMP in (8)), right. CPM=counts per million. ACA=adrenocortical adenomas (benign adrenocortical tumors). Line at mean with 95% confidence interval (CI).
F-G. Representative images and scoring of tissue microarray (TMA) of human adult ACA (n=74) and primary ACC (n=74). TMA stained for EZH2 and H3K27me3 by immunohistochemistry (IHC). EZH2 quantified on 0–4 scale (%positive nuclei) by 2 independent observers. H3K27me3 quantified by MATLAB (20). EZH2 low=ACC with below median EZH2 expression (≤25% nuclei EZH2+), EZH2 high=ACC with above median EZH2 expression (>25% nuclei EZH2+). EZH2 mRNA/protein are correlated (Spearman r=0.5117, p<0.01, not shown). F, bar=100 μm. G, line at mean with 95% CI.
H. Disease-free (after R0/RX resection in patients without metastatic disease at diagnosis) and overall survival (all patients) stratified by ACC EZH2 expression.
Broadly, Wnt-dependent tumorigenesis is associated with dedifferentiation, including in CIMP-high neoplasia (13–16). Given these observations, one would expect CIMP-high ACC to exhibit dedifferentiation. In striking contrast, across ACC, these cancers exhibit the strongest differentiation, with clinical glucocorticoid production and high expression of zF-defining genes (8,9). This suggests that aberrant epigenetic programming in ACC stabilizes a paradoxical differentiated, Wnt-active, and rapidly proliferative cellular state (Figure 1B, Supp Fig 1). Given advances enabling rapid prospective molecular subtyping of ACC (8), the homogeneous clinical and intriguing molecular characteristics of this class, and the abysmal lack of therapies, we investigated the epigenetic underpinnings of CIMP-high ACC.
MATERIALS AND METHODS
Human samples
All human samples used in this study were obtained with written informed consent from the University of Michigan (UM) and Faculdade de Medicina da Universidade de Sao Paulo (FMUSP) (8). Studies were conducted in accordance with the Declaration of Helsinki and study protocols were approved by UM and FMUSP Institutional Review Boards. Tissue microarrays (TMAs) were developed and provided by FMUSP.
In vitro and molecular studies
Cell culture.
Cell lines were obtained from ATCC unless stated otherwise, cultured under standard sterile conditions, and maintained in a humidified tissue culture incubator with 5% CO2 at 37°C. NCI-H295R (RRID:CVCL_0458) were cultured in DMEM/F12 (Gibco, 11330-032) supplemented with 10% Nu serum (Corning, 35500), 1% ITS-X (Gibco, 51500-056), and 1% penicillin/streptomycin (Gibco, 15140-122). Y1 (RRID:CVCL_0585) were cultured in high glucose DMEM (Invitrogen, 11995-065), supplemented with 2.5% fetal bovine serum (Sigma-Aldrich, F2442-500ML), 7.5% horse serum (Invitrogen, 16050122), and 1% penicillin/streptomycin. ATC7L were a generous gift from A. Lefrançois-Martinez and A. Martinez (GReD, CNRS, Inserm, Université Clermont-Auvergne, Clermont-Ferrand, France) and cultured in DMEM/F12 supplemented with 2.5% fetal bovine serum, 2.5% horse serum, 1% ITS-X, and 1% penicillin/streptomycin. All cell lines were routinely screened (every 3–5 passages and/or each experimental plating) for microbial contamination by DAPI staining or e-Myco Mycoplasma PCR Detection Kit (Bulldog Bio, 2523348). Cells were discarded after 20–25 passages, or when exponential growth was no longer evident (whichever came first).
Pharmacological experiments.
NCI-H295R were plated at 400,000 or 800,000 cells/well in 6 well plates or scaled accordingly for other well sizes. 18–24 hours after plating, media was changed for media containing an EZH2 inhibitor (EZH2i; EPZ-6438, EED226, or GSK126), and/or PRI-724, or vehicle (DMSO). Concentration of vehicle was constant for all wells in an experiment. Media was replaced with fresh drug-containing media every 24 hours. After 96 hours of drug treatment, cells were harvested for genomic DNA (gDNA), RNA, protein or cell viability. Alternatively, after 96 hours of EZH2i, cells were treated with 10 μM forskolin for 48 hours, after which time media was harvested for steroid measurement and cells were harvested for protein or RNA. Alternatively, NCI-H295R were plated and treated with 10 μM forskolin for 48 hours, then harvested for RNA. NCI-H295R were essentially treated as above and harvested for RNA-seq, methylation array, ChIP-seq, or ATAC-seq; additional details provided in Supplementary Methods. RNA-seq table provided in Supp Table 1.
Y1 and ATC7L were treated and harvested as above except cells were plated at 250,000 cells/well or 600,000 cells/well in 6 well plates or scaled accordingly. Alternatively, Y1 and ATC7L were treated for durations indicated with 2.5–40 mM LiCl or equivalent volume of vehicle (water; % vehicle was constant for all doses), media containing 20% Wnt3a conditioned medium (CM) or 20% parental medium. Wnt3a CM or parental medium was derived from L cells (17).
siRNA experiments.
NCI-H295R were plated at 800,000 cells per well in 6 well plates (or scaled accordingly) in antibiotic-free media. 18–24 hours after plating, media was changed for fresh antibiotic-free media containing 50 nM siRNA (Thermo Fisher, Negative Control #1, s4916, s4917, or s4918) and transfection reagent (Mirus TransitX2, MIR 6000). Transfection was repeated 72 hours after first transfection and cells were harvested at 144 hours after first transfection for desired endpoint readouts.
Nucleic acid extraction and quantification.
Nucleic acid extraction and quantification was performed as described (8) with optional nuclease treatments, using the DNeasy Blood & Tissue Kit (Qiagen, 69504), RNeasy Plus Mini Kit (Qiagen, 74134), or TRI reagent (Sigma, 93289) with RNeasy kit (Qiagen).
Targeted gene expression and G0S2 methylation analysis.
cDNA synthesis and qPCR was performed as described (8) or with iScript cDNA Synthesis Kit (Bio-Rad, 1708841), Power SYBR Green qPCR Mastermix (Invitrogen, 4367659), and QuantStudio 6 Flex Real-Time PCR System (Applied Biosystems, 4489826). SYBR qPCR primers are detailed in Supp Table 2; Actb or PPIB were housekeeping genes. G0S2 methylation was measured as described (8). In patient samples, measurement of BUB1B and GUSB expression, and G0S2 methylation was previously performed and reported (8); EZH2 expression was measured by TaqMan Gene Expression Assays (Applied Biosystems, hs00544830_m1). In murine tissue, bulk gene expression was measured by TaqMan (Applied Biosystems) for the following genes: Actb (Mm02619580_g1), Lef1 (Mm00550265_m1). Expression levels were calculated using the ΔCt method (8).
Protein extraction, quantification, and analysis.
At endpoint, cells were washed and protein lysates were collected in whole-cell nuclear lysis buffer (50 mM Tris-HCl pH 8.1, 10 mM EDTA, 1% SDS in ultrapure H2O, adapted from (18)), supplemented with protease inhibitors (Roche, 04693159001) and phosphatase inhibitors (Roche, 04906845001). Alternatively, cytoplasmic and nuclear extracts were prepared using the ActiveMotif Nuclear Complex Co-IP Kit (ActiveMotif, 54001). Protein extracts were quantified by BCA (Thermo, PI23227). Samples were reduced in bromophenol blue and reducing-agent containing buffers and then analyzed by standard SDS-PAGE, Coomassie staining, and western blot procedures. Alternatively, nuclear lysates were analyzed by size-exclusion chromatography. Additional details are in Supplementary Methods.
Nuclear complex immunoprecipitation (co-IP).
Nuclear co-IP was performed using the Nuclear Complex Co-IP Kit (ActiveMotif, 54001) and Protein G Agarose Columns (ActiveMotif, 53039) according to manufacturer’s protocol (detailed in Supplementary Methods). Co-IPs were evaluated by mass spectrometry (IP-MS) or eluted and evaluated by Coomassie staining and/or western blot.
Viability assays and calculations.
Viability was measured using alamarBlue (Invitrogen, DAL1025) per manufacturer instructions, and coefficient of drug interaction calculated as detailed in Supplementary Methods.
2D Clonogenicity.
At endpoint, cells were washed with PBS and viable cells (identified by Trypan blue exclusion) were plated at 1,000 cells/well in 6 well plates in 3 mL standard media. Cells were maintained under standard conditions without media changes. 4 weeks after plating, colonies were washed, fixed in 4% PFA for 15 minutes, stained with crystal violet staining solution (0.1% crystal violet, 5% ethanol in water), and washed with water. After drying overnight, plates were imaged with LI-COR Odyssey imaging system at 700 nm. Colonies were counted using the “Analyze Particles” tool in Fiji (RRID:SCR_002285).
Murine studies
All animal procedures were approved by Boston Children’s Hospital’s Institutional Animal Care and Use Committee. Control (ASCre/+), PCreAS/+, BCreAS/+, BPCreAS/+ transgenic mice were previously described (19). 7 to 8-week-old NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) immunodeficient male mice (RRID:IMSR_JAX:005557) were obtained from Jackson laboratories and housed in Boston Children’s Hospital’s specific-pathogen free facility. The BCH-ACC3A cell line was derived from a BPCreAS/+ male mouse harboring a 538 mg primary ACC tumor and documented lung metastasis. The primary tumor was dissociated using Tumor Dissociation Kit, mouse (Miltenyi Biotec, 130-096-730), and serially transplanted in NSG mice for 6 months. Cells were then cultivated in vitro in DMEM/F12 supplemented with 2% Nu serum, 1% ITS (Corning, 25-800-CR), 2 mM L-glutamine and 1% penicillin/streptomycin. 5 × 104 cells were resuspended in PBS mixed with Matrigel Basement Membrane Matrix (BD Biosciences) at 1:1 ratio and injected into subcutaneous flank tissue of NSG mice. Tumor length (L) and width (W) were measured with a digital caliper; volumes were calculated using formula L*W2*(π/6) where L<W. When subcutaneous tumors achieved ~100 mm3, mice were randomized to 200 mg/kg EPZ-6438 or equivalent volume of vehicle (1% DMSO, 0.5% NaCMC, 0.1% Tween-80 in water) daily by oral gavage delivered as 10μL/g mouse.
Immunohistochemistry (IHC), RNA in situ hybridization, and proximity ligation assay
IHC and RNA in situ hybridization.
For TMAs, EZH2 staining was performed using the NovoLink Max Polymer Detection System (Leica, RE7159), Tris-EDTA, pH 9.0 (Spring Bioscience, PMB4-235), Cas Block (Invitrogen, 00-8120), post-primary block (Leica, RE7159) and Novolink Polymer (Leica, RE7161). H3K27me3 staining was performed and quantified as described (20). For murine tissues, IHC was performed with VECTASTAIN Elite ABC-HRP Kit, Peroxidase (Rabbit IgG) (Vector Labs, PK-6101) with or without M.O.M. (Mouse on Mouse) Immunodetection Kit – Basic (Vector Laboratories, BMK-2022), and SIGMAFAST™ 3,3′-Diaminobenzidine (DAB) tablets (Sigma-Aldrich, D4168-50SET); or ImmPRESS HRP Horse Anti-Rabbit IgG Polymer Detection Kit (Vector Labs, MP-7401) and ImmPACT DAB EqV (Vector Labs, SK-4103) or as described (21,22). DAB signal was quantified using a homemade macro in Fiji. H3K27me3 signal on allografts was quantified by MATLAB (RRID:SCR_001622) (21,22). Immunofluorescence was performed as described (19,23). For immunocytochemistry, cells were plated on sterilized and lysine-coated glass cover slips. At endpoint, cells were fixed and multiplex immunocytochemistry was performed with ImmPRESS Kits (Vector Labs, MP-7401, MP-7402) and Tyramide conjugated to Alexa Fluor 488 or 555 (Thermo Fisher, B40953, B40955). RNA in situ hybridization was performed with RNAscope 2.5 HD Assay – BROWN (Advanced Cell Diagnostics, 322310) and associated reagents (Advanced Cell Diagnostics, 322335, 322335, 310091, 322001, 322331, 564431, 312471, 310043), and quantified using a custom Fiji macro. Additional details are supplied in Supplementary Methods.
Proximity Ligation Assay (PLA).
Briefly, PLA was performed with Duolink Detection Reagents Brightfield Kit (Sigma-Aldrich, DUO92012), associated wash buffers (Sigma-Aldrich, DUO82047-4L), and probes (Sigma-Aldrich, DUO92002, DUO92004), omitting counterstain step. For mouse samples, blocking was performed using a custom 1:1 mixture of blocking buffers prepared from VECTASTAIN Elite ABC-HRP Kit, Peroxidase (Rabbit IgG) (Vector Labs, PK-6101) and the M.O.M. Immunodetection Kit – Basic (Vector Laboratories, BMK-2022). Signal was quantified using a custom Fiji macro. Protocol is further detailed in Supplementary Methods.
Generation and analysis of high-throughput data
Immunoprecipitation/mass spectrometry (IP-MS).
IP-MS was performed by MS Bioworks (Ann Arbor, MI) per their standard pipeline as detailed in Supplementary Methods.
LC-MS/MS.
Steroids were extracted from culture medium and quantitated using liquid chromatography and tandem mass spectrometry as described (24). Protein was harvested from corresponding cells. Normalized steroid output is the ratio of steroid concentration measured in the media (in pg/mL) to total protein content in the sample (in μg).
RNA-seq.
RNA-seq was performed by the University of Michigan Advanced Genomics Core, who prepared libraries from total mRNA using NEBNext PolyA Ultra II RNA Library Prep Kit for Illumina and sequenced at 2×50 bp using an Illumina NovaSeq-6000. Reads were processed and analyzed using STAR (RRID:SCR_004463), featureCounts (RRID:SCR_012919), RNA-SeQC (RRID:SCR_005120), edgeR (RRID:SCR_012802), and limma (RRID:SCR_010943). BAM files for Y1 RNA-seq were generated according to the same pipeline from fastq files downloaded from DDBJ/EMBL/GenBank DRA000853. ACC-TCGA RNA-seq was analyzed as described (8) and using independent component analysis. GSVA (RRID:SCR_021058) was used to calculate gene signature scores. Additional details including citations for packages, gene signature lists, validation, and microarray datasets and pipelines are provided in Supplementary Methods.
ATAC-seq.
ATAC-seq was performed as essentially as described (25), and libraries were sequenced by the University of Michigan Advanced Genomics Core at 2×150 bp using a NovaSeq 6000. Reads were aligned using bowtie2 (RRID:SCR_016368). Peaks were called and analyzed using genrich (26), diffbind (RRID:SCR_012918), diffTF, and HOMER (RRID:SCR_010881). BigWig files were generated with deepTools (RRID:SCR_016366), visualized using JBR browser (27) or Signac (RRID:SCR_021158). ACC-TCGA ATAC-seq BigWig files were downloaded from TCGA Genomic Data Commons (RRID:SCR_014514), and signal quantified using deepTools. Further details including citations for packages are provided in Supplementary Methods.
ChIP-seq.
ChIP-seq was performed by Active Motif Services (Carlsbad, CA) with Drosophila melanogaster spike-in controls for all conditions. Reads were aligned to human and fly genomes using bowtie2. Peaks were called, annotated, and analyzed using SPAN and the JBR browser (27), ChIPseeker (RRID:SCR_021322), HOMER, and ROSE (RRID:SCR_017390) (28,29). Overlap between peak sets was computed with bedtools (RRID:SCR_006646). BigWig files were generated and quantified as above. Several datasets were downloaded from ENCODE (RRID:SCR_006793) for analysis. These and additional details including citations for packages and databases in Supplementary Methods.
Methylation arrays.
Regions targeted for differential methylation in CIMP-high vs. non-CIMP-high ACC were identified using DMRcate and published in (8), filtered, and evaluated for enrichment with curated collections by GSEA (RRID:SCR_003199). Otherwise, extracted gDNA was subject to 850k array profiling performed by the University of Michigan Advanced Genomics Core or Diagenode Epigenomic Services. Data was processed, annotated, and analyzed with methylAid (RRID:SCR_002659), minfi (RRID:SCR_012830), ChIPseeker, conumee (30) and limma, detailed and cited in Supplementary Methods.
Analysis of single-cell RNA-seq (scRNA-seq) and single-cell ATAC-seq (scATAC-seq).
Gene expression matrices of human fetal, neonatal and adult adrenal scRNA-seq data (31) were downloaded from GEO (GSE134355) and filtered, normalized, batch-corrected, integrated, scaled, and UMAP clustered using Seurat (RRID:SCR_016341) with CCA integration algorithm. Pseudotime trajectory analysis was performed with Monocle3 (RRID:SCR_018685). ScATAC-seq was processed, integrated and plotted with Signac and rtracklayer (RRID:SCR_021325). Adult adrenal scATAC-seq peak matrices and fragment files were downloaded from GEO (GSM5047828) (32). Fetal adrenal scATAC-seq peak matrices and fragment files were downloaded from Descartes Human Chromatin Accessibility During Development Atlas (33) (descartes.brotmanbaty.org). Specific pipeline details and citations in Supplementary Methods.
Identification of enhancer/gene links.
Promoter-other contact tables from adrenal promoter capture Hi-C (pcHi-C) were downloaded from GEO (GSE86189) (34). Putative active enhancers of a given gene were identified by overlapping adrenal pcHi-C contact tables with NCI-H295R baseline H3K27ac peaks. Overlaps were computed using bedtools. The set of active enhancers was overlapped with the consensus SF1/β-catenin peak set to identify genes putatively regulated by active SF1/β-catenin enhancers. Otherwise, active enhancers were manually inspected for epitope of interest.
DepMap analysis.
CRISPR dependency scores (DepMap 22Q4) across all genes and cancer cell lines were downloaded from the DepMap portal (RRID:SCR_017655) (35). Dependency scores for all genes were correlated with each other using the correlate function from corrr (36). Matrix was ranked in descending order, subsetted by gene of interest, and plotted using ggplot2 (RRID:SCR_014601).
Quantification and statistical analysis
Statistical information including number of replicates and statistical tests performed to compare experimental groups are described in methods above, Supplement, figures and figure legends. P-value or adjusted p-value<0.05 was considered significant for all analyses. Statistical analyses were performed using GraphPad Prism (RRID:SCR_002798) or R (RRID:SCR_001905). Unsupervised hierarchical clustering was performed with pheatmap (RRID:SCR_016418).
Data availability
Requests for resources and reagents should be directed to corresponding authors Antonio Marcondes Lerario (alerario@umich.edu) and Gary D. Hammer (ghammer@umich.edu). The original RNA-seq, methylation array, ChIP-seq, and ATAC-seq data generated in this study have been deposited in GEO: GSE205283. The publicly available data analyzed in this study were obtained from TCGA Genomic Data Commons at https://gdc.cancer.gov/ and https://portal.gdc.cancer.gov/legacy-archive; GEPIA at http://gepia.cancer-pku.cn/; GEO at https://www.ncbi.nlm.nih.gov/geo/ using identifiers GSE68889, GSE109578, GSE134355, GSE86189, GSE10927, or GSM5047828; ENCODE at https://www.encodeproject.org/ using identifiers ENCLB278KZX, ENCLB060VHC, ENCLB923ALY, ENCLB245VYO, ENCLB872HUE, ENCLB191XGX, ENCLB281DGJ, ENCLB670CFC, ENCLB382MNY, ENCLB198CGW, ENCLB172SDF, ENCLB735YID, ENCFF050CUG, ENCFF671NVD, ENCFF639HFP, ENCFF998IEU, ENCFF202US, ENCFF495GNE, ENCFF154IMT, ENCFF662NTZ, ENCFF342DVJ, ENCFF680AKW, ENCFF456EQK, ENCFF725EMZ, ENCFF009PDQ, ENCFF612HRW, ENCFF015TOT, ENCFF441TOI, ENCFF218TIK, ENCFF342EGR, ENCFF250ASJ, ENCFF495YGS, or ENCFF019XYA; DDBJ/EMBL/GenBank at https://www.ddbj.nig.ac.jp/ using identifier DRA000853; Descartes Human Chromatin Accessibility During Development Atlas at http://descartes.brotmanbaty.org/; 3DIV at http://3div.kr/; and DepMap at https://depmap.org/portal/.
RESULTS
DNA hypermethylation in CIMP-high ACC is pathologically directed to PRC2 targets
DNA hypermethylation is a powerful predictor of survival in ACC; hypermethylation of the G0S2 CpG island (CpGi) alone captures all features of CIMP-high (8). To illuminate mechanisms enabling aberrant epigenetic patterning, we performed GSEA on genes with promoter hypermethylation in CIMP-high tumors from The Cancer Genome Atlas study on ACC (ACC-TCGA; (9)). As expected, we observed significant enrichment for embryonic targets of the PRC2 (Figure 1C), including HOX clusters, GATA3, PAX6, and CIMP-high biomarker G0S2 (Supp Fig 2A), mirroring other CIMP-high cancers.
PRC2 targets may gain DNA methylation in mammalian tissues with aging (14,15,37,38). To determine if CIMP-high methylation reflects tissue origin, we profiled the DNA methylome of fetal and adult adrenals (Figure 1D). We observed PRC2 target CpGi that acquire minimal methylation during adult differentiation acquire indiscriminate methylation in non-CIMP-high ACC, and are targeted for methylation in CIMP-high ACC. ACC possess exquisitely high purity ((9), Supp Fig 2B), suggesting aberrant CpGi methylation originates specifically from cancer cells.
Unlike with G0S2 (8), promoter hypermethylation did not lower gene expression (Supp Fig 2C). Given PRC2 targets bear H3K27me3 and exhibit low expression in normal tissue, these data suggest PRC2 target DNA methylation reflects an epigenetic class switch (H3K27me3 exchanged for alternative repressive marks, e.g. DNA methylation and/or H3K9me3 (16,39)), or that PRC2 collaborates with DNA methyltransferases (DNMTs) to write DNA methylation at H3K27me3 sites (40).
PRC2 is catalytically active and required for sustained proliferation in CIMP-high ACC
We observed EZH2 is upregulated in a cell-cycle-dependent manner in CIMP-high ACC (Figure 1E; Supp Fig 1E, 2D–F), as expected (41). EZH2 is nuclear, coupled to high H3K27me3, and predictive of poor clinical outcomes (Figure 1F–H; Supp Fig 2G–H). These data are consistent with prior studies (18,42,43), and also suggest EZH2 remains catalytically active on histone substrates in CIMP-high ACC. As EZH2 requires PRC2 incorporation to possess catalytic activity (44–47), this suggested a major role of EZH2 in ACC involves PRC2.
We then examined if the catalytic function of EZH2 was required for CIMP-high ACC proliferation. Here, we primarily used mainstay human ACC cell line, NCI-H295R, with driver alterations in TP53, RB1 and CTNNB1 (encoding β-catenin) (Supp Fig 3A). We performed multiplatform profiling of NCI-H295R cells and demonstrated it is a bona fide model of CIMP-high ACC (Figure 2A–C). We also characterized the murine Y1 and ATC7L ACC cell lines, which harbor genetic alterations in cell cycle machinery characteristic of CIMP-high with variable zF differentiation and Wnt/β-catenin activation (Supp Fig 3A–G); however, both lines express G0s2 (Supp Fig 3D).
Figure 2: DNA methylation is propagated independently of PRC2 in CIMP-high ACC.
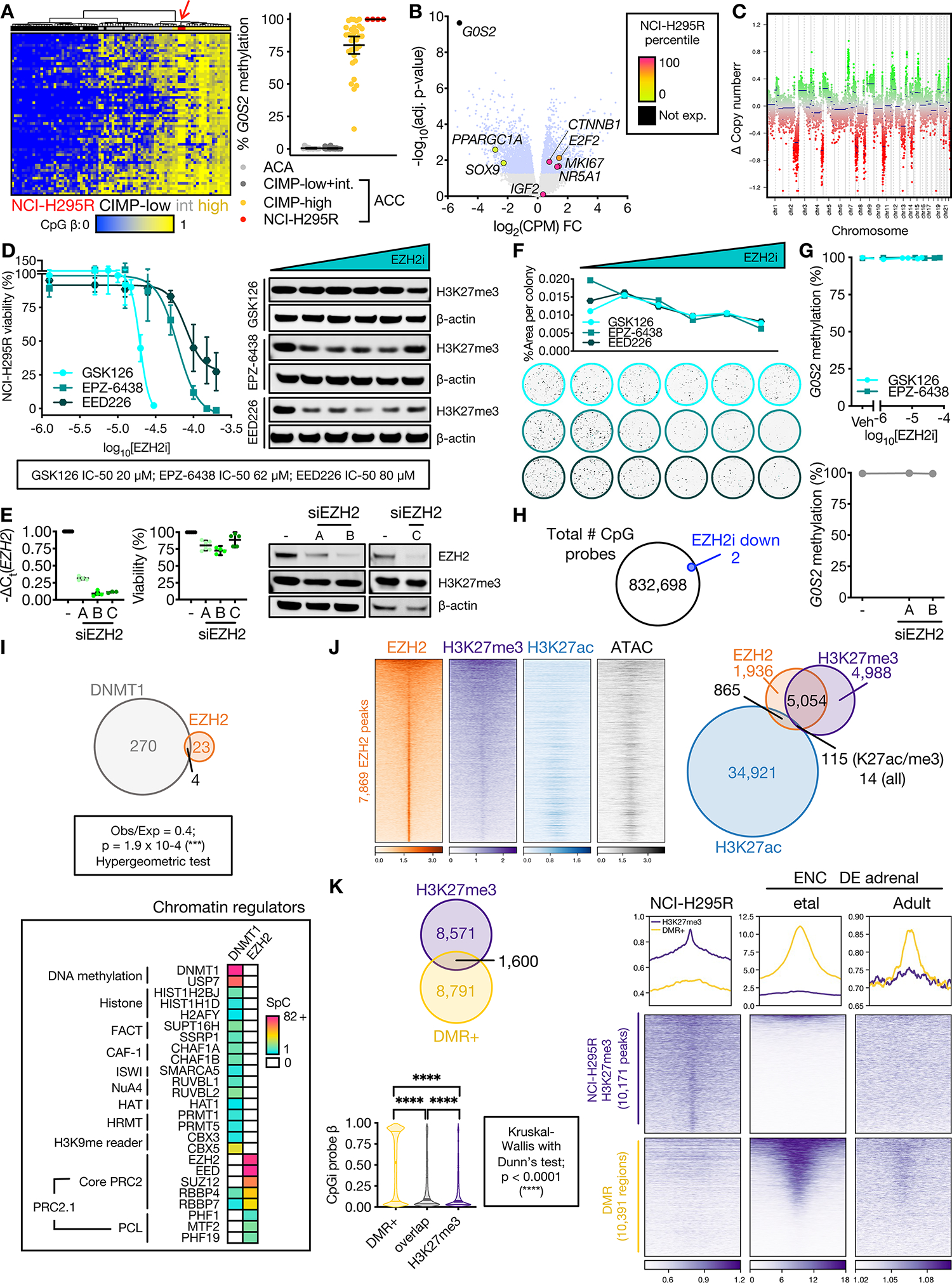
A. Left, heatmap of methylation at probes (rows) that define ACC-TCGA CIMP groups. Columns are ACC-TCGA samples classified by CIMP or baseline NCI-H295R cell line (red arrow, n=3, Illumina 850k array). Unsupervised hierarchical clustering with ward.D2 algorithm, Euclidean distance. Right, targeted assessment of G0S2 methylation in ACA (n=14), ACC stratified by CIMP-status (n=49 non-CIMP-high, n=33 CIMP-high), and baseline NCI-H295R, n=4; line at mean with 95% CI. ACA+ACC data from (8).
B. Volcano plot on RNA-seq data from CIMP-high vs. non CIMP-high ACC from ACC-TCGA. Light blue dots correspond to differentially expressed genes (adj. p-value=Benjamini-Hochberg-corrected p-value<0.05). Named genes are color-coded by NCI-H295R gene expression percentile (calculated from baseline RNA-seq). IGF2 is overexpressed in 90% of ACC, not differentially expressed across CIMP classes.
C. Total CpG signal across NCI-H295R baseline methylome was summed to predict DNA content, chromosomal segments and copy number, demonstrating “noisy” chromosomal signature characteristic of CIMP-high. In euploidy, Δ Copy number = 0.
D. Left, viability curves for NCI-H295R treated with different classes of EZH2i for 96 hours (EPZ-6438, GSK126 are SAM-competitive EZH2i, EED226 is an allosteric EZH2i; n=4 each), x-axis is log10 of the drug concentration in M. Data shown as mean with SEM. Right, western blot measuring H3K27me3 relative to β-actin loading control shown right, n>3; doses tested, GSK126: 0 (Vehicle), 1.25 μM, 5 μM, 7.5 μM, 15 μM, 20 μM (IC-50); EPZ-6438: 0, 1.25 μM, 12.5 μM, 25 μM, 50 μM, 62 μM (IC-50); EED226: 0, 1.25 μM, 12.5 μM, 25 μM, 50 μM, 80 μM (IC-50). Across replicates and all EZH2i, H3K27me3 levels measured by western blot (corrected by loading control and normalized to vehicle) are negatively linearly correlated with EZH2i concentration (Pearson test; p < 0.0001, r = −0.5117).
E. NCI-H295R transfected with EZH2 siRNA (siEZH2 A, B, or C) or scrambled negative control (–), and harvested at 144 hours to assess viability (line at mean with 95% CI) and EZH2/H3K27me3/β-actin by western blot (right, n≥2). NCI-H295R doubling time is ~60 hours; time points selected adequate to measure replication dilution of H3K27me3.
F. NCI-H295R were pre-treated with different classes of EZH2i for 96 hours as in D (right), and viable cells were plated at colony forming density in regular (EZH2i-free) medium, grown for 4 weeks. Top, colony area (quantified with Fiji) vs. EZH2i pre-treatment doses. Bottom, representative images of crystal-violet stained colonies at increasing EZH2i doses. Representative experiment shown, n=2.
G. G0S2 methylation after increasing doses of EZH2i (n=3, top) or EZH2 siRNA (n=1, bottom). Data represented as mean with SEM for each condition. Veh=Vehicle.
H. Venn diagram of total number of CpG probes in NCI-H295R methylome and those which were differentially methylated following EZH2i (EPZ-6438 at the IC-50 dose). No probes gained methylation after EZH2i.
I. Top, Venn diagram of unique proteins retrieved by DNMT1 IP-MS and EZH2 IP-MS on NCI-H295R nuclear lysates. Bottom, peptides retrieved from DNMT1 or EZH2 IP-MS mapping to known chromatin regulators. SpC=spectral counts.
J. Left, heatmap of EZH2, H3K27me3, and H3K27ac ChIP-seq and accessibility (ATAC=ATAC-seq) signal in baseline NCI-H295R at EZH2 peaks, ranked by EZH2 signal. Centered at peak +/− 3 kb window. Right, Venn diagram of EZH2, H3K27me3, and H3K27ac peaks.
K. Top, Venn diagram of baseline NCI-H295R H3K27me3 peaks and regions targeted for hypermethylation in CIMP-high ACC (DMR+). Bottom, violin plot of average CpG island methylation level (β) in DMR+, DMR+/H3K27me3 overlap regions, and H3K27me3 peaks; lines at median and quartiles.
L. Profile plot and heatmap of H3K27me3 signal at regions annotated as baseline NCI-H295R H3K27me3 peaks and DMR+ in NCI-H295R and ENCODE fetal and adult adrenal ChIP-seq. Centered at peak/DMR +/− 3 kb window.
We treated ACC cell lines with S-adenosyl-L-methionine (SAM)-competitive or allosteric EZH2/PRC2 inhibitors (EZH2i). EZH2i induced dose-dependent cell death preceded by H3K27me3 depletion (Figure 2D, Supp Fig 3H). In contrast, EZH2 knockdown induced minimal loss of viability and mild H3K27me3 depletion, suggesting H3K27me3 rather than EZH2 itself is essential for proliferation, Figure 2E. Strikingly, transient EZH2i exposure diminished 2D colony formation and survival in a dose-dependent manner (Figure 2F), suggesting EZH2i induces heritable epigenomic changes that diminish sustained proliferation potential.
DNA hypermethylation excludes PRC2 and is associated with aberrant H3K27me3 deposition
We next examined if PRC2 target DNA hypermethylation is directed by catalytically active EZH2 (40). EZH2i and EZH2 knockdown did not change DNA methylation at the G0S2 locus or genome-wide (Figure 2G–H). As PRC2 may direct DNMTs through protein-protein interactions (40,48), we performed EZH2-directed complex immunoprecipitation paired with mass spectrometry (IP-MS) and DNMT1 IP-MS on NCI-H295R nuclear lysates. We proceeded with DNMT1 as this is the predominant DNMT in this line, like EZH2 is the predominant H3K27 methyltransferase (Supp Fig 3I). DNMT1 also exhibits cell-cycle-dependent upregulation in ACC and cancer (Supp Fig 1E, 2D).
We observed EZH2 binds no DNMTs, with virtually no overlap between EZH2 and DNMT1 interactomes (Figure 2I). DNMT1 binds many chromatin-bound proteins including the HP1 family of H3K9me readers, representing a conserved DNMT1 mode (49), and consistent with a model in which H3K9me3, rather than H3K27me3, instructs CIMP-high DNA methylation (16). EZH2 is assembled in PRC2.1, a canonical PRC2 assembly defined by association with PCL accessory proteins that target preferential PRC2 recruitment to unmethylated CpGi (Figure 2I, (50,51)).
We next examined EZH2 recruitment genome-wide relative to H3K27me3, active chromatin measured by H3K27 acetylation (H3K27ac), and accessible chromatin. H3K27me3/H3K27ac were mutually exclusive, and most EZH2 peaks co-localized with broad, inaccessible H3K27me3 domains (Figure 2J). Regions targeted for hypermethylation in CIMP-high ACC (DMR+) and H3K27me3 peaks exhibited minimal overlap, and DNA methylation levels of H3K27me3 peaks were substantially lower than those of DMR+ (Figure 2K). EZH2 and H3K27me3 were excluded from the hypermethylated G0S2 locus (Supp Fig 3J). In fetal and adult adrenals, we observed strong H3K27me3 deposition at DMR+, and reduced deposition at NCI-H295R H3K27me3 peaks (Figure 2L). In ACC-TCGA, we observed reduced accessibility of both DMRcate regions and NCI-H295R H3K27me3 in CIMP-high compared to non-CIMP-high ACC (Supp Fig 3K). These observations suggest DNA hypermethylation in CIMP-high ACC is propagated independently of PRC2, leads to epigenetic class switching, PRC2 eviction and recruitment to novel sites for H3K27me3 catalysis.
EZH2i disrupts EZH2 recruitment, wipes H3K27me3, and reverses CIMP-high-defining transcriptional programs
We then treated NCI-H295R cells with EZH2i (EPZ-6438) at the IC-50 dose (Figure 2D), and performed chromatin immunoprecipitation sequencing (ChIP-seq). At baseline EZH2 and H3K27me3 sites, EZH2i decreased EZH2 and H3K27me3 while increasing chromatin accessibility. Surprisingly, EZH2i triggered EZH2 displacement to active and accessible chromatin without new H3K27me3 deposition (Figure 3A). EZH2i failed to restore expression of hypermethylated genes, like G0S2, which was undetectable by RNA-seq at baseline and after EZH2i. However, EZH2i restored expression lowly expressed genes including unmethylated PRC2 targets like FOXF1 (Figure 3B; Supp Fig 4A–B), suggesting catalytically active EZH2 restrains gene expression in CIMP-high ACC. Intriguingly, EZH2i globally disrupted gene expression, with more than half the transcriptome classified as differentially expressed (Figure 3C).
Figure 3: EZH2i disrupts EZH2 recruitment genome-wide, restrains zF differentiation, and reverses the CIMP-high molecular state.
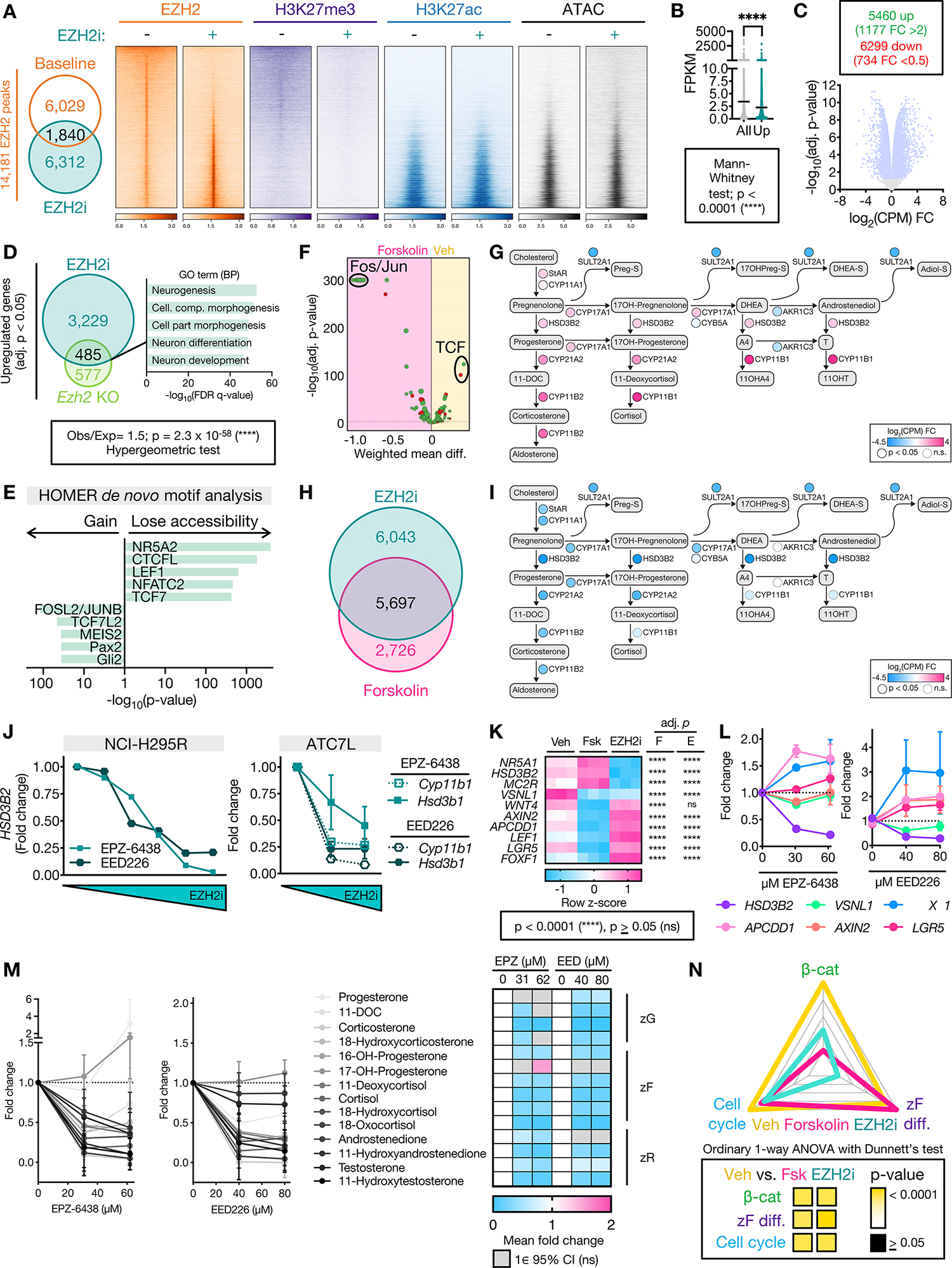
A. Left, Venn diagram of NCI-H295R EZH2 peaks at baseline (vehicle-treated, EZH2i−) and after EZH2i (EZH2i+=EPZ-6438 at the IC-50 dose). Right, heatmap of EZH2, H3K27me3, H3K27ac signal in union set of EZH2 peaks at baseline and after EZH2i. Heatmap ranked by ratio of EZH2 signal at baseline to after EZH2i. Centered at peak +/− 3 kb window.
B. Average baseline NCI-H295R expression (fragments per kilobase of transcript per million mapped reads, FPKM) of all genes in the transcriptome compared to baseline FPKM of genes induced by EZH2i (Up).
C. Top, number of differentially expressed genes (adj. p-value<0.05) in EZH2i- vs. vehicle-treated NCI-H295R, FC=fold change. Bottom, corresponding volcano plot. Light blue dots correspond to differentially expressed genes.
D. Left, Venn diagram of genes upregulated by EZH2i in NCI-H295R with genes upregulated in mouse model of SF1-driven Ezh2 deficiency (Ezh2 KO, (52)). Right, 5 most significant gene sets resulting from GSEA on overlap genes using the GO (BP=Biological Processes) gene set.
E. HOMER motif analysis on differentially accessible peaks from NCI-H295R EZH2i compared to baseline ATAC-seq. NR5A2 shares same motif as NR5A1 (SF1).
F. DiffTF integrating RNA-seq and ATAC-seq from NCI-H295R treated with forskolin to induce zF differentiation vs. vehicle. Negative and positive weighted mean difference reflect transcription factor signal stronger in forskolin- or vehicle-treated cells, respectively.
G. Steroidogenesis diagram depicting impact of forskolin on expression of zonally expressed steroidogenic enzymes in NCI-H295R by RNA-seq.
H. Venn diagram of differentially expressed genes (compared to baseline) in NCI-H295R treated with EZH2i or forskolin by RNA-seq (Supp Table 1).
I. Steroidogenesis diagram depicting impact of EZH2i on enzyme expression in NCI-H295R by RNA-seq.
J. Fold change in expression of steroidogenic enzymes in ACC cell lines treated with increasing doses of EZH2i measured by qPCR. NCI-H295R, n=1. ATC7L, n=2–3 for all points. Data shown as mean with SEM. Concentrations tested were: NCI-H295R, as in Figure 2D; ATC7L - 0, 42 μM, 84 μM EPZ-6438 (IC-50) and 0, 53 μM, 107 μM EED226 (IC-50).
K. Heatmap of gene expression of NCI-H295R at baseline (vehicle, Veh), following forskolin (Fsk) treatment or following EZH2i with adj. p-value for comparison to Veh shown right, by RNA-seq. FOXF1 is a PRC2 target (Supp Fig 4B). Per Supp Fig 3B, “zF genes”= HSD3B2, MC2R and remainder are “zG genes”.
L. NCI-H295R were treated with indicated doses of EZH2i followed by forskolin. Gene expression measured by qPCR, n=3. Data shown as mean with SEM.
M. LC-MS/MS analysis of media from NCI-H295R treated as in L to measure zone-specific steroid output. Left, line graph depicting fold change of normalized steroid output relative to vehicle. Data shown as mean and 95% CI. Right, heatmap displaying mean fold change. If the null value (1) falls in the 95% CI of the mean for each treatment group, change in steroid output is considered insignificant (ns) and fold change is not displayed.
N. ZF differentiation, Wnt, and cell cycle scores for NCI-H295R at baseline (Veh) or treated with forskolin (Fsk) or EZH2i, calculated and graphed as in Figure 1B.
ACC exhibit a spectrum of zF differentiation, Wnt/β-catenin activation and proliferation, with CIMP-high ACC at the relative maxima of these poles (Figure 1B; Supp Fig 1, 3B–G). In the mouse model of SF1-driven Ezh2 ablation, mice develop glucocorticoid insufficiency due to a failure of the zF to differentiate and proliferate in response to ACTH (52). Despite H3K27me3 deposition at several new sites in CIMP-high ACC (Figure 2L; Supp Fig 2K), EZH2i derepressed >50% of the genes induced in this mouse model including neuronal programs (Figure 3D). This was accompanied by marked epigenetic changes, where EZH2i (1) restored chromatin accessibility of programs silenced in steroidogenic adrenocortical cells (e.g. those driven by PAX2 and GLI), (2) disrupted accessibility of targets of canonical Wnt/β-catenin transcription factors TCF/LEF, and (3) reduced accessibility of putative SF1 and CTCFL targets (Figure 3E). Based on these findings, we hypothesized EZH2i disrupts adrenocortical differentiation.
To evaluate this, we treated NCI-H295R cells with forskolin, a zF differentiation agent that induces the PKA/cAMP signaling cascade downstream of ACTH (53,54). Forskolin administration (1) increased expression of zF differentiation genes, (2) diminished expression and accessibility of TCF targets, and (3) induced expression of zonally expressed steroidogenic enzymes (Figure 3F–G; Supp Fig 4C). Strikingly, EZH2i disrupted ~70% of genes differentially expressed after forskolin treatment (Figure 3H), and potently downregulated steroidogenic enzymes (Figure 3I). Suppression of steroidogenic enzymes was dose-dependent and observed with different classes of EZH2i across ACC cell lines (Figure 3J). Moreover, EZH2i pretreatment followed by forskolin administration diminished both forskolin-induced silencing of canonical Wnt targets and induction of steroidogenic enzymes, ultimately restraining steroid output (Figure 3K–M).
These observations are consistent with a role for EZH2 in programming the cellular response to ACTH/PKA in CIMP-high ACC, though not solely by dysregulating expression of PKA signaling components (52). Though EZH2i induced few canonical Wnt targets (Figure 3E, K), EZH2i reversed all three core modules of CIMP-high ACC (Figure 3N), while forskolin induced differentiation at the expense of cell cycle and Wnt activation (Figure 3F, K, N). Observing that EZH2i disrupted a spectrum of transcriptional programs (Figure 3C), including those governed by EZH2 in the normal adrenal (Figure 3D), led us to explore if the effects of EZH2i result from an alternative EZH2 role.
EZH2 binds transcriptional coactivator β-catenin in an off-chromatin complex
EZH2 IP-MS revealed several non-PRC2 partners, including nuclear receptors known to regulate adrenocortical biology (55), as well as β-catenin (Figure 4A), constitutively active in NCI-H295R due to the p.S45P mutation (Supp Fig 3A). Given β-catenin’s abundance in the EZH2 interactome (Figure 4A), possible role in the chromatin response to EZH2i (Figure 3E) and its well-established role in adrenocortical differentiation and tumorigenesis, we elected to focus our studies on EZH2/β-catenin.
Figure 4: Nuclear pools of off-chromatin (EZH2-bound) and on chromatin (SF1-bound) β-catenin direct upper zF differentiation in CIMP-high ACC.
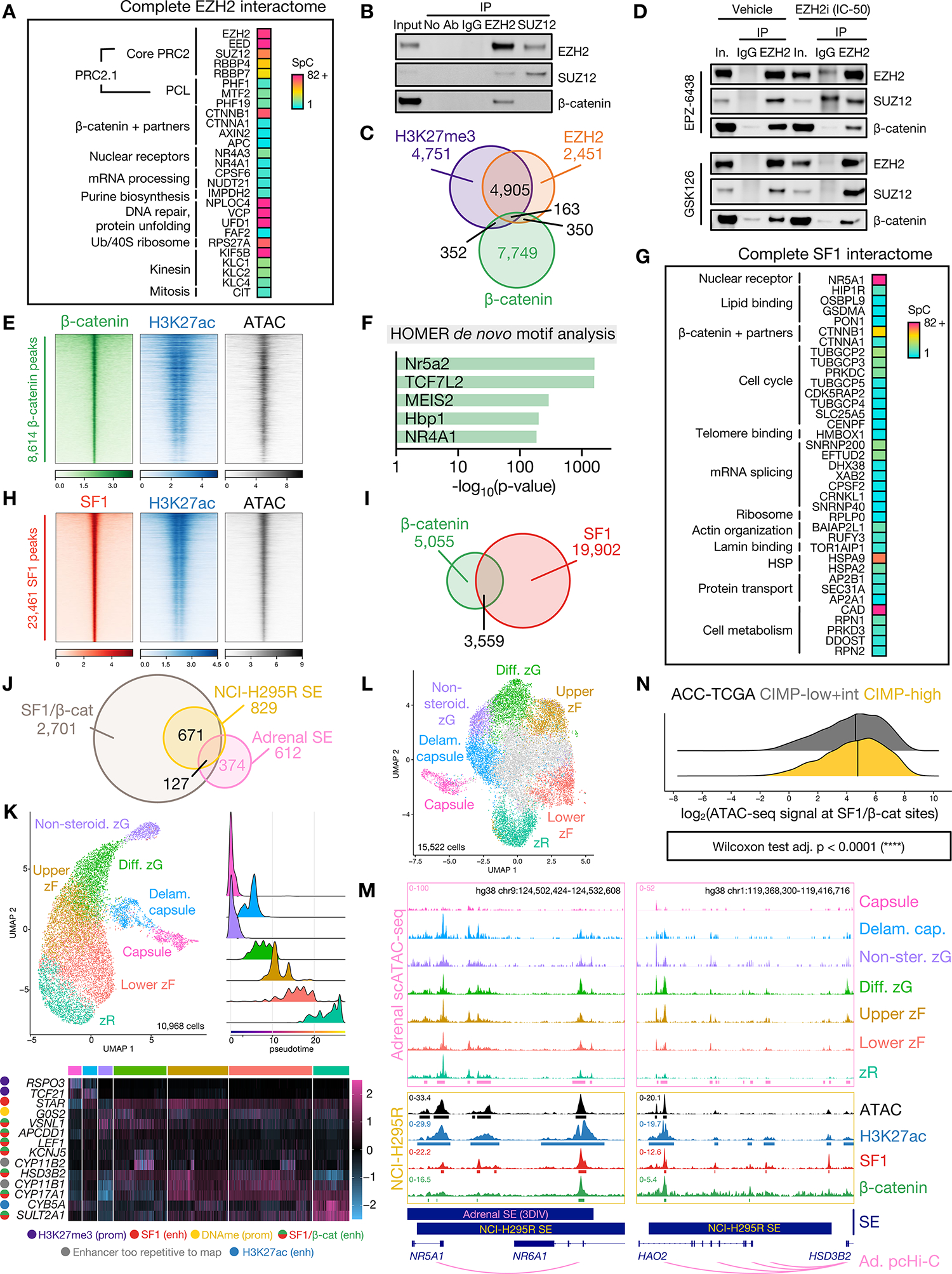
A. Peptides retrieved from EZH2 IP-MS on NCI-H295R nuclear lysates.
B. Representative western blot of NCI-H295R nuclear co-IP, detecting EZH2, SUZ12 and β-catenin. Lanes are 10% input, negative control co-IP (no antibody, IgG), EZH2 IP and SUZ12 IP. n>5 (EZH2 IP), n=2 (SUZ12 IP).
C. Venn diagram of H3K27me3, EZH2, and β-catenin ChIP-seq peaks in baseline NCI-H295R.
D. Representative western blot of EZH2 IP in vehicle- (left) or EZH2i-treated (right, IC-50 dose) NCI-H295R. Lanes are 10% input, negative control IgG IP, EZH2 IP. Higher molecular weight band in EPZ-6438 IgG lane in EZH2/SUZ12 blots is non-specific and emerges when using the same antibody species for IP and western.
E. Heatmap of β-catenin, H3K27ac, and ATAC signal in baseline NCI-H295R at β-catenin peaks, ranked by β-catenin signal. Centered at peak +/− 3 kb window.
F. HOMER motif analysis on baseline NCI-H295R β-catenin peaks.
G. Peptides retrieved from SF1 IP-MS on NCI-H295R nuclear lysates.
H. Heatmap of SF1, H3K27ac and ATAC signal in baseline NCI-H295R at SF1 peaks, ranked by SF1 signal. Centered at peak +/− 3 kb window.
I. Venn diagram of baseline NCI-H295R SF1 and β-catenin peaks.
J. Venn diagram of baseline NCI-H295R SF1/β-catenin peaks, baseline NCI-H295R super-enhancers (SE), and physiologic adrenal SE called by 3DIV on ENCODE samples.
K. Single-cell RNA-seq (scRNA-seq) data from fetal, neonatal and adult human adrenal (31) was integrated and analyzed with comparison to reference markers to identify populations comprising the corticocapsular unit. Top left, scRNA-seq UMAP. Non-steroid.=Non-steroidogenic, Diff.=Differentiated, Delam.=Delaminating. Top right, scRNA-seq pseudotime analysis (origins set for fetal and adult adrenal populations in the capsule and non-steroidogenic zG). Bottom, heatmap of scaled expression of lineage-defining genes across scRNA-seq with epigenetic regulation in NCI-H295R shown left. Prom=promoter, enh=enhancer, DNAme=DNA methylation. Gene regulation by active (H3K27ac only) and SF1- or SF1/β-catenin-bound enhancers was identified by overlap of ChIP-seq with adrenal promoter capture Hi-C (pcHi-C (34)).
L. Single-cell ATAC-seq (scATAC-seq) data from fetal and adult human adrenal (32,33) was integrated and analyzed with comparison to scRNA-seq and reference markers to identify analogous populations comprising the corticocapsular unit. Cells colored in grey in UMAP plot are likely cortical given accessibility within NR5A1 locus though possess ambiguous classification.
M. Example adrenal scATAC-seq and NCI-H295R ATAC, SF1, β-catenin, and H3K27ac tracks across the NR5A1 and HSD3B2 loci in baseline NCI-H295R. Adrenal scATAC peak calls are depicted by pink bars at the bottom of top window, and NCI-H295R peak calls are depicted by bars below each track. 3DIV annotation of adrenal super-enhancers (SE), and NCI-H295R baseline SE are shown by bars below window. Promoter/enhancer contacts from adrenal (Ad.) pcHi-C (34) are depicted below gene annotations.
N. Ridge plot of chromatin accessibility signal at SF1/β-catenin co-targets in ACC-TCGA ATAC-seq samples (n=9, (65)). Line at median.
We were unable to purify endogenous β-catenin-containing transcriptional complexes by β-catenin IP-MS, due to β-catenin’s strong affinity for contaminating adherens junctions (56), Supp Fig 4D. However, EZH2, SUZ12, and β-catenin co-eluted with histones bearing K27 modifications by size exclusion chromatography (SEC) (Supp Fig 4E). While EZH2 IP consistently retrieved both SUZ12 (a core PRC2 member) and β-catenin, SUZ12 IP did not retrieve β-catenin (Figure 4B). EZH2/H3K27me3/β-catenin possess minimal overlap on chromatin (Figure 4C), and EZH2i did not disrupt nuclear EZH2/β-catenin (Figure 4D, Supp Fig 4F). Together, these data suggest EZH2/β-catenin is a nuclear but off-chromatin complex that may compete for β-catenin binding to chromatin and participate in the epigenetic response to EZH2i, in contrast to its role in other contexts (57).
EZH2i reversed the transcriptional and epigenomic programs that define CIMP-high ACC (Figure 3E, 3N). Given CIMP-high are Wnt-active and β-catenin is a major EZH2 binding partner spared by EZH2i, we next investigated β-catenin’s role on chromatin.
SF1/β-catenin regulates a super-enhancer-driven zF differentiation program in ACC
In NCI-H295R cells, β-catenin colocalized with active and accessible chromatin regions genome-wide (Figure 4E). Surprisingly, β-catenin peaks exhibited substantial enrichment for the SF1 motif, comparable to enrichment for TCF/LEF motifs (Figure 4F). This was consistent with SEC demonstrating co-elution of SF1, β-catenin, and H3K27ac in ACC cell lines (Supp Fig 4E). By SF1 IP-MS, we observed that p.S45P β-catenin is a major SF1 binding partner in NCI-H295R cells (Figure 4G).
We and others previously reported an SF1/β-catenin complex, thought to regulate gene expression but without a clear global role (58–60). ChIP-seq for SF1 in NCI-H295R cells revealed that, as expected, SF1 binds active and accessible chromatin regions (Figure 4H). There was substantial overlap between SF1 and β-catenin recruitment (Figure 4I), and SF1/β-catenin sites encompassed predominantly distal regions (Supp Fig 4G), suggesting enhancer regulation.
A special class of enhancers, super-enhancers (SE), are defined by high density H3K27ac and possess occupancy by lineage-defining transcription factors, driving cell-of-origin programs in development and disease (28,29,61–64). We observed that >90% of normal adrenal SE remain active enhancers in NCI-H295R cells (Supp Fig 4H); however, the vast majority of NCI-H295R SE are novel (Figure 4J). Strikingly, virtually all NCI-H295R SE possess SF1/β-catenin, representing a major departure from normal adrenal (Figure 4J).
To understand the genomic architecture and physiologic relevance of these SE, we examined promoter capture Hi-C, single-cell RNA-seq, and single-cell ATAC-seq from normal adrenals (31–34), and integrated these studies with our epigenomics data. We observed SF1/β-catenin SE and enhancers contact promoters of many genes critical for zone-specific and adrenocortical steroidogenic identity, including HSD3B2 and NR5A1 itself (Figure 4K–M). These enhancers are present in normal adrenal (Figure 4M, Supp Fig 4I), and more accessible in outer cortex, consistent with nuclear localization of β-catenin in these regions (Figure 1A, 4L–M). Genes that retain promoter H3K27me3 in NCI-H295R cells (e.g. capsular genes RSPO3 and TCF21) are also silenced in the cortex (Figure 4K).
To extend these observations to ACC, we analyzed ACC-TCGA ATAC-seq (65) and observed CIMP-high have increased accessibility of SF1/β-catenin co-targets (Figure 4N). Collectively, these data suggest epigenetic programming in CIMP-high ACC maintains a differentiation state resembling the upper zF through SF1/β-catenin chromatin regulation that co-opts physiologic programming.
EZH2/β-catenin and SF1/β-catenin are selected for through all stages of adrenocortical neoplasia
Members of our team recently developed an autochthonous mouse model of zF-differentiated ACC, driven by combined β-catenin gain-of-function (GOF)/p53 loss-of-function (LOF) in cells expressing adrenocortical enzyme Cyp11b2, BPCreAS/+ (19). β-catenin GOF causes adrenal hyperplasia, though single hit β-catenin GOF or p53 LOF is insufficient for ACC (19,23). Genetically, BPCreAS/+ is similar to CIMP-high ACC, and exhibits selective expansion, unrestricted growth, and dissemination of Wnt-active, rapidly cycling cells with high EZH2 expression and autonomous glucocorticoid production (Supp Fig 5A, Figure 5A–B, (19)).
Figure 5: Nuclear EZH2/β-catenin and SF1/β-catenin complexes persist through adrenocortical neoplastic evolution.
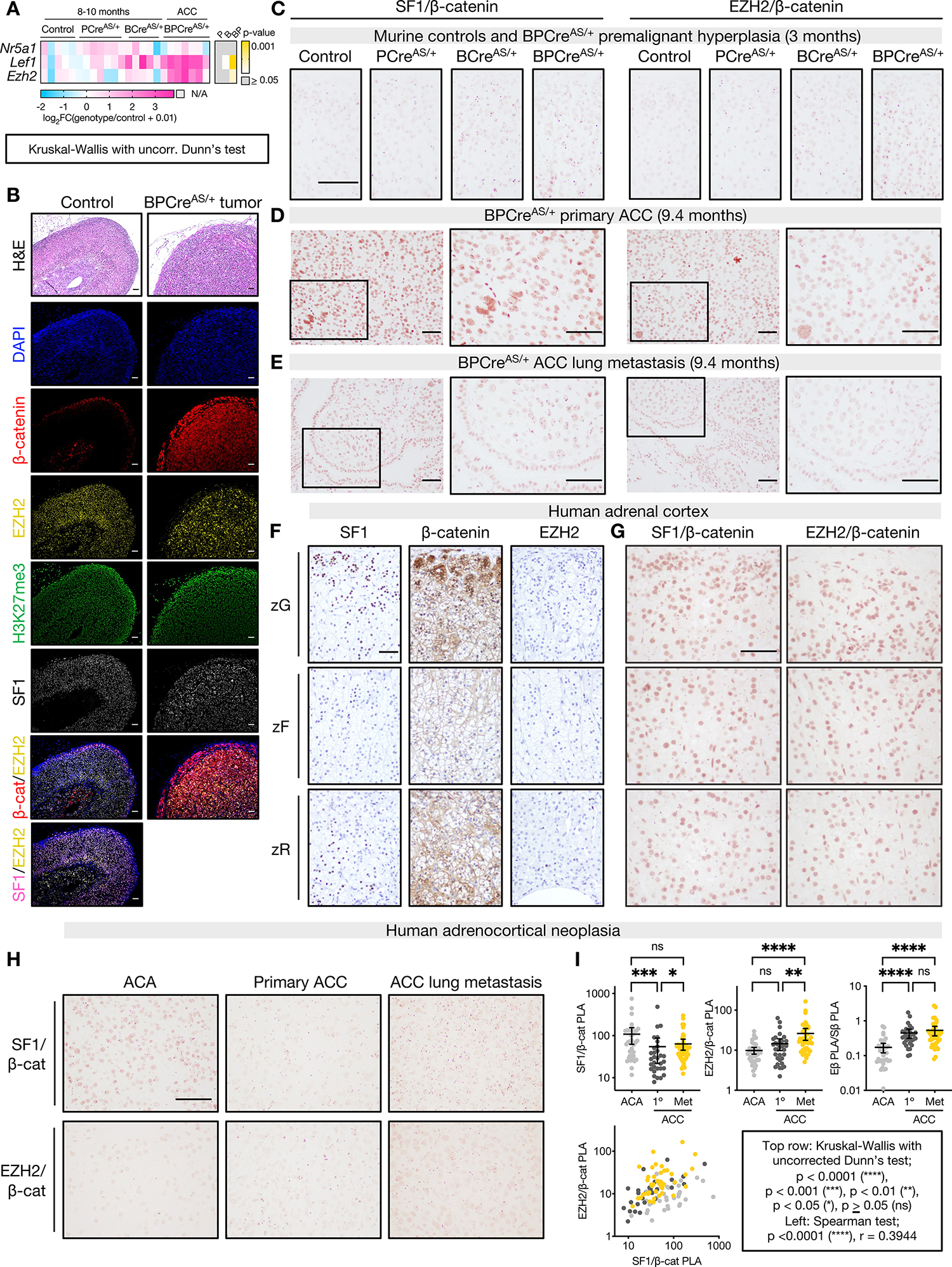
A. Left, heatmap of gene expression measured by qPCR in adrenals from control mice (ASCre/+), mice with p53 LOF (PCreAS/+), mice with β-catenin GOF (BCreAS/+), or ACC from combined p53 LOF/β-catenin GOF (BPCreAS/+). Right, p-value for each genotype compared to control.
B. Top row, representative hematoxylin and eosin (H&E) staining of control adult mouse adrenal and BPCreAS/+ primary tumor (10-month-old). Rows 2–6, immunofluorescence staining nuclei (DAPI), β-catenin, EZH2, H3K27me3, or SF1. Rows 7–8, colocalization of β-catenin or SF1 and EZH2. Bar=50 μm.
C-E. Representative images of SF1/β-catenin (left) and EZH2/β-catenin (right) proximity ligation assay (PLA) performed on 3-month-old adrenals (control, n=4; PCreAS/+, n=3; BCreAS/+, n=3; BPCreAS/+, n=4), BPCreAS/+ primary ACC (n=5), BPCreAS/+ lung metastases (n=3). PLA signal are subnuclear pink dots. Bar=100 μm.
F-G. Representative images of SF1, β-catenin, and EZH2 IHC or SF1/β-catenin and EZH2/β-catenin PLA in human adult adrenal cortex (n=2). Bar=100 μm.
H. Representative images of SF1/β-catenin and EZH2/β-catenin PLA in a TMA of human adult ACA (n=39) and primary (n=34) and metastatic (n=35) ACC.
I. Normalized TMA PLA signal, quantified by Fiji. Each sample is represented by a point. Top right, “Eβ PLA/Sβ PLA” refers to ratio of EZH2/β-catenin PLA signal to SF1/β-catenin PLA signal. Top row, line at mean with 95% CI.
To determine if EZH2/β-catenin and SF1/β-catenin complexes participate in tumorigenesis, we optimized a technique to detect protein-protein interactions in situ (proximity ligation assay, PLA). We observed that β-catenin-containing complexes are nuclear, and zonally distributed in control mice (Figure 5C, Supp Fig 5B–C), mirroring the Wnt/β-catenin signaling gradient (Figure 1A). We also observed transformation and metastatic seeding of cells uniformly expressing EZH2/β-catenin and SF1/β-catenin complexes (Figure 5D–E, Supp Fig 5D).
Given the persistence of EZH2/β-catenin and SF1/β-catenin complexes throughout murine carcinogenesis and the increased accessibility of SF1/β-catenin co-targets in CIMP-high ACC (Figure 4N), we speculated that these complexes would accompany human carcinogenesis. We applied PLA to the normal human adrenal cortex, benign adrenocortical tumors, and primary and metastatic ACC. In the human adrenal, we observed abundant SF1/β-catenin complexes following the Wnt/β-catenin gradient, and infrequent EZH2/β-catenin complexes reflecting the rarity of EZH2 expression (Figure 5F–G). In human adrenocortical tumors, we observed retention of β-catenin-containing complexes through metastatic disease (Figure 5H–I), with increased abundance of EZH2/β-catenin complexes relative to SF1/β-catenin complexes in malignancy (Figure 5I) mirroring increased EZH2 and decreased NR5A1 expression in cancer (Figure 1G, Supp Fig 5E). These data demonstrate that persistence of EZH2/β-catenin and SF1/β-catenin complexes is conserved across murine and human adrenocortical carcinogenesis, suggesting programs correlated with or coordinated by these complexes are subject to positive selection through all phases of CIMP-high ACC evolution.
EZH2i erases SF1/β-catenin-dependent transcriptional and epigenetic programming
The presence of EZH2/β-catenin and SF1/β-catenin in vivo was compelling, given EZH2/β-catenin is an off-chromatin complex that persists with EZH2i (Figure 4B–D, Supp Fig 4F). We also identified that EZH2i reverses zF differentiation (Figure 3), coordinated by SF1/β-catenin in CIMP-high ACC (Figure 4E–N). Furthermore, nearly 40% of genes putatively regulated by SF1/β-catenin enhancers are downregulated by EZH2i (Figure 6A).
Figure 6: EZH2i evicts SF1 and β-catenin genome-wide, disrupting enhancer programming in CIMP-high ACC.
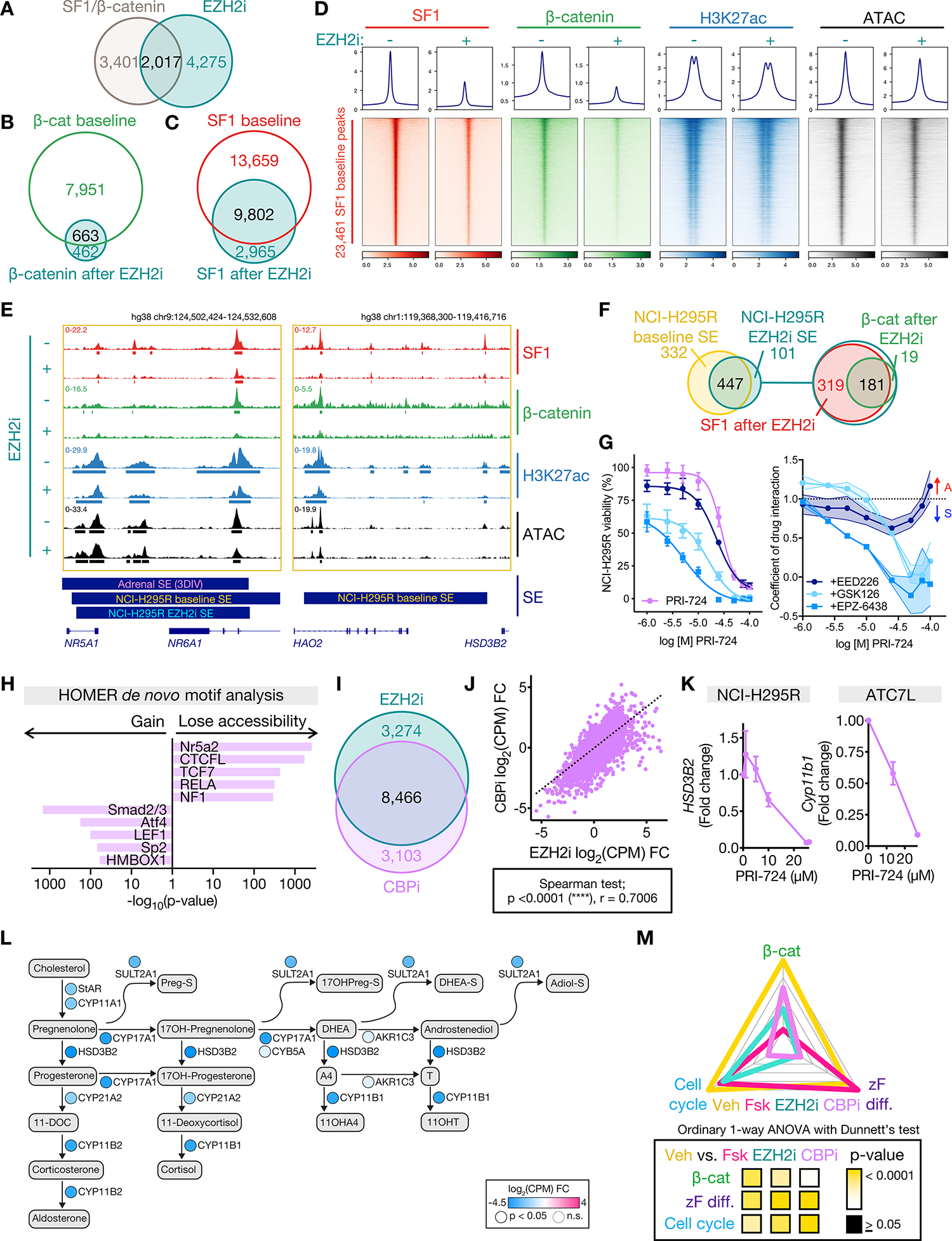
A. Venn diagram of genes putatively regulated by active SF1/β-catenin-bound enhancers and downregulated by EZH2i.
B-C. Venn diagram of NCI-H295R β-catenin or SF1 peaks at baseline (vehicle-treated) and after EZH2i.
D. Profile plot and heatmap of NCI-H295R SF1, β-catenin, H3K27ac, and ATAC signal at SF1 peaks at baseline (EZH2i−) and after EZH2i (EZH2i+), ranked by baseline SF1 signal. Centered at peak +/− 3 kb window.
E. Example SF1, β-catenin, H3K27ac ChIP-seq, and accessibility (ATAC-seq) tracks across the NR5A1 and HSD3B2 loci in NCI-H295R at baseline (as in Figure 4M) or after EZH2i. Peak calls depicted by bars below each track. 3DIV annotation of adrenal SE, NCI-H295R baseline and EZH2i SE shown by bars below window.
F. Left, Venn diagram of NCI-H295R SE at baseline and after EZH2i. Right, Venn diagram of NCI-H295R EZH2i SE with SF1 EZH2i peaks and β-catenin EZH2i peaks.
G. Left, viability curves for NCI-H295R treated with increasing concentrations of CBP inhibitor (CBPi) PRI-724 +/− different EZH2i (GSK126, EPZ-6438 or EED226) at the IC-50 dose. Right, coefficient of drug interaction (CDI) for viability. CDI>1 represents antagonism (A), CDI <1 represents synergy (S). Data are represented by mean with SEM (whiskers or error bands). CBPi, n=9; CBPi + GSK126, n=3; CBPi + EPZ-6438, n=3; CBPi + EED226, n=3.
H. HOMER motif analysis on differentially accessible peaks from NCI-H295R CBPi (IC-50) compared to baseline ATAC-seq.
I. Venn diagram of differentially expressed genes (compared to baseline) in NCI-H295R treated with EZH2i or CBPi (IC-50) measured by RNA-seq (Supp Table 1).
J. Scatterplot of change in gene expression (compared to baseline) in NCI-H295R treated with CBPi vs. EZH2i.
K. Fold change in expression of steroidogenic enzymes in ACC cell lines treated with increasing doses of CBPi measured by qPCR. NCI-H295R, n=2. ATC7L, n=3. Data shown as mean with SEM.
L. Steroidogenesis diagram depicting impact of CBPi on enzyme expression in NCI-H295R by RNA-seq.
M. ZF differentiation, Wnt, and cell cycle scores for NCI-H295R at baseline (Veh) or treated with forskolin (Fsk), EZH2i, or CBPi calculated and graphed as in Figure 3N.
Strikingly, EZH2i evicted SF1 and β-catenin genome-wide, decreasing H3K27ac and accessibility at SF1 sites (Figure 6B–D). We speculated EZH2i expunges β-catenin from chromatin via accumulation of EZH2/β-catenin (Figure 4D; Supp Fig 4F) induced by EZH2 eviction from H3K27me3 domains (Figure 3A). As EZH2 and SF1 do not directly interact (Figures 4A, 4G), it was difficult to rationalize why EZH2i disrupted global SF1 programming even at regions not annotated as β-catenin co-targets (Figure 6C–D). However, EZH2i disrupted SF1/β-catenin recruitment to prototype SE including those regulating NR5A1 (encoding SF1) and HSD3B2, resulting in decreased gene expression and accessibility (Figure 3J–K, 6E). Indeed, EZH2i downgraded half of all baseline SE (Figure 6F).
To assess the extent to which EZH2i genomic changes result directly from β-catenin-dependent SE disruption, we treated ACC cell lines with a specific inhibitor of β-catenin binding to CBP (CBPi PRI-724 (66)), an H3K27 acetyltransferase required for enhancer activity (67,68). CBPi is unlikely to directly alter EZH2 recruitment; however, CBPi induced dose-dependent loss of viability synergistic with EZH2i in all ACC cell lines (Figure 6G; Supp Fig 5F). We observed redundant and highly correlated effects of CBPi and EZH2i on the NCI-H295R epigenome (Figure 6H) and transcriptome (Figure 6I–J), including dose-dependent dedifferentiation (Figure 6K–L). Like EZH2i, CBPi reverses all core modules that define CIMP-high ACC (Figure 6M). Together, these data suggest SF1/β-catenin enhancer programming is a central CIMP-high susceptibility.
EZH2i hinders tumor growth, proliferation, and SF1/β-catenin-dependent differentiation in vivo
To determine if differentiation programs targeted by EZH2i represent a viable therapeutic strategy in zF-differentiated ACC bearing SF1/β-catenin and EZH2/β-catenin complexes, we developed a subcutaneous allograft model using a cell line (BCH-ACC3A) derived from BPCreAS/+ ACC with metastatic potential, Figure 7A. Recipient mice were treated with vehicle or EZH2i, which was well tolerated (Supp Fig 5G). EZH2i-treated mice exhibited diminished tumor growth (Figure 7B) with decreased H3K27me3 deposition (Figure 7C) and proliferation (Figure 7D, Supp Fig 5H). Tumors from EZH2i-treated mice also exhibited dedifferentiation (Figure 7E–F) with a reduction in SF1/β-catenin complexes (Figure 7G) and retention of EZH2/β-catenin complexes (Figure 7H), recapitulating the molecular features we observed in vitro (Figures 3–4, 6; Supp Fig 4F, 6). Taken together, these studies point to zF differentiation as a targetable epigenetic vulnerability selected for in CIMP-high ACC and provide proof-of-principle support for efficacy of dedifferentiating therapies in ACC treatment.
Figure 7: EZH2i hinders ACC growth, proliferation, and differentiation in vivo.
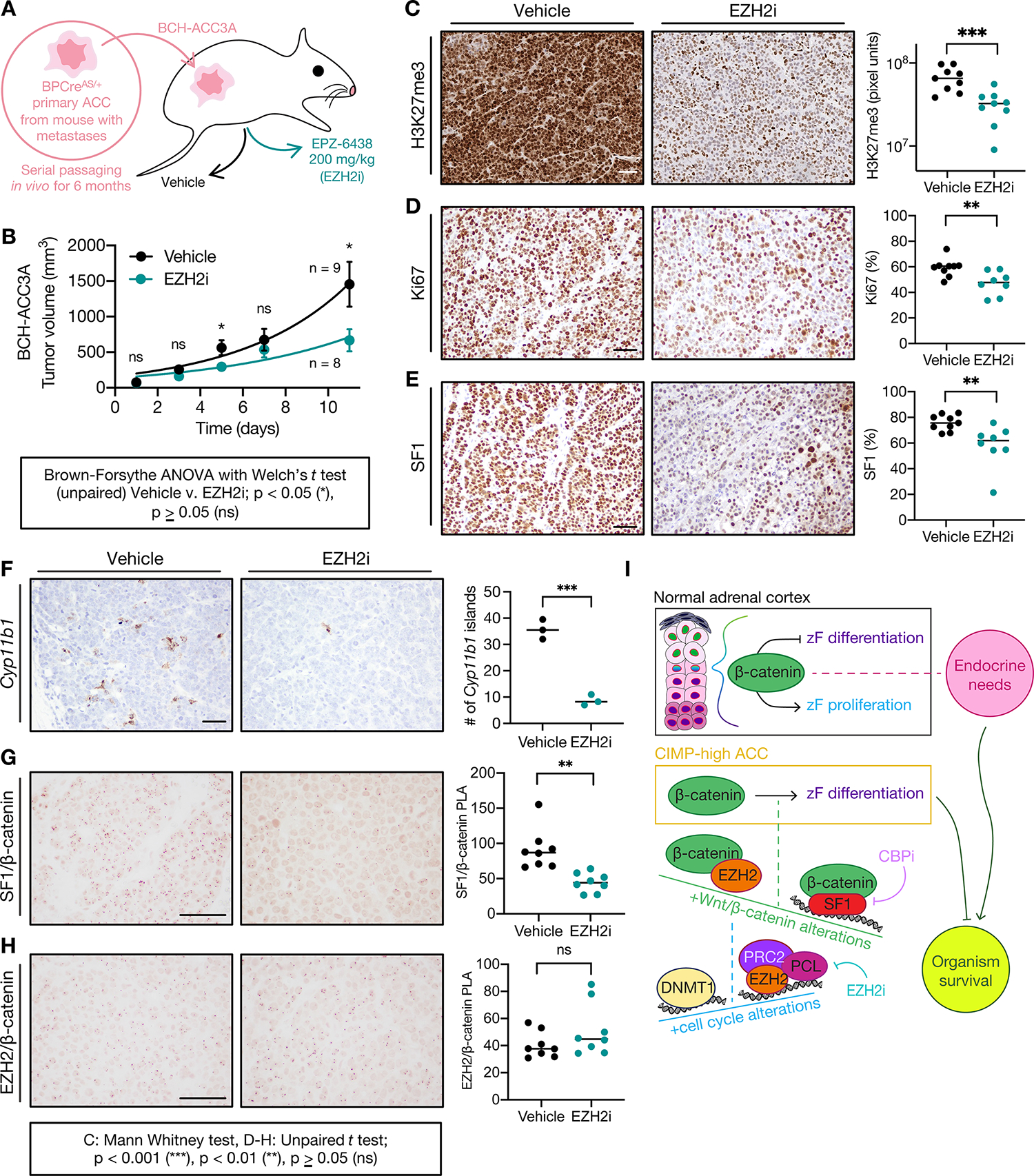
A. Derivation of BCH-ACC3A cell line and subcutaneous NSG mouse allograft model, randomized to vehicle or EZH2i treatment at tumor volume 100 mm3.
B. Tumor growth across treatment groups; data shown as mean with SEM.
C. H3K27me3 IHC across treatment groups. Left, bar=50 μm; right, each sample represented by 3 points, H3K7me3 quantified by MATLAB, line at median.
D-H. Ki67 IHC, SF1 IHC, Cyp11b1 RNA in situ hybridization, SF1/β-catenin PLA or EZH2/β-catenin PLA across treatment groups. Left, bar=100 μm; right, each sample is represented by a point and % nuclear signal, Cyp11b1 islands, or normalized PLA signal quantified by Fiji, line at median.
I. Model: In the upper zF of the normal adrenal cortex, β-catenin restrains zF differentiation or permits zF proliferation depending on endocrine demands (systemic need for glucocorticoids and flux through ACTH). This homeostasis is required for organism survival. In CIMP-high ACC, β-catenin drives zF differentiation through SF1/β-catenin hijacking of genome-wide SE. SF1/β-catenin’s actions on chromatin are limited by EZH2/β-catenin, an off-chromatin complex that completes for β-catenin binding. EZH2/β-catenin abundance is limited by on chromatin EZH2 and PRC2 catalytic activity. PRC2 remains catalytically active in CIMP-high ACC despite displacement by CpGi hypermethylation (written by DNA methyltransferases like DNMT1). Recurrent Wnt pathway and cell cycle alterations in CIMP-high ACC promote the formation of β-catenin-containing and EZH2-containing complexes. Ultimately, β-catenin-dependent zF differentiation is required for sustained ACC proliferation at the cost of organism survival. This program is erased by ACC dedifferentiating agents like EZH2i or CBPi, representing a promising therapeutic avenue.
DISCUSSION
ACC is exquisitely rare and outcomes remain dismal. CIMP-high ACC is prevalent, invariably metastatic, and lethal (8,9). Molecularly, CIMP-high ACC is defined by abnormal DNA methylation and paradoxical activation of cell cycle, Wnt/β-catenin signaling, and zF differentiation. We discovered that abnormal CpGi hypermethylation, in addition to serving as a pathognomonic marker of aggressive disease, displaces EZH2/PRC2 to novel sites. CIMP-high DNA hypermethylation is uniform, with many targets possessing binary and complete methylation (8). These data, consistent with literature examining etiology and emergence of CIMP-high (14,15), suggest acquisition of this signature is an early selection event in adrenocortical carcinogenesis.
We reconcile the convergence of zF differentiation and Wnt/β-catenin activation in ACC by discovering uniform SF1/β-catenin control of CIMP-high SE, including an SE that regulates expression of SF1 itself. Selection for zF differentiation is counterintuitive given the many events other cancers acquire to harness proliferation potential at the expense of differentiation (69). Our data suggest that the cell originating CIMP-high ACC is one which relies on β-catenin and zF differentiation for sustained proliferation, perhaps a transit-amplifying ACTH-responsive cell of the upper zF which fails to proliferate in the setting of congenital adrenocortical EZH2 deficiency or β-catenin ablation (52,70). This is further supported by the demonstrations that combined β-catenin/cell cycle GOF generates zF-differentiated glucocorticoid producing ACC (19), and adrenocortical deficiency of negative Wnt pathway regulator ZNRF3 induces zF hyperplasia with eventual malignant transformation (71,72).
We identify a series of protein complexes, EZH2/β-catenin and SF1/β-catenin, that shuttle β-catenin off and on chromatin. These complexes are zonally distributed and exist in the presence of active Wnt signaling, stabilized either by genetic alteration or physiologic Wnt ligands (Figure 5). They are conserved across murine and human adrenocortical carcinogenesis, and selected for throughout ACC evolution. EZH2/β-catenin formation is a necessary but costly consequence of Wnt and cell cycle activation in CIMP-high, destabilizing the SF1/β-catenin program. CIMP-high ACC may thus require high PRC2 catalytic activity to restrain EZH2/β-catenin in the setting of CpGi hypermethylation, in contrast to other tissues that use CIMP-high to select for PRC2 loss of function with malignancy (20). The EZH2/β-catenin/SF1 triangulation therefore creates a dependence of zF differentiation on PRC2, illuminating an intrinsic tissue-specific vulnerability in CIMP-high ACC with therapeutic significance in vitro and in vivo (Figure 7I).
Repressive epigenetic modifiers are often modeled as complexes that maintain stemness. PRC2 is critical for embryonic pluripotency and gastrulation (4), and in many cancers restrains differentiation for sustained proliferation potential (5), in apparent contrast to our work. A perhaps more nuanced interpretation is that PRC2/H3K27me3 deposition facilitates cell state transitions required for accurate differentiation. Indeed, our studies reveal that catalytically active PRC2 stabilizes a pro-proliferative differentiation state in CIMP-high ACC, by limiting EZH2’s interaction with a transcriptional coactivator core to adrenocortical cell type specification, β-catenin.
The Wnt/β-catenin pathway is recurrently activated by genetic alteration in ~20% of cancer and >50% of CIMP-high ACC (cBioPortal (RRID:SCR_014555), (8–10)). Efforts to target this pathway clinically have failed, due to life-limiting on-target toxicities in Wnt-dependent organs with rapid turnover (66). Here, we discover that selection for active β-catenin in ACC is dependent on maintenance of a tissue specific program: SF1-dependent zF differentiation. Given the paucity of organs that require both programs for homeostatic renewal, this opens a large therapeutic window for targeting oncogenic β-catenin. Do alternative context-specific nuclear receptors bind β-catenin in other cancers? A paradigm geared towards tissue-specific disruption of oncogenic programs will be essential to combat cancers that rely on differentiation for growth and dissemination, like ACC.
This study has several important limitations. We rely heavily on small molecules, in part because we seek to demonstrate how pharmacologic tools unveil vulnerabilities in disease. While EZH2i induces a reduction in H3K27me3 at low doses, the doses of EZH2i that induce loss of viability in vitro exceed the effective doses of EZH2i in EZH2-mutant lymphomas for which EZH2i has been indicated (73). At such high doses, it is possible that EZH2i also targets EZH1 (74), though this appears unlikely in our system for multiple reasons (Supp Fig 3I, 6), including our demonstration of concordant findings in vivo, where our dosing regimen yields steady state plasma levels in the low micromolar range (73). Even still, many unanswered questions about Polycomb biology remain. How does PRC2 deposit H3K27me3 at novel sites in CIMP-high tumors? Does aberrant epigenetic patterning lead to selection for specific PRC2 assemblies in cancer? Are the therapeutic actions of EZH2i in cancer limited to its PRC2-targeting effects? Do Polycomb-independent complexes like EZH2/β-catenin participate in normal tissue homeostasis?
Our work illuminates how derailed epigenetic programs advantage cancer cells by maintaining a permissive chromatin environment for context-specific sustained proliferation. Here, we demonstrated through mechanistic and preclinical studies that dedifferentiation using epigenetic agents already FDA approved for other cancers (75) represents a promising avenue for CIMP-high ACC. As we move forward to discover new classes of therapies to equip all patients fighting this disease, it will be crucial to understand how aberrant epigenetic patterning emerges in the adrenal cortex and forges subtype-specific routes to malignancy.
Supplementary Material
SIGNIFICANCE.
Oncogenic β-catenin can use tissue-specific partners to regulate cellular differentiation programs that can be reversed by epigenetic therapies, identifying epigenetic control of differentiation as a viable target for β-catenin-driven cancers.
ACKNOWLEDGEMENTS
This work is supported by the University of Michigan Rogel Cancer Center (grant to G.D. Hammer, scholarship to D.R. Mohan), The Drew O’Donoghue Fund (to G.D. Hammer and D.R. Mohan), the Cissell-Roell Innovation Fund (to G.D. Hammer, A.M. Lerario, and D.R. Mohan), the NIH through the University of Michigan’s Cancer Center Support Grant (5 P30 CA46592), FAPESP (2020/02988-1 to SKNM) and CNPq Universal (422140/2016-3 to S.K.N. Marie). I. Finco, C.R. LaPensee, A.M. Lerario, and G.D. Hammer are/were supported by R01 DK062027 (grant to G.D. Hammer). D.R. Mohan, C.R. LaPensee, T. Else, R.J. Auchus, A.M. Lerario, and G.D. Hammer are supported by the US Department of Defense (CA180750 to G.D. Hammer and CA180751 to G.D. Hammer, T. Else, and R.J. Auchus). D.R. Mohan is/was also supported by the University of Michigan Medical Scientist Training Program (T32 GM7863), the University of Michigan Doctoral Program in Cancer Biology, and the University of Michigan Rackham Graduate School. D.T. Breault and K.S. Borges are supported by R01 DK123694. J. Rege was supported by American Heart Association grant 20CDA35320016. A.L. Solon and R. Ohi were supported by NIH grant R01 GM086610. E.R. Lawlor was supported by NIH grant R01 CA215981. A.A. Apfelbaum was supported by F31 CA247104. The authors would like to express their deepest gratitude to the patients, families, and advocates who have contributed essentially and immeasurably to advancing our understanding of ACC. The authors would also like to thank: Yulan Chu, for assistance with RNAscope. Pat O’Day, for extraction of steroids and analysis of LC-MS/MS data. Amy Blinder, for assistance with human adrenal samples. Eric Wang, Trey Weaver, Emilia Pinto, for critical reading of this manuscript. Tom Wilson, Andy Muntean, Sundeep Kalantry, for reading components of this work and always providing thoughtful, frank feedback. Preeti Mohan, for her constructive input throughout project development.
Footnotes
CONFLICT OF INTEREST STATEMENT
D.R. Mohan, A.M. Lerario, and G.D. Hammer are inventors on three pending patent applications describing compositions and methods for treating or characterizing cancer. G.D. Hammer reports unrelated personal fees from Radionetics and Orphagen Pharmaceuticals for consultation on projects outside the scope of this work. The remaining authors declare no competing interests.
REFERENCES
- 1.Flavahan WA, Gaskell E, Bernstein BE. Epigenetic plasticity and the hallmarks of cancer. Science 2017;357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, et al. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet 2013;45:592–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Easwaran H, Johnstone SE, Van Neste L, Ohm J, Mosbruger T, Wang Q, et al. A DNA hypermethylation module for the stem/progenitor cell signature of cancer. Genome Res 2012;22:837–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deevy O, Bracken AP. PRC2 functions in development and congenital disorders. Development 2019;146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schuettengruber B, Bourbon HM, Di Croce L, Cavalli G. Genome Regulation by Polycomb and Trithorax: 70 Years and Counting. Cell 2017;171:34–57 [DOI] [PubMed] [Google Scholar]
- 6.Widschwendter M, Fiegl H, Egle D, Mueller-Holzner E, Spizzo G, Marth C, et al. Epigenetic stem cell signature in cancer. Nat Genet 2007;39:157–8 [DOI] [PubMed] [Google Scholar]
- 7.Baylin SB, Jones PA. Epigenetic Determinants of Cancer. Cold Spring Harb Perspect Biol 2016;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mohan DR, Lerario AM, Else T, Mukherjee B, Almeida MQ, Vinco M, et al. Targeted Assessment of G0S2 Methylation Identifies a Rapidly Recurrent, Routinely Fatal Molecular Subtype of Adrenocortical Carcinoma. Clin Cancer Res 2019;25:3276–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zheng S, Cherniack AD, Dewal N, Moffitt RA, Danilova L, Murray BA, et al. Comprehensive Pan-Genomic Characterization of Adrenocortical Carcinoma. Cancer Cell 2016;29:723–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Assie G, Letouze E, Fassnacht M, Jouinot A, Luscap W, Barreau O, et al. Integrated genomic characterization of adrenocortical carcinoma. Nat Genet 2014;46:607–12 [DOI] [PubMed] [Google Scholar]
- 11.Nusse R, Clevers H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017;169:985–99 [DOI] [PubMed] [Google Scholar]
- 12.Lerario AM, Mohan DR, Hammer GD. Update on Biology and Genomics of Adrenocortical Carcinomas: Rationale for Emerging Therapies. Endocrine Reviews 2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schwitalla S, Fingerle AA, Cammareri P, Nebelsiek T, Göktuna SI, Ziegler PK, et al. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell 2013;152:25–38 [DOI] [PubMed] [Google Scholar]
- 14.Tao Y, Kang B, Petkovich DA, Bhandari YR, In J, Stein-O’Brien G, et al. Aging-like Spontaneous Epigenetic Silencing Facilitates Wnt Activation, Stemness, and Braf. Cancer Cell 2019;35:315–28.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vaz M, Hwang SY, Kagiampakis I, Phallen J, Patil A, O’Hagan HM, et al. Chronic Cigarette Smoke-Induced Epigenomic Changes Precede Sensitization of Bronchial Epithelial Cells to Single-Step Transformation by KRAS Mutations. Cancer Cell 2017;32:360–76.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ohm JE, McGarvey KM, Yu X, Cheng L, Schuebel KE, Cope L, et al. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat Genet 2007;39:237–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shibamoto S, Higano K, Takada R, Ito F, Takeichi M, Takada S. Cytoskeletal reorganization by soluble Wnt-3a protein signalling. Genes Cells 1998;3:659–70 [DOI] [PubMed] [Google Scholar]
- 18.Drelon C, Berthon A, Mathieu M, Ragazzon B, Kuick R, Tabbal H, et al. EZH2 is overexpressed in adrenocortical carcinoma and is associated with disease progression. Hum Mol Genet 2016;25:2789–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Borges KS, Pignatti E, Leng S, Kariyawasam D, Ruiz-Babot G, Ramalho FS, et al. Wnt/β-catenin activation cooperates with loss of p53 to cause adrenocortical carcinoma in mice. Oncogene 2020;39:5282–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bayliss J, Mukherjee P, Lu C, Jain SU, Chung C, Martinez D, et al. Lowered H3K27me3 and DNA hypomethylation define poorly prognostic pediatric posterior fossa ependymomas. Sci Transl Med 2016;8:366ra161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Panwalkar P, Clark J, Ramaswamy V, Hawes D, Yang F, Dunham C, et al. Immunohistochemical analysis of H3K27me3 demonstrates global reduction in group-A childhood posterior fossa ependymoma and is a powerful predictor of outcome. Acta Neuropathol 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chung C, Sweha SR, Pratt D, Tamrazi B, Panwalkar P, Banda A, et al. Integrated Metabolic and Epigenomic Reprograming by H3K27M Mutations in Diffuse Intrinsic Pontine Gliomas. Cancer Cell 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pignatti E, Leng S, Yuchi Y, Borges KS, Guagliardo NA, Shah MS, et al. Beta-Catenin Causes Adrenal Hyperplasia by Blocking Zonal Transdifferentiation. Cell Rep 2020;31:107524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wright C, O’Day P, Alyamani M, Sharifi N, Auchus RJ. Abiraterone acetate treatment lowers 11-oxygenated androgens. Eur J Endocrinol 2020;182:413–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nature Methods 2017;14:959–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gaspar JM. 2018. Genrich: detecting sites of genomic enrichment. <https://github.com/jsh58/Genrich>. [Google Scholar]
- 27.Shpynov O, Dievskii A, Chernyatchik R, Tsurinov P, Artyomov MN. Semi-supervised peak calling with SPAN and JBR Genome Browser. Bioinformatics 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 2013;153:307–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013;153:320–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hovestadt V, Zapatka M. conumee: Enhanced copy-number variation analysis using Illumina DNA methylation arrays. R package version 1.9.02020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Han X, Zhou Z, Fei L, Sun H, Wang R, Chen Y, et al. Construction of a human cell landscape at single-cell level. Nature 2020;581:303–9 [DOI] [PubMed] [Google Scholar]
- 32.Zhang K, Hocker JD, Miller M, Hou X, Chiou J, Poirion OB, et al. A single-cell atlas of chromatin accessibility in the human genome. Cell 2021;184:5985–6001.e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Domcke S, Hill AJ, Daza RM, Cao J, O’Day DR, Pliner HA, et al. A human cell atlas of fetal chromatin accessibility. Science 2020;370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jung I, Schmitt A, Diao Y, Lee AJ, Liu T, Yang D, et al. A compendium of promoter-centered long-range chromatin interactions in the human genome. Nat Genet 2019;51:1442–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, et al. Defining a Cancer Dependency Map. Cell 2017;170:564–76.e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuhn M, Jackson S, Cimentada J. corrr: Correlations in R. 2022. [Google Scholar]
- 37.Chaligne R, Gaiti F, Silverbush D, Schiffman JS, Weisman HR, Kluegel L, et al. Epigenetic encoding, heritability and plasticity of glioma transcriptional cell states. Nat Genet 2021;53:1469–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci U S A 1999;96:8681–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gal-Yam EN, Egger G, Iniguez L, Holster H, Einarsson S, Zhang X, et al. Frequent switching of Polycomb repressive marks and DNA hypermethylation in the PC3 prostate cancer cell line. Proc Natl Acad Sci U S A 2008;105:12979–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Viré E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C, et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 2006;439:871–4 [DOI] [PubMed] [Google Scholar]
- 41.Bracken AP, Pasini D, Capra M, Prosperini E, Colli E, Helin K. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J 2003;22:5323–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ip JC, Pang TC, Glover AR, Soon P, Zhao JT, Clarke S, et al. Immunohistochemical validation of overexpressed genes identified by global expression microarrays in adrenocortical carcinoma reveals potential predictive and prognostic biomarkers. Oncologist 2015;20:247–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tabbal H, Septier A, Mathieu M, Drelon C, Rodriguez S, Djari C, et al. EZH2 cooperates with E2F1 to stimulate expression of genes involved in adrenocortical carcinoma aggressiveness. British Journal of Cancer 2019;121:384–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cao R, Zhang Y. SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol Cell 2004;15:57–67 [DOI] [PubMed] [Google Scholar]
- 45.Pasini D, Bracken AP, Jensen MR, Lazzerini Denchi E, Helin K. Suz12 is essential for mouse development and for EZH2 histone methyltransferase activity. EMBO J 2004;23:4061–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Margueron R, Justin N, Ohno K, Sharpe ML, Son J, Drury WJ, et al. Role of the polycomb protein EED in the propagation of repressive histone marks. Nature 2009;461:762–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Montgomery ND, Yee D, Chen A, Kalantry S, Chamberlain SJ, Otte AP, et al. The murine polycomb group protein Eed is required for global histone H3 lysine-27 methylation. Curr Biol 2005;15:942–7 [DOI] [PubMed] [Google Scholar]
- 48.Neri F, Krepelova A, Incarnato D, Maldotti M, Parlato C, Galvagni F, et al. Dnmt3L antagonizes DNA methylation at bivalent promoters and favors DNA methylation at gene bodies in ESCs. Cell 2013;155:121–34 [DOI] [PubMed] [Google Scholar]
- 49.Catania S, Dumesic PA, Pimentel H, Nasif A, Stoddard CI, Burke JE, et al. Evolutionary Persistence of DNA Methylation for Millions of Years after Ancient Loss of a De Novo Methyltransferase. Cell 2020;180:263–77.e20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li H, Liefke R, Jiang J, Kurland JV, Tian W, Deng P, et al. Polycomb-like proteins link the PRC2 complex to CpG islands. Nature 2017;549:287–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Perino M, van Mierlo G, Karemaker ID, van Genesen S, Vermeulen M, Marks H, et al. MTF2 recruits Polycomb Repressive Complex 2 by helical-shape-selective DNA binding. Nat Genet 2018;50:1002–10 [DOI] [PubMed] [Google Scholar]
- 52.Mathieu M, Drelon C, Rodriguez S, Tabbal H, Septier A, Damon-Soubeyrand C, et al. Steroidogenic differentiation and PKA signaling are programmed by histone methyltransferase EZH2 in the adrenal cortex. Proc Natl Acad Sci U S A 2018;115:E12265–E74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Seamon KB, Padgett W, Daly JW. Forskolin: unique diterpene activator of adenylate cyclase in membranes and in intact cells. Proc Natl Acad Sci U S A 1981;78:3363–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xing Y, Edwards MA, Ahlem C, Kennedy M, Cohen A, Gomez-Sanchez CE, et al. The effects of ACTH on steroid metabolomic profiles in human adrenal cells. J Endocrinol 2011;209:327–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bassett MH, White PC, Rainey WE. A role for the NGFI-B family in adrenal zonation and adrenocortical disease. Endocr Res 2004;30:567–74 [DOI] [PubMed] [Google Scholar]
- 56.Yakulov T, Raggioli A, Franz H, Kemler R. Wnt3a-dependent and -independent protein interaction networks of chromatin-bound β-catenin in mouse embryonic stem cells. Mol Cell Proteomics 2013;12:1980–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hoffmeyer K, Junghans D, Kanzler B, Kemler R. Trimethylation and Acetylation of β-Catenin at Lysine 49 Represent Key Elements in ESC Pluripotency. Cell Rep 2017;18:2815–24 [DOI] [PubMed] [Google Scholar]
- 58.Gummow BM, Winnay JN, Hammer GD. Convergence of Wnt signaling and steroidogenic factor-1 (SF-1) on transcription of the rat inhibin alpha gene. J Biol Chem 2003;278:26572–9 [DOI] [PubMed] [Google Scholar]
- 59.Mizusaki H, Kawabe K, Mukai T, Ariyoshi E, Kasahara M, Yoshioka H, et al. Dax-1 (dosage-sensitive sex reversal-adrenal hypoplasia congenita critical region on the X chromosome, gene 1) gene transcription is regulated by wnt4 in the female developing gonad. Mol Endocrinol 2003;17:507–19 [DOI] [PubMed] [Google Scholar]
- 60.Hossain A, Saunders GF. Synergistic cooperation between the beta-catenin signaling pathway and steroidogenic factor 1 in the activation of the Mullerian inhibiting substance type II receptor. J Biol Chem 2003;278:26511–6 [DOI] [PubMed] [Google Scholar]
- 61.Pott S, Lieb JD. What are super-enhancers? Nat Genet 2015;47:8–12 [DOI] [PubMed] [Google Scholar]
- 62.Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-André V, Sigova AA, et al. Super-enhancers in the control of cell identity and disease. Cell 2013;155:934–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hnisz D, Schuijers J, Lin CY, Weintraub AS, Abraham BJ, Lee TI, et al. Convergence of developmental and oncogenic signaling pathways at transcriptional super-enhancers. Mol Cell 2015;58:362–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zamudio AV, Dall’Agnese A, Henninger JE, Manteiga JC, Afeyan LK, Hannett NM, et al. Mediator Condensates Localize Signaling Factors to Key Cell Identity Genes. Mol Cell 2019;76:753–66.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Corces MR, Granja JM, Shams S, Louie BH, Seoane JA, Zhou W, et al. The chromatin accessibility landscape of primary human cancers. Science 2018;362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kahn M Can we safely target the WNT pathway? Nat Rev Drug Discov 2014;13:513–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Merika M, Williams AJ, Chen G, Collins T, Thanos D. Recruitment of CBP/p300 by the IFN beta enhanceosome is required for synergistic activation of transcription. Mol Cell 1998;1:277–87 [DOI] [PubMed] [Google Scholar]
- 68.Raisner R, Kharbanda S, Jin L, Jeng E, Chan E, Merchant M, et al. Enhancer Activity Requires CBP/P300 Bromodomain-Dependent Histone H3K27 Acetylation. Cell Rep 2018;24:1722–9 [DOI] [PubMed] [Google Scholar]
- 69.LaFave LM, Kartha VK, Ma S, Meli K, Del Priore I, Lareau C, et al. Epigenomic State Transitions Characterize Tumor Progression in Mouse Lung Adenocarcinoma. Cancer Cell 2020;38:212–28.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Finco I, Lerario AM, Hammer GD. Sonic Hedgehog and WNT Signaling Promote Adrenal Gland Regeneration in Male Mice. Endocrinology 2018;159:579–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Warde KM, Liu L, Smith LJ, Lohman BK, Stubben CJ, Ekiz HA, et al. Senescence-Induced Immune Remodeling Facilitates Metastatic Adrenal Cancer in a Sex-Dimorphic Manner. bioRxiv 2022:2022.04.29.488426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wilmouth JJ, Olabe J, Garcia-Garcia D, Lucas C, Guiton R, Roucher-Boulez F, et al. Sexually dimorphic activation of innate antitumor immunity prevents adrenocortical carcinoma development. Sci Adv 2022;8:eadd0422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sneeringer CJ, Scott MP, Kuntz KW, Knutson SK, Pollock RM, Richon VM, et al. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc Natl Acad Sci U S A 2010;107:20980–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yamagishi M, Hori M, Fujikawa D, Ohsugi T, Honma D, Adachi N, et al. Targeting Excessive EZH1 and EZH2 Activities for Abnormal Histone Methylation and Transcription Network in Malignant Lymphomas. Cell Rep 2019;29:2321–37.e7 [DOI] [PubMed] [Google Scholar]
- 75.Morschhauser F, Tilly H, Chaidos A, McKay P, Phillips T, Assouline S, et al. Tazemetostat for patients with relapsed or refractory follicular lymphoma: an open-label, single-arm, multicentre, phase 2 trial. Lancet Oncol 2020;21:1433–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Requests for resources and reagents should be directed to corresponding authors Antonio Marcondes Lerario (alerario@umich.edu) and Gary D. Hammer (ghammer@umich.edu). The original RNA-seq, methylation array, ChIP-seq, and ATAC-seq data generated in this study have been deposited in GEO: GSE205283. The publicly available data analyzed in this study were obtained from TCGA Genomic Data Commons at https://gdc.cancer.gov/ and https://portal.gdc.cancer.gov/legacy-archive; GEPIA at http://gepia.cancer-pku.cn/; GEO at https://www.ncbi.nlm.nih.gov/geo/ using identifiers GSE68889, GSE109578, GSE134355, GSE86189, GSE10927, or GSM5047828; ENCODE at https://www.encodeproject.org/ using identifiers ENCLB278KZX, ENCLB060VHC, ENCLB923ALY, ENCLB245VYO, ENCLB872HUE, ENCLB191XGX, ENCLB281DGJ, ENCLB670CFC, ENCLB382MNY, ENCLB198CGW, ENCLB172SDF, ENCLB735YID, ENCFF050CUG, ENCFF671NVD, ENCFF639HFP, ENCFF998IEU, ENCFF202US, ENCFF495GNE, ENCFF154IMT, ENCFF662NTZ, ENCFF342DVJ, ENCFF680AKW, ENCFF456EQK, ENCFF725EMZ, ENCFF009PDQ, ENCFF612HRW, ENCFF015TOT, ENCFF441TOI, ENCFF218TIK, ENCFF342EGR, ENCFF250ASJ, ENCFF495YGS, or ENCFF019XYA; DDBJ/EMBL/GenBank at https://www.ddbj.nig.ac.jp/ using identifier DRA000853; Descartes Human Chromatin Accessibility During Development Atlas at http://descartes.brotmanbaty.org/; 3DIV at http://3div.kr/; and DepMap at https://depmap.org/portal/.