

Abstract
Background:
Atrial fibrillation (AF), the most common arrhythmia, is associated with the downregulation of FKBP5 (encoding FK506-binding protein 5, FKBP5). However, the function of FKBP5 in the heart remains unknown. Here, we elucidate the consequences of cardiomyocyte (CM)-restricted loss of FKBP5 on cardiac function and AF development and study the underlying mechanisms.
Methods:
Right-atrial samples from patients with AF were used to assess the protein levels of FKBP5. A CM-specific FKBP5 knockdown (cKD) mouse model was established by crossbreeding Fkbp5flox/flox mice with Myh6MerCreMer/+ mice. Cardiac function and AF inducibility were assessed by echocardiography and programmed intracardiac stimulation. Histology, optical mapping, cellular electrophysiology, and biochemistry were employed to elucidate the proarrhythmic mechanisms due to loss of CM FKBP5.
Results:
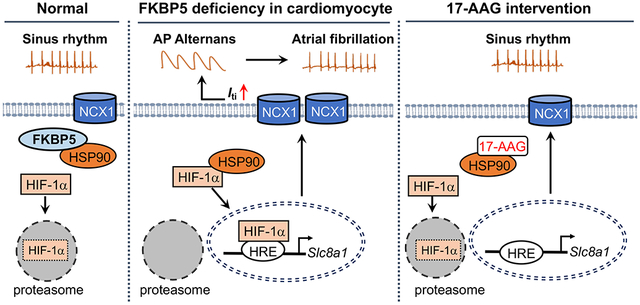
FKBP5 protein levels were lower in atrial lysates of patients with paroxysmal AF (pAF) or long-lasting persistent (chronic) AF (cAF). cKD mice exhibited increased AF inducibility and duration compared with control mice. Enhanced AF susceptibility in cKD mice was associated with the development of action potential alternans and spontaneous Ca2+ waves, and increased protein levels and activity of the Na+/Ca2+-exchanger 1 (NCX1), mimicking the cellular phenotype of cAF patients. FKBP5-deficiency enhanced transcription of Slc8a1 (encoding NCX1) via transcription factor hypoxia-inducible-factor 1α (HIF-1α). In vitro studies revealed that FKBP5 negatively modulated the protein levels of HIF-1α by competitively interacting with heat-shock-protein 90 (HSP90). Injections of the HSP90 inhibitor 17-AAG normalized protein levels of HIF-1α and NCX1, and reduced AF susceptibility in cKD mice. Furthermore, the atrial CM-selective knockdown of FKBP5 was sufficient to enhance AF arrhythmogenesis.
Conclusions:
This is the first study to demonstrate a role for the FKBP5-deficiency in atrial arrhythmogenesis and to establish FKBP5 as a negative regulator of HIF-1α in cardiomyocytes. Our results identify a potential molecular mechanism for the pro-arrhythmic NCX1 upregulation in cAF patients.
Keywords: Atrial fibrillation, FKBP5, NCX1, HIF-1α, HSP90
Subject Terms: Atrial Fibrillation, Basic Science Research, Gene Therapy, Ion Channels/Membrane Transport, Pathophysiology
Graphical Abstract
INTRODUCTION
Atrial fibrillation (AF) is the most common arrhythmia, and its prevalence is increasing.1, 2 Although progression of paroxysmal AF (pAF) to long-lasting persistent (‘chronic’) AF (cAF) is a major clinical problem, the molecular mechanisms underlying AF progression remain poorly understood. Identifying the key molecules and associated mechanisms contributing to the development of persistent AF forms is a crucial step towards developing a therapeutic strategy to prevent AF progression and maintenance. A recent transcriptome analysis in atrial samples of pAF patients identified a downregulation of FKBP5 (encoding FK506-binding protein 5, FKBP5).3 FKBP5 (also known as FKBP51) is a 51-kDa protein with co-chaperone activities.4-6 The known functions of FKBP5 include the regulation of steroid hormone receptors, inhibition of apoptosis in cancer cells, promotion of Akt1 dephosphorylation and inhibition of the Akt1 pathways, to name a few.7-9 Previous studies have linked genetic variants in FKBP5 to psychological disorders.10-12 However, the role of FKBP5 in the heart has not been elucidated. In particular, it is not known whether dysregulation of FKBP5 in cardiomyocytes (CMs) is involved in atrial arrhythmogenesis, which was the object of the present study.
To elucidate the role of FKBP5-deficiency in AF development, we established a CM-specific FKBP5 knockdown (cKD) mice. We found that FKBP5-deficiency increases AF-inducibility. The arrhythmogenesis in cKD mice is associated with action potential (AP) alternans and enhanced Na+/Ca2+-exchanger type-1 (NCX1) expression. As an essential regulator of Ca2+ homeostasis within CMs, NCX1 protein levels were previously reported to be increased in sheep and patients with cAF.13-16 However, the molecular mechanism underlying the increased protein level of NCX1 in cAF patients remains elusive. Here, we discovered that FKBP5-deficiency upregulates the transcription of Slc8a1 (encoding NCX1) in CMs by promoting transcription-factor (TF) hypoxia-inducible factor 1-alpha (HIF-1α) signaling. We then validated that HIF-1α can directly bind to the promoter of Slc8a1, and that FKBP5 inhibits the accumulation of HIF-1α and prevents its nuclear translocation. Taken together, our results identify that FKBP5-deficiency in CMs plays a role in atrial arrhythmogenesis, making it a potential important target for treatment of AF.
METHODS
Data Availability
The detailed experimental materials, methods, and data supporting the findings of this study are available within the article and its Supplemental Material. Raw data are available from the corresponding author upon reasonable request. The Major Resources Table is provided in the Supplemental Material.
Human atrial samples
Right atrial appendages were collected from patients undergoing open-heart surgery for coronary bypass grafting and/or valve replacement. Control patients with normal sinus rhythm (NSR) and patients with pAF or cAF provided informed consent prior to surgery. All experimental protocols were approved by the Human Ethics Committee of the Medical Faculty of the University Duisburg-Essen (approval number AZ:12-5268-BO) and were performed in accordance with the Declaration of Helsinki. Patient demographics and characteristics are listed in Supplementary Table S1 - S5.
Animal studies
All studies involving mice were performed according to protocols approved by the Institutional Animal Care and Use Committee at Baylor College of Medicine and conformed to the Guide for the Care and Use of Laboratory Animals published by National Institutes of Health. The Fkbp5floxl/flox mice were generated by crossbreeding the Fkbp5tm1a(KOMP)Wtsi mice (purchased from Knockout Mouse Project) with FLP transgenic mice (The Jackson Laboratory). Afterwards, the Fkbp5flox/flox mice were crossbred with inducible CM-specific Cre mice (Myh6MCM/+)17 to generate Myh6MCM/+;Fkbp5flox/flox mice, of which the offspring maintained the Mendelian ratio. At the age of 6 weeks, Myh6MCM/+; Fkbp5flox/flox (cKD) and Myh6+/+; Fkbp5flox/flox (as control, Ctl) in both sexes were injected with tamoxifen (50mg/kg, i.p.) for 5 days to induce the CM-specific knockdown of Fkbp5. All strains were maintained on a C57Bl6/J genetic background.
Programmed intracardiac stimulation
Programmed intracardiac stimulation was performed to assess AF-inducibility.18 To determine whether AF-inducibility was reproducible, mice were subjected to the same atrial burst-pacing protocol for 3 times, and only mice in which AF (>=1 second) could be induced by burst-pacing in at least 2 out of 3 attempts were considered as AF-positive. The incidence of inducible AF was calculated as the percentage of the AF-positive mice divided by the total number of mice studied. For quantification of AF duration, we capped the duration at 5 minutes for any episodes longer than 5 minutes. The experimenter was blinded to genotype or viral gene transfer status of mice.
Ca2+ imaging
Atrial CMs were isolated and incubated in KB buffer. Ca2+ imaging was performed in cells loaded with the Ca2+-indicator Cal-520®-AM (4 μmol/L) as previously described.19 Once steady-state Ca2+ transients (CaTs) were observed, pacing was stopped and sarcoplasmic reticulum (SR) Ca2+ load was estimated as the CaT amplitude induced by perfusion with Tyrode’s solution containing 10 mmol/L caffeine. The CaT decay was calculated with pClamp. NCX activity was estimated as the decay of caffeine-induced CaT [K(Caff-CaT)]. Sarcoplasmic/endoplasmic reticulum Ca2+/ATPase (SERCA) activity was estimated as the difference between the decay of pacing-induced CaT and the decay of caffeine-induced CaT [K(pacing-CaT) – K(Caff-CaT)].20
Patch-clamp recordings
The whole-cell patch-clamp method was employed to record total membrane current in atrial CMs. Patch-pipette resistances were 1–3 MΩ before sealing. Seal-resistances were 1-10 GΩ. Series resistance and cell capacitance were compensated. The pipette solution contained (in mM) 125 CsOH, 8 TEACl, 12 TEAOH, 1 MgCl2, 10 HEPES, and 0.02 EGTA. 4 mM Na2-ATP was added fresh, and pH was adjusted to 7.2. The external solution contained (in mM) 94 NaCl, 51 NaOH, 1 MgCl2, 1 CaCl2, 10 HEPES, 2 probenecid and 5 CsCl. The pH was adjusted to 7.4. 4-aminopyridine (5 mmol/L) and BaCl2 (0.1 mmol/L) were added to the bath solution to block K+-currents. Experiments were performed at room temperature. Data were acquired with an Axopatch 200B amplifier (Axon Instruments, Inc.) with the Axopatch’s filter set at 10 kHz. The function of the forward-mode NCX was assessed as the caffeine (10 mmol/L)-induced transient inward current (Iti), which was sampled at 200 kHz with a holding potential at −80 mV. Large conductance endogenous channel activities were subtracted during data Analysis.21
Adeno-associated virus (AAV) production and delivery
Adeno-associated virus 9 (AAV9) was used to express an atrial CM-selective Cre (AAV9-ANF-Cre) as described22. 6 weeks old Fkbp5flox/flox mice of both sexes were injected with one dose of AAV9-ANF-Cre or AAV9-ANF-Flag virus (5x1011 GC/mouse) via retro-orbital route. For the intervention study, 6 weeks old Fkbp5−/− mice of both sexes were injected with one dose of AAV9-ANF-FKBP5 or AAV9-ANF-Flag virus (1x1012 GC/mouse) via retro-orbital route.
Statistics
Data were presented as the mean±SEM. The representative images and western blots were chosen based on the resemblance to the average values. Equal number of samples per group was included in the pilot studies. If the difference showed a trending significance, additional samples were added to reach statistical significance. Statistical analyses were performed in GraphPad Prism 9. Normality was evaluated with D’Agostino & Pearson test. ROUT analyses were applied to identify outliers, which were excluded from group comparison analyses. Mann-Whitney tests were used for data where normality cannot be assumed. Student’s t-tests were used to compare data between two groups maintaining normal distributions. Fisher’s exact test was used to compare categorical data. ANOVA followed by Tukey’s comparison tests were used for multiple comparisons. For dataset with multiple measures taken from the same animal, hierarchical statistical approach with the multilevel mixed model was performed with R, as described previously.23 A p-value less than 0.05 was considered statistically significant.
RESULTS
Downregulation of FKBP5 in atria of patients with cAF
We recently discovered that FKBP5 mRNA levels were downregulated in patients with pAF.3 To determine whether the altered FKBP5 protein level is associated with AF development and correlates with AF progression, Western blots were performed with the atrial samples of patients with NSR, pAF, or cAF (Supplementary Table S1 & Table S2). In pAF, FKBP5 protein levels were non-significantly reduced by 15% (p=0.0709 vs NSR) (Figure 1A). FKBP5 protein levels were significantly reduced by 27% in cAF patients (p=8.41x10−3 vs NSR) (Figure 1B), suggesting that the reduction of FKBP5 in atria could play a role in cAF patients.
Figure 1. Reduced FKBP5 protein levels in patients with AF and FKBP5-cKD mice.

(A-B) Representative Western blots and quantification of FKBP5 protein in atrial tissue of paroxysmal AF (pAF, A) and chronic AF (cAF, B) compared with NSR patients. (C) Representative Western blots and quantification of FKBP5 protein in atrial cardiomyocytes (CMs) of cAF patients compared with NSR patients. (D) The schematic diagram showed the development of the FKBP5-cKD and Ctl mice. (E) mRNA levels of Fkbp5 in atria and ventricles of Ctl and cKD mice. (F) Western blots and quantification of FKBP5 protein levels in atria and ventricles of Ctl and cKD mice. p-values were determined using unpaired Student’s t-test in A, B, and C, and Mann-Whitney test in E and F.
FKBP5-deficiency enhances AF susceptibility
The whole-body FKBP5 knockout (Fkbp5−/−) mice were susceptible to pacing-induced AF (Supplementary Figure S1). Although other studies have demonstrated a role of FKBP5 in immune cells,24, 25 no statistically significant difference was observed for the protein levels of the macrophage marker CD68 in atria of Fkbp5−/− mice (Supplementary Figure S1) and atria of AF patients18, making it unlikely that the atrial phenotype is due to a selective FKBP5-deficiency in macrophages. On the other hand, the protein levels of FKBP5 were significantly reduced by almost 30% in human atrial CM samples of cAF patients (p=0.0474 vs NSR) (Figure 1C, Supplementary Table S3), suggesting that the loss of CM FKBP5 could contribute to atrial arrhythmogenesis. Thus, to elucidate the CM-specific deficiency of FKBP5 in AF development, we developed the FKBP5-cKD model (Figure 1D). Three weeks post tamoxifen injections, qPCR and Western blots confirmed that the levels of Fkbp5 mRNA and FKBP5 protein were significantly reduced in both atria and ventricles (Figure 1E, F) of cKD mice compared to Ctl mice. To determine whether FKBP5-deficiency affects atrial arrhythmogenesis, programmed intracardiac stimulation was performed in the age-matched Ctl and cKD mice 1-month after tamoxifen injection. While the RR-, QRS-, QTc-intervals, sinus node recovery time, and atrioventricular effective recovery period were comparable between Ctl and cKD mice (Supplementary Table S6), the incidence of inducible AF was much higher in cKD compared to Ctl mice (58.3% vs 6.3%, p=9.08x10−4) (Figure 2A-B). The duration of pacing-induced AF episodes was generally longer in cKD than in Ctl mice (Figure 2C), with a few episodes longer than 5 minutes. Of note, cKD mice did not exhibit an increased susceptibility to pacing-induced ventricular tachycardia (Supplementary Table S7). To examine whether ventricular dysfunction could contribute to the increased AF susceptibility in cKD mice, echocardiography was performed one day prior to the induction. The results showed that left ventricular (LV) ejection fraction (LVEF%) was mildly reduced (70.9±1.6% vs 61.4±2.6%, p=9.18x10−3) and the end-systolic diameter (ESD) of LV was trending to increase in cKD mice compared with Ctl (Figure 2D-F), while the end-diastolic diameter (EDD) and wall thickness of LV were comparable between Ctl and cKD mice (Figure 2G & Supplementary Table S8). There was no statistically significant difference in left atrial (LA) dimensions between Ctl and cKD mice (Figure 2H-I). Additionally, both Picrosirius red staining (Figure 2J-K) and Masson’s trichrome staining (Supplementary Figure S2) showed that atrial and ventricular fibrosis were comparable between Ctl and cKD mice (Figure 2J-K). No statistically significant difference was found for the protein levels of the macrophage marker CD68 and the fibrotic markers collagen-I and MMP9 in the atria of cKD mice, although the levels of α-smooth muscle actin were higher in cKD mice (Supplementary Figure S3). These results establish that CM-specific FKBP5-deficiency promotes the development of a pro-arrhythmic substrate for AF unrelated to structural and fibrotic remodeling.
Figure 2. CM-specific FKBP5-deficiency enhances AF susceptibility.

(A) Representative simultaneous recordings of surface ECG and intracardiac electrograms in Ctl and cKD mice, suggestive of sinus rhythm in Ctl and AF in cKD mice after pacing. (B) The incidence and (C) the duration of pacing-induced AF in Ctl and cKD mice. (D) Representative M-mode echocardiography recording in Ctl and cKD mice. (E-G) The quantification of LVEF%, ESD, and EDD in Ctl and cKD mice. (H) Representative long-axis echocardiography recording to assess left atria (LA) in Ctl and cKD mice. (I) The quantification of LA area. (J) Representative Picrosirius Red staining in whole hearts (i), atria (ii) and ventricles (iii) of Ctl and cKD mice. (K) The quantification of fibrosis areas in atria and ventricles of Ctl and cKD mice. p-values were determined with Fisher’s exact test in B, Mann-Whitney test in C and E, and unpaired Student’s t-test in F.
FKBP5-deficiency promotes action potential alternans
To elucidate the nature of the pro-arrhythmic substrate, we then performed optical mapping on perfused hearts as previously described.18, 26 We found that atrial-effective-refractory period (AERP) and AP duration (APD at 20%, 50%, 70%, and 90% repolarization) at 10 Hz pacing were comparable between Ctl and cKD mice (Figure 3A-C). The level of APD dispersion was not significantly different between Ctl and cKD mice (Supplementary Figure S4). Atrial conduction velocity (CV) in atria was trending to increase at 10 Hz pacing in cKD mice but was not significantly different at 15 Hz pacing between cKD and Ctl (Figure 3D). Protein levels of connexin-40, connexin-43, and Na+-channel Nav1.5-subunit were comparable between Ctl and cKD mice (Supplementary Figure S5). However, the coefficient of variation (CoV) of CV was significantly higher in cKD mice than in Ctl (Figure 3E), suggesting an increase in conduction heterogeneity in cKD atria. Interestingly, at 15 Hz pacing, AP alternans occurred more often in the atria of cKD mice than in Ctl mice (46.2% vs 7.1%, p=0.0328) (Figure 3F-G), and they appeared to concordant (Supplementary Figure S6). It is established that a higher susceptibility to alternans is associated with enhanced AF vulnerability.27 Indeed, the incidence of the pacing-induced atrial tachycardia was increased in cKD mice compared with Ctl mice (61.5% vs 21.4%, p=0.0412) (Figure 3H-I, Supplementary Video S1).
Figure 3. CM-specific FKBP5-deficiency promotes arrhythmogenic alternans.

(A) Representative activation maps in Ctl and cKD mice. (B) Quantification of APD and (C) AERP at 10 Hz pacing. (D) CV at 10 Hz and 15 Hz pacing. (E) Coefficient of variation (CoV) of CV at 10 Hz and 15 Hz pacing. (F) Representative examples of AP alternans in cKD mice evoked by 15 Hz pacing, but not by 10 Hz pacing. (G) Incidence of AP alternans. (H) Representative traces of normal rhythm in Ctl and atrial tachycardia in cKD mice after pacing. (I) Incidence of pacing-induced atrial arrhythmia ex vivo. p-values were determined with unpaired Student’s t-test in D (with Welch’s correction) and E, and Fisher’s exact test in G and I.
FKBP5-deficiency increases the protein levels of NCX1
The development of AP alternans could be due to Ca2+-handling abnormalities,28, 29 which are also known to promote cellular delayed afterdepolarizations (DADs)-mediated triggered activity in patients with pAF and cAF, as well as patients developing postoperative AF.14, 20, 30 To determine whether abnormal Ca2+ handling may contribute to atrial arrhythmogenesis in cKD mice, we performed Ca2+-imaging studies in atrial CMs. After 1 Hz pacing, susceptibility to spontaneous Ca2+ waves (SCaWs) was higher in atrial CMs from cKD than CMs from Ctl mice (Figure 4A-B). The frequency of SCaWs was also greater in atrial CMs of cKD than those of Ctl mice (Figure 4C). Because the development of SCaWs is often attributed to the enhanced SR Ca2+ release via ryanodine receptor type-2 (RyR2) channels, we recorded Ca2+ sparks as an indicator of RyR2 function.31 Surprisingly, the frequency of Ca2+ sparks (CaSF) was comparable in atrial CMs of Ctl and cKD mice (Supplementary Figure S7). To determine the function of SERCA and NCX, we next assessed the decay of the pacing-induced and caffeine-induced CaTs as previously described.20 While no statistically significant difference was found in SERCA activity (Figure 4D), NCX function was higher in atrial CMs of cKD mice (Figure 4E). The amplitude of the caffeine-induced CaT, an indicator of SR Ca2+ load, was not significantly different (Figure 4F). Similarly, there were not statistically significant differences in the protein levels of major Ca2+-handling proteins such as RyR2, α-subunit of L-type Ca2+-channel (Cav1.2), SERCA2a and plasma membrane Ca2+ ATPase (PMCA) between groups. However, the total NCX1 protein was increased by 40% in atria and 30% in ventricles of cKD (p=0.0492, p=0.0734 vs Ctl mice) (Supplementary Figure S8).
Figure 4. CM-specific FKBP5-deficiency leads to the increased activity of NCX1.

(A) Representative traces of the 1 Hz-pacing induced Ca2+ transients (CaTs), followed by baseline recording and the caffeine (10 mmol/L) induced CaTs in atrial CMs of Ctl and cKD mice. Red arrows pointed to the spontaneous Ca2+ waves (SCaWs) in the atrial CM of cKD mice. (B) Incidence and (C) frequency of SCaWs in atrial CMs of Ctl and cKD mice. (D) Quantification of relative SERCA activity. (E) Quantification of relative NCX function. (F) SR Ca2+ load. (G) Western blots and (H) quantification of NCX1 in cytosol- and membrane-fractions of atrial tissue of Ctl and cKD mice. GAPDH and caveolin 3 (Cav3) were used as control for the cytosol- and membrane-fractions, respectively. (I) Western blots and quantification of NCX1 in cytosol (Cyto)- and membrane (Mem)-fractions of atrial tissue of NSR and cAF patients. GAPDH and Gβ were used as control for the cytosol- and membrane-fractions, respectively. (J) Representative recording of Iti elicited by the rapid application caffeine in atrial CMs. (K) Quantification of the current density of Iti. p-values were determined with Fisher’s exact test in B, Mann-Whitney test in C, H, and I, and multilevel mixed model in E and K.
NCX1 is an essential regulator of Ca2+ homeostasis within CMs.32-34 In its forward-mode, NCX extrudes one Ca2+ ion in exchange for three Na+ ions, producing a depolarizing transient-inward current (INCX). Overactive forward-mode NCX increases the propensity for proarrhythmic DADs.14, 20, 31 Increased NCX activity is also associated with the development of arrhythmogenic alternans,28, 29 as well as AP prolongation and conduction disturbances,35 thereby promoting a substrate for AF development. NCX1 protein levels are increased in sheep and patients with cAF.13, 14, 16 However, the molecular mechanism underlying the increased protein levels of NCX1 in cAF patients remains elusive. To assess the amount of NCX1 protein at the plasma membrane, we separated the sarcolemma and cytosol of atrial tissues of Ctl and cKD mice and assessed NCX1 protein levels by Western blots. Compared to Ctl mice, the atrial NCX1 protein level was increased by 28% in the cytosolic fraction and by 55% in the membrane faction of cKD mice (Figure 4G-H). Similarly, in human atrial tissues from NSR and cAF patients, NCX1 in the membrane fraction was increased by more than 4-folds, while the NCX1 in the cytosolic fraction was trending to increase in cAF patients (Figure 4I, Supplementary Table S4). Thus, NCX1 protein accumulated at the sarcolemma of both cAF patients and cKD mice. To further evaluate the transport function of NCX1, we performed whole-cell patch clamping to record Iti during the application of 10 mM caffeine in atrial CMs (Figure 4J). We found that the amplitude of Iti was higher in atrial CMs of cKD mice compared with Ctl mice (Figure 4K), consistent with previous findings in cAF patients.14 These results suggest that the overactive NCX1 could contribute to AP alternans and the enhanced AF susceptibility in cKD mice.
FKBP5-deficiency enhances the HIF-1α-mediated transcription of Slc8a1
Our data suggest that the higher amplitude of NCX-mediated Iti in cKD mice likely results from an increase in NCX1 protein levels. To further dissect the molecular mechanisms of enhanced NCX1 function due to FKBP5-deficiency, we assessed the transcript levels of SLC8A1 and Slc8a1 (encoding NCX1) in atrial tissues of cAF patients and cKD mice. We found that NCX1 mRNA levels were upregulated in both cAF patients and cKD mice compared to their respective controls (Figure 5A-B, Supplementary Table S5). To investigate how FKBP5-deficiency enhances Slc8a1 transcription, we analyzed the promoter of cardiac Slc8a1 in silico. Previous studies reported that Slc8a1 has different transcriptional variants in CMs and neurons, determined by the differences in promoter regions. HIF-1α is a TF known to modulate Slc8a1 transcription in neurons, and its activity is enhanced in AF patients;36-38 however, the binding site of HIF-1α within the cardiac Slc8a1-promoter region was unknown.39 In silico analysis revealed 6 regions (P1-6) containing one or multiple canonical HIF-1α-response elements (HREs) within the 2-kb of the cardiac Slc8a1-promoter region (Figure 5C). To examine the interaction between HIF-1α and these 6 HRE regions, ChIP-PCR was performed with atrial tissue of Ctl and cKD mice. To prevent the rapid degradation of HIF-1α ex vivo, we incubated the cardiac tissue in oxygenated KB buffer (normoxia) or KB buffer pre-conditioned in a hypoxia chamber (2% O2 overnight) for 2-hours before crosslink. The results revealed that the interactions between HIF-1α and P5- and P6-regions were enhanced in atria of cKD compared to Ctl mice under normoxia, which was further exacerbated by hypoxia challenge (Figure 5D). To verify the direct interaction between HIF-1α and P5- or P6-regions, we performed EMSA with P5- and P6-probes, respectively. This study revealed a dose-dependent shift of P6-probe upon interacting with HIF-1α protein (Figure 5E), which was eliminated with the mutant probe. These findings confirmed that HIF-1α directly regulates the transcription of cardiac Slc8a1.
Figure 5. CM-specific FKBP5-deficiency enhances the HIF-1α-mediated transcription of Slc8a1.

(A-B) Increased SLC8A1 mRNA level in atrial tissue of cAF patients (A) and cKD mice (B). (C) Schematic diagram showed the 6 HREs in the promoter region (P1-P6) of cardiac Slc8a1. (D) ChIP-qPCR showed that the interaction between HIF-1α and the P5- and P6-regions of Slc8a1-promoter were increased in cKD tissue, which was further exacerbated under hypoxia. (E) EMSA results confirmed the direct binding between HIF-1α protein and the P6-probe. p-values were determined with Mann-Whitney test in A and B, and two-way ANOVA with Tukey’s comparison in D.
FKBP5 negatively regulates HIF-1α protein level
To determine whether FKBP5 regulates HIF-1α activity, we first evaluated HIF-1α protein levels in atrial lysates of cKD mice. Western blots showed that HIF-1α levels were increased in cKD mice (vs Ctl mice, p=6.99x10−3, Figure 6A), suggesting that FKBP5 could negatively impact HIF-1α activity. To demonstrate this relationship, nuclear translocation of HIF-1α was assessed by immunostaining in HEK293 cells overexpressing GFP-tagged FKBP5. Compared to cells expressing control vector (pcDNA.Gfp), the nuclear accumulation of HIF-1α was reduced in cells overexpressing FKBP5 (pcDNA.Fkbp5.Gfp) (Figure 6B). Consistently, overexpression of FKBP5 in H9C2 cells (rat CM cell line) reduced the hypoxia-induced increase in nuclear HIF-1α protein (Figure 6C-D), as well as the expression of Slc8a1 (Figure 6E) and other downstream targets of HIF-1α such as Stc2 (encoding Stanniocalcin-2, STC-2) and Bnip3 (encoding BCL2 interacting protein 3, BNIP3) (Supplementary Figure S9). CoCl2-treated cells were used as positive control. The protein levels of HIF-1α are regulated by heat-shock proteins (HSPs) such as HSP90. When HSP90 interacts with HIF-1α, it can stabilize HIF-1α and prevent it from proteosome-mediated degradation.40, 41 HSP90 is also a known binding partner of FKBP542, 43 and genetic ablation of FKBP5 reduces the interaction between HSP90 and FKBP5 (Supplementary Figure S10A). Co-immunoprecipitation revealed that the interaction between HSP90 and HIF-1α was increased in atrial samples of cKD mice (Supplementary Figure S10B), but impaired when FKBP5 was overexpressed in H9C2 cells (Figure 6F). These data suggest that HIF-1α competes with FKBP5 for stabilization by HSP90. To further validate this hypothesis, we then treated the H9C2 cells with either the selective HSP90 inhibitor 17-AAG (1 μmol/L) or the proteasome inhibitor MG-132 (5 μmol/L) for 3-hours, respectively. Compared to vehicle-treated cells, 17-AAG reduced HIF-1α levels in the control vector transfected H9C2 cells but failed to further decrease the HIF-1α protein levels in FKBP5-overexpressing H9C2 cells (Figure 6G-H). Most important, the proteasome inhibitor MG-132 strongly increased the HIF-1α protein levels in the FKBP5-overexpressing H9C2 cells, confirming that the negative FKBP5 regulation of the HSP90/HIF-1α complex occurs upstream of the proteasome-degradation system (Figure 6G-H).
Figure 6. FKBP5 negatively regulates HIF-1α.

(A) Increased HIF-1α protein level in the atria of cKD mice revealed by Western blots. (B) Immunostaining of HIF-1α in the HEK 293 cells transfected with pcDNA.Gfp (control) or pcDNA.Fkbp5.Gfp. (C) Representative Western blots with the cytosol- or nuclear-fractions of H9C2 cells transfected with pcDNA (control) or pcDNA.Fkbp5 (FKBP5-OE) vectors. (D) Relative level of the nuclear HIF-1α protein normalized to Histone H3 in H9C2 cells. (E) Relative mRNA levels of Slc8a1 in H9C2 cells treated with pcDNA vector (control) or pcDNA.Fkbp5 (FKBP5-OE). (F) Co-immunoprecipitation of HSP90 (IP) and HIF-1α (Western blots) in H9C2 cells treated with pcDNA (control) or pcDNA.Fkbp5 (FKBP5-OE) vectors. (G) Representative Western blots and (H) quantification of HIF-1α protein levels in the pcDNA or pcDNA.Fkbp5 vector transfected H9C2 cells, in the presence of vehicle (Veh), 17-AAG (HSP90 inhibitor), or MG (MG-132, proteasome inhibitor). p-values were determined with Mann-Whitney test in A and D, Shapiro-Wilk test and the unpaired Student’s t-test in E, and two-way ANOVA with Tukey’s comparison in H.
Since FKBP5-deficiency increases HSP90-mediated HIF-1α stabilization, which in turn upregulates NCX, we determined whether inhibition of HSP90 with 17-AAG prevents atrial arrhythmogenesis in cKD mice. Two weeks after tamoxifen injections in cKD mice, we treated the cKD mice with either vehicle or 17-AAG (50mg/kg, i.p., once every two days) for two weeks (Figure 7A). Echocardiography studies showed that 17-AAG did not affect the LA size, LVEF%, LV diameters, and ECG parameters in cKD mice (Figure 7B-F, Supplementary Table S9 & Table S10). However, the upregulation of HIF-1α and NCX1 proteins was attenuated by 17-AAG in atria of cKD mice (vs vehicle) (Figure 7G-H). Most importantly, 17-AAG prevented the induction of AF (75% vs 0%, p=0.0476, Figure 7I-K) in cKD compared to the vehicle-treated cKD mice. These results support the hypothesis that the CM-specific loss of FKBP5 enhances the HSP90-mediated stabilization of HIF-1α, increases its interaction with cardiac Slc8a1, and promotes NCX1-mediated atrial arrhythmogenesis.
Figure 7. HIF-1α inhibition prevents AF inducibility in FKBP5 cKD mice.

(A) Timeline of HIF-1α inhibition studies in cKD mice. (B-C) Representative long-axis echocardiography images (B) and quantification (C) of left atrial (LA) area in cKD mice treated with vehicle (control) or 17-AAG. (D-F) Representative M-mode echocardiography images (D) and quantification of LVEF% (E) and diameters (F) of left ventricles in cKD mice treated with vehicle (control) or 17-AAG. (G-H) Representative Western blots (G) and quantification (H) of protein levels of NCX1, HIF-1α and HSP90 in atria of cKD mice treated with vehicle (control) or 17-AAG. (I-K) Representative recordings of surface and intracardiac electrograms (I) and the incidence (J) and duration (K) of pacing-induced AF in cKD mice treated with vehicle (control) or 17-AAG. p-values were determined with Mann-Whitney test in H and K, and Fisher’s exact test in J.
Atrial CM-selective FKBP5-deficiency enhances AF susceptibility
We recently developed an AAV9 viral vector that could achieve atrial CM-selective gene targeting.22 To determine whether atrial CM-selective knockdown of FKBP5 is sufficient to promote AF, we injected the Fkbp5flox/flox mice with the AAV9 expressing the atrial CM-selective Cre-recombinase (AAV9-ANF-Cre, aKD) or AAV9-ANF-Flag (as Ctl, Figure 8A).22 4 weeks after injection, the Fkbp5 mRNA level was decreased by 50%, while Slc8a1 mRNA level was increased 3-fold in atria, but not in ventricles of aKD mice (Figure 8B-C), recapitulating the atrial phenotype of cKD mice. Similarly, FKBP5 protein level was decreased by 50%, while NCX1 protein was increased by 40% in atria, but not in ventricles of aKD mice (Figure 8D-F), quantitatively matching the results in cKD mice (Figure 4H). The protein level of HIF-1α was increased by 40% in atria of aKD mice (vs Ctl atria, Figure 8G). The incidence of pacing-induced AF was significantly higher in aKD mice than in Ctl mice (60.0% vs 18.8%, p=0.0290, Figure 8H). Conversely, restoring the protein levels of FKBP5 in atrial CMs of Fkbp5−/− mice with the AAV9-ANF-FKBP5 virus (1x1012, GC/mouse) showed a trend of reduced AF-inducibility compared to the Fkbp5−/− mice receiving the AAV9-ANF-Flag control virus (33% vs 81.8%, p=0.072, Supplementary Figure S11A-C & Table S11). The levels of HIF-1α and NCX1 were significantly reduced by 44% (p=0.0158) and by 48% (p=2.16x10−3), respectively, in the atria of Fkbp5−/− mice by the injection of AAV9-ANF-FKBP5 virus compared with the control virus injected Fkbp5−/− mice (Supplementary Figure S11D-G). These results not only establish that atrial deficiency of FKBP5 is sufficient to enhance AF arrhythmogenesis, but also validate the ‘FKBP5 - HIF-1α - NCX1 - AF’ regulatory axis in the atrium.
Figure 8. Atrial CM-selective knockdown of FKBP5 enhances AF susceptibility.

(A) Schematic diagram showed the designs to knockdown FKBP5 in an atrial CM-selective manner using AAV9-ANF-Cre virus injected Fkbp5flox/flox mice (aKD). AAV9-ANF-Flag virus injected Fkbp5flox/flox mice used as control (Ctl) (B) Relative mRNA levels of Fkbp5 and Slc8a1 in atria and ventricles of Ctl and mice. (C) Representative Western blots and (D) quantification of FKBP5 and NCX1 proteins in atria and ventricles of Ctl and aKD mice. (E) Representative Western blots and quantification of HIF-1α protein level in the atria of Ctl and aKD mice. (F) Representative recording and the incidence of pacing-induced AF in Ctl and aKD mice. p-values were determined with unpaired Student’s t-test in B, Mann-Whitney test in C, E, F, and G, and Fisher’s exact test in H.
DISCUSSION
FKBP5 is a newly discovered gene in AF patients by RNA-seq.3 Although FKBP5 has been studied to certain extent in neurons,43, 44 its function in the heart is largely unknown. By establishing the FKBP5-cKD and FKBP5-aKD mouse models, we demonstrate a role of atrial CM-selective downregulation of FKBP5 in AF pathogenesis. Prior to this study, it was shown that FKBP5 is a co-chaperone of HSP90, which interacts with HIF-1α to prevent its proteasomal degradation in different cell systems.41-43 Here we demonstrate that the loss of FKBP5 strengthens the HSP90-mediated stabilization of HIF-1α and prevents its degradation in CMs. The increased level of HIF-1α in CMs subsequently upregulates the transcription of Slc8a1 and elevates NCX1 protein levels, thereby causing arrhythmogenic SCaWs and AP alternans that enhance the susceptibility to AF. Inhibition of HSP90 by the small molecule 17-AAG restored the protein homeostasis of HIF-1α and attenuated the FKBP5-deficiency induced upregulation of NCX1 and the related atrial arrhythmogenicity (see Graphical Abstract). Thus, our study demonstrates a role of FKBP5-deficiency in atrial arrhythmogenesis, establishes FKBP5 as a negative regulator of HIF-1α in CMs, and identifies a molecular mechanism for the pro-arrhythmic NCX1 upregulation in cAF patients.
Because FKBP5 is a co-chaperone protein, it is very likely that FKBP5 can affect many aspects of cell biology. In this study, we elucidated how FKBP5 modulates the transcription of a key CM transporter (i.e., NCX1) via its negative regulation of HIF-1α. It remains possible that FKBP5 could modulate the function of ion channels in CMs via other mechanisms, such as protein trafficking or protein degradation pathways. For example, when we measured the NCX1 in cytosol and membrane separately, we found the NCX1 protein increased more strongly in sarcolemma, especially in the samples of cAF patients. This altered distribution of NCX1 protein in different cellular compartment implies that the NCX1 trafficking could be enhanced during AF and due to loss of FKBP5. In agreement, previous work has reported that FKBP5 could affect the membrane trafficking of glucose transporter type-4.45 Thus, whether NCX1 trafficking is influenced by FKBP5 requires direct demonstration. A recent study revealed that FKBP5 is a modulator of inflammatory signaling in human fibroblasts25. We have found an increased level of fibrosis in cardiac tissue of the whole-body Fkbp5−/− mice (Supplementary Figure S12), which could be due to the FKBP5-deficiency in cardiac fibroblasts and the associated inflammatory responses therein. The increased interstitial fibrosis can impair conduction and facilitate the development of macro- or micro-reentrant circuits, in favor of the maintenance of AF.46 It is also possible that activated fibroblasts directly modify cardiomyocyte function, thereby increasing cellular triggered activity. The mechanisms through which FKBP5 signaling in fibroblasts promotes fibrotic remodeling and atrial arrhythmogenesis require detailed characterization in future studies.
Abnormalities in Ca2+-handling proteins can alter the electrophysiological properties of CMs and contribute to atrial arrhythmogenesis.30 NCX-mediated DADs are one well-established mechanism of ectopic (triggered) activity.47, 48 Enhanced NCX activity can also alter APD, and inhibition of NCX activity can attenuate the occurrence of arrhythmogenic alternans.28, 29, 49, 50 The increased NCX1 protein levels are a well-known molecular signature in cAF patients;13-16 however, the underlying molecular underpinnings have not been established prior to the current study. Here, we reveal that the increased NCX1 protein levels can be partially attributed to enhanced transcription. Although the transcriptional modulations of NCX1 have been studied extensively in patients and animal models of heart failure,51, 52 little is known about which TFs are involved in NCX1 transcription in AF. Through in silico analysis, ChIP-PCR, and EMSA experiments, our study confirms that HIF-1α directly upregulates the transcription of cardiac Slc8a1, which is further amplified by FKBP5-deficiency. Previous work demonstrated that an increase in ventricular NCX1 can improve the pressure overload-induced ventricular dysfunction in mice.53 In our FKBP5-cKD model, NCX1 protein was increased in both atria and ventricles. Although NCX1 protein levels were higher in ventricles, cKD mice did not exhibit an increased susceptibility to pacing-induced ventricular arrhythmias. This could be attributed to the different ‘source-sink’ relationship and the distinct CM ultrastructure and Ca2+ handling principles in the atria compared to the ventricles. In order to develop arrhythmias, ventricles require more cellular triggered activities and electrical remodeling to overcome the ‘sink’.
HIF-1α is an O2-sensing TF. Under normoxia, O2 promotes the prolyl hydroxylase domain protein-mediated hydroxylation of HIF-1α, a process crucial for the proteasome-mediated degradation of HIF-1α.54 When the O2 supply is reduced (e.g., under hypoxia), HIF-1α can escape the proteasome-degradation pathway, translocate into nucleus, bind to HREs, and subsequently induce transcription of target genes.55 The interaction with the chaperone HSP90 is required for the stability of HIF-1α protein, especially in the nascent process.56 Increased level of HIF-1α protein has been associated with AF.36, 37, 57 However, its precise role in atrial arrhythmogenesis has not been explored. AF is associated with tissue hypoxia. The irregular high-frequency excitation and contraction during AF increases atrial energy demand and O2 expenditure.38 The imbalance between energy demand and O2 supply during AF could cause cellular hypoxia in CMs.38 In this study, we show for the first time that the loss of CM FKBP5 increases HIF-1α protein levels. In contrary, overexpression of FKBP5 in H9C2 cells (rat CM cell line) hinders the hypoxia-induced nuclear translocation of HIF-1α, which is associated with a reduced interaction between HSP90 and HIF-1α. The reduction of HIF-1α protein levels due to FKBP5 overexpression can be attenuated by proteasome inhibition in H9C2 cells. These results not only prove that FKBP5 is a negative regulator of HIF-1α signaling, but also establish the existence of a ‘FKBP5 - HSP90 - HIF-1α’ protein homeostasis axis in CMs. Promotion of HIF-1α degradation with a HSP90 inhibitor such as 17-AAG (also known as tanespimycin) has been proposed as an anticancer therapeutic strategy.40 Because tanespimycin is well tolerated in patients and derailed proteostasis is involved in long-lasting persistent AF,58, 59 we suggest that HSP90 inhibition could constitute a new anti-AF option that improves atrial proteostasis in AF patients.
HIF-1α can regulate a variety of targets and affects multiple cellular processes such as metabolism, cell survival, angiogenesis, etc.60 Here, we have confirmed that two known targets of HIF-1α - Stc2 and Binp3 were downregulated in H9C2 cells with FKBP5 overexpression, where the hypoxia-induced activation of HIF-1α was impaired. STC-2 can influence the insulin-like growth factor pathway via its interaction with pregnancy-associated plasma protein-A.61 Recent studies suggest that STC-2 might also be a good biomarker for AF associated with mitral regurgitation and myocardial infarction.61, 62 Meanwhile, BNIP3 was reportedly increased in heart failure with reduced EF (HFrEF) patients.63 Knockdown of BNIP3 in a murine model of HFrEF can improve glycolysis and mitochondrial metabolism and restore the SR-mitochondrial Ca2+ homeostasis.63-65 Thus, future work should study whether enhanced HIF-1α signaling contributes to atrial remodeling via modulation of STC-2, BNIP3, or other HIF-1α targets.
Our study has limitations. First, in the patient biopsy samples, we were unable to detect difference in the HIF-1α protein level, which could be due to the rapid degradation of HIF-1α when the tissue was excised from patients. However, an upregulation of HIF-1α protein in AF patients has been documented in previous reports.37, 38 FKBP4 (also known as FKBP52) is the paralog of FKBP5 and has usually opposite functions to FKBP5.66 Whether FKBP4 could counteract some consequences of FKBP5-deficiency for atrial arrhythmogenesis requires further investigation. HIF-1α is usually considered to mediate acute cellular metabolic adaptation to the rapid changes in O2 concentrations; whereas HIF-2α is considered to mediate adaptations to the long-term lower O2 concentrations.67 Although they share the same binding motif (RCGTG), their targeted genes are only partially overlapped.68 Whether HIF-2α participates in the regulation of Slc8a1 remains to be elucidated in future works. For many experiments we used multiple cell lines (H9C2, HEK293, etc.); further experiments are required to definitively prove and validate the role of the ‘FKBP5 - HSP90 - HIF-1α’ in native CMs. Although we did not observe changes in APD and AERP in cKD mice, we cannot rule out that ionic remodeling may affect resting membrane potential. It was reported recently that the altered levels of the circulating microRNA-199a-5p (miR-199a) in HFrEF patients with AF affects the expression of NCX1, L-type Ca2+ channel, and connexin-40 in HL-1 cells 69. Interestingly, we have previously shown that FKBP5 was a putative target of miR-199a and miR-199a was upregulated in atrial samples of pAF patients, in which FKBP5 mRNA levels were downregulated.3 Future studies are needed to assess whether the circulating miR-199a enhances atrial arrhythmogenesis via modulation of FKBP5. Finally, the current study used young adult mice only. Given the association between aging and the increased AF risk, future work should evaluate the role of FKBP5-deficiency for AF promotion in advanced age.
In summary, this work establishes a role for FKBP5-deficiency in atrial arrhythmogenesis. Our data unveil the mechanistic link between FKBP5 downregulation and upregulation of NCX1 in the context of AF. We also discovered that FKBP5 is a negative regulator of HIF-1α, which is highly sensitive to high atrial rate during atrial tachycardia and AF. Our study will foster future investigations about the precise role of FKBP5 in cardiac physiology and pathophysiology and is expected to promote the development of novel therapeutic strategy targeting FKBP5 in heart.
Supplementary Material
Novelty and Significance.
What is known?
Downregulation of FKBP5 (encoding FK506-binding protein 5, FKBP5) mRNA is found in patients with paroxysmal AF.
Upregulation of Na+/Ca2+-exchanger type-1 (NCX1) is a well-established molecular signature in patients with long-lasting persistent (chronic) AF.
It is known that hypoxia-inducible factor 1-alpha (HIF-1α) level is upregulated in AF patients.
What new information does this article contribute?
The cardiomyocyte-specific loss of FKBP5 enhances the vulnerability to AF by promoting arrhythmogenic alternans, which is associated with enhanced expression and activity of NCX1.
FKBP5 is a negative regulator of HIF-1α by competing with HIF-1α-stabilizing HSP90. FKBP5-deficiency upregulates the transcription of Slc8a1 by enhancing the activity of HIF-1α.
HSP90 inhibitor 17-AAG can prevent atrial arrhythmogenesis by normalizing HIF-1α activity in the context of FKBP5-deficiency.
In this study, we delineate the function of the newly discovered FKBP5 protein in heart and in atrial cardiomyocytes. We found that FKBP5-deficiency increases the susceptibility to AF induction by causing action potential alternans and an enhancement of NCX1 expression and function. Although increases in NCX1 function were previously reported in AF paradigms, the underlying molecular mechanism was unclear. Here, we demonstrate that FKBP5-deficiency upregulates the transcription of Slc8a1 (encoding NCX1) in cardiomyocytes by enhancing transcription-factor HIF-1α signaling. We show that HIF-1α directly interacts with the promoter of Slc8a1 and that FKBP5 inhibits the accumulation of HIF-1α and prevents its nuclear translocation. Our results reveal that FKBP5-deficiency in cardiomyocytes, as observed in AF patients, promotes atrial arrhythmogenesis, pointing to FKBP5 as potential novel target for the treatment of AF.
ACKNOWLEDGEMENTS
We thank the technical support from the Mouse Metabolism and Phenotyping Core (MMPC) and Human Tissue Acquisition and Pathology (HTAP) core at Baylor College of Medicine and Ramona Löcker, Annette Kötting-Dosch, and Simone Olesch for technical assistance.
FUNDING SOURCES
This study is supported by grants from National Institutes of Health [R01HL136389 and R01HL163277 to N.L. and D.D., R01HL147108 to N.L. and X.H.T.W., R01HL131517 to D.D., R01HL089598 to X.H.T.W. and D.D., R01-HL160992 to X.H.T.W. and D.D., UM1HG006348, R01DK114356, S10OD032380 to MMPC, and P30CA125123 to HTAP], the European Union [large-scale network project MAESTRIA No. 965286 to D.D.], the Netherlands Organization for Scientific Research [NWO/ZonMWVidi 09150171910029 to J.H.], and American Heart Association [23POST1013888 to Y.Y. and EIA 936111 to N.L.].
Non-standard Abbreviations and Acronyms
- AAV
adeno-associated virus
- AERP
atrial effective refractory period
- aKD
atrial CM-selective knockdown
- AP
action potential
- APD
action potential duration
- BNIP3
BCL2 interacting protein 3
- cAF
long-lasting persistent (‘chronic’) atrial fibrillation
- CaSF
frequency of Ca2+ sparks
- CaT
Ca2+ transient
- Cav1.2
α-subunit of L-type Ca2+-channel
- cKD
cardiomyocyte-specific knockdown
- CM
cardiomyocyte
- CoV
coefficient of variation
- Ctl
control
- CV
conduction velocity
- DAD
delayed afterdepolarization
- EDD
end-diastolic diameter
- ESD
end-systolic diameter
- FKBP5
FK506 binding protein 5
- FKBP4
FK506 binding protein 4
- GC
genome copy
- HFrEF
heart failure with reduced ejection fraction
- HIF-1α
hypoxia-inducible factor 1-alpha
- HRE
HIF-1α-response element
- HSP
heat-shock protein
- LA
left atrial
- LV
left ventricular
- LVEF
left ventricular ejection fraction
- NCX1
Na+/Ca2+ exchanger type-1
- NSR
normal sinus rhythm
- pAF
paroxysmal AF
- PMCA
plasma membrane Ca2+ ATPase
- RyR2
ryanodine receptor type-2
- SCaW
spontaneous Ca2+ wave
- SERCA
sarcoplasmic/endoplasmic reticulum Ca2+/ATPase
- SR
sarcoplasmic reticulum
- STC-2
Stanniocalcin-2
- TF
transcription factor
Footnotes
Conflict of interest statement: None.
REFERENCES
- 1.Jackson SL, Tong X, Yin X, George MG and Ritchey MD. Emergency Department, Hospital Inpatient, and Mortality Burden of Atrial Fibrillation in the United States, 2006 to 2014. Am J Cardiol. 2017;120:1966–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schnabel RB, Yin X, Gona P, Larson MG, Beiser AS, McManus DD, Newton-Cheh C, Lubitz SA, Magnani JW, Ellinor PT, et al. 50 year trends in atrial fibrillation prevalence, incidence, risk factors, and mortality in the Framingham Heart Study: a cohort study. Lancet. 2015;386:154–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chiang DY, Zhang M, Voigt N, Alsina KM, Jakob H, Martin JF, Dobrev D, Wehrens XH and Li N. Identification of microRNA-mRNA dysregulations in paroxysmal atrial fibrillation. International journal of cardiology. 2015;184C:190–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cioffi DL, Hubler TR and Scammell JG. Organization and function of the FKBP52 and FKBP51 genes. Curr Opin Pharmacol. 2011;11:308–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zannas AS, Balsevich G and Gassen NC. The emerging role of FKBP5 in the regulation of metabolism and body weight. Surg Obes Relat Dis. 2016;12:1560–1561. [DOI] [PubMed] [Google Scholar]
- 6.Fries GR, Gassen NC and Rein T. The FKBP51 Glucocorticoid Receptor Co-Chaperone: Regulation, Function, and Implications in Health and Disease. Int J Mol Sci. 2017;18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pei H, Li L, Fridley BL, Jenkins GD, Kalari KR, Lingle W, Petersen G, Lou Z and Wang L. FKBP51 affects cancer cell response to chemotherapy by negatively regulating Akt. Cancer Cell. 2009;16:259–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schulke JP, Wochnik GM, Lang-Rollin I, Gassen NC, Knapp RT, Berning B, Yassouridis A and Rein T. Differential impact of tetratricopeptide repeat proteins on the steroid hormone receptors. PLoS One. 2010;5:e11717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Romano S, D'Angelillo A, Pacelli R, Staibano S, De Luna E, Bisogni R, Eskelinen EL, Mascolo M, Cali G, Arra C, et al. Role of FK506-binding protein 51 in the control of apoptosis of irradiated melanoma cells. Cell Death Differ. 2010;17:145–57. [DOI] [PubMed] [Google Scholar]
- 10.Calabro M, Crisafulli C, Di Nicola M, Colombo R, Janiri L and Serretti A. FKBP5 Gene Variants May Modulate Depressive Features in Bipolar Disorder. Neuropsychobiology. 2019;78:104–112. [DOI] [PubMed] [Google Scholar]
- 11.Criado-Marrero M, Gebru NT, Gould LA, Smith TM, Kim S, Blackburn RJ, Dickey CA and Blair LJ. Early Life Stress and High FKBP5 Interact to Increase Anxiety-Like Symptoms through Altered AKT Signaling in the Dorsal Hippocampus. Int J Mol Sci. 2019;20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferrer A, Costas J, Labad J, Salvat-Pujol N, Segalas C, Urretavizcaya M, Real E, de Arriba-Arnau A, Alonso P, Crespo JM, et al. FKBP5 polymorphisms and hypothalamic-pituitary-adrenal axis negative feedback in major depression and obsessive-compulsive disorder. J Psychiatr Res. 2018;104:227–234. [DOI] [PubMed] [Google Scholar]
- 13.El-Armouche A, Boknik P, Eschenhagen T, Carrier L, Knaut M, Ravens U and Dobrev D. Molecular determinants of altered Ca2+ handling in human chronic atrial fibrillation. Circulation. 2006;114:670–80. [DOI] [PubMed] [Google Scholar]
- 14.Voigt N, Li N, Wang Q, Wang W, Trafford AW, Abu-Taha I, Sun Q, Wieland T, Ravens U, Nattel S, et al. Enhanced sarcoplasmic reticulum Ca2+ leak and increased Na+-Ca2+ exchanger function underlie delayed afterdepolarizations in patients with chronic atrial fibrillation. Circulation. 2012;125:2059–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lenaerts I, Bito V, Heinzel FR, Driesen RB, Holemans P, D'Hooge J, Heidbuchel H, Sipido KR and Willems R. Ultrastructural and functional remodeling of the coupling between Ca2+ influx and sarcoplasmic reticulum Ca2+ release in right atrial myocytes from experimental persistent atrial fibrillation. Circ Res. 2009;105:876–85. [DOI] [PubMed] [Google Scholar]
- 16.Schotten U, Greiser M, Benke D, Buerkel K, Ehrenteidt B, Stellbrink C, Vazquez-Jimenez JF, Schoendube F, Hanrath P and Allessie M. Atrial fibrillation-induced atrial contractile dysfunction: a tachycardiomyopathy of a different sort. Cardiovasc Res. 2002;53:192–201. [DOI] [PubMed] [Google Scholar]
- 17.Sohal DS, Nghiem M, Crackower MA, Witt SA, Kimball TR, Tymitz KM, Penninger JM and Molkentin JD. Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circ Res. 2001;89:20–5. [DOI] [PubMed] [Google Scholar]
- 18.Yao C, Veleva T, Scott L, Cao S, Li L, Chen G, Jeyabal P, Pan X, Alsina KM, Abu-Taha I, et al. Enhanced Cardiomyocyte NLRP3 Inflammasome Signaling Promotes Atrial Fibrillation. Circulation. 2018;138:2227–2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scott L Jr., Fender AC, Saljic A, Li L, Chen X, Wang X, Linz D, Lang J, Hohl M, Twomey D, et al. NLRP3 inflammasome is a key driver of obesity-induced atrial arrhythmias. Cardiovasc Res. 2021;117:1746–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Voigt N, Heijman J, Wang Q, Chiang DY, Li N, Karck M, Wehrens XHT, Nattel S and Dobrev D. Cellular and molecular mechanisms of atrial arrhythmogenesis in patients with paroxysmal atrial fibrillation. Circulation. 2014;129:145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kienitz MC, Bender K, Dermietzel R, Pott L and Zoidl G. Pannexin 1 constitutes the large conductance cation channel of cardiac myocytes. J Biol Chem. 2011;286:290–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ni L, Scott L, Campbell HM, Pan X, Alsina KM, Reynolds JO, Philippen LE, Hulsurkar M, Lagor WR, Li N, et al. Atrial-Specific Gene Delivery Using an Adeno-Associated Viral Vector. Circ Res. 2019;124:256–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sikkel MB, Francis DP, Howard J, Gordon F, Rowlands C, Peters NS, Lyon AR, Harding SE and MacLeod KT. Hierarchical statistical techniques are necessary to draw reliable conclusions from analysis of isolated cardiomyocyte studies. Cardiovasc Res. 2017;113:1743–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wada K, Misaka T, Yokokawa T, Kimishima Y, Kaneshiro T, Oikawa M, Yoshihisa A and Takeishi Y. Blood-Based Epigenetic Markers of FKBP5 Gene Methylation in Patients With Dilated Cardiomyopathy. J Am Heart Assoc. 2021;10:e021101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zannas AS, Jia M, Hafner K, Baumert J, Wiechmann T, Pape JC, Arloth J, Kodel M, Martinelli S, Roitman M, et al. Epigenetic upregulation of FKBP5 by aging and stress contributes to NF-kappaB-driven inflammation and cardiovascular risk. Proc Natl Acad Sci U S A. 2019;116:11370–11379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.O'Shea C, Holmes AP, Yu TY, Winter J, Wells SP, Correia J, Boukens BJ, De Groot JR, Chu GS, Li X, et al. ElectroMap: High-throughput open-source software for analysis and mapping of cardiac electrophysiology. Sci Rep. 2019;9:1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pearman CM, Madders GWP, Radcliffe EJ, Kirkwood GJ, Lawless M, Watkins A, Smith CER, Trafford AW, Eisner DA and Dibb KM. Increased Vulnerability to Atrial Fibrillation Is Associated With Increased Susceptibility to Alternans in Old Sheep. J Am Heart Assoc. 2018;7:e009972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Szlovak J, Tomek J, Zhou X, Toth N, Veress R, Horvath B, Szentandrassy N, Levijoki J, Papp JG, Herring N, et al. Blockade of sodiumcalcium exchanger via ORM-10962 attenuates cardiac alternans. J Mol Cell Cardiol. 2021;153:111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wan X, Cutler M, Song Z, Karma A, Matsuda T, Baba A and Rosenbaum DS. New experimental evidence for mechanism of arrhythmogenic membrane potential alternans based on balance of electrogenic I(NCX)/I(Ca) currents. Heart Rhythm. 2012;9:1698–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang X, Chen X, Dobrev D and Li N. The crosstalk between cardiomyocyte calcium and inflammasome signaling pathways in atrial fibrillation. Pflugers Arch. 2021;473:389–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li N, Wang T, Wang W, Cutler MJ, Wang Q, Voigt N, Rosenbaum DS, Dobrev D and Wehrens XH. Inhibition of CaMKII phosphorylation of RyR2 prevents induction of atrial fibrillation in FKBP12.6 knockout mice. Circ Res. 2012;110:465–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ginsburg KS, Weber CR and Bers DM. Cardiac Na+-Ca2+ exchanger: dynamics of Ca2+-dependent activation and deactivation in intact myocytes. J Physiol. 2013;591:2067–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shattock MJ, Ottolia M, Bers DM, Blaustein MP, Boguslavskyi A, Bossuyt J, Bridge JH, Chen-Izu Y, Clancy CE, Edwards A, et al. Na+/Ca2+ exchange and Na+/K+-ATPase in the heart. J Physiol. 2015;593:1361–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Neco P, Rose B, Huynh N, Zhang R, Bridge JH, Philipson KD and Goldhaber JI. Sodium-calcium exchange is essential for effective triggering of calcium release in mouse heart. Biophys J. 2010;99:755–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gussak G, Marszalec W, Yoo S, Modi R, O’Callaghan C, Aistrup GL, Cordeiro JM, Goodrow R, Kanaporis G, Blatter LA, et al. Triggered Ca2+ Waves Induce Depolarization of Maximum Diastolic Potential and Action Potential Prolongation in Dog Atrial Myocytes. Circ Arrhythm Electrophysiol. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abe I, Teshima Y, Kondo H, Kaku H, Kira S, Ikebe Y, Saito S, Fukui A, Shinohara T, Yufu K, et al. Association of fibrotic remodeling and cytokines/chemokines content in epicardial adipose tissue with atrial myocardial fibrosis in patients with atrial fibrillation. Heart Rhythm. 2018;15:1717–1727. [DOI] [PubMed] [Google Scholar]
- 37.Gramley F, Lorenzen J, Jedamzik B, Gatter K, Koellensperger E, Munzel T and Pezzella F. Atrial fibrillation is associated with cardiac hypoxia. Cardiovasc Pathol. 2010;19:102–11. [DOI] [PubMed] [Google Scholar]
- 38.Liu Y, Bai F, Liu N, Ouyang F and Liu Q. The Warburg effect: A new insight into atrial fibrillation. Clin Chim Acta. 2019;499:4–12. [DOI] [PubMed] [Google Scholar]
- 39.Valsecchi V, Pignataro G, Del Prete A, Sirabella R, Matrone C, Boscia F, Scorziello A, Sisalli MJ, Esposito E, Zambrano N, et al. NCX1 is a novel target gene for hypoxia-inducible factor-1 in ischemic brain preconditioning. Stroke. 2011;42:754–63. [DOI] [PubMed] [Google Scholar]
- 40.Yu T, Tang B and Sun X. Development of Inhibitors Targeting Hypoxia-Inducible Factor 1 and 2 for Cancer Therapy. Yonsei Med J. 2017;58:489–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tang X, Chang C, Hao M, Chen M, Woodley DT, Schonthal AH and Li W. Heat shock protein-90alpha (Hsp90alpha) stabilizes hypoxia-inducible factor-1alpha (HIF-1alpha) in support of spermatogenesis and tumorigenesis. Cancer Gene Ther. 2021;28:1058–1070. [DOI] [PubMed] [Google Scholar]
- 42.Galigniana NM, Ballmer LT, Toneatto J, Erlejman AG, Lagadari M and Galigniana MD. Regulation of the glucocorticoid response to stress-related disorders by the Hsp90-binding immunophilin FKBP51. J Neurochem. 2012;122:4–18. [DOI] [PubMed] [Google Scholar]
- 43.Sabbagh JJ, Cordova RA, Zheng D, Criado-Marrero M, Lemus A, Li P, Baker JD, Nordhues BA, Darling AL, Martinez-Licha C, et al. Targeting the FKBP51/GR/Hsp90 Complex to Identify Functionally Relevant Treatments for Depression and PTSD. ACS Chem Biol. 2018;13:2288–2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gaali S, Kirschner A, Cuboni S, Hartmann J, Kozany C, Balsevich G, Namendorf C, Fernandez-Vizarra P, Sippel C, Zannas AS, et al. Selective inhibitors of the FK506-binding protein 51 by induced fit. Nat Chem Biol. 2015;11:33–7. [DOI] [PubMed] [Google Scholar]
- 45.Balsevich G, Häusl AS, Meyer CW, Karamihalev S, Feng X, Pöhlmann ML, Dournes C, Uribe-Marino A, Santarelli S, Labermaier C, et al. Stress-responsive FKBP51 regulates AKT2-AS160 signaling and metabolic function. Nat Commun. 2017;8:1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nattel S, Heijman J, Zhou L and Dobrev D. Molecular Basis of Atrial Fibrillation Pathophysiology and Therapy: A Translational Perspective. Circ Res. 2020;127:51–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dobrev D and Wehrens XHT. Calcium-mediated cellular triggered activity in atrial fibrillation. J Physiol. 2017;595:4001–4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Heijman J, Voigt N, Nattel S and Dobrev D. Cellular and molecular electrophysiology of atrial fibrillation initiation, maintenance, and progression. Circ Res. 2014;114:1483–99. [DOI] [PubMed] [Google Scholar]
- 49.Burashnikov A and Antzelevitch C. Late-phase 3 EAD. A unique mechanism contributing to initiation of atrial fibrillation. Pacing Clin Electrophysiol. 2006;29:290–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Szabo B, Sweidan R, Rajagopalan CV and Lazzara R. Role of Na+:Ca2+ exchange current in Cs(+)-induced early afterdepolarizations in Purkinje fibers. J Cardiovasc Electrophysiol. 1994;5:933–44. [DOI] [PubMed] [Google Scholar]
- 51.Gudmundsson H, Curran J, Kashef F, Snyder JS, Smith SA, Vargas-Pinto P, Bonilla IM, Weiss RM, Anderson ME, Binkley P, et al. Differential regulation of EHD3 in human and mammalian heart failure. J Mol Cell Cardiol. 2012;52:1183–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Menick DR, Li MS, Chernysh O, Renaud L, Kimbrough D, Kasiganesan H and Mani SK. Transcriptional pathways and potential therapeutic targets in the regulation of Ncx1 expression in cardiac hypertrophy and failure. Adv Exp Med Biol. 2013;961:125–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ujihara Y, Iwasaki K, Takatsu S, Hashimoto K, Naruse K, Mohri S and Katanosaka Y. Induced NCX1 overexpression attenuates pressure overload-induced pathological cardiac remodelling. Cardiovasc Res. 2016;111:348–61. [DOI] [PubMed] [Google Scholar]
- 54.Fallah J and Rini BI. HIF Inhibitors: Status of Current Clinical Development. Curr Oncol Rep. 2019;21:6. [DOI] [PubMed] [Google Scholar]
- 55.Semenza GL. Hypoxia-Inducible Factor 1 and Cardiovascular Disease. Annual Review of Physiology. 2014;76:39–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xia Y, Choi HK and Lee K. Recent advances in hypoxia-inducible factor (HIF)-1 inhibitors. Eur J Med Chem. 2012;49:24–40. [DOI] [PubMed] [Google Scholar]
- 57.Ogi H, Nakano Y, Niida S, Dote K, Hirai Y, Suenari K, Tonouchi Y, Oda N, Makita Y, Ueda S, et al. Is structural remodeling of fibrillated atria the consequence of tissue hypoxia? Circ J. 2010;74:1815–21. [DOI] [PubMed] [Google Scholar]
- 58.Li N and Brundel B. Inflammasomes and Proteostasis Novel Molecular Mechanisms Associated With Atrial Fibrillation. Circ Res. 2020;127:73–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Henning RH and Brundel B. Proteostasis in cardiac health and disease. Nat Rev Cardiol. 2017;14:637–653. [DOI] [PubMed] [Google Scholar]
- 60.Shohet RV and Garcia JA. Keeping the engine primed: HIF factors as key regulators of cardiac metabolism and angiogenesis during ischemia. J Mol Med (Berl). 2007;85:1309–15. [DOI] [PubMed] [Google Scholar]
- 61.Cediel G, Rueda F, Oxvig C, Oliveras T, Labata C, de Diego O, Ferrer M, Aranda-Nevado MC, Serra-Gregori J, Nunez J, et al. Prognostic value of the Stanniocalcin-2/PAPP-A/IGFBP-4 axis in ST-segment elevation myocardial infarction. Cardiovasc Diabetol. 2018;17:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yu J, Zhou Y, Li L, Zhang K, Gao L, He X and Dong H. Identification of Biomarkers Related to Atrial Fibrillation With Mitral Regurgitation. Am J Med Sci. 2021;361:319–326. [DOI] [PubMed] [Google Scholar]
- 63.Chaanine AH, Joyce LD, Stulak JM, Maltais S, Joyce DL, Dearani JA, Klaus K, Nair KS, Hajjar RJ and Redfield MM. Mitochondrial Morphology, Dynamics, and Function in Human Pressure Overload or Ischemic Heart Disease With Preserved or Reduced Ejection Fraction. Circ Heart Fail. 2019;12:e005131. [DOI] [PubMed] [Google Scholar]
- 64.Chaanine AH, Gordon RE, Kohlbrenner E, Benard L, Jeong D and Hajjar RJ. Potential role of BNIP3 in cardiac remodeling, myocardial stiffness, and endoplasmic reticulum: mitochondrial calcium homeostasis in diastolic and systolic heart failure. Circ Heart Fail. 2013;6:572–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chaanine AH, Higgins L, Lauterboeck L, Markowski T, Yang Q and Delafontaine P. Multiomics Approach Reveals an Important Role of BNIP3 in Myocardial Remodeling and the Pathogenesis of Heart Failure with Reduced Ejection Fraction. Cells. 2022;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wu B, Li P, Liu Y, Lou Z, Ding Y, Shu C, Ye S, Bartlam M, Shen B and Rao Z. 3D structure of human FK506-binding protein 52: implications for the assembly of the glucocorticoid receptor/Hsp90/immunophilin heterocomplex. Proc Natl Acad Sci U S A. 2004;101:8348–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Smythies JA, Sun M, Masson N, Salama R, Simpson PD, Murray E, Neumann V, Cockman ME, Choudhry H, Ratcliffe PJ, et al. Inherent DNA-binding specificities of the HIF-1alpha and HIF-2alpha transcription factors in chromatin. EMBO Rep. 2019;20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Downes NL, Laham-Karam N, Kaikkonen MU and Yla-Herttuala S. Differential but Complementary HIF1alpha and HIF2alpha Transcriptional Regulation. Mol Ther. 2018;26:1735–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Garcia-Elias A, Tajes M, Yanez-Bisbe L, Enjuanes C, Comin-Colet J, Serra SA, Fernandez-Fernandez JM, Aguilar-Agon KW, Reilly S, Marti-Almor J, et al. Atrial Fibrillation in Heart Failure Is Associated with High Levels of Circulating microRNA-199a-5p and 22-5p and a Defective Regulation of Intracellular Calcium and Cell-to-Cell Communication. Int J Mol Sci. 2021;22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The detailed experimental materials, methods, and data supporting the findings of this study are available within the article and its Supplemental Material. Raw data are available from the corresponding author upon reasonable request. The Major Resources Table is provided in the Supplemental Material.