

Abstract
POU3F3 variants cause developmental delay, behavioral problems, hypotonia and dysmorphic features. We investigated the phenotypic and genetic landscape, and genotype-phenotype correlations in individuals with POU3F3-related disorders.
We recruited unpublished individuals with POU3F3 variants through international collaborations and obtained updated clinical data on previously published individuals. Trio exome sequencing or single exome sequencing followed by segregation analysis were performed in the novel cohort. Functional effects of missense variants were investigated with 3D protein modelling.
We included 28 individuals (five previously published) from 26 families carrying POU3F3 variants; 23 de novo and one inherited from an affected parent. Median age at study inclusion was 7.4 years. All had developmental delay mainly affecting speech, behavioral difficulties, psychiatric comorbidities and dysmorphisms. Additional features included gastrointestinal comorbidities, hearing loss, ophthalmological anomalies, epilepsy, sleep disturbances and joint hypermobility. Autism, hearing and eye comorbidities, dysmorphisms were more common in individuals with truncating variants, whereas epilepsy was only associated with missense variants. In silico structural modeling predicted that all (likely) pathogenic variants destabilize the DNA-binding region of POU3F3.
Our study refined the phenotypic and genetic landscape of POU3F3-related disorders, it reports the functional properties of the identified pathogenic variants, and delineates some genotype-phenotype correlations.
Keywords: POU3F3, Neurodevelopmental disorder, Autism, Cupped ears, Epilepsy
Graphical Abstract
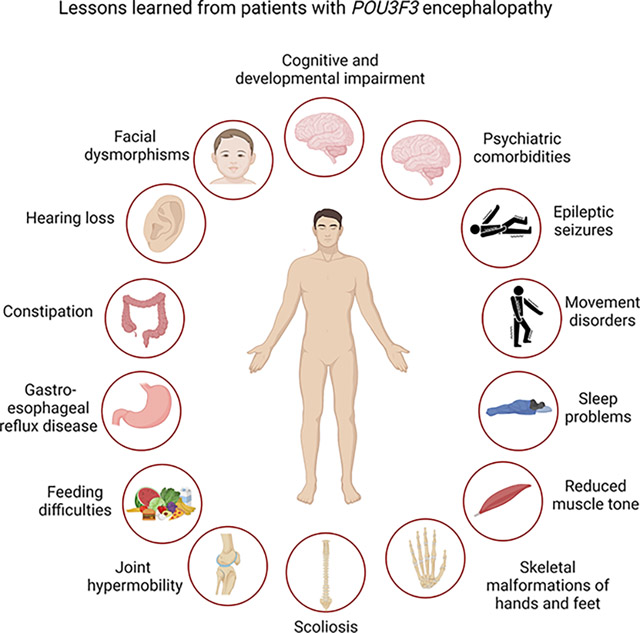
Clinicians should suspect POU3F3-related disorder when a patient presents with developmental delay mainly affecting speech, psychiatric comorbidities and facial dysmorphisms. Additional features include gastrointestinal comorbidities, hearing loss, epilepsy, hypotonia, sleep disturbances, skeletal malformations and joint hypermobility.
1. Introduction
The transcription factor Class III POU family of transcription factors (POU3F3 is an important regulator of neural development1 and is implicated in the cortical neuronal migration, upper-layer specification and production, and neurogenesis2. POU3F3 is composed by two functional domains needed for the site-specific binding to its target genes: the POU-specific (POU-S) domain and the homeobox domain (POU-H)3. Heterozygous pathogenic variants in POU3F3 are associated with a rare neurodevelopmental disorder named Snijders Blok-Fisher Syndrome (OMIM #618604)2. Only 20 individuals with this syndrome have been reported to date2,4. Although autosomal dominant inheritance has been reported in one family with an affected mother and child2, most of the variants occur de novo. Clinical manifestations include mild-to-severe intellectual disability (ID), developmental delay (DD) mainly affecting speech, and to a lesser extent motor skills, congenital hypotonia, autistic and dysmorphic features4. Epilepsy is reported in only three published individuals2,4.
Our knowledge on the phenotypic spectrum, the genetic landscape, genotype-phenotype correlations, and natural history of POU3F3-related disorders, remains limited. To fill in these gaps, we describe herein 23 novel and five previously published individuals, thereby more than doubling the number of individuals with a POU3F3-related disorder2,4. This enables us to further deep-phenotype the spectrum of manifestations including the epileptology, refine the genetic landscape, and to identify genotype-phenotype correlations in affected children as well as in adults.
2. Material and methods
2.1. Cohort analysis
Individuals with a POU3F3-related disorder were recruited either through caregivers, an international network of epilepsy and genetic centers, or through matchmaking approaches including DECIPHER5 and GeneMatcher6. Medical information including birth parameters, epilepsy, (video-)electroencephalograms (EEGs), developmental histories, brain magnetic resonance imaging (MRI) and physical examinations were collected from local healthcare providers and/or families of affected individuals using a standardized questionnaire. We also contacted the healthcare providers of previously published individuals and asked for updated information2,4. Published individuals were identified by searching PubMed (Medline) through July 2022 using the search string “POU3F3” AND “Disorder”. References were manually searched to find relevant publications.
Developmental and cognitive level was, when possible, based on evaluations or formal assessments by referring physicians, and classified according to the Diagnostic and Statistical Manual of Mental Disorders 5th edition7. DD was classified as mild, moderate, severe or profound in children aged 5 years or younger. Children who were at least 6 years old were classified as having mild, moderate, severe or profound ID. Seizures, epilepsies and, when possible, epilepsy syndromes were classified using the 2017 International League Against Epilepsy classifications8,9 and the 2022 Report of the ILAE Task Force on Nosology and Definitions of epilepsy syndromes10. Individuals were classified as having a developmental and epileptic encephalopathy (DEE) if there was evidence that seizures and/or interictal EEG abnormality negatively impacted on development (i.e., epileptic encephalopathy) or, alternatively, intellectual disability and epilepsy (ID+E) if they had developmental impairment and seizures but no evidence of an epileptic encephalopathy8. EEG recordings in individuals with seizures, when available, were reviewed by an epileptologist (G.R.).
2.2. Genetic analysis
POU3F3 variants were identified using single or trio exome sequencing by research or diagnostic laboratories. Whenever possible, segregation analysis was performed in parents. Genetic testing was performed as a diagnostic tool due to cognitive and/or developmental impairment. Variants were annotated using the NM_ 006236 (GRCh37/hg19) transcript of POU3F3. Impact of POU3F3 variants was evaluated by SIFT (sorting intolerant from tolerant)11, PolyPhen-2 (polymorphism phenotyping-v2)12, CADD v1.6 (combined annotation dependent depletion)13 and their frequency in Genome Aggregation Database (gnomAD) v2.1.1 14. Potential splicing effects were assessed using SpliceAI15, transcript inferred pathogenicity score16 and human splicing finder17. Variants were classified according to the 2015 American College of Medical Genetics and Genomics guidelines18.
2.3. Standard protocol approvals, registrations, and individual consents
The study was conducted in agreement with the Declaration of Helsinki and approved by the local ethics committees; the main part being from region of Sjaelland/Denmark (number SJ-91). Authors consented each individual using research protocols approved by the local human research ethics committees. Written informed consent and permission to share individuals’ images were obtained by the parents or legal guardian.
2.4. In silico structural modeling
For visualization of missense variants within the three-dimensional protein structure, the AlphaFold protein structure AF-P20264-F119,20 of POU3F3 was downloaded and processed in the PyMol software by Schrödinger. The DNA molecule from the PDB file 2XSD (POU-domain bound with DNA)21 was aligned to the AlphaFold structure by the respective PyMol algorithm. Variant positions were manually inspected, highlighted and amino acid changes were simulated with the PyMol wizard Mutagenesis. Cluster analysis was performed with the webtool of mutation3d22, based on the ModBase Model 2xsd (ID: EN ENSP00000501036.1).
3. Results
3.1. Individuals and families
The cohort included 28 individuals (17 males and 11 females) from 26 families harboring heterozygous POU3F3 variants (Table S1); 23 individuals were novel, whereas five were retrieved from the literature (individuals #8, #15, #18, #22, #28)2,4. The median age at last follow-up was 7.4 years (age range: 0.8 months - 39 years).
3.2. Phenotypic analysis
An overview of phenotypes is provided in Table S1. We detected no gender related clinical differences.
3.2.1. Global developmental delay/intellectual disability:
Of the 28 individuals, developmental assessment was possible in 27 that were older than 2 years; all exhibited cognitive and/or developmental impairment. A single individual was 8 months old at last evaluation leaving the neurodevelopmental trajectories uncertain. When last assessed, global developmental impairment was rated as either profound (n=2/27; 7.4%), severe (n=2/27; 7.4%), moderate (n=10/27; 37%), or borderline-mild (n=13/27; 48.1%). In addition, 2/27 (7.4%) were non-ambulant, 23/27 (85.2%) walked at a median age of 22 months (range: 12 – 38 months) while the age of walking was unavailable for two individuals. Six (22.2%) individuals had developmental regression, three of them in relation to a period with a high seizure burden. Speech delay and/or difficulties were observed in all individuals; of the 26 individuals older than 3 years, three (11.5%) were nonverbal.
Among the 24 verbal individuals, seven (29.0%) had no speech difficulties, six (25.0%) presented with dysarthria, two (8.3%) could communicate using single words, nine (37.5%) could pronounce short sentences, two (8.3%) had difficulties with pronunciation not further specified and four (16.7%) had both difficulties in expressive language and comprehension.
In individuals older than 6 years, IQ was assessable in 8/20 (40.0%) and ranged from 35–81.
At last follow-up 24 individuals went to school; 19 (79.2%) attended a special needs school while four (16.7%) received specific educational support in the setting of a regular school. A single child (4.2%) was homeschooled.
3.2.2. Behavioral issues:
Attention-deficit/hyperactivity disorder (ADHD) was reported in 5/27 (18.5%) and aggressive behavior in 10/27 (37.0%) individuals. Autism or autistic traits were observed in 15/24 (62.5%). Individual #14 was too young to be evaluated for psychiatric and behavioral comorbidities.
3.2.3. Neurological findings:
Three out of twenty-eight (10.7%) individuals had ataxia of either trunk or limbs, 2/28 (7.1%) showed limb spasticity, 1/28 (3.6%) showed tremor and 5/28 (17.9%) exhibited unclassified difficulties of balance and coordination not further specified, and/or wide based gait. Movement disorders such as chorea and choreo-athetosis were observed in 2/28 (7.1%).
Sleep disturbances, such as disorders in initiating and maintaining sleep, sleep-wake transition disorders and disorders of excessive somnolence, were described in 10/26 (38.5%).
3.2.4. Epilepsy:
Four (14.3%) out of twenty-eight (individuals #10, novel, #154, #17 and #182) experienced seizures that began at a mean age of 18 months (range: 9 months to 6 years). Epilepsy onset was characterized by atonic (#10 and #17), tonic (#15), or tonic–clonic seizures (#18) occurring daily or several times per day. During follow-up, the epilepsy disorder evolved to a Lennox Gastaut syndrome (LGS) (#15 and #18), epilepsy with myoclonic-astatic seizures (#10) and to a further unclassified DEE (#17). Individual #10 had sleep-related myoclonic seizures. In addition, focal seizure such as gelastic (#15 and #18), dacrystic or autonomic (#18) were also reported. Status epilepticus occurred in 2/4 (#17 and #18). No triggers of seizures, including fever, were reported.
EEG was available in 8/28 (28.6%) and showed diffuse spike-slow wave complexes, background slowing (Fig.1A) (#15), fast recruiting rhythmic activities associated with tonic seizures (#15, #18) (Fig. 1B), and generalized spike-waves and polyspike-waves (#10, #15 and #17) (Fig.1C–D). Sleep EEG physiological graphoelements (K complexes and sleep spindles) were poorly represented.
Figure 1A-B; C-D.
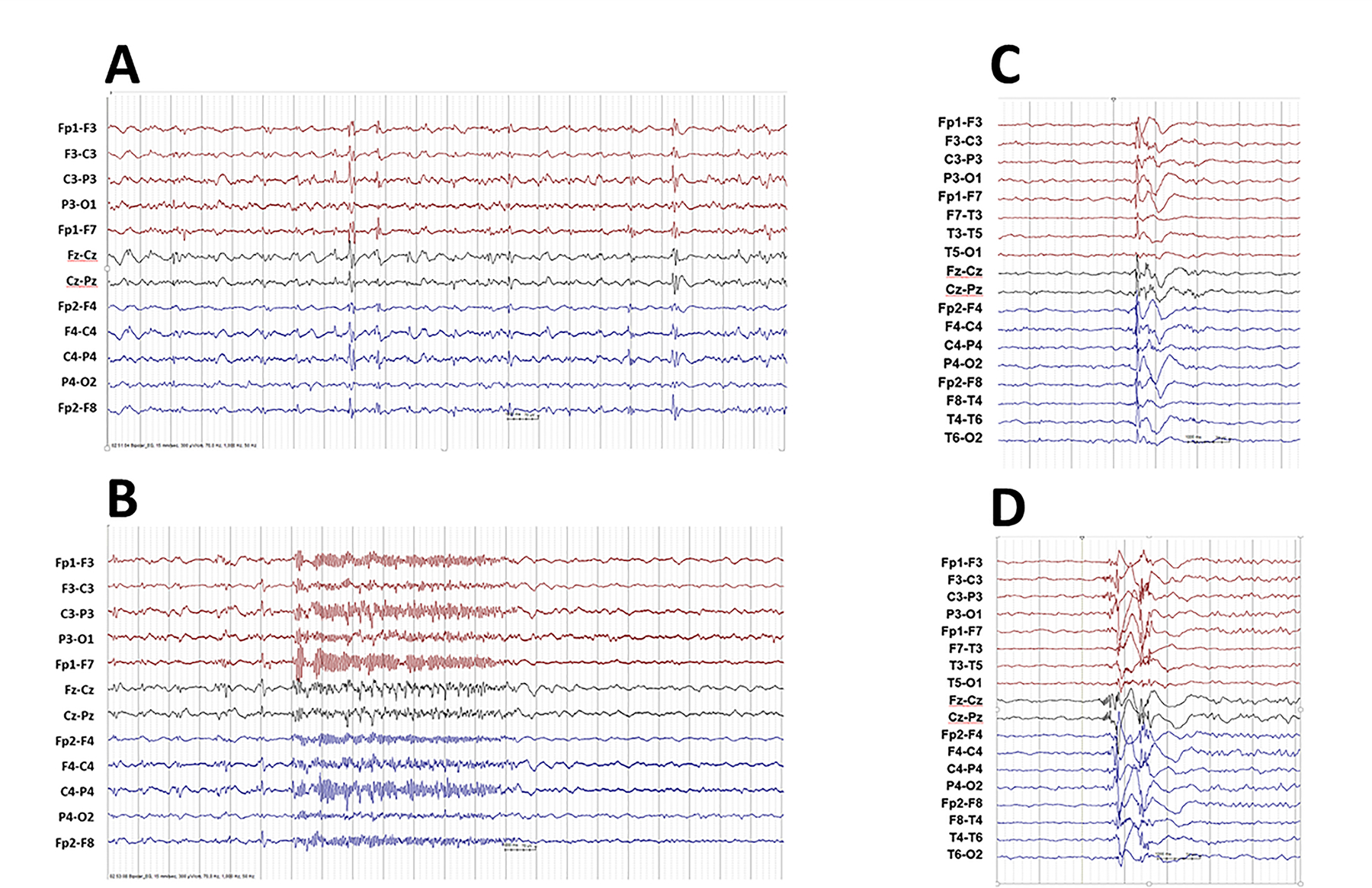
A-B. Individual 15. Interictal EEG showing runs of irregular delta activities with diffuse spike-slow wave complexes and background slowing. Ictal EEG showing diffuse recruiting rhythmic fast discharge associated with a tonic seizure.
C-D. Individual 10. EEG recorded during sleep showed generalized epileptiform discharge of polyspike-and-slow-wave (C) originating from right frontal /centro-frontal followed by alpha-activity with discrete right-sided prevalence, and (D) originating from the vertex followed by alpha-activity with discrete right-sided prevalence.
In #10, seizures were well controlled by sulthiame and zonisamide. The remaining three individuals were refractory to multiple anti-seizure medications, even though lamotrigine and clobazam were effective in reducing the frequency of atonic seizures in individual #17. The combination of clobazam, valproate, lamotrigine and cannabidiol reduced the frequency of tonic and bilateral tonic-clonic seizures in #15, and >50% seizure reduction was achieved with ketogenic diet. Individual #18 showed a partial response to steroids and vagal nerve stimulation.
3.2.5. Dysmorphisms:
Facial dysmorphisms were reported in all individuals (Figure 2). These included cupped and/or prominent and often low-set ears (23/28; 82.1%), full/bulbous nasal tip (7/28; 25.0%), full lips (6/28; 21.4%), epicanthic folds (6/28; 21.4%), hypertelorism (5/28; 17.9%), open-mouth appearance (4/28; 14.3%), up slanting palpebral fissures (4/28; 14.3%), thick ear helices (3/28; 10.7%), periorbital fullness (3/28; 10.7%), short (3/28; 10.7%) or smooth (2/28; 7.1%) philtrum, arched (3/28; 10.7%) and/or thick (2/28; 7.1%) eyebrows, down slanting palpebral fissures (2/28; 7.1%), and prominent nasal bridge (2/28; 7.1%). Microcephaly (head circumference ≤ 3rd percentile) was present at birth in 1/9 (11%) and in 4/20 (20%) individuals at follow-up, where 2/20 (10%) had a head circumference on the 3th-10th percentile. No correlation was found between microcephaly and neuroradiological findings.
Figure 2.

Facial features of 11 individuals with a pathogenic POU3F3 variant, showing cupped and/or prominent and often low-set ears. Other overlapping features are full lips, bulbous nasal tip, narrow palpebral fissures, hypertelorism, and epicanthal folds. Ind 5: 8 m. Ind 11: 3 y 4 m. Ind 13: 15 y 0 m. Ind 18: 9 y 0 m. Ind 19: 10 y 0 m. Ind 20: 8 y 0 m. Ind 21: at birth; 4 y 0 m. Ind 23: 7 y 9 m. Ind 25: 3 y 0 m. Ind 26: 6 y 0 m. Ind 28: 3,5 m; 3 y 0 m; 6 y 0m. Ind: Individual. m: months. y: years
Abnormalities of the hands and feet were detected in 10/28 (35.7%) individuals: pes planus (3/10; 30.0%), syndactyly (3/10; 30.0%), brachydactyly (3/10; 30.0%), short toes (3/10; 30.0%), short broad feet with sandal gaps (2/10; 20%), broad toes (2/10; 20%), clinodactyly (2/10; 20%), tapering fingers (2/10; 20%).
3.2.6. Other findings:
Joint hypermobility was reported in 12/26 (46.1%) individuals. Scoliosis was noted in 4/28 (14.3%) and ranged from mild to severe. A sacral dimple and Perthes disease were observed in 1/28 (3.6%). Hearing loss was reported in 6/28 (21.4%), mainly conductive associated to recurrent otitis. One out of 28 (3.6%) presented with obstructive sleep apneas and was treated with nocturnal continuous positive airway pressure.
Common gastro-intestinal disturbances were chronic constipation, affecting 10/28 (35.7%), followed by gastroesophageal reflux disease in 4/28 (14.3%). Eleven out of 28 (39.3%) newborns showed feeding difficulties, requiring nasogastric tube feeding in 4/28 (14.3%); two also required gastrostomy. Drooling was observed twice (7.1%), and gastroptosis in a single individual (3.6%). One out of 28 (3.6%) presented with Hirschsprung disease. Spontaneously resolved unilateral vesicoureteral reflux was described in #9 (1/28; 3.6%). Cryptorchidism was reported in 3/17 (17.6%) males.
Capillary hemangiomas were reported in #15 (1/28; 3.6%). Finally, #7 (1/28; 3.6%) was found to have edema of a lower extremity of unknown etiology.
3.2.7. Neuroimaging:
MRI of the brain was available in 14 individuals and was reported as abnormal in four participants (29.6%). Anomalies included cerebral atrophy (#17 and #18), abnormal corpus callosum (#6 and #8), and hypoplasia of olfactory bulbs (#8).
3.3. Analysis of variants
POU3F3 variant details are presented in Table S1. Among the 26 families included in the study, 24 different heterozygous POU3F3 variants were detected and 19 were novel. None were detected in gnomAD (v2.1.1)23 (last accessed November 2022). The variant occurred de novo in 23 individuals although parental testing was not available in three individuals (#3, #5 and #12). Individuals #1 and #2 inherited the variant from their affected mother (#3). The maternal grandmother of #1 and #2 had cupped ears but maternal grandparents were not available for testing. All variants were either single-nucleotide or del-ins variants; these included 12 truncating, 10 missense and two in-frame deletion variants. Recurrent variants included p.Glu414*, identified in two individuals, one of which was previously published2, and p.Pro113Hisfs*14, identified in another two individuals. We detected two different missense variants affecting the same amino acid residues; p.Gln340Leu and p.Gln340Pro. All variants were predicted to be (likely) damaging by in silico prediction tools.
A dual genetic diagnosis was found in two individuals; #20 was diagnosed with Klinefelter syndrome (XXY karyotype), while #18 was diagnosed with a familial hypertrophic cardiomyopathy, due to a maternally inherited MYBPC3 variant.
Linear representation of POU3F3 indicating the location of both novel and all previously published variants2,4, compared with variants in gnomAD23, is shown in Figure 3A–B. Novel variants include 10 truncating variants (blue), seven missense variants and two in-frame deletions (orange) while the published variants include 12 truncating variants, five missense variants, and one in-frame deletion.
Figure 3A-B-C.
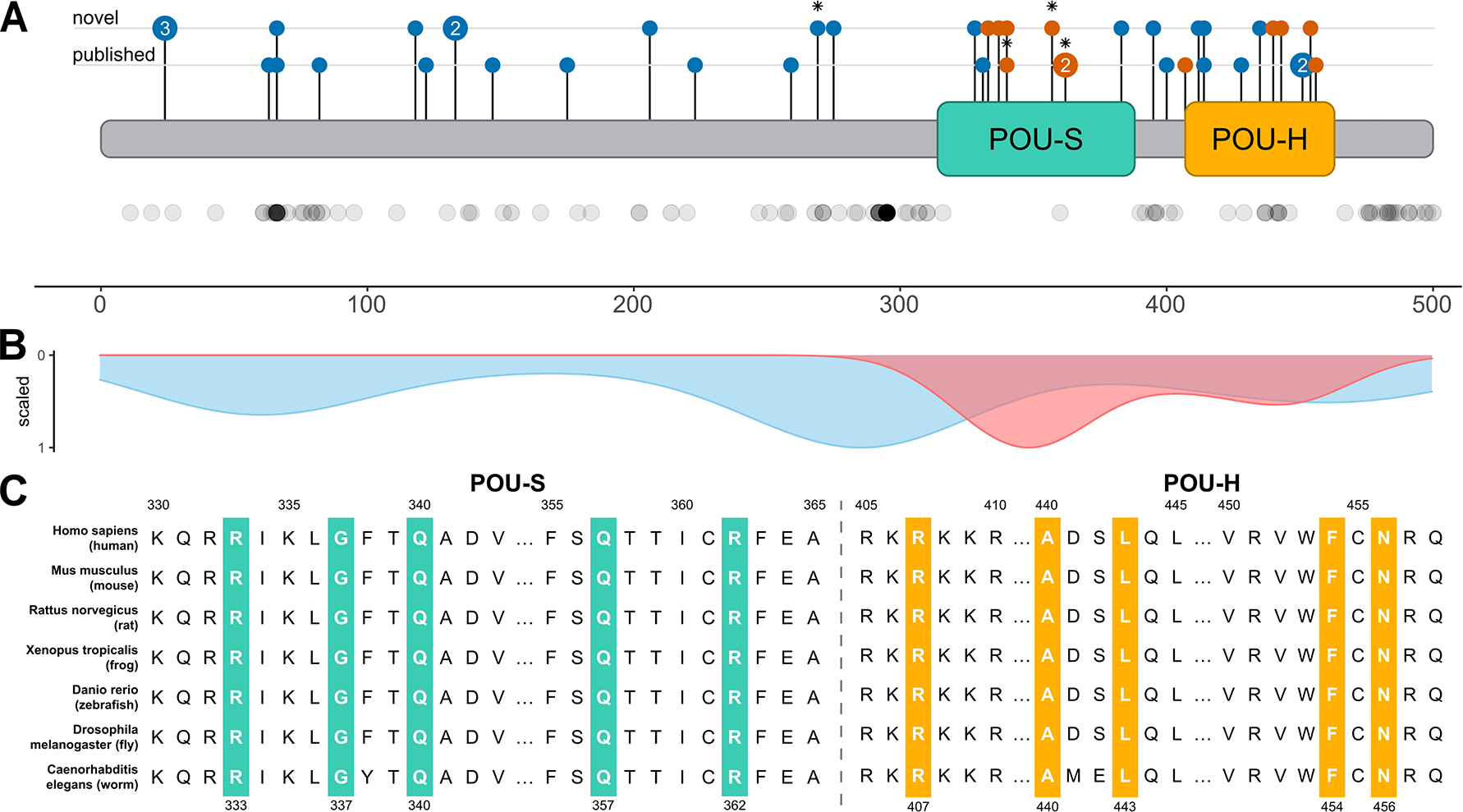
Characterization of POU3F3 variants
A. Linear overview of the POU3F3-protein (blue) with the POU-specific (POU-S, green) and Homeobox (POU-H, yellow) domains. Published (lower line) and novel (upper line) variants are shown at their respective location within the linear protein. Truncating variants in blue, missense variants in orange. Variants, which were identified in individuals with seizures, are marked with an asterisk. Below the linear protein model, every dot represents one missense variant in gnomAD. An overview with variant specifics per individual is provided in Table S1.
B. Density plot of all missense variants (de novo pathogenic or likely pathogenic variants in orange and variants present in gnomAD in green).
C. Conservation of amino acids of all missense variants throughout evolution. All missense variants are included and colored according to their functional domain position: green and yellow designate the functional domains of POU-specific (POU-S) and Homeobox (POU-H), respectively.
All (likely) pathogenic non-truncating variants were located in functionally important regions of the protein, the POU-H and the POU-S domains, which are highly conserved sites across species. All missense variants substituted highly conserved amino acids (Figure 3C). Spatial proximity analysis with mutation3D revealed two clusters of affected amino acid positions. The first cluster includes positions 333, 337, 340, 357 and 362 with a significant p-value of 0.00143, while the second was calculated with a significant p-value of 0.0138 for positions 440, 443, 454 and 456.
Furthermore, the p.Trp383_Glu385del in-frame deletion removed three amino acids from within the POU-S domain of the encoded protein, while the p.Glu430del involved the POU-H domain. The published p.Gln331_Lys335del in-frame deletion removed five amino acids from within the POU-specific domain of the encoded protein. Instead, nonsense/frameshift variants were distributed widely across POU3F3.
3.4. In silico structural modeling
In silico structural modelling was performed on 43 variants; 28 found in the cohort analyzed in our study and 15 in already published individuals.
As transcription factor, POU3F3 employs POU-S and POU-H to bind with high affinity to a specific octamer (50-ATGCAAAT-30) or closely related DNA sequences in the enhancers and promoters of several different target genes. The structure of this complex protein is shown in Figure 3A indicating the positions of the missense variants detected in our cohort and extended to individuals reported in the literature2,4.
We used three-dimensional protein modeling (Figure 4A–D. Figure S1A–H) to further investigate the potential functional impact of the identified non-truncating variants. This analysis indicated that all pathogenic or likely pathogenic variants were predicted to destabilize the DNA-binding region of POU3F3, which spans amino acids 316–466.
Figure 4A-B; C-D.
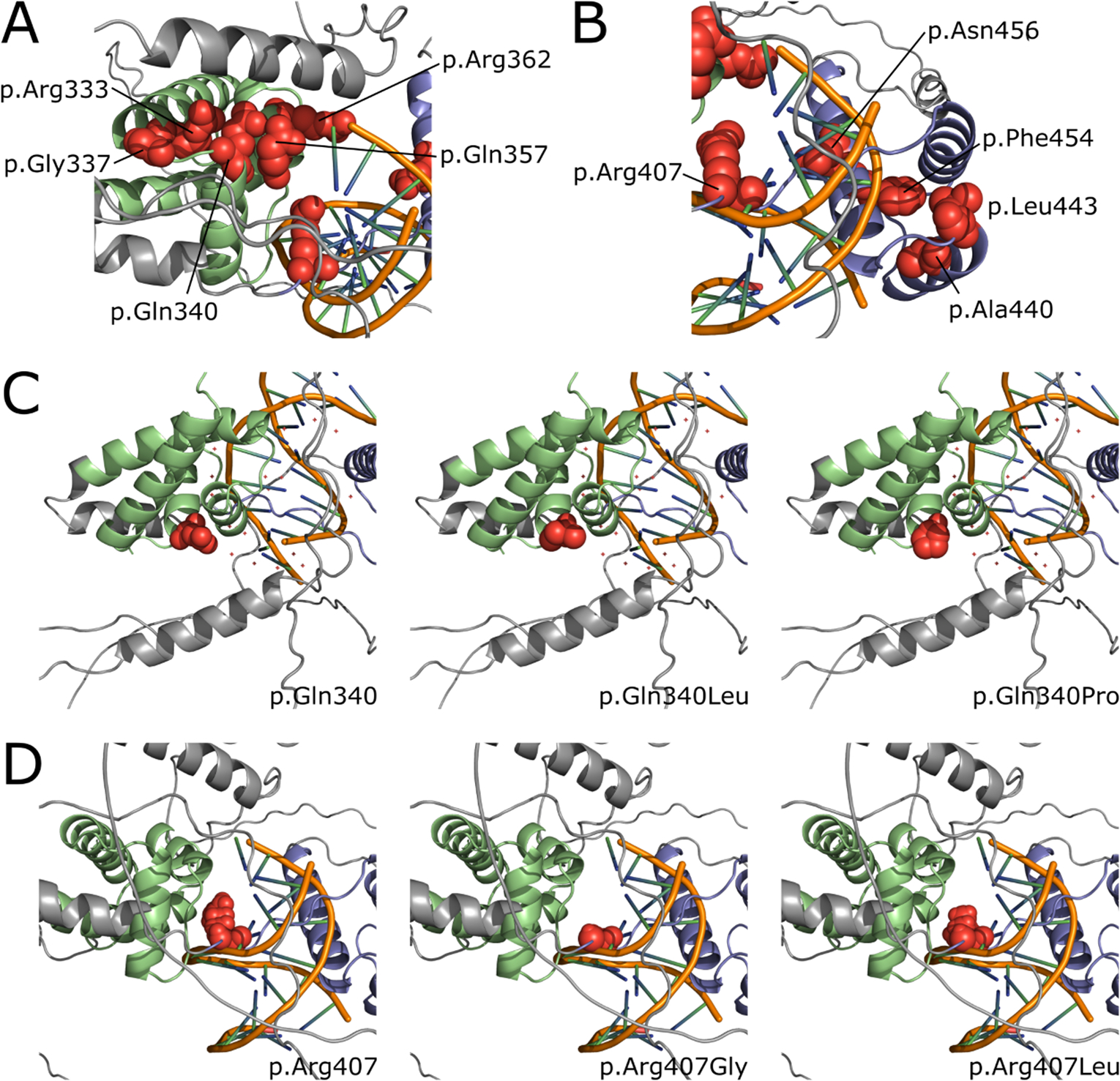
A-B. Representative three dimensional overview of the two POU3F3 domains with amino acid positions (highlighted as spheres in red) affected by disease-associated missense variants within the cohort and literature (based on AlphaFold structure aligned with DNA molecule). Panel A represents the POU-S domain in green and panel B the POU-H domain in blue with the DNA molecule in orange in the middle. The rest of the POU3F3 protein is colored in grey.
C-D. Two chosen examples of amino acid changes at positions 340 (C) and 407 (D). Left panels show unchanged amino acid structure, while each of the right panels show structural consequences of respective amino acid change.
3.5. Genotype-phenotype correlation
We merged data on all 43 individuals reported so far; 23 novel, five previously published individuals with updated information and 15 from the literature without updated information2,4. We compared the features associated with the non-truncating (in frame deletion/missense, 16 individuals) as well as truncating (27 individuals) variants. For a detailed overview see Table 1.
Table 1.
Genotype–phenotype correlations in this series and individuals of the literature.
| Non-truncating Variants | Truncating Variants | |||||
|---|---|---|---|---|---|---|
| Published | Unpublished | Total | Published | Unpublished | Total | |
|
| ||||||
| General | ||||||
| Number of individuals | 7 | 9 | 16 | 13 | 14 | 27 |
| Number of variants | 6 | 9 | 15 | 12 | 10 | 22 |
| M/F | 2/5 | 6/3 | 8/8 | 10/3 | 7/7 | 17/10 |
| Median age (y) | 11.5 y (range 1.9–20 y) |
6 y (range 0.8–39 y) |
8 y (range 0.8–39 y) |
10 y (range 2.2–41 y) |
7.8 y (n.11) (range 4–29 y) |
8.9 y (range 2.2–41 y) |
| Development | ||||||
| DD and/or ID | 7/7 | 8/9 | 15/16 (93.7%) | 12/13 | 14/14 | 26/27 (96.3%) |
| Borderline or mild | 2/7 | 5/9 | 7/16 (43.7%) | 5/13 | 7/14 | 12/27 (44.4%) |
| Moderate | 3/7 | 2/9 | 5/16 (31.2%) | 6/13 | 6/14 | 12/27 (44.4%) |
| Severe | 2/7 | 0/9 | 2/16 (12.5%) | 1/13 | 1/14 | 2/27 (7.4%) |
| Profound | 0/7 | 1/9 | 1/16 (6.2%) | 0/13 | 0/14 | 0/27 (0%) |
| Unable to classify | 0/7 | 1/9 | 1/16 (6.3%) | 1/13 | 0/14 | 1/27 (3.7%) |
| Speech delay/impairment | 7/7 | 8/9 | 15/16 (93.7%%) | 13/13 | 14/14 | 27/27 (100%) |
| Unable to classify | 0/7 | 1/9 | 1/16 (6.3%) | 0/13 | 0/14 | 0/27 (0%) |
| Behaviour | ||||||
| Autism or autistic features | 3/7 | 3/9 | 6/16 (37.5%) | 7/13 | 8/14 | 15/27 (55.5%) |
| Unable to classify | 0/7 | 1/9 | 1/16 (6.2%) | 0/13 | 1/14 | 1/27 (3.7%) |
| Other behavioral/psychiatric disturbances | 3/7 | 5/9 | 8/16 (50.0%) | 7/13 | 5/14 | 12/27 (44.4%) |
| Unable to classify | 0/7 | 2/9 | 2/16 (12.5%) | 0/13 | 0/14 | 0/27 (0%) |
| Neurological | ||||||
| Epilepsy | 3/7 | 1/9 | 4/16 (25.0%) | 0/13 | 1/14 | 1/27 (3.7%) |
| Ataxia or poor balance | 2/7 | 2/9 | 4/16 (25.0%) | 3/13 | 1/14 | 4/27 (14.8%) |
| Sleep problems | 2/7 | 2/9 | 4/16 (25.0%) | 4/13 | 5/14 | 9/27 (33.3%) |
| Unable to classify | 1/7 | 2/9 | 3/16 (18.7%) | 4/13 | 1/14 | 5/27 (18.5%) |
| Imaging | ||||||
| Abnormal brain MRI | 3/7 | 1/9 | 4/16 (25.0%) | 4/13 | 1/14 | 5/27 (18.5%) |
| Normal | 2/7 | 2/9 | 4/16 (25.0%) | 3/13 | 5/14 | 8/27 (29.6%) |
| Not performed | 2/7 | 4/9 | 6/16 (37.5%) | 6/13 | 7/14 | 13/27 (48.1%) |
| Unable to evaluate | 0/7 | 2/9 | 2/16 (12.5%) | 0/13 | 1/12 | 1/27 (3.7%) |
| Facial features | ||||||
| Cupped and/or low-set ears | 4/7 | 7/9 | 11/16 (68.7%) | 13/13 | 11/14 | 24/27 (88.9%) |
| Full lips | 3/7 | 2/9 | 5/16 (31.2%) | 6/13 | 3/14 | 9/27 (30.0%) |
| Full nasal tip | 2/7 | 2/9 | 4/16 (25.0%) | 1/13 | 4/14 | 5/27 (18.5%) |
| Smooth philtrum | 1/7 | 1/9 | 2/16 (12.5%) | 1/13 | 1/14 | 2/27 (18.5%) |
| Hypertelorism | 3/7 | 2/9 | 5/16 (31.2%) | 0/13 | 3/14 | 3/27 (11.1%) |
| Slanted palpebral fissures | 1/7 | 3/9 | 4/16 (25.0%) | 1/13 | 3/14 | 4/27 (14.8%) |
| Epicanthus | 1/7 | 2/9 | 3/16 (18.7%) | 2/13 | 5/14 | 7/27 (25.9%) |
| Orthopedic | ||||||
| Distal skeletal anomalies | 4/7 | 2/9 | 6/16 (37.5%) | 6/13 | 7/14 | 13/27 (48.1%) |
| Sensory | ||||||
| Bilateral hearing loss (perceptive or mixed) | 0/7 | 1/9 | 1/16 (6.2%) | 2/13 | 1/14 | 3/27 (11.1%) |
| Conductive hearing loss | 0/7 | 1/9 | 1/16 (6.2%) | 1/13 | 4/14 | 5/27 (18.5%) |
| Vision problems | 2/7 | 2/9 | 4/16 (25.0%) | 8/13 | 5/14 | 13/27 (48.1%) |
| Recurrent variants | p.(Arg362Leu) | - | p.(Arg451Leufs*185) | p.Ser24* p.Pro133Hisfs*14 |
||
DD: developmental disorder; F: female; ID: intellectual disability; M: male; y: year.
The median age of individuals featuring non-truncating variants was 8.0 years; for those with truncating variant was 8.9 years. All individuals presented ID and/or mild-to-severe DD and speech impairment, except #14 that was too young to be evaluated. The ID severity of the individual with the p.Arg451Leufs*185 variant2 was unknown.
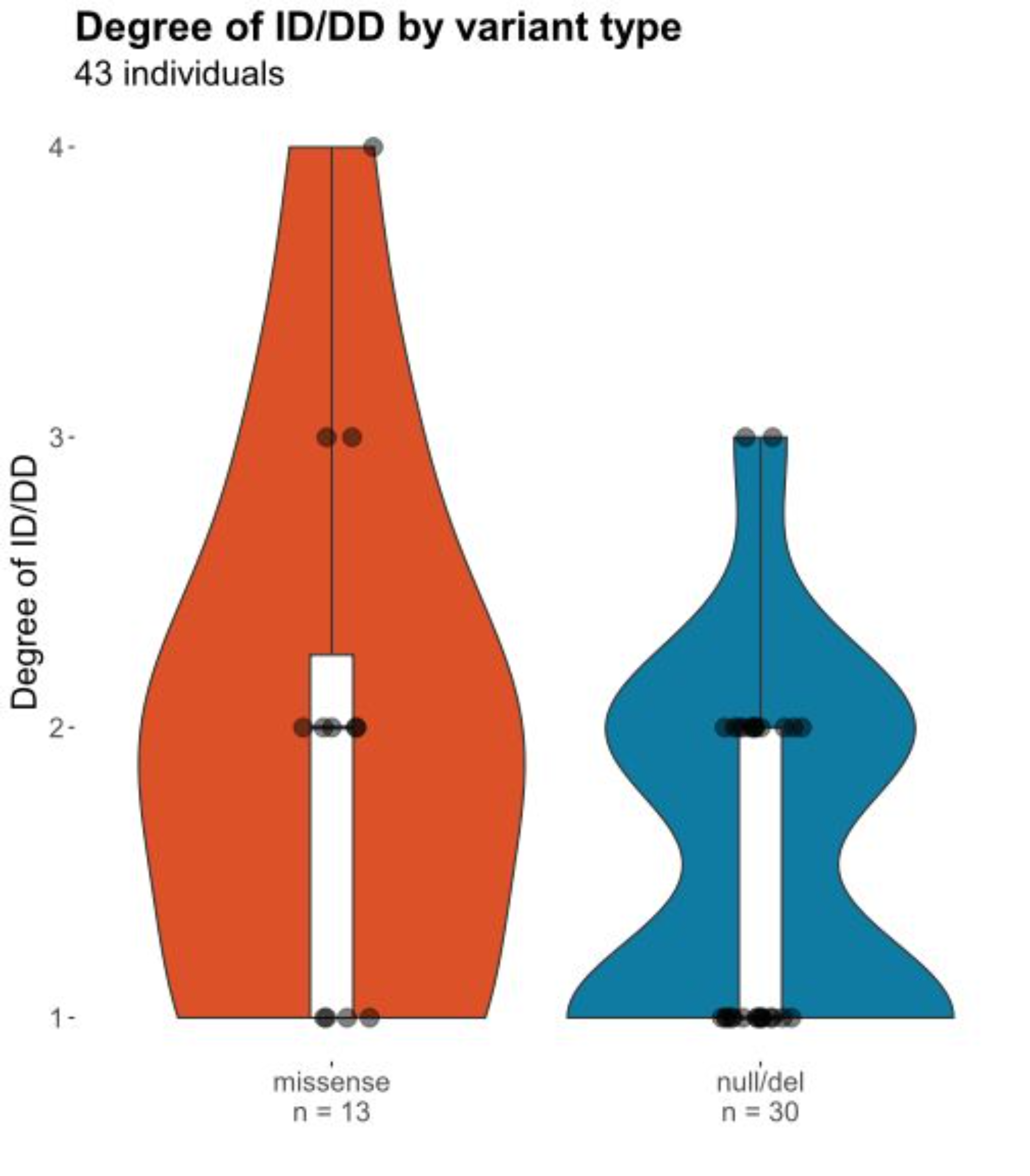
Mild ID was observed in 7/15 (46.7%) individuals with non-truncating and in 12/26 (46.2%) with truncating variants, moderate ID in 5/15 (33.3%) with truncating and in 12/26 (46.2%) individuals with non-truncating variants, severe ID in 2/15 (13.3%) with truncating and in 2/26 (7.6%) with non-truncating variants. One out of 15 (6.7%) with a missense variant showed a profound ID. See Figure S2 for a violin plot of levels DD/ID2,4.
Five out of sixteen (31.2%) individuals with missense variants remained nonverbal, whereas speech was absent in only one out of sixteen (6.2%) with truncating variants. Sleep problems, behavioral and psychiatric features did not differ in the two groups (Table 1).
Epilepsy was more frequent in the non-truncating variant group, occurring in 4/16 (25%), while it was reported in only one patient amongst those with a truncating variant. Epilepsy was associated with p.Arg362Leu (2 individuals), p.Glu269*, p.Gln357Arg and p.Gln340Leu (each in a single individual), respectively.
Those with missense variants in the POU-S domain presented with the most severe phenotype, characterized by severe developmental encephalopathy with generalized or multifocal epilepsy.
Facial features such as cupped or prominent ears, eye anomalies (up/down-slanting and narrow palpebral fissures, hypertelorism, epicanthic folds, ptosis, periorbital fullness), depressed or broad nasal bridge and bulbous nasal tip overlapped between individuals carrying the truncating or non-truncating variants, as well as anomalies of the extremities, such as broad thumbs, short digits, and flat feet, and skeletal malformations, mostly deformations of the knees, feet, and spine did not differ between the two groups.
Visual disturbances were observed in 4/16 (25%) in non-truncating group versus 13/27 (48.1%) in truncating variant group. In addition, bilateral hearing loss was more frequent in this latter group [1/16 (6.2%) versus 5/27 (18.5%)).
4. Discussion
We report 23 novel individuals with POU3F3-related neurodevelopmental disorder, thereby doubling the number of previously described individuals. We also present follow-up data on five previously reported individuals2,4. Overall, the main phenotypic features include neurodevelopmental disorders such as developmental delay, cognitive impairment, behavioral problems, dysmorphic features, psychiatric comorbidities, and seizures. The most prevalent dysmorphic feature were the cupped and often low-set ears. Our study showed that developmental delay could range from a normal or borderline-normal development to profoundly disabled individuals that are nonverbal and non-ambulant. No difference was observed in the degree of DD/ID when comparing participants with truncating and non-truncating variants. Although the number of adults in was low (5/28; 17.8%), no regression or worsening of symptoms was reported in adolescence or adulthood. We found no apparent evolution of epilepsy, motor, behavioral and intellectual impairment when comparing symptoms in childhood and adolescence to those reported in adults. Assessment of more adults with POU3F3 variants will enable us to better delineate the natural history especially in adolescence and adulthood.
Epilepsy in POU3F3-related disorders starts in early childhood but seems to be an uncommon feature so far only reported in five individuals. All presented with generalized epilepsy, i.e. myoclonic astatic epilepsy (individual #10), with DEE both atonic and tonic seizures (#17), and LGS (in three previously reported individuals)2,4. One of the individuals with LGS was not included in our analysis because of lack of information. LGS seems to be a recurrent POU3F3-related epilepsy syndrome but more individuals are needed to support this observation. Sleep disturbances, such as disorders in initiating and maintaining sleep, sleep-wake transition disorders and disorders of excessive somnolence, were prevalent features. This was in agreement with previous reports2,4.
To investigate genotype-phenotype correlations, the comparison between individuals with non-truncating and truncating variants was extended also to individuals reported in the literature2,4. The distinctive genotype-phenotype differences were predominance of autistic features, conductive hearing loss and vision problems, and cupped and/or low-set ears in individuals featuring truncating variants, whereas epilepsy was described in individuals with non-truncating variants in the POU-H and POU-S domain, except for one individual (Table 1). In a recent genome-wide meta-analysis of over 29.000 people with epilepsy24, the investigation of genetic generalized epilepsy uncovered a total of 25 independent genome-wide significant signals across 22 loci, including POU3F3. Generalized seizures, not responding to anti-seizure medications, seem to be the prevalent seizure type. More individuals and further studies are needed to further clarify the role of POU3F3 in epilepsy and to understand the mechanisms underlying POU3F3-related epileptic disorders. Our data suggest that epilepsy is more prevalent in individuals with missense variants, however, we are currently unable to explain this observation. We speculate if this observed clinical difference may be linked to a different functional effects of loss-of-function versus missense variants. While the loss-of-function variants are expected to cause symptoms due to haploinsufficiency, the missense variants may perhaps act though a gain of function mechanism, however future functional tests are needed to prove this hypothesis. A better understanding of the underlying pathophysiology, alongside functional testing of missense variants in affected individuals and in vitro drug trials might provide useful information for developing targeted approaches or for drug repurposing.
In this study, nonsense and frameshift variants, predicted to truncate POU3F3, were common. For most genes, the post-transcriptional surveillance system of nonsense-mediated mRNA decay (NMD) aids to prevent translation of abnormal truncated versions of proteins25. In mammalian cells this surveillance system is closely related to pre-mRNA splicing26. POU3F3 is an intronless gene, therefore abnormal transcripts with truncating variants are insensitive to NMD, thus they will be expressed. As already observed by Snijders Blok et al.2, truncating variants were distributed widely across POU3F3and are consequently expected to produce truncated proteins of different sizes.
Ten individuals in our cohort carried missense variants that were all either in the POU-S domain or in the POU-H domain (Figure 3A). Analysis of spatial proximity along with reported variant locations showed significant clustering of missense variants within two clusters with nearly complete overlap with these domains. Both functional domains, connected via a flexible linker, are required for site-specific DNA binding with high affinity2. For all missense variants and the in-frame deletions, highly conserved residues are affected (Figure 3C). We complemented the evaluation of missense variants with in silico structural protein modeling. The manual evaluation of variant location reveals that most of the affected amino acids are within β-sheet motifs in close proximity to the interacting DNA (Figure 4A–B). Detailed review of every distinct amino acid indicates that changes in size and charge may disturb the binding of the DNA molecule, even though further data will be needed to predict specific variant effects regarding functional changes for the interaction with the DNA molecule.
In conclusion, POU3F3-related disorder should be considered in individuals with mild-to-severe DD or ID, language delay and/or speech disorders, combined with psychiatric comorbidities and dysmorphic features. POU3F3 should be included in commercial gene panels for individuals with DD/ID and epilepsy. A small proportion of individuals may even have normal or borderline normal development and pathogenic variants can be inherited from affected parents. Our data suggest, that seizures and absence of speech is more frequently observed in the non-truncating group while ophthalmological comorbidities and hearing loss is more prevalent in individuals with truncating variants. However, more affected individuals are needed to support such findings. Finally, our study suggests a minimum surveillance and follow-up protocol for individuals with POU3F3-related disorders, such as: i) to monitor nutritional status and signs of ongoing feeding problems, gastroesophageal reflux disease and constipation; ii) to evaluate individuals for seizures, changes in muscle tone, movement disorders, poor coordination and sleep problems; iii) to monitor developmental milestones, behavioral problems, and educational needs; and iv) to monitor for audiologic, ophthalmological and orthopedic comorbidities.
Supplementary Material
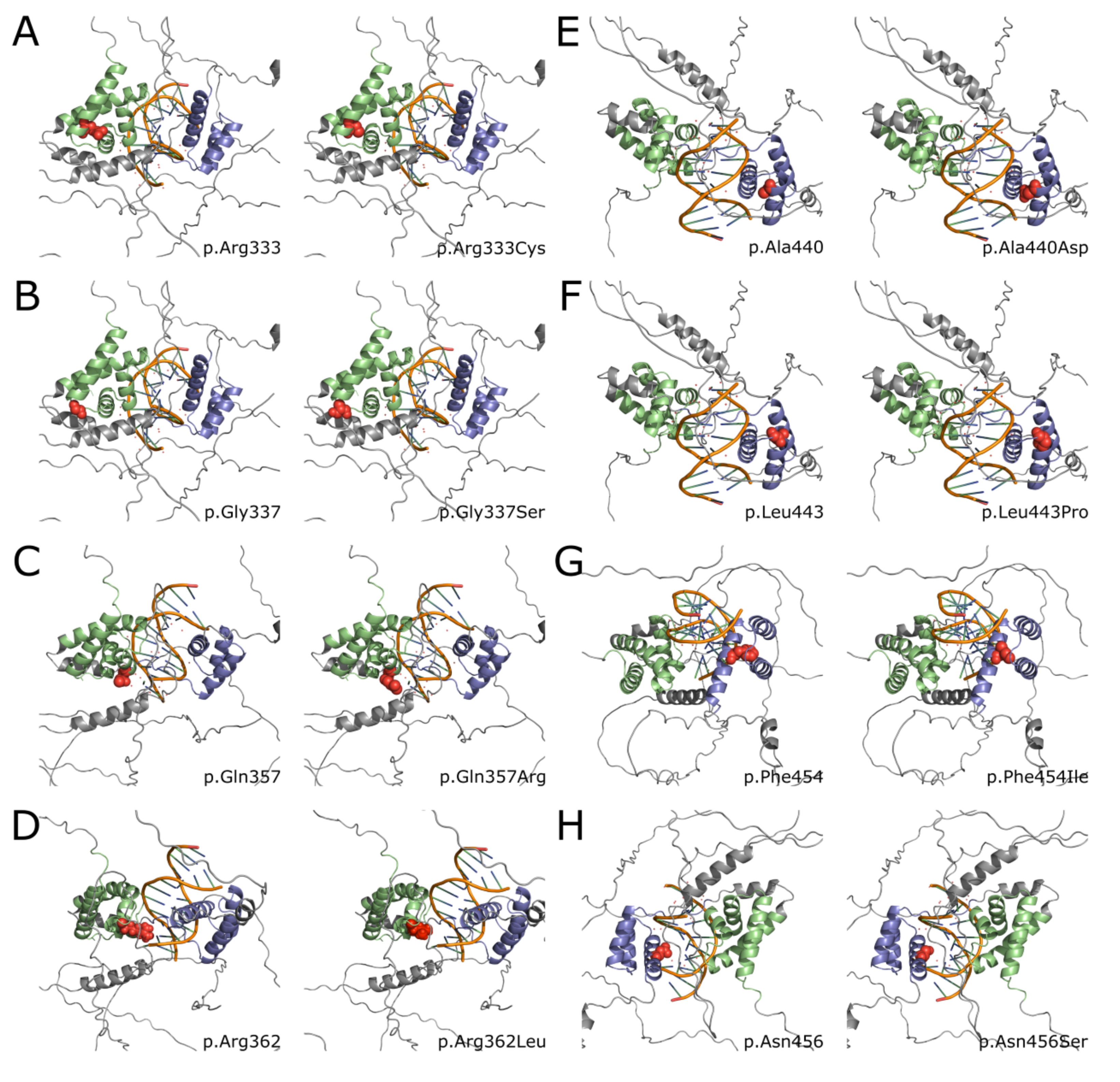
Figure S1A-H. Left panels show unchanged amino acid structure of all affected positions from the cohort. Right panels show structural consequences of respective amino acid change.
{kind=link}
Figure S2. Severity of ID/DD. Comparison of severity of ID/DD in carriers of missense/indel (red) and null (truncating) variants (blue). Degree of ID/DD 1: borderline/mild; 2: moderate; 3 severe; 4: profound.
{kind=link}
Table S1. Clinical and genetic characteristics of the POU3F3 cohort. DD: developmental disorder; F: female; FU: follow-up; ID: intellectual disability; M: male; m: month; MRI: magnetic resonance imaging; NA: not available; N: no; y: year; Y: yes.
Acknowledgments
The authors would like to thank the families for their participation.
This study makes use of data generated by the DECIPHER community. A full list of centers who contributed to the generation of the data is available from https://deciphergenomics.org/about/stats and via email from contact@deciphergenomics.org. Funding for the DECIPHER project was provided by Wellcome. This study was also, in part, generated within the European Reference Network ITHACA (NBP). This work was in part supported by the Telethon Foundation, Telethon Undiagnosed Diseases Program (TUDP, GSP15001 to Nicola Brunetti-Pierri). Siddharth Srivastava is funded by NIH-NINDS (K23NS119666). Allan Bayat is funded by a BRIDGE - Translational Excellence Program grant funded by the Novo Nordisk Foundation, grant agreement number: NNF20SA0064340.
Footnotes
Potential conflicts of interest
Nothing to report.
Ethical statement
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Data availability
All methods and data are available on request.
References
- 1.Castro DS, Skowronska-Krawczyk D, Armant O, et al. Proneural bHLH and Brn proteins coregulate a neurogenic program through cooperative binding to a conserved DNA motif. Dev Cell. 2006; 11(6):831–44. 10.1016/j.devcel.2006.10.006 [DOI] [PubMed] [Google Scholar]
- 2.Snijders Blok L, Kleefstra T, Venselaar H, et al. De Novo Variants Disturbing the Transactivation Capacity of POU3F3 Cause a Characteristic Neurodevelopmental Disorder. Am J Hum Genet. 2019;105(2):403–412. 10.1016/j.ajhg.2019.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jin W, Wang L, Zhu F, et al. Critical POU domain residues confer Oct4 uniqueness in somatic cell reprogramming. Sci Rep. 2016;6:20818. 10.1038/srep20818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Torun D, Arslan M, Yüksel Z. Coexistence of severe developmental delay, epilepsy, and hemangioma in Snijders Blok-Fisher syndrome suggests the presence of a POU3F3-related SNIBFIS endophenotype: A case report. Am J Med Genet A. 2021;185(5):1554–1560. 10.1002/ajmg.a.62135 [DOI] [PubMed] [Google Scholar]
- 5.Firth HV, Richards SM, Bevan AP, et al. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans using Ensembl Resources. Am J Hum Genet. 2009; 84:524–533. 10.1016/j.ajhg.2009.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat. 2015; 36(10):928–30. 10.1002/humu.22844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.American Psychiatric Association. (2022). Diagnostic and statistical manual of mental disorders (5th ed., text rev.). 10.1176/appi.books.9780890425596 [DOI]
- 8.Scheffer IE, Berkovic S, Capovilla G, et al. ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017; 58:512–521. 10.1111/epi.13709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fisher RS, Cross JH, French JA, et al. Operational classification of seizure types by the International League Against Epilepsy: position paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017; 58:522–530. 10.1111/epi.13670 [DOI] [PubMed] [Google Scholar]
- 10.Wirrell EC, Nabbout R, Scheffer IE, et al. Methodology for classification and definition of epilepsy syndromes with list of syndromes: Report of the ILAE Task Force on Nosology and Definitions. Epilepsia. 2022; 63(6):1333–1348. 10.1111/epi.17237 [DOI] [PubMed] [Google Scholar]
- 11.Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function Nucleic Acids Res. Nucleic Acids Res. 2003;31:3812–3814. 10.1093/nar/gkg509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu H, He X, Liu X, et al. A novel missense variant in cathepsin C gene leads to PLS in a Chinese patient: A case report and literature review. Mol Genet Genomic Med. 2021: e1686. 10.1002/mgg3.1686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rentzsch P, Schubach M, Shendure J, Kircher M. CADD-Splice-improving genome-wide variant effect prediction using deep learning-derived splice scores. Genome Med. 2021;13(1):31. 10.1186/s13073-021-00835-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–443. 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell. 2019;176:535–548 e524. 10.1016/j.cell.2018.12.015 [DOI] [PubMed] [Google Scholar]
- 16.Gelfman S, Wang Q, McSweeney KM, et al. Annotating pathogenic non-coding variants in genic regions. Nat Commun. 2017; 8:236. 10.1038/s41467-017-00141-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Desmet FO, Hamroun D, Lalande M, et al. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009; 37:e67. 10.1093/nar/gkp215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;7:405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596(7873):583–589. 10.1038/s41586-021-03819-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Varadi M, Anyango S, Deshpande M, et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022;50(D1):D439–D444. 10.1093/nar/gkab1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jauch R, Choo SH, Ng CK, Kolatkar PR. Crystal structure of the dimeric Oct6 (POU3f1) POU domain bound to palindromic MORE DNA. Proteins. 2011;79(2):674–7. 10.1002/prot.22916 [DOI] [PubMed] [Google Scholar]
- 22.Meyer MJ, Lapcevic R, Romero AE, et al. mutation3D: Cancer Gene Prediction Through Atomic Clustering of Coding Variants in the Structural Proteome. Hum Mutat. 2016;37(5):447–56. 10.1002/humu.22963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Genome Aggregation Database (gnomAD v.2.1.1),POU3F3. Accessed 3 January 2023. https://gnomad.broadinstitute.org/gene/ENSG00000198914?dataset=gnomad_r2_1
- 24.International League Against Epilepsy Consortium on Complex Epilepsies, Berkovic SF, Cavalleri GL, Koeleman BPC. Genome-wide meta-analysis of over 29,000 people with epilepsy reveals 26 loci and subtype-specific genetic architecture. MedRxiv 2022. 6 8.22276120. 10.1101/2022.06.08.22276120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holbrook JA, Neu-Yilik G, Hentze MW, Kulozik AE. Nonsense-mediated decay approaches the clinic. Nat Genet. 2004; 36:801–808. 10.1038/ng1403 [DOI] [PubMed] [Google Scholar]
- 26.Chang YF, Imam JS, Wilkinson MF. The nonsense-mediated decay RNA surveillance pathway. Annu Rev Biochem. 2007; 76:51–74. 10.1146/annurev.biochem.76.050106.093909 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1A-H. Left panels show unchanged amino acid structure of all affected positions from the cohort. Right panels show structural consequences of respective amino acid change.
Figure S2. Severity of ID/DD. Comparison of severity of ID/DD in carriers of missense/indel (red) and null (truncating) variants (blue). Degree of ID/DD 1: borderline/mild; 2: moderate; 3 severe; 4: profound.
Table S1. Clinical and genetic characteristics of the POU3F3 cohort. DD: developmental disorder; F: female; FU: follow-up; ID: intellectual disability; M: male; m: month; MRI: magnetic resonance imaging; NA: not available; N: no; y: year; Y: yes.
Data Availability Statement
All methods and data are available on request.