

Abstract
Background:
Antithrombin, protein C (PC) and protein S (PS) are circulating natural-anticoagulant proteins that regulate hemostasis and of which partial deficiencies are causes of venous thromboembolism. Previous genetic association studies involving antithrombin, PC, and PS were limited by modest sample sizes or by being restricted to candidate genes. In the setting of the Cohorts for Heart and Aging Research in Genomic Epidemiology consortium, we meta-analyzed across ancestries the results from 10 genome-wide association studies (GWAS) of plasma levels of antithrombin, PC, PS free and PS total.
Methods:
Study participants were of European and African ancestries and genotype data were imputed to TOPMed, a dense multi-ancestry reference panel. Each of 10 studies conducted a GWAS for each phenotype and summary results were meta-analyzed, stratified by ancestry. Analysis of antithrombin included 25,243 European ancestry (EA) and 2,688 African ancestry (AA) participants, PC analysis included 16,597 EA and 2,688 AA participants, PSF and PST analysis included 4,113 and 6,409 EA participants. We also conducted transcriptome-wide association analyses (TWAS) and multi-phenotype analysis to discover additional associations. Novel GWAS and TWAS findings were validated by in vitro functional experiments. Mendelian randomization was performed to assess the causal relationship between these proteins and cardiovascular outcomes.
Results:
GWAS meta-analyses identified 4 newly associated loci: 3 with antithrombin levels (GCKR, BAZ1B, and HP-TXNL4B) and 1 with PS levels (ORM1-ORM2). TWAS identified 3 newly associated genes: 1 with antithrombin level (FCGRT), 1 with PC (GOLM2), and 1 with PS (MYL7). In addition, we replicated 7 independent loci reported in previous studies. Functional experiments provided evidence for the involvement of GCKR, SNX17, and HP genes in antithrombin regulation.
Conclusions:
The use of larger sample sizes, diverse populations, and a denser imputation reference panel allowed the detection of 7 novel genomic loci associated with plasma antithrombin, PC, and PS levels.
Summary:
Using cross-ancestry GWAS and TWAS methods, we report 7 novel associations for antithrombin, PC, and PS plasma levels: 4 novel loci regulating antithrombin plasma levels, 2 novel loci regulating PS plasma levels, and 1 novel locus regulating PC plasma levels. Post-GWAS analyses and functional work suggest both SNX17 and GCKR are regulators of antithrombin on the chromosome 2 locus and validate an AA-specific HP gene locus. MR analyses provided evidence implicating low antithrombin levels in VTE risk and low PC levels in VTE and CAD risk. Overall, our findings identified novel pathways regulating the main anticoagulant proteins in hemostasis and strengthen their implication on disease outcomes.
Graphical Abstract:
INTRODUCTION
Antithrombin, protein C (PC), and protein S (PS) are circulating anticoagulant proteins, and low levels or low activity of these proteins are associated with the risk of venous thromboembolism (VTE)1–4. Genetic variants in the protein-coding genes for antithrombin, PC, and PS (SERPINC1, PROC, and PROS1, respectively)5–7 have been studied for decades, and rare mutations have been associated both with low protein levels and with risk of VTE5,8–11. There have been at least 6 agnostic genome-wide association studies (GWAS) for antithrombin, PC, and PS, with sample sizes ranging from 351 (antithrombin) to 13,968 (PC). For antithrombin, no additional genome-wide significant loci beyond SERPINC1 were identified12,13. For PC, significant loci at the GCKR and BAZ1B genes had been identified in European ancestry (EA) populations14,15, and the CELSR2-PSRC1-SORT1, PROC and PROCR loci were identified in both EA and African-ancestry (AA) populations13,15–17. For PS, no genome-wide significant associations have been found. In this report, using larger sample sizes, diverse populations, and a denser imputation reference panel, we sought to identify novel genomic loci associated with plasma antithrombin, PC, and PS levels.
METHODS
The data that support the findings of this study are available from the corresponding author upon reasonable request. GWAS summary statistics are accessible through dbGAPs.
Overview
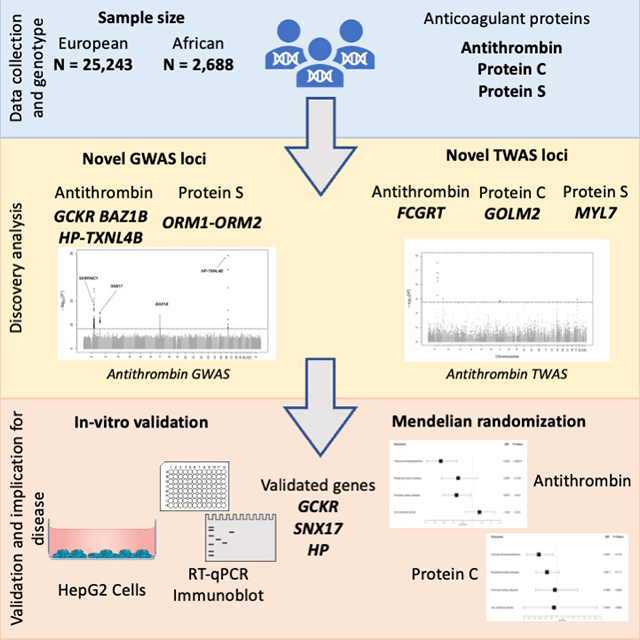
We used densely imputed genotypes to perform cross-ancestry (antithrombin and PC) and EA-only (PS) GWAS meta-analyses and attempted replication of the lead variants using available summary data from a proteomics-based study18. This was followed by a multi-phenotype analysis and transcriptome-wide association analyses (TWAS) in EA individuals. For characterization and prioritization of genes, we used colocalization and fine-mapping analyses, and novel GWAS findings were functionally interrogated. Last, we conducted Mendelian randomization (MR) analyses to assess causal relationships with cardiovascular clinical events. Figure 1 is a schematic summarizing our approach.
Figure 1.

Schematic view of the analysis’s workflow.
Study Design and Participating Studies
The setting for the meta-analysis was the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium Hemostasis Working Group19. We included data from 10 studies from the US and Europe that measured 1 or more of the 3 natural anticoagulants in plasma, by antigen or activity methods. Study details including genotype and phenotype measurement, study design, population, and baseline time are found in Supplementary Tables S1, S2, and Supplementary Materials13,15,20–32. In total, 27,606 EA and 2,688 AA participants were included: 25,243 EA and 2,688 AA participants for antithrombin; 16,597 EA and 2,688 AA participants for PC; and 4,113 and 6,409 EA participants for PS. There were no AA participants with PS measures and too few non-EA/non-AA individuals with other measurements to perform meaningful association analyses. All studies were approved by appropriate research ethics committees and all participants provided informed consent.
Discovery Analysis
Study-Specific Genome-Wide Association Analyses
Each study imputed measured genotypes to the Trans-Omic for Precision Medicine (TOPMed) reference panel before association analyses33. Study-specific quality control was implemented before the analysis. Details about genotyping platforms and specific quality control parameters can be found in Supplementary Table S2. Each study followed a common analysis plan that required performing linear regression within each ancestry group, adjusting for sex, age, principal components, and study-specific variables, which included a kinship matrix when necessary to account for family structure. Details of the measures of the 3 natural anticoagulants and on regression methods can be found in Supplementary Table S2, and Supplementary Methods.
Population-Specific and Cross-Ancestry Meta-Analysis
Quality control across studies was conducted using EasyQC34. Details of meta-analysis quality control can be found in Supplementary Materials. We meta-analyzed study-level summary results using METAL software, first by phenotype measure (antigen or activity), then by ancestry. Only variants appearing in at least 2 studies were retained in the final meta-analyses. Cross-ancestry meta-analyses were conducted on those phenotypes that included EA and AA participants (antithrombin and PC). No other ancestries groups were available. Meta-analyses were performed by 2 analysts in parallel and compared for consistency.
The significance threshold35 was set at 5 × 10-9. A locus was defined as 1 Mb upstream and downstream of the variant with the lowest p-value. Genome-wide significant variants with MAF < 1%, present in 2 studies or fewer, or with inconsistent beta directions between studies were not considered.
Conditional Analysis
We performed approximate conditional and joint analyses for all variants with MAF > 1% using summary statistics from ancestry-specific meta-analyses (see Supplementary Methods).
Replication
We sought for replication of associations for the identified lead variants in an external dataset, using available summary data from DeCODE Genetics (see Supplementary Methods).
Transcriptome-Wide Association Analyses
We used GWAS results and S-PrediXcan and S-MultiXcan36,37 to perform transcriptome-wide analyses for each phenotype within the EA populations in order to infer significant associations between the cis component of gene expression and the phenotypes. See detailed methods in Supplementary Methods.
Multi-Phenotype Meta-Analysis
We jointly analyzed the 4 meta-analyses results (cross-ancestry meta-analyses for antithrombin and PC, and the 2 EA PS meta-analyses) using a multi-phenotype method implemented in the metaUSAT R package 1.1738. Significant multi-phenotype associations were defined as any genome-wide significant lead variants in the multivariate analysis (p-valuesmultivariate for the lead variant < 5 × 10−9), that were also nominally significant in a least 2 of the phenotypes individually (p-valueunivariate < 0.005)39. Additionally, we considered novel variants to be those that were not genome-wide significant for any of the 4 phenotypes individually, or that had not been associated with antithrombin, PC, PS free or total in a previous GWAS for antithrombin, PC, PS free or total. Lead variants for each phenotype found in the discovery (Table 1) were queried using the HaploR R package v4.0.6 to extract functional annotations and biological information (Supplementary Table S4). Further details are reported in Supplementary Methods.
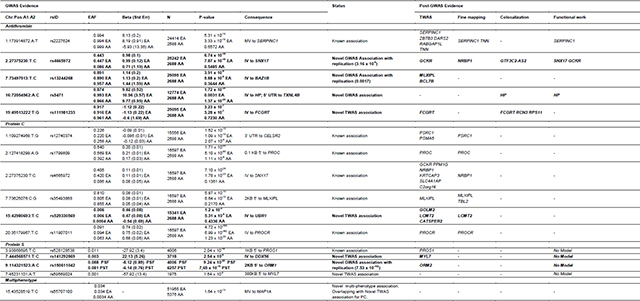
Table 1.
GWAS and post-GWAS evidence of candidate genes. A1: Effect Allele; A2: Other Allele; AA: African ancestry; EA: European ancestry; IV: Intronic Variant; MV: missense variant; 3’UTR: 3 Prime Untranslated Region; 5’UTR: 5 Prime Untranslated Region; PST: protein S total; PSF: protein S free.
|
Characterization and Prioritization of Candidate Loci
Fine-Mapping and Colocalization
In all GWAS associated loci, we performed fine- mapping using FOCUS, and colocalization using COLOC and HyPrColoc. Detailed methods for fine-mapping and colocalization can be found in the Supplementary Methods.
Gene Prioritization
For all genes within 1 Mb of the lead variant from novel GWAS associated loci, we selected the closest gene to the lead variant, genes that had a TWAS significant p-value on the prioritized tissues, genes that had fine-mapping posterior inclusion probability (PIP) equal or higher than 0.95, genes that had a conditional probability of colocalization (CPC) equal or higher than 0.8, and genes with relevant functional annotations in HaploReg. Additionally, we selected all TWAS significant genes in the 5 prioritized tissues. Finally, to qualify for in vitro functional validation, prioritized genes were additionally required to be expressed in liver (>10 transcripts per million in GTeX) and in HepG2 cells (>10nTPM, Human Protein Atlas). Figure 2 shows gene prioritization steps, and Supplementary Table S5 shows the specific genes selected for each phenotype.
Figure 2.

Prioritization steps of functional analysis. GWAS: Genome wide association study; TWAS: Transcriptome wide association study; PIP: Posterior inclusion probability; CPC: Conditional probability of colocalization; TPM: Transcripts per million.
In vitro Functional Validation
Functional validation of prioritized candidates was performed by in vitro silencing of candidate genes in a liver-derived hepatoblastoma (HepG2) cell expression system. Briefly, HepG2 cells were reverse-transfected with small interfering RNA (siRNA) against candidate genes. Cells were counted, and target proteins and genes were characterized by immunoblot of cell supernatants and RT-qPCR, respectively. Details on cell culture, transfection, RNA extraction, RT-qPCR, and immunoblotting methods can be found in Supplementary Methods.
Mendelian Randomization
Two-sample summary statistics-based MR was used to assess the association of genetically determined levels of antithrombin and PC with the risk of venous thromboembolism (VTE; 42,078 cases and 424,073 controls)40,41, peripheral artery disease (PAD; 31,307 cases and 211,753 controls )42, coronary artery disease (CAD; 60,801 cases and 123,504 controls)43, and ischemic stroke (IS; 60,341 cases and 454,450 controls)44. We used instrumental variants from our GWAS results as main analyses. Additional details on methods are reported on Supplementary Methods. Given the small proportion of variance explained by the identified PS variants we did not investigate PS (PSfree and PStotal) in MR analyses because of insufficient number of genetic instruments. We then additionally validated our analyses using weights derived from deCODE genetics data18.
RESULTS
Antithrombin activity (% or IU/mL*100, n = 26,999) or antigen (IU/mL*100; n = 932) was measured in 9 studies, PC activity (% or IU/mL*100; n = 6,734) or antigen (μg/mL; n = 12,551) was measured in 8 studies, PS Total (PStotal) activity (% or IU/mL*100 or IU/mL; n = 5,045 ) or antigen (IU/mL or μg/dL; n = 1,363) was measured in 7 studies, and PS Free (PSfree) activity (μg/dL or %; n = 1,998) or antigen (IU/mL*100; n = 2,115) was measured in 6 studies. See Supplementary Table S6.
Antithrombin
GWAS:
The antithrombin meta-analysis include 25,243 EA and 2,688 AA participants. After quality control and filtering, 80,168,840 variants remained in the meta-analysis. All λGC for individual GWAS were 1.04 or below for all chromosomes. Additional details about quality control are provided in Supplementary Table S7 and Supplementary Material. Manhattan plots for the overall cross-ancestry meta-analyses are shown in Figure 3. A quantile-to-quantile plot (QQ plot) of p-values for these variants is presented in Supplementary Figure S1 and Manhattan plots for the EA and AA population specific analyses are available at Supplementary Figure S2.
Figure 3.

Manhattan plots for discovery meta-analyses of GWAS (up) and TWAS (down) results. (A) Antithrombin (B) Protein C (C) Protein S Free (D) Protein S Total. Dots represent all allelic variants (GWAS) or genes (TWAS) sorted by chromosome and position throughout the X-axis.
Y-axis report inverse log transformed p-value for the associations.
In total, variants in 4 loci, exceeded the established genome-wide significance level in the cross-ancestry analysis, 2 loci in the EA-specific analysis and 1 locus in the AA-specific analysis. Forest plots for significant variants can be found in Supplementary Figure S3. Loci at SNX17-GCKR-NRBP1 (2p23.3), MLXIPL-BAZ1B-BCL7B (7q11.23) and HP-TXNL4B (16q22.2) were new associations. The association at HP-TXNL4B (16q22.2) was only found in the AA population. Lead variants in the cross-ancestry meta-analysis in each region are listed in Table 1 along with the meta-analysis p-value, ancestry specific p-value, effect allele frequency (EAF), beta estimates, and closest gene.
No significant heterogeneity was found in the direction or magnitude of beta coefficients for any of the lead variants associated with antithrombin, within or between ancestries. Conditional analyses using the population specific meta-analyses (Supplementary Table S8), identified no additional independent variants on SNX17-GCKR-NRBP1 and HP-TXNL4B surrounding regions. On chromosome 1 locus (SERPINC1), we found 1 variant (rs182221508, MAF = 0.0017) intronic to RABGAP1L gene (600 kb upstream the lead variant), that was independent from the lead missense variant rs2227624 on SERPINC1 gene.
Supplementary Table S9 shows the lead variants with the strongest associations in the EA and AA meta-analyses. There was 1 significant locus in the AA population specific analysis at chromosomal position 16q22.2 (HP-TXNL4B), which also appeared in cross-ancestry analysis. In the EA-specific population analysis, the results reflected cross-ancestry findings at 1q25.1 (SERPINC1) and 2p23.3 (SNX17-GCKR-NRBP1), with a different lead variant on chromosome 2: rs4665972, located in an intronic region of SNX17, was the lead variant in the cross-ancestry analysis, while rs11127048,150 kb upstream rs4665972 and located in an intergenic region between SNX17 and GCKR genes was the lead variant in the EA-specific analysis. We did not find significant signals at 7q11.23 in the EA-specific analysis. The proportion of variance explained by the independent lead variants was 1.4% in EA and 4.3% in AA, of the total antithrombin variance.
All lead variants from GWAS were replicated in the deCODE summary results derived from SOMAscan measures of these anticoagulants (all p-values <0.05/11 = 4.5×10−3), except for the lead variant of the chromosome 16 locus, that was specific for the AA population and was not present in the DeCODE data (Table 1 and Supplementary Table S10).
TWAS:
TWAS analyses identified associated genes in 4 different loci (Figure 3A). Associations on chromosomes 1 (SERPINC1), 2 (GKCR) and 7 (MLXIPL), identified by the strongest associated gene in the TWAS, matched associated loci found in the GWAS. Additionally, the FCGRT gene represented a new association on chromosome 19. The smallest GWAS p-value for this region approached significance and was for a rare intronic variant (rs111981233) in FCGRT gene (Figure 3 and Supplementary Table S11) that was replicated in the DeCODE study (Table 1 and Supplementary Table S10).
Fine Mapping:
EA-specific fine-mapping results prioritized the SERPINC1 gene on chromosome 1 and the NRBP1 gene on chromosome 2. Given that FOCUS only prioritizes GWAS hits at TWAS risk loci, loci on chromosomes 16 (only GWAS) or 19 (only TWAS) could not be further explored for gene prioritization. In addition, after correcting for LD and pleiotropic effects, none of genes in chromosome 7 locus was included in the credible set, suggesting a regulation mechanism that does not involve gene expression (Supplementary Table S12).
Colocalization:
We obtained 2 significant colocalizations in lead variants located in the new antithrombin loci (Conditional Probability of Colocalization (CPC) > 0.8) and gene expression of nearby genes. On chromosome 2, GTF3C2-AS2 (at SNX17-GCKR-NRBP1 locus) gene expression in artery tibial tissue colocalized with antithrombin plasma levels and on chromosome 16 locus, HP gene expression in liver and whole blood also colocalized with antithrombin plasma regulation (Supplementary Table S13).
Functional Validation:
We selected 1–3 genes per locus for functional analysis (4 genes total) based on the gene prioritization criteria described above (Figure 2, Supplementary Table S5): SNX17, GCKR, NRBP1 (Chr 2), and HP (Chr 16). Genes in the newly associated locus on chromosome 7, and the gene associated in TWAS on chromosome 19 were not included because no expression was detected in HepG2 cells for of the selected genes (BAZ1B, BCL7B, FCGRT) or for biological implausibility (MLXIPL). We transfected HepG2 cells with siRNA against each candidate gene and confirmed that target gene expression was significantly reduced (p-value<0.005). We then characterized effects of the gene knockdowns on cell count; NRBP1 silencing significantly reduced the cell count and was removed from the screen (Supplementary Figure 4). Finally, we quantified antithrombin expression by immunoblot of cell supernatants and SERPINC1 expression by RT-qPCR. As expected, control experiments showed that treatment of HepG2 cells with lipofectamine (alone) or siRNA against PROC did not significantly alter antithrombin (protein) or SERPINC1 (gene) expression, whereas silencing SERPINC1 significantly suppressed antithrombin and SERPINC1 expression (Figure 4A–B). Quantification of immunoblots revealed that silencing GCKR enhanced, whereas silencing SNX17 and HP suppressed, antithrombin protein production (Figure 4A). The GCKR-dependent increase in antithrombin was associated with a significant increase in SERPINC1 expression, suggesting GCKR negatively regulates antithrombin gene expression (Figure 4B). The SNX17-dependent loss of antithrombin was associated with a trend toward decreased SERPINC1 expression (p-value<0.09), suggesting SNX17 positively regulates antithrombin gene expression (Figure 4B). Interestingly, HP-dependent loss of antithrombin was not accompanied by a significant decrease in SERPINC1 expression (Figure 4B) suggesting effects of HP on antithrombin production are manifested via a post-transcriptional mechanism.
Figure 4.

Knockdown of GCKR, SNX17, and HP alter antithrombin production in HepG2 cells. A) Antithrombin secreted into the culture supernatant was detected by immunoblot and quantified by densitometry. B) SERPINC1 expression and C) PROC expression were measured by RT-qPCR using the ΔΔCt method to compare mRNA abundance and 18S as the reference gene. Bars and error bars indicate mean and standard error of the mean; Numbers indicate biological replicates; Statistical comparisons were performed by one-way ANOVA and Šidák’s multiple comparisons tests.
MR analysis:
For the main analyses, we used 4 genetic instruments derived from our GWAS data (Supplementary Table S14) to investigate the association between antithrombin levels and VTE and PAD, and 3 to investigate its association with CAD and IS. We detected a significant deleterious effect of genetically determined low antithrombin levels and risk of VTE (IVW OR 0.84 [0.72–0.97], p-value: 0.015; Figure 5A). Sensitivity analyses showed consistent effect in size and direction with MR Egger, MR weighted median, and MR weighted mode (Supplementary Table S15 and Supplementary Figure S5). Leave-one-out sensitivity analyses showed homogeneity of effects among the instruments. No significant results were found for the association of genetically determined antithrombin levels with IS, CAD or PAD (Figure 5A and Supplementary Figure S5).
Figure 5.

Forest plot showing inverse variance weighted mendelian randomization results for multiple outcomes using antithrombin (A) and protein C (B) as exposure. Squares indicate OR (95% CI).
Additional analyses using weights derived from deCODE genetics data confirmed the same trends although with non-significant results due to weaker instruments (Supplementary Table S15).
Protein C
GWAS:
The PC meta-analysis included 16,597 EA and 2,688 AA participants. After quality control, 72,929,079 variants were included. All λGC for individual GWAS were 1.04 or below for autosomal chromosomes (1.18 for X chromosome). Additional details about quality control are provided in Supplementary Table S7 and Supplementary Material.
Manhattan and QQ-plots showing the cross-ancestry meta-analysis results are presented in Figure 3 and Supplementary Figure S1, respectively. We identified 5 regions associated with PC levels. All loci, located near CELSR2-PRSC1 (1p13.3), PROC (2q14.3), SNX17-GCKR-NRBP1 (2p23.3), MLXIPL-TBL2 (7q11.23), and PROCR (20q11.22) genes, have been previously reported to be associated with PC. Coefficients, p-values, ancestry stratified EAF and p-values, and closest genes are listed in Table 1. Forest plots of significant signals found in the GWAS analysis can be found at Supplementary Figure S6.
In the conditional analysis at 1p13.3 (CELSR2-PRSC1), 2p23.3 (SNX17-GCKR-NRBP1), and 7q11.23 (MLXIPL-TBL2) loci in the EA population, no additional independent variants were identified (Supplementary Table S8). Within the PROC locus on chromosome 2, an additional independent variant (rs74392719, MAF = 0.01, 300 bases upstream of the lead variant) was identified in the EA population, located within the PROC gene. Finally, an additional independent variant (rs6060300, MAF = 0.2, 13 kb upstream of the lead variant) was found in the EA population, intronic to PROCR.
No significant heterogeneity was found in the direction or magnitude of beta coefficients for any of the lead variants associated with PC, within or between ancestries. AA and EA population-specific results are shown in Supplementary Table S9 and Supplementary Figure S2. The AA population analysis had findings at 2q14.3 (PROC) and 20q11.22 (PROCR); the EA population analysis recapitulated all the candidate loci found in cross-ancestry analysis. The proportion of variance explained by the identified independent variants was 12.7 % in EA and 7.4% in AA. The lead variants at the PROCR locus (rs11907011 and rs867186) alone explain 9.5% and 9% of the total variance in the EA and AA meta-analyses, respectively. All lead variants from GWAS were replicated in the deCODE data (all p-values <0.05/11 = 4.5×10−3; Table 1 and Supplementary Table S10).
TWAS:
For PC levels, TWAS (Figure 3B) identified associated genes at 6 loci, matching all loci found in the cross-ancestry and EA GWAS, of which, the most significant based on TWAS z-score values were PSRC1 (chromosome 1, CELSR2-PRSC1 locus), GCKR (chromosome 2, SNX17-GCKR-NRBP1 locus), PROC (chromosome 2), MLXIPL (chromosome 7, MLXIPL-TBL2 locus) and PROCR (chromosome 20). Additionally, 3 new associations with PC were found in 1 locus on chromosome 15 for GOLM2, LCMT2 and CATSPER2 genes (Table 1 and Supplementary Table S11).
Fine Mapping:
Fine-mapping results for PC prioritized the PSRC1 gene on chromosome 1, NRBP1 and PROC on chromosome 2 (SNX17-GCKR-NRBP1 and CELSR2-PRSC1 locus, respectively), MLXIPL and TBL2 on chromosome 7 (MLXIPL-TBL2 locus), and PROCR on chromosome 20. (Supplementary Table S12).
Functional Validation:
Following the prioritization criteria described on Figure 2, a novel gene identified in the TWAS analysis (GOLM2) was selected as a candidate gene for functional validation. Transfection of HepG2 cells with siRNA against the GOLM2 gene did not alter the cell count (Supplementary Figure S4). PC could not be detected in the media; however, we quantified effects of GOLM2 silencing on PROC RNA expression by RT-qPCR. As expected, control experiments showed that treatment of HepG2 cells with lipofectamine (alone) or siRNA against SERPINC1 did not significantly alter PROC (gene) expression, whereas silencing PROC significantly suppressed PROC expression (Figure 4C). However, silencing GOLM2 did not significantly alter PROC expression (Figure 4C).
MR analysis:
For PC, 4 variants were initially selected as genetic instruments for the main analyses, using GWAS summary results (Supplementary Table S14). After examination of pleiotropic effects, the variant at the PROCR gene (rs1799809) was excluded to avoid violations of MR assumptions. Moreover, additional evidence indicates that this variant is strongly associated with several hemostasis and thrombosis phenotypes and has opposite effect directions for venous and arterial thrombosis reflecting distinct pleiotropic biological mechanisms17,45. Details of selected genetic instruments can be found in Supplementary Table S14. There was a significant deleterious effect of genetically determined lower PC levels on VTE and CAD risk (VTE IVW OR:0.83 (0.76–0.92), p-value: < 0.001; CAD IVW OR: 0.92 (0.84–0.99), p-value: 0.031; Figure 5B). Sensitivity analyses showed consistent significant associations (Supplementary Table S15 and Supplementary Figure S5). No significant associations were found between genetically determined PC with PAD or IS (Figure 5B and Supplementary Figure S5).
Results using instruments and weights derived from deCODE genetics data replicated a causal association of lower PC on increased VTE risk but could not replicate the effect on CAD (Supplementary Table S15).
Protein S
GWAS:
The PS meta-analysis included 4,113 EA individuals in PSfree analyses and 6,408 EA individuals in PStotal analyses. A total of 19,791,246 variants were investigated in the analysis of PSfree and 25,365,467 in the analysis of PStotal. All λGC for individual GWAS were 1.04 or below for autosomal chromosomes (1.19 for X chromosome). Additional details about quality control are provided in Supplementary Table S7 and Supplementary Material. Manhattan and QQ-plots describing the main results are shown in Figures 2C/D and Supplementary Figure S1 for PSfree and PStotal, and main associated variants are listed in Table 1. Forest plots of significant signals for PSfree and PStotal can be found in Supplementary Figure S7.
We identified 1 novel genome-wide significant locus associated with PSfree and PStotal near ORM1 and ORM2 genes (9q32) and a known association located near PROS1 gene (3q11.1) for PSfree. The lead variant at PROS1 locus (rs121918472, EA p-value = 2.04 × 10−16, PSfree EAF (G) = 0.0108) was a missense variant located in the protein S coding gene PROS1. In our analysis, this variant was associated with PSfree level, but genome-wide significance was not observed in PStotal (PStotal p-value = 2 × 10−4) although there was a consistent direction of effect.
Nominally significant heterogeneity p-values were detected in the ORM1/ORM2 locus lead variant (PStotal Heterogeneity p-value = 0.03), indicating minor differences between the 2 measurement methods. No additional independent variants were found with conditional analyses (Supplementary Table S8). The variance explained by the identified variants in PSfree is 6% of the total variance of PSfree while the variance explained by the unique identified variant in PStotal is 1% of the phenotypic variance for PStotal.
Variants at both loci replicated in the deCODE data (all p-values <0.05/11 = 4.5×10−3; Table 1 and Supplementary Table S10).
TWAS:
PSfree TWAS results recapitulated the 2 significant GWAS associations at chromosomes 3 (PROS1) and 9 (ORM2) and additionally revealed a new association at MYL7 gene on chromosome 7 (Figure 3 and Supplementary Table S11).
Fine Mapping:
Fine-mapping results did not prioritize any genes for PSfree or PStotal.
Colocalization:
There was a significant colocalization for both PS phenotypes and ORM2 gene expression in liver. (Supplementary Table S13).
Functional validation:
Since we were unable to detect PS production in the HepG2 expression system, we were unable to perform functional validation for PS candidates.
MR analysis:
Given the small proportion of variance explained by the limited number of genetic instruments (< 3), we did not investigate PS (PSfree and PStotal) in MR analyses.
Antithrombin, Protein C and S Multi-phenotype Analysis
Multi-phenotype analyses between antithrombin, PC, PSfree and PStotal revealed 1 additional novel GWAS association close to the MAP1A gene46, on chromosome 15 (Table 1), previously found in the PC TWAS (GOLM2-LCMT2-CATSPER2 locus). The lead variant is a missense variant on the MAP1A gene (rs55707100, p-value = 1.64 × 10−13, EAF EA [T] = 0.03, EAF AA [T] = 0.0042) that was nominally associated in the GWAS for antithrombin and PC individually (antithrombin p-value = 1.04 × 10−6, PC p-value = 4.76 × 10−8) and was not significantly associated to either of the PS phenotypes (PStotal p-value = 0.2717, PSfree p-value = 0.9937). The colocalization results were significant (CPC > 0.8) between antithrombin and PC, suggesting the existence of a common variant as regulator of both phenotypes. However, functional validation by silencing GOLM2 gene in this locus did not significantly alter expression of PROC or SERPINC1 in HepG2 cells (Figures 4B and 4C), suggesting any potential co-regulatory effect is not mediated in the production of these proteins.
DISCUSSION
We performed GWAS for 3 natural anticoagulant hemostasis phenotypes (antithrombin, PC, and PS [PStotal and PSfree]) using larger sample sizes and better imputation panels than previously reported and detected 4 novel associations: 3 loci for antithrombin (SNX17-GCKR-NRBP1, MLXIPL-BAZ1B-BCL7B, and HP-TXNL4B) and 1 locus for PS (ORM1-ORM2). For 3 genes within the newly associated loci with antithrombin (SNX17, GCKR, and HP), in vitro gene silencing in liver-derived cells provided functional evidence. Using TWAS methods, we detected 3 additional novel associations that did not reach significance in individual GWAS: FCGRT for antithrombin; GOLM2 for PC; and MYL7 for PS. Using MR, we also identified a causal relationship of antithrombin and PC levels with VTE, and of PC levels with CAD. This investigation elucidated genetic regulation of the anticoagulant pathway and provides new information that could identify therapeutic targets in VTE prevention or treatment.
Additionally, we replicated 7 known loci5,13,15–17 for PC; and 1 for PS7. Two of the known PC loci, also had novel associations with antithrombin, demonstrating some genetic overlap between different anticoagulant proteins. This was also reflected in the multi-phenotype analysis results.
Characterization of Novel Loci
Antithrombin-associated Loci
More than 45 rare variants within the SERPINC1 gene have already been described using non-GWAS approaches47. Our lead variant, rs2227624, is a known missense variant causing a Val to Glu amino-acid substitution that leads to antithrombin deficiency48,49 and increases risk of VTE50.
On chromosome 2, lead variants in locus SNX17-GCKR-NRBP1 differed by ancestry. In the cross-ancestry analysis, rs4665972 was in an intronic region of SNX17 whereas, in the EA-specific analysis, the lead variant (rs11127048) was located in an intergenic region between the SNX17 and GCKR genes. Neither rs4665972 nor rs11127048 were significant in AA population suggesting that these variants are tagging an association within a large LD block in EA population. Consistent with this observation, conditional results indicate that the lead variant (rs4665972) is the only independent variant on this locus. Given limited power in the AA-specific analysis, we could not refine the region with AA data (Supplementary Figure S12). Functional validation in liver-derived cells suggests that SNX17 positively, and GCKR negatively, alter plasma antithrombin levels via effects on SERPINC1 expression.
SNX17 is a regulator of low density lipoprotein (LDL) receptors51 and has not been previously associated to antithrombin levels but has been associated with CAD52,53. GCKR is a highly pleiotropic gene that has been found significantly associated to multiple phenotypes18. We and others have also reported genetic associations with several hemostatic factors, including PC14,15, Factor VII (FVII)54, Factor XI (FXI)55 and C-reactive protein (CRP)56,57 in previous GWAS meta-analyses. In previous candidate gene studies58–60, variant rs1260326 in GCKR was found to be related to multiple cardiometabolic traits, including total and LDL cholesterol, fasting plasma glucose, liver fat content and metabolic syndrome, suggesting that GCKR might act as a broad regulator of hepatocyte function.61
On chromosome 7, the lead variant (rs13244268) was located in an intronic region of BAZ1B gene and was only significant in the EA population. This gene has been previously associated with PC14,15 and in our PC meta-analysis, but not with antithrombin. rs13244268 was also found significant in bivariate and univariate GWAS of CRP and high-density lipoprotein62. TWAS results confirmed an association between BCL7B and MLXIPL genes in this locus and antithrombin levels. Given the differences in LD blocks observed for this region in different populations, we sought to confirm the most plausible candidate genes in this locus with in vitro silencing studies in liver cells. Within the 3 candidate genes in the region (BAZ1B, MLXIPL and BCL7B), BAZ1B presented low expression in liver, and MLXIPL and BCL7B were not prioritized for functional validation due to biological implausibility. Since no candidates in this region passed pre-selection for functional work, the elucidation of this locus and its role on antithrombin regulation warrants further investigation.
The lead variant on HP-TXNL4B locus (rs5471) is in an intronic region of the TXNL4B gene and 5’ UTR of the HP gene and was only significant in the AA population. Colocalization results performed using cross-ancestry data, suggested the existence of a common regulatory variant between HP gene expression and antithrombin levels in liver and whole blood, and suggested that higher expression of HP in liver and blood were associated with higher levels of antithrombin in plasma. In the same direction, functional validation in HepG2 cells suggested a significant reduction of antithrombin levels upon HP silencing through as-yet unidentified post-transcriptional mechanism. HP codes for haptoglobin (Hp), which serves as a binding protein of hemoglobin, and affects the release of hemoglobin from red blood cells63. Its phenotype Hp2–2 was identified as a potential regulator of inflammation and reverse cholesterol transportation and has been suggested to have higher prevalence in VTE patients64–66. Overall, previous evidence suggests a potential role of Hp in the inflammation-induced thrombosis, and our results suggest HP is a potential direct regulator of antithrombin production.
Finally, TWAS results suggested a novel locus associated to antithrombin levels on the FCGRT gene. Colocalization results suggested the existence of a common regulatory variant between antithrombin levels and the expression of FCGRT, RPS11 and RCN3 in the aorta, tibial artery, and whole blood. In GWAS analysis, rs111981233 (intronic to FCGRT) nearly reached genome-wide significance levels. FCGRT encodes a receptor that binds immunoglobulin G and transfers immunoglobulin G antibodies from mother to fetus across the placenta67 and previous studies demonstrate that FCGRT is also expressed in the liver68. Additional work is needed to further elucidate the role of this gene in antithrombin regulation.
Protein C-associated Loci
We found 5 loci associated with PC in the present GWAS meta-analysis, all of which had been previously described. In addition, 3 genes (GOLM2, LCMT2, and CATSPER2) were associated in a novel locus on chromosome 15 in the TWAS analysis. GOLM2, the only gene with significant expression in the liver, encodes for a transmembrane protein predicted to colocalize in the Golgi apparatus with no known function; Interestingly, a variant near this locus was significant in the PC-antithrombin multi-phenotype GWAS analysis, and colocalization results suggested the existence of a common variant between antithrombin and PC. Our functional validation suggest GOLM2 does not regulate PC transcription, although our experiments did not allow us to assess a post-transcriptional role of GOLM2 on PC, or a role in clearance.
Protein S-associated Loci
For PS, genome-wide associations found at the ORM1-ORM2 locus represented novel findings for both PSfree and PStotal, and colocalization analysis suggests the existence of a common regulatory variant between PSfree and PStotal levels and ORM2 expression in liver. ORM1 is responsible for encoding acute phase plasma protein orosomucoid (ORM, also known as α1-acid-glycoprotein, AGP), which is increased with acute inflammation69. Previous genetic results suggested that ORM1 was associated with thrombin generation potential70 and the discovery was further confirmed with in vitro experiments. ORM1 has also been associated with cell-free DNA levels in plasma, a surrogate marker of neutrophil extracellular traps that contribute to immunothrombosis71. Moreover, AGPs encoded by the ORM1 and ORM2 genes strongly bind to the vitamin K antagonist warfarin that reaches circulation, suggesting that these genes could be relevant in regulating the response to oral anticoagulation72. Supporting this hypothesis, ORM1, ORM2 and PROC were nominally associated with warfarin dose requirement in a study of candidate gene analysis with 201 patients73. This is interesting, since it is widely known that one of the challenges in oral anticoagulation is the wide variation in response among patients74. Confirming novel genomic regulators of anticoagulant response could help explain the mechanisms of action of these drugs and move towards a personalized treatment based on genomic background.
MYL7, associated with PS levels in the TWAS analyses, is the gene coding for myosin light chain 7 protein, and was previously related to calcium ion binding activity75,76. Variants in this gene have been associated with fasting glucose levels and type II diabetes77,78 probably for their proximity to the glucokinase (GCK) gene, which lies 1.9 kb upstream of MYL7, and is essential for producing glucose-6-phosphate.
Implication for disease outcomes:
Several of the identified loci have been previously associated with cardiovascular disease outcomes (Supplementary Table S16).
The present MR results confirm a causal relationship between genetically determined plasma levels of antithrombin and PC with VTE events, and for PC with CAD outcomes. Specifically, we observed a 19% VTE risk increase per 1 SD decrease in antithrombin plasma levels, a 20% VTE risk increase per 1 SD decrease of PC plasma levels, and a 9% CAD risk increase per 1 SD decrease in PC plasma levels. Our findings of a causal relationship of antithrombin and PC with VTE agree with previous epidemiological studies that report an increased VTE risk in individuals with deficiencies of these anticoagulants4,79,80. The causal relationship between PC and CAD was also reported in previous epidemiological and MR studies.81,82 Overall, these results support previous data suggesting that antithrombin and PC are relevant proteins that regulate the risk of VTE, confirmed the causal association between PC levels and CAD, and corroborated that intervention in the anticoagulant system could be considered for VTE or CAD prevention83,84.
Strengths and limitations:
A major strength of this study is in the modestly large sample size, including around 30,000 individuals, compared with more limited studies in the previous discovery efforts. Additionally, the TOPMed imputation panel, provides better imputation quality for low-frequency variants compared with previous panels, which increases our power to detect rare variation. However, the present study was not designed to provide a detailed evaluation of rare variation within coding genes, and some rare variants within these genes were excluded from the analyses if they were present in less than 2 studies. Larger studies combined with whole genome sequencing data will help identify novel rare (familial) associations for these phenotypes and may provide better instruments that will improve the power for MR studies.
Inclusion of AA individuals has allowed the identification of novel associated loci for antithrombin in this population. We were not able to perform discovery in other ancestry populations since measures of phenotypes and genotypes were not available to us. Investing in these measures in other ancestry groups may increase the number of discoveries and provide more global insights. There is a recent debate 85,86 on transferability of results from GWAS studies to non-European populations, given the overwhelming majority of GWAS results in EA populations for most phenotypes. Although our sample was predominantly of EA, we were able to observe differences in LD blocks between EA and AA ancestry groups, which allowed us to detect novel associations in variants with lower frequency in the EA population, and to refine loci where the linkage blocks differed between ancestries. Ancestry-differences in genetic studies are usually not due to environmental or socials factors when populations stratification is properly accounted for as these factors cannot confound genetic associations. However, some of the follow-up methods (TWAS, approximate conditional analyses) depend on population reference panels and were limited to the EA population.
Additionally, sex-stratified analyses were not included in this work, and may have provided insights into possible sex-specific regulators of natural anticoagulants.
Finally, to reduce the risk of false positives, we used a stringent significance threshold (5 × 10−9), sought replication of the main findings in an external study, and provided additional post-GWAS evidence for our novel findings. We included functional validation using in vitro silencing to provide evidence for causality of candidate genes and help understand the biological mechanism. We believe this strengthens the credibility of our results. However, liver cell-derived expression system is only able to assess effects of candidate genes on synthetic mechanisms (e.g., transcription, translation), and is not able to assess potential effects on protein stability and/or clearance. Thus, genes that did not demonstrate an effect, as well as genes that were not selected for testing in this system, could regulate circulating anticoagulant protein expression via synthesis-independent mechanisms.
Supplementary Material
Supplementary Table S1: Phenotype measurements and sample size in selected cohorts
Supplementary Table S2: Detailed Description of cohort characteristics
Supplementary Table S3: Number of genes tested in TWAS by tissues
Supplementary Table S4: Functional annotations for candidate variants
Supplementary Table S5: Genes selected through prioritization process in functional study
Supplementary Table S6: Participants and cohorts count of all specific meta-analyses
Supplementary Table S7: Number of variants considered for each specific cohort
Supplementary Table S8: Additional independent variants from conditional analyses
Supplementary Table S9: Ancestry specific results of antithrombin and protein C meta-analyses
Supplementary Table S10: Summary statistics results for the selected lead variants in deCODE genetics dataset
Supplementary Table S11: Significant results from TWAS S-MultiXcan analyses
Supplementary Table S12: Prioritized genes in fine-mapping analyses
Supplementary Table S13: Colocalization results in candidate novel loci
Supplementary Table S14: Selected genetic instruments for Mendelian randomization analyses
Supplementary Table S15: MR analysis results and replication in DeCODE
Supplementary Table S16: Associations found between cardiovascular disease phenotypes
Supplementary Figure S1: Q-Q Plots of meta-analyses
Supplementary Figure S2: Manhattan plots of population specific meta-analysis
Supplementary Figure S3: Forest plots of lead variants
Supplementary Figure S4: Cell count of tested genes in the functional study
Supplementary Figure S5: Forest plots of MR analysis results on selected outcomes
Supplementary Figure S6: Forest plots of significant loci in protein C cross-ancestry meta-analysis
Supplementary Figure S7: Forest plots of significant loci in Protein S free and total meta-analyses
Supplementary Figure S8: uncropped blots (provided in a separate file)
HIGHLIGHTS.
Using cross-ancestry GWAS and TWAS methods, we report 4 novel loci regulating antithrombin plasma levels, 2 novel loci regulating PS plasma levels, and 1 novel locus regulating PC plasma levels.
Post-GWAS analyses and functional work suggest both SNX17 and GCKR are regulators of antithrombin on the chromosome 2 locus and validate an AA-specific HP gene locus.
MR analyses provide evidence implicating low antithrombin levels in VTE risk and low PC levels in VTE and CAD risk.
ACKNOWLEDGMENTS
We would also like to thank MEGASTROKE, deCODE genetics, CARDIoGRAMplusC4D and Million Veterans Program for making their data publicly available. The MEGASTROKE project received funding from sources specified at https://www.megastroke.org/acknowledgments.html. Appendix 1 contains a list with all authors that contributed to MEGASTROKE project (https://www.megastroke.org/authors.html). Data on coronary artery disease has been contributed by CARDIoGRAMplusC4D investigators and have been downloaded from http://www.cardiogramplusc4d.org. Data from deCODE genetics was accessed through deCODE genetics web page at decode.com. VTE and PAD data from de Million Veteran Program were accessed through dbGAPs (phs001672 v7.p1).
SOURCES OF FUNDING
This study is supported in part by the National Heart, Lung, and Blood Institute grant HL134894, HL139553, HL126974 and HL141291-S1A; infrastructure for the CHARGE Consortium is supported in part by HL105756. The views expressed in this manuscript are those of the authors and do not necessarily represent the views of the National Heart, Lung, and Blood Institute; the National Institutes of Health; or the US Department of Health and Human Services. G. Temprano-Sagrera is supported by the Pla Estratègic de Recerca i Innovació en Salut (PERIS) grant from the Catalan Department of Health for junior research personnel (SLT017/20/000100). P de Vries is supported by American Heart Association grant 17POST33350042. M. Sabater-Lleal is supported by a Miguel Servet contract from the Instituto de Salud Carlos III (ISCIII) Spanish Health Institute (CP17/00142) and co-financed by the European Social Fund, and acknowledges funding from the CERCA Programme/Generalitat de Catalunya. Sources of funding for the specific studies can be found in the online-only Data Supplement.
ABBREVIATIONS
- TOPMed
Trans-Omic for Precision Medicine
- PC
protein C
- PS
protein S
- VTE
venous thromboembolism
- CAD
coronary artery disease
- PAD
peripheral artery disease
- IS
ischemic stroke
- GWAS
genome-wide association study
- TWAS
transcription-wide association study
- EA
European ancestry
- AA
African ancestry
- eQTL
expression quantitative trait locus
Footnotes
DISCLOSURES
None
REFERENCES
- 1.Broekmans AW, Veltkamp JJ, Bertina RM. Congenital protein C deficiency and venous thromboembolism. A study of three Dutch families. N Engl J Med. 1983;309:340–344. doi: 10.1056/nejm198308113090604 [DOI] [PubMed] [Google Scholar]
- 2.Griffin JH, Evatt B, Zimmerman TS, Kleiss AJ, Wideman C. Deficiency of protein C in congenital thrombotic disease. J Clin Invest. 1981;68:1370–1373. doi: 10.1172/jci110385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schwarz HP, Fischer M, Hopmeier P, Batard MA, Griffin JH. Plasma protein S deficiency in familial thrombotic disease. Blood. 1984;64:1297–1300. [PubMed] [Google Scholar]
- 4.Folsom AR, Aleksic N, Wang L, Cushman M, Wu KK, White RH. Protein C, antithrombin, and venous thromboembolism incidence: a prospective population-based study. Arterioscler Thromb Vasc Biol. 2002;22:1018–1022. doi: 10.1161/01.atv.0000017470.08363.ab [DOI] [PubMed] [Google Scholar]
- 5.Antón AI, Teruel R, Corral J, Miñano A, Martínez-Martínez I, Ordóñez A, Vicente V, Sánchez-Vega B. Functional consequences of the prothrombotic SERPINC1 rs2227589 polymorphism on antithrombin levels. Haematologica. 2009;94:589–592. doi: 10.3324/haematol.2008.000604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shamsher MK, Chuzhanova NA, Friedman B, Scopes DA, Alhaq A, Millar DS, Cooper DN, Berg LP. Identification of an intronic regulatory element in the human protein C (PROC) gene. Hum Genet. 2000;107:458–465. doi: 10.1007/s004390000391 [DOI] [PubMed] [Google Scholar]
- 7.Leroy-Matheron C, Duchemin J, Levent M, Gouault-Heilmann M. Genetic modulation of plasma protein S levels by two frequent dimorphisms in the PROS1 gene. Thromb Haemost. 1999;82:1088–1092. [PubMed] [Google Scholar]
- 8.El-Galaly TC, Severinsen MT, Overvad K, Steffensen R, Vistisen AK, Tjønneland A, Kristensen SR. Single nucleotide polymorphisms and the risk of venous thrombosis: results from a Danish case-cohort study. Br J Haematol. 2013;160:838–841. doi: 10.1111/bjh.12132 [DOI] [PubMed] [Google Scholar]
- 9.Grundy CB, Chisholm M, Kakkar VV, Cooper DN. A novel homozygous missense mutation in the protein C (PROC) gene causing recurrent venous thrombosis. Hum Genet. 1992;89:683–684. doi: 10.1007/bf00221963 [DOI] [PubMed] [Google Scholar]
- 10.Millar DS, Grundy CB, Bignell P, Mitchell DC, Corden D, Woods P, Kakkar VV, Cooper DN. A novel nonsense mutation in the protein C (PROC) gene (Trp-29-->term) causing recurrent venous thrombosis. Hum Genet. 1993;91:196. doi: 10.1007/bf00222726 [DOI] [PubMed] [Google Scholar]
- 11.Wu D, Zhong Z, Chen Y, Ding H, Yang M, Lian N, Huang Z, Zhang Q, Zhao J, Deng C. Analysis of PROC and PROS1 single nucleotide polymorphisms in a thrombophilia family. Clin Respir J. 2019;13:530–537. doi: 10.1111/crj.13055 [DOI] [PubMed] [Google Scholar]
- 12.de la Morena-Barrio ME, Buil A, Antón AI, Martínez-Martínez I, Miñano A, Gutiérrez-Gallego R, Navarro-Fernández J, Aguila S, Souto JC, Vicente V, et al. Identification of antithrombin-modulating genes. Role of LARGE, a gene encoding a bifunctional glycosyltransferase, in the secretion of proteins? PLoS One. 2013;8:e64998. doi: 10.1371/journal.pone.0064998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oudot-Mellakh T, Cohen W, Germain M, Saut N, Kallel C, Zelenika D, Lathrop M, Trégouët DA, Morange PE. Genome wide association study for plasma levels of natural anticoagulant inhibitors and protein C anticoagulant pathway: the MARTHA project. Br J Haematol. 2012;157:230–239. doi: 10.1111/j.1365-2141.2011.09025.x [DOI] [PubMed] [Google Scholar]
- 14.Tang W, Basu S, Kong X, Pankow JS, Aleksic N, Tan A, Cushman M, Boerwinkle E, Folsom AR. Genome-wide association study identifies novel loci for plasma levels of protein C: the ARIC study. Blood. 2010;116:5032–5036. doi: 10.1182/blood-2010-05-283739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pankow JS, Tang W, Pankratz N, Guan W, Weng LC, Cushman M, Boerwinkle E, Folsom AR. Identification of Genetic Variants Linking Protein C and Lipoprotein Metabolism: The ARIC Study (Atherosclerosis Risk in Communities). Arterioscler Thromb Vasc Biol. 2017;37:589–597. doi: 10.1161/atvbaha.116.308109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Munir MS, Weng LC, Tang W, Basu S, Pankow JS, Matijevic N, Cushman M, Boerwinkle E, Folsom AR. Genetic markers associated with plasma protein C level in African Americans: the atherosclerosis risk in communities (ARIC) study. Genet Epidemiol. 2014;38:709–713. doi: 10.1002/gepi.21868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Athanasiadis G, Buil A, Souto JC, Borrell M, López S, Martinez-Perez A, Lathrop M, Fontcuberta J, Almasy L, Soria JM. A genome-wide association study of the Protein C anticoagulant pathway. PLoS One. 2011;6:e29168. doi: 10.1371/journal.pone.0029168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ferkingstad E, Sulem P, Atlason BA, Sveinbjornsson G, Magnusson MI, Styrmisdottir EL, Gunnarsdottir K, Helgason A, Oddsson A, Halldorsson BV, et al. Large-scale integration of the plasma proteome with genetics and disease. Nat Genet. 2021;53:1712–1721. doi: 10.1038/s41588-021-00978-w [DOI] [PubMed] [Google Scholar]
- 19.Psaty BM, O’Donnell CJ, Gudnason V, Lunetta KL, Folsom AR, Rotter JI, Uitterlinden AG, Harris TB, Witteman JC, Boerwinkle E. Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium: Design of prospective meta-analyses of genome-wide association studies from 5 cohorts. Circ Cardiovasc Genet. 2009;2:73–80. doi: 10.1161/circgenetics.108.829747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Investigators A The Atherosclerosis risk in COMMUNIT (ARIC) study: design and objectives. American journal of epidemiology. 1989;129:687–702. [PubMed] [Google Scholar]
- 21.Pattaro C, Gögele M, Mascalzoni D, Melotti R, Schwienbacher C, De Grandi A, Foco L, D’Elia Y, Linder B, Fuchsberger C, et al. The Cooperative Health Research in South Tyrol (CHRIS) study: rationale, objectives, and preliminary results. J Transl Med. 2015;13:348. doi: 10.1186/s12967-015-0704-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fried LP, Borhani NO, Enright P, Furberg CD, Gardin JM, Kronmal RA, Kuller LH, Manolio TA, Mittelmark MB, Newman A, et al. The Cardiovascular Health Study: design and rationale. Ann Epidemiol. 1991;1:263–276. doi: 10.1016/1047-2797(91)90005-w [DOI] [PubMed] [Google Scholar]
- 23.Desch K, Li J, Kim S, Laventhal N, Metzger K, Siemieniak D, Ginsburg D. Analysis of informed consent document utilization in a minimal-risk genetic study. Annals of internal medicine. 2011;155:316–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Souto JC, Almasy L, Borrell M, Blanco-Vaca F, Mateo J, Soria JM, Coll I, Felices R, Stone W, Fontcuberta J, et al. Genetic susceptibility to thrombosis and its relationship to physiological risk factors: the GAIT study. Genetic Analysis of Idiopathic Thrombophilia. Am J Hum Genet. 2000;67:1452–1459. doi: 10.1086/316903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Psaty BM, Heckbert SR, Koepsell TD, Siscovick DS, Raghunathan TE, Weiss NS, Rosendaal FR, Lemaitre RN, Smith NL, Wahl PW, et al. The risk of myocardial infarction associated with antihypertensive drug therapies. Jama. 1995;274:620–625. [PubMed] [Google Scholar]
- 26.Tomaschitz A, Pilz S, Ritz E, Meinitzer A, Boehm BO, März W. Plasma aldosterone levels are associated with increased cardiovascular mortality: the Ludwigshafen Risk and Cardiovascular Health (LURIC) study. European heart journal. 2010;31:1237–1247. [DOI] [PubMed] [Google Scholar]
- 27.Antoni G, Oudot-Mellakh T, Dimitromanolakis A, Germain M, Cohen W, Wells P, Lathrop M, Gagnon F, Morange PE, Tregouet DA. Combined analysis of three genome-wide association studies on vWF and FVIII plasma levels. BMC Med Genet. 2011;12:102. doi: 10.1186/1471-2350-12-102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vázquez-Santiago M, Vilalta N, Cuevas B, Murillo J, Llobet D, Macho R, Pujol-Moix N, Carrasco M, Mateo J, Fontcuberta J, et al. Short closure time values in PFA-100® are related to venous thrombotic risk. Results from the RETROVE Study. Thromb Res. 2018;169:57–63. doi: 10.1016/j.thromres.2018.07.012 [DOI] [PubMed] [Google Scholar]
- 29.Mills JL, Carter TC, Scott JM, Troendle JF, Gibney ER, Shane B, Kirke PN, Ueland PM, Brody LC, Molloy AM. Do high blood folate concentrations exacerbate metabolic abnormalities in people with low vitamin B-12 status? The American journal of clinical nutrition. 2011;94:495–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pattaro C, Gögele M, Mascalzoni D, Melotti R, Schwienbacher C, De Grandi A, Foco L, D’elia Y, Linder B, Fuchsberger C. The Cooperative Health Research in South Tyrol (CHRIS) study: rationale, objectives, and preliminary results. Journal of translational medicine. 2015;13:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Psaty BM, Heckbert SR, Atkins D, Lemaitre R, Koepsell TD, Wahl PW, Siscovick DS, Wagner EH. The risk of myocardial infarction associated with the combined use of estrogens and progestins in postmenopausal women. Arch Intern Med. 1994;154:1333–1339. [PubMed] [Google Scholar]
- 32.Desch KC, Ozel AB, Siemieniak D, Kalish Y, Shavit JA, Thornburg CD, Sharathkumar AA, McHugh CP, Laurie CC, Crenshaw A, et al. Linkage analysis identifies a locus for plasma von Willebrand factor undetected by genome-wide association. Proc Natl Acad Sci U S A. 2013;110:588–593. doi: 10.1073/pnas.1219885110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kowalski MH, Qian H, Hou Z, Rosen JD, Tapia AL, Shan Y, Jain D, Argos M, Arnett DK, Avery C, et al. Use of >100,000 NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium whole genome sequences improves imputation quality and detection of rare variant associations in admixed African and Hispanic/Latino populations. PLoS Genet. 2019;15:e1008500. doi: 10.1371/journal.pgen.1008500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Winkler TW, Day FR, Croteau-Chonka DC, Wood AR, Locke AE, Mägi R, Ferreira T, Fall T, Graff M, Justice AE, et al. Quality control and conduct of genome-wide association meta-analyses. Nat Protoc. 2014;9:1192–1212. doi: 10.1038/nprot.2014.071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pulit SL, de With SA, de Bakker PI. Resetting the bar: Statistical significance in whole-genome sequencing-based association studies of global populations. Genet Epidemiol. 2017;41:145–151. doi: 10.1002/gepi.22032 [DOI] [PubMed] [Google Scholar]
- 36.Barbeira AN, Dickinson SP, Bonazzola R, Zheng J, Wheeler HE, Torres JM, Torstenson ES, Shah KP, Garcia T, Edwards TL, et al. Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat Commun. 2018;9:1825. doi: 10.1038/s41467-018-03621-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barbeira AN, Pividori M, Zheng J, Wheeler HE, Nicolae DL, Im HK. Integrating predicted transcriptome from multiple tissues improves association detection. PLoS Genet. 2019;15:e1007889. doi: 10.1371/journal.pgen.1007889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ray D, Boehnke M. Methods for meta-analysis of multiple traits using GWAS summary statistics. Genet Epidemiol. 2018;42:134–145. doi: 10.1002/gepi.22105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Benjamin DJ, Berger JO, Johannesson M, Nosek BA, Wagenmakers EJ, Berk R, Bollen KA, Brembs B, Brown L, Camerer C, et al. Redefine statistical significance. Nat Hum Behav. 2018;2:6–10. doi: 10.1038/s41562-017-0189-z [DOI] [PubMed] [Google Scholar]
- 40.Lindström S, Wang L, Smith EN, Gordon W, van Hylckama Vlieg A, de Andrade M, Brody JA, Pattee JW, Haessler J, Brumpton BM, et al. Genomic and transcriptomic association studies identify 16 novel susceptibility loci for venous thromboembolism. Blood. 2019;134:1645–1657. doi: 10.1182/blood.2019000435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Klarin D, Busenkell E, Judy R, Lynch J, Levin M, Haessler J, Aragam K, Chaffin M, Haas M, Lindström S, et al. Genome-wide association analysis of venous thromboembolism identifies new risk loci and genetic overlap with arterial vascular disease. Nat Genet. 2019;51:1574–1579. doi: 10.1038/s41588-019-0519-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Klarin D, Lynch J, Aragam K, Chaffin M, Assimes TL, Huang J, Lee KM, Shao Q, Huffman JE, Natarajan P, et al. Genome-wide association study of peripheral artery disease in the Million Veteran Program. Nat Med. 2019;25:1274–1279. doi: 10.1038/s41591-019-0492-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nikpay M, Goel A, Won HH, Hall LM, Willenborg C, Kanoni S, Saleheen D, Kyriakou T, Nelson CP, Hopewell JC, et al. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet. 2015;47:1121–1130. doi: 10.1038/ng.3396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Malik R, Chauhan G, Traylor M, Sargurupremraj M, Okada Y, Mishra A, Rutten-Jacobs L, Giese AK, van der Laan SW, Gretarsdottir S, et al. Multiancestry genome-wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat Genet. 2018;50:524–537. doi: 10.1038/s41588-018-0058-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stacey D, Chen L, Stanczyk PJ, Howson JM, Mason AM, Burgess S, MacDonald S, Langdown J, McKinney H, Downes K. Elucidating mechanisms of genetic cross-disease associations at the PROCR vascular disease locus. Nature communications. 2022;13:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Temprano-Sagrera G, Sitlani CM, Bone WP, Martin-Bornez M, Voight BF, Morrison AC, Damrauer SM, de Vries PS, Smith NL, Sabater-Lleal M. Multi-phenotype analyses of hemostatic traits with cardiovascular events reveal novel genetic associations. J Thromb Haemost. 2022;20:1331–1349. doi: 10.1111/jth.15698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Manderstedt E, Lind-Halldén C, Halldén C, Elf J, Svensson PJ, Dahlbäck B, Engström G, Melander O, Baras A, Lotta LA, et al. Classic Thrombophilias and Thrombotic Risk Among Middle-Aged and Older Adults: A Population-Based Cohort Study. J Am Heart Assoc. 2022;11:e023018. doi: 10.1161/jaha.121.023018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dürr C, Hinney A, Luckenbach C, Kömpf J, Ritter H. Genetic studies of antithrombin III with IEF and ASO hybridization. Hum Genet. 1992;90:457–459. doi: 10.1007/bf00220477 [DOI] [PubMed] [Google Scholar]
- 49.Daly M, O’Meara A, Hallinan FM. Identification and characterization of a new antithrombin III familial variant (AT Dublin) with possible increased frequency in children with cancer. Br J Haematol. 1987;65:457–462. doi: 10.1111/j.1365-2141.1987.tb04150.x [DOI] [PubMed] [Google Scholar]
- 50.Downes K, Megy K, Duarte D, Vries M, Gebhart J, Hofer S, Shamardina O, Deevi SVV, Stephens J, Mapeta R, et al. Diagnostic high-throughput sequencing of 2396 patients with bleeding, thrombotic, and platelet disorders. Blood. 2019;134:2082–2091. doi: 10.1182/blood.2018891192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stockinger W, Sailler B, Strasser V, Recheis B, Fasching D, Kahr L, Schneider WJ, Nimpf J. The PX-domain protein SNX17 interacts with members of the LDL receptor family and modulates endocytosis of the LDL receptor. Embo j. 2002;21:4259–4267. doi: 10.1093/emboj/cdf435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhao D, Li X, Liang H, Zheng N, Pan Z, Zhou Y, Liu X, Qian M, Xu B, Zhang Y, et al. SNX17 produces anti-arrhythmic effects by preserving functional SERCA2a protein in myocardial infarction. Int J Cardiol. 2018;272:298–305. doi: 10.1016/j.ijcard.2018.07.025 [DOI] [PubMed] [Google Scholar]
- 53.Yang J, Villar VAM, Rozyyev S, Jose PA, Zeng C. The emerging role of sorting nexins in cardiovascular diseases. Clin Sci (Lond). 2019;133:723–737. doi: 10.1042/cs20190034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smith NL, Chen MH, Dehghan A, Strachan DP, Basu S, Soranzo N, Hayward C, Rudan I, Sabater-Lleal M, Bis JC, et al. Novel associations of multiple genetic loci with plasma levels of factor VII, factor VIII, and von Willebrand factor: The CHARGE (Cohorts for Heart and Aging Research in Genome Epidemiology) Consortium. Circulation. 2010;121:1382–1392. doi: 10.1161/circulationaha.109.869156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sennblad B, Basu S, Mazur J, Suchon P, Martinez-Perez A, van Hylckama Vlieg A, Truong V, Li Y, Gådin JR, Tang W, et al. Genome-wide association study with additional genetic and post-transcriptional analyses reveals novel regulators of plasma factor XI levels. Hum Mol Genet. 2017;26:637–649. doi: 10.1093/hmg/ddw401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Folsom AR, Lutsey PL, Astor BC, Cushman M. C-reactive protein and venous thromboembolism. A prospective investigation in the ARIC cohort. Thromb Haemost. 2009;102:615–619. doi: 10.1160/th09-04-0274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ellis J, Lange EM, Li J, Dupuis J, Baumert J, Walston JD, Keating BJ, Durda P, Fox ER, Palmer CD, et al. Large multiethnic Candidate Gene Study for C-reactive protein levels: identification of a novel association at CD36 in African Americans. Hum Genet. 2014;133:985–995. doi: 10.1007/s00439-014-1439-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Santoro N, Zhang CK, Zhao H, Pakstis AJ, Kim G, Kursawe R, Dykas DJ, Bale AE, Giannini C, Pierpont B, et al. Variant in the glucokinase regulatory protein (GCKR) gene is associated with fatty liver in obese children and adolescents. Hepatology. 2012;55:781–789. doi: 10.1002/hep.24806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Petit JM, Masson D, Guiu B, Rollot F, Duvillard L, Bouillet B, Brindisi MC, Buffier P, Hillon P, Cercueil JP, et al. GCKR polymorphism influences liver fat content in patients with type 2 diabetes. Acta Diabetol. 2016;53:237–242. doi: 10.1007/s00592-015-0766-4 [DOI] [PubMed] [Google Scholar]
- 60.Yeh KH, Hsu LA, Teng MS, Wu S, Chou HH, Ko YL. Pleiotropic Effects of Common and Rare GCKR Exonic Mutations on Cardiometabolic Traits. Genes (Basel). 2022;13. doi: 10.3390/genes13030491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kitamoto A, Kitamoto T, Nakamura T, Ogawa Y, Yoneda M, Hyogo H, Ochi H, Mizusawa S, Ueno T, Nakao K. Association of polymorphisms in GCKR and TRIB1 with nonalcoholic fatty liver disease and metabolic syndrome traits. Endocrine journal. 2014:EJ14–0052. [DOI] [PubMed] [Google Scholar]
- 62.Ligthart S, Vaez A, Võsa U, Stathopoulou MG, de Vries PS, Prins BP, Van der Most PJ, Tanaka T, Naderi E, Rose LM, et al. Genome Analyses of >200,000 Individuals Identify 58 Loci for Chronic Inflammation and Highlight Pathways that Link Inflammation and Complex Disorders. Am J Hum Genet. 2018;103:691–706. doi: 10.1016/j.ajhg.2018.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wejman JC, Hovsepian D, Wall JS, Hainfeld JF, Greer J. Structure of haptoglobin and the haptoglobin-hemoglobin complex by electron microscopy. J Mol Biol. 1984;174:319–341. doi: 10.1016/0022-2836(84)90341-3 [DOI] [PubMed] [Google Scholar]
- 64.Landis RC, Philippidis P, Domin J, Boyle JJ, Haskard DO. Haptoglobin Genotype-Dependent Anti-Inflammatory Signaling in CD163(+) Macrophages. Int J Inflam. 2013;2013:980327. doi: 10.1155/2013/980327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Asleh R, Miller-Lotan R, Aviram M, Hayek T, Yulish M, Levy JE, Miller B, Blum S, Milman U, Shapira C, et al. Haptoglobin genotype is a regulator of reverse cholesterol transport in diabetes in vitro and in vivo. Circ Res. 2006;99:1419–1425. doi: 10.1161/01.Res.0000251741.65179.56 [DOI] [PubMed] [Google Scholar]
- 66.Vormittag R, Vukovich T, Mannhalter C, Minar E, Schönauer V, Bialonczyk C, Hirschl M, Pabinger I. Haptoglobin phenotype 2–2 as a potentially new risk factor for spontaneous venous thromboembolism. Haematologica. 2005;90:1557–1561. [PubMed] [Google Scholar]
- 67.Mikulska JE, Pablo L, Canel J, Simister NE. Cloning and analysis of the gene encoding the human neonatal Fc receptor. Eur J Immunogenet. 2000;27:231–240. doi: 10.1046/j.1365-2370.2000.00225.x [DOI] [PubMed] [Google Scholar]
- 68.Pyzik M, Rath T, Kuo TT, Win S, Baker K, Hubbard JJ, Grenha R, Gandhi A, Krämer TD, Mezo AR, et al. Hepatic FcRn regulates albumin homeostasis and susceptibility to liver injury. Proc Natl Acad Sci U S A. 2017;114:E2862–e2871. doi: 10.1073/pnas.1618291114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dente L, Pizza MG, Metspalu A, Cortese R. Structure and expression of the genes coding for human alpha 1-acid glycoprotein. Embo j. 1987;6:2289–2296. doi: 10.1002/j.1460-2075.1987.tb02503.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rocanin-Arjo A, Cohen W, Carcaillon L, Frère C, Saut N, Letenneur L, Alhenc-Gelas M, Dupuy AM, Bertrand M, Alessi MC, et al. A meta-analysis of genome-wide association studies identifies ORM1 as a novel gene controlling thrombin generation potential. Blood. 2014;123:777–785. doi: 10.1182/blood-2013-10-529628 [DOI] [PubMed] [Google Scholar]
- 71.Lopez S, Martinez-Perez A, Rodriguez-Rius A, Viñuela A, Brown AA, Martin-Fernandez L, Vilalta N, Arús M, Panousis NI, Buil A, et al. Integrated GWAS and Gene Expression Suggest ORM1 as a Potential Regulator of Plasma Levels of Cell-Free DNA and Thrombosis Risk. Thromb Haemost. 2022;122:1027–1039. doi: 10.1055/s-0041-1742169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Otagiri M, Maruyama T, Imai T, Suenaga A, Imamura Y. A comparative study of the interaction of warfarin with human alpha 1-acid glycoprotein and human albumin. J Pharm Pharmacol. 1987;39:416–420. doi: 10.1111/j.2042-7158.1987.tb03412.x [DOI] [PubMed] [Google Scholar]
- 73.Wadelius M, Chen LY, Eriksson N, Bumpstead S, Ghori J, Wadelius C, Bentley D, McGinnis R, Deloukas P. Association of warfarin dose with genes involved in its action and metabolism. Hum Genet. 2007;121:23–34. doi: 10.1007/s00439-006-0260-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bourgeois S, Jorgensen A, Zhang EJ, Hanson A, Gillman MS, Bumpstead S, Toh CH, Williamson P, Daly AK, Kamali F, et al. A multi-factorial analysis of response to warfarin in a UK prospective cohort. Genome Med. 2016;8:2. doi: 10.1186/s13073-015-0255-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Morano I, Hofmann F, Zimmer M, Rüegg JC. The influence of P-light chain phosphorylation by myosin light chain kinase on the calcium sensitivity of chemically skinned heart fibres. FEBS Lett. 1985;189:221–224. doi: 10.1016/0014-5793(85)81027-9 [DOI] [PubMed] [Google Scholar]
- 76.Himpens B, Matthijs G, Somlyo AV, Butler TM, Somlyo AP. Cytoplasmic free calcium, myosin light chain phosphorylation, and force in phasic and tonic smooth muscle. J Gen Physiol. 1988;92:713–729. doi: 10.1085/jgp.92.6.713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vujkovic M, Keaton JM, Lynch JA, Miller DR, Zhou J, Tcheandjieu C, Huffman JE, Assimes TL, Lorenz K, Zhu X, et al. Discovery of 318 new risk loci for type 2 diabetes and related vascular outcomes among 1.4 million participants in a multi-ancestry meta-analysis. Nat Genet. 2020;52:680–691. doi: 10.1038/s41588-020-0637-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chung RH, Chiu YF, Wang WC, Hwu CM, Hung YJ, Lee IT, Chuang LM, Quertermous T, Rotter JI, Chen YI, et al. Multi-omics analysis identifies CpGs near G6PC2 mediating the effects of genetic variants on fasting glucose. Diabetologia. 2021;64:1613–1625. doi: 10.1007/s00125-021-05449-9 [DOI] [PubMed] [Google Scholar]
- 79.Mahmoodi BK, Brouwer JL, Ten Kate MK, Lijfering WM, Veeger NJ, Mulder AB, Kluin-Nelemans HC, Van Der Meer J. A prospective cohort study on the absolute risks of venous thromboembolism and predictive value of screening asymptomatic relatives of patients with hereditary deficiencies of protein S, protein C or antithrombin. J Thromb Haemost. 2010;8:1193–1200. doi: 10.1111/j.1538-7836.2010.03840.x [DOI] [PubMed] [Google Scholar]
- 80.Pabinger I, Kyrle PA, Heistinger M, Eichinger S, Wittmann E, Lechner K. The risk of thromboembolism in asymptomatic patients with protein C and protein S deficiency: a prospective cohort study. Thromb Haemost. 1994;71:441–445. [PubMed] [Google Scholar]
- 81.Schooling CM, Zhong Y. Plasma levels of the anti-coagulation protein C and the risk of ischaemic heart disease. A Mendelian randomisation study. Thromb Haemost. 2017;117:262–268. doi: 10.1160/th16-07-0518 [DOI] [PubMed] [Google Scholar]
- 82.O’Connor NT, Broekmans AW, Bertina RM. Protein C values in coronary artery disease. Br Med J (Clin Res Ed). 1984;289:1192. doi: 10.1136/bmj.289.6453.1192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wessler S, Gaston LW. Anticoagulant therapy in coronary artery disease. Circulation. 1966;34:856–864. doi: 10.1161/01.cir.34.5.856 [DOI] [PubMed] [Google Scholar]
- 84.Wilbur J, Shian B. Deep Venous Thrombosis and Pulmonary Embolism: Current Therapy. Am Fam Physician. 2017;95:295–302. [PubMed] [Google Scholar]
- 85.Kuchenbaecker K, Telkar N, Reiker T, Walters RG, Lin K, Eriksson A, Gurdasani D, Gilly A, Southam L, Tsafantakis E, et al. The transferability of lipid loci across African, Asian and European cohorts. Nat Commun. 2019;10:4330. doi: 10.1038/s41467-019-12026-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Evans DS, Avery CL, Nalls MA, Li G, Barnard J, Smith EN, Tanaka T, Butler AM, Buxbaum SG, Alonso A, et al. Fine-mapping, novel loci identification, and SNP association transferability in a genome-wide association study of QRS duration in African Americans. Hum Mol Genet. 2016;25:4350–4368. doi: 10.1093/hmg/ddw284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–2191. doi: 10.1093/bioinformatics/btq340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yang J, Ferreira T, Morris AP, Medland SE, Madden PA, Heath AC, Martin NG, Montgomery GW, Weedon MN, Loos RJ, et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat Genet. 2012;44:369–375, s361–363. doi: 10.1038/ng.2213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88:76–82. doi: 10.1016/j.ajhg.2010.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, Abecasis GR. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45:580–585. doi: 10.1038/ng.2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Barbeira AN, Bonazzola R, Gamazon ER, Liang Y, Park Y, Kim-Hellmuth S, Wang G, Jiang Z, Zhou D, Hormozdiari F, et al. Exploiting the GTEx resources to decipher the mechanisms at GWAS loci. Genome Biol. 2021;22:49. doi: 10.1186/s13059-020-02252-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gamazon ER, Wheeler HE, Shah KP, Mozaffari SV, Aquino-Michaels K, Carroll RJ, Eyler AE, Denny JC, Nicolae DL, Cox NJ, et al. A gene-based association method for mapping traits using reference transcriptome data. Nat Genet. 2015;47:1091–1098. doi: 10.1038/ng.3367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Urbut SM, Wang G, Carbonetto P, Stephens M. Flexible statistical methods for estimating and testing effects in genomic studies with multiple conditions. Nat Genet. 2019;51:187–195. doi: 10.1038/s41588-018-0268-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mancuso N, Freund MK, Johnson R, Shi H, Kichaev G, Gusev A, Pasaniuc B. Probabilistic fine-mapping of transcriptome-wide association studies. Nat Genet. 2019;51:675–682. doi: 10.1038/s41588-019-0367-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.nygcesearch/ IDetect. https://bitbucket.org/nygcresearch/ldetect. Accessed April 06.
- 97.Giambartolomei C, Vukcevic D, Schadt EE, Franke L, Hingorani AD, Wallace C, Plagnol V. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS genetics. 2014;10:e1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Foley CN, Staley JR, Breen PG, Sun BB, Kirk PD, Burgess S, Howson JM. A fast and efficient colocalization algorithm for identifying shared genetic risk factors across multiple traits. Nature communications. 2021;12:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Deloukas P, Kanoni S, Willenborg C, Farrall M, Assimes TL, Thompson JR, Ingelsson E, Saleheen D, Erdmann J, Goldstein BA, et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet. 2013;45:25–33. doi: 10.1038/ng.2480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent Estimation in Mendelian Randomization with Some Invalid Instruments Using a Weighted Median Estimator. Genet Epidemiol. 2016;40:304–314. doi: 10.1002/gepi.21965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hartwig FP, Davey Smith G, Bowden J. Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. Int J Epidemiol. 2017;46:1985–1998. doi: 10.1093/ije/dyx102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44:512–525. doi: 10.1093/ije/dyv080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cushman M, Cornell ES, Howard PR, Bovill EG, Tracy RP. Laboratory methods and quality assurance in the Cardiovascular Health Study. Clin Chem. 1995;41:264–270. [PubMed] [Google Scholar]
- 104.Souto JC, Almasy L, Borrell M, Garí M, Martínez E, Mateo J, Stone WH, Blangero J, Fontcuberta J. Genetic determinants of hemostasis phenotypes in Spanish families. Circulation. 2000;101:1546–1551. doi: 10.1161/01.cir.101.13.1546 [DOI] [PubMed] [Google Scholar]
- 105.Blondon M, van Hylckama Vlieg A, Wiggins KL, Harrington LB, McKnight B, Rice KM, Rosendaal FR, Heckbert SR, Psaty BM, Smith NL. Differential associations of oral estradiol and conjugated equine estrogen with hemostatic biomarkers. J Thromb Haemost. 2014;12:879–886. doi: 10.1111/jth.12560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Stone N, Pangilinan F, Molloy AM, Shane B, Scott JM, Ueland PM, Mills JL, Kirke PN, Sethupathy P, Brody LC. Bioinformatic and genetic association analysis of microRNA target sites in one-carbon metabolism genes. PloS one. 2011;6:e21851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Trégouët DA, Morange PE. What is currently known about the genetics of venous thromboembolism at the dawn of next generation sequencing technologies. British journal of haematology. 2018;180:335–345. [DOI] [PubMed] [Google Scholar]
- 108.Folsom AR, Tang W, Weng LC, Roetker NS, Cushman M, Basu S, Pankow JS. Replication of a genetic risk score for venous thromboembolism in whites but not in African Americans. J Thromb Haemost. 2016;14:83–88. doi: 10.1111/jth.13193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wolford BN, Zhao Y, Surakka I, Wu K-HH, Yu X, Richter C, Bhatta L, Brumpton BM, Desch K, Thibord F. Multi-ancestry GWAS for venous thromboembolism identifies novel loci followed by experimental validation in zebrafish. MedRxiv. 2022. [Google Scholar]
- 110.Thibord F, Klarin D, Brody JA, Chen M-H, Levin MG, Chasman DI, Goode EL, Hveem K, Teder-Laving M, Martinez-Perez A. Cross-Ancestry Investigation of Venous Thromboembolism Genomic Predictors. Circulation. 2022: 10.1161/CIRCULATIONAHA.1122.059675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Howard AD, Wang X, Prasad M, Sahu AD, Aniba R, Miller M, Hannenhalli S, Chang Y-PC. Allele-specific enhancers mediate associations between LCAT and ABCA1 polymorphisms and HDL metabolism. PloS one. 2019;14:e0215911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rizk NM, El-Menyar A, Egue H, Souleman Wais I, Mohamed Baluli H, Alali K, Farag F, Younes N, Al Suwaidi J. The Association between Serum LDL Cholesterol and Genetic Variation in Chromosomal Locus 1p13.3 among Coronary Artery Disease Patients. Biomed Res Int. 2015;2015:678924. doi: 10.1155/2015/678924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Arvind P, Nair J, Jambunathan S, Kakkar VV, Shanker J. CELSR2–PSRC1–SORT1 gene expression and association with coronary artery disease and plasma lipid levels in an Asian Indian cohort. Journal of Cardiology. 2014;64:339–346. [DOI] [PubMed] [Google Scholar]
- 114.Pan LA, Chen YC, Huang H, Zhang L, Liu R, Li X, Qiang O, Zeng Z. G771C Polymorphism in the MLXIPL Gene Is Associated with a Risk of Coronary Artery Disease in the Chinese: A Case-Control Study. Cardiology. 2009;114:174–178. doi: 10.1159/000226610 [DOI] [PubMed] [Google Scholar]
- 115.Qin J, Tian J, Liu G, Zhang Y, Tian L, Zhen Y, Zhang H, Xu J, Sun X, Fang H. Association between 1p13 polymorphisms and peripheral arterial disease in a Chinese population with diabetes. J Diabetes Investig. 2018;9:1189–1195. doi: 10.1111/jdi.12804 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1: Phenotype measurements and sample size in selected cohorts
Supplementary Table S2: Detailed Description of cohort characteristics
Supplementary Table S3: Number of genes tested in TWAS by tissues
Supplementary Table S4: Functional annotations for candidate variants
Supplementary Table S5: Genes selected through prioritization process in functional study
Supplementary Table S6: Participants and cohorts count of all specific meta-analyses
Supplementary Table S7: Number of variants considered for each specific cohort
Supplementary Table S8: Additional independent variants from conditional analyses
Supplementary Table S9: Ancestry specific results of antithrombin and protein C meta-analyses
Supplementary Table S10: Summary statistics results for the selected lead variants in deCODE genetics dataset
Supplementary Table S11: Significant results from TWAS S-MultiXcan analyses
Supplementary Table S12: Prioritized genes in fine-mapping analyses
Supplementary Table S13: Colocalization results in candidate novel loci
Supplementary Table S14: Selected genetic instruments for Mendelian randomization analyses
Supplementary Table S15: MR analysis results and replication in DeCODE
Supplementary Table S16: Associations found between cardiovascular disease phenotypes
Supplementary Figure S1: Q-Q Plots of meta-analyses
Supplementary Figure S2: Manhattan plots of population specific meta-analysis
Supplementary Figure S3: Forest plots of lead variants
Supplementary Figure S4: Cell count of tested genes in the functional study
Supplementary Figure S5: Forest plots of MR analysis results on selected outcomes
Supplementary Figure S6: Forest plots of significant loci in protein C cross-ancestry meta-analysis
Supplementary Figure S7: Forest plots of significant loci in Protein S free and total meta-analyses
Supplementary Figure S8: uncropped blots (provided in a separate file)