


Abstract
Type II topoisomerases are essential enzymes that are also the primary cellular targets for a number of important anticancer drugs. These drugs act by increasing levels of topoisomerase II-mediated DNA cleavage. Recent studies indicate that endogenous forms of DNA damage, such as abasic sites and base mismatches, also stimulate the DNA scission activity of the enzyme. To extend our understanding of how type II topoisomerases react to DNA damage, the effects of abasic sites, and oxidized and alkylated bases on DNA cleavage mediated by human topo-isomerase IIα and β were determined. Based on experiments that incorporated random abasic sites into plasmid DNA, human type II enzymes can locate lesions even within a background of several thousand undamaged base pairs. As determined by experiments that utilized site-specific forms of DNA lesions, oxidized or monoalkylated purines that allow base pairing and induce little distortion in the double helix have modest effects on topoisomerase II-mediated DNA cleavage. In contrast, 1,N6-ethenoadenine, a bulky lesion that disrupts base pairing, enhanced DNA cleavage ~10-fold. 1,N6-Ethenoadenine is the first lesion found to rival the stimulatory effects of apurinic sites on the DNA scission activity of eukaryotic type II topoisomerases.
INTRODUCTION
Type II topoisomerases are essential enzymes that interconvert topological forms of DNA by making transient double-stranded breaks in the backbone of the genetic material (1–3). These enzymes play important roles in a number of fundamental nuclear processes, including DNA replication and recombination (1,4). In addition, they are required for proper chromosome structure and segregation (1,4). Lower eukaryotes, such as yeast and Drosophila, contain only a single species of topoisomerase II. However, vertebrates contain two closely related isoforms of the enzyme, topoisomerase IIα and topoisomerase IIβ (1–7). Levels of topoisomerase IIα increase dramatically during periods of rapid cell proliferation (8). Consequently, it is believed to be the isoform responsible for untangling daughter chromosomes during mitosis (4,9). Topoisomerase IIβ appears to be present in all cell types, independent of growth conditions, and is believed to be involved in a number of ongoing nuclear processes (2,4,7,10). Despite the fact that either topoisomerase IIα or β can complement the loss of type II activity in yeast (11), these two isoforms appear to have non-overlapping functions in vertebrate cells (4,7,9). Furthermore, both are essential to development in vertebrate species (4,7,12).
In addition to their indispensable physiological functions, type II topoisomerases are the targets for some of the most active anticancer drugs currently in clinical use (2,3,13–17). These agents kill cells in an unusual fashion. They do not act by blocking the catalytic activity of topoisomerase II; rather, they dramatically increase levels of enzyme-mediated DNA cleavage (13–16,18). Consequently, anticancer drugs convert type II topoisomerases into potent toxins that generate high numbers of double-stranded breaks in the genomes of treated cells (2,3,13–17). As a result of their actions, anticancer drugs targeted to type II topoisomerases are referred to as ‘topoisomerase II poisons’ (2,3,13–17,19).
The unusual cytotoxic mechanism of anticancer agents suggests that they may represent exogenous counterparts of cellular factors that enhance topoisomerase II-mediated DNA cleavage as part of DNA recombination, mutagenesis or cell death pathways (20–22). Since drugs act at the topoisomerase II–DNA interface (23–27), interactions between the enzyme and nucleic acid lesions were characterized to determine whether various forms of DNA damage could act as endogenous topoisomerase II poisons. Previous studies were restricted primarily to Drosophila topoisomerase II and human topoisomerase IIα. Furthermore, they focused on DNA lesions generated by spontaneous physiological events, such as the generation of abasic (apurinic/apyrimidinic) sites, deamination of cytosine residues or the incorporation of mismatched bases (20,21,28–31). In all cases, spontaneous DNA lesions were found to be position-specific topoisomerase II poisons. Incorporation of a single lesion stimulated enzyme-mediated double-stranded DNA scission. However, for stimulation to occur, damage had to be located within the 4-base stagger that separates the scissile bonds on the opposite strands of the double helix (21,22,28–30).
Because of the potential ramifications of these findings, it is important to determine whether other forms of DNA damage are capable of acting as topoisomerase II poisons. It is also important to determine whether topoisomerase IIβ is affected by DNA lesions. Therefore, the present work addresses the effects of environmental damage on DNA scission mediated by human topoisomerase IIα and β. Results indicate that both human type II topoisomerases can locate DNA lesions in a background of several thousand undamaged base pairs. Oxidized and monoalkylated DNA adducts that allow base pairing had only modest effects on enzyme-mediated DNA scission; however, 1,N6-ethenoadenine, a highly mutagenic DNA adduct that disrupts base pairing, was a potent topoisomerase II poison.
MATERIALS AND METHODS
Materials
Human topoisomerase IIα and β were purified as previously described (30) from Saccharomyces cerevisiae containing the respective genes in the inducible overexpression plasmids YEpWOB6 and YEpWOBeta (32). Bacteriophage T4 polynucleotide kinase was from New England Biolabs (Beverly, MA); Escherichia coli uracil DNA glycosylase was from United States Biochemical (Cleveland, OH); [γ-32P]ATP (~6000 Ci/mmol) was from Amersham (Arlington Heights, IL); deoxyuridine, 8-oxo-2′-deoxyguanosine, 8-oxo-2′-deoxyadenosine, O6-methyl-2′-deoxyguanosine, N6-methyl-2′-deoxyadenosine and 1,N6-etheno-2′-deoxyadenosine phosphoramidites were from Glen Research (Sterling, VA); and tetrahydrofuran phosphoramidite was from Cruachem (Dulles, VA). All other chemicals were analytical reagent grade.
Generation of random abasic sites in plasmid DNA
Abasic sites were generated in negatively supercoiled pBR322 plasmid DNA by incubation in 25 mM sodium citrate, pH 4.8, 250 mM KCl at 70°C as described previously (20,33). Reactions were stopped by the addition of 1 M Tris–HCl, pH 7.9, at 4°C followed by buffer exchange into 10 mM Tris–HCl, pH 7.9, 0.1 mM EDTA in a Bio-Spin 6 chromatography column (Bio-Rad, Richmond, CA). Under the conditions employed, approximately one abasic site per plasmid was generated every 22 min (20,33).
Topoisomerase II-mediated cleavage of plasmid DNA
pBR322 plasmid DNA was cleaved by a protocol similar to that described by Corbett et al. (34). Reaction mixtures containing 150 nM human topoisomerase IIα or β and 10 nM negatively supercoiled pBR322 DNA in 20 µl of 50 mM Tris–HCl, pH 7.9, 100 mM KCl, 10 mM MgCl2, 0.5 mM EDTA and 2.5% glycerol were incubated at 37°C for 10 min. Reactions were stopped by the addition of 2 µl of 5% SDS followed by 2 µl of 250 mM EDTA. Proteinase K was added (2 µl of 0.8 mg/ml) and reactions were incubated for 30 min at 45°C to digest the type II topoisomerase. Samples were mixed with 2 µl of 30% sucrose, 0.5% bromophenol blue and 0.5% xylene cyanole FF in 10 mM Tris–HCl, pH 7.9. They were then heated at 70°C for 2 min and subjected to electrophoresis on a 1% agarose gel in 40 mM Tris–acetate, pH 8.3, 2 mM EDTA containing 0.5 µg/ml ethidium bromide. Cleavage was monitored by the conversion of negatively supercoiled plasmids to linear molecules. DNA bands were visualized by UV light, photographed through Kodak 23A and 12 filters with Polaroid type 665 positive/negative film, and quantitated by scanning photographic negatives with an E-C apparatus model EC910 scanning densitometer in conjunction with Hoefer GS-370 Data System software. The intensity of bands in the negative was proportional to the amount of DNA present.
Preparation of oligonucleotides
Experiments utilized one of two double-stranded 40mer oligonucleotides, designated Site I and II, and a double-stranded 42mer oligonucleotide, designated Site III. The top strand of the 40mer of Site I corresponds to residues 87–126 of pBR322 (35,36). The sequences of the top and bottom oligonucleotides were 5′-TGAAATCTAACAATG↓CGCTCATCGTCATCC-TCGGCACCGT-3′ and 5′-ACGGTGCCGAGGATGACGA-TG↓AGCGCATTGTTAGATTTCA-3′, respectively. The points of topoisomerase II-mediated DNA cleavage are denoted by arrows (36,37). Oligonucleotides were prepared on an Applied Biosystems DNA synthesizer. Top or bottom strand Site I oligonucleotides containing uracil, 8-oxoguanine, or O6-methylguanine were synthesized in a similar manner utilizing the corresponding phosphoramidite. Apurinic sites were generated by treating uracil-containing double-stranded oligonucleotides with uracil DNA glycosylase as previously described (21). Following this treatment, samples were prepared for topoisomerase II assays by the addition of KCl (100 mM final concentration) and MgCl2 (5 mM final concentration) (21).
The top strand of the Site II 40mer oligonucleotide corresponds to residues 1072–1111 of the MLL oncogene (38–40), which, together with its complementary oligonucleotide, were prepared as described above. The sequences of the top and bottom oligonucleotides were 5′-GCCTGGGTGACAAA-GC↓AAAACACTGTCTCCAAAAAAAATT-3′ and 5′-AATT-TTTTTTGGAGACAGTG↓TTTTGCTTTGTCACCCAGGC-3′, respectively. The points of topoisomerase II-mediated DNA cleavage are denoted by arrows (30,39). Top strand Site II oligonucleotides containing the abasic site analog tetrahydrofuran were prepared by Cruachem. Those containing 8-oxoadenine, N6-methyladenine or 1,N6-ethenoadenine were prepared on an Applied Biosystems DNA synthesizer utilizing the corresponding phosphoramidite.
The top strand of the Site III 42mer oligonucleotide corresponds to residues 1050–1091 of the MLL oncogene (31,38,39), which, together with its complementary oligonucleotide, were prepared as described above. The sequences of the top and bottom oligonucleotides were 5′-ATGATTGTACCACTGC-AG↓TCCAGCCTGGGTGACAAAGCAAAA-3′ and 5′-TTT-TGCTTTGTCACCCAGGC↓TGGACTGCAGTGGTACAA-TCAT-3′, respectively. The points of topoisomerase II-mediated DNA cleavage are denoted by arrows (30,39). Top and bottom strand Site III oligonucleotides containing 1,N6-ethenoadenine were prepared on an Applied Biosystems DNA synthesizer utilizing the corresponding phosphoramidite.
When appropriate, single-stranded oligonucleotides were labeled on their 5′-termini with [32P]phosphate (21). Oligonucleotides were purified by electrophoresis on polyacrylamide gels and annealed as described previously (21,36). The length and purity of all oligonucleotides was confirmed by polyacrylamide gel electrophoresis. The presence of adducted bases was confirmed by mass spectrometry. Oligonucleotides were prepared for mass spectrometry by polyacrylamide gel purification followed by desalting using a G-25 Sephadex column and a Millipore µ-C18 Zip Tip. Samples were eluted onto a sample plate using an 8:1 mixture of 50 mg/ml 3-hydroxypicolinic acid:50 mg/ml ammonium citrate in 50% acetonitrile. Mass spectral analyses were done using a Voyager Elite 2 MALDI-TOF system and software from PerSeptive Biosystems.
Topoisomerase II-mediated cleavage of oligonucleotides
Topoisomerase II-mediated DNA cleavage reactions were carried out as previously described by Kingma et al. (30). Assay mixtures contained 150 nM human topoisomerase IIα or β and 100 nM double-stranded Site I, II or III oligonucleotide in 20 µl of 10 mM HEPES–HCl, pH 7.9, 0.1 mM EDTA, 50 mM NaCl, 50 mM KCl, 5 mM MgCl2 and 2.5% glycerol. Reactions were incubated for 10 min at 37°C and stopped with 2 µl of 10% SDS followed by 2 µl of 250 mM EDTA. Cleavage products were digested with proteinase K (2 µl of a 1.6 mg/ml solution) for 20 min at 37°C, precipitated twice with ethanol and resuspended in 5 µl of 40% formamide, 8.5 mM EDTA, 0.02% bromophenol blue and 0.02% xylene cyanol FF. Products were resolved by electrophoresis in a denaturing 7 M urea, 14% polyacrylamide sequencing gel in 100 mM Tris-borate, pH 8.3, and 2 mM EDTA. Gels were fixed in 10% methanol and 10% acetic acid for 10 min and dried. Reaction products were visualized and quantified using a Molecular Dynamics PhosphorImager system.
Topoisomerase II-mediated DNA religation
DNA religation assays were carried out by a modification (40) of the procedure described by Osheroff and Zechiedrich (41). DNA cleavage/religation equilibria were established with 100 nM duplex Site I or II oligonucleotide and 150 nM topoisomerase II per reaction in 10 mM Tris–HCl, pH 7.9, 0.1 mM EDTA, 100 mM KCl, 5 mM CaCl2 and 2.5% glycerol. Topoisomerase II–DNA cleavage complexes were trapped by the addition of EDTA (final concentration, 6 mM). NaCl was added (final concentration, 500 mM) to prevent recleavage. Religation was initiated by the addition of MgCl2 (0.1 mM final concentration) and terminated by the addition of 2 µl of 10% SDS at times up to 60 s. Samples were analyzed as described in the preceding section. The apparent first order rate of DNA religation was determined by quantifying the loss of the DNA cleavage product.
RESULTS
Randomly located abasic sites stimulate plasmid DNA cleavage mediated by human type II topoisomerases
Abasic sites are the most commonly formed DNA lesions in the cell (42–45). It is estimated that approximately 10 000 abasic sites are created by spontaneous hydrolysis per mammalian cell per day (42–46). These lesions are also generated by a variety of environmental insults and cellular events, including oxidation, ionizing radiation, DNA reactive chemicals and base excision repair (43,45,47). As a result, some human tissues contain steady-state levels of abasic sites that are as high as 200 000 per genome (48,49). Thus, under typical cellular conditions, abasic sites may occur as frequently as once every 20 000 bp. Under conditions of environmental stress, the frequency of abasic sites may be three times higher (49).
To determine whether human type II topoisomerases can locate DNA lesions in a background of undamaged nucleotides, their ability to cleave pBR322 molecules that contain randomly located abasic sites was assessed. Abasic sites were generated by heating the plasmid at acidic pH. Under the conditions employed, the vast majority of lesions constitute a mixed population of apurinic (the predominant lesion) and apyrimidinic sites (42,44,45). The major contaminating lesion is deaminated cytosine; however, its rate of formation is only 0.1% that of apurinic site formation (50).
As seen in Figure 1, both topoisomerase IIα and β can detect the presence of random abasic sites within a pBR322 molecule that is nearly 4400 bp in length. A single abasic lesion per plasmid nearly doubled levels of enzyme-mediated DNA scission and the presence of approximately five lesions per plasmid increased cleavage 4–5-fold. As a comparison, more than 2000 molecules of the anticancer drug etoposide were required per plasmid to double cleavage mediated by either human topoisomerase II isoform (not shown). These results are similar to those found for Drosophila topoisomerase II (20) and demonstrate that the human type II enzymes can locate DNA lesions situated within several thousand base pairs of undamaged DNA.
Figure 1.

Randomly generated abasic sites stimulate plasmid DNA cleavage mediated by human type II topoisomerases. Data represent the average of two independent experiments and are expressed relative to cleavage observed with pBR322 containing 0 abasic sites, which was set to 1. Assays utilized either topoisomerase IIα (closed circles) or topoisomerase IIβ (open circles).
Positional preference of human topoisomerase IIβ for apurinic sites
Vertebrate species contain two distinct isoforms of topoisomerase II, α and β (1–7). Although these enzymes differ in their physiological regulation and function, both appear to be essential to survival (1,4,7,12). Previous studies demonstrated that apurinic sites are position-specific poisons of human topoisomerase IIα, and stimulate enzyme-mediated DNA cleavage only when they are located within the four base pairs that separate the two scissile bonds (30,40). Similar studies have yet to be carried out for human topoisomerase IIβ. Therefore, the effects of apurinic sites on DNA scission mediated by the β isoform were determined. Two independent oligonucleotides, each containing a single centrally located topoisomerase II cleavage site, were used for these experiments. The first, designated Site I, was derived from plasmid pBR322 and contains three guanine residues within the cleavage overhang (Fig. 2, left) (35,36). The second, designated Site II, was derived from the MLL gene at chromosomal band 11q23 and is located proximal to a leukemic breakpoint. This site contains a run of four adenine residues within the cleavage overhang (Fig. 2, right) (39,40). Both oligonucleotides have been used previously to assess the positional specificity of human topoisomerase IIα toward DNA lesions (30,40).
Figure 2.
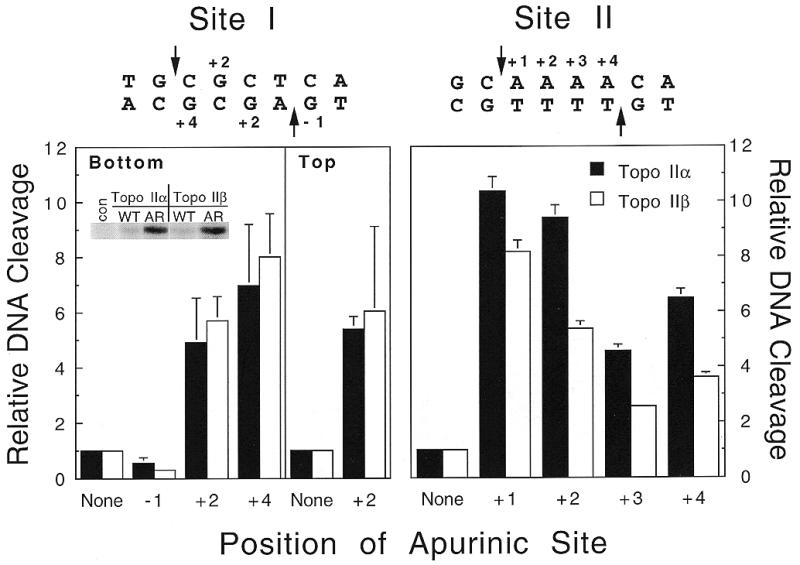
Apurinic sites act as position-specific poisons of human topoisomerase IIβ. Topoisomerase II-mediated DNA cleavage was monitored with Site I oligonucleotides (left) containing single apurinic sites at bottom strand positions –1, +2 or +4, or top strand position +2, or with Site II oligonucleotides (right) at top strand positions +1, +2, +3 or +4. Levels of cleavage were calculated relative to that of the wild-type sequence (None, set to 1). Reactions contained either topoisomerase IIα (closed bars) or topoisomerase IIβ (open bars). Inset, a representative gel comparing the amount of cleavage product seen with wild-type Site I substrate (WT) or with substrate containing an apurinic site at the bottom +4 position (AR). Data represent the averages of three to four independent experiments and standard deviations are indicated by error bars.
As seen in Figure 2, apurinic sites located between the scissile bonds stimulated DNA cleavage mediated by human topoisomerase IIβ. Although some differences in levels of cleavage enhancement were observed, the positional specificity of the β isoform toward these lesions paralleled that of topoisomerase IIα.
Effects of oxidized purines on oligonucleotide cleavage mediated by human type II topoisomerases
The eukaryotic genome is under the constant threat of oxidative stress, both from endogenous and exogenous sources (45,51). One of the most common oxidative lesions is 8-oxoguanine (51,52). It is estimated that as many as 100 000 8-oxoguanine adducts are formed each day in rat cells (53,54). Normal human tissues contain approximately 7500 of these adducts per genome (at steady state) and levels in smokers are ~50% higher (52,55–57).
Because of the prevalence of oxidative lesions in cellular DNA, their effects on DNA cleavage mediated by human topoisomerase IIα and β were assessed. Since purines are more reactive towards oxidation than pyrimidines, the model adducts utilized for this study were 8-oxoguanine and 8-oxoadenine (structures are shown in Fig. 3). Neither of these lesions induces significant perturbations in the double helix and both can form Watson–Crick base pairs (58,59). However, 8-oxoguanine and 8-oxoadenine are mutagenic because they allow polymerase misinsertion (60–62).
Figure 3.

Structures of DNA lesions. The structures of the DNA lesions utilized in this study are shown next to those of the unmodified bases.
At best, these oxidative lesions were modest poisons of human topoisomerase IIα or β (Fig. 4). For the most part, DNA cleavage levels were at or below baseline for oligonucleotides that contained 8-oxoguanine. DNA scission rose slightly (~1.5-fold) for 8-oxoguanine:adenine mispairs, the mutagenic mismatch associated with this lesion (60,61; not shown). However, cleavage was less than observed for oligonucleotides containing the corresponding guanine:adenine mispair.
Figure 4.

Effects of oxidized purines on the DNA cleavage activity of human type II topoisomerases. Individual DNA substrates containing 8-oxoguanine at the indicated positions of the Site I oligonucleotide (left) or 8-oxoadenine at the indicated positions of the Site II oligonucleotide (right) were characterized. DNA cleavage enhancement was calculated relative to the level of scission observed for the wild-type substrate (None, set to 1). Reactions contained either topoisomerase IIα (closed bars) or topoisomerase IIβ (open bars). Data represent the averages of three independent experiments and standard deviations are indicated by error bars.
The effects of 8-oxoadenine on topoisomerase II-mediated DNA scission were slightly greater. When the adduct was inserted at the +4 position of the 11q23 cleavage site (Site II), scission was 2–3-fold higher than wild-type levels.
Effects of alkylated purines on oligonucleotide cleavage mediated by human type II topoisomerases
Alkylated bases are generated by many cellular processes or exposure to a number of industrial chemicals (44,51,63). O6-methylguanine is one of the most mutagenic lesions that result from alkylation (51,64,65). It is present in normal human tissue, and levels may be as high as approximately 20 000 per genome in individuals with a history of smoking or occupational exposure to alkylating agents (51,66)
Initial studies on the effects of alkylated bases on topoisomerase II-mediated DNA cleavage focused on O6-methylguanine and N6-methyladenine (see Fig. 3 for structures). As seen with the oxidative lesions, these adducts induce little distortion in DNA and form base pairs with their appropriate partners (67,68). O6-methylguanine induces mutations because it frequently directs the misinsertion of thymine (51,69).
Once again, adducts that did not significantly alter the structure of the double helix had little effect on DNA cleavage mediated by topoisomerase IIα or β (Fig. 5). O6-Methylguanine enhanced scission 2–3-fold, while no enhancement was observed with N6-methyladenine. Together with the above results, these findings support the hypothesis that the ability of lesions to stimulate topoisomerase II-mediated DNA cleavage is related to their ability to distort the double helix (21,22,31).
Figure 5.

Effects of alkylated purines on the cleavage activity of human type II topoisomerases. Individual DNA substrates containing O6-methylguanine at the indicated positions of the Site I oligonucleotide (left) or N6-methyladenine at the indicated positions of the Site II oligonucleotide (right) were characterized. DNA cleavage enhancement was calculated relative to the level of scission observed for the wild-type substrate (None, set to 1). Reactions contained either topoisomerase IIα (closed bars) or topoisomerase IIβ (open bars). Data represent the averages of three independent experiments and standard deviations are indicated by error bars.
To further address this hypothesis, the effects of 1,N6-ethenoadenine (shown in Fig. 3) on DNA cleavage of the Site II oligonucleotide by human topoisomerase IIα and β were determined. This adduct is formed by products of lipid peroxidation or exposure to industrial carcinogens such as vinyl chloride (51,70,71). 1,N6-Ethenoadenine does not base pair with thymine and forces the base opposite to adopt a non-planar conformation (72).
In marked contrast to lesions that allow proper base pairing, 1,N6ethenoadenine profoundly affected topoisomerase II-mediated DNA cleavage (Fig. 6). DNA scission by human topoisomerase IIα was increased ~10-fold, while that by topoisomerase IIβ was increased up to 7-fold. This enhancement was similar to those observed for apurinic sites (Fig. 2). Moreover, the positional specificity of human type II topoisomerases for 1,N6-ethenoadenine was similar to that of apurinic lesions.
Figure 6.

1,N6-Ethenoadenine stimulates the cleavage activity of human type II topoisomerases. Individual DNA substrates containing 1,N6-ethenoadenine at the indicated positions of the Site II (left) or Site III (right) oligonucleotides were characterized. DNA cleavage enhancement was calculated relative to the level of scission observed for the wild-type substrate (None, set to 1). Reactions contained either topoisomerase IIα (closed bars) or topoisomerase IIβ (open bars). Data represent the averages of two or three independent experiments and standard deviations are indicated by error bars.
Because of the large effect of 1,N6-ethenoadenine on topoisomerase II-mediated DNA cleavage, the ability of this lesion to enhance scission was assessed using an additional DNA cleavage sequence (Site III) from the MLL oncogene. This site contains two adenine residues located at the +4 positions on the top and bottom strands, respectively (Fig. 6). Substitution of either of these adenine residues with 1,N6-ethenoadenine resulted in a substantial increase in DNA cleavage mediated by topoisomerase IIα or β (Fig. 6). Taken together, these results indicate that 1,N6-ethenoadenine is a potent position-specific poison of human type II topoisomerases.
Effects of lesions on DNA religation mediated by human topoisomerase IIα
Since covalent topoisomerase II–DNA cleavage complexes are formed in an equilibrium reaction, there are two possible mechanisms (that are not mutually exclusive) by which poisons can increase levels of enzyme-generated DNA breaks. They can block the ability of topoisomerase II to religate cleaved DNA molecules or increase the formation of cleavage complexes (2,3). Some poisons, such as the anticancer drugs etoposide and amsacrine, strongly inhibit enzyme-mediated DNA religation (2,3,13). In contrast, others such as quinolones and genistein have little effect on religation and therefore are presumed to act primarily by enhancing the formation of enzyme–DNA cleavage complexes (2,3,13).
Previous studies indicate that abasic sites and base mismatches have little effect on DNA religation mediated by Drosophila topoisomerase II or human topoisomerase IIα (20,21,29,40). Consequently, these lesions are believed to act primarily by the latter mechanism. To extend these findings, the present study characterized rates of religation for a broader spectrum of DNA lesions. Oligonucleotides contained the adducted bases in the +4 position of topoisomerase II DNA cleavage overhang, because this position represents the location at which the most DNA cleavage enhancement was observed for the lesions employed.
Human topoisomerase IIα was used as the experimental model for this study. Results are shown in Table 1. A plot of DNA religation versus time for 1,N6-ethenoadenine (which displayed the most dramatic stimulation of enzyme-mediated DNA cleavage) is shown in Figure 7. For the most part, religation rates calculated for the lesion-containing oligonucleotides were comparable to (or faster than) those determined for the wild-type oligonucleotides. The only exception was the 8-oxoadenine substrate, which was ligated at a rate that was ~30% slower than wild type. This decrease is not significant compared to the effects of drugs such as etoposide, which diminish religation rates by nearly an order of magnitude (73,74). These findings indicate that the oxidized and alkylated bases used in the present study do not inhibit DNA religation mediated by human topoisomerase IIα. Therefore, it is likely that 1,N6-ethenoadenine lesions increase levels of DNA breaks by enhancing the rate of formation of enzyme–DNA cleavage complexes.
Table 1. Rates of topoisomerase IIα-mediated religation of oligonucleotides containing DNA lesions at the +4 position.
| Oligonucleotide | Lesion | Religation rate (s–1) |
|---|---|---|
| Site I | None | 0.035 |
| Apurinic | 0.069 | |
| 8-oxoguanine | 0.075 | |
| |
O6-methylguanine |
0.044 |
| Site II | None | 0.046 |
| Apurinic | 0.063 | |
| 8-oxoadenine | 0.032 | |
| N6-methyladenine | 0.046 | |
| 1,N6-ethenoadenine | 0.046 |
Figure 7.

Effect of 1,N6-ethenoadenine on the religation activity of human topoisomerase IIα. Religation of the Site II oligonucleotide containing a single 1,N6-ethenoadenine lesion at the top +4 position (closed squares) was compared to religation seen with wild-type substrate (closed circles) or with substrate containing a +4 apurinic site (open circles). Data represent the averages of two or three independent experiments.
DISCUSSION
The human genome is constantly subjected to a variety of endogenous and environmental insults (45,51). These insults generate numerous forms of DNA damage, including abasic sites, mismatches, adducted bases, strand breaks and crosslinks (45,51). In an effort to maintain genomic integrity, the cell has evolved numerous repair systems to correct this DNA damage (45,75–78).
DNA lesions are deleterious to cell survival because they induce mutations in the genetic material and interfere with fundamental nuclear processes such as DNA replication and transcription (45,76,79–81). Beyond these effects, recent studies indicate that some lesions also act as position-specific poisons of eukaryotic topoisomerase I or II (21,28–30,82–84). If these enzymes interact with DNA lesions before they are repaired, the resulting stimulation of topoisomerase-mediated DNA cleavage has the capacity to convert strand-specific damage to permanent breaks in the double helix. Given the potential physiological ramifications of such an event, it is important to understand how topoisomerase I and II react to a broad spectrum of DNA damage.
Previous studies, which focused primarily on Drosophila topoisomerase II and human topoisomerase IIα, characterized interactions of the type II enzyme with abasic sites and base mismatches (20,21,28–30). The present work extends these studies by determining the effects of abasic lesions on human topoisomerase IIβ and those of oxidized and alkylated bases on the DNA cleavage activity of both human isoforms. For all lesions examined, similar results were found for both enzymes. Data indicate that adducts that allow base pairing and induce little distortion in the double helix have modest effects on DNA cleavage mediated by human type II topoisomerases. In contrast, 1,N6-ethenoadenine, a lesion which disrupts base pairing, is a potent topoisomerase II poison. This adduct increased DNA cleavage by an order of magnitude, and is the first lesion found to rival the stimulatory effects of apurinic sites.
The present work underscores differences between the response of type I and type II topoisomerases to individual forms of DNA damage. For example, 1,N6-ethenoadenine markedly inhibited DNA religation by topoisomerase I (83), but had no effect on religation mediated by topoisomerase IIα. In addition, the response of topoisomerase I to 8-oxoguanine was dramatically different from that of the type II enzyme. This oxidative lesion was a potent topoisomerase I poison (84), but had marginal (if any) effects on DNA scission mediated by topoisomerase II. The basis for these differences is not known. However, they point to fundamental distinctions between the mechanisms used by type I and type II topoisomerases to recognize and interact with their DNA substrates.
It is not yet known whether type II topoisomerases interact with lesions that exist in cellular DNA. To do so would require the enzyme to locate DNA damage within a considerable excess of undamaged nucleotides. To this point, Drosophila (20) and human type II topoisomerases can detect a single abasic site located within a background of >4000 bp. This number represents the size of the plasmid employed (pBR322) rather than a true upper limit for the detection of DNA damage. Although future experiments will be required to establish the actual scanning range of the enzyme, the observation that some human tissues contain as many as one abasic site for every 20 000 bp of genomic DNA (48,49) strongly suggests that type II topoisomerases have the potential to locate DNA damage in a physiological setting.
Finally, it should be noted that many of the lesions employed in the present study are converted to apurinic sites by the initial step of base excision repair pathways (45,47,77,85–87). Thus, even if these lesions have little direct influence on topoisomerase II-mediated DNA cleavage, their presence in the genome still has the potential to poison the enzyme indirectly.
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to Dr Ole Westergaard and Dr Anni H. Andersen for providing constructs used in the overexpression and purification of human topoisomerase IIβ. We thank the Vanderbilt University School of Medicine Mass Spectrometry Research Center for the use of their facilities and James N. Riggins for his help in oligonucleotide mass spectral analysis. We also thank Dr D. Andrew Burden and Mr John M. Fortune for critical reading of the manuscript. This work was supported by grant GM53960 from the National Institutes of Health. M.S. was a trainee under National Institutes of Health Grant 5 T32 CA09582.
REFERENCES
- 1.Wang J.C. (1996) Annu. Rev. Biochem., 65, 635–692. [DOI] [PubMed] [Google Scholar]
- 2.Burden D.A. and Osheroff,N. (1998) Biochim. Biophys. Acta, 1400, 139–154. [DOI] [PubMed] [Google Scholar]
- 3.Fortune J.M. and Osheroff,N. (2000) Prog. Nucleic Acids Res. Mol. Biol., 64, 221–253. [DOI] [PubMed] [Google Scholar]
- 4.Nitiss J.L. (1998) Biochim. Biophys. Acta, 1400, 63–81. [DOI] [PubMed] [Google Scholar]
- 5.Drake F.H., Zimmerman,J.P., McCabe,F.L., Bartus,H.F., Per,S.R., Sullivan,D.M., Ross,W.E., Mattern,M.R., Johnson,R.K. and Crooke,S.T. (1987) J. Biol. Chem., 262, 16739–16747. [PubMed] [Google Scholar]
- 6.Drake F.H., Hofmann,G.A., Bartus,H.F., Mattern,M.R., Crooke,S.T. and Mirabelli,C.K. (1989) Biochemistry, 28, 8154–8160. [DOI] [PubMed] [Google Scholar]
- 7.Austin C.A. and Marsh,K.L. (1998) Bioessays, 20, 215–226. [DOI] [PubMed] [Google Scholar]
- 8.Isaacs R.J., Davies,S.L., Sandri,M.I., Redwood,C., Wells,N.J. and Hickson,I.D. (1998) Biochim. Biophys. Acta, 1400, 121–137. [DOI] [PubMed] [Google Scholar]
- 9.Grue P., Grasser,A., Sehested,M., Jensen,P.B., Uhse,A., Straub,T., Ness,W. and Boege,F. (1998) J. Biol. Chem., 273, 33660–33666. [DOI] [PubMed] [Google Scholar]
- 10.Holden J.A., Rolfson,D.H. and Wittwer,C.T. (1992) Oncol. Res., 4, 157–166. [PubMed] [Google Scholar]
- 11.Jensen S., Redwood,C.S., Jenkins,J.R., Andersen,A.H. and Hickson,I.D. (1996) Mol. Gen. Genet., 252, 79–86. [PubMed] [Google Scholar]
- 12.Yang X., Li,W., Prescott,E.D., Burden,S.J. and Wang,J.C. (2000) Science, 287, 131–134. [DOI] [PubMed] [Google Scholar]
- 13.Corbett A.H. and Osheroff,N. (1993) Chem. Res. Toxicol., 6, 585–597. [DOI] [PubMed] [Google Scholar]
- 14.Osheroff N., Corbett,A.H. and Robinson,M.J. (1994) In Liu,L. (ed.), Advances in Pharmacology. Academic Press, New York, NY, Vol. 29, pp. 105–126. [DOI] [PubMed]
- 15.Froelich-Ammon S.J. and Osheroff,N. (1995) J. Biol. Chem., 270, 21429–21432. [DOI] [PubMed] [Google Scholar]
- 16.Pommier Y. (1997) In Teicher,B.A. (ed.), Cancer Therapeutics: Experimental and Clinical Agents. Humana Press, Totowa, NJ, Vol. I, pp. 153–174.
- 17.Hande K.R. (1998) Biochim. Biophys. Acta, 1400, 173–184. [DOI] [PubMed] [Google Scholar]
- 18.Chen A.Y. and Liu,L.F. (1994) Annu. Rev. Pharmacol. Toxicol., 34, 191–218. [DOI] [PubMed] [Google Scholar]
- 19.Kreuzer K.N. and Cozzarelli,N.R. (1979) J. Bacteriol., 140, 424–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kingma P.S., Corbett,A.H., Burcham,P.C., Marnett,L.J. and Osheroff,N. (1995) J. Biol. Chem., 270, 21441–21444. [DOI] [PubMed] [Google Scholar]
- 21.Kingma P.S. and Osheroff,N. (1997) J. Biol. Chem., 272, 1148–1155. [DOI] [PubMed] [Google Scholar]
- 22.Kingma P.S. and Osheroff,N. (1998) Biochim. Biophys. Acta, 1400, 223–232. [DOI] [PubMed] [Google Scholar]
- 23.Corbett A.H., Hong,D. and Osheroff,N. (1993) J. Biol. Chem., 268, 14394–14398. [PubMed] [Google Scholar]
- 24.Freudenreich C.H. and Kreuzer,K.N. (1994) Proc. Natl Acad. Sci. USA, 91, 11007–11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Osheroff N., Corbett,A.H., Elsea,S.H. and Westergaard,M. (1994) Cancer Chemother. Pharmacol., 34, S19–S25. [DOI] [PubMed] [Google Scholar]
- 26.Elsea S.H., Westergaard,M., Burden,D.A., Lomenick,J.P. and Osheroff,N. (1997) Biochemistry, 36, 2919–2924. [DOI] [PubMed] [Google Scholar]
- 27.Kwok Y., Zeng,Q. and Hurley,L.H. (1998) Proc. Natl Acad. Sci. USA, 95, 13531–13536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bigioni M., Zunino,F., Tinelli,S., Austin,C.A., Willmore,E. and Capranico,G. (1996) Biochemistry, 35, 153–159. [DOI] [PubMed] [Google Scholar]
- 29.Kingma P.S. and Osheroff,N. (1997) J. Biol. Chem., 272, 7488–7493. [DOI] [PubMed] [Google Scholar]
- 30.Kingma P.S., Greider,C.A. and Osheroff,N. (1997) Biochemistry, 36, 5934–5939. [DOI] [PubMed] [Google Scholar]
- 31.Cline S.D., Jones,W.R., Stone,M.P. and Osheroff,N. (1999) Biochemistry, 38, 15500–15507. [DOI] [PubMed] [Google Scholar]
- 32.Wasserman R.A., Austin,C.A., Fisher,L.M. and Wang,J.C. (1993) Cancer Res., 53, 3591–3596. [PubMed] [Google Scholar]
- 33.Cabral Neto J.B., Gentil,A., Caseira Cabral,R.E. and Sarasin,A. (1992) J. Biol. Chem., 267, 19718–19723. [PubMed] [Google Scholar]
- 34.Corbett A.H., Zechiedrich,E.L., Lloyd,R.S. and Osheroff,N. (1991) J. Biol. Chem., 266, 19666–19671. [PubMed] [Google Scholar]
- 35.Sutcliffe J.G. (1978) Cold Spring Harbor Symp. Quant. Biol., 43, 77–90. [DOI] [PubMed] [Google Scholar]
- 36.Corbett A.H., Zechiedrich,E.L. and Osheroff,N. (1992) J. Biol. Chem., 267, 683–686. [PubMed] [Google Scholar]
- 37.Sander M. and Hsieh,T. (1983) J. Biol. Chem., 258, 8421–8428. [PubMed] [Google Scholar]
- 38.Gu Y., Alder,H., Nakamura,T., Schichman,S.A., Prasad,R., Canaani,O., Saito,H., Croce,C.M. and Canaani,E. (1994) Cancer Res., 54, 2326–2330. [PubMed] [Google Scholar]
- 39.Felix C.A., Lange,B.J., Hosler,M.R., Fertala,J. and Bjornsti,M.-A. (1995) Cancer Res., 55, 4287–4292. [PubMed] [Google Scholar]
- 40.Kingma P.S. and Osheroff,N. (1998) J. Biol. Chem., 273, 17999–18002. [DOI] [PubMed] [Google Scholar]
- 41.Osheroff N. and Zechiedrich,E.L. (1987) Biochemistry, 26, 4303–4309. [DOI] [PubMed] [Google Scholar]
- 42.Lindahl T. and Nyberg,B. (1972) Biochemistry, 11, 3610–3618. [DOI] [PubMed] [Google Scholar]
- 43.Loeb L.A. and Preston,B.D. (1986) Annu. Rev. Genet., 20, 201–230. [DOI] [PubMed] [Google Scholar]
- 44.Lindahl T. (1993) Nature, 362, 709–715. [DOI] [PubMed] [Google Scholar]
- 45.Friedberg E.C., Walker,G.C. and Siede,W. (1995) DNA Repair and Mutagenesis, 2nd Edn. American Society for Microbiology Press, Washington DC.
- 46.Nakamura J., Walker,V.E., Upton,P.B., Chiang,S.Y., Kow,Y.W. and Swenberg,J.A. (1998) Cancer Res., 58, 222–225. [PubMed] [Google Scholar]
- 47.Wilson S.H. (1998) Mutat. Res., 407, 203–215. [DOI] [PubMed] [Google Scholar]
- 48.Nakamura J. and Swenberg,J.A. (1999) Cancer Res., 59, 2522–2526. [PubMed] [Google Scholar]
- 49.Atamna H., Cheung,I. and Ames,B.N. (2000) Proc. Natl Acad. Sci. USA, 97, 686–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Drake J.W. and Baltz,R.H. (1976) Annu. Rev. Biochem., 45, 11–37. [DOI] [PubMed] [Google Scholar]
- 51.Marnett L.J. and Burcham,P.C. (1993) Chem. Res. Toxicol., 6, 771–785. [DOI] [PubMed] [Google Scholar]
- 52.Beckman K.B. and Ames,B.N. (1997) J. Biol. Chem., 272, 19633–19636. [DOI] [PubMed] [Google Scholar]
- 53.Fraga C.G., Shigenaga,M.K., Park,J.W., Degan,P. and Ames,B.N. (1990) Proc. Natl Acad. Sci. USA, 87, 4533–4537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Park E.M., Shigenaga,M.K., Degan,P., Korn,T.S., Kitzler,J.W., Wehr,C.M., Kolachana,P. and Ames,B.N. (1992) Proc. Natl Acad. Sci. USA, 89, 3375–3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Asami S., Hirano,T., Yamaguchi,R., Tomioka,Y., Itoh,H. and Kasai,H. (1996) Cancer Res., 56, 2546–2549. [PubMed] [Google Scholar]
- 56.Fraga C.G., Motchnik,P.A., Wyrobek,A.J., Rempel,D.M. and Ames,B.N. (1996) Mutat. Res., 351, 199–203. [DOI] [PubMed] [Google Scholar]
- 57.Yamaguchi R., Itoh,H. and Kasai,H. (1997) Carcinogenesis, 18, 1763–1766. [DOI] [PubMed] [Google Scholar]
- 58.Guschlbauer W., Duplaa,A.M., Guy,A., Teoule,R. and Fazakerley,G.V. (1991) Nucleic Acids Res., 19, 1753–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lipscomb L.A., Peek,M.E., Morningstar,M.L., Verghis,S.M., Miller,E.M., Rich,A., Essigmann,J.M. and Williams,L.D. (1995) Proc. Natl Acad. Sci. USA, 92, 719–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cheng K.C., Cahill,D.S., Kasai,H., Nishimura,S. and Loeb,L.A. (1992) J. Biol. Chem., 267, 166–172. [PubMed] [Google Scholar]
- 61.Wood M.L., Esteve,A., Morningstar,M.L., Kuziemko,G.M. and Essigmann,J.M. (1992) Nucleic Acids Res., 20, 6023–6032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kamiya H., Miura,H., Murata-Kamiya,N., Ishikawa,H., Sakaguchi,T., Inoue,H., Sasaki,T., Masutani,C., Hanaoka,F., Nishimura,S. and Ohtsuka,E. (1995) Nucleic Acids Res., 23, 2893–2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kyrtopoulos S.A. (1998) Mutat. Res., 405, 135–143. [DOI] [PubMed] [Google Scholar]
- 64.Newbold R.F., Warren,W., Medcalf,A.S.C. and Amos,J. (1980) Nature, 283, 596–599. [DOI] [PubMed] [Google Scholar]
- 65.Kaina B., Ziouta,A., Ochs,K. and Coquerelle,T. (1997) Mutat. Res., 381, 227–241. [DOI] [PubMed] [Google Scholar]
- 66.Kang H., Konishi,C., Kuroki,T. and Huh,N. (1993) Environ. Health Perspect., 99, 269–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gaffney B.L. and Jones,R.A. (1989) Biochemistry, 28, 5881–5889. [DOI] [PubMed] [Google Scholar]
- 68.Leonard G.A., Thomson,J., Watson,W.P. and Brown,T. (1990) Proc. Natl Acad. Sci. USA, 87, 9573–9576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Singer B. and Dosanjh,M.K. (1990) Mutat. Res., 233, 45–51. [DOI] [PubMed] [Google Scholar]
- 70.Swenberg J.A., Fedtke,N., Ciroussel,F., Barbin,A. and Bartsch,H. (1992) Carcinogenesis, 13, 727–729. [DOI] [PubMed] [Google Scholar]
- 71.el Ghissassi F., Barbin,A., Nair,J. and Bartsch,H. (1995) Chem. Res. Toxicol., 8, 278–283. [DOI] [PubMed] [Google Scholar]
- 72.Kouchakdjian M., Eisenberg,M., Yarema,K., Basu,A., Essigmann,J. and Patel,D.J. (1991) Biochemistry, 30, 1820–1828. [DOI] [PubMed] [Google Scholar]
- 73.Osheroff N. (1989) Biochemistry, 28, 6157–6160. [DOI] [PubMed] [Google Scholar]
- 74.Robinson M.J. and Osheroff,N. (1991) Biochemistry, 30, 1807–1813. [DOI] [PubMed] [Google Scholar]
- 75.Demple B. and Harrison,L. (1994) Annu. Rev. Biochem., 63, 915–948. [DOI] [PubMed] [Google Scholar]
- 76.Sancar A. (1995) Annu. Rev. Genet., 29, 69–105. [DOI] [PubMed] [Google Scholar]
- 77.Wood R.D. (1996) Annu. Rev. Biochem., 65, 135–167. [DOI] [PubMed] [Google Scholar]
- 78.Yu Z., Chen,J., Ford,B.N., Brackley,M.E. and Glickman,B.W. (1999) Environ. Mol. Mutagen., 33, 3–20. [DOI] [PubMed] [Google Scholar]
- 79.Hanawalt P.C. (1998) Mutat. Res., 400, 117–125. [DOI] [PubMed] [Google Scholar]
- 80.Wang D., Kreutzer,D.A. and Essigmann,J.M. (1998) Mutat. Res., 400, 99–115. [DOI] [PubMed] [Google Scholar]
- 81.Eckert K.A. and Opresko,P.L. (1999) Mutat. Res., 424, 221–236. [DOI] [PubMed] [Google Scholar]
- 82.Pourquier P., Ueng,L.M., Kohlhagen,G., Mazumder,A., Gupta,M., Kohn,K.W. and Pommier,Y. (1997) J. Biol. Chem., 272, 7792–7796. [DOI] [PubMed] [Google Scholar]
- 83.Pourquier P., Bjornsti,M.-A. and Pommier,Y. (1998) J. Biol. Chem., 273, 27245–27249. [DOI] [PubMed] [Google Scholar]
- 84.Pourquier P., Ueng,L.-M., Fertala,J., Wang,D., Park,H.-J., Essigmann,J.M., Bjornsti,M.-A. and Pommier,Y. (1999) J. Biol. Chem., 274, 8516–8523. [DOI] [PubMed] [Google Scholar]
- 85.Oesch F., Adler,S., Rettelbach,R. and Doerjer,G. (1986) IARC Sci. Publ., 373–379. [PubMed] [Google Scholar]
- 86.Tchou J., Kasai,H., Shibutani,S., Chung,M.H., Laval,J., Grollman,A.P. and Nishimura,S. (1991) Proc. Natl Acad. Sci. USA, 88, 4690–4694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dosanjh M.K., Chenna,A., Kim,E., Fraenkel-Conrat,H., Samson,L. and Singer,B. (1994) Proc. Natl Acad. Sci. USA, 91, 1024–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]