

Abstract
Background:
Primary ciliary dyskinesia (PCD) is typically an autosomal recessive disease characterized by recurrent infections of the lower respiratory tract, frequent and severe otitis media, chronic rhinosinusitis, neonatal respiratory distress, and organ laterality defects. While severe lower respiratory tract infections and bronchiectasis are common in Inuit, PCD has not been recognized in this population.
Methods:
We report a case series of seven Inuit patients with PCD identified by genetic testing in three Canadian PCD centers.
Results:
Patients ranged from 4 to 59 years of age (at time of last evaluation) and originated in the Qikiqtaaluk region (Baffin Island, n = 5), Nunavut, or Nunavik (northern Quebec, n = 2), Canada. They had typical features of PCD, including neonatal respiratory distress (five patients), situs inversus totalis (four patients), bronchiectasis (four patients), chronic atelectasis (six patients), and chronic otitis media (six patients). Most had chronic rhinitis. Genetic evaluation demonstrated that all had homozygous pathogenic variants in DNAH11 at NM_001277115.1:c.4095+2C>A.
Conclusions:
The discovery of this homozygous DNAH11 variant in widely disparate parts of the Nunangat (Inuit homelands) suggests this is a founder mutation that may be widespread in Inuit. Thus, PCD may be an important cause of chronic lung, sinus, and middle ear disease in this population. Inuit with chronic lung disease, including bronchiectasis or laterality defects, should undergo genetic testing for PCD. Consideration of including PCD genetic analysis in routine newborn screening should be considered in Inuit regions.
Keywords: bronchiectasis, ciliary motility disorders, Inuits, Kartagener syndrome
1 |. INTRODUCTION
Primary ciliary dyskinesia (PCD) is predominantly an autosomal recessive, chronic disease characterized by recurrent infections of the lower respiratory tract, frequent and severe otitis media, chronic rhinosinusitis, neonatal respiratory distress with atelectasis, and, particularly in males, infertility.1,2 The clinical findings in PCD are a result of genetically determined abnormalities in ciliary ultrastructure, resulting in dysmotile respiratory cilia that are unable to effectively perform normal mucociliary clearance. To date, 51 genes involved in ciliary ultrastructure and function have been associated with PCD.3 The estimated prevalence of PCD is between 1:10,000 and 1:40,000, though a recent study suggests it may be as high as 1 per 7600 births.2,4 Of those with PCD, approximately 50% are born with situs inversus totalis (SIT), a mirror‐image reversal of abdominal and thoracic organs. Accurate PCD diagnosis is difficult as numerous clinical tests can be used, including ciliary transmission electron microscopy (TEM), nasal nitric oxide measurement, high‐speed videomicroscopy analysis of ciliary motion, immunofluorescent staining of ciliary proteins, and genetic testing, with varying degrees of success outside of highly specialized centers, and no single test is completely sensitive.
Bronchiectasis and chronic suppurative lung disease are unusually common in Inuit populations from Nunavut, Canada, and Alaska, in the United States, with an estimated prevalence of 202 per 100,000 children.5,6 The etiology of this increased prevalence is incompletely understood. Several large series have suggested that bronchiectasis in these populations is most often the sequelae of severe viral respiratory infections occurring early in life, but there have not been any contemporary publications investigating PCD in Indigenous peoples.5,6 An early Alaskan report failed to diagnose PCD in a large case series,7 and a similar but smaller study of Canadian First Nations children also failed to detect PCD patients.8 However, these studies only used ciliary TEM, and did not utilize PCD genetic testing, which is now routinely suggested for PCD diagnosis in North America.9
The Inuit are one of three constitutionally recognized Indigenous Peoples of Canada, alongside the First Nations and the Metis. The Inuit are the Indigenous Peoples of the Arctic. The Inuit homeland, referred to as Inuit Nunangat, consists of four Arctic sub‐regions, including Nunavut, Nunavik (Arctic Quebec), the Inuvialuit Settlement of Northwest Territory, and Nunatsiavut (Labrador).10 We report a case series of Inuit patients from two of these regions (Nunavut and Nunavik) with genetically confirmed PCD, all with the same homozygous, pathogenic variant in the dynein axonemal heavy chain 11 (DNAH11) gene. As PCD caused by variants in DNAH11 results in normal ciliary ultrastructure, past reliance on TEM analysis in Inuit cohorts failed to clinically detect this form of PCD. These are the first reported patients with PCD in this population.
2 |. METHODS
All but one patient were initially seen for chronic respiratory disease through the respiratory clinics at the McGill University Health Centre (MUHC—Montreal, Canada), the Children’s Hospital of Eastern Ontario (CHEO—Ottawa, Canada), or the Hospital for Sick Children (HSC—Toronto, Canada). Clinical information was obtained from routine medical evaluation or research protocols, and all patients underwent multigene panel testing in clinical laboratories at Blueprint Genetics (https://blueprintgenetics.com/), Invitae (https://www.invitae.com), Fulgent Genetics (https://www.fulgentgenetics.com/), or Ambry Genetics (https://ambrygen.com). One patient was enrolled in the Genetic Disorders of Mucociliary Clearance Consortium (GDMCC) 5902 genetic research protocol (NCT00323167) and one in both the Care4Rare Canada whole exome sequencing protocol (https://www.care4rare.ca/) as well as the Silent Genomes Precision Diagnosis sequencing protocol (https://www.bcchr.ca/silent-genomes-project), performing research targeted Sanger sequencing, multigene panels whole exome sequencing or whole genome sequencing, using similar methods to those previously described.11–14 Nasal nitric oxide testing was performed per standardized protocol, as previously described,15 for patients seen at the MUHC or HSC. Ciliary TEM was performed on a case‐by‐case basis in the clinical laboratory of the MUHC, HSC, CHEO, or in the central GDMCC research laboratory, as previously described.16–18
3 |. ETHICS STATEMENT
Community consultations were carried out and support was obtained from the Nunavut Tunngavik Inc., Qikiqtani Inuit Association (QIA), and the Department of Health, Government of Nunavut. Following these consultations, we obtained ethics approval from the CHEO Research Ethics Board and Nunavut Research Institute (License 01 004 22N‐M). All patients and/or their families provided written informed consent. Additionally, protocols for human studies were approved by the Institutional Review Board at the University of North Carolina, the McGill University Health Centre Research Institute, Children’s Hospital of Eastern Ontario Research Institute, University of British Columbia, and the Hospital for Sick Children, and studies were performed in compliance with the Ethics regulations.
4 |. RESULTS—PATIENTS
4.1 |. Extended family A
4.1.1 |. Case A1
A female born at 34 + 1 weeks, from west Qikiqtaaluk region (Baffin Island), Nunavut developed respiratory distress after birth, requiring hospitalization for 2 months, and intermittent oxygen therapy. The family history was notable for a mother with bronchiectasis. Abdominal ultrasound and echocardiogram completed in the first 2 days of life revealed SIT. TEM revealed normal ciliary ultrastructure. She had multiple episodes of shifting atelectasis and chronic sinusitis (Table 1).
TABLE 1.
Phenotypic characteristics of Inuit patients with PCD from homozygous DNAH11 variants at c.4095+2C>A.
| Case | Age (at last assessment) | Neonatal Respiratory Distress | Situs | Chronic otitis | Chronic wet cough | Chronic rhinosinusitis | Chronic Atelectasisa | Bronchiectasisb | FEV1% predicted | TEM | nNO (nL/min) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| A1 | 10 years | ✓ | SIT | ✓ | ✓ | ✓ | ✓ | ✓ | 73% | Normal | 5.6 |
| A2 | 10 years | ✓ | SS | ✓ | ✓ | ✓ | ✓ | Not seen on CT at 4 years of age | 114% | Normal | ND |
| A3 | 4 years | ✓ | SA | No | ✓ | ✓ | ✓ | No CT | ND | Normal | ND |
| 4 | 13 years | ✓ | SIT | ✓ | ✓ | +/− | ✓ | ✓ | 71% | Normal | ND |
| 5 | 11 years | No | SS | ✓ | No | No | No | No CT | ND | ND | ND |
| D6 | 56 years | ✓ | SIT | ✓ | ✓ | ✓ | ✓ | ✓ | 77% | Normal | 23 |
| D7 | 59 years | Unknown | SS | ✓ | ✓ | ✓ | ✓ | ✓ | 107% | ND | 8.9 |
Note: Summary of typical PCD phenotypic findings among patients.
Abbreviations: FEV1%, percent predicted of forced expiratory volume in 1 s; ND, not done; nNO, nasal nitric oxide; SA, situs ambiguus; SIT, situs inversus totalis; SS, situs solitus; TEM, transmission electron microscopy.
On chest radiography or chest computed tomography.
On chest computed tomography.
She was admitted at 3 years of age with pneumonia and atelectasis and a chest computerized tomography (CT) scan revealed bronchiectasis in the left upper, left middle, right lingula, and right lower lobe (Figure 1). At 4 years of age, she was found to have bilateral serous otitis media and bilateral mild hearing loss. Respiratory cultures in the first 10 years of life grew Streptococcus pneumoniae and Haemophilus influenzae. Nasal nitric oxide was markedly reduced at 5.6 nL/min.
FIGURE 1.
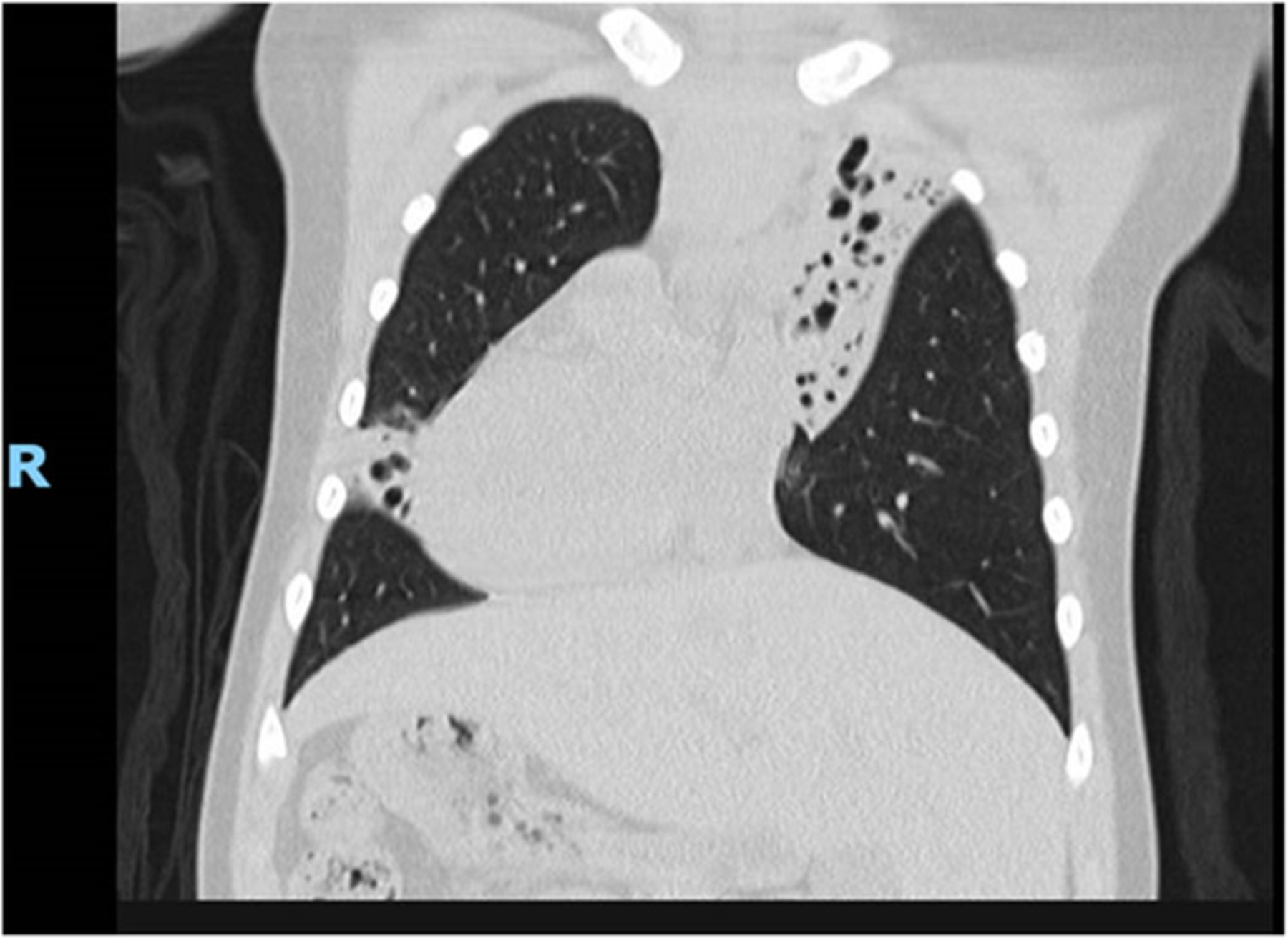
Coronal CT chest demonstrating dextrocardia and severe bronchiectasis in the left hemithorax (upper lobe) and right‐sided lingula. This patient also has known bronchiectasis in the left‐sided middle lobe and right‐sided lower lobe (not pictured here). CT, computerized tomography.
Clinical molecular genetic testing revealed a homozygous pathogenic variant (DNAH11; NM_001277115.1:c.4095+2C>A), in keeping with a diagnosis of PCD. No other pathogenic variants were identified. This splicing mutation occurs in the canonical splice donor site of intron 22 of the 82 exons of DNAH11. It is predicted to affect splicing of the gene’s full‐length transcript, most likely resulting in a frameshift with premature truncation, and is therefore considered to result in loss of protein expression (Supporting Information: Figure 1). This allele is not represented in gnomAD (gnomad.broadinstitute.org) nor in the ExAC database (exac.broadinstitute.org), which are comprised of data from >125,000 and >60,000 individuals, respectively. SpliceAI predicts loss of the donor site with high confidence (0.96; https://spliceailookup.broadinstitute.org), a prediction further supported by MaxEntScan. This variant is classifiable as likely pathogenic both by the American College of Medical Genetics and Genomics criteria (very strong evidence of pathogenicity—meets PVS1 criteria and moderate evidence of pathogenicity, based on PM2 criteria, strong evidence of pathogenicity based on PS4 criteria, by applying the evidence from this case series), commercial laboratories reporting this variant to ClinVar (variation Id:580732 and dbSNP Id: rs532007878), and by in‐house prediction bioinformatic analysis.19,20 We did not apply criterion PP5 (supporting evidence of pathogenicity) to avoid potential double‐counting of evidence, in case our patients overlapped with those referenced in ClinVar.21
4.1.2 |. Case A2
The younger brother of Case 1 was admitted to the neonatal intensive care unit (NICU) with neonatal respiratory distress and atelectasis despite being born at term. An echocardiogram and abdominal ultrasound confirmed normal organ arrangement (situs solitus), including the presence of a normal spleen. He developed chronic otitis media within the first 2 years of life, and myringotomy tube placement was performed three times before 5 years of age. At 4 years of age, he had a pediatric intensive care unit admission for septic shock, when persistent consolidation/atelectasis of the right middle lobe was noted on chest radiography, though a CT scan did not reveal bronchiectasis. At 5 years of age, TEM revealed normal ciliary ultrastructure. By 10 years of age, he had chronic sputum production and chronic nasal discharge. Spirometry was normal, with a forced expiratory volume in 1 s (FEV1) of 114% predicted, and sputum cultures grew Moraxella catarrhalis. Molecular genetic testing showed the presence of the same homozygous pathogenic variant (DNAH11; c.4095+2C>A) as his sibling.
4.1.3 |. Case A3
A 3‐month‐old male from north Qikiqtaaluk region was referred for dextrocardia noted on chest radiography during a prior hospitalization for bronchiolitis. The patient was born at term via Cesarean section and had respiratory distress at 7 min of life with desaturations and dyspnea, which required initial treatment with continuous positive airway pressure and supplementary oxygen for approximately 1 h. There was no family history of chronic lung disease though a third cousin (Case 1, above) was known to have SIT. An echocardiogram revealed dextrocardia with a right‐sided aortic arch, interruption of the inferior vena cava with hemiazygos continuation to the left superior vena cava, and likely left atrial isomerism. Abdominal ultrasound showed situs inversus abdominalis with poly-splenia in the right upper quadrant, all consistent with situs ambiguus. Normal hepatic and splenic uptake of radionucleotide tracer was seen on liver‐spleen scan. A nasal ciliary biopsy for TEM showed normal ciliary ultrastructure.
The patient had a second hospitalization for bronchiolitis at 5 months of age. Over his first 4 years of life, he developed chronic rhinitis and had one recorded episode of otitis media. Interval chest radiographs revealed moderate bilateral perihilar, peribronchial thickening with worsening over time in addition to the development of a chronic area of atelectasis in the left middle lobe. Sputum culture grew H. influenzae at 4 years of age. Subsequent clinical genetic testing of the patient demonstrated homozygous, pathogenic variants in DNAH11 (c.4095+2C>A), thus confirming the suspected diagnosis of PCD.
4.2 |. Extended family B
4.2.1 |. Case 4
A female from west Qikiqtaaluk region was born via spontaneous vaginal delivery at 36 weeks gestation and transferred to NICU after onset of respiratory distress at 3 days of age. Following transfer, she required invasive ventilation for 3 weeks during which time she was found to have SIT. Family history was significant for a maternal cousin who also had SIT.
After discharge, she had recurrent episodes of respiratory distress necessitating multiple hospital admissions in the first 2 years of life. Following this, she had no further reported episodes of pneumonia or bronchitis, although she did have a chronic wet, productive cough and frequent nasal congestion. An evaluation for immunodeficiency was unremarkable. Sputum and bronchial cultures obtained between ages 11 and 13 years showed heavy growth of H. influenzae, S. pneumoniae, and M. catarrhalis. Chest CT performed at 11 years of age showed bronchiectasis in the left upper lobe, with little interval change on a repeat CT at age 13. Spirometry at 16 years of age revealed an FEV1 of 71% predicted. There was a marked bronchodilator response (21% increase in FEV1), and while she had been prescribed inhaled bronchodilators and inhaled corticosteroids, she used them infrequently. Magnetic resonance imaging of the sinuses revealed thickening and fluid within the paranasal sinuses and mastoid air cells, although she did not clinically have acute sinusitis. She had bilateral, chronic middle ear effusions and mild, bilateral conductive hearing loss. Nasal brushing revealed normal ciliary structure on TEM analysis. Genetic testing showed homozygous, pathogenic variants in DNAH11 at c.4095+2C>A.
4.3 |. Extended family C
4.3.1 |. Case 5
A female infant from the north Qikiqtaaluk region had recurrent pneumonias in early childhood. She was born at 41 weeks gestation via uncomplicated delivery and had no respiratory problems in the newborn period. She had a history of bilateral recurrent otitis media and examination revealed clear rhinorrhea and frequent cough. Abdominal ultrasound was normal without evidence of organ laterality issues. The patient also had a history of spasticity, developmental delay with microcephaly, and was gastrostomy‐tube fed due to gastroesophageal reflux disease and oral aspiration. To investigate her neurologic issues, whole exome sequencing was performed through the Care4Rare Canada protocol at age 11, but no cause was found. She then enrolled in The Silent Genomes Project (a Canadian initiative working toward equitable accuracy and opportunity in precision genomics for Indigenous populations22; https://www.bcchr.ca/silent-genomes-project), which performed whole genome sequencing. This found homozygous pathogenic deletions of exon 4 (c.225+976_294+326del, p.(?)) in the AP4S1 gene, resulting in a diagnosis of AP4S1‐associated hereditary spastic paraplegia. This whole genome sequencing revealed that she was also homozygous for the likely pathogenic c.4095+2C>A variant in the DNAH11 gene. There was no prior ciliary assessment via electron microscopy or nasal nitric oxide measurement.
4.4 |. Extended family D
4.4.1 |. Case D6
A 56‐year‐old Inuit woman from the east coast of Ungava Bay in Nunavik, Quebec was referred for recurrent lower respiratory tract infections (Table 1). She was born at term and required neonatal critical care assistance for respiratory distress. Her paternal grandfather was of Scottish origin with the remainder of her ancestry being Inuit. She had active tuberculosis that was successfully treated when she was 5 years of age, as well as atrioventricular block, conductive hearing loss requiring amplification, and SIT. She reported chronic nasal congestion, chronic suppurative cough, and dyspnea on exertion since early childhood. She had several children who were easily conceived without fertility assistance. CT scan revealed bronchiectasis in the right‐sided lingula and bilateral lower lobes, with complete left middle lobe collapse (Figure 2). Pulmonary function testing showed mild airflow obstruction (FEV1 77% predicted). Sinus CT showed underdeveloped paranasal sinuses with polyposis and pansinusitis (Figure 3). Nasal nitric oxide levels were reduced at 23 nL/min, and clinical genetic testing revealed homozygous, pathogenic variants at c.4095+2C>A in the DNAH11 gene.
FIGURE 2.
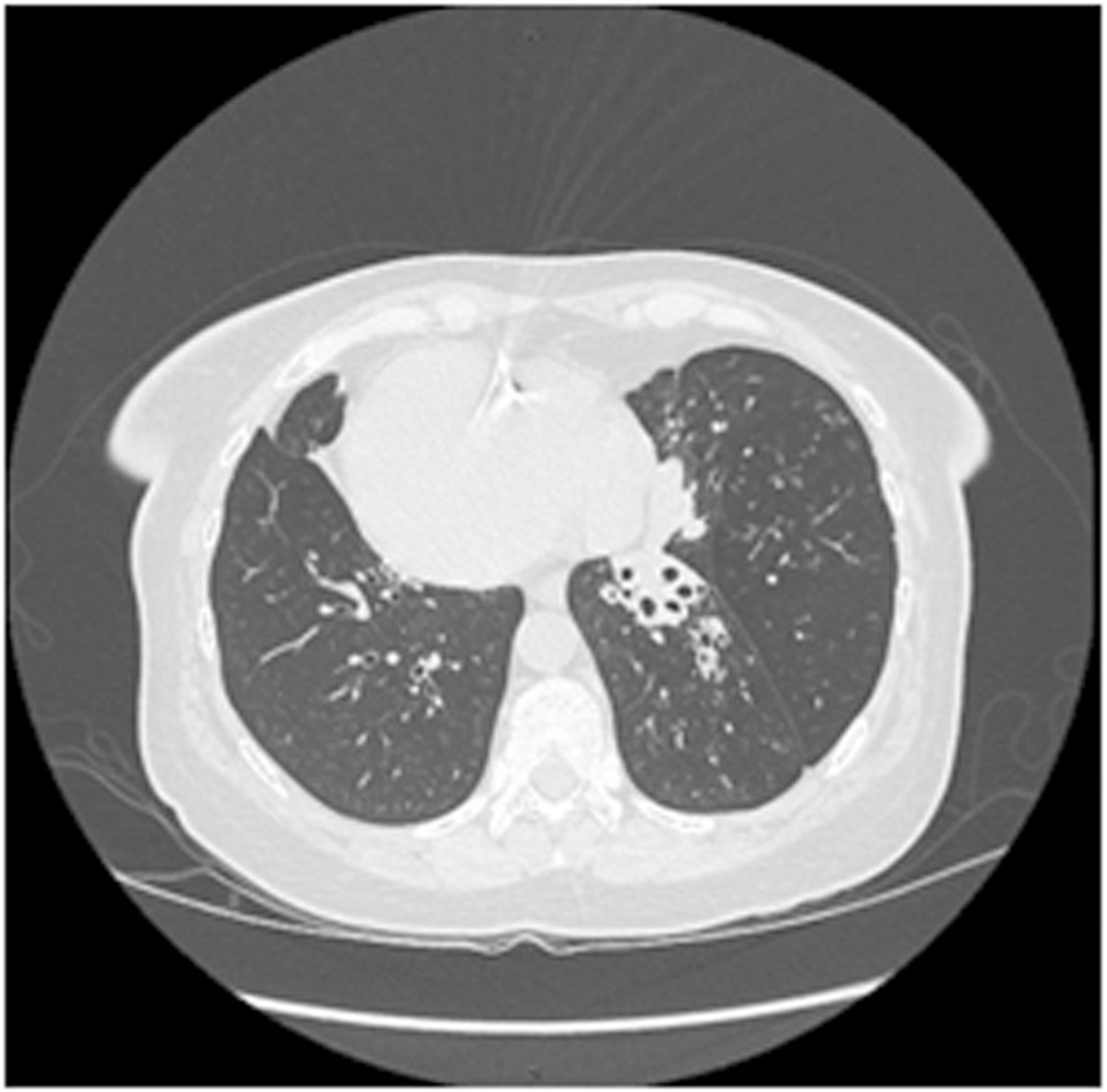
CT chest of adult patient D6 with situs inversus totalis, showing bilateral basilar bronchiectasis with complete collapse of the anatomic right middle lobe (on the left side). CT, computerized tomography.
FIGURE 3.
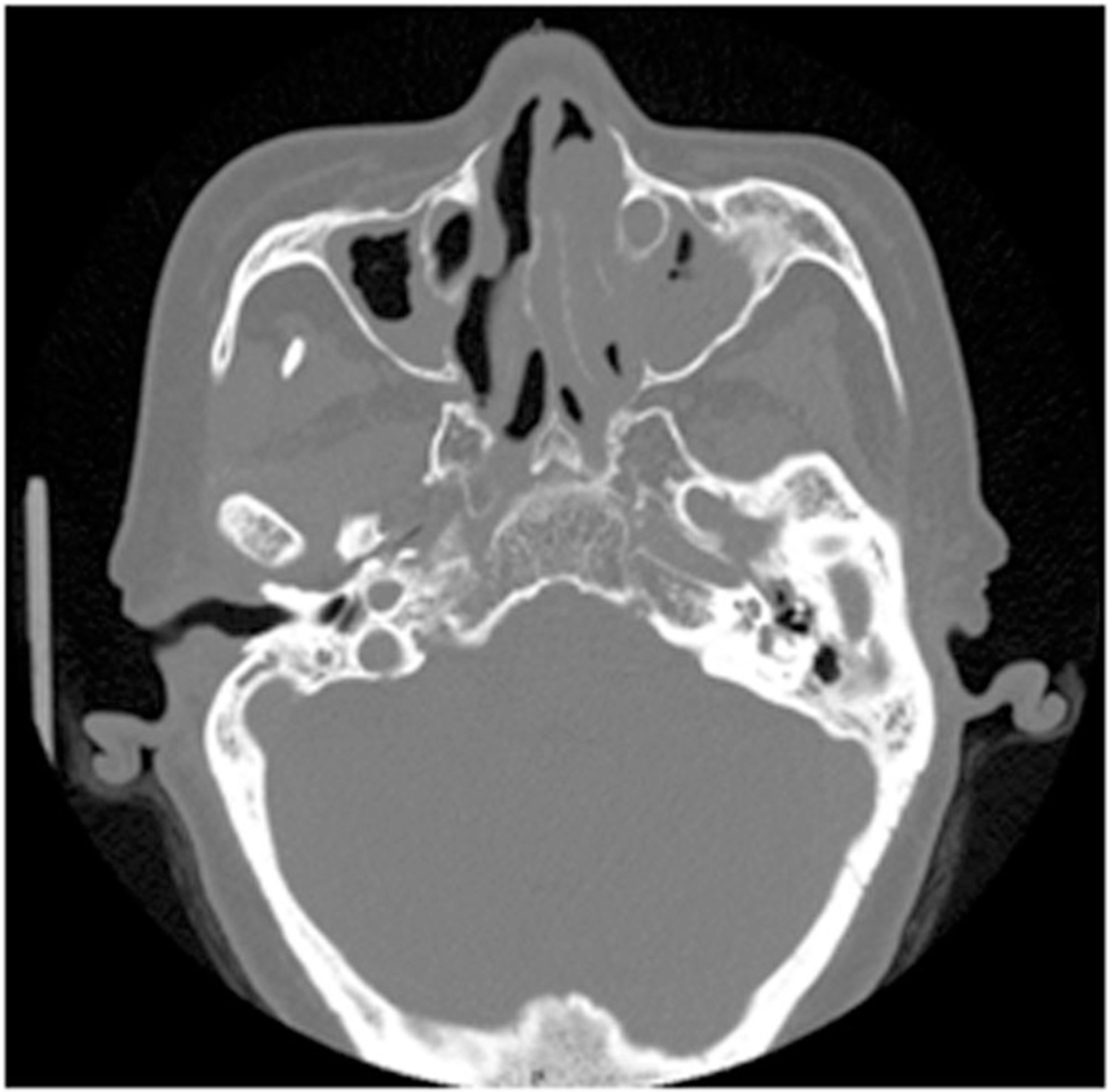
CT sinus of adult patient D6, showing hypoplastic maxillary sinuses, maxillary sinusitis, and left‐sided polypoid tissue. CT, computerized tomography.
4.4.2 |. Case D7
The 59‐year‐old sister of Case 6 was referred for recurrent pneumonias, year‐round wet cough and nasal congestion since early childhood, chronic sinusitis with two past polypectomies, permanent hearing loss from recurrent otitis media in childhood, and right middle/lower lobe bronchiectasis discovered at age 56. However, unlike her sister, this patient had normal organ arrangement (situs solitus). She easily conceived and delivered two healthy children without fertility assistance. Nasal nitric oxide was low at 8.9 nL/min, and she had overall normal lung function with an FEV1 of 107% predicted. Targeted genetic testing revealed homozygous likely pathogenic variants at c.4095+2C>A in the DNAH11 gene.
5 |. DISCUSSION
The patients in this series demonstrate that PCD can occur in Inuit, there is clinical heterogeneity of Inuit patients with identical pathogenic variants, and PCD should be considered in Inuit presenting with bronchiectasis or other symptoms suggestive of PCD. This recognition of PCD in Inuit is particularly important since the Indigenous population of Canada experiences a higher frequency of chronic respiratory disease and pulmonary infections compared to the nonindigenous community, particularly in infancy.23 In Inuit children, bronchiectasis is common with most cases appearing to follow severe viral respiratory infections occurring early in life, such as respiratory syncytial virus or adenovirus,5,6 although a recent case series describes a group of Inuit infants primarily from the west Qikiqtaaluk region with idiopathic oral aspiration,24 which may be another significant cause of bronchiectasis in this population. Social determinants of health in Inuit communities, such as overcrowding and reduced ventilation in houses, play an important role in the development of severe lower respiratory tract infection early in life; however, our study demonstrates that genetic conditions also need to be considered as a cause of chronic respiratory disease in Inuit patients.25
To date, PCD has not been described in the Canadian Indigenous populations and was thought to be rare due to the absence of abnormal electron microscopy (EM) findings when studied.7,8 Moreover, otitis media has been less useful for raising the clinical suspicion of PCD in this population, as recurrent otitis media is common in Indigenous children.26 This lack of past PCD recognition may also stem from the clinical heterogeneity that we report in Inuit PCD patients, despite having the identical, disease‐associated, pathogenic genetic variant. As observed previously in PCD,27 organ laterality defects appear to be randomly distributed among Inuit with PCD.
An increasing number of genetic variants causing PCD continue to be described in the literature,28,29 some of which cause PCD despite normal appearing ciliary ultrastructure on TEM.13,30 Given their normal ciliary ultrastructure, these variants run the risk of remaining undiagnosed when investigated via TEM alone. In fact, it is now well recognized that TEM will fail to detect up to 30% of PCD patients.31 The DNAH11 gene encodes a member of the axonemal dynein heavy chain family and contributes to the assembly of respiratory cilia; however, standard TEM does not have the resolution necessary to detect an outer dynein arm ultrastructural defect. Furthermore, mutations in the DNAH11 gene are associated with abnormal ciliary beat pattern on high‐speed videomicroscopy analysis,32 showing hyperkinetic beating patterns and a reduced waveform amplitude.30,33 However, outside of specialized PCD centers, both TEM and high‐speed videomicroscopy analysis are difficult to reliably perform, while genetic testing is quite feasible to perform in most medical facilities. Our use of genetic analysis, and in three patients, nasal NO testing, highlight the importance of these two modalities for the diagnosis of PCD, in accordance with recent American Thoracic Society guidelines.9
The homozygous DNAH11 variant at c.4095+2C>A in this series, to our knowledge, has not previously been reported in the literature, nor has this allele been previously appreciated in large public data sets, including gnomAD and TOPMed. Variant classification criteria from the American College of Medical Genetics and Genomics19 predict this canonical splice‐site variant to be pathogenic, likely causing aberrant transcript leading to nonsense‐mediated RNA decay. Data is emerging that some splice variants may be associated with a somewhat‐milder PCD phenotype34; this may be supported by our case series where patients had significant CT evidence of bronchiectasis, but among those who underwent lung function testing, values were largely normal, even in adults.34,35 Further analysis of the splicing defect through RNAseq, quantitative transcript analysis, and protein expression in patients with this variant are needed to understand if residual functional protein causes these milder phenotypes.
Previous reports estimated the prevalence of bronchiectasis in Inuit children in the Qikiqtaaluk region to be approximately 1/500 (202 per 100,000).5 Here, we report 5 affected PCD patients from a population of approximately 20,000 persons in the Qikiqtaaluk region, of whom 80% are Inuit,36 and where 43% of the population falls in the pediatric age group.37 This indicates a minimum prevalence of 1/1400 and a carrier frequency for this DNAH11 variant that could be at least 1 in 19. This estimate needs to be tempered by the recognition that several of our patients were related, and a more accurate determination will require a larger, population‐based study. In conjunction with Inuit organizations and public health officials, population‐level analysis on the impact of DNAH11 is planned within Nunavut, utilizing the template of a previous study where respiratory outcomes of Inuit children are linked to genotype.38,39 The surprisingly high Inuit PCD prevalence is most likely explained by a founder effect, given the history of the Inuit, the small population, the lack of admixture, and our findings of 4 affected families that are geographically separate. This may also be explained through distant relatedness. Parental consanguinity is another possible explanation, yet to our knowledge, none of our subjects resulted from parental consanguineous relationships. Furthermore, consanguinity is not commonly practiced amongst Inuit. However, nearly all Inuit communities are small and geographically isolated, with populations ranging mainly between 400 and 3000 individuals per community.40 As travel between communities in Nunavut and Nunavik is possible essentially only by air, the possibility of distant relatedness within each of these communities may be increased. Finally, to date, there is no evidence for selection of this variant within the population, but further studies are required to examine this.
These data strongly suggest that inherited PCD is a considerable cause of respiratory disease in Canadian Inuit, and given that we have observed this variant in disparate portions of the Inuit Nunangat, it may be due to a founder effect. This estimated minimum carrier frequency is notably higher than the 1 in 27 frequency of F508del, the commonest variant causing cystic fibrosis in white individuals.41 Founder effects have been reported for other PCD mutations, such as in Volendam, North Holland,42 and Puerto Ricans of Hispanic descent.43 Understanding the prevalence of PCD caused by variants in DNAH11 as well as discovering possible variants in other PCD genes are important steps towards improved delivery of care to affected Inuit patients. Specifically, early identification of PCD in the Inuit community will enable care providers to pursue early interventions such as audiological evaluation, sputum surveillance, and respiratory syncytial virus prophylaxis. The potential that carriers of this DNAH11 variant may be present with regular background frequency, the availability of a simple and accurate genetic test, the knowledge of the natural history of the disease, and the availability of effective early interventions suggest that newborn screening for this disorder should be strongly considered in Canadian Inuit.
Our results indicate that the etiology of more frequent respiratory disease and bronchiectasis in Inuit may be partly accounted for by a DNAH11 founder variant causing PCD. These findings suggest that genetic evaluation for PCD should be included in the investigation of bronchiectasis in Inuit individuals, as well as in Inuit with an organ laterality defect or recurrent oto‐sino‐respiratory infections.
Supplementary Material
ACKNOWLEDGMENTS
The authors are grateful to the patients/families for their participation in this study. The authors thank the investigators and coordinators of the Genetic Disorders of Mucociliary Clearance Consortium which is part of the Rare Disease Clinical Research Network, Dr. Jaclyn Stonebraker and Elizabeth Schecterman MLT for technical assistance, and Kelli Sullivan MPH and Nicole Capps DNP for help with specimen accrual and coordination. The authors wish to thank Dr. Letizia Gardin for their assistance in identifying patients. The authors wish to acknowledge the Silent Genomes Indigenous Rare Disease Diagnosis Steering Committee for reviewing this manuscript and for helpful suggestions. Investigators of Silent Genomes Precision Diagnosis Consortium involved in this study (Patient number 5) include Dr. Laura Arbour, Dr. Kym Boycott, Mireille Cloutier, Karen Jacob, Sarah McIntosh, Anna Lehman, and Dr. Jill Mwenifumbo. A portion of this work was performed under the Care4Rare Canada Consortium funded by Genome Canada and the Ontario Genomics Institute (OGI‐147), the Canadian Institutes of Health Research, Ontario Research Fund, Genome Alberta, Genome British Columbia, Genome Quebec, and Children’s Hospital of Eastern Ontario Foundation. Funding support for research was provided to MRK, MAZ, SD, and AJS by US NIH/ORDR/NACTS/NHLBI Grant U54HL096458; and to MRK, MAZ by US NIH/NHLBI Grant R01HL071798. The Genetic Disorders of Mucociliary Clearance Consortium (U54HL096458) is part of the National Center for Advancing Translational Sciences (NCATS) Rare Diseases Clinical Research Network (RDCRN) and supported by the RDCRN Data Management and Coordinating Center (DMCC) (U2CTR002818). RDCRN is an initiative of the Office of Rare Diseases Research (ORDR) funded through a collaboration between NCATS and National Heart, Lung, and Blood Institute (NHLBI). The contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institute of Health. Additional work was performed by investigators of the Silent Genomes Precision Diagnosis Consortium funded by Genome Canada and Genome BC (275SIL), Canadian Institutes of Health Research (GP1‐155868), BC Children’s Hospital Research Institute, and BC Children’s Hospital Foundation, Illumina (in‐kind).
Funding information
Genome Canada; Ontario Genomics Institute, Grant/Award Number: OGI‐147; Canadian Institutes of Health Research, Grant/Award Number: GP1‐155868; Ontario Research Fund; Genome Alberta; Genome British Columbia, Grant/Award Number: 275SIL; Genome Quebec; Children’s Hospital of Eastern Ontario Foundation; US NIH/ORDR/NACTS/NHLBI, Grant/Award Number: U54HL096458; US NIH/NHLBI, Grant/Award Number: R01HL071798; RDCRN Data Management and Coordinating Center, Grant/Award Number: U2CTR002818; NCATS and National Heart, Lung, and Blood Institute (NHLBI)
Footnotes
Adam J. Shapiro and Thomas A. Kovesi should be considered co‐senior authors of this paper.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflicts of interest.
SUPPORTING INFORMATION
Additional supporting information can be found online in the Supporting Information section at the end of this article.
DATA AVAILABILITY STATEMENT
Research data are not shared.
REFERENCES
- 1.Mullowney T, Manson D, Kim R, Stephens D, Shah V, Dell S. Primary ciliary dyskinesia and neonatal respiratory distress. Pediatrics 2014;134(6):1160–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kuehni CE, Frischer T, Strippoli MPF, et al. Factors influencing age at diagnosis of primary ciliary dyskinesia in European children. Eur Respir J 2010;36(6):1248–1258. [DOI] [PubMed] [Google Scholar]
- 3.O’Connor MG, Horani A, Shapiro AJ. Progress in diagnosing primary ciliary dyskinesia: the North American perspective. Diagnostics 2021;11(7):1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hannah WB, Seifert BA, Truty R, et al. The global prevalence and ethnic heterogeneity of primary ciliary dyskinesia gene variants: a genetic database analysis. Lancet Respir Med 2022;10(5):459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Das L, Kovesi TA. Bronchiectasis in children from Qikiqtani (Baffin) Region, Nunavut, Canada. Ann Am Thorac Soc 2015;12(1):96–100. [DOI] [PubMed] [Google Scholar]
- 6.Singleton R, Morris A, Redding G, et al. Bronchiectasis in Alaska Native children: causes and clinical courses. Pediatr Pulmonol 2000; 29(3):182–187. [DOI] [PubMed] [Google Scholar]
- 7.Fleshman JK, Wilson JF, Cohen JJ. Bronchiectasis in Alaska Native children. Arch Environ Health 1968;17(4):517–523. [DOI] [PubMed] [Google Scholar]
- 8.Rubin BK, Kumar V. Chronic lung disease in Canadian Aboriginal children is not caused by abnormal cilia. Can Respir J 1997;4(4):211–214. [Google Scholar]
- 9.Shapiro AJ, Davis SD, Polineni D, et al. Diagnosis of primary ciliary dyskinesia. An official American Thoracic Society clinical practice guideline. Am J Respir Crit Care Med 2018;197(12):e24–e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crown‐Indigenous Relations and Northern Affairs Canada. Indigenous Peoples and Communities 2022. Accessed October 18, 2022. https://www.rcaanc-cirnac.gc.ca/eng/1100100013785/1529102490303
- 11.Davis SD, Ferkol TW, Rosenfeld M, et al. Clinical features of childhood primary ciliary dyskinesia by genotype and ultrastructural phenotype. Am J Respir Crit Care Med 2015;191(3):316–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Backman K, Mears WE, Waheeb A, et al. A splice site and copy number variant responsible for TTC25‐related primary ciliary dyskinesia. Eur J Med Genet 2021;64(5):104193. [DOI] [PubMed] [Google Scholar]
- 13.Knowles MR, Leigh MW, Carson JL, et al. Mutations of DNAH11 in patients with primary ciliary dyskinesia with normal ciliary ultrastructure. Thorax 2012;67(5):433–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elliott AM, Adam S, du Souich C, et al. Genome‐wide sequencing and the clinical diagnosis of genetic disease: the CAUSES study. HGG Adv 2022;3(3):100108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shapiro AJ, Dell SD, Gaston B, et al. Nasal nitric oxide measurement in primary ciliary dyskinesia. A technical paper on standardized testing protocols. Ann Am Thorac Soc 2020;17(2):e1–e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shapiro AJ, Kaspy K, Daniels MLA, et al. Autosomal dominant variants in FOXJ1 causing primary ciliary dyskinesia in two patients with obstructive hydrocephalus. Mol Genet Genomic Med 2021;9(7):e1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Olin JT, Burns K, Carson JL, et al. Diagnostic yield of nasal scrape biopsies in primary ciliary dyskinesia: a multicenter experience. Pediatr Pulmonol 2011;46(5):483–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.MacCormick J, Robb I, Kovesi T, Carpenter B. Optimal biopsy techniques in the diagnosis of primary ciliary dyskinesia. J Otolaryngol 2002;31(1):013. [DOI] [PubMed] [Google Scholar]
- 19.Nykamp K, Anderson M, Powers M, et al. Sherloc: a comprehensive refinement of the ACMG‐AMP variant classification criteria. Genet Med 2017;19(10):1105–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dilliott AA, Farhan SMK, Ghani M, et al. Targeted next‐generation sequencing and bioinformatics pipeline to evaluate genetic determinants of constitutional disease. J Vis Exp 2018;134:57266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17(5): 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Appelbaum PS, Burke W, Parens E, et al. Is there a way to reduce the inequity in variant interpretation on the basis of ancestry. Am J Hum Genet 2022;109(6):981–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Evers SE, Rand CG. Morbidity in Canadian Indian and non‐Indian children in the first year of life. Can Med Assoc J 1982;126(3): 249–252. [PMC free article] [PubMed] [Google Scholar]
- 24.Farrow S, Agarwal A, Saban J, Scott D, Barrowman N, Kovesi T. Oral aspiration, type 1 laryngeal cleft, and respiratory tract infections in Canadian inuit children. Pediatr Pulmonol 2019;54(11):1837–1843. [DOI] [PubMed] [Google Scholar]
- 25.Kovesi T, Gilbert NL, Stocco C, et al. Indoor air quality and the risk of lower respiratory tract infections in young Canadian Inuit children. Can Med Assoc J 2007;177(2):155–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ayukawa H, Bruneau S, Proulx JF, Macarthur J, Baxter J. Otitis media and hearing loss among 12‐16‐year‐old Inuit of Inukjuak, Quebec, Canada. Int J Circumpolar Health 2004;63(Suppl 2):S312–S314. [DOI] [PubMed] [Google Scholar]
- 27.Rumman N, Jackson C, Collins S, Goggin P, Coles J, Lucas JS. Diagnosis of primary ciliary dyskinesia: potential options for resource‐limited countries. Eur Respir Rev 2017;26(143):160058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kurkowiak M, Ziętkiewicz E, Witt M. Recent advances in primary ciliary dyskinesia genetics. J Med Genet 2015;52(1):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zariwala MA, Omran H, Ferkol TW. The emerging genetics of primary ciliary dyskinesia . Proc Am Thorac Soc 2011;8(5):430–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schwabe GC, Hoffmann K, Loges NT, et al. Primary ciliary dyskinesia associated with normal axoneme ultrastructure is caused by DNAH11 mutations. Hum Mutat 2008;29(2):289–298. [DOI] [PubMed] [Google Scholar]
- 31.Kouis P, Yiallouros PK, Middleton N, Evans JS, Kyriacou K, Papatheodorou SI. Prevalence of primary ciliary dyskinesia in consecutive referrals of suspect cases and the transmission electron microscopy detection rate: a systematic review and meta‐analysis. Pediatr Res 2017;81(3):398–405. [DOI] [PubMed] [Google Scholar]
- 32.Bartoloni L, Blouin JL, Pan Y, et al. Mutations in the DNAH11 (axonemal heavy chain dynein type 11) gene cause one form of situs inversus totalis and most likely primary ciliary dyskinesia. Proc Natl Acad Sci U S A 2002;99(16):10282–10286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pifferi M, Michelucci A, Conidi ME, et al. New DNAH11 mutations in primary ciliary dyskinesia with normal axonemal ultrastructure. Eur Respir J 2010;35(6):1413–1416. [DOI] [PubMed] [Google Scholar]
- 34.Ostrowski LE, Yin W, Smith AJ, et al. Expression of a truncated form of ODAD1 associated with an unusually mild primary ciliary dyskinesia phenotype. Int J Mol Sci 2022;23(3):1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yiallouros PK, Kouis P, Pirpa P, et al. Wide phenotypic variability in RSPH9‐associated primary ciliary dyskinesia: review of a case‐series from Cyprus. J Thorac Dis 2019;11(5):2067–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Association QI. About the Qikiqtani Region 2022. Accessed September 10, 2022. https://www.qia.ca/about-qikiqtani/
- 37.Canada. Census Profile, 2016 Census Nunavut [Territory] and Canada [Country] 2021. Accessed September 10, 2022. https://www12.statcan.gc.ca/census-recensement/2016/dp-pd/prof/details/Page.cfm?Lang=E&Geo1=PR&Code1=62&Geo2=&Code2=&Data=Count&SearchText=Nunavut&SearchType=Begins&SearchPR=01&B1=All&GeoLevel=PR&GeoCode=62
- 38.Collins SA, Sinclair G, McIntosh S, et al. Carnitine palmitoyltransferase 1A (CPT1A) P479L prevalence in live newborns in Yukon, Northwest Territories, and Nunavut. Mol Gen Metab 2010;101(2‐3): 200–204. [DOI] [PubMed] [Google Scholar]
- 39.Collins SA, Edmunds S, Akearok GH, et al. Association of the CPT1A p.P479L metabolic gene variant with childhood respiratory and other infectious illness in Nunavut. Front Pediatr 2021;9:678553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Canada. Population and Dwelling Counts: Canada, Provinces and Territories, and Census Subdivisions (Municipalities) – Geographic Name: Nunavut Accessed February 4, 2023. https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=9810000202&pickMembers%5B0%5D=1.5434
- 41.Zvereff VV, Faruki H, Edwards M, Friedman KJ. Cystic fibrosis carrier screening in a North American population. Genet Med 2014;16(7):539–546. [DOI] [PubMed] [Google Scholar]
- 42.Onoufriadis A, Paff T, Antony D, et al. Splice‐site mutations in the axonemal outer dynein arm docking complex gene CCDC114 cause primary ciliary dyskinesia. Am J Hum Genet 2013;92(1):88–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Daniels MLA, Leigh MW, Davis SD, et al. Founder mutation in RSPH4A identified in patients of Hispanic descent with primary ciliary dyskinesia. Hum Mutat 2013;34(10):1352–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Research data are not shared.