

Abstract
Background:
The epidermal growth factor receptor (EGFR)-K745_E746insIPVAIK and others with XPVAIK amino-acid insertions are exon 19 insertion mutations, which, at the structural modeling level, resemble EGFR tyrosine kinase inhibitor (TKI)-sensitizing mutants. An important unmet need is the characterization of therapeutic windows plus clinical outcomes of exon 19 XPVAIK amino-acid insertion mutations to available EGFR TKIs.
Methods:
We used preclinical models of EGFR-K745_E746insIPVAIK and more typical EGFR mutations (exon 19 deletion, L858R, L861Q, G719S, A763_Y764insFQEA, other exon 20 insertion mutations) to probe representative 1st (erlotinib), 2nd (afatinib), 3rd generation (osimertinib), and EGFR exon 20 insertion active (mobocertinib) TKIs. We also compiled outcomes of EGFR exon 19 insertion mutated lung cancers—from our institution plus the literature—treated with EGFR TKIs.
Results:
Exon 19 insertions represented 0.3–0.8% of all EGFR kinase domain mutation in two cohorts (n=1772). Cells driven by EGFR-K745_E746insIPVAIK had sensitivity to all classes of approved EGFR TKIs when compared to cells driven by EGFR-WT in proliferation assays and at the protein level. However, the therapeutic window of EGFR-K745_E746insIPVAIK driven cells was most akin to those of cells driven by EGFR-L861Q and EGFR-A763_Y764insFQEA than the more sensitive patterns seen with cells driven by an EGFR exon 19 deletion or EGFR-L858R. The majority (69.2%, n=26) of patients with lung cancers harboring EGFR-K745_E746insIPVAIK and other mutations with rare XPVAIK amino-acid insertions responded to clinically available EGFR TKIs (including icotinib, gefitinib, erlotinib, afatinib and osimertinib), with heterogeneous periods of progression-free survival. Mechanisms of acquired EGFR TKI resistance of this mutant remained underreported.
Conclusions:
This is the largest preclinical/clinical report to highlight that EGFR-K745_E746insIPVAIK and other mutations with exon 19 XPVAIK amino-acid insertions are rare but sensitive to clinically available 1st, 2nd, and 3rd generation as well as EGFR exon 20 active TKIs; in a pattern that mostly resembles the outcomes of models with EGFR-L861Q and EGFR-A763_Y764insFQEA mutations. These data may help with the off-label selection of EGFR TKIs and clinical expectations of outcomes when targeted therapy is deployed for these EGFR mutated lung cancers.
Keywords: lung cancer, EGFR exon 19 insertion, K745_E746insIPVAIK, afatinib, osimertinib, erlotinib, mobocertinib
Introduction:
Precision oncology is one of the cornerstones of the management of advanced lung cancer1,2. Epidermal growth factor receptor (EGFR) kinase domain mutations are some of the most prevalent driver oncogenes linked to approved oral targeted therapies3. However, there is significant heterogeneity among the common (exon 19 deletions/indels, L858R), less common (exon 20 insertions, L861Q, G719X, S768I) and rare (A763_Y764insFQEA, delE709_T710insX, E709X, kinase domain duplications, kinase domain fusions) EGFR mutants as it relates to preclinical sensitivity to EGFR tyrosine kinase inhibitors (TKIs) plus clinical outcomes4–15. As of 2023, 1st generation reversible (gefitinib, erlotinib), 2nd generation irreversible (afatinib, dacomitinib), 3rd generation covalent mutation-selective (osimertinib) and EGFR exon 20 insertion mutation active (mobocertinib) EGFR TKIs have completed the long haul of clinical development into regulatory approval in the United States16–18.
One of the least studied rare subtypes of EGFR mutations are EGFR exon 19 insertions centered around amino-acids K745 to E746 with usually in-frame addition of six amino-acids to the structure of the kinase domain (Fig. 1)19. Prior reports have disclosed that these mutants seldom exceed 0.5–1% of all identified EGFR mutations (Fig.1A)4,20, with the most common variant the nucleotide changes that lead to the exact amino-acid insertions of EGFR-K745_E746insIPVAIK (Fig.1B)19. EGFR-I744_K745insKIPVAI generates the identical protein. Other reported variants include EGFR-K745_E746insTPVAIK and EGFR-K745_E746insVPVAIK19. Limited structural modeling reports have proposed a mechanism of kinase activation that hinges on the EGFR-L747 to EGFR-P747 amino-acid structural change that occurs with the aforementioned mutants that both activates the kinase domain and creates a therapeutic window to EGFR TKIs in relation to wild-type (WT) EGFR (Fig.1C)19.
Figure 1. EGFR exon 19 deletions in context of other genomic aberrations.
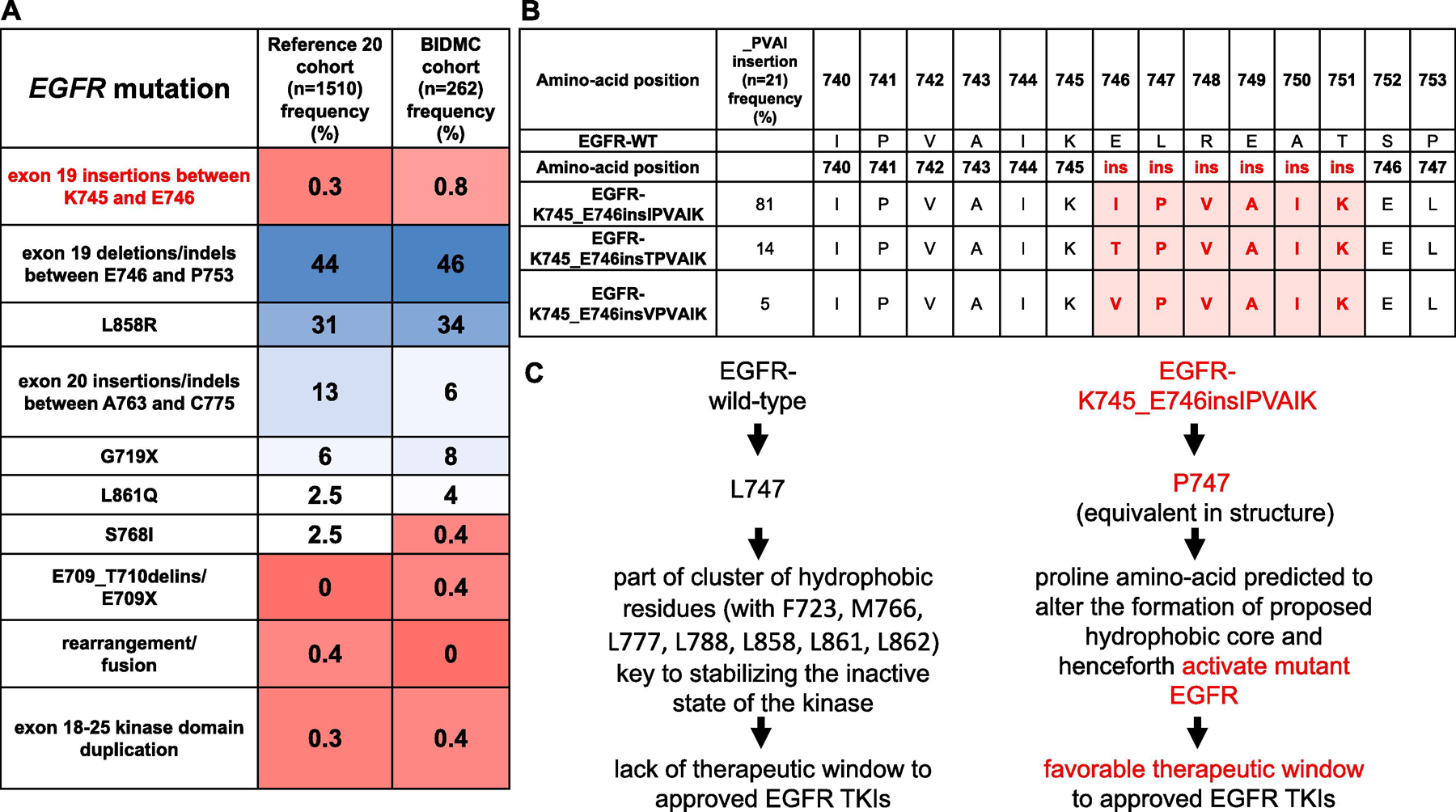
(A) Frequency of common EGFR mutations and EGFR-K747_E746insIPVAIK and other variants in two separate cohorts. The larger cohort represents data from the commercial vendor Foundation Medicine20 and the smaller from our institutional cases from Beth Israel Deaconess Medical Center (BIDMC) between years 2004 to 2021. (B) Display of amino acid sequence of wild-type EGFR compared to the different variants of EGFR-K745_E746ins_XPVAIK mutations based on reference 19. Frequency of the different XPVAIK insertions curated from the COSMIC database is displayed. (C) Theoretical mechanism of the sensitivity of the EGFR-K747_E746insIPVAIK mutation that details the change that may allow for sensitivity to approved EGFR TKIs based on structural modeling from reference 19. There is no published crystal structure for this mutant. The proposed mechanism of activation depicted is similar to mechanisms described for EGFR-A763_Y764insFQEA and EGFR-L861Q.
However, important unmet preclinical and clinical needs include the full characterization of therapeutic windows of EGFR-exon 19 XPVAIK amino-acid insertions to all classes of available EGFR TKIs and the compilation of clinical outcomes with the same inhibitors when given to patients with lung cancers harboring these mutations. In this report, we provide a comprehensive characterization of EGFR exon 19 insertion mutations to close the aforementioned gap of knowledge.
Materials and Methods:
Drugs
Erlotinib, afatinib, osimertinib (LC Laboratories) and mobocertinib (MedChemExpress) were dissolved in dimethyl sulfoxide (DMSO, Fisher Scientific) at 10 mM and stored at −80°C before dilutions.
EGFR gene sequencing
RNA was extracted using Direct-zol RNA MiniPrep Plus Kit (Zymo Research). cDNA was transcribed from 1 μg of total RNA using High Capacity cDNA RT kit (Applied Biosystem). cDNA was used as a template for subsequent PCR amplifications of EGFR genes. EGFR exons 18 to 21 were amplified and sequenced. National Center for Biotechnology Information (NCBI) reference sequence NM 005228.5 was used as a reference for EGFR sequence analysis.
Cell lines and reagents
Ba/F3 murine cells and Bosc23 cells were kindly gifted by Dr. Daniel G. Tenen at Beth Israel Deaconess Medical Center (USA). The parental Ba/F3 cells were maintained in Roswell Park Memorial Institute-1640 (RPMI) medium (Corning) supplemented with 10% fetal bovine serum (FBS) (Corning), 1% penicillin and streptomycin (PS) (Corning), and 5% myelomonocytic leukemia, macrophage-like, Balb/C Mouse cells (WEHI)-conditioned medium as the source of interleukin-3 (IL-3). In the case of EGFR-WT driven Ba/F3 cells, 10 ng/mL of EGF (PeproTech) was added. Bosc23 cells were maintained in Dulbecco’s modified eagle medium (DMEM) (Corning) supplemented with 10% FBS and 1% PS. All cells were grown at 37°C in a humidified atmosphere with 5% CO2 and tested for absence of mycoplasma contamination (MycoAlert Mycoplasma Detection Kit, Lonza) prior to experiments (initiated within the initial 1 to 4 passages).
EGFR mutant constructs
EGFR exon 18 to exon 21 mutant constructs were introduced into the human EGFR WT sequence in the context of the MigR1 retrovirus vector (Addgene) using Q5® Site-Directed Mutagenesis Kits (New England BioLabs) as described previously7,8,21,22. The mutant EGFR constructs used in this study were K745_E746insIPVAIK, delL747_P753insS (exon 19 deletion), L858R, G719S, L861Q, A763_Y764insFQEA and V769_D770insASV. G719S and L861Q mutant constructs were kindly gifted from Dr. Hiroyuki Yasuda at Keio University (Japan)23. The oligonucleotide sequences used to K745_E746insIPVAIK are CGTCGCTATCAAGGAATTAAGAGAAGCAAC (forward) and GGAATTTTAATAGCGACGGGAATTTTAAC (reverse), using NEB changer (New England BioLabs). The resulting constructs were confirmed by nucleotide sequencing.
Generation of EGFR mutant driven Ba/F3 cell lines
Bosc23 cells were transfected with 15 μg of EGFR WT and mutant constructs using TransIT-X2® Dynamic Delivery System (Mirus Bio). The supernatant of Bosc23 cells containing MigR1 retrovirus was collected 2 days after transfection. Retronectin (Takara) was applied to Falcon petri dishes (Fisher Scientific). To infect Ba/F3 cells, the retrovirus and Ba/F3 cells were incubated for 24 hours on these plates. The infection was repeated twice. To select EGFR expressing Ba/F3 cells, green fluorescent protein (GFP) positive cells were sorted by FACSAria (Beckton Dickinson BioSciences). The sorted cells were grown in RPMI with 10% FBS. All EGFR mutants were able to drive Ba/F3 proliferation in the absence of IL-3. IL-3 independent Ba/F3 cells were used for further experiments, as described in prior reports7,8,21,22. After establishment, sequencing analysis was performed to confirm the mutant EGFR.
Cell proliferation assays
Cell viability was determined by CellTiter 96 aqueous one solution proliferation kit (Promega) for Ba/F3 cells. 10,000 cells were plated in 96-well plates and then treated in RPMI with indicated EGFR TKIs at 10 different concentrations for 3 days. A one solution reagent containing a mix of a tetrazolium compound (MTS) and an electron coupling reagent (PES) was added at the end of the 72-hour period and incubated for 2 hours. The number of living cells in each well was measured by using a 96-well plate reader to record the absorbance at 490 nm, which directly correlated to the number of living cells oxidizing the MTS solution. Inhibitory proliferation curves and the 50% inhibitory concentration (IC50) were generated using GraphPad Prism 7 (GraphPad Software). Preclinical therapeutic window was calculated using logarithm of IC50 of EGFR mutants compared to EGFR-WT with values below zero indicating sensitivity (i.e., favorable therapeutic window) and values above zero indicating resistance (i.e., unfavorable therapeutic window) to each EGFR TKI tested.
Immunoblotting
Cells were treated with indicated EGFR TKIs for 6 hours at various concentrations. Cytoplasmic proteins were isolated via cell lysis for western blotting, as detailed in prior reports7,8,21,22. Protein extracts were then subjected to electrophoresis on 7.5% to 10% SDS polyacrylamide gels, with 30 to 45 mg of protein being added to each well. Samples were transferred to polyvinylidene difluoride membranes (MilliporeSigma), which were blocked then with 5% nonfat milk in phosphate-buffered saline with tween (PBST). Primary incubation with antibodies for the proteins of interest in a mix of 5% bovine serum albumin (BSA) and 0.02% sodium azide in PBST was performed overnight at 4º C. Total EGFR (Santa Cruz Biotechnology), β-actin (Cell Signaling) antibodies, phospho-EGFR antibody (pY1068) (Cell Signaling), total ERK (Cell Signaling) and phospho-ERK (Cell Signaling) were at 1:1000 dilution. Total AKT (Cell Signaling) and phospho-AKT (Cell Signaling) were at 1:500. Secondary incubation was performed for 2 hours at room temperature using horseradish peroxidase- (HRP) conjugated goat-anti-rabbit IgG and goat-anti-mouse IgG (Bio-Rad) antibodies at 1:5000 dilution in 5% nonfat milk. Membrane films were exposed and developed using enhanced chemiluminescence (ECL) Prime Western Blotting detection reagent Pierce ECL Western Blotting Substrate, 500 mL Kit (Thermo Scientific). Chemiluminescence assays for detecting the target proteins were performed using an Amersham Imager 600 (GE Healthcare).
Clinical and tumor genomic data collection
The frequency of EGFR mutations was calculated using three separate cohorts of cases, including our own institutional database (Fig.1). Genotype frequency, clinical, radiographic and survival outcomes used for this study were obtained from an ongoing Institutional Review Board approved protocol at Beth Israel Deaconess Medical Center. Additional genotype-inhibitor data were obtained through a literature review of studies published in PubMed and other databases, as well as oncology meeting abstracts using the search field “EGFR exon 19 insertion” plus “IPVAIK”. Response evaluation criteria in solid tumors (RECIST) was used, when provided. Progression-free survival (PFS) and overall survival (OS) were calculated in months, from the time of initiation of an EGFR TKI, when provided.
Results:
Frequency and EGFR mutation patterns for exon 19 insertions in lung cancer
We analyzed a large cohort from a commercial sequencing vendor20, our own institutional database of EGFR mutated lung cancer and the Catalogue Of Somatic Mutations In Cancer (COSMIC) compendium catalogue24. EGFR exon 19 insertions between amino-acids K745 and E746 were reported in less than 0.8% of all EGFR mutated lung cancer cases (Fig.1A). The COSMIC catalogue (21 mutants) enumerated that the majority (81%) of EGFR exon 19 PVAIK insertions were of nucleotide sequences that generated equivalent mutants to EGFR-K745_E746insIPVAIK, while less common variants included EGFR-K745_E746insTPVAIK (14%) and EGFR-K745_E746insVPVAIK (5%) (Fig.1B). In view of these findings, we generated preclinical models driven by EGFR-K745_E746insIPVAIK for further experimentation.
Preclinical characterization of EGFR-K745_E746insIPVAIK and its pattern of sensitivity to EGFR TKIs in comparison to other EGFR mutants
Cells driven by EGFR-K745_E746insIPVAIK had sensitivity to all classes of representative EGFR TKIs tested (erlotinib, afatinib, osimertinib, mobocertinib) when compared to cells driven by EGFR-WT in proliferation assays (Fig.2A); thus, these cells displayed a favorable therapeutic window (IC50 below that of EGFR-WT) to EGFR TKIs. However, the preclinical therapeutic window to all drugs tested was less robust than that of cells driven by an EGFR-exon 19 deletion (the indel EGFR-delL747_P753insS) or EGFR-L858R by 1 to 2 or more logarithms of mutant IC50/WT IC50 (Fig.2A). To better highlight these differences, the dose-dependent inhibition of proliferation for cells harboring EGFR-K745_E746insIPVAIK or EGFR-delL747_P753insS treated with erlotinib or osimertinib consistently disclosed more than 10 to 20-fold higher doses of drug needed to fully inhibit EGFR-K745_E746insIPVAIK dependent cells (Fig.2B). To further highlight both the EGFR TKI sensitivity of EGFR exon 19 insertion positive cells and the less robust patterns of inhibition when compared to EGFR-delL747_P753insS positive cells, we studied the intracellular signaling response to TKI therapy (Fig.2C). The active form of EGFR, as measured by one of the many phosphorylated epitopes of the protein, was inhibited by sub-micromolar doses of EGFR TKIs but again EGFR-K745_E746insIPVAIK dependent cells required 10-fold higher concentrations to achieve similar levels of inhibition (Fig.2C). The same pattern was observed with downstream signaling cascades mediated by the mitogen-activated protein kinase (ERK1 and ERK2) and the PI3K-AKT pathways (Fig. 2C).
Figure 2. Ba/F3 system isogenic preclinical models of EGFR mutations to probe EGFR inhibitors.
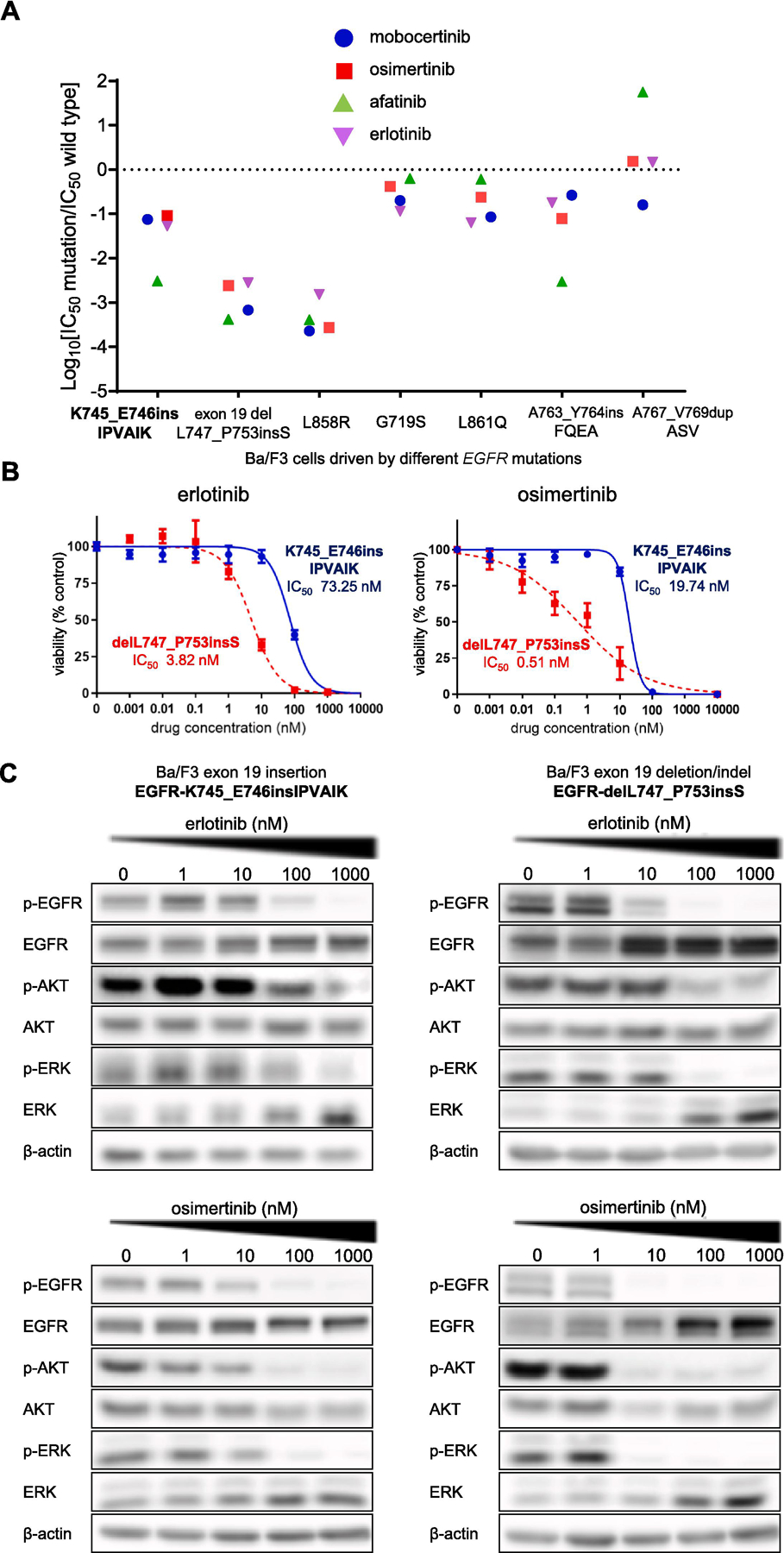
(A) Therapeutic window of different EGFR-TKIs to EGFR mutants. Cells were plated at a density of 10,000 cells per well (96-well plates) and grown over 3 days after treatment. Logarithm of the 50% inhibitory concentration (IC50) of EGFR mutants compared to EGFR-WT is plotted with 3 separate experiments used to generate IC50. Values below zero (0) indicate sensitivity, while values above 0 indicate resistance to EGFR-TKIs. The therapeutic window of Ba/F3 cells with EGFR-K745_E746insIPVAIK are contrasted with other mutations.
(B) Dose-response proliferation assays (percent viability) for patient-derived lung cancer cell lines harboring EGFR-K745_E746insIPVAIK and EGFR-delL747_P753insS after exposure to increasing concentrations of EGFR TKIs. Three separate experiments were used to generate IC50, and standard deviations are depicted in vertical bars. (C) Western blotting of Ba/F3 cells driven by EGFR-K745_E746insIPVAIK and EGFR-delL747_P753insS. Cells were treated with the EGFR TKIs for 6 hours at the indicated ascending concentrations. pEGFR, phosphorylated EGFR at position 1068, total EGFR, pAKT, phosphorylated AKT, total AKT, phosphorylated ERK, total ERK and β-actin (loading control) are exhibited.
The preclinical therapeutic window of our model driven by EGFR-K745_E746insIPVAIK was more similar to the pattern we observed for cells with EGFR-A763_Y764insFQEA for all EGFR TKIs tested or cells with EGFR-L861Q for erlotinib, osimertinib and mobocertinib (Fig.2A). The pattern was also not that dissimilar to that of EGFR-G719S dependent cells for erlotinib, osimertinib and mobocertinib (Fig.2A). Cells with one of the most common EGFR exon 20 insertion mutants (A767_V769dupASV) had unfavorable therapeutic windows to erlotinib, afatinib and mobocertinib, as expected from our prior experience and the structural conformation of this mutant’s kinase5,7,8,22,23. Using the recent structure-function analysis of preclinical subgroups of EGFR mutations to predict their putative clinical response to various EGFR TKIs, both EGFR-L861Q and EGFR-A763_Y764insFQEA mutants are considered to cluster as “classical-like” mutations11—a subgrouping that would also apply to EGFR-K745_E746insIPVAIK and presumably to other exon 19 XPVAIK insertion mutants.
Clinical responses to EGFR TKIs in patients with EGFR exon 19 insertion mutated lung cancer
To better understand if the aforementioned preclinical results with EGFR-K745_E746insIPVAIK would translate into responses to approved EGFR TKIs in the clinical sphere, we concluded an extensive literature search to identify 23 separate individuals25–38 and added 3 unpublished cases from our institutional cohort of EGFR mutated lung cancer (Table 1). These 26 patients with tumors harboring EGFR exon 19 insertion mutations (that lead to amino-acid changes that represent either EGFR-K745_E746insIPVAIK, EGFR-K745_E746insTPVAIK or EGFR-K745_E746insVPVAIK) were treated with 1st generation (icotinib, gefitinib, erlotinib; n=20), 2nd generation (afatinib; n=3) and 3rd generation (osimertinib; n=3) EGFR TKIs. The clinical and pathologic characteristics were similar to those of other EGFR mutated lung cancers4–15, with predominance of adenocarcinoma histology and never smokers (Table 1).
Table 1.
Clinical, pathological characteristics and response to EGFR TKIs of patients with tumors harboring EGFR exon 19 insertion mutations: K745_E746insIPVAIK and others.
| Case number | Ref. (number) | EGFR mutation/histology | EGFR TKI (line of therapy) | Dose | Response RECI ST | Percent change target lesion(s) | PFS (months) | OS(months from start of EGFR TKI) | sex/age(years)/ethnicity/PS | smoking history (pack-years) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 25 | I740_K745dupI PVAIK*/adenocarcinoma | Icotinib (1st) | NA | PR | NA | 13 | NA | M/74/Asian/NA | NA |
| 2 | 31 | I744_K745insKIPVAI*/adenocarcinoma | Gefitinib (1st) | 250 mg | PR | − 43.7% | 4 | 10 | M/68/Asian/NA | 50 |
| 3 | 31 | I744_K745insKIPVAI*/adenocarcinoma | Gefitinib (1st) | 250 mg | SD | NA | 5 | NA | F/51/Asian/NA | 0 |
| 4 | 33 | I744_K745insKIPVAI*/adenocarcinoma | Gefitinib (1st) | NA | PR | NA | 5 | 9 | F/42/Asian/NA | 0 |
| 5 | 36 | I744_K745insKIPVAI*/adenocarcinoma | Gefitinib (1st) | 250 mg | PR | -70% | 5 | NA | F/56/white/NA | 0 |
| 6 | 36 | I744_K745insKIPVAI*/adenocarcinoma | Gefitinib (2nd) | 250 mg | SD | NA | >9 | NA | F/48/white/NA | 0 |
| 7 | 36 | I744_K745insKIPVAI*/adenocarcinoma | Gefitinib (1st) | 250 mg | SD | NA | >11 | NA | F/54/white/NA | 0 |
| 8 | 27 | I744_K745insKIPVAI*/adenocarcinoma | Gefitinib (1st) | 250 mg | PR | NA | 12 | 18 | F/48/white/2 | 0 |
| 9 | 33 | I744_K745insKIPVAI*/adenocarcinoma | Gefitinib (2nd) | NA | SD | NA | 22 | 32 | M/46/Asian/NA | 15 |
| 10 | 37 | I744_K745insKIPVAI*/adenocarcinoma | Gefitinib (1st) | NA | SD | NA | 24 | NA | F/37/Asian/NA | 0 |
| 11 | 33 | I744_K745insKIPVAI*/adenocarcinoma | Erlotinib (2nd) | NA | PD | NA | 2 | 11 | F/47/Asian/NA | 0 |
| 12 | 26 | delI744_K745in sKIPVAI*/adenocarcinoma | Erlotinib (2nd) | 150 mg | PR | NA | 7 | 9 | F/36/white/NA | NA |
| 13 | current BIDM C | K739_I744dup KIPVAI*/adenocarcinoma | Erlotinib (1st) | 150 mg | PR | − 30.4% | 7 | 11 | F/59/white/3 | 0 |
| 14 | current BIDM C | K739_I744dup KIPVAI*/adenocarcinoma | Erlotinib (2nd) | 100 mg | PR | − 31.2% | 8 | 12 | F/68/white/0 | 10 |
| 15 | 34 | K745_E746insI PVAIK/adenocarcinoma | Erlotinib (1st) | 150 mg | PR | NA | 9 | NA | F/39/Arab/3 | 0 |
| 16 | 37 | E746_L747insV PVAIK/adenocarcinoma | Erlotinib (1st) | NA | PR | NA | 16 | NA | F/60/Asian/NA | 0 |
| 17 | 29 | I740_K745dupI PVAIK*/adenocarcinoma | Erlotinib (3rd) | 150 mg | PR | NA | 16 | >48 | F/39/white/NA | 0 |
| 18 | 28 | K745_E746insI PVAIK/adenocarcinoma | Erlotinib (1st) | 150 mg | SD | NA | 18 | 24 | F/55/Asian/1 | 0 |
| 19 | 19 | K745_E746ins PVAIK/adenocarcinoma | Erlotinib (1st) | NA | PR | NA | 19 | NA | NA | NA |
| 20 | 19 | K745_E746insIPVAIK/adenocarcinoma | Erlotinib (1st) | NA | PR | NA | 50 | NA | NA | NA |
| 21 | 35 | K745_E746insIPVAIK/adenocarcinoma | Afatinib (1st) | 40 mg | PD | NA | 1 | 4 | F/75/Asian/1 | NA |
| 22 | 30 | I740_K745dupIPVAIK*/adenocarcinoma | Afatinib (2nd) | 30 mg | PR | NA | 13 | 21 | M/63/Asian/NA | 45 |
| 23 | 19 | K745_E746insI PVAIK/adenocarcinoma | Afatinib (1st) | NA | PR | NA | 14 | NA | NA | NA |
| 24 | 38 | I740_K745dupI PVAIK*/NSCLC NOS | Osimertinib (1st)+bevacizumab | NA | PR | NA | >1 | NA | F/58/Asian/NA | NA |
| 25 | 38 | I740_K745dupI PVAIK*/NSCLC NOS | Osimertinib (1st) | NA | PR | NA | >2 | NA | F/67/Asian/NA | 0 |
| 26 | current BIDM C | K745_E746insI PVAIK/adenocarcinoma | Osimertinib (1st) | 80 mg | PR | − 31.6% | 9 | >13 | F/73/white/0 | 2 |
EGFR, epidermal growth factor receptor; PS, ECOG performance status; TKI, tyrosine kinase inhibitor; RECIST, Response evaluation in solid tumors version 1.1; PR, partial response; SD, stable disease; PD, progressive disease; PFS, progression-free survival; OS, overall survival; NSCLC NOS, non-small-cell lung cancer not otherwise specified; NA, not available; +, ongoing survival for PFS or OS. For OS, it was assumed survival was ongoing (>) when report did not specify otherwise. When extrapolating from written or graphic data from publications, we rounded response change or months to nearest full value. * indicates mutations that have the same amino-acid sequence as EGFR-K745_E746insIPVAIK
The majority of patients responded to EGFR TKIs. For 1st generation EGFR TKIs, 13 out of 20 (65%) of cases had a partial response (PR). For the 2nd generation TKI afatinib, 2 out of 3 (66%) had a PR. For the 3rd generation TKI osimertinib, 3 out of 3 (100%) had a PR (Table 1). Very few cases had progressive disease as the best tumor assessment (Table 1). The periods of tumor burden control for cases with PR and stable disease (SD) on EGFR TKI therapy were variable. For icotinib, gefitnib and erlotinib, the PFS varied from 4 to 50 months (Table 1). For afatinib and osimertinib, data on PFS was more limited but some cases had PFS that equaled or exceed 9 months (Table 1).
The calculated median PFS for the 26 cases receiving any type of EGFR TKI was 12 months (95% confidence interval [CI], 8–18 months). The calculated median OS for the whole cohort after initiation of an EGFR TKI was 12.5 months (95% CI, 9–19 months).
Mechanisms of acquired EGFR TKI resistance of this mutant remained underreported in the literature review and our compilation of cases without a genomic aberration identified in the few rebiopsy reports despite the growing use of clinical rebiopsy samples in academic centers32. As examples, our internal case 14 (Table 1) did not have on-target EGFR resistant mutations at time of progression tumor rebiopsy and our internal case 26 (Table 1) of EGFR-K745_E746insIPVAIK mutated lung adenocarcinoma that responded to osimertinib for 9 months lacked identifiable new on-target or off-target genomic aberrations at liquid and tumor rebiopsy.
Discussion:
To the best of our knowledge, the current report represents the largest preclinical and clinical report to highlight that EGFR-K745_E746insIPVAIK and other EGFR mutations with rare exon 19 XPVAIK amino-acid insertions (Fig.1B) are sensitive to clinically available 1st, 2nd, and 3rd generation, as well as EGFR exon 20 active TKIs (Fig.2A), and are responsive to icotinib, gefitinib, erlotinib, afatinib or osimertinib in patients with lung cancer (Table 1). We were able to determine that these EGFR exon 19 insertion changes are relatively uncommon, accounting for less than 1% of all cases of EGFR genomic aberrations in lung cancer (Fig.1A). The main amino-acid changes cluster into an insertion of around 6 amino-acids between the location of EGFR-K745 and EGFR-E746 of the WT EGFR structure, with insertions of IPVAIK (close to 80% of cases), TPVAIK (less than 15%) or VPVAIK (5% of cases) following the WT EGFR IPVAIK amino-acid stack of I740 to K745 (Fig.2B). Most comprehensive genomic profiling assays used for lung cancer genotyping1,39 should be capable of identifying all the aforementioned mutations.
The mechanism of activation of these primary EGFR exon 19 insertions is glanced at from structural modeling efforts in lieu of a known crystal structural report. The current understanding of the structure-function of EGFR-K745_E746insIPVAIK and other mutants is anchored on the EGFR-P747 amino-acid equivalent structural change from WT EGFR-L747 of the mutants (Fig.1C), which is predicted to alter the formation of the hydrophobic core of the EGFR kinase domain (involving amino-acids EGFR-F723, −M766, −L777, −L788, −L858, −L861 and −L862 that are key to stabilizing the inactive kinase state) and henceforth activate the mutant EGFR in a manner that generates a favorable therapeutic window to most approved EGFR TKIs19. This mechanism of activation shares similarities with the proposed structure-function models currently understood to explain EGFR activation and favorable therapeutic windows to EGFR TKIs of the EGFR-A763_Y764insFQEA exon 20 mutant and of the EGFR-L861Q exon 21 mutant5. Within the proposed structure-function subgrouping to better classify in vitro sensitivity of structurally-defined categories of EGFR mutants, there are a few clusters including classical-like (exon 19 deletions/indels, L858R, L861Q, A763_Y764insFQEA) with broad sensitivity to multiple generations of EGFR-directed TKIs, P-loop αC-helix compressing (PACC) mutants (such as G719X and S768I) with most sensitivity to 2nd generation irreversible EGFR TKIs, T790M-like hydrophobic core mutants (EGFR-T790M plus other mutations) and exon 20 loop insertions11. Our preclinical data would support EGFR exon 19 insertions (EGFR-K745_E746insIPVAIK) as another subgroup that clusters within the classical-like cohort.
It is notable that EGFR structure-function models and preclinical cell line models have translated remarkably faithfully to clinical outcomes. The most sensitive preclinical mutations with the lowest inhibitory concentrations and widest therapeutic windows—exon 19 indels and L858R (Fig.2A)—also have the best clinical outcomes. Patients with advanced EGFR-exon 19 indels or EGFR-L858R mutated lung adenocarcinoma treated with 1st, 2nd and 3rd generation EGFR TKIs have radiographic response rates (i.e., ORR) that exceed 60–80% and that can be prolonged (PFS times that exceed 12–18 months)40,41,17. The most robust overall survival with monotherapy has been achieved with use of 1st line osimertinib with a reported median OS of more than 3 years42. The next best annotated mutations are EGFR-L861Q and EGFR-G719X. These have intermediate preclinical sensitivity patterns (Fig.2A) and less stellar clinical outcomes. Patients with advanced EGFR-G719X mutated lung adenocarcinoma have the best reported clinical activity with 2nd generation EGFR TKIs such as afatinib (with ORR of >60%, PFS >12 months and OS >24 months) with more limited activity of 1st or 3rd generation TKIs (with ORR of <55% and PFS <9 months)43–45. For patients with EGFR-L861Q mutated lung adenocarcinoma, the ORR exceeds 50–70% (PFS >8 months and OS >17 months) to 1st, 2nd and 3rd generation EGFR TKIs43–45—with the most robust activity seen with osimertinib43. The data for the unique exon 20 insertion mutant EGFR-A763_Y764insFQEA is not that dissimilar to EGFR-L861Q. The largest clinical series compilation disclosed ORR >60%, PFS>5 months and OS>20 months to all classes of EGFR TKIs21. Unlike these latter cohorts of classical-like and PACC mutants, typical EGFR exon 20 insertion mutations do not have a therapeutic window to 1st, 2nd and 3rd generation EGFR TKIs or clinical responses to these drugs21; with only modest preclinical and clinical activity to EGFR exon 20 insertion active TKIs such as mobocertinib8,18,22. Our database (Table 1) would support that the preclinical clustering of EGFR exon 19 insertions into classical-like mutants best matches a clinical pattern seen with EGFR-L861Q or EGFR-A763_Y764insFQEA. Indeed, we see ORR >65% to 1st, 2nd and 3rd generation EGFR TKIs and PFS times more than 11 months for cases with EGFR-K745_E746insIPVAIK and similar mutated lung adenocarcinomas (Table 1).
The one area that our large database is unable to address are mechanism of resistance to EGFR TKIs in EGFR exon 19 insertion mutated lung cancer, as none of the cases either reported rebiopsy specimens or identified a new genetic/epigenetic event. It is well known from most classical-like and PACC EGFR mutated lung cancers that the three most common mechanisms of resistance include: on-target resistance (i.e., EGFR-T790M to 1st/2nd generation EGFR TKIs or EGFR-C797S to 3rd generation EGFR TKIs), off-target resistance (i.e., amplification, mutation or another mechanism of activation of bypass driver oncogenes) or epigenetic histological transformation46–50. One can only speculate that EGFR exon 19 insertion mutated lung cancers would display similar patterns of resistance.
Conclusion:
Ours is the largest preclinical and clinical report of EGFR-K745_E746insIPVAIK and other mutations with exon 19 XPVAIK amino-acid insertions. We are able to conclude that these mutants are relatively rare (<1% of all EGFR mutations) but sensitive to clinically available 1st, 2nd, and 3rd generation as well as EGFR exon 20 active TKIs. The preclinical and clinical pattern mostly resembles the outcomes of EGFR-L861Q or EGFR-A763_Y764insFQEA mutated lung cancers. We believe the data provide here may help with the off-label selection of EGFR TKIs in real-world settings and in the definition of clinical expectations of outcomes when targeted therapy is deployed for EGFR exon 19 insertion mutated lung cancers.
Highlights.
EGFR-K745_E746insIPVAIK and others with XPVAIX amino-acid insertions are exon 19 insertion mutations (<0.8% of all EGFR mutations), which, at the structural modeling level, resemble EGFR inhibitor-sensitizing mutants
An important unmet need is the characterization of therapeutic windows plus clinical outcomes of exon 19 XPVAIX amino-acid insertion mutations to available EGFR TKIs.
Preclinical models of EGFR-K745_E746insIPVAIK had sensitivity to all classes of approved EGFR TKIs when compared to cells driven by EGFR-WT in proliferation assays and at the protein level but therapeutic windows most akin to EGFR-L861Q and EGFR-A763_Y764insFQEA than the more sensitive patterns seen with cells driven by an EGFR exon 19 deletion or EGFR-L858R
This is the largest preclinical/clinical report to highlight that EGFR-K745_E746insIPVAIK and other mutations with exon 19 XPVAIX amino-acid insertions are rare but sensitive to clinically available 1st, 2nd, and 3rd generation as well as EGFR exon 20 active TKIs
Acknowledgements/Funding:
This work was funded in part through National Institutes of Health (NIH)/National Cancer Institute (NCI) grants R37 CA218707 (to D. B. Costa), R01 CA240257 (to S. S. Kobayashi) plus Department of Defense LC170223 (to S. S. Kobayashi)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure of Potential Conflicts of interest: DR reports receiving personal fees (consulting fees and honoraria) from TelaDoc Health, DynaMed, and Astra Zeneca; nonfinancial support (institutional research support) from Bristol-Myers Squibb, Novocure, and Abbvie/Stemcentrx; all outside the submitted work. PVL reports personal fees (consulting fees) from Gala Therapeutics, Galvanize Therapeutics, Intuitive Surgical, and Ruby Robotics; all outside the submitted work. SSK reports research support from Boehringer Ingelheim, MiRXES, Johnson&Johnson, and Taiho Therapeutics, as well as personal fees (honoraria) from AstraZeneca, Boehringer Ingelheim, Bristol Meyers Squibb, Chugai Pharmaceutical, and Takeda Pharmaceuticals plus royalties from Life Technologies; all outside the submitted work. DBC reports receiving consulting fees and honoraria from Takeda/Millennium Pharmaceuticals, AstraZeneca, Pfizer, Blueprint Medicines, and Janssen, institutional research support from Takeda/Millennium Pharmaceuticals, AstraZeneca, Pfizer, Merck Sharp and Dohme, Merrimack Pharmaceuticals, Bristol Myers Squibb, Clovis Oncology, Spectrum Pharmaceuticals, Tesaro and Daiichi Sankyo, and consulting fees from Teladoc and Grand Rounds by Included Health plus royalties from Life Technologies; all outside the submitted work. No other conflict of interest is reported.
CRediT author statement:
Daniel B. Costa: conceptualization, resources, data curation, writing original draft, writing review & editing, visualization, supervision and funding acquisition
William Shaffer: resources, data curation, writing original draft, writing reviewed & editing and visualization
Ikei S. Kobayashi: resources, data curation, writing original draft, writing reviewed & editing and visualization
Daniel Sentana-Lledo: resources, data curation and writing review & editing
Shriram Sundararaman: resources, data curation and writing review & editing
Deepa Rangachari: resources, data curation and writing review & editing
Paul A. VanderLaan: resources, data curation and writing review & editing
Meghan D. Lee: resources, data curation and writing review & editing
Susumu S. Kobayashi: resources, data curation, writing review & editing, visualization, supervision and funding acquisition
References:
- 1.VanderLaan PA, Rangachari D, Costa DB. The rapidly evolving landscape of biomarker testing in non-small cell lung cancer. Cancer Cytopathol 2021;129(3):179–181. doi: 10.1002/cncy.22334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.VanderLaan PA, Rangachari D, Majid A, et al. Tumor biomarker testing in non-small-cell lung cancer: A decade of change. Lung Cancer Amst Neth 2018;116:90–95. doi: 10.1016/j.lungcan.2018.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jorge SEDC Kobayashi SS, Costa DB. Epidermal growth factor receptor (EGFR) mutations in lung cancer: preclinical and clinical data. Braz J Med Biol Res Rev Bras Pesqui Medicas E Biol 2014;47(11):929–939. doi: 10.1590/1414-431X20144099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Costa DB. Kinase inhibitor-responsive genotypes in EGFR mutated lung adenocarcinomas: moving past common point mutations or indels into uncommon kinase domain duplications and rearrangements. Transl Lung Cancer Res 2016;5(3). doi: 10.21037/tlcr.2016.06.04 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yasuda H, Park E, Yun CH, et al. Structural, biochemical, and clinical characterization of epidermal growth factor receptor (EGFR) exon 20 insertion mutations in lung cancer. Sci Transl Med 2013;5(216):216ra177. doi: 10.1126/scitranslmed.3007205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sehgal K, Rangachari D, VanderLaan PA, Kobayashi SS, Costa DB. Clinical Benefit of Tyrosine Kinase Inhibitors in Advanced Lung Cancer with EGFR-G719A and Other Uncommon EGFR Mutations. The Oncologist 2021;26(4):281–287. doi: 10.1002/onco.13537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jorge SE, Lucena-Araujo AR, Yasuda H, et al. EGFR Exon 20 Insertion Mutations Display Sensitivity to Hsp90 Inhibition in Preclinical Models and Lung Adenocarcinomas. Clin Cancer Res 2018;24(24):6548–6555. doi: 10.1158/1078-0432.CCR-18-1541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kobayashi IS, Viray H, Rangachari D, Kobayashi SS, Costa DB. EGFR-D770>GY and Other Rare EGFR Exon 20 Insertion Mutations with a G770 Equivalence Are Sensitive to Dacomitinib or Afatinib and Responsive to EGFR Exon 20 Insertion Mutant-Active Inhibitors in Preclinical Models and Clinical Scenarios. Cells 2021;10(12):3561. doi: 10.3390/cells10123561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yasuda H, Kobayashi S, Costa DB. EGFR exon 20 insertion mutations in non-small-cell lung cancer: preclinical data and clinical implications. Lancet Oncol 2012;13(1):e23–31. doi: 10.1016/S1470-2045(11)70129-2 [DOI] [PubMed] [Google Scholar]
- 10.Kobayashi S, Canepa HM, Bailey AS, et al. Compound EGFR mutations and response to EGFR tyrosine kinase inhibitors. J Thorac Oncol Off Publ Int Assoc Study Lung Cancer 2013;8(1):45–51. doi: 10.1097/JTO.0b013e3182781e35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robichaux JP, Le X, Vijayan RSK, et al. Structure-based classification predicts drug response in EGFR-mutant NSCLC. Nature 2021;597(7878):732–737. doi: 10.1038/s41586-021-03898-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown BP, Zhang YK, Kim S, et al. Allele-specific activation, enzyme kinetics, and inhibitor sensitivities of EGFR exon 19 deletion mutations in lung cancer. Proc Natl Acad Sci 2022;119(30):e2206588119. doi: 10.1073/pnas.2206588119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robichaux JP, Elamin YY, Tan Z, et al. Mechanisms and clinical activity of an EGFR and HER2 exon 20–selective kinase inhibitor in non–small cell lung cancer. Nat Med 2018;24(5):638–646. doi: 10.1038/s41591-018-0007-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Low JL, Lim SM, Lee JB, Cho BC, Soo RA. Advances in the management of non-small-cell lung cancer harbouring EGFR exon 20 insertion mutations. Ther Adv Med Oncol 2023;15:17588359221146132. doi: 10.1177/17588359221146131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.VanderLaan PA, Rangachari D, Mockus SM, et al. Mutations in TP53, PIK3CA, PTEN and other genes in EGFR mutated lung cancers: Correlation with clinical outcomes. Lung Cancer Amst Neth 2017;106:17–21. doi: 10.1016/j.lungcan.2017.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jänne PA, Yang JCH, Kim DW, et al. AZD9291 in EGFR Inhibitor–Resistant Non–Small-Cell Lung Cancer. N Engl J Med 2015;372(18):1689–1699. doi: 10.1056/NEJMoa1411817 [DOI] [PubMed] [Google Scholar]
- 17.Soria JC, Ohe Y, Vansteenkiste J, et al. Osimertinib in Untreated EGFR-Mutated Advanced Non–Small-Cell Lung Cancer. N Engl J Med 2018;378(2):113–125. doi: 10.1056/NEJMoa1713137 [DOI] [PubMed] [Google Scholar]
- 18.Riely GJ, Neal JW, Camidge DR, et al. Activity and Safety of Mobocertinib (TAK-788) in Previously Treated Non-Small Cell Lung Cancer with EGFR Exon 20 Insertion Mutations from a Phase I/II Trial. Cancer Discov 2021;11(7):1688–1699. doi: 10.1158/2159-8290.CD-20-1598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He M, Capelletti M, Nafa K, et al. EGFR Exon 19 Insertions: A New Family of Sensitizing EGFR Mutations in Lung Adenocarcinoma. Clin Cancer Res 2012;18(6):1790–1797. doi: 10.1158/1078-0432.CCR-11-2361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Konduri K, Gallant JN, Chae YK, et al. EGFR Fusions as Novel Therapeutic Targets in Lung Cancer. Cancer Discov 2016;6(6):601–611. doi: 10.1158/2159-8290.CD-16-0075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vasconcelos PENS, Gergis C, Viray H, et al. EGFR-A763_Y764insFQEA Is a Unique Exon 20 Insertion Mutation That Displays Sensitivity to Approved and In-Development Lung Cancer EGFR Tyrosine Kinase Inhibitors. JTO Clin Res Rep 2020;1(3):100051. doi: 10.1016/j.jtocrr.2020.100051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vasconcelos PENS Kobayashi IS, Kobayashi SS Costa DB. Preclinical characterization of mobocertinib highlights the putative therapeutic window of this novel EGFR inhibitor to EGFR exon 20 insertion mutations. JTO Clin Res Rep 2021;2(3):100105. doi: 10.1016/j.jtocrr.2020.100105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Udagawa H, Hasako S, Ohashi A, et al. TAS6417/CLN-081 Is a Pan-Mutation-Selective EGFR Tyrosine Kinase Inhibitor with a Broad Spectrum of Preclinical Activity against Clinically Relevant EGFR Mutations. Mol Cancer Res MCR 2019;17(11):2233–2243. doi: 10.1158/1541-7786.MCR-19-0419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cosmic. COSMIC - Catalogue of Somatic Mutations in Cancer Accessed October 15, 2022. https://cancer.sanger.ac.uk/cosmic
- 25.Zhu N, Dong C, Weng S, Yuan Y, Yuan Y. A Patient of Advanced NSCLC with a New EGFR Exon 19 Insertion Mutation and its Response to EGFR-TKIs. J Coll Physicians Surg--Pak JCPSP 2019;29(12):S126–S128. doi: 10.29271/jcpsp.2019.12.S126 [DOI] [PubMed] [Google Scholar]
- 26.Pas TD, Toffalorio F, Manzotti M, et al. Activity of Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitors in Patients with Non-small Cell Lung Cancer Harboring Rare Epidermal Growth Factor Receptor Mutations. J Thorac Oncol 2011;6(11):1895–1901. doi: 10.1097/JTO.0b013e318227e8c6 [DOI] [PubMed] [Google Scholar]
- 27.Kozlov V, Karpov I, Kovalenko S, Shamanin V. Adenocarcinoma of the Lung with Rare Insertion Mutation in EGFR Exon 19 That Had Partial Response to Gefitinib: A Case Report. Exp Oncol 2017;39(2):155–156. doi: 10.31768/2312-8852.2017.39(2):155-156 [DOI] [PubMed] [Google Scholar]
- 28.Chan AWH, Tong JHM, Lo SH, To KF. An Uncommon Insertion Mutation in Exon 19 of EGFR Showed Stable Disease after TKI Treatment. J Thorac Oncol 2013;8(12):e107–e108. doi: 10.1097/JTO.0b013e3182a471e0 [DOI] [PubMed] [Google Scholar]
- 29.Tiseo M, Bersanelli M, Perrone F, et al. Different clinical effects upon separate inhibition of coexisting EGFR and PI3KCA mutations in a lung adenocarcinoma patient. Lung Cancer 2015;87(2):204–206. doi: 10.1016/j.lungcan.2014.12.008 [DOI] [PubMed] [Google Scholar]
- 30.Shan BB, Li Y, Zhao C, An XQ, Zhang QM. Efficacy of EGFR-TKI sequential therapy in patients with EGFR exon 19 insertion-positive non-small-cell lung cancer: A case report. World J Clin Cases 2022;10(6):1883–1888. doi: 10.12998/wjcc.v10.i6.1883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Park J, Kondo C, Shimizu J, Horio Y, Yoshida K, Hida T. EGFR Exon 19 Insertions Show Good Response to Gefitinib, but Short Time to Progression in Japanese Patients. J Thorac Oncol 2014;9(2):e10–e11. doi: 10.1097/JTO.0b013e3182a735cc [DOI] [PubMed] [Google Scholar]
- 32.Redig AJ, Costa DB, Taibi M, et al. Prospective Study of Repeated Biopsy Feasibility and Acquired Resistance at Disease Progression in Patients With Advanced EGFR Mutant Lung Cancer Treated With Erlotinib in a Phase 2 Trial. JAMA Oncol 2016;1 2(9):1240–2. doi: 10.1001/jamaoncol.2016.1304 [DOI] [PubMed] [Google Scholar]
- 33.Lin YT, Liu YN, Wu SG, Yang JCH, Shih JY. Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor-sensitive Exon 19 Insertion and Exon 20 Insertion in Patients With Advanced Non-Small-cell Lung Cancer. Clin Lung Cancer 2017;18(3):324–332.e1. doi: 10.1016/j.cllc.2016.12.014 [DOI] [PubMed] [Google Scholar]
- 34.Agbarya A, Melamed-Frank M, Kaidar-Person O, et al. Getting out of a wheelchair: an uncommon insertion mutation in exon 19 of EGFR responsive to erlotinib. SpringerPlus 2014;3(1):507. doi: 10.1186/2193-1801-3-507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhu Y cai, Du K qi, Wang W xian, et al. Lung adenocarcinoma patient with EGFR 19 exon insert mutation and its response to icotinib. Lung Cancer 2018;121:101–104. doi: 10.1016/j.lungcan.2018.04.019 [DOI] [PubMed] [Google Scholar]
- 36.Iyevleva AG, Mitiushkina NV, Karaseva NA, et al. Lung Carcinomas with EGFR Exon 19 Insertions Are Sensitive to Gefitinib Treatment. J Thorac Oncol 2014;9(4):e31–e33. doi: 10.1097/JTO.0000000000000106 [DOI] [PubMed] [Google Scholar]
- 37.Su J, Zhong W, Zhang X, et al. Molecular characteristics and clinical outcomes of EGFR exon 19 indel subtypes to EGFR TKIs in NSCLC patients. Oncotarget 2017;8(67):111246–111257. doi: 10.18632/oncotarget.22768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu J, Jiang Q, Xu H, Liu A, Huang L. Two Patients Having NSCLC With Novel Duplication Mutation in Their EGFR Gene (p.I740_K745dupIPVAIK) and Their Response to Osimertinib. J Thorac Oncol 2020;15(4):e49–e51. doi: 10.1016/j.jtho.2019.11.026 [DOI] [PubMed] [Google Scholar]
- 39.Sheikine Y, Rangachari D, McDonald DC, et al. EGFR Testing in Advanced Non-Small-Cell Lung Cancer, A Mini-Review. Clin Lung Cancer 2016;17(6):483–492. doi: 10.1016/j.cllc.2016.05.016 [DOI] [PubMed] [Google Scholar]
- 40.Paz-Ares L, Tan EH, O’Byrne K, et al. Afatinib versus gefitinib in patients with EGFR mutation-positive advanced non-small-cell lung cancer: overall survival data from the phase IIb LUX-Lung 7 trial. Ann Oncol Off J Eur Soc Med Oncol 2017;28(2):270–277. doi: 10.1093/annonc/mdw611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 2012;13(3):239–246. doi: 10.1016/S1470-2045(11)70393-X [DOI] [PubMed] [Google Scholar]
- 42.Ramalingam SS, Vansteenkiste J, Planchard D, et al. Overall Survival with Osimertinib in Untreated, EGFR-Mutated Advanced NSCLC N Engl J Med 2020;382(1):41–50. doi: 10.1056/NEJMoa1913662 [DOI] [PubMed] [Google Scholar]
- 43.Bar J, Peled N, Schokrpur S, et al. UNcommon EGFR Mutations: International Case Series on Efficacy of Osimertinib in Real-Life Practice in First-LiNe Setting (UNICORN). J Thorac Oncol 2023;18(2):169–180. doi: 10.1016/j.jtho.2022.10.004 [DOI] [PubMed] [Google Scholar]
- 44.Cho JH, Lim SH, An HJ, et al. Osimertinib for Patients With Non-Small-Cell Lung Cancer Harboring Uncommon EGFR Mutations: A Multicenter, Open-Label, Phase II Trial (KCSG-LU15–09). J Clin Oncol Off J Am Soc Clin Oncol 2020;38(5):488–495. doi: 10.1200/JCO.19.00931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang JCH, Sequist LV, Geater SL, et al. Clinical activity of afatinib in patients with advanced non-small-cell lung cancer harbouring uncommon EGFR mutations: a combined post-hoc analysis of LUX-Lung 2, LUX-Lung 3, and LUX-Lung 6. Lancet Oncol 2015;16(7):830–838. doi: 10.1016/S1470-2045(15)00026-1 [DOI] [PubMed] [Google Scholar]
- 46.Niederst MJ, Sequist LV, Poirier JT, et al. RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat Commun 2015;6(1):6377. doi: 10.1038/ncomms7377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nguyen KSH, Kobayashi S, Costa DB. Acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancers dependent on the epidermal growth factor receptor pathway. Clin Lung Cancer 2009;10(4):281–289. doi: 10.3816/CLC.2009.n.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rangachari D, To C, Shpilsky JE, et al. EGFR-Mutated Lung Cancers Resistant to Osimertinib through EGFR C797S Respond to First-Generation Reversible EGFR Inhibitors but Eventually Acquire EGFR T790M/C797S in Preclinical Models and Clinical Samples. J Thorac Oncol Off Publ Int Assoc Study Lung Cancer 2019;14(11):1995–2002. doi: 10.1016/j.jtho.2019.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kobayashi S, Boggon TJ, Dayaram T, et al. EGFR Mutation and Resistance of Non–Small-Cell Lung Cancer to Gefitinib. N Engl J Med 2005;352(8):786–792. doi: 10.1056/NEJMoa044238 [DOI] [PubMed] [Google Scholar]
- 50.Oxnard GR, Hu Y, Mileham KF, et al. Assessment of Resistance Mechanisms and Clinical Implications in Patients With EGFR T790M-Positive Lung Cancer and Acquired Resistance to Osimertinib. JAMA Oncol 2018;4(11):1527–1534. doi: 10.1001/jamaoncol.2018.2969 [DOI] [PMC free article] [PubMed] [Google Scholar]