

Abstract
Purpose of review:
To highlight advances in congenital hyperinsulinism (HI), including newly described molecular mechanisms of disease, novel therapeutic interventions, and improved understanding of long-term outcomes.
Recent findings:
Important advances have been made elucidating the molecular mechanisms responsible for HI. Non-coding variants in HK1 have been found to cause aberrant hexokinase expression. Inactivating mutations in SLC25A36 have been identified in children with features of the hyperinsulinism hyperammonemia syndrome. Low-level mosaic mutations in known HI genes have been detected in cases of “genetic testing negative” HI. Identification and localization of focal HI lesions remains a priority, since focal HI can be cured with surgery. Use of 68Ga-NODAGA-exendin-4 PET has been proposed to localize focal lesions. Additional studies are needed before this technique replaces 18F-DOPA PET as standard of care. Treatment options for children with diffuse HI remain limited. The long-acting somatostatin analog, lanreotide, was shown to significantly improve glycemic control in a large series of children with HI. New therapies are under development, with promising preliminary results. Long-term quality of life and neurodevelopmental outcomes remain suboptimal.
Summary:
Advanced genetic and epigenomic analytic techniques have uncovered novel molecular mechanisms of HI. Development of new drugs holds promise to improve long-term outcomes for individuals with HI.
Keywords: Insulin, beta-cell, hexokinase, exendin, hypoglycemia
Introduction
Congenital hyperinsulinism (HI) is the most common cause of persistent hypoglycemia in infants and young children. Hypoglycemia in HI results from dysregulated insulin secretion and is biochemically characterized by suppressed ketones and free fatty acids (1). Risk of neurologic impairment in unrecognized or inadequately treated HI is high, owing to the combined insult of hypoglycemia and lack of alternate brain fuels. Prompt diagnosis and treatment of HI is thus critical to improve long-term outcomes. Many ground-breaking developments have been made since cases of HI were first described by Dr. Irvine McQuarrie in 1954 (2). Despite these advances, long-term outcomes remain suboptimal, and gaps persist in diagnosis and treatment. While over 20 genes have been described in association with HI, the cause of HI remains unknown in over 20% of all cases, and up to 50% of diazoxide-responsive cases (3). Now 40 years since its Food and Drug Administration (FDA) approval, diazoxide remains the only FDA approved medication for HI. More recently, novel discoveries have been made in the molecular mechanisms of HI, technological advances and new therapeutics have emerged to improve management, and new strategies have been employed to understand long-term outcomes. These advances will be reviewed.
Background
HI represents a heterogenous group of disorders that can be classified by etiology (perinatal stress-induced HI, monogenic defects in the insulin secretion pathway, syndromic), histopathology (diffuse, focal, atypical), or responsiveness to medical therapy. Assessing response to diazoxide is a critical starting point toward optimizing HI care (Figure 1). Focal HI accounts for 50% of diazoxide-unresponsive cases and can be cured by limited surgical resection (4). Clinical phenotyping thus informs which children benefit from expedited genetic testing and imaging to identify and localize a focal lesion preoperatively. For diazoxide unresponsive cases due to diffuse disease, second-line medical management options used in clinical practice to date have included continuous intragastric dextrose and the somatostatin analogues octreotide and lanreotide (Figure 2). For medically refractory diffuse cases, near-total (98%) pancreatectomy is required. However, this procedure is palliative, not curative, and is complicated by exocrine pancreatic insufficiency and post-pancreatectomy diabetes underscoring the critical need for new treatments (5).
Fig 1.
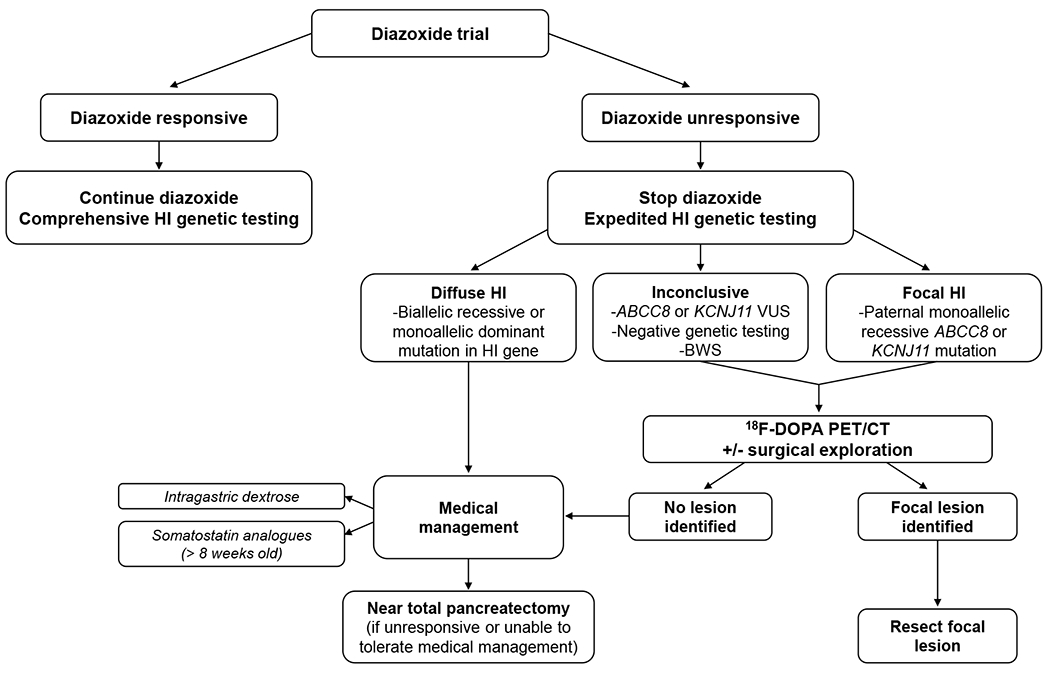
Management Algorithm for Congenital Hyperinsulinism.
After the diagnosis of hyperinsulinism has been made, the next step is to assess whether diazoxide is effective treatment. Responsiveness to diazoxide is established by showing that the cardinal feature of HI, hypoketotic hypoglycemia, has been reversed. In practice, this means demonstrating the ability to fast and generate hyperketonemia (beta-hydroxybutyrate >1.8 mmol/L) prior to developing hypoglycemia (plasma glucose <50-60 mg/dL). If responsive, diazoxide treatment is continued and comprehensive genetic testing is obtained. For patients unresponsive to diazoxide, expedited genetic testing is obtained to differentiate diffuse and focal forms of HI. 18F-DOPA PET/CT is performed when genetic testing is suggestive of possible focal disease, to localize the focal lesion preoperatively. For patients with diffuse, diazoxide-unresponsive disease, intensive medical therapy is initiated, with near-total pancreatectomy reserved for medically unresponsive cases.
18F-DOPA PET/CT 6-fluoro-L-3,4-dihydroxyphenylalanine positron emission tomography/computed tomography, BWS Beckwith-Wiedemann syndrome, HI hyperinsulinism, VUS variant of uncertain significance
Source: Original
Fig 2.

Diagram of the beta-cell insulin secretion pathway, highlighting genetic causes of congenital hyperinsulinism and therapeutic targets.
Glucose enters the beta-cell via glucose transporters, predominantly glucose transporter 1 (GLUT-1), and is phosphorylated by glucokinase (GCK), the glucose sensor of the beta-cell. Hexokinase 1 (HXK1) has a higher affinity for glucose than glucokinase and is not normally expressed in the beta-cell. Further metabolism of glucose-6-phosphate leads to an increase in the ATP-to-ADP ratio, closure of the KATP channel beta-cell membrane depolarization, opening of voltage-dependent calcium channels, calcium influx, and insulin secretion. Amino acids stimulate insulin secretion through glutamine-mediated amplification of glucagon-like peptide 1 receptor (GLP-1R) signaling. In addition, leucine stimulates insulin secretion by activating glutamate dehydrogenase (GDH), increasing the oxidation of glutamate to alpha-ketoglutarate, thereby increasing the ATP-to-ADP ratio, triggering insulin secretion. GDH is allosterically inhibited by guanine triphosphate and short-chain 3-OH acyl-CoA dehydrogenase (SCHAD). Monocarboxylate transporter 1 (MCT1) is not normally present on the beta-cell. Known sites of defects associated with congenital hyperinsulinism are highlighted in bold and include the KATP channel subunits, SUR1 (sulfonylurea receptor) and Kir6.2 (inwardly rectifying potassium channel), GCK, HK1, GDH, SCHAD, PNC2 (pyrimidine nucleotide carrier 2), HNF4a (hepatic nuclear transcription factor 4 alpha), HNF1a (hepatic nuclear transcription factor 1 alpha), FOXA2 (forkhead box A2), MCT1, and UCP2 (uncoupling protein 2). Therapeutic agents currently used to treat hyperinsulinism are underlined and therapies under investigation are italicized. Diazoxide suppresses insulin secretion by activating the KATP channel. Somatostatin suppresses insulin secretion downstream of calcium entry. ATP, adenosine triphosphate; ADP, adenosine diphosphate; Ca, calcium; cAMP, cyclic adenosine monophosphate; G6P, glucose-6-phosphate
Source: Original
Molecular Mechanisms of Disease
An expanding area of focus in the field of HI is the application of advanced genetic diagnostic tools to uncover novel mechanisms of disease.
The initial reports of HI (then termed “idiopathic hypoglycemia of infancy”) described by McQuarrie detailed a pedigree of five children born to three siblings with infantile onset symptomatic hypoglycemia. A linkage analysis performed a few years ago pointed at the possibility that aberrant expression of hexokinase 1 (encoded by HK1) was the molecular mechanism in this family, but until now the genetic etiology remained unconfirmed (6). Hexokinase 1, a glucose phosphorylating enzyme that has markedly higher affinity for glucose than glucokinase, is normally selectively silenced in pancreatic beta-cells, thereby ensuring appropriate glucose sensing and preventing insulin release when plasma glucose is low (Figure 2). Recently, Wakeling et al. performed whole genome sequencing on 135 individuals with genetic testing negative HI and identified non-coding variants within a regulatory region of HK1 intron 2 that co-segregated with disease in families (7). Using epigenomic analysis, single-nuclei ATAC-seq and Hi-C, they demonstrated that these variants encompass a region of chromatin accessibility within a regulatory domain for the HK1 promoter. These variants disrupted transcription factor binding sites for FOXA2, NKX2 and NFAT families, suggesting that continuous transcription binding is necessary to maintain silencing of HK1 in beta-cells. Notably, inactivating mutations in FOXA2, have been established to cause a syndrome of HI and hypopituitarism (8, 9). Additionally, they found that hexokinase 1 was expressed and colocalized with insulin, but not glucagon, in pancreatic tissue resected from individuals with non-coding variants. In contrast, hexokinase 1 was not expressed in islets isolated from controls, confirming that these variants result in inappropriate HK1 expression within beta-cells, presumably resulting in insulin secretion at inappropriately low glucose thresholds. These findings highlight the potential role of other, yet undiscovered, non-coding variants responsible for HI by disruption of beta-cell gene silencing. Over 60 genes selectively silenced in beta-cells have been described, paving new avenues for the discovery of novel HI genes.
Two different mechanisms have recently been uncovered to underly cases of HI with clinical features of the hyperinsulinism hyperammonemia (HIHA) syndrome, but negative genetic testing for such. HIHA syndrome is caused by dominant mutations in GLUD1, encoding glutamate dehydrogenase, that diminish sensitivity of this enzyme to allosteric inhibition by guanine triphosphate (GTP). The syndrome is clinically characterized by both fasting and protein-induced hypoglycemia and persistent hyperammonemia. Seizures, characteristically atypical absence, and neurobehavioral impairments are also frequently observed.
Shahroor et al., Jasper et al., and Safran, et al., each reported findings of biallelic inactivating mutations in the SLC25A36 gene as a novel cause of HIHA syndrome (10–12). SLC25A36 encodes pyrimidine nucleotide carrier 2 (PNC2), a mitochondrial nucleotide carrier that transports pyrimidine, as well as guanine, nucleotides across the inner mitochondrial membrane (Figure 2). In functional analysis, Shahroor et al., showed that knockdown of SLC25A36 expression in HeLa cells caused a reduction in mitochondrial GTP content, likely resulting in disinhibition of glutamate dehydrogenase (12). A total of seven affected individuals have been reported to date, each of whom had HI – often with history suggestive of protein-induced hypoglycemia – as well as hyperammonemia, and seizures. Many also had short stature, which has not previously been reported in association with HIHA syndrome. Neurocognitive function was reportedly normal in all but one individual, who had intellectual disability and attention deficit hyperactivity disorder, but who also had comorbid suboptimally treated primary hypothyroidism (10). Supplementation with uridine, as a pyrimidine replacement, in this child resulted in normalized thyroid function, and improved glycemic control, growth, and behavior. Combined, these findings expand the role of mitochondrial transporters in the regulation of insulin secretion.
In addition to the discovery of novel genes, another advance has been the use of next generation sequencing at high coverage depth to detect mosaic mutations in known HI genes are a cause of “genetic testing negative” HI. Boodhansingh et al. reported three patients with clinical features of HIHA syndrome all of whom had negative mutation analysis in peripheral blood DNA for GLUD1 as well as other known HI genes (13). Two patients had protein-induced, diazoxide-responsive HI and hyperammonemia, and one patient was the mother of a child with presumed de novo GLUD1 mutation, who herself had a history of HI treated with pancreatectomy as a child. Low-level mosaicism for known disease-causing GLUD1 mutations were detected in each patient with percent mosaicism ranging from 2.7% to 10.4%. In the one patient for whom pancreatic tissue was available, the GLUD1 mutation was detected at higher percent mosaicism in pancreatic DNA (17.9%-28.9%).
Boodhansingh et al. also identified low-level mosaic mutations in known HI genes as responsible for some cases of diazoxide-unresponsive HI with atypical pancreatic histology and negative genetic testing (14). This atypical histological form, also termed Localized Islet Nuclear Enlargement (LINE)-HI, is characterized by islet nucleomegaly, like diffuse HI. However, unlike diffuse HI, in which the entire pancreas is affected, nucleomegaly is confined to an isolated region of pancreas, and partial pancreatectomy may be curative in some cases. They performed next generation sequencing on pancreatic specimens from eight children with LINE-HI and found disease-causing mutations ranging from 2.2% to 10.3% mosaicism in six children (n=3 ABCC8, n=3 GCK). These findings build on prior reports by Houghton et al. and Henquin et al. in which mosaic mutations (combined mosaic ABCC8 mutation and mosaic chromosome 11 paternal uniparental disomy, and mosaic GCK mutation, respectively) were detected in affected pancreatic tissue, but not in normal tissue or peripheral blood DNA in two children with LINE-HI (15, 16). Both Boodhansingh et al. and Henquin et al. demonstrated increased immunostaining for hexokinase 1 in affected, but not normal pancreatic tissue in some cases of LINE-HI. Notably, this work preceded the identification of disease-causing variants in HK1 intron 2. While the use of highly sensitive next-generation sequencing offers an attractive approach to uncover the molecular mechanisms of “genetic testing negative” HI, broad use hinges upon the development of methods to both detect and validate low-level mosaic mutations.
Imaging
Imaging with 6-fluoro-L-3,4-dihydroxyphenylalanine positron emission tomography/computed tomography (18F-DOPA PET/CT) is the standard-of-care for identification and localization of lesions in suspected focal cases of HI (Figure 1). While this imaging modality has >90% sensitivity for detection of focal lesions – based upon pooled analysis across all studies to date in which histopathological confirmation of disease was performed – focal lesions are still missed in some cases (17). Accurate identification of a focal lesion is paramount, since 97% of focal HI patients can be cured by surgical resection (4). An alternative radiotracer targeting the glucagon-like peptide-1 (GLP-1) receptor, 68Ga-NODAGA-exendin-4 PET/CT, has been shown to detect insulinomas with high sensitivity in adults (18). These findings have garnered interest in the application of this tracer for detection of focal HI, particularly since exendin-4 offers molecular specificity for GLP-1 receptors on pancreatic beta-cells (Figure 2), and 68Ga-NODAGA-exendin-4 PET/CT is effective at lower radiation doses than 18F-DOPA PET/CT (19).
Boss et al. compared the performance of 68Ga-NODAGA-exendin-4 PET/CT and 18F-DOPA PET/CT in 19 patients with HI (8 prospectively enrolled, 11 retrospectively analyzed) who met criteria for imaging (Figure 1) (20). Fourteen patients with histologically confirmed focal HI were included in the analysis. The authors reported increased sensitivity of 68Ga-NODAGA-exendin-4 PET/CT (100%, 95% CI: 77–100%) compared to 18F-DOPA PET/CT (71%, 95% CI: 42–92%) when images were interpreted by unblinded providers. When interpreted by a blinded nuclear medicine expert, sensitivity of the two modalities was more similar (93%, 95% CI: 66-100% vs. 86%, 95% CI: 57-98%). Notably, this study was subject to several forms of bias (21, 22). Observer bias was likely introduced due to the inclusion of retrospective data. Overestimation of imaging sensitivity was possible, as the 4 patients in whom a focal lesion was not identified on imaging did not undergo surgery and thus did not have histologic confirmation of diffuse disease. The order of scanning was not explicitly clear. Consequently, information bias may have resulted if knowledge of the 18F-DOPA PET/CT results influenced interpretation of the 68Ga-NODAGA-exendin-4 scan. Pending replication in rigorously designed studies of 68Ga-NODAGA-exendin-4 PET/CT in HI, 18F-DOPA PET/CT performed by experts in multidisciplinary HI centers remains best clinical practice.
Management
Preventing and treating hypoglycemia is critical to improve neurodevelopmental outcomes in HI. Intermittent glucose monitoring via fingerstick is current standard-of-care for most patients. However, this approach is susceptible to missed episodes of hypoglycemia, particularly overnight, and does not provide detailed information on glycemic trends. Increasingly, there has been interest in the use of continuous glucose monitoring (CGM) to improve glycemic control in HI. Several small studies in infants and children with HI have reported low positive predictive value (reported range: 14.8-16.0%), but high negative predictive value (reported range: 99.1-99.6%), for CGM detection of hypoglycemia with sensitivities ranging 54-73% (23–25). Given the limited study of CGM in HI, questions regarding the utility and impact of its use have persisted. In a pilot study of 10 children with HI, Worth et al. reported that short-term use of an algorithm to detect weekly patterns in hypoglycemia frequency (HYPO-CHEAT) resulted in reduced time below range (hypoglycemia) from 7.1% to 5.4% (26). However, in a related study, this same group found that CGM alone had insufficient accuracy for use as a standalone tool in clinical practice. Following development of an error grid tailored for use in HI, akin to the Clarke and Parkes error grids developed for use in diabetes mellitus (27, 28), the authors found that 15% of CGM errors posed moderate risk of patient harm (29). Despite these limitations, patients and families with HI have reported overall favorable experiences with CGM use, citing advantages of reassurance, decreased fingersticks, and opportunities to detect trends as outweighing the drawbacks of suboptimal accuracy and alarm fatigue (30).
While diazoxide remains the only FDA-approved medication for HI, 60% of HI cases are unresponsive to this first-line therapy. Somatostatin analogues have long been used off-label as second-line treatment. Cuff et al. recently reported on 54 children treated with lanreotide, the largest series to date (31). They found lanreotide resulted in significantly longer fasting tolerance, compared to fasting tolerance prior to lanreotide initiation. Lanreotide also permitted simplification of treatment regimen – 42% of children were able to discontinue other treatments and were managed with lanreotide monotherapy.
New therapies for HI are under investigation with promising results (Figure 2). In clinical trials, dasiglucagon, a soluble glucagon analog administered via continuous subcutaneous infusion, was shown to decrease intravenous glucose requirements and reduce time spent in hypoglycemia in infants with HI (32, 33). Phase 2 studies with exendin-(9–39) (also known as avexitide), a GLP-1 receptor antagonist, demonstrated a reduction in likelihood of hypoglycemia during fasting and during oral protein challenge, up to 84% and 82%, respectively at higher doses, in children with KATP-HI (34). An allosteric inhibitor of the insulin receptor (RZ358) resulted in a statistically and clinically significant reduction of time spent in hypoglycemia (35). Phase 2 studies of a long-acting glucagon analog (HM15136) and preclinical studies of a selective nonpeptide somatostatin receptor 5 agonist are currently underway (36, 37).
Outcomes
Over half a century since HI was first described, neurodevelopmental outcomes have not considerably improved. The reported prevalence of neurodevelopmental differences in children with HI ranges 26-48% in different cohorts from around the world (38). Sigal et al. showed that children with perinatal stress-induced HI (in whom hypoglycemia exposure is limited to the first months of life) experience similar rates of adverse neurodevelopment (39). Rapid advances in the monitoring and therapeutic landscape for HI holds promise to improve these outcomes, and new approaches to assess long-term impact have emerged. Using a novel magnetic resonance imaging technique, glutamate weighted chemical exchange saturation transfer, Rosenfeld et al. found abnormal patterns of brain glutamate in patients with HIHA syndrome. Their findings open the door for the development of neuroimaging biomarkers to assess neurological involvement and response to investigational therapies in HI (40). Roeper et al. performed the first systematic evaluation of psychosocial well-being for caregivers of children with HI (41). The authors found high rates of caregiver anxiety and depression. Moreover, they reported an association between child neurodevelopmental differences and both caregiver mental health and quality of life outcomes. The development of an HI Global Registry (HIGR) has provided a framework to comprehensively evaluate natural history, including quality of life outcomes, in patients with HI (42).
Conclusion:
Continued progress has been made elucidating the molecular mechanisms responsible for HI. These advances have opened new avenues for the discovery of novel HI genes. New therapeutic targets have been identified and directed therapies for HI are under investigation. Currently, HI remains associated with high levels of morbidity, particularly adverse neurodevelopmental outcomes. Recent developments hold promise to tailor treatment, and improve long-term outcomes for individuals with HI.
Key Points:
While the genetic cause of hyperinsulinism remains unknown in over 20% of patients, discovery efforts continue to uncover novel molecular mechanisms responsible for disease. Understanding the genetic etiology of hyperinsulinism is crucial to tailor management and may guide development of new therapies.
Additional studies evaluating the performance of 68Ga-NODAGA-exendin-4 PET/CT for localization of focal hyperinsulinism lesions are needed. Currently, 18F-DOPA PET/CT performed by experts in multidisciplinary hyperinsulinism centers remains best clinical practice.
Continuous glucose monitors have suboptimal accuracy compared to fingerstick glucose measurements in children with hyperinsulinism but may still provide benefit.
Improved therapies are needed to reduce long-term complications, particularly the neurodevelopmental sequelae, of hyperinsulinism. New therapies under development include a GLP-1 receptor antagonist, short- and long-acting glucagon analogues, a selective non-peptide somatostatin receptor agonist, and an inhibitor of the insulin receptor.
Financial Support and Sponsorship
This work was supported by National Institutes of Health grants R01-DK056268 and R01-DK098517 (D.D.D.L), and Administrative Supplements to Support Emerging Physician Scientists to Develop Research Expertise in Diabetes, Endocrinology and Metabolic Diseases R01-DK056268 (E.R.).
Conflicts of Interest
Diva D. De Leon has served as principal investigator for industry-sponsored trials from Zealand Pharma, Hanmi Pharmaceuticals and Rezolute. She has received research funding from Twist Bioscience and Crinetics Pharmaceuticals. Diva D. De Leon has received consulting fees from Zealand Pharma, Crinetics Pharmaceuticals, Hanmi Pharmaceutical and Eiger Biopharmaceuticals. Diva D. De Leon is a named inventor in patents # USA Patent Number 9,616,108, 2017, USA Patent Number 9,821,031, 2017, Europe Patent Number EP 2120994, 2018, and Europe Patent Number EP2818181, 2019. These funders played no role in the completion of this manuscript.
References and recommended reading:
- 1.Stanley CA, Baker L. Hyperinsulinism in infancy: diagnosis by demonstration of abnormal response to fasting hypoglycemia. Pediatrics. 1976;57(5):702–11. [PubMed] [Google Scholar]
- 2.Quarrie Mc. Idiopathic spontaneously occurring hypoglycemia in infants; clinical significance of problem and treatment. AMA Am J Dis Child. 1954;87(4):399–428. [PubMed] [Google Scholar]
- 3.Snider KE, Becker S, Boyajian L, Shyng SL, MacMullen C, Hughes N, et al. Genotype and phenotype correlations in 417 children with congenital hyperinsulinism. J Clin Endocrinol Metab. 2013;98(2):E355–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adzick NS, De Leon DD, States LJ, Lord K, Bhatti TR, Becker SA, et al. Surgical treatment of congenital hyperinsulinism: Results from 500 pancreatectomies in neonates and children. J Pediatr Surg. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lord K, Radcliffe J, Gallagher PR, Adzick NS, Stanley CA, De Leon DD. High Risk of Diabetes and Neurobehavioral Deficits in Individuals With Surgically Treated Hyperinsulinism. J Clin Endocrinol Metab. 2015;100(11):4133–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pinney SE, Ganapathy K, Bradfield J, Stokes D, Sasson A, Mackiewicz K, et al. Dominant form of congenital hyperinsulinism maps to HK1 region on 10q. Horm Res Paediatr. 2013;80(1):18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **7.Wakeling MN, Owens NDL, Hopkinson JR, Johnson MB, Houghton JAL, Dastamani A, et al. Non-coding variants disrupting a tissue-specific regulatory element in HK1 cause congenital hyperinsulinism. Nat Genet. 2022;54(11):1615–20. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study describes the identification of variants in intron 2 of the HK1 gene as a new molecular etiology of congenital hyperinsulinism.
- 8.Giri D, Vignola ML, Gualtieri A, Scagliotti V, McNamara P, Peak M, et al. Novel FOXA2 mutation causes Hyperinsulinism, Hypopituitarism with Craniofacial and Endoderm-derived organ abnormalities. Hum Mol Genet. 2017;26(22):4315–26. [DOI] [PubMed] [Google Scholar]
- 9.Vajravelu ME, Chai J, Krock B, Baker S, Langdon D, Alter C, et al. Congenital Hyperinsulinism and Hypopituitarism Attributable to a Mutation in FOXA2. J Clin Endocrinol Metab. 2018;103(3):1042–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *10.Jasper L, Scarcia P, Rust S, Reunert J, Palmieri F, Marquardt T. Uridine Treatment of the First Known Case of SLC25A36 Deficiency. Int J Mol Sci. 2021;22(18). [DOI] [PMC free article] [PubMed] [Google Scholar]; This is the first report of a novel mutation in SLC25A36 causing a clinical phenotype similar to the hyperinsulinism hyperammonemia syndrome.
- *11.Safran A, Proskorovski-Ohayon R, Eskin-Schwartz M, Yogev Y, Drabkin M, Eremenko E, et al. Hyperinsulinism / hyperammonemia syndrome caused by biallelic SLC25A36 mutation. J Inherit Metab Dis. 2023. [DOI] [PubMed] [Google Scholar]; This study describes the clinical characteristics of four children with hyperinsulinism hyperammonemia syndrome due to inactivating mutations in SLC25A36 and summarizes the seven cases reported to date.
- **12.Shahroor MA, Lasorsa FM, Porcelli V, Dweikat I, Di Noia MA, Gur M, et al. PNC2 (SLC25A36) Deficiency Associated With the Hyperinsulinism/Hyperammonemia Syndrome. J Clin Endocrinol Metab. 2022;107(5):1346–56. [DOI] [PubMed] [Google Scholar]; This study reports findings from two siblings with inactivating mutations in SLC25A36 and provides functional evidence, via knockdown of SLC25A36 expression in HeLa cells, of the molecular mechanism responsible for the hyperinsulinism hyperammonemia phenotype.
- *13.Boodhansingh KE, Rosenfeld E, Lord K, Adzick NS, Bhatti T, Ganguly A, et al. Mosaic GLUD1 Mutations Associated with Hyperinsulinism Hyperammonemia Syndrome. Horm Res Paediatr. 2022;95(5):492–8. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study describes the use of next-generation sequencing with high depth of coverage to detect mosiac GLUD1 mutations in three patients with clinical features of the hyperinsulinsim hyperammonemia syndrome but negative clinical genetic testing.
- *14.Boodhansingh KE, Yang Z, Li C, Chen P, Lord K, Becker SA, et al. Localized islet nuclear enlargement hyperinsulinism (LINE-HI) due to ABCC8 and GCK mosaic mutations. Eur J Endocrinol. 2022;187(2):301–13. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study describes the phenotype and genotype features of 12 children with localized islet nuclear enlargement hyperinsulinism.
- 15.Houghton JA, Banerjee I, Shaikh G, Jabbar S, Laver TW, Cheesman E, et al. Unravelling the genetic causes of mosaic islet morphology in congenital hyperinsulinism. J Pathol Clin Res. 2020;6(1):12–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Henquin JC, Sempoux C, Marchandise J, Godecharles S, Guiot Y, Nenquin M, et al. Congenital hyperinsulinism caused by hexokinase I expression or glucokinase-activating mutation in a subset of beta-cells. Diabetes. 2013;62(5):1689–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.States LJ, Davis JC, Hamel SM, Becker SA, Zhuang H. (18)F-6-Fluoro-l-Dopa PET/CT Imaging of Congenital Hyperinsulinism. J Nucl Med. 2021;62(Suppl 2):51S–6S. [DOI] [PubMed] [Google Scholar]
- 18.Antwi K, Fani M, Heye T, Nicolas G, Rottenburger C, Kaul F, et al. Comparison of glucagon-like peptide-1 receptor (GLP-1R) PET/CT, SPECT/CT and 3T MRI for the localisation of occult insulinomas: evaluation of diagnostic accuracy in a prospective crossover imaging study. Eur J Nucl Med Mol Imaging. 2018;45(13):2318–27. [DOI] [PubMed] [Google Scholar]
- 19.Boss M, Buitinga M, Jansen TJP, Brom M, Visser EP, Gotthardt M. PET-Based Human Dosimetry of (68)Ga-NODAGA-Exendin-4, a Tracer for beta-Cell Imaging. J Nucl Med. 2020;61(1):112–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *20.Boss M, Rottenburger C, Brenner W, Blankenstein O, Prasad V, Prasad S, et al. (68)Ga-NODAGA-Exendin-4 PET/CT Improves the Detection of Focal Congenital Hyperinsulinism. J Nucl Med. 2022;63(2):310–5. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study prospectively enrolled 8 patients and retrospectively analyzed data from 11 patients with suspected focal hyperinsulinism to compare the performance of 18F-DOPA PET/CT and 68Ga-NODAGA-exendin-4 PET/CT for the detection and localization of focal lesions.
- *21.Banerjee I, Sajjan R, Estebanez MS, Dunne MJ, Mohnike K, Mohnike W, et al. (68)Ga-NODAGA-Exendin-4 PET Scanning for Focal Congenital Hyperinsulinism: Need for Replication. J Nucl Med. 2022;63(3):493. [DOI] [PubMed] [Google Scholar]; This Letter to the Editor summarizes methodological concerns that limit interpretation of findings from the investigation of 68Ga-NODAGA-exendin-4 PET/CT for the localization of focal hyperinsulinism.
- 22.Prasad V, Boss M, Rottenburger C, Brenner W, Blankenstein O, Prasad S, et al. Reply: (68)Ga NODAGA-Exendin-4 PET Scanning for Focal Congenital Hyperinsulinism: Need for Replication. J Nucl Med. 2022;63(3):493–4. [DOI] [PubMed] [Google Scholar]
- 23.Rayannavar A, Elci OU, Mitteer L, De Leon DD. Continuous Glucose Monitoring Systems: Are They Useful for Evaluating Glycemic Control in Children with Hyperinsulinism? Horm Res Paediatr. 2019;92(5):319–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *24.Vijayanand S, Stevenson PG, Grant M, Choong CS, Davis EA, Abraham MB. The utility of continuous glucose monitoring systems in the management of children with persistent hypoglycaemia. J Pediatr Endocrinol Metab. 2021;34(12):1567–72. [DOI] [PubMed] [Google Scholar]; Retrospective analyses of paired Dexcom G4 continuous glucose monitor and fingerstick glucose data from 40 children with hypoglycemia were performed in this study to evaluate the accuracy of continuous glucose monitor use in hypoglycemic disorders. Parent experience was evaluated via questionnaires.
- *25.Win M, Beckett R, Thomson L, Thankamony A, Beardsall K. Continuous Glucose Monitoring in the Management of Neonates With Persistent Hypoglycemia and Congenital Hyperinsulinism. J Clin Endocrinol Metab. 2022;107(1):e246–e53. [DOI] [PMC free article] [PubMed] [Google Scholar]; Retrospective study assessing the clinical utility of real-time continuous glucose monitor use in 14 neonates with persistent hypoglycemia, including neonates with congenital hyperinsulinism.
- 26.Worth C, Nutter PW, Dunne MJ, Salomon-Estebanez M, Banerjee I, Harper S. HYPO-CHEAT’s aggregated weekly visualisations of risk reduce real world hypoglycaemia. Digit Health. 2022;8:20552076221129712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clarke WL, Cox D, Gonder-Frederick LA, Carter W, Pohl SL. Evaluating clinical accuracy of systems for self-monitoring of blood glucose. Diabetes Care. 1987;10(5):622–8. [DOI] [PubMed] [Google Scholar]
- 28.Parkes JL, Slatin SL, Pardo S, Ginsberg BH. A new consensus error grid to evaluate the clinical significance of inaccuracies in the measurement of blood glucose. Diabetes Care. 2000;23(8):1143–8. [DOI] [PubMed] [Google Scholar]
- *29.Worth C, Dunne MJ, Salomon-Estebanez M, Harper S, Nutter PW, Dastamani A, et al. The hypoglycaemia error grid: A UK-wide consensus on CGM accuracy assessment in hyperinsulinism. Front Endocrinol (Lausanne). 2022;13:1016072. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study details the development and performance of a new error grid to evaluate the clinical utility of continuous glucose monitors in children with hyperinsulinism.
- 30.Auckburally SH, Worth C, Salomon-Estebanez M, Nicholson J, Harper S, Nutter PW, et al. Families’ Experiences of Continuous Glucose Monitoring in the Management of Congenital Hyperinsulinism: A Thematic Analysis. Front Endocrinol (Lausanne). 2022;13:894559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *31.Cuff H, Lord K, Ballester L, Scully T, Stewart N, De Leon DD. The Use of Lanreotide in the Treatment of Congenital Hyperinsulinism. J Clin Endocrinol Metab. 2022;107(8):e3115–e20. [DOI] [PubMed] [Google Scholar]; This large case series describes the safety and effectiveness of lanreotide in 54 children with congenital hyperinsulinism.
- 32.Banerjee I, De Leon DD, Kendall DM, Birch S, Bøge E, Ivkovic J, et al. Dasiglucagon treatment over 21 days in infants with congenital hyperinsulinism results in glycaemic stability and reduces requirement for intravenous glucose: 60th Annual Meeting of the European Society for Paediatric Endocrinology (ESPE). Hormone Research in Paediatrics. 2022;95(suppl 2)(2). [Google Scholar]
- 33.De Leon DD, Banerjee I, Kendall DM, Birch S, Bøge E, Ivkovic J, et al. Dasiglucagon Significantly Reduces Requirement for Intravenous Glucose in Children with Congenital Hyperinsulinism ages 7 Days to 12 Months: 60th Annual Meeting of the European Society for Paediatric Endocrinology (ESPE). Hormone Research in Paediatrics. 2022;95(suppl 2)(2). [Google Scholar]
- *34.Stefanovski D, Vajravelu ME, Givler S, De Leon DD. Exendin-(9-39) Effects on Glucose and Insulin in Children With Congenital Hyperinsulinism During Fasting and During a Meal and a Protein Challenge. Diabetes Care. 2022;45(6):1381–90. [DOI] [PMC free article] [PubMed] [Google Scholar]; This open-label, crossover study evaluated the effect of three different dosing regimens of exendin-(9-39) versus vehicle on fasting and protein-induced hypoglycemia in 16 children with hyperinsulinism.
- 35.Demirbilek H, Melikyan M, Galcheva S, Dastamani A, Thornton P, De Leon D, et al. Results from a Global, Multi-Center, Phase 2b Study (RIZE) in Congenital Hyperinsulinism: Characterization of a High Unmet Treatment Need and Glycemic Response to RZ358: 60th Annual Meeting of the European Society for Paediatric Endocrinology (ESPE). Hormone Research in Paediatrics. 2022;95(suppl 2)(2). [Google Scholar]
- 36.Heo YH, Kim JK, Lee JS, Lee SH, Shin SH, Choi IY, et al. A novel glucagon analog with an extended half-life, HM15136, normalizes glucose levels in rodent models of congenital hyperinsulinism. Sci Rep. 2022;12(1):16765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao J, Wang S, Hee Kim S, Han S, Rico-Bautista E, Sturchler E, et al. Discovery of 4-(3-aminopyrrolidinyl)-3-aryl-5-(benzimidazol-2-yl)-pyridines as potent and selective SST5 agonists for the treatment of congenital hyperinsulinism. Bioorg Med Chem Lett. 2022;71:128807. [DOI] [PubMed] [Google Scholar]
- 38.Banerjee I, Salomon-Estebanez M, Shah P, Nicholson J, Cosgrove KE, Dunne MJ. Therapies and outcomes of congenital hyperinsulinism-induced hypoglycaemia. Diabet Med. 2019;36(1):9–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *39.Sigal WM, Alzahrani O, Guadalupe GM, Guzman H, Radcliffe J, Thomas NH, et al. Natural history and neurodevelopmental outcomes in perinatal stress induced hyperinsulinism. Front Pediatr. 2022;10:999274. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study combined retrospective chart review, caregiver interview, and neurodevelopmental assessments to describe natural history and neurodevelopmental outcomes in perinatal stress-induced hyperinsulinism.
- *40.Rosenfeld E, Nanga RPR, Lucas A, Revell AY, Thomas A, Thomas NH, et al. Characterizing the neurological phenotype of the hyperinsulinism hyperammonemia syndrome. Orphanet J Rare Dis. 2022;17(1):248. [DOI] [PMC free article] [PubMed] [Google Scholar]; This cross-sectional study details the neurological phenotype of 12 patients with HIHA syndrome through use of neurocognitive assessments, electroencephalogram, and glutamate weighted chemical exchange saturation transfer magnetic resonance imaging.
- 41.Roeper M, Hoermann H, Salimi Dafsari R, Koestner F, Mayatepek E, Kummer S, et al. Anxiety, depression, and quality of life in parents of children with congenital hyperinsulinism. Eur J Pediatr. 2022;181(7):2779–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **42.Pasquini TLS, Mesfin M, Schmitt J, Raskin J. Global Registries in Congenital Hyperinsulinism. Front Endocrinol (Lausanne). 2022;13:876903. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper describes the development of a global registry for congenital hyperinsulinism and summarizes patient/caregiver-reported data on the medical and day-to-day experience of living with this congenital hyperinsulinism.