

Abstract
CO2 is produced during aerobic respiration. Normally, levels of CO2 in the blood are tightly regulated but pCO2 can rise (hypercapnia, pCO2 > 45 mmHg) in patients with lung diseases e.g. Chronic Obstructive Pulmonary Disease (COPD). Hypercapnia is a risk factor in COPD but may be of benefit in the context of destructive inflammation. The effects of CO2 per se, on transcription, independent of pH change are poorly understood and warrant further investigation.
Here we elucidate the influence of hypercapnia on monocytes and macrophages through integration of state-of-the-art RNA-sequencing, metabolic and metabolomic approaches. THP-1 monocytes and IL4 polarised primary murine macrophages were exposed to 5% CO2 Vs 10% CO2 for up to 24 h in pH- buffered conditions.
In hypercapnia, we identified ~370 differentially expressed genes (DEGs) under basal and ~1889 DEGs under LPS-stimulated conditions in monocytes. Transcripts relating to both mitochondrial and nuclear-encoded gene expression were enhanced in hypercapnia in basal and LPS-stimulated cells. Mitochondrial DNA content was not enhanced, but acylcarnitine species and genes associated with fatty acid metabolism were increased in hypercapnia. Primary macrophages exposed to hypercapnia also increased activation of genes associated with fatty acid metabolism and reduced activation of genes associated with glycolysis. Thus, hypercapnia elicits metabolic shifts in lipid metabolism in monocytes and macrophages under pH buffered conditions. These data indicate that CO2 is an important modulator of monocyte transcription that can influence immunometabolic signalling in immune cells in hypercapnia. These immunometabolic insights may be of benefit in the treatment of patients experiencing hypercapnia.
Keywords: hypercapnia, carbon dioxide, CO2, mitochondria, acylcarnitine, monocyte, gene expression, RNA-Seq
Introduction
Life on planet Earth has evolved around a background of changing CO2 concentrations 1 and organisms have developed a range of sensing and adaptive mechanisms. Humans have also evolved mechanisms to tightly regulate the levels of CO2 in the blood through acute chemosensation of elevated CO2 and adjustment of breathing, mainly via central chemoreceptors in the brain stem 2, 3. Despite the acute neuronal responses to CO2 being well described, there is still a relative dearth of knowledge relating to the transcriptional responses elicited under conditions of elevated CO2 which is a focus of this study. Normocapnia in humans equates to 35–45 mmHg CO2 in the circulation; however, tissue CO2 levels will differ depending on the physiological context. Patients with lung diseases e.g. COPD, cystic fibrosis, and acute respiratory distress syndrome (ARDS) can experience hypercapnia as evidenced by chronically elevated circulating pCO2 levels > 45mmHg 4, 5. Furthermore, local microenvironments such as the hypoxic core of solid tumours experience significantly higher than normal pCO2 levels 6. Hypercapnia is a risk factor with increased mortality in COPD 7 and is associated with higher intensive care unit (ICU) mortality in ARDS 4. Hypercapnia elicits a worse outcome in the context of bacterial infections 8, impairs wound healing 9, 10, promotes muscle wasting 11 and increases airway smooth muscle contractility 12. However, intriguingly, therapeutic hypercapnia is associated with improved prognosis in some settings e.g. after one-lung ventilation in lung lobectomy patients 13. Thus, hypercapnia has been likened to a ‘double-edged sword’ where any potential beneficial effects must be balanced against known deleterious effects 14, 15. Here we contend that limitations in our current knowledge in relation to the transcriptional response of immune cells to elevated CO2 is a major impediment to better predicting the outcome of elevated CO2 exposure in an immune context e.g. infection/ inflammation.
Thus, the objective of this project is to use an unbiased next-generation Ribonucleic acid (RNA)-sequencing approach to determine the impact of 10% CO2 (akin to that observed in patients with chronic respiratory disease) on transcription in monocytes. Additionally, we have buffered pH changes under these elevated CO2 conditions to decipher the effects of CO2 per se on monocytes as opposed to hypercapnic acidosis.
Monocytes are used as a model because their recruitment to the lung is important in COPD 16, a disease where hypercapnia is a prominent feature. Patients with severe COPD have increased numbers of total circulating monocytes and non-classical patrolling monocytes, compared to normal subjects and patients with less severe COPD. M2 macrophages which we also study here are enhanced in COPD 17. This study is not intended to directly model COPD (which is one of several lung diseases associated with hypercapnia), but focuses on CO2–dependent responses in monocytes and macrophages for the reasons outlined above.
We believe that the experimental approaches taken in this study can develop our understanding of the complex milieu of cellular signalling events that predispose to an advantageous suppression of inflammation in certain contexts versus a deleterious immunosuppression in others. To our knowledge this is the first study of its kind to use next-generation RNA sequencing integrated with metabolomic analysis to examine the cellular response to elevated CO2 in pH-buffered conditions in immune cells.
Results
RNA-seq: Effect of hypercapnia on monocytes in the basal state
We performed a principal component analysis (PCA) sample similarity analysis to investigate in a non-biased manner the robustness of our buffered hypercapnia experimental stimulus. Data in Figure 1a demonstrates separation of our normocapnia and hypercapnia monocyte replicates by PC1. 5% CO2 treated monocyte replicates in red clustered to the right-hand side of the graph, and the 10% CO2 treated monocyte replicates in turquoise blue clustered to the left-hand side of the graph. This indicates that our relatively modest 10% CO2 buffered hypercapnia protocol is sufficient to robustly segregate hypercapnia treated versus normocapnia control samples. We next examined the differentially expressed genes between normocapnia and hypercapnia using RNA-seq analysis. In Figure 1b, Log2 -fold change and adjusted p-value are depicted in a volcano plot. Rather than select an arbitrary fold change cut-off, we included all transcripts with an adjusted P-value of ≤ 0.05. This was because several transcripts of interest e.g. Dynein Light Chain LC8-Type 1 (DYNLL1) were modestly changed in terms of fold change (Log2 -fold change of 0.607) but to an extremely high degree of statistical confidence (P-adj 6.86E-06). 370 genes met this threshold with comparable numbers of upregulated (196 green) and downregulated (184 red) transcripts demonstrating sensitivity to hypercapnia. A list of the 8 most differentially expressed genes (up and down) is shown in Figure 1c with solute carrier family 16 Member 9 (SLC16A9); a pH-independent carnitine efflux transporter 18, 19 and a long noncoding RNA gene (AC048341.1 also known as MIRLET7IHG) being the most differentially upregulated and downregulated genes respectively. Supplementary figure 1a displays the differential expression data in a different way highlighting the top 50 most significantly affected transcripts (as determined by P-adj) ordered by expression level (log2 of transcript per million (TPM) values). We next performed gene ontology (GO) enrichment analysis to identify CO2-sensitive biological pathways/processes using the PANTHER database. The GO terms highlighted by this analysis related primarily to mitochondrial function, protein folding and ribosomal biogenesis. The top 20 GO terms from this analysis, ranked by fold enrichment, are displayed in Figure 1d.
Figure 1. RNA-seq analysis of THP-1 monocytes exposed to buffered hypercapnia for 4 h.
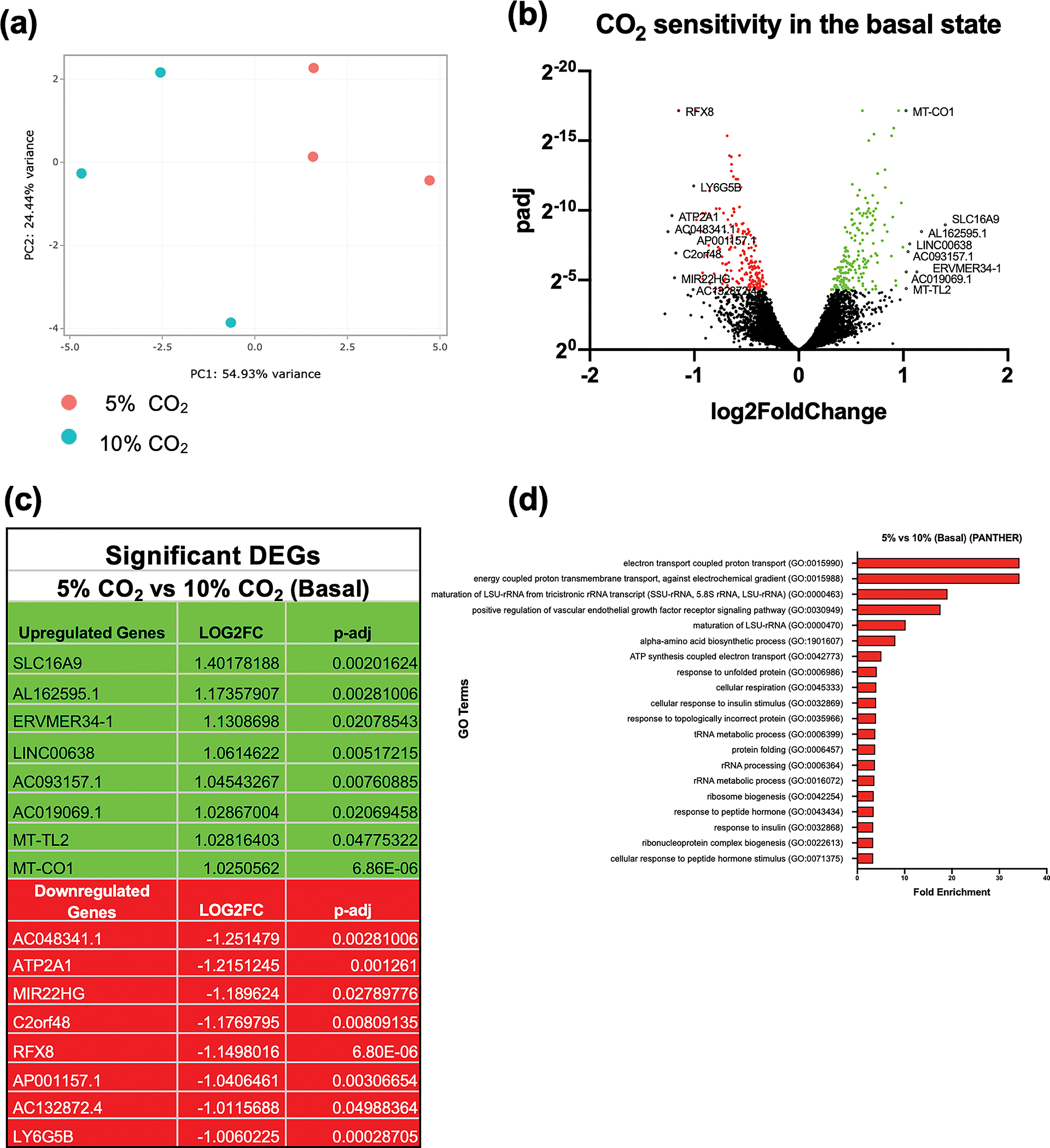
Principal component analysis of THP-1 cells exposed to 5% CO2 (red) or 10% CO2 (blue) for 4 h. The x-axis (PC1) is the vector that displays the most variance between samples and the y-axis (PC2) displays the second most. The percentage of total variance per principal component is shown on the axis label. Data are representative of n = 3 independent experiments (a). Volcano plot of differential expression between 5% and 10% CO2 in the basal state (4 h). The cut off for significance (P-adj < 0.05) is shown with a solid line on the y-axis. Significantly upregulated genes are shown in green and downregulated genes are shown in red (b). List of the 8 most significantly differentially expressed genes (up (green) and down (red)) in hypercapnia in the basal state. Includes all genes with a Log2FC of ± 1 and P-adj of < 0.05 (c). The top gene ontology terms associated with significantly differentially expressed genes in hypercapnia in the basal state (10% CO2 for 4 h) using a cut off value for significance of P-adj < 0.05. Terms are ranked by fold enrichment (d). Data are representative of n = 3 independent experiments in all cases (a–d).
RNA-seq: Effect of LPS stimulation on monocytes.
To examine the response of monocytes to hypercapnia in the context of inflammation we next exposed cells to buffered hypercapnia in the presence of the pro-inflammatory mediator lipopolysaccharide (LPS). LPS induced a strong transcriptional effect in both normocapnia and hypercapnia, with classical pro-inflammatory genes demonstrating markedly enhanced expression e.g. ICAM1, CXCL2, IL1A, TLR4, IL-6, CXCL8 (Supplementary figure 2, Supplementary tables 7, 8). Raw data relating to the LPS response in normocapnia (5% CO2) have previously been deposited in the GEO repository, accession number GSE 178391) 20. These data validate our RNA-sequencing approach in THP-1 monocytes.
RNA-seq: Effect of hypercapnia in the presence of LPS on monocytes.
Similar to the basal state, buffered hypercapnia was a robust stimulus in LPS-treated monocytes as indicated by PCA analysis Figure 2a. We next examined the differentially expressed genes between normocapnia and hypercapnia in the presence of LPS. 1889 genes met this threshold, with comparable numbers of upregulated (946 green) and downregulated (943 red) transcripts demonstrating sensitivity to hypercapnia Figure 2b. A list of the 10 most differentially expressed genes (up and down) is shown in Figure 2c with RNA component of signal recognition particle 7SL2 (RN7SL2) and Scavenger receptor class F member 2 (SCARF2) being the most differentially upregulated and downregulated genes respectively. Supplementary figure 1b displays the differential expression data in a different way, highlighting the top 50 most significantly affected transcripts (as determined by P-adj) ordered by expression level (Log2 of transcript per million (TPM)) values. We next performed gene ontology (GO) enrichment analysis, to identify CO2-sensitive biological pathways/processes in the presence of LPS using the PANTHER database (Figure 2d). The GO terms highlighted in this analysis related largely to protein localisation and telomeres. Some other terms related to glucocorticoid receptor signalling and protein folding. The top 20 GO terms from this analysis, ranked by fold enrichment, are displayed in Figure 2d. While there are clearly more differentially expressed transcripts in hypercapnia in the presence of LPS (1889); compared to in the basal state (370); we found a high proportion of genes (254) that were sensitive to elevated CO2 regardless of LPS (Figure 2e). We propose that this subset of transcripts represent particularly robustly CO2 sensitive genes e.g. Mitochondrially Encoded Cytochrome C Oxidase 1 (MT-CO1), ATPase Sarcoplasmic/Endoplasmic Reticulum Ca2+ Transporting 1 (ATP2A1) Pyrroline-5 Carboxylate Reductase 1 (PYCR1) and Receptor Interacting Serine/Threonine Kinase I (RIPK1). Additionally, most genes that are regulated by CO2 in both the basal and stimulated state, respond to CO2 in the same direction (Figure 2f).
Figure 2. RNA-seq analysis of THP-1 monocytes exposed to buffered hypercapnia for 4 h in the presence of LPS.
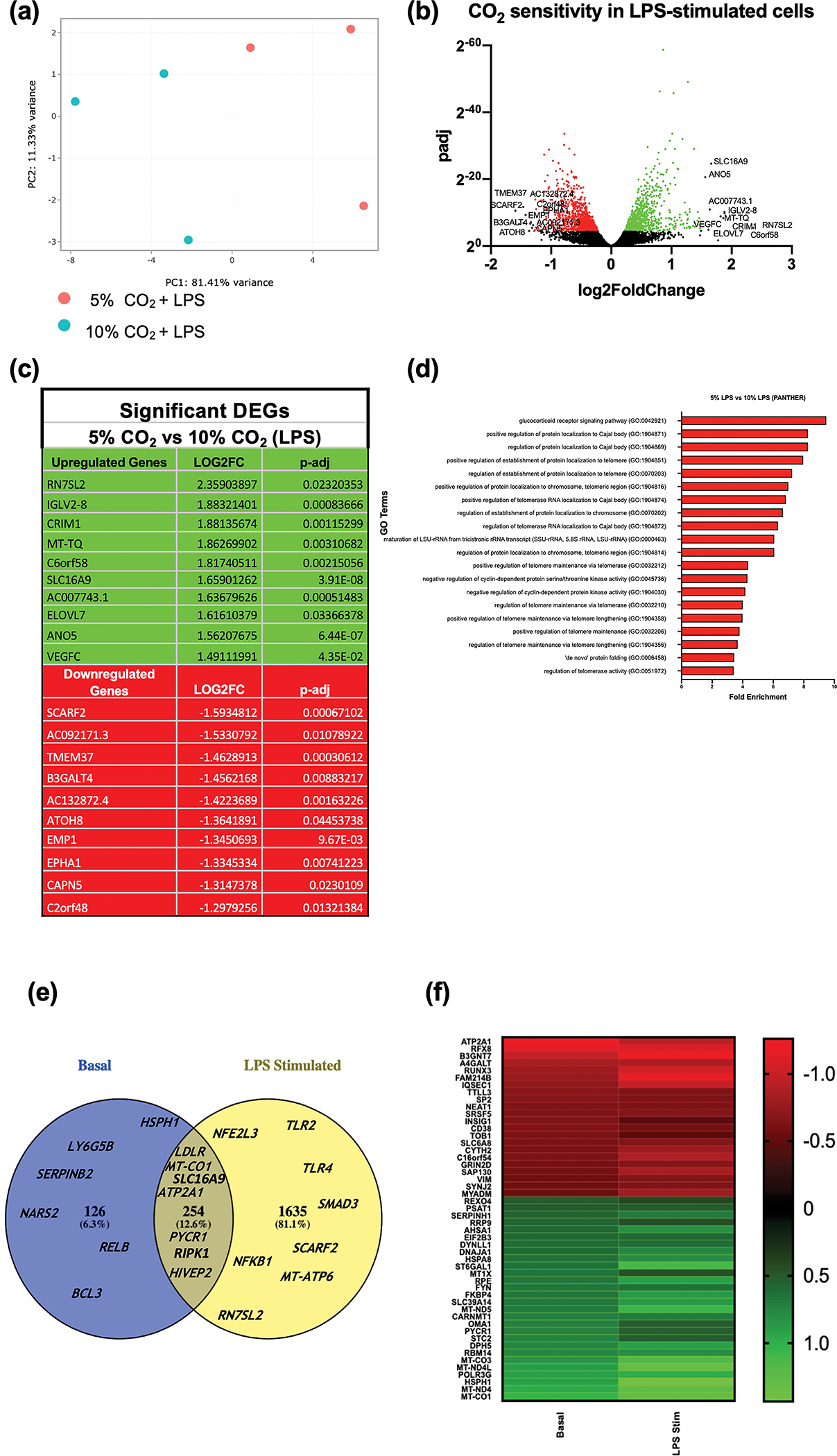
Principal component analysis of THP-1 cells exposed to 5% CO2 (red) or 10% CO2 (blue) (4 h) in an LPS stimulated state (2.5 μg mL−1 for 2 h). The x-axis (PC1) is the vector that displays the most variance between samples and the y-axis (PC2) displays the second most. The percentage of total variance per principal component is shown on the axis label (a). Volcano plot of differential expression between 5% and 10% CO2 (4 h) in an LPS stimulated state (2.5 μg mL−1 for 2 h). The cut off for significance (P-adj < 0.05) is shown with a solid line on the y-axis. Significantly upregulated genes are shown in green and downregulated genes are shown in red (b). List of significant differentially expressed genes in hypercapnia in LPS stimulated state. Includes top 10 most differentially expressed genes up and down ranked by Log2FC with a P-adj of < 0.05 (c). The top gene ontology terms associated with significantly differentially expressed genes in hypercapnia (4 h) in an LPS stimulated state (2.5 μg mL−1 for 2 h) using a cut off value for significance of P-adj < 0.05. Terms are ranked by fold enrichment (d). Venn diagram representing the number of genes which are commonly and exclusively sensitive to CO2 in THP-1 cells in the basal (blue) and LPS stimulated (yellow) state. Selected representative examples are included for each group (e). Heat map of the top 50 common differentially expressed genes between THP-1 cells exposed to 5% and 10% CO2 in the basal and LPS stimulated state. The top 50 genes were selected by significance (P-adj) in the basal state and then ranked by differential expression level (Log2FC) in the basal state for visualisation. All genes had a P-adj < 0.05 (f). Data are representative of n = 3 independent experiments in all cases (a–f).
RNA-seq: Effect of hypercapnia on mitochondrial gene expression in monocytes.
Based on the data in Figure 1 and 2, there was a clear signature for CO2 -dependent changes in genes related to the mitochondria and oxidative phosphorylation in both the basal and LPS-stimulated state. To better understand the effect of hypercapnia on mitochondria we examined our RNA-seq data for mitochondrially encoded genes which were differentially expressed in 10% CO2 compared to 5% CO2 in both the basal and LPS-stimulated states. Of the 37 mitochondrially encoded genes, 35 were detected in our experiments. A heat-map of the expression (in TPM) of the 35 detected mitochondrial genes is shown in Figure 3a. In the basal state, 9 mitochondrial genes were significantly differentially expressed in hypercapnia, 7 oxidative phosphorylation (OXPHOS) protein subunits and 2 transfer (t)RNAs. In the LPS stimulated state, 18 mitochondrial genes were significantly differentially expressed, 10 OXPHOS protein subunits and 8 tRNAs. These significant DEGs are highlighted in Figure 3b. These data are consistent with information presented in Figure 2f highlighting concordant responses of genes in both the basal and the LPS-stimulated states. In both the basal and LPS stimulated states, several (but not all) mitochondrial genes which produce subunits of Complex I of the OXPHOS pathway were significantly upregulated by hypercapnia. Mitochondrially Encoded NADH: Ubiquinone Oxidoreductase Core Subunit 4 (MT-ND4) (Figure 3c), MT-ND4L, MT-ND5 and MT-ND6 were all upregulated in both the basal and stimulated state, while MT-ND1 was only significantly upregulated in the presence of LPS. MT-ND2 and MT-ND3 (Figure 3d) were not significantly differentially expressed in hypercapnia in either the basal or LPS stimulated state. The mitochondria encode a single subunit from Complex III, MT-CYB. This gene was not significantly affected by hypercapnia in either the basal or LPS stimulated states. All 3 of the mitochondrially encoded Complex IV subunits (MT-CO1 (Figure 3e), MT-CO2 and MT-CO3) were upregulated in 10% CO2 compared to 5% CO2 in both the basal and LPS stimulated states.
Figure 3. Monocyte mitochondrial gene expression in hypercapnia.
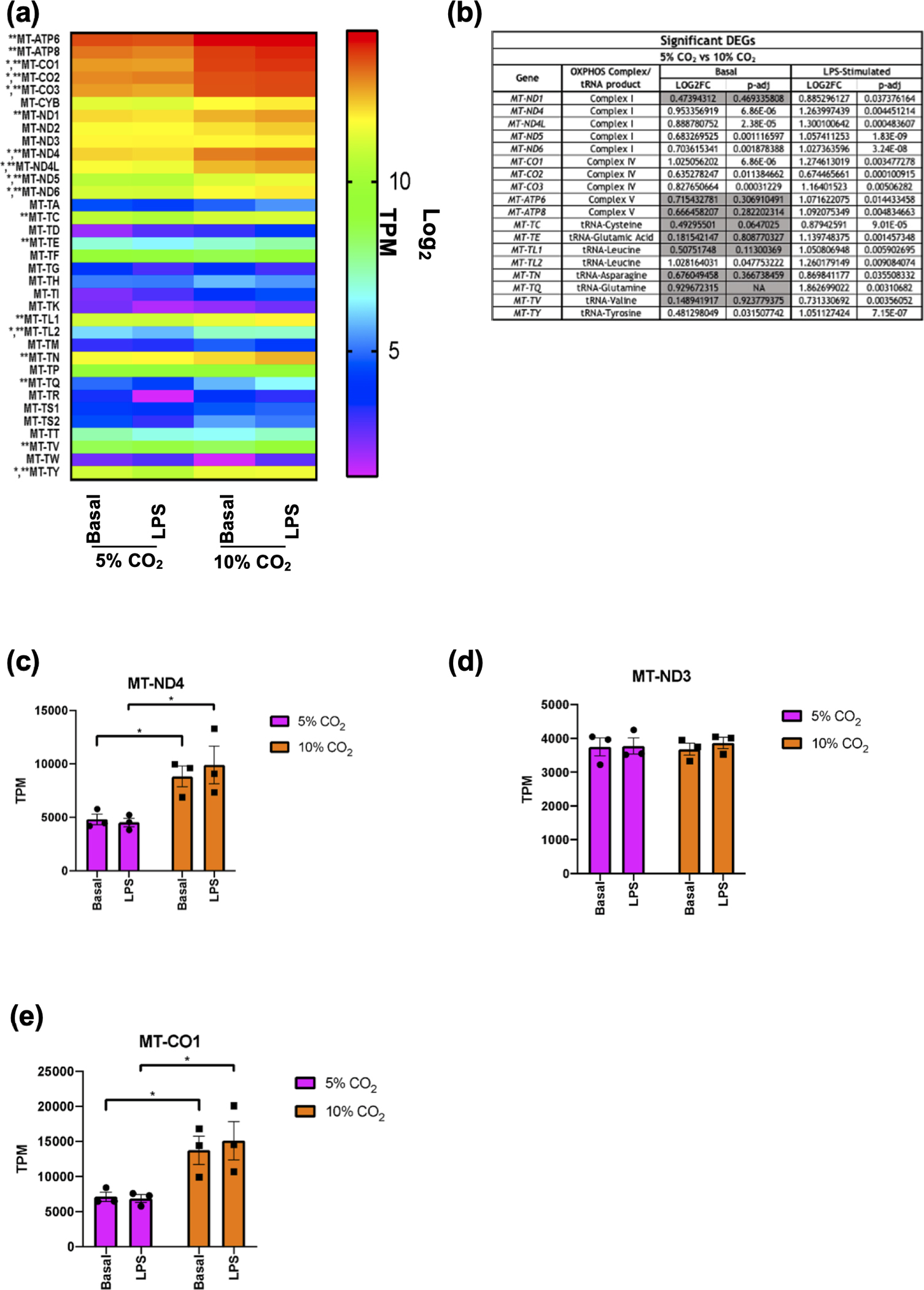
Heat map of all detected mitochondrially-encoded genes in THP-1 cells exposed to 5% or 10% CO2 for 4 h ± LPS (2.5 μg mL−1) for 2 h. Expression is displayed as Log2 of TPM. Significantly differentially expressed genes in hypercapnia in the basal state are denoted by (*). Significantly differentially expressed genes in hypercapnia the LPS stimulated state are denoted by (**) (a). List of significant differentially expressed mitochondrial genes in hypercapnia in basal and LPS stimulated states. Non-significant results in the basal state are shown in grey (b). Raw TPM values extracted from RNA-seq data for MT-ND4 (c), MT-ND3 (d), and MT-CO1 (e) in THP-1 cells in the basal (unstimulated) and LPS stimulated (2.5 μg mL−1 for 2 h) state in both 5% (pink) and 10% (orange) CO2 conditions (4 h). Data shown as mean ± SEM for n = 3 independent experiments. Significant differential expression from individual comparisons using DESeq2 analysis is denoted by (*).
Thus, we observed that several mitochondrially encoded genes were upregulated in hypercapnia. However, there are also many nuclear encoded genes whose protein product is localised in the mitochondria. To gain a more global view of the effect of short-term hypercapnia on mitochondria in monocytes, we used MitoCarta3.0, a database of proteins localised to the mitochondria. We analysed our expanded DEG lists of CO2-sensitive genes in both the basal and LPS stimulated states and compared them to the list of genes in MitoCarta3.0. Of the 1136 genes in this database, 46 were differentially expressed in hypercapnia in the basal state (Supplementary Figure 3a), while 149 were differentially expressed in hypercapnia in the LPS stimulated state (Supplementary figure 3b). Notably, in the presence of LPS, Transcription Factor A, Mitochondrial (TFAM) (a key regulator of mitochondrial transcription) was upregulated by hypercapnia, while Isocitrate dehydrogenase (NADP(+)) 2 (IDH2) (a catalytic enzyme in the TCA cycle) was downregulated. In both the basal and LPS-stimulated cells, the MitoCarta3.0 analyses highlighted 2 groups of genes of interest. (i) Nuclear-encoded mitochondrial tRNA synthetases (Supplementary figure 4a, b) e.g. Glutamyl-TRNA Synthetase 2, Mitochondrial (EARS2) and (ii) mitochondrial ribosome or “mitoribosome” proteins (Supplementary figure 4c, d) e.g. mitochondrial ribosomal protein L 15 (MRPL15). Taken together these data support the idea that 4 h of hypercapnia exposure is a robust cell stimulus capable of selectively increasing the transcription of mitochondrial genes, with both mitochondrial and nuclear encoded transcripts significantly increased.
Effect of hypercapnia on mitochondrial content and mitochondrial function in monocytes.
To investigate the impact of hypercapnia on mitochondrial function and metabolism in monocytes we performed a range of cellular assays. To determine the impact of elevated CO2 on mitochondrial mass we used a quantitative polymerase chain reaction (qPCR)-based approach to compare the ratio of mitochondrial deoxyribonucleic acid (DNA) associated MT-ND4 to the DNA associated with a nuclear encoded gene beta-2-microglobulin B2M. In principle if there is an increase in the ratio of MT-ND4 DNA: B2M DNA this is an indication of increased mitochondrial number or increased mitochondrial mass per cell. Figure 4a demonstrates that there was no significant change in the ratio of MT-ND4: B2M DNA at 2, 4, or 24 h of exposure to 10% CO2 ± LPS. In addition, using a different approach, we measured the expression of a mitochondrial membrane lipid, cardiolipin using the fluorescent dye nonyl-acridine orange (NAO). In response to 10% CO2 over 24 h, THP-1 cells demonstrated significantly less NAO-dependent fluorescence compared to 5% CO2 controls (Figure 4b).
Figure 4. Monocyte mitochondrial DNA and function.
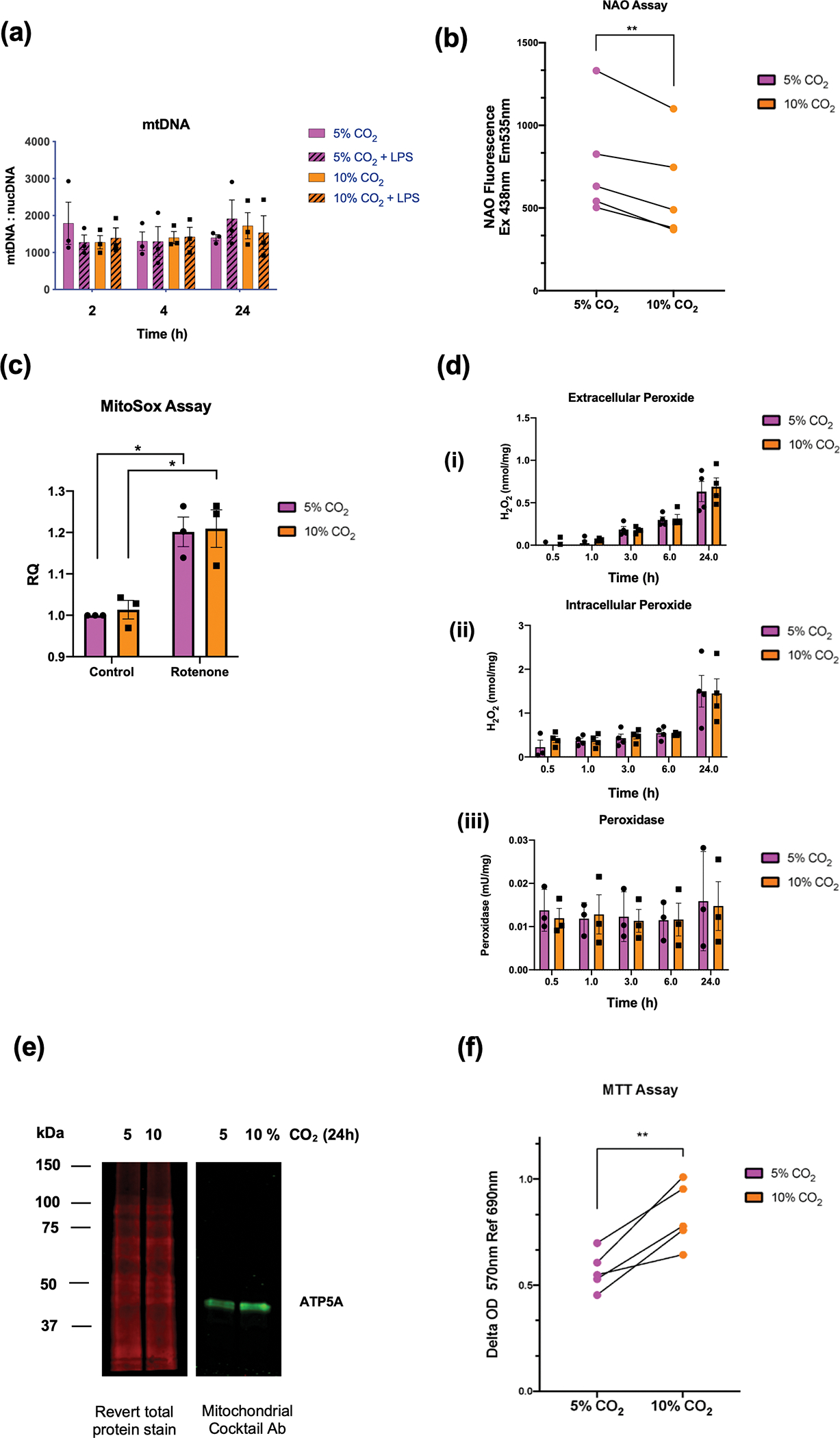
mtDNA content of THP-1 cells untreated or stimulated with LPS (2.5 μg mL−1) for 2, 4 and 24 h in 5% (pink) or 10% (orange) CO2. mtDNA content was determined as a ratio of MT-ND4 to B2M. Data shown as mean ± SEM for n = 3 independent experiments (a). Nonyl acridine Orange Fluorescence readings were obtained using a CLARIOstar plate reader (Ex. 438 nm EM 535 nm) from THP-1 monocytes exposed to 5% or 10% CO2 for 24 h. Data are representative of n = 5 independent experiments. Data shown are the mean values for each individual n-number. Statistical analysis was performed using a paired t-test. Significance values ** denotes a P-value < 0.01 (b). Relative MitoSox fluorescence at 675–715 nm in THP-1 cells exposed to 5% or 10% CO2 for 4 h ± rotenone (1 μM) for 2 h. Fluorescence is normalised to an unstimulated control at 5% CO2. Data are shown as mean ± SEM and representative of n = 3 individual experiments. Statistical analysis was performed using a 2-way ANOVA followed by Sidak’s multiple comparison’s test. Significance values * denotes a P-value < 0.05 (c) Measurement of (i) extracellular peroxide, (ii) intracellular peroxide and (iii) peroxidase activity in THP-1 cells cultured in buffered phenol-free DMEM media at 5% and 10% CO2 for up to 24 h. Concentrations were normalised to a standard curve. Data are shown as mean ± SEM and is representative of n = 3 or n = 4 individual experiments, with all conditions containing at least 2 replicates (d). Western blot analysis of mitochondrial lysates from THP-1 cells exposed to 5 or 10% CO2 for 24 h. Lysates were probed using a revert total protein stain and imaged in the 700 nm channel or incubated with a mitochondrial cocktail primary antibody followed by a fluorescent secondary mouse antibody and imaged in the 800 nm channel on a Li-COR imaging system. Image is representative of n = 4 independent experiments (e). MTT assay was performed on THP-1 monocytes exposed to 5% or 10% CO2 for 24 h with MTT addition to the media for the final 2 h. Formazan production was determined on a CLARIOstar plate reader at 570 nm Ref 690 nm. Data are representative of n = 5 independent experiments. Data shown are the mean value for each individual n-number. Statistical analysis was performed using a paired t-test. Significance values ** denotes a P-value < 0.01 (f).
To determine mitochondrial superoxide production, we employed the superoxide fluorescent indicator MitoSOX. There was no difference in the relative fluorescence between 5% and 10% CO2 at 4 h; however, rotenone treatment did result in a statistically significant increase in fluorescence regardless of the CO2 treatment (Figure 4c). We next used Amplex Red to measure extracellular and intracellular hydrogen peroxide, as well as intracellular peroxidase activity in THP-1 cells in 5% and 10% CO2 from 0.5–24 h. Extracellular peroxide was measured in the culture media. We observed a time dependent change in extracellular peroxide, increasing up to 24 h; however, there was no apparent CO2-dependent effect (Figure 4d (i)). Intracellular peroxide level was measured in cell lysates. There was an increase in intracellular peroxide at 24 h; however, there was no difference between 5% and 10% CO2 (Figure 4d (ii)). Intracellular peroxidase activity was also measured in cell lysates; however, again there was no difference between 5% and 10% CO2 (Figure 4d (iii)). Taken together we observed an increase in extracellular peroxide and intracellular peroxide up to 24 h of exposure, with no effect of elevated CO2 evident. We next exposed THP-1 cells to 5% and 10% CO2 and performed mitochondrial enrichment. Using an antibody capable of measuring oxidative phosphorylation proteins, we observed an increased expression of the nuclear encoded mitochondrial ATP-synthase subunit ATP5A in our mitochondrial enriched fractions (Figure 4e and quantified in Supplementary figure 4e).
In order to determine the level reductase activity in our monocytes exposed to normocapnia and hypercapnia, we employed a 3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide (MTT) assay where MTT is converted to formazan by intracellular reductases. Monocytes were exposed to normocapnia or hypercapnia for 22h followed by addition of MTT to the media for a further 2h. Formazan detection at 570 nm was significantly higher in cells exposed to elevated levels of CO2 indicative of enhanced cellular reductase activity in hypercapnia (Figure 4f). We also performed real time cell metabolic analysis using Seahorse technology but observed no significant CO2-dependent difference in basal oxygen consumption rate, maximal respiration, basal extracellular acidification or glycolysis (Supplementary figure 5). However, due to the nature of this assay, the metabolic analysis must take place at ambient CO2 levels (0.04%), which is a limitation when seeking to compare the effect of elevated CO2 on metabolic activity. Taken together, the data from Figure 4 suggest that despite consistent CO2-dependent changes in mitochondrial gene expression (after 4 h), this does not appear to be associated with increased mitochondrial content as measured by mitochondrial DNA: nuclear DNA ratio or Nonyl-acridine orange assay. Mitochondrial oxidant activity was also not significantly different in hypercapnia using Amplex Red and MitoSox assays; however, we did observe a CO2-dependent increase in ATP5A protein expression in mitochondrial fractions and a significant increase in cellular reductase activity using the MTT assay.
Effect of hypercapnia on acylcarnitine expression and regulation in monocytes
We next took a LC-MS approach to examine the functional metabolic consequences of elevated levels of CO2 on monocytes. Given a reported role for CO2 in lipid metabolism and β-oxidation in skeletal muscle 21 and the important role of β-oxidation in cells of leukemic origin 22 we first focused our analysis on a targeted metabolomic screen of 40 acylcarnitine species following 4 h of exposure to 5% or 10% CO2 ± LPS. Intriguingly, of the detected acylcarnitine species in our cell lysates, several were statistically significantly increased under conditions of elevated CO2 (regardless of LPS treatment). At 4 h C3, C4, C5, C14, C16 and C16:1 were significantly different in hypercapnia in both the basal and LPS-stimulated states (Figure 5a & Supplementary figure 6). In general, the longer acylcarnitine species were increased at 10% CO2 compared to controls. Re-examination of our RNA-seq data revealed several transcripts associated with β-oxidation and carnitine shuttling to be significantly differentially expressed at 10% CO2 +LPS e.g. Acyl-CoA synthetase (ACSL) genes 1, 4 and 5 which are involved in the addition of acyl groups to long chain fatty acids at the outer mitochondrial membrane, and short chain acyl-COA dehydrogenase (ACADS) a key enzyme in the β-oxidation cascade. qPCR analysis of ACADS over a longer time course (up to 24 h) revealed it to be differentially expressed at 10% CO2 with and without LPS (Figure 5b, c), most notably at 24 h of exposure. Taken together, the data in Figure 5 & Supplementary figure 6 reveals a marked change in monocyte lipid metabolism in response to elevated levels of CO2 that is evidenced by significant differences in several acylcarnitine species and associated changes in specific genes related to lipid metabolism. These functional data are concordant with changes in the expression of the acylcarnitine transporter SLC16A9 in Figures 1b, c, 2b, c. Given that the data above linked CO2-sensitive genes associated with fatty acid metabolism (RNA-seq & qPCR) with functional changes in metabolites (liquid chromatography with tandem mass spectrometry (LC-MS/MS)), we re-examined our RNA-seq data to identify robustly CO2-sensitive metabolic genes that are rate limiting in the regulation of amino acids on our LC-MS/MS screen. PYCR1 is a mitochondrial enzyme that catalyses the NAD(P)H- dependent conversion of pyrroline-5 carboxylate to proline. PYCR1 mRNA was significantly increased in response to elevated CO2 in both the basal and LPS-stimulated state in our RNA-seq experiment (Figure 2e, Supplementary table 9). qPCR validation of PYCR1 expression was performed in separate samples where again there is evidence of enhanced PYCR1 expression in hypercapnia in both the basal and LPS-stimulated state (Figure 5d, e). Intriguingly, our LC-MS/MS data revealed a specific and corresponding CO2-dependent decrease in the cellular levels of the amino acid proline in both the basal and LPS-stimulated state (Figure 5f & Supplementary figure 6). Taken together these data reveal CO2-dependent modulation of lipid metabolism and amino acid metabolism in terms of actual metabolite concentration and the expression of key transcripts involved in the expression of those metabolites.
Figure 5. LC-MS/MS analysis of acylcarnitine species in monocytes exposed to hypercapnia.
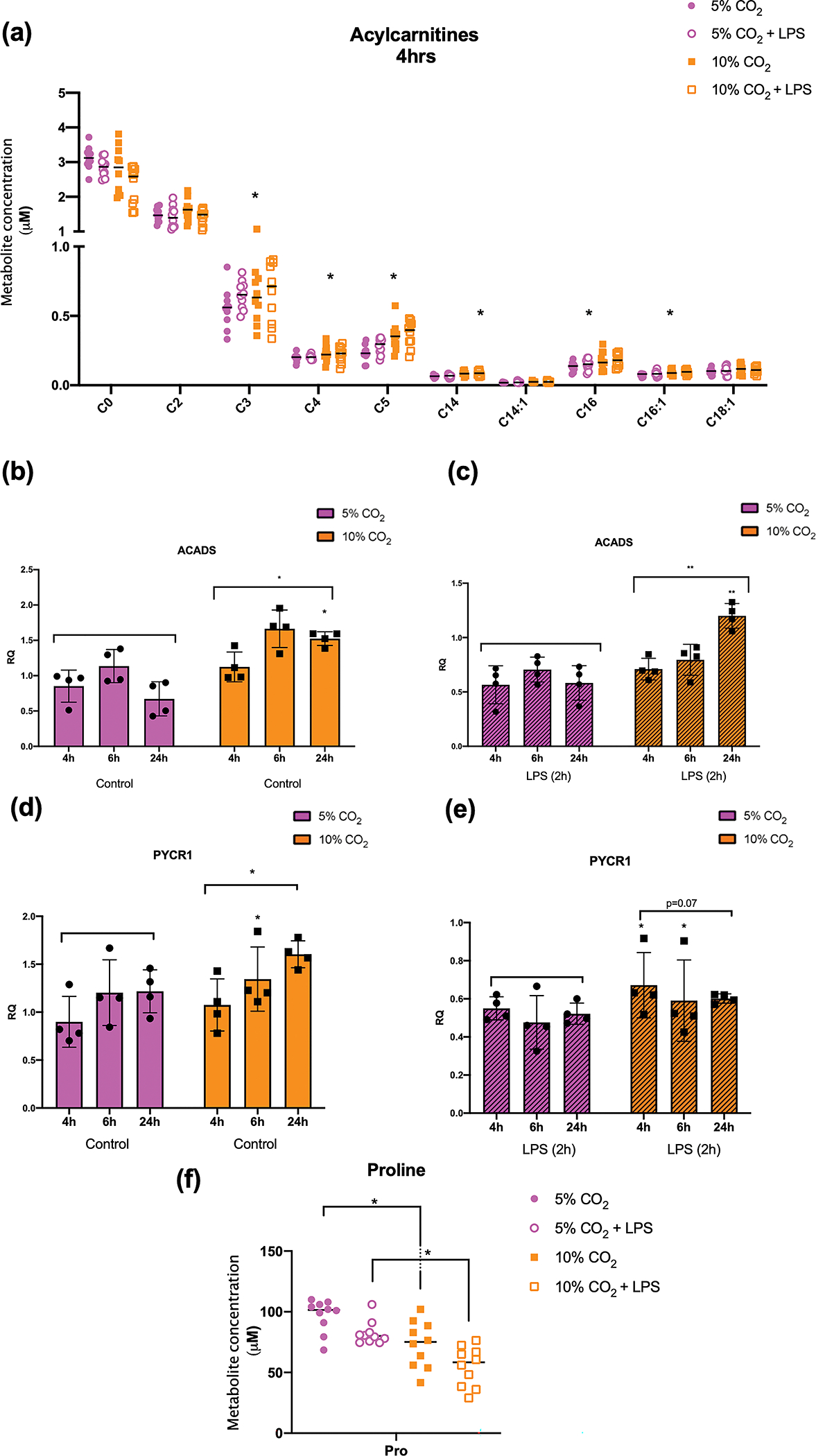
Detected acylcarnitine species from THP-1 cells exposed to 5% or 10% CO2 for 4 h (a). Data shown are median values and individual data points for n = 10 independent experiments. Statistical analysis was performed using one-way ANOVA followed by a Fisher’s LSD post-hoc test. Significance values * denotes a FDR of < 0.05. RT-qPCR of cDNA generated from THP-1 cells exposed to 5% or 10% CO2 for 4, 6 or 24 h in the basal state (b, d) or in the presence of LPS (2.5 μg mL−1) (c, e) using SYBR green primers targeted to ACADS and PYCR1. Data shown are mean ± SD for n = 4 independent experiments. Statistical analysis was performed using 2- way ANOVA followed by Sidak’s multiple comparisons test. Significance values * denotes a P-value of < 0.05, ** denotes a P-value < 0.01. Proline metabolite concentration from THP-1 cells exposed to 5% or 10% CO2 for 4 h. Data shown are median values and individual data points for n = 10 independent experiments. Statistical analysis was performed using one-way ANOVA followed by a Fisher’s LSD post-hoc test. Significance values * denotes a FDR of < 0.05.
Effect of hypercapnia on polarized primary murine macrophages
To further investigate the immunometabolic consequences of hypercapnia in a primary immune cell background we generated primary bone marrow derived macrophages (BMDMs) and differentiated them towards an M2 phenotype using IL4. Here we focus on the expression of inflammatory and metabolic genes in IL4 stimulated BMDMs under conditions of buffered hypercapnia. The polarising effect of IL4 stimulation on BMDMs is illustrated in Supplementary figure 7. Using targeted transcriptomic analysis, we identified 192 genes that were differentially expressed when IL4 polarised BMDMs were exposed to hypercapnia versus normocapnia for 24 h in pH-buffered media. Gene set analysis revealed pathways that were upregulated and down regulated in response to elevated CO2 in primary IL4 stimulated BMDMs (Figure 6a). Absolute mRNA levels for a selection of CO2-sensitive exemplar genes have been presented. These exemplar genes correspond to several upregulated pathways (red) “Lysosomal degradation’ (Hexosaminidase Subunit beta, HEXB), ‘Fatty Acid Synthesis’ (Fatty Acid Binding Protein 5, FABP5), ‘Fatty Acid Oxidation’ (Carnitine Palmitoyltransferase 1A, CPT1a), Figure 6b, c, d respectively, and selected downregulated pathways (blue) ‘KEAP1/NRF2’ (NAD(P) H Quinone Dehydrogenase 1, NQO1), ‘Glycolysis’ (Aldolase, Fructose-Bisphosphate A, ALDOA) and ‘Cytokine & Chemokine Signaling’ (Tumor Necrosis Factor, TNF), Figure 6e, f, g, respectively.
Figure 6. IL4 polarised BMDM response to hypercapnia.
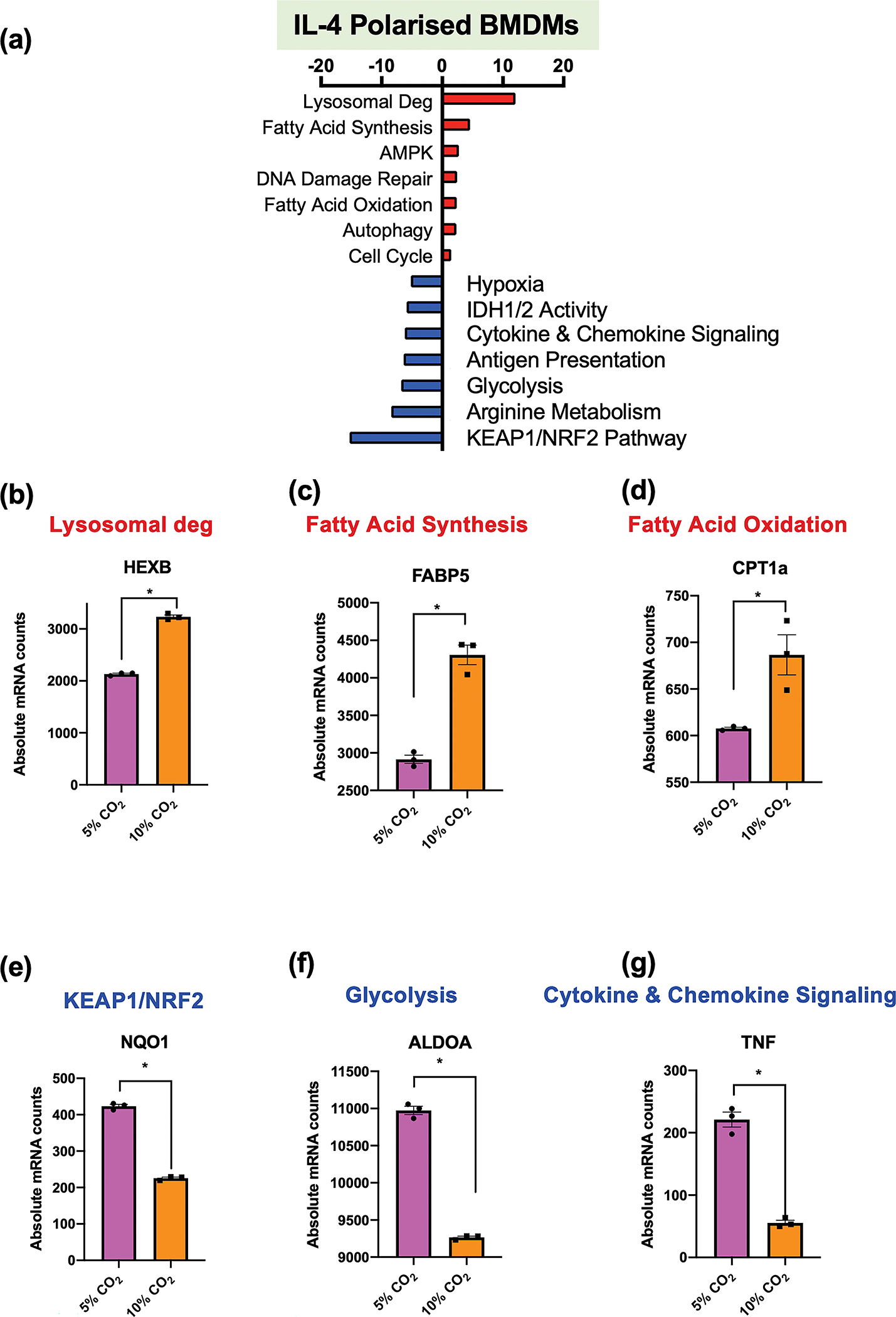
Bone marrow-derived macrophages (BMDMs) were exposed to 5% (pink) or 10% CO2 (orange) for 6h followed by IL4 (100 ng mL−1) stimulation for an additional 18h. Absolute mRNA levels and differential gene expression were assessed by a metabolic and inflammatory gene panel on the NanoString nCounter platform. Gene set enrichment analysis is presented (a). Absolute mRNA levels were measured for HEXB (b), FABP5 (c) CPT1a (d), NQO1 (e), ALDOA (f) and TNF (g). These are exemplar genes for pathways that are enhanced (red) or repressed (blue) in the NanoString gene set analysis. Data shown are mean ± SEM for n = 3 independent experiments. Significant differential expression from individual comparisons as determined by NanoString nCounter analysis, P-value ≤ 0.05 (*).
Discussion
This study has for the first time used next-generation RNA-sequencing to investigate the transcriptional response to CO2 in immune cells. Using pH buffered conditions and modest degrees of hypercapnia, we observe significant transcriptional changes in monocytes exposed to 10% CO2, resulting in comprehensive changes to mitochondrial associated genes and mitochondrial metabolism. CO2-dependent changes in immunometabolism observed in monocytes are subsequently supported and validated in primary BMDMs polarized with IL4.
Monocyte infiltration / activation and elevated levels of CO2 are key components in the pathophysiology of hypercapnic COPD. To directly interrogate the influence of CO2 levels on monocyte function under pH -buffered conditions, we initially exposed monocytes exposed to 5% CO2 or 10% CO2 for 4 h. Previous studies have used higher concentrations of CO2, up to 20% 23 and/or much longer duration of exposure, 3–6 days 24, 25. However, we chose to utilise a modest and more clinically relevant concentration of CO2 26 to determine a physiological role for CO2-dependent transcriptional regulation in a focused fashion. It is technically challenging to separate hypercapnia from pH under human physiologic conditions, thus the purpose of this study is to dissect the specific role of CO2 on monocyte gene expression and function. To our knowledge this is the first study of its kind to use next-generation RNA sequencing to examine the cellular response to elevated CO2 in pH-buffered conditions in immune cells.
PCA analysis (Figures 1a and 2a) shows clear separation of the 5% and 10% CO2 samples in relation to PC1. The volcano plot (Figure 1b) further reveals that 10% CO2 is a modest but robust stimulus in monocytes and that hypercapnia + LPS results in a much more pronounced transcriptional response (Figure 2b). These data are consistent with the idea that hypercapnia can serve as a microenvironmental modulator capable of modifying immune responses. Enriched GO terms in the basal state relate to muscle contraction (e.g. ATP2A1), mitochondrial function (e.g. MT-CO1), calcium signalling and transmembrane transport (Supplementary table 5). These data align well with current literature as these pathways and processes have all been previously identified as showing some sensitivity to CO2. For example, high CO2 has been shown to downregulate skeletal muscle protein anabolism 27 and cause mitochondrial dysfunction 24. In the presence of LPS, GO terms associated with the most differentially expressed genes (Log2FC > 1 or < −1) again revealed transcripts related to mitochondrial activity, angiogenesis and developmental processes (Supplementary table 6), which is also consistent with the current literature. Taken together there is evidence for a core CO2 sensitive cohort of genes that is regulated by CO2 in both the basal and stimulated states and respond to CO2 in a congruent and consistent manner (Figure 2f). This core cohort of CO2 sensitive targets is enriched in genes associated with mitochondrial metabolism, calcium signaling and muscle contraction.
While several inflammatory genes e.g. Toll- Like Receptor 4 (TLR4) were differentially expressed (Figure 2e and Supplementary figure 8) in hypercapnia, ‘immune signaling’ was not amongst the most prominent GO terms (Figure 1d & 2d) as has been reported in similar models 28. There was however a particularly strong signature for alterations in mitochondrial function, mainly in relation to oxidative phosphorylation. Mitochondrially encoded genes were among the top upregulated genes in both the basal and LPS-stimulated states (Figures 1c, 2c). Furthermore, GO terms related to mitochondrial function, specifically the electron transport chain, were among the top terms for both comparisons. From the literature chronic exposure to hypercapnia causes mitochondrial dysfunction in lung epithelium and fibroblasts 24. In rats, acute 20% CO2 inhaled for 10 min improved mitochondrial function and upregulated the expression of the mitochondrial biogenesis regulators peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) and mitochondrial transcription factor a (TFAM) 29. Interestingly in this study, TFAM was increased in hypercapnia in the presence of LPS at 4 h (Supplementary figure 3b, Supplementary table 4). To our knowledge this study is the first to demonstrate a distinct upregulation of mitochondrial encoded genes in hypercapnia. Unbiased RNA-sequencing revealed that almost all the mitochondrial mRNA, and several mitochondrial tRNA transcript levels were higher in 10% CO2 compared to 5% CO2, suggesting an increase in mitochondrial transcription in hypercapnia. Interestingly, in both the basal and LPS states, nuclear encoded mitochondrial tRNA synthetases (Supplementary figure 4a, b) and mitoribosome proteins (Supplementary figure 4c, d) were significantly upregulated. Given both sets of these proteins are involved in the translation of mitochondrial mRNA, these data provide further evidence that mitochondrial transcription and transcripts relating to the translation of mitochondrial proteins are increased in a CO2-dependent manner.
Mitochondria play a central bioenergetic role in cells. Regulation of mitochondria occurs not only at a transcriptional level with dynamic regulation of cellular mitochondrial content also critical to the maintenance of functional mitochondria 30. The data in Figures 1–3 and Supplementary figure 4 indicating enhanced mitochondrial gene expression suggests that mitophagy is unlikely under these conditions. With our observations of increased mitochondrial gene expression in hypercapnia, we investigated the effect of hypercapnia on the mitochondrial content of monocytes for up to 24 h. We observed no CO2-dependent difference between the mtDNA content of monocytes using a qPCR-based technique (Figure 4a). Using the fluorescent dye NAO to stain the mitochondrial membrane lipid cardiolipin, we observed a statistically significant decrease in NAO-dependent fluorescence following 24 h of exposure to 10% CO2 (Figure 4b). Altered cardiolipin levels in response to cellular stress have the potential to affect multiple pathways 31, thus reduced cardiolipin following exposure to 10% CO2 is further suggestive of a CO2-dependent shift in mitochondrial homeostasis. The ROS O2− and subsequently H2O2 are produced by electron leakage during OXPHOS, primarily from Complex I and Complex III. Our data suggested that mitochondrial ROS production may be altered in our model. However, the data (Figure 4c, d) demonstrate that hypercapnia does not significantly affect mitochondrial ROS production in monocytes. It is possible; however, that there are small differences in oxidant production below the threshold of sensitivity of our assays. Looking at mitochondrial protein expression, ATP5A was relatively abundant in our mitochondrial extracts in comparison to other proteins involved in oxidative phosphorylation and was enhanced in response to hypercapnia (Figure 4e). We next investigated cellular reductase activity in monocytes exposed to elevated CO2 using MTT yellow tetrazole dye. MTT conversion is proposed to occur intracellularly and likely reflects changes in several cytosolic sub-compartments including the mitochondria 32. Interestingly, we observed an increase in MTT conversion to formazan in monocytes exposed to elevated CO2 for 24 h (Figure 4f).
Taken together the data in Figure 4 do not support the concept of wholesale increases in mitochondrial mass under conditions of elevated CO2. Thus, we believe that our observed transcriptional increase in mitochondrial subunit expression represents an early adaptive response to the metabolic stress of elevated CO2, that subsequently becomes modified following more prolonged exposure. For this reason, we next used a LC-MS/MS targeted screen of acylcarnitine species and amino acids to probe specific changes in metabolite levels in hypercapnia.
Metabolomic analysis of acylcarnitines permits a quantitative evaluation of mitochondrial lipid metabolism under conditions of elevated CO2. Immunometabolism is central to immune cell phenotype and function, with M1 ‘proinflammatory’ and M2 ‘anti-inflammatory’ macrophages having distinct metabolic profiles. In general, M1 macrophages are considered to be more glycolytic while M2 macrophages use more oxidative phosphorylation and β-oxidation33. This is, however, an oversimplification of a complex topic 34, but the metabolic context of the cell is clearly intrinsically linked to its physiological role and inflammatory phenotype. Notably, we observed several statistically significantly increased acylcarnitine species in cells from hypercapnic cells compared to normocapnic controls (Figure 5a and Supplementary figure 6a). Acylcarnitines are intermediates in the transport of long-chain acyl (CoA) into the mitochondria for β-oxidation. They can also be exported and released into the plasma of organisms 35 by acylcarnitine efflux proteins e.g. SLC16A9 18. In the context of this cell-based experiment we are examining intracellular acylcarnitine species. Altered acylcarnitine levels are associated with several factors including inherited genetic defects, exercise, fasting, insulin resistance, obesity and cardiovascular disease 36. Skeletal muscle cells exposed to chronically high levels of CO2 demonstrated increased levels of specific acylcarnitine species (Ceco et al. 2020).21 Altered lipid metabolism is also associated with COPD and lung health 37.
The shuttling of fatty acids into the mitochondria involves several key steps38 and incomplete β-oxidation is associated with elevated serum acylcarnitine levels 39. A number of transporters are linked to acylcarnitine transport across the plasma membrane including organic cation/ carnitine transporter (OCTN) family members 40, and SLC16A9 18. Interestingly, in addition to observing a CO2-dependent shift in acylcarnitine species and a key transporter (SLC16A9) in our model, we also observed changes in gene expression in specific steps within the fatty acid metabolism pathway. E.g. ACSL1,4 & 5 were all significantly increased in THP-1s in hypercapnia in the presence of LPS and transcripts associated with CPT1a were enhanced in IL4 polarised BMDMs (Figure 6d). ACADS and was significantly different in response to CO2 in the presence of LPS (Supplementary table 4), and a significant increase in the transcript levels of this key β-oxidation enzyme was determined by qPCR over a longer time course (24 h) (Figure 5b, c). Furthermore, the acylcarnitine transporter SLC16A9 is amongst the most differentially enhanced transcripts in response to elevated CO2 in both the basal and LPS-stimulated state (Figures 1b, c, 2b, c) suggesting a highly coordinated CO2-dependent shift in acylcarnitine processing. Notably, SLC16A9 is described as a pH-independent transporter 18 which further supports our contention that our responses are elicited by CO2 per se in our buffered hypercapnia system.
We also observe CO2-dependent transcriptional changes and altered metabolite levels with respect to amino acid metabolism. PYCR1 gene expression was enhanced in both our RNA-seq and qPCR experiments and this was associated with a highly significant CO2-dependent decrease in the cellular levels of proline in both the basal and LPS stimulated states (Figure 5f and Supplementary figure 6b). Proline can be synthesised intracellularly using a number of substrates including arginine and glutamine. PYCR1 catalyses the final conversion of Pyrroline-5 carboxylate (P5C) to proline. Proline is an important structural amino acid linked with collagen deposition and also has roles as an anti-oxidant and in immune responses 41. Proline metabolism is also linked to the development of ‘trained’ macrophages which pre-dispose to allergic asthma42. The full consequence of reduced proline levels in monocytes exposed to hypercapnia remains to elucidated but may be relevant in wound healing and in determining immune cell phenotype. Our data relating to PYCR1 and proline, in addition to our data relating to ACADs and acylcarnitine clearly suggests a coordinated transcriptomic and metabolomic response to acute hypercapnia under pH buffered conditions.
There are certain limitations associated with using a cancer- derived THP-1 monocyte model in our study. To verify the main findings from our THP-1 monocyte experiments in a primary macrophage model we generated BMDMs and polarised them with IL4 to generate ‘M2-like’ macrophages. M2 macrophages or ‘alternative macrophages’ are generally characterized as producing low levels of inflammatory cytokines and elevated levels of β-oxidation. M2- related genes are enhanced in alveolar macrophages of COPD smokers compared to healthy smoker controls 43 and the ratio of M2/M1 phenotype macrophages is increased in a mouse COPD model 17. Here, the IL4-polarised BMDMs elicited a marked transcriptional response to elevated CO2 (24 h) in pH- buffered media (Figure 6). Interestingly key pathways e.g. fatty acid metabolism, that were identified in our unbiased THP-1 RNA-seq experiments were also enhanced in IL4 polarised BMDMs (Figure 6a, c, d). Genes associated with glycolysis (ALDOA) and cytokine signaling (TNF) were reduced (Figure 6f, g) supporting the notion of a CO2-dependent shift in immunometabolism in IL4-polarised BMDMs. These data from primary macrophages validate several key findings from our THP-1 experiments. Data in Supplementary figure 8 directly compares RNA-seq TPM data from THP-1 monocytes with absolute mRNA counts from IL4 polarised BMDMs for selected genes. TLR4 and SLC16A3 (monocarboxylate transporter 3) are comparably decreased in hypercapnia in both models, while NFKB1 is CO2 sensitive in both models but is different in pattern (which could be explained by different duration of CO2 exposure). In addition to TLR4 and SLC16A3, another 10 genes were CO2 sensitive (in the same direction) in both models (ALDOA, GLUL, STK11, TK2 (downregulated), ASNS, BCL2L1, FNIP2, ITGB1, CCND1, CBR4 (upregulated). Taken together, the IL4- polarised BMDM data is highly supportive and validates key CO2 sensitive pathways (e.g. lipid metabolism) and transcripts (e.g. TLR4) identified in our THP-1 experiments. In order to validate a CO2-sensitive transcript from our NanoString experiment, NFKB2 (which encodes the NF-κB family member p100); we next examined p100 protein expression in THP-1 cells exposed to hypercapnia under the same experimental conditions (buffered hypercapnia for 24 h). NFKB2 mRNA expression is significantly decreased in IL-4 primed BMDMs exposed to 10% CO2 for 24 h under buffered conditions and p100 protein expression is also significantly decreased under the same conditions in THP-1 cells (Supplementary figure 9). These data further validate our parallel experimental approach using THP-1 monocytes and IL-4 polarised BMDMs. Further validation of our RNA-seq data is provided in Supplementary figure 10 where RIPK1 is reduced in response to elevated CO2 on the mRNA and protein level in THP-1 monocytes. RIPK1 regulates cellular decisions between pro-survival/ cell death signaling in the NF-κB pathway and is emerging as a potential therapeutic target in multiple inflammatory diseases 44.
Cellular metabolic changes in immune cells are fluid to allow cellular metabolism to dynamically change in response to the emerging needs of the cell in response to a changing environment. These changes can represent alterations in cell fate decisions as well as effector function 45. A classic example of this is the differentiation of M1 macrophages to a more glycolytic phenotype in response to LPS. This primes the cells for pro-inflammatory effector function and ensures that this is possible through glycolysis-dependent macromolecule generation via the pentose phosphate pathway. Thus, immune cells are capable of dramatic reprogramming of cellular metabolism in response to environmental signals e.g. bacterial products, cytokines, hypoxia and here we provide evidence of metabolic shifts in response to elevated CO2 levels. The position within the immune cell’s life cycle where the environmental signal occurs is also of importance, as cell differentiation can be affected in uncommitted cells while effector function can be modified in mature differentiated cells. Given that monocyte infiltration, M2 macrophage polarisation and hypercapnia are co-incident in COPD, our data provide novel insights into the immunometabolic landscape of hypercapnic lung disease. Hypercapnia and acidosis have previously been reported to attenuate M1 macrophage differentiation and migration 46indicative of an attenuated inflammatory immune response. Here we build on these studies to demonstrate that CO2 per-se causes a marked immunometabolic shift in monocytes and macrophages that further attenuates pro-inflammatory signaling. A CO2-dependent suppression of transcripts associated with glycolysis in both monocytes and IL-4 polarised macrophages suggests a further deviation from pro-inflammatory M1-like ‘classic’ signaling. Suppression of TLR4 transcripts in both cell types is congruent with this. Enhancement of transcripts associated with fatty acid oxidation and lipid metabolism furthermore points away from pro-inflammatory type signaling and more towards that of an immunosuppressive phenotype, where fatty acid synthesis can fuel oxidative phosphorylation in M2 macrophages 47. Furthermore, in monocytes there is evidence of a potential CO2-dependent shift away from expression of chemotactic factors e.g. CX3CR1 and CXCR3), while at the same time promoting the expression of more secretory factors e.g. IL12B (Supplementary table 4). This microenvironmental shift in cellular signaling capacity could be of benefit in the context of a destructive inflammatory milieu, but may be less responsive to pathogenic challenges through reduced innate signaling (e.g. reduced TLR4) (Supplementary figure 8). These scenarios are highly relevant to lung pathologies including COPD. Furthermore, an enhanced understanding of how CO2 affects metabolism and immune signaling in monocytes and macrophages may reveal new opportunities for therapeutic hypercapnia, where the anti-inflammatory effects of CO2 can be of benefit 13.
Conclusions
Taken together this study has for the first time used next-generation RNA-sequencing to investigate the transcriptional response to CO2 under pH buffered conditions in immune cells. Hypercapnia alone elicits a modest but robust and significant transcriptional response that is enhanced in the presence of the pro-inflammatory stimulus LPS. The transcriptional response of monocytes to hypercapnia revealed a novel and marked change in gene expression in relation to mitochondria and mitochondrial associated genes, suggesting that the elevated level of CO2 in this model is a metabolic stressor. Despite widespread changes in mitochondrial gene expression in our RNA-seq experiments, our data indicates that significant changes in mitochondrial ROS production or mitochondrial mass are not associated with the CO2 response under these conditions. Mitochondrial function / activity; however, was significantly altered. Cellular reductase activity was enhanced under hypercapnic conditions, and we observed a marked change in acylcarnitine species as early as 4 h post CO2 exposure indicating a CO2-dependent change in lipid metabolism. Proline levels were also significantly reduced under these conditions. These changes in lipid metabolism and amino acid metabolism were also associated with altered transcription of specific genes associated with mitochondrial lipid processing e.g. ACADS, and proline biosynthesis e.g. PYCR1. Importantly, many of the key observations observed in THP-1 monocytes were also evident in IL4 polarised primary BMDMs e.g. increased expression of genes associated with fatty acid metabolism (Supplementary figure 11). These data help us to understand the immunomodulatory effects of hypercapnia on monocytes and macrophages and may provide insight into future therapeutics for patients experiencing hypercapnia.
Methods
Cell culture
THP-1 monocytes
The immortalised cell line THP-1 was chosen as a suitable substitute for primary monocytic cells. THP-1 cells originated from a 1-year-old male with acute monocytic leukaemia. They have been well described and demonstrate characteristic monocyte functions such as phagocytosis 48, inflammatory cytokine production and differentiation capacity. In addition, THP-1 cells respond to lipopolysaccharide stimulation (LPS) 49–51 and differentiated THP-1 macrophages have shown an altered immune response to elevated CO2 28, 46, 52. We have recently extensively characterized the shNT version of these cells described by Phelan et al. 2021 where monocytes have been stably transduced with a non-target short hairpin RNA. In this study both wild-type and shNT-THP1s were employed as some of these studies were carried out as part of a wider study20. We have previously published that shNT-THP-1s phenocopy the wild type THP-1s 53, 54. THP-1 cells were maintained in RPMI 1640 medium supplemented with 10% Foetal Bovine Serum and 1% Penicillin-Streptomycin. Cells were maintained at a density of 2 × 105-1 × 106 cells mL−1 and subcultured twice per week. For subculture, cells were counted and media containing the appropriate number of cells was transferred to a T75 tissue culture flask containing fresh media, to a total of 20–30 mL. All procedures and treatments prior to cell lysis were performed in a Class II biological safety cabinet or a CO2 chamber. shNT-THP1 monocytes were used in Figures 1–3, 4a, c, d, 5, as well as in Supplementary figures 1, 2, 4a–d, 6, 8a–c, 10a. Untransformed THP-1 monocytes were used in Figure 4b, e–f and Supplementary figures 4e, 5, 9a–b, 10b, c.
Primary murine macrophages
Primary bone marrow-derived macrophages (BMDMs) were isolated and identified as previously described 46, 55 from healthy wild type black 6 mice at about 5–20 weeks of age. Briefly, mice were euthanised by cervical dislocation by trained and licenced animal handlers in the UCD Biomedical Facility. We received the mice post-mortem. Both femurs and tibias were in toto excised and washed in sterile PBS (Sigma-Aldrich/Merck; #D8537). Epiphyses were removed and medullary cavity was flushed with cold sterile PBS using a 27G needle. The flow-through was collected in a 50-mL tube (Greiner Bio-One International; #210270). The cell suspension was centrifuged at 500g for 10 min, and the supernatant was discarded. After lysing of erythrocytes and repeated centrifugation cells were resuspended in RPMI-1640 (Merck) with 10% FCS, 1% Penicillin/Streptomycin, 2mM L-Glutamine and 10 ng mL−1 mM-CSF. Pooled cells of one mouse were seeded in one T-75 flask (Greiner Bio-One International; #660175-120G) and split after 48 h. After 7 days in culture, cells were harvested and identified by F4/80 immunostaining as described previously46.
Primary murine macrophage polarisation
After isolation, differentiation, and identification of BMBMs, murine macrophages were further polarized to analyse the effect of CO2 on macrophage polarization. To induce macrophage polarization, 5 × 105 cells per well were seeded in 12-well plates (Greiner Bio-One International #665180) and placed within gas chambers at different CO2 concentrations (5% versus 10% CO2). After 6 h of CO2 treatment primary murine macrophages were polarised into immuno-modulatory (M2)-like macrophages using interleukin4 (IL4; 100 ng mL−1; Sigma-Aldrich/Merck; #I1020-5UG), for additional 18 h. At the end of the experiments, cells were harvested for RNA isolation.
Hypercapnic exposures
All hypercapnic exposures were performed in humidified environmental chambers (Coy Laboratories) at 37°C, at 5% or 10% CO2. All experiments were performed in buffered, pre-equilibrated DMEM media 56 or phenol-free DMEM (MitoSox experiments). For most THP-1 experiments, cells were used at a density of 750,000 cells mL−1 of media. We have recently demonstrated that intracellular pH does not change significantly in PMA treated THP-1s cultured under these buffered hypercapnia conditions46. Representative media pH, pCO2 and pO2 measurements are included in Supplementary table 1.
RNA-seq
Total RNA was extracted from cultured cells using the E.Z.N.A. Total RNA Kit I. All lysates were homogenised by pipetting 10x up and down using a 21-gauge needle and a 1ml syringe prior to column extraction. RNA concentration and purity were determined using a NanoDrop 2000 spectrometer. RNA clean-up was performed (if required) using ethanol precipitation. Subsequent Qubit results determined a RIN score > 9.5 for all samples. cDNA library preparation was performed with polyA selection using Illumina HiSeq, 2×150 bp configuration, single index, per lane. ~350 M raw paired-end reads per lane. Library preparation and sequencing were performed by GeneWiz (Germany). Raw data quality was evaluated with FastQC. Sequence reads were trimmed using Trimmomatic v.0.36 and subsequently mapped to the Homo sapiens GRCh38 reference genome using the STAR aligner v.2.5.2b. Unique gene hit counts were calculated by using featureCounts from the Subread package v.1.5.2. Downstream differential expression analysis was performed using DESeq2 57. P-values and log2 fold changes were generated using the Wald test. Significant differentially expressed genes (DEGs) were called as genes with an adjusted P-value (P-adj) < 0.05 (Supplementary tables 3, 4, 7). Initial stringent GO analysis was performed using GeneSCF v1.1-p2 to generate GO graphs where a Fisher Exact test was used to determine P-values (Supplementary tables 5, 6, 8 and Supplementary figure 2d). Principal component analysis and read count distribution analysis was performed by GeneWiz. Volcano plots of differentially expressed genes were generated using GraphPad Prism, with log2FC on the x-axis and adjusted P-value on the y-axis. Significant differentially expressed genes (DEGs) were determined as any genes from the DESeq2 workflow, which had an adjusted P-value (P-adj) < 0.05. Nested comparisons for multiple groups were completed by cross referencing lists of DEGs from pairwise comparisons to generate secondary lists of either common or specific DEGs. Gene ontology (GO) analysis was performed on each list of DEGs. This analysis was completed by the PANTHER Classification system (pantherdb.org) using statistical overrepresentation tests. The background reference list for these comparisons was generated by compilation of all genes expressed to any degree in any of the experimental conditions (raw TPM values). Raw data from the RNA-seq experiment is displayed as mean TPM (± SEM) to best visualise expression changes between several groups. Statistical comparisons between individual groups used DESeq2 analysis of normalised counts. The bulk RNA-seq data presented in the study are deposited in the Gene Expression Omnibus (GEO) repository, accession number GSE206333.
Measurement of mtDNA content by qPCR
Cells were collected in 15-mL centrifuge tubes using a pipette and cell scraper as required and centrifuged for 5 min at 300g. DNA extraction was performed using the QIAamp DNA mini kit (Qiagen) according to the manufacturer’s instructions. DNA concentrations were measured using a nanodrop spectrophotometer. A second elution was performed using 100 μL Buffer AE to maximise DNA yield. DNA was stored at −20°C.
DNA was normalised to 20 ng μL−1 and measured again on the nanodrop. A serial dilution was performed to dilute the DNA to 0.5 ng mL−1. Sybr Green qPCR was performed on the extracted DNA as described above using specific primers for the mitochondrially encoded (MT-ND4) and the nuclear encoded beta-2 microglobulin (B2M) and cycle threshold (CT) values determined. Primer details are included in Supplementary table 2.
ΔCT was calculated as CTnucDNA – CTmtDNA or CTB2M -CTMT-ND4. mtDNA copy number per cell was calculated as 2 × 2ΔCT. This method assumes that there are two copies of the nuclear gene B2M per (diploid) cell and one copy of the mitochondrial gene MT-ND4 per molecule of mtDNA. The calculation used gives the number of copies of MT-ND4 relative to B2M and can therefore be used to infer the amount of mtDNA per cell. Similar methods have been used to assess mtDNA copy number in multiple species, including the use of B2M to represent nucDNA in humans58,59. The use of this qPCR method to assess mitochondrial biogenesis in THP-1 cells has also been demonstrated previously49.
NAO Assay
The (nonyl acridine orange) NAO assay uses the chemical dye acridine orange 10-nonyl bromide (Invitrogen) to measure mitochondrial mass. NAO binds to cardiolipin, a phospholipid specifically present on the mitochondrial membrane 60. Briefly, approximately 100,000 THP-1 monocyte cells/ well in 200 μL of media were seeded on flat-bottomed 96-well plates in either 5% or 10% CO2 buffered media. Treatments were carried out in sextuplicate. Cells were incubated for 24 h. Subsequently, the suspension of THP-1 cells was centrifuged at 1500g for 5 min (brake 1) and media was carefully aspirated. 5 μM of NAO dissolved in PBS and 100 μL was added to each well in a 96-well plate. Cells were incubated at 37°C and 5% CO2 for 30 min in the dark. The plate was centrifuged again, and the dye removed. Cells were resuspended in 100 μL PBS and fluorescence measured at (Ex/Em 485/538 nm) on a CLARIOstar plate reader (BMG Labtech). Background was removed by subtracting negative control (cells without NAO dye) value from all samples.
Western blot analysis
Whole cell protein lysates were prepared using either whole cell lysis buffer (150 mM NaCl, 25 mM Tris pH8, 1 mM EDTA, 1% NP-40) supplemented with protease inhibitor cocktail (Sigma-Aldrich, P2714) or precipitated from RNA extraction flow-through and suspended in 1M HEPES, 1% SDS. Mitochondrial lysates were prepared using a mitochondrial isolation kit (Thermo Scientific) according to the manufacturer’s instructions, with isolated mitochondria lysed in 2% CHAPs in TBS. Lysates were quantified using the DC Protein Assay kit before SDS-PAGE on the Biorad mini-protean system using TGX pre-cast gels. Wet transfer was performed onto nitrocellulose membranes and reversibly stained with Revert 700 total protein stain (Li-COR). Membranes were imaged at 700 nm, washed in Revert 700 wash solution (Li-COR) and destained in Revert 700 destaining solution (Li-Cor) prior to immunoblotting with the MitoProfile™ Total OXPHOS Human WB Antibody Cocktail (ab110411, Abcam) (1:650–1:1000 dilution), p100/p52 antibody (CST4882) (1:1000 dilution) or RIP antibody (CST3493) 1:1000 dilution. Fluorescent secondary goat anti-mouse or anti-rabbit antibody incubations were performed (DyLight ™ 800, Invitrogen) and imaged at 800 nm (1:2000 dilution) Membranes were imaged using a LiCor Odyssey Clx imager and analysed using EmperiaStudio image analysis software.
MitoSox Superoxide assay
THP-1 cells were suspended in 1 μM MitoSOX™ working solution or DMSO vehicle (for unstained cells), at a concentration of 1 million cells mL−1. Cells were incubated in a 37°C water bath for 20 minutes, protected from light, with gentle agitation every 5 min. Cells were centrifuged at 1200rpm and pellets were washed with warm PBS 3 times (with centrifugation between washes), before dividing cells evenly between tubes for re-suspension (~750k cells mL−1) in pre-equilibrated low glucose phenol free (D2902 Sigma Aldrich) 5% CO2 media or 10% CO2 supplemented with 10% Foetal Bovine Serum and 1% Penicillin-Streptomycin in the environmental chambers. A volume of 1 mL of the cell suspension was added per well of a 24-well tissue culture plate, protected from light, with wells of stained and unstained cells immediately transferred to 1.5-mL microcentrifuge tubes to confirm successful staining. Plates were incubated for 4 h ± rotenone (1 μM) for 2h, protected from the light. Following incubation, the cell suspension was analysed in a Cytoflex S flow cytometer (Beckman Coulter). The blue laser (488 nm) was used to excite the fluorophore and emission was measured from 675 to 715 nm. Cells were selected for analysis based on the forward and side scatter and subsequently single cells were selected based on forward scatter area and forward scatter height. Cell loading with MitoSOX was confirmed by an increase in the median fluorescence detected in the 675–715 nm range. Treatment or CO2-dependent changes in fluorescence were calculated by relative fluorescence with respect to the untreated 5% CO2 control at each time point.
Amplex Red H2O2 assay
Following CO2 exposure cells were centrifuged at 1200rpm for 5 min and the media was removed. The media is used to measure extracellular peroxide. Cells were lysed in 1X reaction buffer, 0.1% Triton, briefly pulsing samples on a vortex every 3 min for 15 min on ice. Homogenates were centrifuged for 15 min at max speed (21,000 × G) at 4°C. Supernatants were transferred to a fresh tube and analysed immediately for peroxide and peroxidase. For intracellular H2O2 the lysate was diluted 1 in 5 (25 μL lysate + 100 μL 1X reaction buffer). For peroxidase the lysate was diluted 1 in 2 (60 μL lysate + 60 μL 1X reaction buffer). Protein content was determined using the remaining lysate for normalisation. 50 μL of appropriately diluted standards and samples were loaded in duplicate to a white flat-bottom 96-well plate. Reaction mixtures were prepared as follows: (i) For H2O2: 50 μL of 10 mM Amplex® Red reagent stock solution, 100 μL of 10 U mL−1 HRP stock (31490 Thermofisher) diluted in 1X reaction buffer solution (0.05M sodium phosphate ~pH 7.4) and 4.85 mL of 1X Reaction Buffer (ii) for Peroxidase: 50 μL of 10 mM Amplex® Red reagent stock solution, 500 μL of 20 mM H2O2 stock solution and 4.45 mL of 1X Reaction Buffer. Reaction was initiated by adding 50 μL of reaction mix (peroxide or peroxidase) per well. The plate was incubated at room temperature for 30 min, protected from light. Fluorescence was measured in a CLARIOstar plate reader (BMG Labtech) using an excitation wavelength of 530 nm and emission wavelength of 590 nm. Background was removed by subtracting negative control (cells without Amplex Red dye) value from all samples. Concentration of H2O2/peroxidase was calculated against the standard curve. Calculated concentrations were normalised to total protein content for analysis. All working stocks of reagents were freshly prepared just before the assay was performed.
MTT Assay
The MTT assay exploits the chemical reduction of a yellow tetrazole dye (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) to insoluble formazan by intracellular reductases and NADH in particular. The conversion of MTT to formazan mainly occurs intracellularly at mitochondria as well as other intracellular organelles 32. The MTT assay is occasionally used as an assay of cellular viability as intracellular reductase activity can be proportional to cell viability. However, the assay can also determine augmented or reduced reductase activity linked to enhanced enzymatic activity and/or mitochondrial mass.
Briefly, approximately 100,000 THP-1 monocyte cells/ well in 200 μl of media were seeded on flat-bottomed 96-well plates in either 5% or 10% CO2 buffered DMEM media. Treatments were carried out in sextuplicate. Cells were incubated for 22h ± addition of MTT stock solution (5 mg mL−1) (Sigma Aldrich) at a ratio of media:dye of 3:1 for a further 2h in the dark. Subsequently, the suspension of THP-1 cells was centrifuged (Eppendorf 5810R) at 1500 G for 5 min (brake 1) and the media was carefully aspirated. Following this, 100 μL of DMSO was added to each well to dissolve the produced formazan and incubated at 37°C and 5% CO2 for 30 min. The absorbance was measured at a wavelength of 570 nm Ref 690 nm on a CLARIOstar plate reader (BMG Labtech). Background was removed by subtracting negative control (cells without MTT dye) value from all samples.
LC MS/MS assay
THP-1 monocytes (~750k cells mL−1) were incubated at 37°C with either 5% or 10% CO2 buffered DMEM (high glucose, Sigma D1152 with 10% Foetal Bovine Serum and 1% Penicillin-Streptomycin) media for 4 h, ± LPS (2.5μg mL−1) for the final 2 h. The cell suspension was transferred to a 15-mL tube and any attached cells were collected from the plate with a cell scraper and cold PBS. Cells were pelleted at 1000g for 5 min and washed twice with ice-cold PBS. The final cell pellet was snap-frozen in liquid nitrogen before extraction. Metabolite extractions were performed to isolate a broad range of metabolites from cell pellets (10 technical replicates per condition). Briefly, cells were extracted with ethanol/phosphate buffer (85%v/v). The metabolites were measured using LC-MS/MS analysis and flow injection analysis–tandem mass spectrometry (FIA-MS/MS) analysis based on the p180 Biocrates assay. The data was acquired on a SCIEX QTRAP 6500plus mass spectrometer coupled to SCIEX ExionLC™ Series UHPLC capability as previously described 61. Liquid chromatography was performed using a custom UHPLC column with acetonitrile and water with 0.2% formic acid as mobile phase. The targeted LC-MS platform captures over 120 metabolites giving a comprehensive coverage of multiple metabolites. For the purposes of this study we are focusing on the acylcarnitine species and amino acids. Analysis was performed using a one-way ANOVA followed by a Fisher’s LSD post-hoc test. A false discover rate (FDR) of < 0.05 was deemed significant using the total dataset.
Quantitative Reverse Transcription Polymerase Chain Reaction (RT-qPCR)
RNA was extracted using the EZNA total RNA Kit I (Omega Bio-tek) according to the manufacturer’s instructions. DNase I digestions were performed on all RNA samples intended for use in qPCR, Samples were incubated for 15 min at 20°C in a thermocycler before addition of 1 μL of 25 mM EDTA solution to inactivate the DNase I (ThermoFisher Scientific), and samples were heated to 65°C for 10 min. Complementary DNA (cDNA) was synthesised using Moloney Murine Leukaemia Virus
(M-MLV) Reverse Transcriptase (Promega). Master mixes were prepared using dilute RNA, M-MLV, reaction buffer, dNTPs random primers (ThermoFisher Scientific) and nuclease free water. Control samples lacking the reverse transcriptase (NRT) were used to detect any genomic DNA contamination.
The samples were incubated for 60 min at 37°C for the cDNA synthesis reaction. cDNA samples were used immediately or stored at −20°C.
For qPCR, cDNA samples were diluted 1 in 4 in nuclease free water. Master mixes for SYBR green reactions (Applied Biosystems) were prepared using master mix solutions, forward and reverse primers and nuclease free water for 10 μl reactions in a 384-well plate. The plate was centrifuged briefly to collect samples and remove bubbles. qPCR was performed in an Applied Biosystems RT-PCR machine with the appropriate Applied Biosystems Quant Studio 7 software. For SYBR Green primers, melt curves were generated for each sample for assessment of primer performance. Primer details are included in Supplementary table 2.
Targeted Transcriptomic analysis in primary macrophages
Targeted transcriptomic analysis was performed on RNA derived from primary BMDMs. Total RNA isolation was achieved using trizol (TRI Reagent.; Sigma-Aldrich/Merck). RNA samples were subsequently analysed on the NanoString nCounter. The Metabolic Pathways Panel (XT-CSO-MMP1-12; NanoStringTechnologies, Seattle, USA) was utilised for analysis and encompassed 768 inflammatory and metabolic genes and 20 internal reference genes for data normalisation. Normalisation of absolute mRNA counts was accomplished by using the nSolver software (advanced analysis module version 2.0.134). The log2 transformed output data were analysed using R (version 3.3.2). Genes with normalised expression values below 20 were removed. The remaining genes were utilised for global gene set analysis, pathway score analysis, and individual gene expression analysis.
Real Time Cell Metabolic Analysis
THP-1 cells were exposed to 5% or 10% CO2 for 24 h in buffered DMEM as per previous experiments. 1h prior to termination of experiment cells were counted, removed from the chambers and washed with phenol-free, bicarbonate free serum free seahorse XF DMEM supplemented with glutamine (2mM), glucose (10 mM) and pyruvate (1 mM) (Agilent). Cells were re-suspended in seahorse DMEM and seeded onto Cell-Tak (Corning) adhesive coated seahorse 96-well plates at ~150,000 cells per well, with 8 technical replicates per treatment. Cells were centrifuged on the 96-well plate (500g) for 5 min to promote adhesion. The plate was then incubated for 90 min at 37° C in a non- CO2 incubator prior to running the assay. A seahorse XF Cell Mito Stress Test cartridge that had previously been prepared with water and subsequently assay calibrant was loaded with oligomycin (15 μM), FCCP (10 μM), antimycin A/ rotenone (5 μM) and 2-deoxy d-glucose (500 mM) (Sigma Aldrich) in ports A-D respectively. Assay was run on a Seahorse XF 96 well Analyser (Agilent). Basal oxygen consumption was calculated as: OCR pre-injection (3rd measurement) minus OCR post-rotenone injection. Maximal Respiration was calculated as follows: Maximal OCR post FCCP injection minus OCR post-rotenone/antimycin A injection (3rd measurement). Basal ECAR was calculated as ECAR pre-injection (3rd measurement). Glycolysis was calculated as: ECAR pre-injection (3rd injection) minus ECAR post-2-deoxy-glucose (3rd measurement).
Statistical analysis
Statistical analysis was performed for the RNA-seq and targeted transcriptomic analysis as described above. ANOVA or t-tests were applied as indicated in the figure captions and figures prepared using GraphPad Prism (version8).
Supplementary Material
Acknowledgments
We acknowledge the support of the Conway Institute Transcriptomics Core Facility led by Dr Catherine Moss, the Conway Institute Flow Cytometry Core Facility led by Dr Alfonso Blanco and the Conway Institute Metabolomics Core Facility led by Professor Lorraine Brennan, the UCD Veterinary Sciences Centre Transcriptomics Laboratory supported by Professor Stephen Gordon and Dr John Browne.
Funding
This work was supported by a Science Foundation Ireland (SFI) Career Development award to Eoin Cummins (SFI 15/CDA/3490) which also supported David Phelan and Catarina Mota. Masahiko Shigemura and Jacob Sznajder are supported by the NIH grant: HL-147070. Joanne Masterson and Louise Crowe are supported by SFI award (SFI/FRL/4863). This research was supported in part by The Comprehensive Molecular Analytical Platform (CMAP) under The SFI Research Infrastructure Programme, reference 18/RI/5702. This research was supported in part due to the SFI Research Infrastructure Programme 15/RI/3215.
Footnotes
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Studies involving animal subjects
Ethical review and approval was not required for the animal study because animal tissue was acquired post-mortem from animals that were killed in the UCD Biomedical Facility in accordance with the University Animal Research Ethics Committee’s approval. The policy to make use of post-mortem tissue is in alignment with Clause 27 of Directive 2010/63/EU which states ‘To promote the principle of reduction, Member States should, where appropriate, facilitate the establishment of programmes for sharing the organs and tissue of animals that are killed.’.
Data availability statement
The bulk RNA-seq data presented in the study are deposited in the Gene Expression Omnibus (GEO) repository, accession number GSE206333.
References
- 1.Lehmer OR, Catling DC, Buick R, Brownlee DE, Newport S. Atmospheric CO2 levels from 2.7 billion years ago inferred from micrometeorite oxidation. Sci Adv 2020; 6: eaay4644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nattie E, Li A. Central chemoreceptors: locations and functions. Compr Physiol 2012; 2: 221–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cummins EP, Strowitzki MJ, Taylor CT. Mechanisms and Consequences of Oxygen and Carbon Dioxide Sensing in Mammals. Physiol Rev 2020; 100: 463–488. [DOI] [PubMed] [Google Scholar]
- 4.Nin N, Muriel A, Penuelas O, et al. Severe hypercapnia and outcome of mechanically ventilated patients with moderate or severe acute respiratory distress syndrome. Intensive Care Med 2017; 43: 200–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Phelan DE, Mota C, Lai C, Kierans SJ, Cummins EP. Carbon dioxide-dependent signal transduction in mammalian systems. Interface Focus 2021; 11: 20200033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kikuchi R, Iwai Y, Tsuji T, et al. Hypercapnic tumor microenvironment confers chemoresistance to lung cancer cells by reprogramming mitochondrial metabolism in vitro. Free Radic Biol Med 2019; 134: 200–214. [DOI] [PubMed] [Google Scholar]
- 7.Ahmadi Z, Bornefalk-Hermansson A, Franklin KA, Midgren B, Ekström MP. Hypo- and hypercapnia predict mortality in oxygen-dependent chronic obstructive pulmonary disease: a population-based prospective study. Respiratory research 2014; 15: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Laserna E, Sibila O, Aguilar PR, et al. Hypocapnia and hypercapnia are predictors for ICU admission and mortality in hospitalized patients with community-acquired pneumonia. Chest 2012; 142: 1193–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O’Toole D, Hassett P, Contreras M, et al. Hypercapnic acidosis attenuates pulmonary epithelial wound repair by an NF-κB dependent mechanism. Thorax 2009; 64: 976–982. [DOI] [PubMed] [Google Scholar]
- 10.Bharat A, Angulo M, Sun H, et al. High CO2 Levels Impair Lung Wound Healing. Am J Respir Cell Mol Biol 2020; 63: 244–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jaitovich A, Angulo M, Lecuona E, et al. High CO2 levels cause skeletal muscle atrophy via AMP-activated kinase (AMPK), FoxO3a protein, and muscle-specific Ring finger protein 1 (MuRF1). J Biol Chem 2015; 290: 9183–9194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shigemura M, Lecuona E, Angulo M, et al. Hypercapnia increases airway smooth muscle contractility via caspase-7-mediated miR-133a-RhoA signaling. Sci Transl Med 2018; 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao W, Liu DD, Li D, Cui GX. Effect of Therapeutic Hypercapnia on Inflammatory Responses to One-lung Ventilation in Lobectomy Patients. Anesthesiology 2015; 122: 1235–1252. [DOI] [PubMed] [Google Scholar]
- 14.O’Croinin D, Ni Chonghaile M, Higgins B, Laffey JG. Bench-to-bedside review: Permissive hypercapnia. Crit Care 2005; 9: 51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Curley G, Laffey JG, Kavanagh BP. Bench-to-bedside review: carbon dioxide. Crit Care 2010; 14: 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Corrigan CJ, Kay AB. The roles of inflammatory cells in the pathogenesis of asthma and of chronic obstructive pulmonary disease. Am Rev Respir Dis 1991; 143: 1165–1168; discussion 1175–1166. [DOI] [PubMed] [Google Scholar]
- 17.He S, Xie L, Lu J, Sun S. Characteristics and potential role of M2 macrophages in COPD. Int J Chron Obstruct Pulmon Dis 2017; 12: 3029–3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suhre K, Shin SY, Petersen AK, et al. Human metabolic individuality in biomedical and pharmaceutical research. Nature 2011; 477: 54–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stieglitz LM, Barnighausen T, Leyna GH, et al. Patterns of comorbidity and multimorbidity among middle-aged and elderly women in peri-urban Tanzania. J Multimorb Comorb 2022; 12: 26335565221076254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Phelan DE, Shigemura M, Aldhafiri S, et al. Transcriptional Profiling of Monocytes Deficient in Nuclear Orphan Receptors NR4A2 and NR4A3 Reveals Distinct Signalling Roles Related to Antigen Presentation and Viral Response. Front Immunol 2021; 12: 676644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ceco E, Celli D, Weinberg S, et al. Elevated CO2 Levels Delay Skeletal Muscle Repair by Increasing Fatty Acid Oxidation. Front Physiol 2020; 11: 630910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tcheng M, Roma A, Ahmed N, et al. Very long chain fatty acid metabolism is required in acute myeloid leukemia. Blood 2021; 137: 3518–3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Casalino-Matsuda SM, Wang N, Ruhoff PT, et al. Hypercapnia Alters Expression of Immune Response, Nucleosome Assembly and Lipid Metabolism Genes in Differentiated Human Bronchial Epithelial Cells. Sci Rep 2018; 8: 13508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vohwinkel CU, Lecuona E, Sun H, et al. Elevated CO2 levels cause mitochondrial dysfunction and impair cell proliferation. J Biol Chem 2011; 286: 37067–37076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gates KL, Howell HA, Nair A, et al. Hypercapnia Impairs Lung Neutrophil Function and Increases Mortality in Murine Pseudomonas Pneumonia. Am J Respir Cell Mol Biol 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Quintana JM, Esteban C, Unzurrunzaga A, et al. Predictive score for mortality in patients with COPD exacerbations attending hospital emergency departments. BMC Med 2014; 12: 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Korponay TC, Balnis J, Vincent CE, et al. High CO2 Downregulates Skeletal Muscle Protein Anabolism via AMP-activated Protein Kinase α2-mediated Depressed Ribosomal Biogenesis. Am J Respir Cell Mol Biol 2020; 62: 74–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Casalino-Matsuda SM, Berdnikovs S, Wang N, et al. Hypercapnia selectively modulates LPS-induced changes in innate immune and DNA replication-related gene transcription in the macrophage. Interface Focus 2021; 11: 20200039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chi L, Wang N, Yang W, et al. Protection of Myocardial Ischemia-Reperfusion by Therapeutic Hypercapnia: a Mechanism Involving Improvements in Mitochondrial Biogenesis and Function. J Cardiovasc Transl Res 2019; 12: 467–477. [DOI] [PubMed] [Google Scholar]
- 30.Wang K, Klionsky DJ. Mitochondria removal by autophagy. Autophagy 2011; 7: 297–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dudek J Role of Cardiolipin in Mitochondrial Signaling Pathways. Front Cell Dev Biol 2017; 5: 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berridge MV, Herst PM, Tan AS. Tetrazolium dyes as tools in cell biology: new insights into their cellular reduction. Biotechnol Annu Rev 2005; 11: 127–152. [DOI] [PubMed] [Google Scholar]
- 33.Viola A, Munari F, Sanchez-Rodriguez R, Scolaro T, Castegna A. The Metabolic Signature of Macrophage Responses. Front Immunol 2019; 10: 1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abuawad A, Mbadugha C, Ghaemmaghami AM, Kim DH. Metabolic characterisation of THP-1 macrophage polarisation using LC-MS-based metabolite profiling. Metabolomics 2020; 16: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ribel-Madsen A, Ribel-Madsen R, Brons C, Newgard CB, Vaag AA, Hellgren LI. Plasma acylcarnitine profiling indicates increased fatty acid oxidation relative to tricarboxylic acid cycle capacity in young, healthy low birth weight men. Physiol Rep 2016; 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu G, Hansen JS, Zhao XJ, et al. Liver and Muscle Contribute Differently to the Plasma Acylcarnitine Pool During Fasting and Exercise in Humans. J Clin Endocrinol Metab 2016; 101: 5044–5052. [DOI] [PubMed] [Google Scholar]
- 37.Chen H, Li Z, Dong L, Wu Y, Shen H, Chen Z. Lipid metabolism in chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis 2019; 14: 1009–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Longo N, Frigeni M, Pasquali M. Carnitine transport and fatty acid oxidation. Biochim Biophys Acta 2016; 1863: 2422–2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mai M, Tonjes A, Kovacs P, Stumvoll M, Fiedler GM, Leichtle AB. Serum levels of acylcarnitines are altered in prediabetic conditions. PLoS One 2013; 8: e82459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garrett Q, Xu S, Simmons PA, Vehige J, Flanagan JL, Willcox MD. Expression and localization of carnitine/organic cation transporter OCTN1 and OCTN2 in ocular epithelium. Invest Ophthalmol Vis Sci 2008; 49: 4844–4849. [DOI] [PubMed] [Google Scholar]
- 41.Phang JM, Donald SP, Pandhare J, Liu Y. The metabolism of proline, a stress substrate, modulates carcinogenic pathways. Amino Acids 2008; 35: 681–690. [DOI] [PubMed] [Google Scholar]
- 42.Li H, Ma L, Li W, et al. Proline metabolism reprogramming of trained macrophages induced by early respiratory infection combined with allergen sensitization contributes to development of allergic asthma in childhood of mice. Front Immunol 2022; 13: 977235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shaykhiev R, Krause A, Salit J, et al. Smoking-dependent reprogramming of alveolar macrophage polarization: implication for pathogenesis of chronic obstructive pulmonary disease. J Immunol 2009; 183: 2867–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mifflin L, Ofengeim D, Yuan J. Receptor-interacting protein kinase 1 (RIPK1) as a therapeutic target. Nat Rev Drug Discov 2020; 19: 553–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ganeshan K, Chawla A. Metabolic regulation of immune responses. Annu Rev Immunol 2014; 32: 609–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Strowitzki MJ, Nelson R, Garcia MP, et al. Carbon Dioxide Sensing by Immune Cells Occurs through Carbonic Anhydrase 2-Dependent Changes in Intracellular pH. J Immunol 2022; 208: 2363–2375. [DOI] [PubMed] [Google Scholar]
- 47.Liu Y, Xu R, Gu H, et al. Metabolic reprogramming in macrophage responses. Biomark Res 2021; 9: s40364–020–00251-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ackerman ME, Moldt B, Wyatt RT, et al. A robust, high-throughput assay to determine the phagocytic activity of clinical antibody samples. J Immunol Methods 2011; 366: 8–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Widdrington JD, Gomez-Duran A, Pyle A, et al. Exposure of Monocytic Cells to Lipopolysaccharide Induces Coordinated Endotoxin Tolerance, Mitochondrial Biogenesis, Mitophagy, and Antioxidant Defenses. Front Immunol 2018; 9: 2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ubanako P, Xelwa N, Ntwasa M. LPS induces inflammatory chemokines via TLR-4 signalling and enhances the Warburg Effect in THP-1 cells. PLoS One 2019; 14: e0222614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sharif O, Bolshakov VN, Raines S, Newham P, Perkins ND. Transcriptional profiling of the LPS induced NF-κB response in macrophages. BMC Immunol 2007; 8: 1471–2172–8-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang N, Gates KL, Trejo H, et al. Elevated CO2 selectively inhibits interleukin-6 and tumor necrosis factor expression and decreases phagocytosis in the macrophage. FASEB J 2010; 24: 2178–2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McEvoy C, de Gaetano M, Giffney HE, et al. NR4A Receptors Differentially Regulate NF-κB Signaling in Myeloid Cells. Front Immunol 2017; 8: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Crean D, Cummins EP, Bahar B, Mohan H, McMorrow JP, Murphy EP. Adenosine Modulates NR4A Orphan Nuclear Receptors To Attenuate Hyperinflammatory Responses in Monocytic Cells. J Immunol 2015; 195: 1436–1448. [DOI] [PubMed] [Google Scholar]
- 55.Strowitzki MJ, Ritter AS, Radhakrishnan P, et al. Pharmacological HIF-inhibition attenuates postoperative adhesion formation. Sci Rep 2017; 7: 13151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Keogh CE, Scholz CC, Rodriguez J, Selfridge AC, von Kriegsheim A, Cummins EP. Carbon dioxide-dependent regulation of NF-κB family members RelB and p100 gives molecular insight into CO2-dependent immune regulation. J Biol Chem 2017; 292: 11561–11571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014; 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rooney JP, Ryde IT, Sanders LH, et al. PCR based determination of mitochondrial DNA copy number in multiple species. Methods Mol Biol 2015; 1241: 23–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Furda AM, Bess AS, Meyer JN, Van Houten B. Analysis of DNA damage and repair in nuclear and mitochondrial DNA of animal cells using quantitative PCR. Methods Mol Biol 2012; 920: 111–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Widlansky ME, Wang J, Shenouda SM, et al. Altered mitochondrial membrane potential, mass, and morphology in the mononuclear cells of humans with type 2 diabetes. Transl Res 2010; 156: 15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yin X, Prendiville O, McNamara AE, Brennan L. Targeted Metabolomic Approach to Assess the Reproducibility of Plasma Metabolites over a Four Month Period in a Free-Living Population. J Proteome Res 2022; 21: 683–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The bulk RNA-seq data presented in the study are deposited in the Gene Expression Omnibus (GEO) repository, accession number GSE206333.