

Abstract
The anatomical distribution of most neurodegenerative diseases displays considerable interindividual variations. In contrast, FTLD-TDP type C (TDP-C) shows a consistent predilection for the anterior temporal lobe (ATL). The relatively selective atrophy of ATL in TDP-C patients has highlighted the importance this region for complex cognitive and behavioral functions. This review includes observations on 28 TDP-C cases, 18 with semantic primary progressive aphasia and 10 with other syndromes. Longitudinal imaging allowed the delineation of progression trajectories. At post-mortem examination, the pathognomonic feature of TDP-C consisted of long, thick neurites found predominantly in superficial cortical layers. These neurites may represent dystrophic apical dendrites of Layer III and V pyramidal neurons that are known to play pivotal roles in complex cortical computations. Other types of FTLD-TDP, such as TDP-A and TDP-B are not associated with long dystrophic neurites in the cerebral cortex and do not display similar predilection patterns for ATL. Research is beginning to identify molecular, structural and immunological differences between pathologic TDP-43 in TDP-C versus TDP-A and B. Parallel investigations based on proteomics, somatic mutations, and GWAS are detecting molecular features that could conceivably mediate the selective vulnerability of ATL to TDP-C. Future work will focus on characterizing the distinctive features of the abnormal TDP-C neurites, the mechanisms of neurotoxicity, initial cellular targets within the ATL, trajectory of spread, and the nature of ATL-specific markers that modulate vulnerability to TDP-C.
Graphical Abstract
FTLD-TDP Type C (TDP-C) targets the anterior temporal lobes (ATL) for neurodegeneration. Left-sided disease impairs word comprehension and object naming; right-sided disease impairs comportment and object recognition. Mechanisms mediating the neurotoxicity of TDP-C and the predilection for the ATL are poorly understood. We summarize current knowledge on the nature of the dystrophic neurites in TDP-C, their molecular distinction from inclusions of TDP-A, and potential features of the ATL that may mediate the selective vulnerability to TDP- C but not TDP-A.
INTRODUCTION
Neurodegenerative diseases can have common, uncommon, and idiosyncratic manifestations. Alzheimer’s Disease (AD), for example, is typically seen in the setting of amnestic disorders but can also have atypical clinical presentations such as primary progressive aphasia (PPA) or unique presentations such as hemiplegia.1,2 Each of these syndromes tends to be associated with clinically concordant distributions of neurofibrillary degeneration.2–4 Similar heterogeneity is seen in synucleinopathies and frontotemporal lobar degenerations with either tau or TDP-43 abnormalities (FTLD-tau, FTLD-TDP).5–8 Even in autosomal dominant forms of FTLD, the same mutation may cause distinctly different clinical syndromes in members of the same family, presumably because of differences in the anatomic distribution of neurodegeneration.9,10 FTLD-TDP type C (TDP-C) appears to constitute an exception because of its consistent affinity for the anterior temporal lobe (ATL). The goal of this review is to illustrate the anatomy of this selectivity, its neurobehavioral consequences, its neuropathologic fingerprints, and the features that distinguish TDP-C from FTLD-TDP types not associated with ATL degeneration. For the purposes of this account, the term ‘ATL’ will be used to designate the anterior third of the temporal lobe and will encompass circumpolar regions, the planum polare, rostral parts of the superior temporal, middle temporal, inferior temporal, and fusiform gyri, piriform cortex, periamygdaloid cortex, and rhinal cortices (i.e., perirhinal, transentorhinal, anterior entorhinal).11
THE CONSISTENT AFFINITY OF TDP-C FOR THE ANTERIOR TEMPORAL LOBE DEMONSTRATED IN A PPA COHORT
A cohort of prospectively enrolled and longitudinally investigated participants with PPA offers a model system for illustrating the relationship of TDP-C with ATL. At this writing, the Northwestern PPA Program has recruited 177 right-handed participants at mild stages of impairment, each fulfilling criteria for the root diagnosis of PPA.12 All PPA variants, agrammatic, logopenic, mixed, and semantic, are represented.13 Participants underwent biannual evaluations of object naming, word comprehension, sentence comprehension, repetition, grammaticality, and object recognition. Structural MRI volumes were analyzed quantitatively by the Free Surfer toolkit for depicting cortical thinning (atrophy) on whole brain surface maps, and by voxel-based morphometry (VBM)14 for more detailed illustration of sulcal and gyral atrophy on coronal sections. Methods used for language assessment, FreeSurfer analyses, and neuropathologic diagnoses have been described in previous publications.15,16
Of the 177 participants, 90 came to autopsy and received primary neuropathologic diagnoses of AD (n=34), CBD/PSP (n=18), Pick’s disease (n=8), TDP-A (n=7), TDP-B (n=3), TDP-C (n=18), leukodystrophy with spheroids (n=1), and glial globular glial tauopathy (n=1). Two of the TDP-A cases had pathogenic GRN mutations. General features of this cohort have been reported previously.16 As expected in this group of right-handed PPA patients, leftward asymmetry of atrophy, in vivo as well as at post-mortem, was the common denominator for all except one case. Imaging revealed that the AD, CBD/PSP, Pick, TDP-A and TDP-B groups displayed considerable inter-individual variations of peak atrophy sites despite the common PPA diagnosis.16 For example, some AD cases had initial atrophy confined to the left temporoparietal junction, while others had peak atrophy sites also in the left inferior frontal gyrus and still others in the left ATL. In Pick’s disease some cases displayed equally prominent atrophy in left prefrontal cortex and ATL, whereas others had no significant atrophy in prefrontal areas. In CBD/PSP, some cases had characteristic peak atrophy in left posterior frontal areas and others in the left superior temporal gyrus. In TDP-A peak atrophy sites variably included parietal, frontal and temporal regions of the left hemisphere. One of the TDP-B patients had atrophy confined to the left ATL but the remaining 2 showed no significant cortical atrophy at the conventional 0.05 False Discovery Rate cutoffs.
In contrast to the interindividual heterogeneity observed in all other neuropathologic entities associated with PPA, TDP-C uniformly targeted the ATL as the principal site of initial atrophy (Fig. 1). In 15 cases (83%), significant atrophy at initial encounter was limited to the left ATL, except for minor extension into the immediately adjacent ventral insula and, less consistently, caudal orbitofrontal cortex. In one case, the atrophy at initial examination was more extensive in the right ATL, but the left ATL also had substantial atrophy. Progression was slow. In 10 of the 14 cases where longitudinal imaging was available, the ATL remained the predominant region of peak atrophy for up to a decade after symptom onset.11 In many cases, external inspection of the autopsied brain identified the left ATL as the area of most extensive atrophy, up to 12 years after symptom onset (Fig. 1C).11,17
Figure 1. Selectivity of TDP-C for ATL Shown by Free Surfer and at Autopsy.
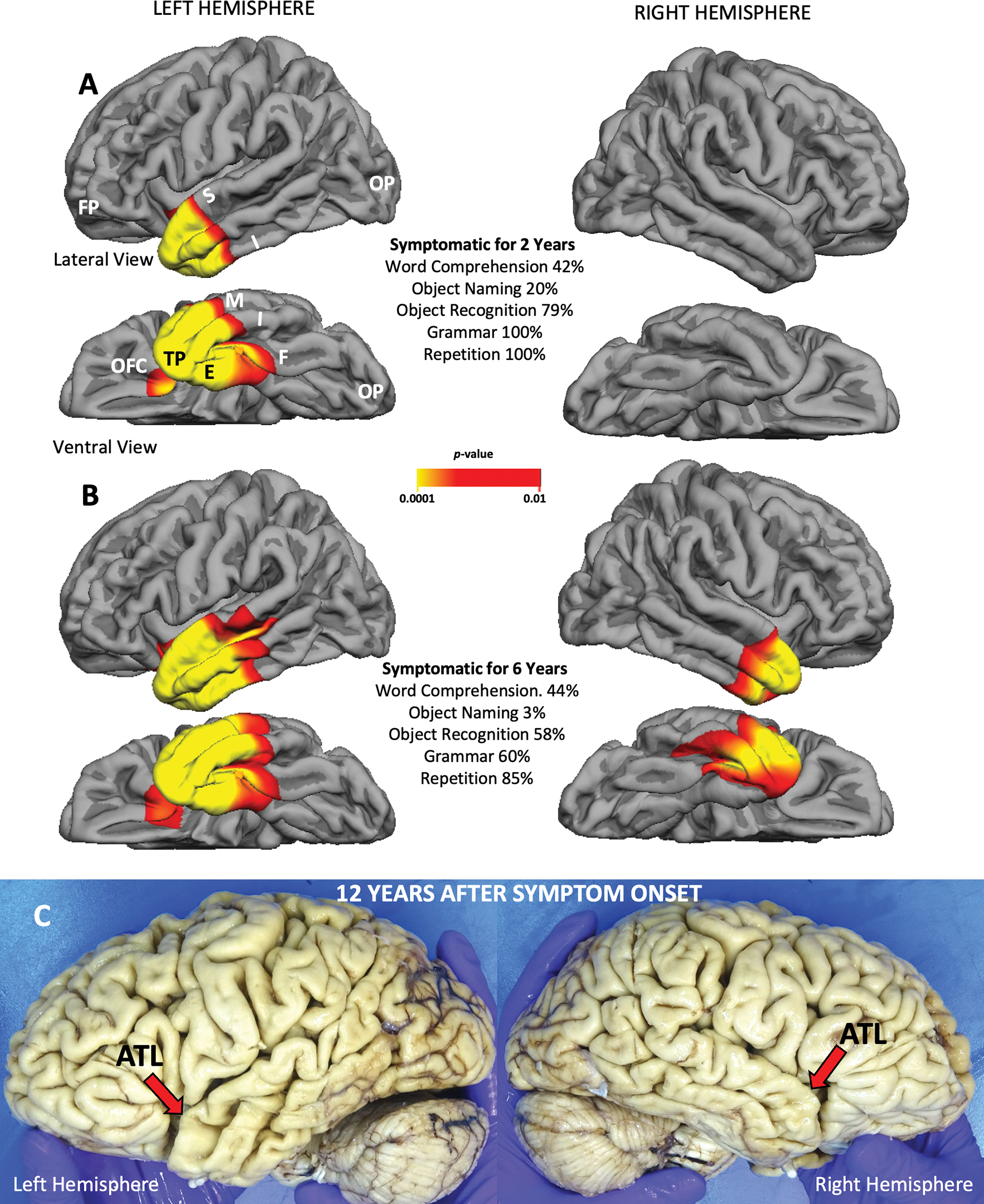
All images are from a semantic PPA patient (right-handed woman) with symptom onset at the age of 59 and TDP-C at autopsy. Significant atrophy on the cortical surface in A and B is shown by the colored areas, at the false discovery rate of 0.05 using the Free Surfer tool kit. A. Atrophy is confined to left ATL and is sufficient to cause severe and isolated impairment of word comprehension and object naming. B. Four years later all significant atrophy is still confined to ATL but has also emerged on the right. Object naming further decreased and non-verbal object recognition started to fail, likely due to bilaterality of ATL atrophy. C. Autopsy specimen of the patient 12 years after symptom onset. The macroscopic neurodegeneration is still strongly selective for ATL. Abbreviations: ATL-anterior temporal lobe; E- entorhinal/perirhinal cortex; F-fusiform gyrus; FP- frontal pole; I-inferior temporal gyrus; M- middle temporal gyrus; OFC- orbitofrontal cortex; OP-occipital pole; TP- temporal pole.
The predilection of TDP-C for the ATL has been described in multiple cohorts.17–23 One study reported that the limbic part of ATL is the most vulnerable to TDP-C and suggested the presence of a medial to lateral progression of neurodegeneration;18 another identified 4 stages of neurodegeneration and included the amygdala in stage 1.23 Our sample is in agreement with the overall caudal progression described in these studies but also adds new anatomical characterization based on imaging of earlier stages of neurodegeneration and the representation of cortical atrophy in greater detail on coronal brain sections. Although the progression illustrated in Figure 2 A–B and summarized in Figure 2C is based on individual patients, none of the remaining 17 PPA and 10 non-PPA cases (see below) with TDP-C displayed substantial deviations from this trajectory in either the left or the right hemisphere. Nonetheless, the progression described below is descriptive and subject to change as more information becomes available.
Figure 2. Progression of Gyral and Sulcal Peak Atrophy Sites Shown by Voxel Based Morphometry.
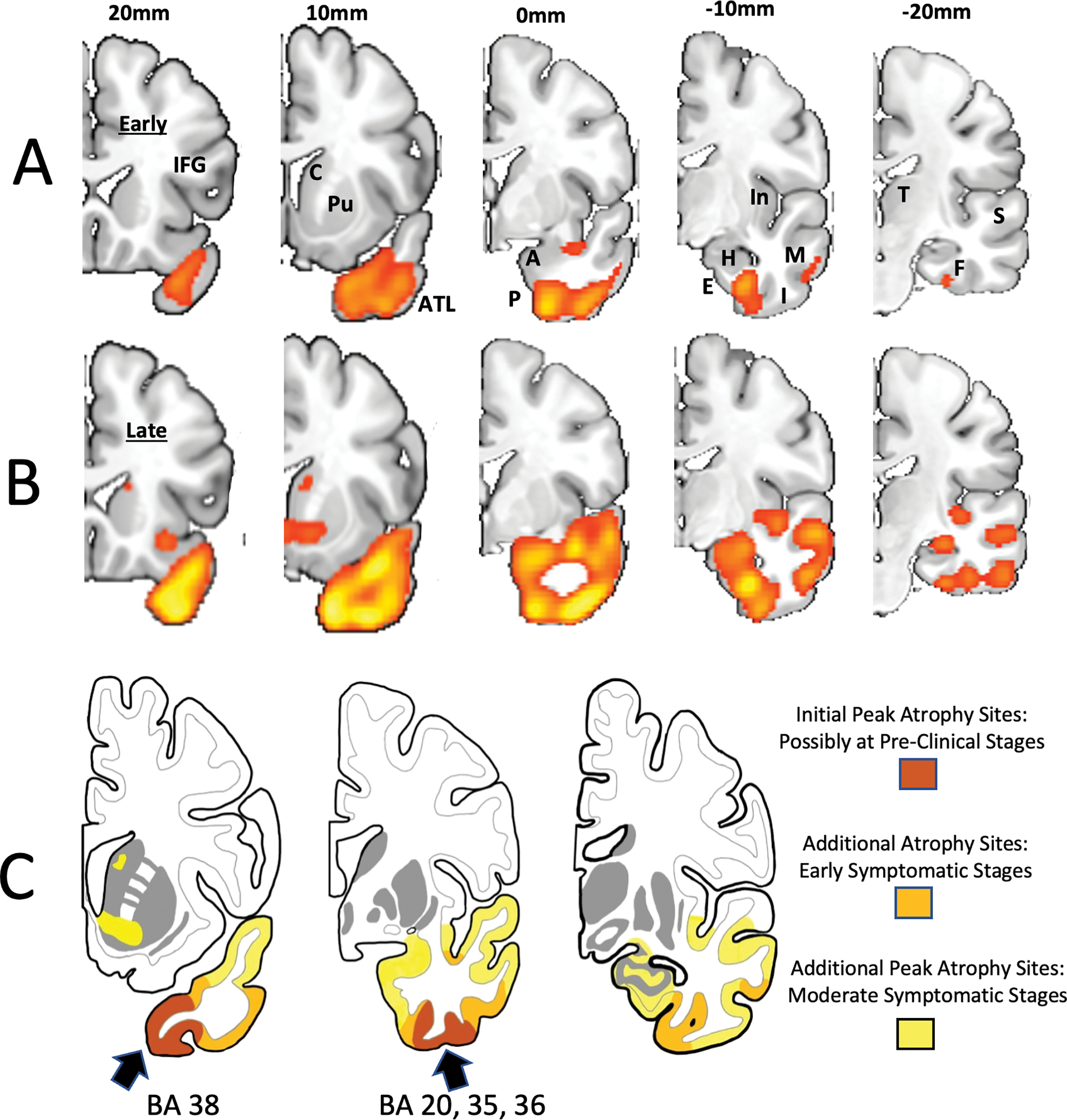
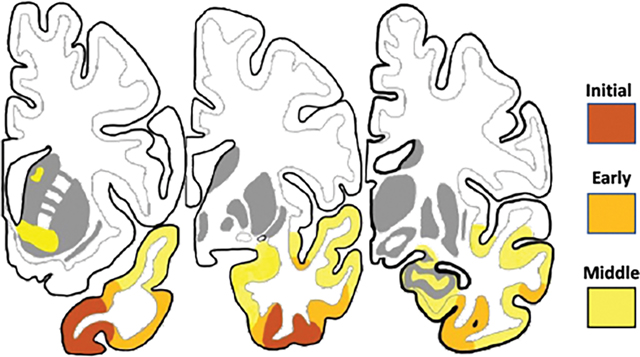
The colored patches on rows A and B indicate areas of cortical volume loss in comparison to a control group at a statistical significance of p<.001 and minimum cluster size of 100 voxels. These are peak atrophy sites. The presence of additional atrophy that fails to reach statistical significance cannot be ruled out. From left to right, each row represents increasingly more caudal coronal sections from the same hemisphere. The mm designations refer to the distance from the anterior commissure (AC) in millimeters, positive numbers indicating locations in front of the AC. A. and B. Left hemisphere of a semantic PPA patient initially seen at an earlier stage of clinical severity than the patient in Figure 1. Images were obtained at an interval of 4 years and represent early (A) and moderate (B) symptomatic stages of neurodegeneration. C. Composite illustration of progressive atrophy. The stage of ‘initial a peak trophy’ is inferred based on emergent right ATL atrophy in PPA patients who had no such atrophy at prior imaging. The progression illustrated in C is approximated to occur over a span of 6–8 years from symptom onset and is based on the examination of 56 hemispheres of the 28 patients with TDP-C, many with longitudinal imaging. The arrows point to the Brodmann areas where initial peak atrophy emerges in our collection of cases. Abbreviations: A- amygdala; AC- anterior commissure; ATL- anterior temporal lobe; BA- Brodmann area; C- caudate; E- entorhinal cortex; F- fusiform gyrus; H- hippocampus; I- inferior temporal gyrus; IFG- inferior frontal gyrus (Broca’s); In- insula; M- middle temporal gyrus; P- perirhinal area; Pu- putamen; S- superior temporal gyrus; T- thalamus.
In TDP-A and TDP-B, mutation carriers have enabled the imaging of pre-clinical disease stages.24 In the absence of known causative mutations, this is not possible in TDP-C. However, the leftward asymmetry of neuropathology and the initial sparing of the right hemisphere in the majority of PPA patients offers an alternative approach. In these patients, longitudinal imaging can capture the very onset of atrophy in the right ATL, a pattern that is likely to mirror the stage of emergent atrophy on the left. Through this reasoning, we identified the ventromedial circumpolar region anterior to the limen insulae (area 38) and its caudal extension into the adjacent perirhinal-fusiform-inferotemporal regions (areas 20, 35, 36) as sites of emerging atrophy (Fig. 2C). Since no symptomatic ATL patient had such limited atrophy on the left, this band of atrophy may well reflect pre-clinical stages of TDP-C. Subsequent progression extended caudally along the ventral surface of the ATL where it encompassed additional perirhinal, entorhinal, fusiform and inferotemporal cortices (Fig. 2A) A patch of ventral insula, which is known to be continuous with medial temporopolar cortex,25 also displayed significant atrophy. The amygdala, primary olfactory cortex, and hippocampus were not among peak atrophy sites at these points in time (Fig. 2A). At a later stage (Fig. 2B), peak atrophy expanded into the amygdala, striatum, uncus, inferior temporal cortex, and lateral temporal cortices, including the middle and superior temporal gyri. Despite the affiliation of ATL with the language network, Broca’s area and Wernicke’s were rarely sites of peak atrophy at this stage, suggesting that atrophy starts spreading by contiguity rather than through network connectivity.26 The colored patches in Figures 1 and 2 identify only areas of statistically significant cortical shrinkage. It is important to keep in mind that there may be other areas of neurodegeneration that fail to meet the statistical threshold as well as areas of cellular pathology that escape detection because they do not cause significant cortical thinning. However, metabolic PET has shown that the pattern of hypometabolism in these patients is generally concordant with the pattern of peak atrophy.27 In the future, the development of PET ligands for TDP-43 deposits may allow a more systematic mapping of progression. Even without the analytical morphometry used in Figures 1 and 2, MRI can also be a highly informative diagnostic tool for differential diagnosis in clinical practice. For example, the MRI pattern of severe ATL thinning in the absence of obvious frontal and parietal atrophy has a nearly 90% probability of being associated with TDP-C (the other 10% being associated with Pick’s disease) and can be used to rule out AD (Fig 3A). More advanced stages of progression are marked by the widening of the collateral sulcus and temporal horn (Fig. 3B) and increasing atrophy of more posterior temporal cortices (Fig. 3C).
Figure 3. MRI Appearance of TDP-C Progression Illustrated by Individual Cases.
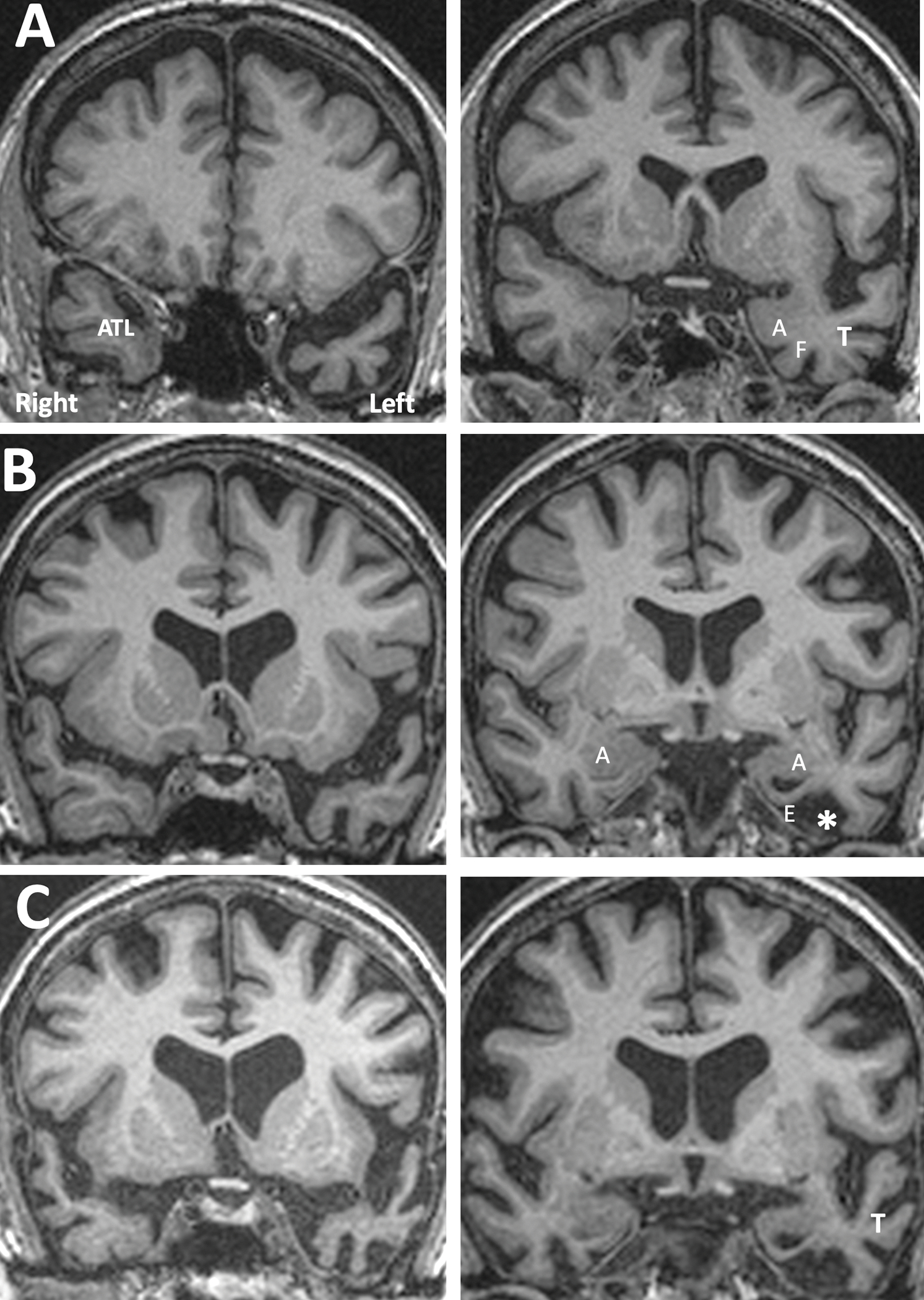
A. Distribution of atrophy at an early symptomatic stage. From a TDP-C semantic PPA patient (right-handed man) with onset at the age of 59. Significant atrophy is confined to the part of the left anterior temporal lobe (ATL) located ahead of the limen insulae. The amygdala, rhinal cortices, the fusiform gyrus, and lateral parts of temporal cortex are relatively preserved. B. Intermediate stage of atrophy from a right-handed woman with symptom onset at 59 (same patient shown in Figure 1). Atrophy of the left ATL has become more extensive. The amygdala has mild atrophy, rhinal cortices are severely thinned, the anteriormost part of the fusiform gyrus has been replaced by cerebrospinal fluid (*), and the temporal horn shows ex vacuo enlargement. C. A more advanced stage of atrophy. There is further neurodegeneration in left ATL, amygdala, rhinal cortices and the fusiform gyrus. Atrophy has now spread to the lateral temporal cortices (T). Abbreviations: A- amygdala; ATL- anterior temporal lobe; E- entorhinal/perirhinal areas; F- fusiform gyrus; T- lateral temporal cortices.
Nearly all 18 TDP-C participants presented with ‘semantic’ impairments, heuristically identified16 by word comprehension scores of ≤ 60% on a subset of difficult items28 in the Peabody Picture Vocabulary Test (PPVT)29 and/or object naming scores of ≤ 40% on the Boston Naming Test (BNT).30 At the early disease stages of these patients, when other language functions are relatively spared, naming impairments of ≤ 40% reflect semantic (i.e., word meaning) rather than retrieval (i.e., word finding) anomia and presage the emergence of more severe single word comprehension failures.28,31,32 The clinical diagnoses were consistent with semantic PPA (svPPA, PPA-S) in 14 of the cases. In the remaining 4 cases, including the one with greater right ATL atrophy, the initial diagnosis of semantic PPA was changed to semantic dementia (SD) upon further review. Both diagnoses entail severe word comprehension impairments as detected by the BNT and PPVT cutoffs mentioned above. The SD diagnosis requires additional non-verbal object recognition deficits at early disease stages, as assessed by the picture format of the Pyramids and Palm Trees Test (PTTp).11,33,34 This distinction between PPA-S and SD is frequently overlooked so the two terms have often been used interchangeably in the literature.
Equally pronounced semantic impairment with pathologies other than TDP-C was present among 7 of the 90 PPA autopsies: 3 with TDP-A, 1 with AD, 2 with Pick’s disease, and 1 with glial globular tauopathy.16 Although atrophy in all 7 included the left ATL, it also encompassed other parts of cortex including the frontal, parietal, and posterior temporal lobes. There were also 2 cases of Pick’s disease and one of TDP-B where peak atrophy was predominantly located within the left ATL but without prominent semantic deficits. Considering the fact that Pick’s disease caused definite semantic impairment only if atrophy extended beyond ATL, the speculative possibility arises that TDP-C may have a distinctive affinity not just for the ATL in general but also for neuronal components somewhat more critical for lexicosemantic functions. There are reports of 4 TDP-B cases (one established at autopsy7 and 3 predicted by C9orf72 mutations35) in whom relatively isolated left ATL atrophy was observed in conjunction with semantic impairment. However, it is important to keep in mind that ATL atrophy is a distinctly rare manifestation of TDP-B36 whereas it is a nearly universal correlate of TDP-C. In fact, only 1 of the 3 TDP-B cases in our cohort of 90 autopsies had selective left ATL atrophy but without prominent semantic impairment.
In addition to the 18 TDP-C cases that were part of the longitudinal PPA research program, the Northwestern ADRC Brain Bank received 10 cases of TDP-C from patients who had declined to participate in the longitudinal investigation or who did not meet diagnostic criteria for PPA. In each of these 10 individuals, ATL was initially the principal area of peak atrophy. Depending on the pattern of asymmetry, and in keeping with multiple reports in the literature, these 10 additional individuals displayed variable combinations of impairment in social conduct, naming, object recognition, person identification, and word comprehension.11,18,37,38 Behavioral variant FTD (bvFTD) has become the most commonly reported TDP-C syndrome associated with right sided ATL atrophy.18 It entails profound loss of empathy, rigidity of comportment, inappropriate social conduct, and bizarre food preferences. This ATL syndrome differs from other types of bvFTD by the co-occurrence of additional naming, word comprehension, and face recognition impairments.
We are not aware of TDP-C cases where imaging at early stages has failed to include the ATL as the principal area of peak atrophy or hypometabolism. Such cases may well be reported in the future but are likely to represent unusual manifestations of TDP-C. All our right-handed TDP-C patients with left ATL atrophy have developed severe semantic deficits. The literature contains an exception to this pattern in the form of a right-handed TDP-C case with nonfluent aphasia and normal word comprehension despite prominent atrophy in the general region of left ATL.39 This unprecedented combination could conceivably reflect an atypical organization of the language network in the affected individual.
SYNAPTIC BACKGROUND OF TDP-C SYNDROMES
The functions of ATL had remained mysterious during the formative century of neurology. Although ATL gained notoriety through the Klüver-Bucy syndrome40, analogous clinical presentations were not identified in humans with any degree of consistency. This state of affairs changed dramatically during the last two decades of the 20th century through increasing awareness of focal ATL atrophies, many of which were subsequently shown to be caused by TDP-C.41,22 The associated syndromes were quite striking. Some patients were unable to understand the meaning of simple words such as ‘pumpkin’ or ‘grass’ at a time when other functions appeared relatively intact; others displayed shocking disregard of common sense and uncharacteristic blunting of empathy, even when they performed normally in cognitive tests of ‘executive function.’ These syndromes have introduced new entities to neurological nosology. The semantic PPA of ATL atrophy is unique. It differs from Wernicke’s Aphasia because of intact language repetition, and from Posterior Transcortical Aphasia because of the much greater severity of the word comprehension impairment with otherwise preserved grammar and basic neurologic function. The behavioral syndrome of right ATL atrophy is equally distinctive because the impairments of social conduct are frequently intermixed with impairments of verbal and non-verbal semantics.37,38,42 Through the systematic neuropsychological investigation and longitudinal imaging of such patients, the ATL has become transformed from an area of unknown function to one that is considered pivotal for word comprehension, social conduct, person identification, and object recognition.11,43,44
Functional and structural imaging of the human brain, when interpreted under the guidance of experimentally established cortico-cortical connections of the macaque, has revealed an anatomical organization that fits the syndromic complexity of ATL. This research shows that the lateral and inferior ATL contain downstream nodes of auditory and visual association pathways that are likely to convey information related to auditory word-forms, visual word-forms, faces, and objects.11,45–48 Interdigitated bands of heteromodal cortex11 mediate the transformation of unimodal percepts into multimodal concepts so that words can be understood, persons identified, and objects recognized. The medial ATL, on the other hand, contains piriform, rhinal, periamygdaloid and uncal areas. They provide gateways into core limbic structures such as the amygdala and hypothalamus, and are likely to play critical roles in the interoceptive modulation of behavior.11 This overall organization is subject to laterality effects so that word comprehension and object naming are strictly controlled by the left hemisphere whereas social conduct, person identification, and object recognition have a more bilateral representation, but with a right-sided bias. This may explain why many semantic PPA patients with unilateral left ATL atrophy also display lesser but definite behavioral aberrations noted above.11
The question may be asked whether this complex synaptology of ATL could be of relevance to selective TDP-C vulnerability. One potentially relevant line of research has suggested that regional transcriptomic patterns may characterize brain areas not only by their location or cytoarchitecture but also by their synaptic position within large-scale information processing hierarchies.49 To be sure, ATL is not the only site of multimodal convergence in the primate brain. Prefrontal and posterior parietal cortices also contain such sites.50 However, ATL is arguably the only major multimodal convergence site that is also in monosynaptic contact with core limbic structures. This distinctive synaptology could conceivably incorporate constitutive transcriptomic patterns that influence neurodegenerative vulnerabilities.
NEUROPATHOLOGY OF TDP-C AND COMPARISON TO OTHER FTLD-TDP TYPES
Human TDP-43 is the source of the abnormal inclusions in all FTLD-TDP types. It is an RNA/DNA binding nuclear protein of 414 amino acids encoded by TARDBP (1.p36.22). It plays an important role in neuronal RNA metabolism, mRNA transport, transcription, differential splicing, stress granule formation, and perhaps also spinogenesis.51,52–55 In FTLD-TDP, abnormally phosphorylated, ubiquitinated, truncated, predominantly C-terminal TDP-43 is deposited in the form of nuclear, cytoplasmic, and neuritic inclusions. Depending on the morphology and location of the inclusions, FTLD-TDP is divided into types A-E.56 TDP- A is characterized by neuronal cytoplasmic inclusions and short dystrophic neurites, can be associated with GRN mutations, and is clinically manifested as bvFTD or non-semantic forms of PPA. FTLD-B shows compact cytoplasmic inclusions with few dystrophic neurites, can be associated with C9orf72 mutations and is manifested as ALS, bvFTD, or both. TDP-D exhibits lentiform neuronal intranuclear inclusions and few dystrophic neurites, is associated with VCP mutations, and includes inclusion body myositis, Paget’s disease of bone, frontotemporal dementia, and ALS. TDP-E is characterized by granulofilamentous neuronal inclusions, neuropil grains and curvilinear oligodendroglial inclusions leading to a rapidly progressive frontotemporal dementia.57 The cortical distribution of atrophy in each of these FTLD-TDP types shows high interindividual variability. TDP-C is different. Its pathognomonic features are the long, thick, dystrophic neurites that tend to be most densely concentrated in upper layers of cortex (Fig. 4A). TDP-C is not known to be associated with gene mutations, triggers the highly distinctive cognitive and behavioral manifestations described above, and displays much greater anatomical uniformity of cortical atrophy than any of the other FTLD-TDP types or most other neuropathologic entities associated with dementia.
Figure 4. TDP-C Neuropathology.
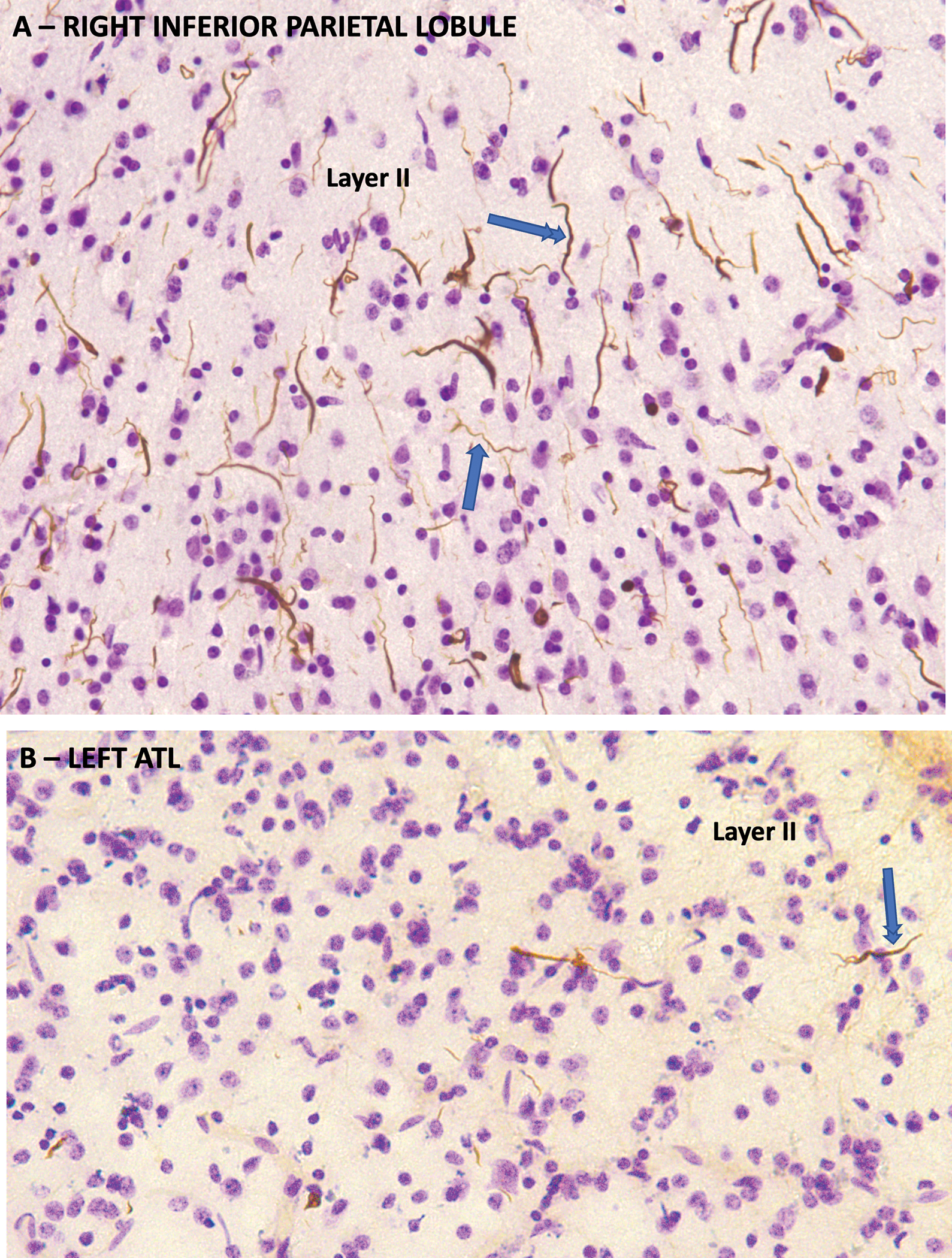
TDP-43 immunohistochemistry counterstained with cresyl violet from the autopsy of a semantic PPA case with symptom onset at the age of 63 and death 10 years later. A. From the right inferior parietal lobule which was relatively spared. Neuronal architecture is mostly preserved. There is a high density of thick, long, dystrophic neurites pathognomonic of TDP-C, many of which have a rectilinear orientation perpendicular to the cortical surface in an orientation consistent with apical dendrites of pyramidal neurons (double arrowhead). Others are wispy and undulating, reminiscent of interneuron dendrites (single arrowhead). B. Same method showing the left ATL, which had advanced neurodegeneration. There is severe gliosis and loss of neurons. The density of dystrophic neurites is much lower than in the inferior parietal lobule.
In AD, atrophy is tightly correlated with the density of neurofibrillary tangles (NFT). This relationship reflects the neurotoxicity of NFTs and their resistance to subsequent clearance.58,59 In TDP-C, the relationship of neurodegeneration to inclusions may deviate from this pattern. For example, quantitative microscopic investigations of 10 autopsies found no consistent overlap between sites of maximal neurodegeneration and sites of maximal TDP-43 inclusions.60 Some cortical areas that appeared normal in neuronal number and cytoarchitecture were shown to have dystrophic neurite densities higher than sites of maximal neurodegeneration such as ATL (Fig. 4B). These studies suggest that the dystrophic neurites of TDP-43, having emerged and reached their highest density and neurotoxic impact in the ATL, undergo gradual clearance from the tissue. However, it is currently unclear whether the abnormal TDP-43 in the dystrophic neurites is the cause or consequence of neurodegeneration. Furthermore, the possibility that the dystrophic neurites emerge simultaneously everywhere in cortex but that they are more toxic to ATL neurons cannot be ruled out. Considering the numerous dystrophic neurites at cortical areas with minimal neuronal abnormality (Fig. 4), there seems to be a long delay before the local neurotoxicity of the abnormal TDP-43 neurites becomes associated with neuronal death. Greater clarity on the trajectory of TDP-43 dystrophic neurites and their relationship to neurodegeneration is unlikely to emerge without in vivo TDP-43 imaging or the availability of autopsy tissue from earlier disease stages.
Full length TDP-43 oligomers and TDP-43 cleavage products from human FTLD-TDP are neurotoxic, while deletion of TDP-43 in the mouse is embryonically fatal.61,62 Neurodegeneration in FTLD-TDP may therefore reflect the loss of normal TDP-43 function as well as the toxicity of the abnormal TDP-43. The relative impact of these two factors may vary across different FTLD-TDP types.52 The identification of distinct molecular forms or strains of the pathological protein, as has been done in tauopathies63 and synucleinopathies8, has not yet been reported for FTLD-TDP. At the present, the biological mechanisms that distinguish TDP-C from other types of FTLD-TDP, or that underlie its unique affinity for the ATL, remain incompletely understood. This will be a challenging question to address because TDP-C, in contrast to TDP-A and TDP-B, is not associated with disease-causing mutations, making it difficult to generate animal models. At the descriptive level, however, features that differentiate TDP-C from the other two most common types, namely TDP-A and TDP-B, are being reported with increasing frequency and offer potential pointers for future research directions.
Along these lines, F-box protein 2 was found to be a marker of astrocytes in TDP-A but not TDP-C.62 Immunohistochemical experiments also showed that an antibody raised against TDP-43 phosphorylated at serine 375 is immunoreactive in tissue from TDP-B and TDP-C but not TDP-A, and that heterogeneous ribonuclear protein hnRNP E2 antibodies bind abnormal TDP-43 inclusions of TDP-C but not TDP-A.64,65 Furthermore, TDP-C aggregates displayed lower polyubiquitination and a more protease resistant C-terminal when compared to either TDP-A or TDP-B.62 Perhaps as a consequence of these factors, pathological aggregates from TDP-A were found to be more neurotoxic and more likely to display seeding activity than those isolated from TDP-C, a finding in keeping with significantly longer survival times in Type C than in Type A.16,57,62 The field of tauopathy has greatly benefited from the structure-based classification of abnormal tau species through cryo-electron microscopy.66 The same methodology has been applied to TDP-B, where abnormal TDP-43 has been shown to have a filamentous structure with a core that spans residues 282–360 arranged in spiral shaped folds. 63 It will be of great interest to see if cryo-electron microscopy can reveal structural details that differentiate TDP-B from TDP-C to explain why TDP-C is the only type of FTLD-TDP with a preponderance of dystrophic neurites. Based on currently available information, it is reasonable to conclude that each type of FTLD-TDP reflects a distinct pathophysiological cascade that converges upon a common proteinopathy. In each type, the abnormal TDP-43 retains some common properties (such as phosphorylation and truncation) but also appears to undergo different post-translational modifications that mediate the distinctive patterns of neurotoxicity.
Immunohistochemical studies suggest that the dystrophic neurites in TDP-C may be of dendritic origin.67 In support of this suggestion, our observations on whole-hemisphere sections processed for TDP-43 immunohistochemistry and counterstained with cresyl violet (Fig. 4) show that a substantial number of the thick layer II neurites have perpendicular orientations reminiscent of apical dendrites ascending from large pyramidal neurons of layers III and V as seen in Golgi preparations of the human cerebral cortex.68 Thinner and less rectilinear TDP-43 neurites are also seen and may represent dendrites of local circuit inhibitory neurons, which are known to display very complex dendritic arborizations throughout cortical layers.69 The possibility of dendritic origin for the dystrophic neurites is of interest because TDP-43 is also thought to be an activity-responsive factor that colocalizes with the post-synaptic dendritic protein PSD-95, and that may participate in synaptogenesis.55,70 Whether all dystrophic neurites are of dendritic origin or if some also represent dystrophic axons or glial processes remains to be established. Axonal origin appears unlikely because the hemispheric white matter and the anterior commissure, which interconnects the ATL on each hemisphere, have no substantial concentration of dystrophic TDP-43 neurites. The possibility that TDP-C causes the degeneration of dendrites of deep pyramidal neurons is of particular interest since these neurons have been singled out for their strategic role in the feedforward-feedback interactions that underlie integrative cortical function.71 Although Pick’s disease also affects large pyramidal neurons of layers III and V, the tauopathy is cytoplasmic and has not been shown to extend into dendrites, at least according to one immunohistochemical investigation addressing this question.72 Such differences in the circuitry targeted by the principal proteinopathy may conceivably help clarify why neurodegeneration confined to ATL consistently undermines word comprehension in TDP-C but not necessarily in Pick’s disease.
Distinctive molecular and structural properties of TDP-C cannot by themselves explain the selective predilection of TDP-C for ATL. Additional constitutive or disease-related factors unique to ATL or to the affected persons are also needed. Such putative factors could include somatic mutations within ATL, transcriptomic fingerprints that resonate with the distinctive pathogenicity of TDP-C, or patient-specific risk factors. As noted above, the unique synaptology of ATL may have distinctive transcriptomic markers that modulate vulnerabilities. Furthermore, GWAS encompassing 4308 controls and 2154 clinically diagnosed FTD cases identified a network of 64 risk genes associated with ‘semantic dementia’, a diagnosis strongly correlated with TDP-C.73 It would be of considerable interest to see whether distinctive expression patterns of these genes can be linked to the ATL of TDP-C cases. Compared to control, AD, and other FTLD datasets, proteomic analyses in 15 patients with TDP-C also found increased cell adhesion proteins of the cadherin-catenin complex at the synaptic junction in samples from TDP-C.74. However, this analysis was limited to the dentate gyrus, which contains mostly Pick-like round TDP-43 inclusions rather than the thick long dystrophic neurites, making its relevance to the cortical ATL degeneration unclear. In another study, somatic variations were explored in fresh frozen middle temporal gyrus and hippocampal tissue from 16 TDP-C patients.21 In 2 cases, somatic variants in the TARDBP gene were identified and linked to potential disruption of normal TDP-43 function. In Alzheimer’s disease, single nucleus RNA sequencing in frozen tissue from temporal cortex has led to the discovery of differentially expressed genes and pathways.75 Similar approaches in TDP-C would be of great interest for addressing the ‘seed versus soil’ question.
CONCLUSIONS
Although we do not yet have a mechanistic explanation of the relationship between TDP-C and ATL, considerable progress has been made. As noted above, a growing list of features differentiates TDP-C from TDP-A and TDP-B. The latter two entities share the proteinopathy but not the predilection for ATL. It is quite likely that additional patient-specific susceptibility factors will emerge through larger GWAS. Evidence is currently much sparser when it comes to the identification of ATL-specific features not shared by other cortical areas or other types of FTLD-TDP. The asymmetric neurodegeneration of PPA is particularly conducive to research along these lines because the more susceptible left ATL can be compared to its counterpart on the right, sidestepping interindividual differences. Additional work is clearly needed to isolate distinguishing features of the abnormal TDP-43 in TDP-C, the mechanisms of neurotoxicity, the initial cellular target within the ATL, the synaptic trajectory of progression, and ATL-specific markers that modulate vulnerability. Although TDP-C is distinctly rare, with an extrapolated prevalence of 1/100,000,76,77, it represents an experiment of nature that stands to shed light on the basic principles of selective vulnerability and the behavioral synaptology of ATL.
ACKNOWLEDGMENT:
R01AG077444; R01AG062566, P30AG072977 from NIH.
Footnotes
CONFLICT OF INTEREST: Nothing to report.
REFERENCES
- 1.Rogalski E, Sridhar J, Rader B, et al. Aphasic variant of Alzheimer’s disease: clinical, anatomic and genetic features. Neurology. 2016;87:1337–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jagust WJ, Davies P, Tiller-Borcich JK, Reed BR. Focal Alzheimer’s disease. Neurology. 1990;40:14–19. [DOI] [PubMed] [Google Scholar]
- 3.Gefen T, Gasho K, Rademaker A, et al. Clinically concordant variations of Alzheimer pathology in aphasic versus amnestic dementia. Brain. 2012;135:1554–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murray ME, Graff-Radford NR, Ross OA, Petersen RC, Duara R, Dickson DW. Neuropathologically defined subtypes of Alzheimer’s disease with distinct clinical characteristics: a retrospective study. The Lancet Neurology. 2011;10:785–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Giannini LAA, Xie SX, McMillan CT, et al. Divergent patterns of TDP-43 and tau pathologies in primary progressive aphasia. Ann Neurol. 2019;85:630–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Irwin DJ, VCairns NJ, Grossman M, et al. Frontotemporal lobar degeneration: defining phenotypic diversity through personalized medicine. Acta Neuropathologica. 2015;129:469–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee DJ, Bigio EH, Rogalski EJ, Mesulam M-M. Speech and language presentations of FTLD-TDP type B neuropathology. J Neuropath Exper Neurol. 2019;79:277–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peng C, Gathagan RJ, Lee VM-Y. Distinct a-synuclein strains and implications for heterogeneity among a-synucleinopathies. Neurobiology of Disease. 2018;109:209–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rademakers R, Baker M, Gass J, et al. Phenotypic variability associated with progranulin haploinsufficiency in patients with the common 1477C-T (Arg493X) mutation: an international initiative. Lancet Neurology. 2007;6:857–868. [DOI] [PubMed] [Google Scholar]
- 10.Bugiani O, Murrell JR, Giaccone G, et al. Frontotemporal dementia and corticobasal degeneration in a family with P301S mutation in tau. J Neuropath Exper Neurol. 1999;58:667–677. [DOI] [PubMed] [Google Scholar]
- 11.Mesulam M-M. Temporopolar regions of the human brain. Brain. 2022;doi: 10.1093/brain/awac339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mesulam M-M. Primary progressive aphasia: A language-based dementia. New Eng J Med. 2003;348:1535–1542. [DOI] [PubMed] [Google Scholar]
- 13.Gorno-Tempini ML, Hillis A, Weintraub S, et al. Classification of primary progressive aphasia and its variants. Neurology. 2011;76:1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scarpazza C, Nichols TE, Seramondi D, Maumet C, Sartori G, Mechelli A. Whenthe single matters more than the group (II): addressing the problem of false positive rates in single case voxel based morphometry using non-parametric statistics. Frontiers in Neuoscience. 2001;10doi:doi: 10.3389/fnins.2016.00006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mesulam M-M, Coventry C, Rader B, et al. Modularity and granularity across the language network- a primary progressive aphasia perspective. Cortex. 2021;141:482–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mesulam M-M, Coventry CA, Bigio EH, et al. Neuropathologic fingerprints of progression, neurodegeneration, and language in primary progressive aphasia. Brain. 2022;145:2133–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kawakatsu S, Kobayashi R, Morioka D, et al. Clinicopathological diversity of semantic dementia: Comparisons of patients with early-onset versus late-onset, left-sided versus right-sided temporal atrophy, and TDP-type A versus type C pathology. Neuropathology. 2022;doi:doi: 10.1111/neup.12859 [DOI] [PubMed] [Google Scholar]
- 18.Borghesani V, Battistella G, Mandelli ML, et al. Regional and hemispheric susceptibility of the temporal lobe FTLD-TDP type C pathology. NeuroImage: Clinical. 2020;28:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rohrer JD, Geser F, Zhou J, et al. TDP-43 subtypes are associated with distinct atrophy patterns in frontotemporal dementia. Neurology. 2010;75:2204–2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Josephs K, Whitwell JL, Murray ME, et al. Corticospinal tract degeneration associated with TDP-43 type C pathology and semantic dementia. Brain. 2013;136:455–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Rooij J, Mol MO, Melhem S, et al. Somatic TARDBP variants as a cause of semantic dementia. Brain. 2020;143:3827–3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mackenzie I, Baborie A, Pickering-Brown S, et al. Heterogeneity of ubiquitin pathology in frontotemporal lobar degeneration: classification and relation to clinical phenotype. Acta Neuropathol. 2006;112:539–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bocchetta M, Espinosa M, Lashley T, Warren JD, Rohrer JD. In vivo staging of frontotemporal lobar degeneration TDP-43 type C pathology. Alzheimer’s Research and Therapy. 2020;12doi: 10.1186/s13195-020-00600-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiskoot LC, Panman JL, Meeter LH, et al. Longitudinal multimodal MRI as prognostic and diagnostic biomarker in presymptomatic familial frontotemporal dementia. Brain. 2019;142:193–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mesulam M-M, Mufson EJ. Insula of the old world monkey. I. Architectonics in the insulo- orbito-temporal component of the paralimbic brain. J Comp Neurol. 1982;212(1):1–22. [DOI] [PubMed] [Google Scholar]
- 26.Jo M, Lee S, Jeon Y-M, Kim SH, Kwon Y, K H-J. The role of TDP-43 propagation in neurodegenerative diseases: integrating insights from clinical and experimental studies. Experimental and Molecular Medicine. 2020;52:1652–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bonakdarpour B, Behn J, Takarabe J, et al. Brain hypometabolism and behavior correlation in primary progressive aphasia. presented at: Conference of the International Society for Frontotemporal Dementia; 2022; Lille, France. [Google Scholar]
- 28.Mesulam M-M, Thompson CK, Weintraub S, Rogalski EJ. The Wernicke conundrum and the anatomy of language comprehension in primary progressive aphasia. Brain. 2015;138:2423–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dunn LA, Dunn LM. Peabody Picture Vocabulary Test-4. Pearson; 2007. [Google Scholar]
- 30.Kaplan E, Goodglass H, Weintraub S. The Boston Naming Test. Lea & Febiger; 1983. [Google Scholar]
- 31.Mesulam M-M, Wieneke C, Hurley RS, et al. Words and objects at the tip of the left temporal lobe in primary progressive aphasia. Brain. 2013;136:601–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Czarnecki K, Duffy JR, Nehl CR, et al. Very early semantic dementia with progressive temporal lobe atrophy. Arch Neurol. 2008;65:1659–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neary D, Snowden JS, Gustafson L, et al. Frontotemporal lobar degeneration. A consensus on clinical diagnostic criteria. Neurology. 1998;51:1546–1554. [DOI] [PubMed] [Google Scholar]
- 34.Howard D, Patterson K. Pyramids and Palm Trees: A Test of Semantic Access From Pictures and Words. Thames Valley Test Company; 1992. [Google Scholar]
- 35.Saracino D, Géraudie A, Remes AM, et al. Primary progressive aphasias associated with C9orf72 expansions; Another side of the story. Cortex. 2021;145:145–159. [DOI] [PubMed] [Google Scholar]
- 36.Simón-Sánchez J, Dopper EGP, Cohn-Hokke PE, et al. The clinical and pathological phenotype of C9ORF72 hexanucleotide repeat expansions. Brain. 2012;135:723–735. [DOI] [PubMed] [Google Scholar]
- 37.Ulugut H, Dijkstra AA, Scarioni M, et al. Right temporal variant frontotemporal dementia is pathologically heterogeneous: a case-series and systematic review Acta Neuropathologica Communications. 2021;9:1–13. doi: 10.1186/s40478-021-01229-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Younes K, Borghesani V, Montembeault M, et al. Right temporal lobe and socioemotional semantics: semantic behavioral variant frontotemporal dementia. Brain. 2022;doi: 10.1093/brain/awac217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Adams-Carr KL, Bocchetta M, Neason M, et al. A case of TDP-43 type C pathology presenting as nonfluent variant primary progressive aphasia. Neurocase. 2019;doi: 10.1080/13554794.2019.1690665 [DOI] [PubMed] [Google Scholar]
- 40.Klüver H, Bucy PC. An analysis of certain effects of bilateral temporal lobectomy in the rhesus monkey, with special reference to “psychic blindness.”. Journal of Psychology. 1938;5:33–54. [Google Scholar]
- 41.Hodges JR, Patterson K, Oxbury S, Funnell E. Semantic dementia. Progressive fluent aphasia with temporal lobe atrophy. Brain. 1992;115:1783–1806. [DOI] [PubMed] [Google Scholar]
- 42.Josephs KA, Whitwell JL, Knopman D, et al. Two distinct subtypes of right temporal variant frontotemporal dementia. Neurology. 2009;73:1443–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Patterson K, Nestor P, Rogers TT. Where do you know what you know? The representation of semantic knowledge in the human brain. Nature Reviews Neuroscience. 2007;8:976–988. [DOI] [PubMed] [Google Scholar]
- 44.Gefen T, Wieneke C, Martersteck AC, et al. Naming vs knowing faces in primary progressive aphasia. A tale of two hemispheres. Neurology. 2013;81:658–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morán MA, Mufson EJ, Mesulam MM. Neural inputs into the temporopolar cortex of the rhesus monkey. J Comp Neurol. 1987;256(1):88–103. [DOI] [PubMed] [Google Scholar]
- 46.Margulies DS, Ghosh SS, Goulas A, et al. Situating the default-mode network along a principal gradient of macroscale cortical organization. Proc Nat Acad Sci (USA). 2016;113:12574–12579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sepulcre J, Sabuncu MR, Yeo TB, Hesheng L, Johnson KA. Stepwise connectivity of the modal cortex reveals the multimodal organization of the human brain. J Neurosci. 2013;32:10649–10661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pascual B, Masdeu JC, Hollenbeck M, et al. Large-scale brain networks of the human left temporal pole: A functional connectivity MRI study. Cereb Cortex. 2013;25:680–702. doi: 10.1093/cercor/bht260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Krienen FM, Yeo TB, Ge T, Buckner RL, Sherwood CC. Transcriptomic profiles of supragranular-enriched genes associated with corticocortical network architecture in the human brain. Proc Nat Acad Sci (USA). 2016;113:E469–E478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mesulam M-M. From sensation to cognition. Brain. 1998;121:1013–1052. [DOI] [PubMed] [Google Scholar]
- 51.de Boer EMJ, Orie VK, Williams T, et al. TDP-43 proteinopathies: a new wave neurodegenerative diseases. Neurodegeneration. 2921;92:86–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fang Y-S, Tsai K-J, Chang Y-J, et al. Full-length TDP-43 forms toxic amyloid oligomers that are present in frontotemporal lobar dementia-TDP patients. Nature Communications. 2014;doi:doi: 10.1038/ncomms5824 [DOI] [PubMed] [Google Scholar]
- 53.Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. [DOI] [PubMed] [Google Scholar]
- 54.Kawakami I, Arai T, Hasegawa M. The basis of clinicopathological heterogeneity in TDP-43 proteinopathy. Acta Neuropathologica. 2019;138:751–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Majumder P, Chen Y-T, Bose JK, et al. TDP-43 regulates the mammalian spinogenesis through translational repression of Rac1. Acta Neuropathologica. 2012;124:231–245. [DOI] [PubMed] [Google Scholar]
- 56.Lee EB, Porta S, Michael Baer G, et al. Expansion of the classification of FTLD-TDP: distinct pathology associated with rapidly progressive frontotemporal degeneration. Acta Neuropathologica. 2017;134:65–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Porta S, Xu Y, Lehr T, et al. Distinct brain-derived TDP-43 strains from FTLD-TDP subtypes induce diverse morphological TDP-43 aggregates and spreading patterns in vitro and in vivo. Neuropath Appl Neurobiol. 2021;47:1033–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gómez-Isla T, Hollister R, West H, et al. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann Neurol. 1997;41:17–24. [DOI] [PubMed] [Google Scholar]
- 59.Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology. 1992;42:631–639. [DOI] [PubMed] [Google Scholar]
- 60.Kawles A, Nishihira Y, Feldman A, et al. Cortical & subcortical pathologic burden and neuronal loss in an autopsy series of FTLD-TDP type C. Brain. 2022;145:1069–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee EB, Lee VM-Y, Trojanowski JQ. Gains or losses: molecular mechanisms of TDP-43-mediated neurodegeneration. Nature Reviews Neuroscience. 2012;13:38–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Laferriére F, Maniecka Z, Pérez-Berlanga M, et al. TDP-43 extracted from frontotemporal lobar degeneration subject brains displays distinct aggregate assemblies and neurotoxic effects reflecting disease progression rates. Nature Neuroscience. 2019;22:65–77. [DOI] [PubMed] [Google Scholar]
- 63.Arseni D, Hasegawa M, Murzin A, et al. Structure of pathological TDP-43 filaments from ALS with FTLD. Nature. 2022;(601):139–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Davidson YS, Robinson AC, Flood L, et al. Heterogeneous ribonuclear protein E2 (hnRNP E2) is associated with TDP-43-immunoreactive neurites in Semantic Dementia but not with other TDP-43 pathologic subtypes of Frontotemporal Lobar Degeneration. Acta Neuropathologica Communications. 2017;5:1–12. doi: 10.1186/s40478-017-0454-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Neumann M, Frick P, Paron F, Kosten J, Buratti E, Mackenzie I. Antibody against TDP-43 phosphorylated at serine 375 suggets conformational differences of TDP-43 aggregates among FTLD-TDP subtypes. Acta Neuropathologica. 2020;140:645–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shi Y, Zhang W, Yang Y, et al. Structure-based classification of tauopathies. bioRxiv. 2021;doi: 10.1101/2021.05.28.446130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Brettschneider J, Del Tredici K, Irwin DJ, et al. Sequential distribution of pTDP-43 pathology in behavioral variant frontotemporal dementia (bvFTD). Acta Neuropathol. 2014;127:423–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Conel JL. The Postnatal Development of The Human Cerebral Cortex. vol 1-7. Harvard University Press; 1955. [Google Scholar]
- 69.Lund JS, Lewis DA. Local circuit neurons of developing and mature macaque prefrontal cortex: Golgi and immunohistochemical characteristics. J Comp Neurol. 1993;328:282–312. [DOI] [PubMed] [Google Scholar]
- 70.Wang I-F, Wu L-S, Chang H-U, Chen JC-K. TDP-43, the signature protein of FTLD-U, is a neuronal activity-responsive factor. J Neurochem. 2008;105:797–806. [DOI] [PubMed] [Google Scholar]
- 71.Aru J, Suzuki M, Larkum ME. Cellular mechanisms of conscious processing. TICS. 2020;24:814–825. [DOI] [PubMed] [Google Scholar]
- 72.Probst A, Tolnay M, Langui D, Goedert M, Spillantini MG. Pick’s disease: hyperphosphorylated tau protein segregates to the somatoaxonal compartment. Acta Neuropathol. 1996;92:588–596. [DOI] [PubMed] [Google Scholar]
- 73.Bonham LW, Steele NZR, Karch CM, et al. Genetic variation across RNA metabolism and cell death gene networks is implicated in the semantic variant of primary progressive aphasia. Scientific Reports. 2019;9:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Moi MO, Miedema SSM, Melhem S, et al. Proteomics of the dentate gyrus reveals semantic dementia specific molecular pathology. Acta Neuropathologica Communications. 2022;10:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Is O, Wang X, Patel T, et al. Pathology-related alterations in gene expression in Alzheimer’s disease: Single cell resolution. Alzheimer’s & Dementia. 2021;doi: 10.1002/alz.053804 [DOI] [Google Scholar]
- 76.Onyike CU, Diehl-Schmid J. The epidemiology of frontotemporal dementia. International Review of Psychiatry. 2013;25:130–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Coyle-Gilchrist TS, Dick KM, Patterson K, et al. Prevalence, characteristics, and survival of frontotemporal lobar degeneration syndromes. Neurology. 2016;86:1736–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]