

Abstract
Although reactive aldehyde species (RASP) are associated with the pathogenesis of many major diseases, there are currently no clinically approved treatments for RASP overload. Conventional aldehyde detox agents are stoichiometric reactants that get consumed upon reacting with their biological targets, which limits their therapeutic efficiency. To achieve longer-lasting detoxification effects, small-molecule intracellular metal catalysts (SIMCats) were used to protect cells by converting RASP into non-toxic alcohols. It was shown that SIMCats were significantly more effective in lowering cell death from the treatment with 4-hydroxynon-2-enal than aldehyde scavengers over a 72 h period. Studies revealed that SIMCats reduced the aldehyde accumulation in cells exposed to the known RASP inducer arsenic trioxide. This work demonstrates that SIMCats offer unique benefits over stochiometric agents, potentially providing new ways to combat diseases with greater selectivity and efficiency than existing approaches.
Keywords: bioorganometallic, catalytic drug, detoxification, metallotherapeutics, reactive aldehyde species
Graphical Abstract
Reactive aldehyde species (RASP) have been implicated in numerous diseases due to their ability to alkylate or crosslink biomolecules. In this work, it was demonstrated that iridium small-molecule intracellular metal catalysts protect cells from RASP-induced cell death with greater efficiency than conventional aldehyde scavengers. A catalyst-based detoxification strategy could offer unique benefits over those relying on stoichiometric reactants.
Introduction
Reactive aldehydes species (RASP) are associated with the pathogenesis of many oxidative-stress related diseases (Scheme 1A).[1–7] RASP are prevalent in the human body and are derived from lipid peroxidation, carbohydrate oxidation, lysine deamination, drug metabolism, and absorption from external sources (e.g., air pollution, smoking, food, drink, etc.). α,β-Unsaturated aldehydes are particularly cytotoxic because they can undergo 1,4-addition with thiols and 1,2-addition with amines in living systems.[6, 8] Their reactions with biomolecules lead to non-specific crosslinking and disruption of cellular function. One of the most well-studied RASP, 4-hydroxynon-2-enal (4-HNE)[9] is found at elevated levels in amyloid β plaques[10–11] and nigral neurons[12] in patients with neurodegenerative disorders. There is growing evidence that 4-HNE accumulate with age and likely contribute to the progression of pulmonary disease,[13] cardiovascular disease,[14] fibrosis,[15] and cancer.[1, 9]
Scheme 1.
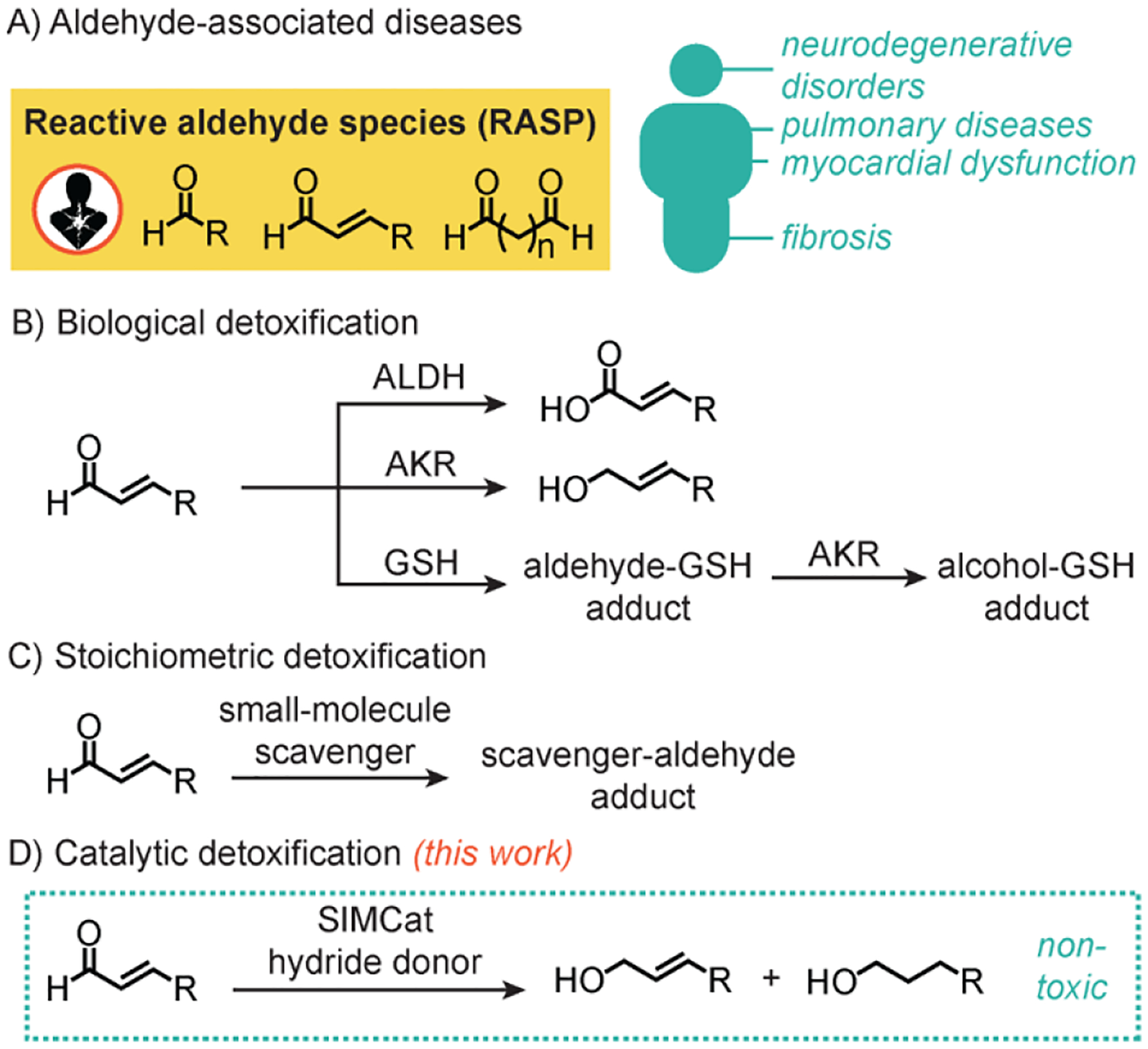
Diseases associated with aldehyde accumulation (A) and various strategies used for detoxification, including biological (B), stoichiometric (C), and catalytic (D) methods. ALDH = aldehyde dehydrogenase; AKR = aldo-keto reductase; GSH = glutathione; SIMCat = small-molecule intracellular metal catalyst.
To prevent RASP accumulation, enzymes such as aldehyde dehydrogenase (ALDH)[16–17] or aldo-keto reductase (AKR)[18–19] convert aldehydes to non-toxic carboxylic acids or alcohols, respectively (Scheme 1B). Alternatively, glutathione (GSH) [8, 20–21] can form adducts with RASP that get further reduced by reductases. Unfortunately, these natural defense systems are impaired in patients with oxidative-stress related illnesses.[9, 22–23]
There are currently no clinically approved treatments for aldehyde overload. Candidates being considered for RASP remediation are small-molecule scavengers that react with aldehydes via nucleophilic addition (Scheme 1C).[24–27] Although a few of these compounds have shown positive effects in clinical trials,[24, 27–29] detox agents capable of achieving catalytic turnover would likely exhibit greater therapeutic efficiency than those that react stoichiometrically.[30–31]
In this work, we demonstrate that iridium small-molecule intracellular metal catalysts (SIMCats),[31–38] which are molecular inorganic complexes that promote chemical reactions in cells, are promising as RASP detox agents (Scheme 1D). These SIMCats operate via a transfer hydrogenation mechanism, which involve transferring H– from a hydride donor to a hydride acceptor,[39–41] and provide significantly longer-lasting cellular protection from aldehydes than conventional small-molecule scavengers. Although other transfer hydrogenation SIMCats have been reported,[39, 42–44] they seek to kill cells rather than to rescue cells. Given the strong link between RASP and numerous diseases, our approach could be broadly useful in the development of more selective disease treatments.
Results and Discussion
Detoxification Strategy and Solution Studies.
Because Ir SIMCats can mimic the function of reductase enzymes,[18] they are ideal candidates for RASP detoxification. We anticipate that cells treated with Ir SIMCats would be capable of promoting transfer hydrogenation between cytotoxic aldehydes and endogenous hydride donors, such as reduced nicotinamide adenine dinucleotide (NADH), to non-toxic alcohols.[40] As long as the SIMCats do not get deactivated inside cells, they could potentially achieve catalytic turnover.
For our studies, we prepared [Cp*Ir(N-phenyl-2-pyridinecarboxamidate)Cl] (Ir1, where Cp* = pentamethylcyclopentadienyl anion)[45] and an electron-rich variant [Cp*Ir(N-(4-methylester)phenyl-2-(4-pyrrolidinyl) pyridinecarboxamidate)Cl] (Ir2) as transfer hydrogenation catalysts (Figure 1A). Ir1 was reported previously to be active inside living cells.[46–47] Although RASP comprise a wide variety of aldehydes, we focused on 4-HNE and 4-oxonon-2-enal (4-ONE) due to their well-established cytotoxicity.[9, 48–49] To determine the Ir SIMcat’s catalytic efficiency, we first tested them in solution by combining 4-HNE, sodium formate (HCOONa),[50] and Ir complex (1 mol% relative to aldehyde) in DMSO/phosphate-buffered saline (PBS) (5:95) and allowing the reactions to stir at 37 °C for 15 h (Figure 1B). Although sodium formate was used as the reductant in these experiments, both HCOONa and NADH were shown to be efficient hydride donors (Table S1, entry 3). Our results revealed that Ir1 afforded non-2-ene-1,4-diol (I) and nonane-1,4-diol (II) in 85 and 7% yield, respectively (Table S1, entry 2). In contrast, Ir2 provided trace amounts of I and 94% yield of II (Figure 1B). Based on our mechanistic studies, the C=O bond in 4-HNE is reduced first prior to reduction of the C=C bond via direct hydrogenation (Figure S4–S6 and Scheme S3). These observations indicate that Ir2 is significantly more active than Ir1 since the former afforded II in substantially greater yield. Our previous work revealed that more electron-rich Ir catalysts dehydrognate formate ions and transfer hydrides faster than their electron-poor counterparts.[51] Under biologically relevant conditions, these Ir SIMCats reduce aldehydes preferentially over ketones.[40]
Figure 1.
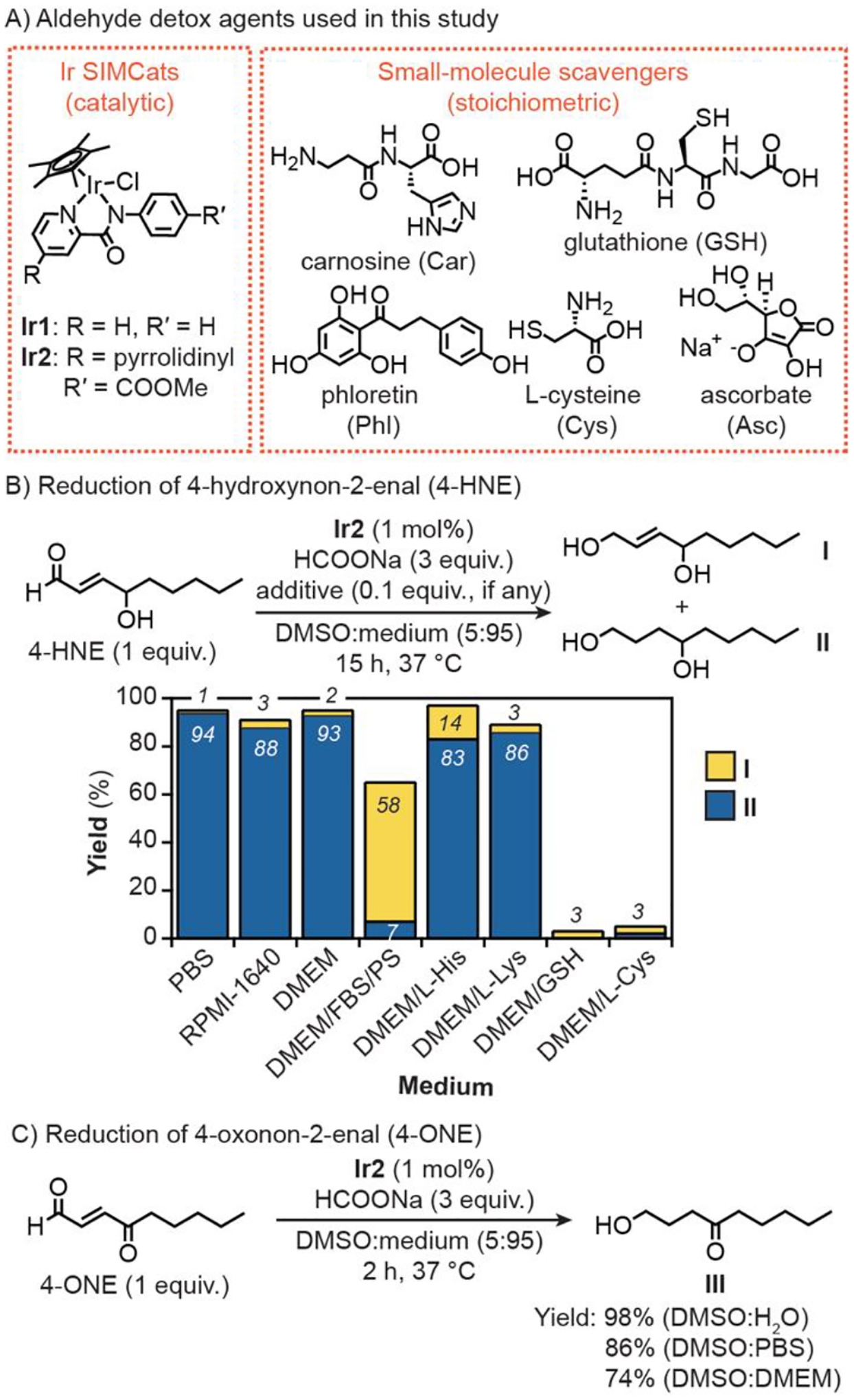
Structures of aldehyde detox agents used in this work (A). Transfer hydrogenation between 4-HNE (50 μmol, part B) or 4-ONE (50 μmol, part C) and sodium formate (150 μmol) catalyzed by Ir2 (0.5 μmol) in various aqueous media (3 mL). PBS = phosphate-buffered saline, RPMI = Roswell Park Memorial Institute, DMEM = Dulbecco’s Modified Eagle Medium, FBS = fetal bovine serum, PS = penicillin-streptomycin, His = L-histidine, Lys = L-lysine.
The catalytic activity of Ir2 was further evaluated in various aqueous mixtures with and without the presence of biological additives. (Figure 1B and Table S1). Similar to reactions in DMSO/PBS, those performed in DMSO and either Roswell Park Memorial Institute-1640 medium (RPMI) or Dulbecco’s Modified Eagle Medium (DMEM) furnished ≥ 88% yield of II. When fetal bovine serum (FBS, 10%) and penicillin streptomycin (PS, 1%) were added to DMEM, which is used as a typical cell growth medium, the activity of Ir2 was lowered, giving more I (58% yield) than II (7% yield). Finally, Ir2 tolerated histidine (His) and lysine (Lys) but was significantly inhibited in the presence of GSH and cysteine (Cys). These results are consistent with other studies showing that half-sandwich metal complexes are susceptible to coordination inhibition by thiols.[40, 52] Thus, it is possible that inside the cell not all Ir SIMCats exist in a catalytically active state. Additionally, our solution studies suggest that the overall reaction rates will be dependent on the intracellular concentrations of Ir SIMCats and aldehydes (Table S2).
Reduction of 4-ONE using Ir2 and HCOONa was performed at 37 °C for 2 h (Figure 1C). We found that greater than 70% yield of the doubly reduced species 1-hydroxynon-4-one (III) was obtained in DMSO mixtures containing either H2O, PBS, or DMEM (5:95). Surprisingly, the singly reduced product 1-oxonon-2-en-4-one was not detected, indicating that hydrogenation of the C=C bond in 4-ONE is highly favorable.
Cell Studies with External RASP.
Next, we conducted biological studies in both mouse fibroblast NIH-3T3 and human neuroblastoma-derived SH-SY5Y cells. These cell lines were selected because the former is robust and easy to propagate and the latter is commonly used in in vitro models of neuronal function,[53–55] which could be useful for studying RASP-associated neurodegeneration. To measure iridium uptake, cells were incubated with either Ir1 or Ir2 (10 μM in NIH-3T3 and 5 μM in SH-SY5Y) for 24 h, washed with fresh DMEM, and then digested in concentrated nitric acid for analysis by inductively coupled plasma-mass spectrometry (ICP-MS). Because iridium is a non-native metal, its presence in cells must be derived from the Ir SIMCats. Our ICP-MS data revealed that there was at least 1.7× more Ir2 than Ir1 in cells under identical treatment conditions (Figure 2A). For example, the Ir concentration in NIH-3T3 cells exposed to 10 μM of Ir1 and Ir2 for 24 h was found to be 198 and 341 ng/106 cells, respectively. The higher cellular uptake of Ir2 with respect to Ir1 is likely due to its greater lipophilicity, which was confirmed by its octanol/water partition coefficient (Log P = 0.7 for Ir1 vs. 1.1 for Ir2). Changing the SIMCat treatment concentration (Table S4) or varying their incubation time (Tables S3–S5 and Figure S8) also impacted the cellular uptake efficiency.
Figure 2.
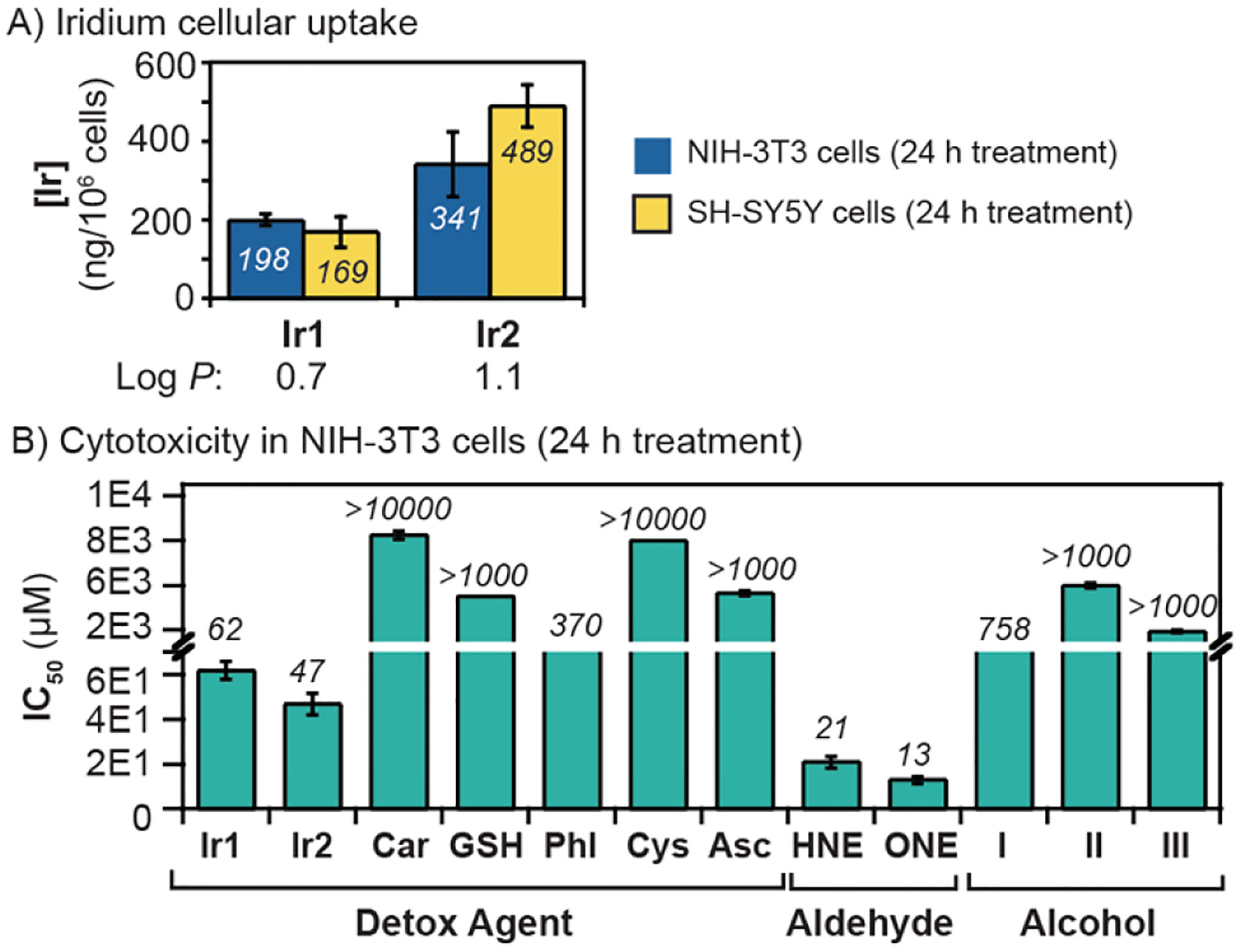
A) Iridium concentration in cells treated with the Ir complexes (10 μM in NIH-3T3 and 5 μM in SH-SY5Y) for 24 h. B) IC50 values (24 h treatment) obtained for various detox agents, aldehydes, and alcohols in NIH-3T3 cells.
Before testing our RASP detoxification strategy in live cells, we assessed the half maximal inhibition concentrations (IC50) of the various detox agents, aldehydes, and expected alcohol products being investigated. Cells were incubated with various concentrations of each compound for 24 h and then commercial Sulforhodamine B assays were used to quantify cell viability. In NIH-3T3 cells (Figure 2B), Ir1 and Ir2 were determined to have IC50 values of 62 and 47 μM, respectively, which suggests that Ir1 is less cytotoxic than Ir2. The addition of sodium formate to cells containing either Ir1 or Ir2 had modest effects on their IC50 values (Figure S12). In the presence of externally added GSH (100 μM), the IC50 value of Ir2 dropped from 47 to 10 μM (Figure S9). We hypothesize that exogenous GSH coordinates to Ir2 so that a new iridium-glutathione species is formed and transported inside the cell. Interactions of GSH with half-sandwich complexes have been studied previously.[52]
For comparison, we also evaluated the cytotoxicity of several conventional aldehyde scavengers, including carnosine (Car),[27] phloretin (Phl),[25] cysteine (Cys),[6] and sodium ascorbate (Asc).[56–58] These detox agents have low cytotoxicity, with IC50 values exceeding 350 μM. Although our Ir SIMCats are more cytotoxic than the aldehyde scavengers, they are catalytic agents and thus, do not likely require high concentrations in cells to achieve their biological effects.
As expected, the aldehydes 4-HNE and 4-ONE caused significant cell death, with IC50 values after 24 h treatment of 21 and 13 μM in NIH-3T3, respectively (Figure 2B). In contrast, their corresponding alcohols (e.g., I, II, and III) are virtually non-toxic (IC50 = > 750 μM). These results suggest that if our Ir SIMCats could reduce RASP fast enough on the biological timescale, we may observe enhanced cellular protection.
Aldehyde detoxification studies were first carried out in NIH-3T3 using externally added RASP. We performed experiments using 20 μM of 4-HNE, which is the concentration typically observed in the plasma of Alzheimer’s disease patients (vs. ~8 μM in healthy individuals).[59] When cells were incubated with 4-HNE for 24 h, their viability was lowered to 48% relative to that of the untreated control (Figure S15). To evaluate whether our Ir SIMCats could protect against RASP-induced cell death, cells were pre-treated with 10 μM of an Ir complex for 3 h prior to being exposed to 4-HNE. Our results showed that cells containing either Ir1 or Ir2 exhibited about 1.4- and 1.6-fold improvement in cell viability, relative to the positive control (i.e., 4-HNE only treatment group) (Figure 3A). The maximum protection achievable is 2.1-fold based on the untreated cell group. Cells that were treated with only Ir1 or Ir2 (10 μM), without 4-HNE, showed no significant cell death. Furthermore, addition of alcohol I or II (100 μM) with Ir2 also did not negatively impact cell viability (Figure S10). Although there are natural hydride sources for our Ir SIMCats inside cells, we wondered whether supplementing cells with additional hydride donors could lead to greater RASP defense. We found that the presence of HCOONa (1 mM) increased the cell viability by an additional ~10% for Ir2 but a statistically insignificant amount for Ir1 (Figure S15), presumably due to the greater activity of the former relative to the latter. Our results suggest that treating NIH-3T3 cells with 10 μM of the Ir SIMCat is the optimal amount since higher concentrations (e.g., 20 μM) resulted in diminished cell protection (Figure S15).
Figure 3.
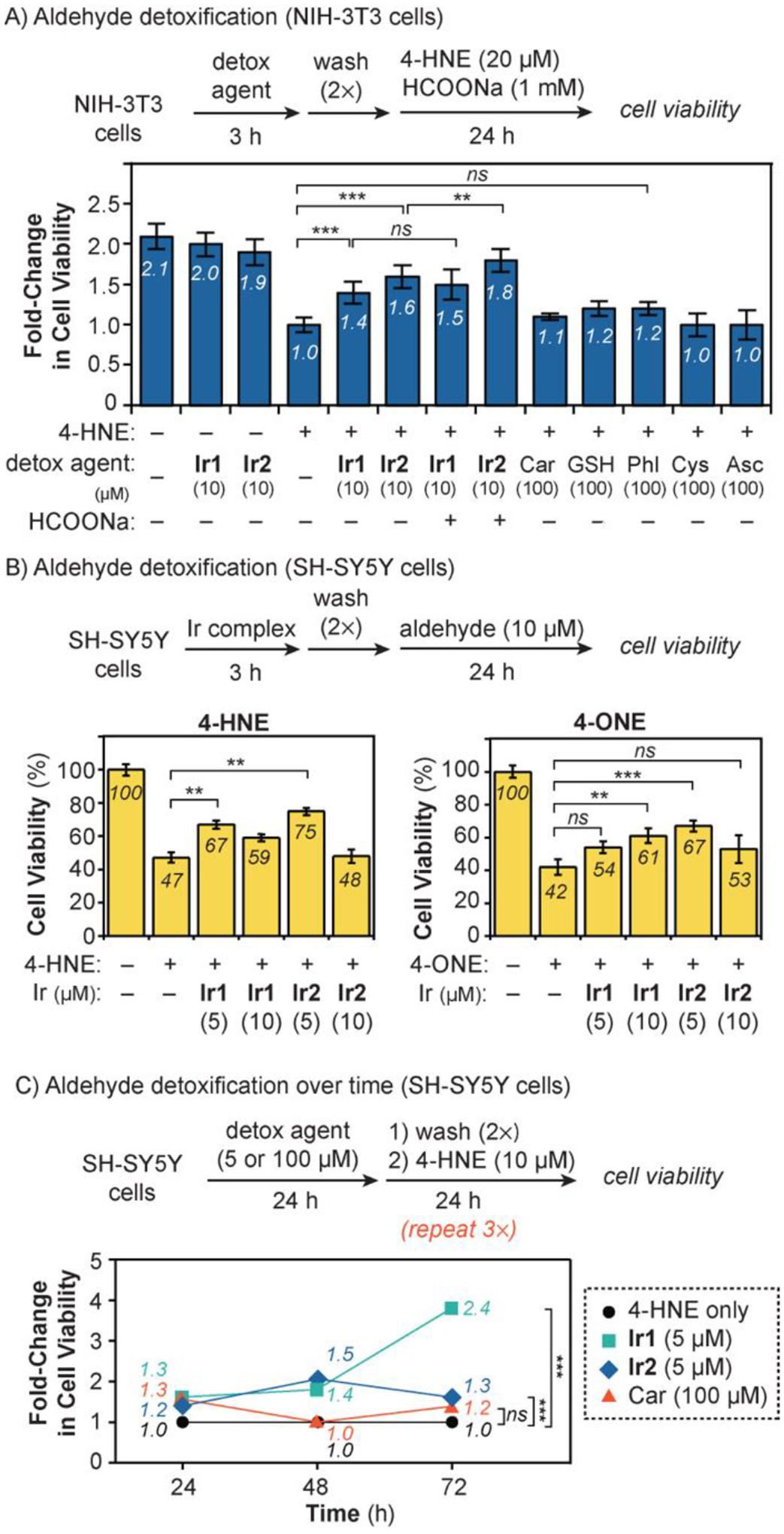
Aldehyde detoxification studies in A) NIH-3T3 with 4-HNE; B) SH-SY5Y with 4-HNE and 4-ONE; and C) SH-SY5Y with consecutive treatments of 4-HNE. The fold change in cell viability was calculated as the ratio of the percentage of viable cells in treated wells divided by the percentage of viable cells in control wells containing only 4-HNE. The data were analyzed using one-way ANOVA. The p-values are indicated as follows: ns = p > 0.05, * = p < 0.05, ** = p < 0.01; and *** = p < 0.001. The error bars in part C were omitted for clarity but are shown in Figures S21 and S23.
For comparison, we performed 4-HNE detoxification studies using conventional small-molecule scavengers in NIH-3T3 cells (Figure 3A). An analogous procedure was used as described in the previous section, except that cells were first combined with 100 μM of Car, GSH, Phl, Cys, or Asc before incubating with 20 μM of 4-HNE. As shown in Figure 3A, cells treated with Car, GSH, and Phl showed improvements in cell viability by 1.1-, 1.2-, and 1.2-fold, respectively, relative to the positive control. However, the use of Cys and Asc did not provide any protection against 4-HNE. Both Ir1 and Ir2 (up to 1.8-fold increase in cell viability) were more effective detox agents than the small-molecule scavengers (up to 1.2-fold increase in cell viability). These results are notable because the amount of Ir SIMCat used to achieve these effects is only one-tenth that of the small-molecule scavengers (i.e., 10 vs. 100 μM, respectively). In the SIMCat experiments, 4-HNE was presumably reduced to I or II but further work is needed to extract and analyze these products from the cells for confirmation. Interestingly, when cells were exposed to both Ir2 (10 μM) and GSH (100 μM), protection against 4-HNE was diminished (Figure S17), most likely due to deactivation of Ir2 by the exogenous thiol.
Because SH-SY5Y cells are highly sensitive to exogeneous species, lower concentrations of detox agents and aldehydes were needed to induce biological responses compared to in NIH-3T3. In these experiments, the viability of SH-SY5Y cells exposed to 10 μM of RASP was measured in the presence and absence of Ir SIMCats (Figure 3B). Cells treated with only 4-HNE or 4-ONE for 24 h exhibited viability of 47 and 42%, respectively, relative to the untreated control. Pre-treatment with Ir1 or Ir2 clearly reduced the extent of cell death caused by RASP damage. For example, 5 μM of Ir2 gave the highest protection of cells against both 4-HNE (75% viability) and 4-ONE (67% viability). In most cases, using more Ir SIMCats (e.g., 10 μM instead of 5 μM) did not result in better outcomes. Although having greater amounts of Ir1 or Ir2 should increase their overall detoxification rates, the iridium complexes could have negative biological effects at higher loading.
Given that many RASP-associated diseases progress slowly,[60–61] we next evaluated how effective our detox agents are as a function of time (Figure 3C). We incubated SH-SY5Y cells with a detox agent (5 μM of Ir SIMCats or 100 μM of small-molecule scavengers) for 24 h, washed with DMEM, and treated with 10 μM of 4-HNE for 24 h. The aldehyde treatment procedure was repeated a total of 3 times and the cell viability was measured after each 24 h period. This protocol ensures a constant level of RASP in cells so that the detox agents’ therapeutic efficiency could be assessed. As shown in Figures 3C, cells treated with Ir1 or Ir2 exhibited protection against 4-HNE continuously up to 72 h. For example, Ir1 increased cell viability by 1.3-, 1.4-, and 2.4-fold relative to the positive control after 24, 48, and 72 h, respectively. Ir2 also showed similar trends, except that after 72 h, it provided less protection (1.3-fold) than Ir1 (2.4-fold). These results revealed that Ir2 is a less effective detox agent than Ir1 despite having greater cell uptake (Figure 2A) and higher intrinsic activity (Table S1). However, because under the same treatment concentration, Ir1 causes less cell death than Ir2 (Figure S11B), enhanced therapeutic effects were observed using the former.
Another surprising result from our time-dependent detoxification study is that conventional RASP scavengers exhibited limited therapeutic lifetimes (Figure S23). For example, although Car increased cell protection by 1.3-fold after 24 h relative to the 4-HNE control, it was ineffective after 48 and 72 h (Figure 3C). Perhaps because Car is a stoichiometric reactant, it is consumed over time and thus, is no longer available to react with 4-HNE after 24 h. In contrast, Ir SIMCats have the potential to be catalytic, so each Ir1 or Ir2 species could continuously detoxify many molecules of 4-HNE over an extended period. However, further studies are needed to establish whether our SIMCats achieve catalytic turnover in cells.
Cell Studies with Endogenous RASP.
We next focused on testing our detoxification strategy against endogenously generated RASP. Although many compounds have been reported to increase aldehyde levels in cells, we selected to use the anti-cancer drug arsenic trioxide (As2O3) due to its clinical relevance.[62–64] Based on literature reports, As2O3 causes oxidative stress in biological systems, which in turn initiates lipid peroxidation to produce aldehydes.[65–67] SH-SY5Y cells were grown in the presence of 3 μM of As2O3 for 4 d to maximize intracellular RASP formation. The cells were then treated with a detox agent for 3 d and their biological changes were evaluated (Figure 4). We observed that cells exposed to only Car (100 μM) or Ir2 (2 μM) had nearly 100% viability, which was expected given that the concentrations of detox agents used were well below their IC50 values. As a positive control, cells introduced to only As2O3 exhibited viability of 24%. When either Car (100 μM) or Ir2 (2 μM) was added to the As2O3-treated cells, their viability increased to 35 and 39%, respectively. Supplementing cells with HCOONa (1 mM) in the presence of Ir2 and As2O3, increased cell viability further to 43%. These results demonstrate that the detox agents are moderately effective in minimizing the cell killing effects of As2O3.
Figure 4.
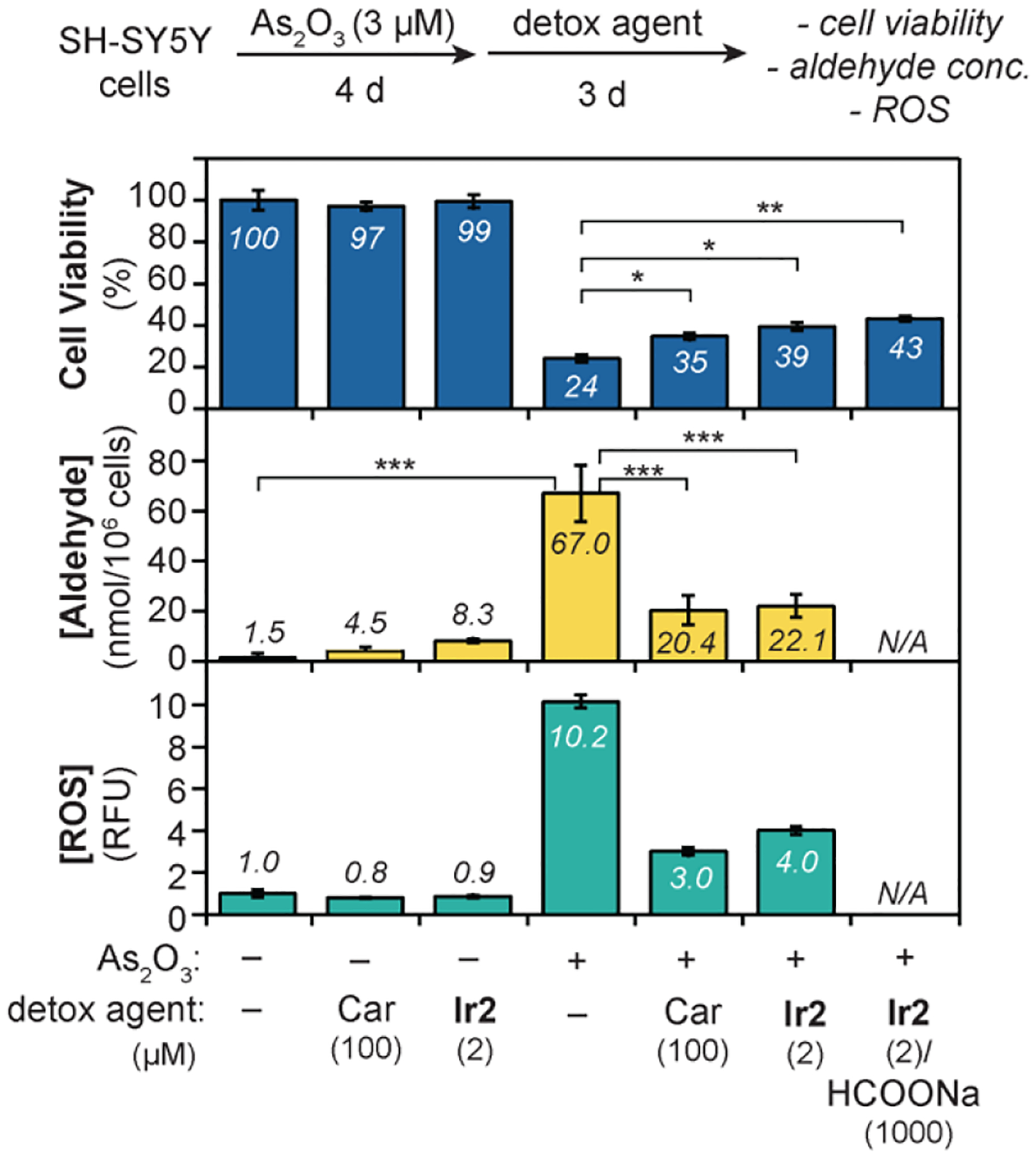
Effect of detox agents on the cell viability, aldehyde concentration, and reactive oxygen species after treatment with As2O3. The data were analyzed using one-way ANOVA. The p-values are indicated as follows: ns = p > 0.05, * = p < 0.05, ** = p < 0.01; and *** = p < 0.001. N/A = not available.
To determine the impact of detox agents on endogenous RASP levels, a commercial colorimetric kit was used to measure aldehyde concentrations in the cell samples above. Because the assay is non-specific, the aldehydes measured represent all RASP, which could include 4-HNE, 4-ONE, and others. We found that SH-SY5Y cells exposed to As2O3 (3 μM) for 4 d produced 67 nmol aldehydes/106 cells (Figure 4, middle). When As2O3-treated cells were exposed to either Car or Ir2, lower amounts of RASP were produced compared to that in the positive control, with concentrations of 20 and 22 nmol aldehydes/106 cells, respectively. In comparison, cells lacking As2O3 have <10 nmol aldehydes/106 cells.
Finally, the reactive oxygen species (ROS) were also measured (Figure 4, bottom). We observed that the As2O3-only treatment group had at least 10-fold greater amounts of ROS than untreated cells or cells containing only detox agents. In the presence of Car or Ir2, the As2O3-exposed cells showed significantly lower levels of ROS. Although further investigations are needed to understand these biological effects, we found strong correlations between cell viability, aldehyde concentration, and ROS levels. Our results are consistent with a mechanism in which As2O3 triggers formation of ROS and lipid peroxidation, leading to an increase in aldehydes.[66] Although the exact role of the detox agents in suppressing RASP and ROS in cells is unclear at this time (e.g., whether they participate directly or indirectly), their cell protecting abilities were clearly demonstrated.
Conclusions
In summary, Ir SIMCats are promising as RASP detox agents in living systems. It was shown that Ir1 and Ir2 were equally or more effective than conventional aldehyde scavengers in minimizing cell death from RASP damage. Our Ir SIMCats are capable of maintaining therapeutic efficacy for at least 72 h, which is substantially longer than stoichiometric aldehyde traps that do not provide any cellular protection after 24 h. Based on studies using As2O3 to induce endogenous RASP formation, our detox agents prevent the accumulation of both aldehydes and reactive oxygen species. Because RASP are implicated in numerous pathological processes, this work offers new defense strategies to combat oxidative stress-associated diseases. In particular, the use of catalytic drugs could have unique benefits over stoichiometric drugs, challenging the one drug molecule per disease target paradigm commonly used in drug discovery.
Experimental Section
General.
All chemicals and reagents were purchased from commercial sources and were used without further purification unless otherwise noted. All air- and water-sensitive manipulations were performed using standard Schlenk techniques or under a nitrogen atmosphere using a glovebox. Anhydrous solvents were obtained from an Innovative Technology solvent drying system saturated with Argon. Compounds 3,4-epoxynonanol (3),[68] [Cp*IrCl2]2,[69] and [Cp*Ir(N-phenyl-2-pyridinecarboxamidate)Cl] (Ir1)[51] were prepared using reported procedures. NIH-3T3 and SH-SY5Y cell lines were obtained commercially from American Type Culture Collection (ATCC).
Physical Methods.
NMR spectra were acquired using JEOL spectrometers (ECA-400, 500, and 600) at room temperature and referenced using residual solvent peaks. All 13C NMR spectra were proton decoupled. Gas chromatography-mass spectrometry was performed using an Agilent 7890 GC/5977A MSD instrument equipped with an HP-5MS capillary column. A Tecan Infinite M200 Pro plate reader was used for absorbance and fluorescence intensity measurements. A Varian 810 instrument was used to acquire inductively coupled plasma-mass spectrometry (ICP-MS) analyses. All biological cell images were obtained using an Olympus IX83 microscope equipped with a 20x air objective.
Synthesis and Characterization
Preparation of Complex Ir2.
In a 50 mL Schlenk flask, 20 mL of ethanol was purged with nitrogen for about 10 min. [Cp*IrCl2]2 (81.2 mg, 0.102 mmol, 1.0 equiv.) and L2 (see the Supporting Information) (66.5 mg, 0.204 mmol, 2.0 equiv.) were added and stirred for 15 min at 80 °C. The reaction mixture was treated with ammonium hexafluorophosphate (84 mg, 0.50 mmol, 2.5 equiv.) and stirred for 40 h at 80 °C. Once the reaction was complete, the ethanol solvent was removed by rotary evaporation and then the solid residual was redissolved in 20 mL of dichloromethane and washed with water (3×20 mL). The organic phase was separated, dried over sodium sulfate, and then evaporated to dryness. The crude product was purified by silica gel column chromatography (100% ethyl acetate, then 10% methanol in ethyl acetate) to obtain a bright yellow solid (57 mg, 83% yield). 1H NMR (CDCl3, 400 MHz): δ 8.04 (d, J = 6.5 Hz, 1H), 7.98 (d, J = 8.3 Hz, 2H), 7.78 (d, J = 8.4 Hz, 2H), 7.18 (d, J = 2.6 Hz, 1H), 6.47 (dd, J = 6.5, 2.7 Hz, 1H), 3.90 (s, 3H), 3.44 (s, 4H), 2.06 (s, 4H), 1.38 (s, 15H) ppm. 13C{1H}[21] NMR (CDCl3, 151 MHz): δ 169.52, 167.54, 153.89, 153.78, 152.68, 148.64, 129.76, 127.28, 125.20, 110.06, 109.26, 85.85, 51.97, 47.72, 25.48, 8.66 ppm. IR: 2921, 1709, 1583, 1511, 1353, 1266, 1165, 1101, 1030, 955, 859, 772, 705 and 510 cm−1. HRMS-ESI(+): calculated for C28H34ClIrN3O3 m/z = 688.1918 [M+H]+, found m/z = 688.1900.
Transfer Hydrogenation Studies in Solution.
4-HNE (7.8 mg, 50 μmol in DMSO, 1.0 equiv.), HCOONa (10.2 mg, 150 μmol, 3.0 equiv.) or NADH (114 mg, 150 μmol, 3.0 equiv.), and iridium catalyst (0.5 μmol in DMSO, 1 mol%.) were combined in 3.0 mL of DMSO:PBS (5:95) (or cell culture media) in a 20 mL glass vial. The vial was capped, and the reaction mixture was stirred at 37 °C for 15 h. At the end of the reaction, pentamethylbenzene was added as an internal standard. The mixture was combined with water, and the product was extracted into diethyl ether and then dried over anhydrous sodium sulfate. The organic products were then analyzed by GC-MS. Quantification of the reduced products was performed using standard calibration curves, which were constructed from solutions containing the authentic samples and pentamethylbenzene as an internal standard. A similar procedure was used for studies of 4-ONE. The reaction mixture was combined with water, and the product was extracted into diethyl ether and then dried over anhydrous sodium sulfate. The products were analyzed and quantified by 1H NMR spectroscopy using 1,3,5-trimethoxybenzene as an internal standard.
ICP-MS Analysis.
Cells were grown in Corning (430167) tissue culture plates at 37°C under a 5% CO2 atmosphere. When ~70% confluence was achieved, the DMEM solution was removed by aspiration and replaced with new medium containing the test compound. At the end of the treatment period, the cells were washed twice with fresh DMEM, detached by treatment with trypsin, and counted using a BIO-RAD TC10 automated cell counter. The trypsinized cells samples were then centrifuged and the supernatant was removed. The cell pellet was washed with fresh DMEM and phosphate-buffered saline by vortexing, centrifuging, and removing the supernatant. The cell pellets were digested using 0.5 mL of 70% metal-free distilled HNO3 at RT overnight. HPLC-grade water (6.5 mL) was added to each sample to obtain a 5% HNO3 solution. The cloudy solutions were then centrifuged to obtain clear samples for ICP-MS analysis. The final concentration of iridium was calculated using the following equation: [Ir] (ng/106 cells) = (total Ir)/(total cells), and total Ir (ng) = [Ir] (in ppb) × 103 × 0.007 (L).
Cytotoxicity Sulforhodamine B (SRB) Assays.
Cells were seeded in a 96-well plate (Corning 3595) and grown at 37 °C in an incubator with a humidified atmosphere containing 5% CO2 to allow them to adhere to the bottom of the wells (~15 h for NIH-3T3 and ~48 h for SH-SY5Y). Stock solutions of the test compounds were prepared in DMSO or DMEM, then diluted in cell culture media (DMEM:F12 (1:1) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin 100X solution) to make a series of desired concentrations. The cell culture medium was then removed and replaced with fresh cell culture media containing the test compounds at different concentrations. The cells were then incubated for a desired amount of time. The solutions were removed by aspiration, and the cells were washed with fresh DMEM before 100 μL of cell culture medium (with no FBS) was added to each well, followed by 50 μL of a fixative reagent (Cytoscan™ SRB Cytotoxicity Assay, G-Biosciences, catalog # 786–213). The 96-well plate was kept at 4 °C for 1 h, then the cells were washed 3 times with distilled water before drying for 2–3 h at 37 °C. A 100 μL solution containing sulforhodamine B (SRB) was then added to each well and the 96-well plate was kept in the dark at RT for 30 min. The cells were then rinsed 4 times with a 1x dye wash solution before drying for 2–3 h at 37 °C. A 200 μL solution of SRB solubilization buffer was added to each well, and mixed by pipetting the mixture up and down to dissolve the dye completely. The absorbance of the 96-well plate was then measured at 510 nm and 565 nm on a Tecan Infinite M200 Pro microplate reader. The cell viability was considered to be proportional to the absorbance measured. The average absorbance value of wells containing only solubilization buffer (background) was subtracted from that of wells containing treated and untreated cells. The percent cell viability was calculated using the following equation: (Aconc/Acontrol)x100%, where Aconc is the absorbance of wells containing cells treated with specific concentrations of the test compound and Acontrol is the absorbance of wells containing untreated cells. The IC50 values were calculated from the sigmoidal curve fit of these data at 50% cell viability.
Aldehyde Detoxification Studies.
Cells were seeded in a 96-well plate (Corning 3595) and incubated at 37 °C with 5% CO2 for ~15 h (with NIH-3T3) or ~48 h (with SH-SY5Y). The cell culture medium was removed by aspiration, and fresh medium containing the iridium complex or aldehyde scavenger was added. At the end of the treatment period, the solution was removed, and cells were washed twice with fresh medium. A solution containing the toxic aldehyde in cell culture medium was then added, and the cells were incubated at 37 °C with 5% CO2 for 24 h. The cell viability was then measured using an SRB assay as described above. The detoxification efficiency of the iridium complex or aldehyde scavenger was determined based on the number of cells alive relative to that of the control. The fold of cells protected was calculated as a ratio of the percentage of viable cells in treated wells divided by the percentage of viable cells in negative control wells (i.e., groups containing only toxic aldehyde but no detoxification agents). The number of cells in positive control wells containing no aldehydes or detoxification agents was considered as 100% cell viability.
Aldehyde Quantification Assays.
SH-SY5Y were cultured in tissue culture plates (Corning 430167) at 37°C under a 5% CO2 atm for at least 2 d to maximize cell adherence. When ~30% confluence was achieved, the solution was removed by aspiration and replaced with fresh cell culture medium containing toxic aldehydes or aldehyde-inducing agents. The cells were then incubated at 37 °C under a 5% CO2 atm for 4 d, washed with DMEM, and incubated with the iridium complexes or aldehyde scavengers for another 3 d. At the end of the treatment period, the medium was removed and the cells were washed twice with fresh DMEM, detached by treating with trypsin, and counted using a BIO-RAD TC10 automated cell counter. The trypsinized cells were then centrifuged and the supernatant was removed. The cell pellets were washed with fresh DMEM and PBS (vortexed, centrifuged, and then the supernatant was removed). The washed cell pellets were mixed with 0.2 mL of 20 mM Tris-HCl (pH 7.4) and ~75 mg of zirconium oxide beads (0.5 mm) and lysed at 4 °C in by Next Advance bullet blender tissue homogenizer BBX24B (speed 8, time 3). These samples were then centrifuged for 10 min at 2,000x g and 4 °C to obtain clear supernatants. In a clear bottom black 96-well plate (Corning 3603), 50 μL of the cell lysate (or blank control, or aldehyde standard) was added into each well, followed by addition of 50 μL of aldehyde blue indicator assay solution (Abcam’s fluorometric aldehyde quantification assay kit, product #ab138882). The plate was then protected from light and incubated at 25 °C for 30 minutes. Next, 25 μL of buffer was added into each well, and gently mixed by pipetting. The fluorescence intensity in each well was measured by exciting at 365 nm and measuring at 435 nm. A set of aldehyde standards (0–1000 μM, triplicated) was used to make the calibration curve. The relative fluorescence unit (RFU) was determined by dividing the integrated fluorescence intensity obtained from each well by the number of viable cells in each well. The fluorescence intensity is proportional to the intracellular concentration of aldehydes.
Reactive Oxygen Species (ROS) Assays.
Cells were seeded in a clear bottom, black 96-well plate (Corning 3603) and incubated in a 5% CO2 humidified incubator (~15 h for NIH-3T3 and ~48 hours for SH-SY5Y). The cells were then incubated with a toxic aldehyde or aldehyde-inducing agent for a certain amount of time. Next, the medium was removed, and the cells were washed with fresh DMEM. Additional cell culture medium containing an aldehyde scavenger or iridium complex was added, and the cells were incubated at 37 °C with 5% CO2. At the end of the treatment period, the medium was removed, and the cells were washed with DMEM. Additional DMEM (with 5% FBS) containing 2, 7-dichlorofluorescein diacetate (DCFDA, Sigma Aldrich, 10 μM) was then added, and the cells were incubated for 45 min at 37°C in the dark. The ROS in each well was determined by exciting the sample at 485 nm and measuring the fluorescence intensity at 535 nm using a Tecan Infinite M200 Pro microplate reader. Cells treated with a 20 μM tert-butyl hydrogen peroxide (TBHP) solution was used as a positive control for ROS. The relative fluorescence unit (RFU) was determined by dividing the integrated fluorescence intensity obtained from each well by the number of viable cells in each well. The fluorescence intensity is proportional to the intracellular concentration of ROS.
Supplementary Material
Acknowledgements
We thank the Welch Foundation (Grant No. E-1894 to L.H.D.) and the National Institutes of Health (Grant No. R01GM129276 to L.H.D.) for supporting this work. We also acknowledge Drs. Lu Yang and Siheng Li for advice on cell work, Dr. Yongjun Gao for assistance with ICP-MS measurements, and Profs. Chengzhi Cai, Tai-Yen Chen, and Michihisa Umetani for use of their instruments.
Footnotes
Supporting information for this article is given via a link at the end of the document.
Supporting Information
Additional references cited within the Supporting Information.[49, 68, 70–72]
References
- [1].O’Brien PJ, Siraki AG, Shangari N, Crit. Rev. Toxicol 2005, 35, 609–662. [DOI] [PubMed] [Google Scholar]
- [2].Fritz KS, Petersen DR, Free Radic. Biol. Med 2013, 59, 85–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Burke ND, Nixon B, Roman SD, Schjenken JE, Walters JL, Aitken RJ, Bromfield EG, Nat. Rev. Urol 2022, 19, 727–750. [DOI] [PubMed] [Google Scholar]
- [4].Ellis EM, Pharmacol. Ther 2007, 115, 13–24. [DOI] [PubMed] [Google Scholar]
- [5].Romero FJ, Bosch-Morell F, Romero MJ, Jareño EJ, Romero B, Marín N, Romá J, Environ. Health Perspect 1998, 106, 1229–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].LoPachin RM, Gavin T, Chem. Res. Toxicol 2014, 27, 1081–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Foret MK, Lincoln R, Do Carmo S, Cuello AC, Cosa G, Chem. Rev 2020, 120, 12757–12787. [DOI] [PubMed] [Google Scholar]
- [8].Witz G, Free Radic. Biol. Med 1989, 7, 333–349. [DOI] [PubMed] [Google Scholar]
- [9].Dalleau S, Baradat M, Guéraud F, Huc L, Cell Death Differ 2013, 20, 1615–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Butterfield DA, Swomley AM, Sultana R, Antioxid. Redox Sign 2012, 19, 823–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Renuka Sanotra M, Huang W-C, Silver S, Lin C-Y, Chang T-C, Nguyen DPQ, Lee C-K, Kao S-H, Chang-Cheng Shieh J, Lin Y-F, Clin. Biochem 2022, 101, 26–34. [DOI] [PubMed] [Google Scholar]
- [12].Yoritaka A, Hattori N, Uchida K, Tanaka M, Stadtman ER, Mizuno Y, P. Natl. Acad. Sci. USA 1996, 93, 2696–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Cho Y, Song M-K, Kim TS, Ryu J-C, Sci. Rep 2018, 8, 10497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Luo J, Hill BG, Gu Y, Cai J, Srivastava S, Bhatnagar A, Prabhu SD, Am. J. Physiol. –Heart C 2007, 293, H3673–H3684. [DOI] [PubMed] [Google Scholar]
- [15].López B, González A, Hermida N, Valencia F, de Teresa E, Díez J, Am. J. Physiol. –Heart C 2010, 299, H1–H9. [DOI] [PubMed] [Google Scholar]
- [16].Ahmed Laskar A, Younus H, Drug Metab. Rev 2019, 51, 42–64. [DOI] [PubMed] [Google Scholar]
- [17].Grünblatt E, Riederer P, J. Neural Transm 2016, 123, 83–90. [DOI] [PubMed] [Google Scholar]
- [18].Barski OA, Tipparaju SM, Bhatnagar A, Drug Metab. Rev 2008, 40, 553–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Li D, Ellis EM, Toxicol. In Vitro 2014, 28, 707–714. [DOI] [PubMed] [Google Scholar]
- [20].Rai SR, Bhattacharyya C, Sarkar A, Chakraborty S, Sircar E, Dutta S, Sengupta R, ChemistrySelect 2021, 6, 4566–4590. [Google Scholar]
- [21].Nguyen HD, Do LH, Curr. Opin. Chem. Biol 2022, 71, 102213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Cho KH, Shim SH, Kim M, Clin. Genet 2018, 93, 721–730. [DOI] [PubMed] [Google Scholar]
- [23].Van Kessel AT, Karimi R, Cosa G, Chem. Sci 2022, 13, 9727–9738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Masato A, Sandre M, Antonini A, Bubacco L, Curr. Neuropharmacol 2021, 19, 1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Zhu Q, Zheng Z-P, Cheng K-W, Wu J-J, Zhang S, Tang YS, Sze K-H, Chen J, Chen F, Wang M, Chem. Res. Toxicol 2009, 22, 1721–1727. [DOI] [PubMed] [Google Scholar]
- [26].Böhme A, Thaens D, Schramm F, Paschke A, Schüürmann G, Chem. Res. Toxicol 2010, 23, 1905–1912. [DOI] [PubMed] [Google Scholar]
- [27].Xie Z, Baba SP, Sweeney BR, Barski OA, Chem. Biol. Interact 2013, 202, 288–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Clark D, Cavanagh B, Shields AL, Karpecki P, Sheppard J, Brady TC, Am. J. Ophthalmol 2021, 230, 60–67. [DOI] [PubMed] [Google Scholar]
- [29].Mandell KJ, Clark D, Chu DS, Foster CS, Sheppard J, Brady TC, J. Ocul. Pharmacol. Ther 2020, 36, 732–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Alonso-delCastro S, Terenzi A, Gurruchaga-Pereda J, Salassa L, Chem.–Eur. J 2019, 25, 6651–6660. [DOI] [PubMed] [Google Scholar]
- [31].Soldevila-Barreda JJ, Sadler PJ, Curr. Opin. Chem. Biol 2015, 25, 172–183. [DOI] [PubMed] [Google Scholar]
- [32].Ngo AH, Bose S, Do LH, Chem.–Eur. J 2018, 24, 10584–10594. [DOI] [PubMed] [Google Scholar]
- [33].Destito P, Vidal C, López F, Mascareñas JL, Chem.–Eur. J 2021, 27, 4789–4816. [DOI] [PubMed] [Google Scholar]
- [34].Seoane A, Mascareñas JL, Eur. J. Org. Chem 2022, e202200118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Madec H, Figueiredo F, Cariou K, Roland S, Sollogoub M, Gasser G, Chem. Sci 2023, 14, 409–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Rebelein JG, Ward TR, Curr. Opin. Biotechnol 2018, 53, 106–114. [DOI] [PubMed] [Google Scholar]
- [37].Soldevila-Barreda JJ, Metzler-Nolte N, Chem. Rev 2019, 119, 829–869. [DOI] [PubMed] [Google Scholar]
- [38].Wang W, Zhang X, Huang R, Hirschbiegel C-M, Wang H, Ding Y, Rotello VM, Adv. Drug Deliv. Rev 2021, 176, 113893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Banerjee S, Sadler PJ, RSC Chem. Biol 2021, 2, 12–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ngo AH, Ibañez M, Do LH, ACS Catal 2016, 6, 2637–2641. [Google Scholar]
- [41].Okamoto Y, Ward TR, Biochemistry 2017, 56, 5223–5224. [DOI] [PubMed] [Google Scholar]
- [42].Soldevila-Barreda JJ, Romero-Canelón I, Habtemariam A, Sadler PJ, Nat. Commun 2015, 6, 6582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Coverdale JPC, Romero-Canelón I, Sanchez-Cano C, Clarkson GJ, Habtemariam A, Wills M, Sadler PJ, Nat. Chem 2018, 10, 347–354. [DOI] [PubMed] [Google Scholar]
- [44].Li J, Guo L, Tian Z, Zhang S, Xu Z, Han Y, Li R, Li Y, Liu Z, Inorg. Chem 2018, 57, 13552–13563. [DOI] [PubMed] [Google Scholar]
- [45].Almodares Z, Lucas SJ, Crossley BD, Basri AM, Pask CM, Hebden AJ, Phillips RM, McGowan PC, Inorg. Chem 2014, 53, 727–736. [DOI] [PubMed] [Google Scholar]
- [46].Bose S, Ngo AH, Do LH, J. Am. Chem. Soc 2017, 139, 8792–8795. [DOI] [PubMed] [Google Scholar]
- [47].Bose S, Nguyen HD, Ngo AH, Do LH, J. Inorg. Biochem 2022, 234, 111877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Andersen C, Grønnemose AL, Pedersen JN, Nowak JS, Christiansen G, Nielsen J, Mulder FAA, Otzen DE, Jørgensen TJD, Biochemistry 2021, 60, 3644–3658. [DOI] [PubMed] [Google Scholar]
- [49].McGrath CE, Tallman KA, Porter NA, Marnett LJ, Chem. Res. Toxicol 2011, 24, 357–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Siek S, Burks DB, Gerlach DL, Liang G, Tesh JM, Thompson CR, Qu F, Shankwitz JE, Vasquez RM, Chambers N, Szulczewski GJ, Grotjahn DB, Webster CE, Papish ET, Organometallics 2017, 36, 1091–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Ngo AH, Do LH, Inorg. Chem. Front 2020, 7, 583–591. [Google Scholar]
- [52].Liu Z, Romero-Canelón I, Qamar B, Hearn JM, Habtemariam A, Barry NPE, Pizarro AM, Clarkson GJ, Sadler PJ, Angew. Chem. Int. Ed 2014, 53, 3941–3946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Agholme L, Lindström T, Kågedal K, Marcusson J, Hallbeck M, J. Alzheimer’s Dis 2010, 20, 1069–1082. [DOI] [PubMed] [Google Scholar]
- [54].Xicoy H, Wieringa B, Martens GJ, Mol. Neurodegener 2017, 12, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Lopez-Suarez L, Awabdh SA, Coumoul X, Chauvet C, NeuroToxicology 2022, 92, 131–155. [DOI] [PubMed] [Google Scholar]
- [56].Nardini M, Finkelstein EI, Reddy S, Valacchi G, Traber M, Cross CE, van der Vliet A, Toxicology 2002, 170, 173–185. [DOI] [PubMed] [Google Scholar]
- [57].Miranda CL, Reed RL, Kuiper HC, Alber S, Stevens JF, Chem. Res. Toxicol 2009, 22, 863–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Kesinger NG, Langsdorf BL, Yokochi AF, Miranda CL, Stevens JF, Chem. Res. Toxicol 2010, 23, 836–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].McGrath L, McGleenon B, Brennan S, McColl D, McIlroy S, Passmore A, QJM 2001, 94, 485–490. [DOI] [PubMed] [Google Scholar]
- [60].Tahami Monfared AA, Byrnes MJ, White LA, Zhang Q, Neurol. Ther 2022, 11, 553–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Masoli JAH, Mensah E, Rajkumar C, Age Ageing 2022, 51, afac179. [DOI] [PubMed] [Google Scholar]
- [62].Maeda H, Hori S, Nishitoh H, Ichijo H, Ogawa O, Kakehi Y, Kakizuka A, Cancer Res 2001, 61, 5432–5440. [PubMed] [Google Scholar]
- [63].Kanzawa T, Kondo Y, Ito H, Kondo S, Germano I, Cancer Res 2003, 63, 2103–2108. [PubMed] [Google Scholar]
- [64].Chen G-Q, Shi X-G, Tang W, Xiong S-M, Zhu J, Cai X, Han Z-G, Ni J-H, Shi G-Y, Jia P-M, Blood 1997, 89, 3345–3353. [PubMed] [Google Scholar]
- [65].Stevens JJ 1, Graham B, Dugo E, Berhaneselassie-Sumner B, Ndebele K, Tchounwou PB, J. Cancer Sci. Ther 2017, 9, 298–306. [PMC free article] [PubMed] [Google Scholar]
- [66].Jomova K, Jenisova Z, Feszterova M, Baros S, Liska J, Hudecova D, Rhodes CJ, Valko M, J. Appl. Toxicol 2011, 31, 95–107. [DOI] [PubMed] [Google Scholar]
- [67].Yu M, Xue J, Li Y, Zhang W, Ma D, Liu L, Zhang Z, Arch. Toxicol 2013, 87, 1025–1035. [DOI] [PubMed] [Google Scholar]
- [68].Gardner HW, Bartelt RJ, Weisleder D, Lipids 1992, 27, 686–689. [Google Scholar]
- [69].Kang JW, Moseley K, Maitlis PM, J. Am. Chem. Soc 1969, 91, 5970–5977. [Google Scholar]
- [70].Bates RW, Díez-Martín D, Kerr WJ, Knight JG, Ley SV, Sakellaridis A, Tetrahedron 1990, 46, 4063–4082. [Google Scholar]
- [71].Guo J, Wang H, Hrinczenko B, Salomon RG, Chem. Res. Toxicol 2016, 29, 1187–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Fuji K, Usami Y, Kiryu Y, Node M, Synthesis 1992, 1992, 852–858. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.