

Abstract
Cancer cell dissemination to sentinel lymph nodes associates with poor patient outcomes, particularly in breast cancer. The process by which cancer cells egress from the primary tumor upon interfacing with the lymphatic vasculature is complex and driven by dynamic interactions between cancer cells and stromal cells, including cancer associated fibroblasts (CAFs). The matricellular protein periostin can distinguish CAF subtypes in breast cancer and is associated with increased desmoplasia and disease recurrence in patients. However, since periostin is secreted, periostin-expressing CAFs are difficult to characterize in situ, limiting our understanding of their specific contribution to cancer progression. Here, we used in vivo genetic labeling and ablation to lineage trace periostin+ cells and characterize their functions during tumor growth and metastasis. Periostin-expressing CAFs were spatially found at periductal and perivascular margins, were enriched at lymphatic vessel peripheries, and were differentially activated by highly-metastatic cancer cells versus poorly-metastatic counterparts. Surprisingly, genetically depleting periostin+ CAFs slightly accelerated primary tumor growth but impaired intratumoral collagen organization and inhibited lymphatic, but not lung, metastases. Periostin ablation in CAFs impaired their ability to deposit aligned collagen matrices and inhibited cancer cell invasion through collagen and across lymphatic endothelial cell monolayers. Thus, highly-metastatic cancer cells mobilize periostin-expressing CAFs in the primary tumor site that promote collagen remodeling and collective cell invasion within lymphatic vessels and ultimately to sentinel lymph nodes.
Introduction
The majority of cancer-associated deaths, including those of breast cancer patients, are caused by metastatic burden rather than primary tumor growth. However, the complex process of cancer cell dissemination and colonization of distant tissues is incompletely understood and largely incurable using existing therapies (1). Metastatic disease is particularly intractable because metastasis is not solely driven by cancer cell-intrinsic properties but is instead a consequence of dynamic crosstalk between cancer cells and other cells in the tumor microenvironment (TME) including vascular cells, immune cells, and cancer-associated fibroblasts (CAFs). A critical early step in the metastatic cascade is the intravasation of cancer cells into blood and lymphatic vessels which serve as routes for cancer cells to spread to secondary sites. Lymphovascular invasion of cancer cells into lymphatic vessels is a predominant method of vascular invasion in breast cancer and is significantly associated with the presence of lymph node metastasis, development of distant metastasis, and decreased disease-free interval and overall survival (2–6). Despite the association of lymphovascular invasion with poor clinical outcomes, the heterotypic cell interactions that drive lymphovascular invasion are not well-characterized. Many studies of lymphovascular invasion focus on paracrine signaling between cancer cells and lymphatic endothelial cells with limited consideration given to the contributions of other auxiliary cells in the TME such as CAFs.
CAFs are an abundant stromal cell in the breast TME and primarily originate from tissue-resident fibroblasts that become persistently activated by a dysregulated “wound-healing” process recapitulated in solid tumors (7). They are important during multiple stages of tumor development though their specific contributions to disease progression are contradictory and context-dependent (8–14). The paradoxical functions of CAFs can be attributed to their extensive molecular diversity both within and across tumor types as revealed by single-cell RNA sequencing studies (15–21). Though CAFs are mainly characterized by expression of contractile proteins such as alpha smooth muscle actin (SMA) and extracellular matrix (ECM) proteins including collagens, single-cell transcriptomic analysis has identified additional subpopulations of CAFs that display expression profiles associated with proliferation, angiogenesis, and immune modulation (15,22). Although these purported functions of CAF subpopulations position them as attractive therapeutic targets, in vivo depletion studies have yielded unexpected results with SMA+ CAF depletion leading to more aggressive, undifferentiated tumors (12,13). These data suggest that CAF subpopulations play opposing roles in the TME, with some CAFs restraining tumor growth whereas others are tumor-supportive. Therefore, functional classification of molecularly-defined CAF subpopulations is necessary to overcome the limitations of broadly targeting CAFs so that specific tumor-promoting and/or metastasis-promoting CAFs can be identified. A challenging but critical first step in this process is identifying reliable biomarkers that distinguish pro-tumorigenic and pro-metastatic CAFs from their counterparts.
A recent single-cell RNA sequencing study demonstrated that periostin, a TGFβ-induced matricellular protein, is expressed by CAF populations in human triple-negative breast cancers (22). As a nonstructural matrix component, periostin directly interacts with other ECM proteins including lysyl oxidase and collagen I, serving as a protein scaffold for mediators of collagen cross-linking and ECM stiffening (23). It is present at low levels in most tissues but induced at sites of inflammation and tissue injury. Periostin’s role in pathologies including cancer have been noted, including its ability to promote immunosuppressive niche formation, mediate interactions between disseminated cancer cells and the ECM, and support cancer stem cell expansion at metastatic sites (24–26). In human breast cancer samples, periostin is associated with a higher density of activated CAFs and stromal desmoplasia. Stromal desmoplasia is a consistent aspect of invasive progression and distinguishes tumors that are “non-progressors” from those that recur and spread (27). Although these studies characterize periostin as a tumor-supportive factor of metastatic disease, its source in the primary TME and the specific contributions of periostin-expressing cells during tumor progression have not been well-studied in vivo. Previous studies of periostin relied on immunostaining to characterize its abundance and distribution in primary and secondary sites. However, given that periostin is a secreted factor, immunostaining is unable to link the source of periostin to specific cell types in the TME. Therefore, we have adapted an in vivo lineage tracing strategy previously used to study activated myofibroblasts in the heart (28) to track, characterize, and ablate periostin-expressing cells in the breast TME. We reveal that highly-metastatic cancer cells mobilize periostin-expressing CAFs in the TME which promote collagen-mediated invasion of cancer cells across the endothelial barrier of nearby lymphatic vessels and ultimately to the proximal lymph node.
Materials and Methods
In vivo animal studies
All animal experiments were approved by the University of Virginia Institutional Animal Care and Use Committee (IACUC) (Protocol #4146) and performed in accordance with the University of Virginia IACUC guidelines for animal handling and care. All tumor studies were performed in 8- to 12-week old female mice with a C57BL/6 genetic background. PostniZSGreen lineage tracing mice were generated by crossing PostnMCM mice (Jax Stock No. 029645) with ZSGreenl/s/l mice (Jax Stock No. 007906). PostniZSGreen mice were injected intraperitoneally (IP) with 75 mg/kg tamoxifen 3 times over the course of 7 days to induce ZSGreen labelling of periostin-expressing cells. In the low- versus highly-metastatic tumor studies, one million EO771mCherry or EO771.LMBmCherry cancer cells were orthotopically injected into the third mammary fat pad of tamoxifen-induced heterozygous PostniZSGreen lineage tracing mice. Tumor volumes were measured with calipers, and tumors were surgically removed when they averaged ~400 mm3. Mice were euthanized and mammary glands, lymph nodes, and lungs were harvested ~2 weeks following tumor resection surgery. For the tail vein model of experimental metastasis, tamoxifen-induced PostniZSGreen mice were injected with one million EO771mCherry or EO771.LMBmCherry cancer cells into their tail vein. Mice were sacrificed and lungs were harvested 2 weeks following tumor cell injection. PostniZSGreen lineage tracing mice were crossed with ROSA-DTA mice (Jax Stock No. 009669) to generate PostnDTA mice that allow for selective depletion of periostin-expressing cells. PostnDTA mice were injected IP with 75 mg/kg tamoxifen 3 times over the course of 7 days to induce depletion of periostin-expressing cells. 500,000 mCherry-labelled EO771.LMB or PyVMT cells were orthotopically injected into the third mammary fat pad of heterozygous tamoxifen-induced (or vehicle-injected control) mice. Tumor volumes were measured with calipers, and tumors were surgically removed when they averaged ~400 mm3. Mice were euthanized and mammary glands, lymph nodes, and lungs were harvested 2 weeks (PyVMT-injected mice) or 4 weeks (EO771.LMB-injected mice) following tumor resection surgery. Postn-Cre:Tgfbr2fl/fl mice were generated by crossing PostniZSGreen mice with Tgfbr2fl/fl mice (Jax Stock No. 012603). Postn-Cre:Tgfbr2fl/fl mice were injected IP with 75 mg/kg tamoxifen 3 times over the course of 7 days to induce Tgfbr2 knockout in periostin-expressing cells. 500,000 mCherry-labelled EO771.LMB cells were orthotopically injected into the third mammary fat pad of tamoxifen-induced Postn-Cre:Tgfbr2fl/fl mice or PostniZSGreen control mice. Tumor volumes were measured with calipers, and tumors were surgically removed when they averaged ~400 mm3. Mice were euthanized and lymph nodes were harvested 3 weeks following tumor resection surgery.
Immunohistology (IHC)
Resected tissues were fixed overnight in 4% PFA/PBS and moved to a 30% sucrose solution for an additional 24 hours before embedding in OCT. Cryosections were DAPI stained and mounted in Vectashield (Vector Labs H-1700-10), then imaged using a Nikon Eclipse Ti-E inverted microscope and NIS-Elements software. Fluorescent populations were quantified using the thresholding function in FIJI. For IHC of immune populations, tumor cryosections were stained using the following primary antibodies: CD3 (Dako A0452), CD68 (Abcam Ab125212), and Foxp3 (Thermo Fisher 14-5773-82) and counterstained with hematoxylin. Positive populations were quantified by thresholding or cell counts.
Cell lines, cell culture, and media
EO771 cells were obtained from CH3 Biostystems (940001) in 2013 and transfected with mCherry. EO771.LMB cells were a gift from Dr. Chad Pecot at UNC Chapel Hill in 2014 and transfected with mCherry. EO771mCherry and EO771.LMBmCherry cells were cultured in 4.5 g/l D-glucose DMEM (HG-DMEM) with 10% FBS. Metastatic PyVMT cells were a gift from Dr. Melanie Rutkowski at UVA in 2021. They were transfected with mCherry and cultured in RPMI1640 with 2mM L-Glutamine, 10% FBS, 0.5% sodium pyruvate, and 0.09% β-mercaptoethanol. MDA-MB-231 human breast cancer cells were purchased from ATCC (HTB-26) in 2014 and transfected with mCherry. MDA-MB-231mCherry cells were cultured in HG-DMEM with 10% FBS. Primary human breast CAFs were isolated and gifted to us by Dr. Melissa Troester at UNC Chapel Hill (29) in 2013 and were cultured on 0.5% gelatin-coated plates in HG-DMEM with 20% FBS and 10 ng/mL bFGF. Primary human lymphatic endothelial cells were purchased from Cell Biologics (H-6092) in 2021 and were cultured in Complete Human Endothelial Cell Medium (Cell Biologics H1168) on 0.5% gelatin-coated plates. HUVECs were purchased from Lonza (C2517A) in 2014 and cultured in Endothelial Cell Growth Media (R&D Systems CCM027) on 1% gelatin-coated plates. Primary mouse mammary fibroblasts and CAFs were isolated from mammary glands and primary tumors by digesting tissues in a cocktail of collagenase/dispase/DNAse for 40 minutes with shaking at 37 °C, filtering through a 100 µm filter, and seeding cells on 0.5% gelatin-coated plates in HG-DMEM with 20% FBS and 10 ng/mL bFGF. All cell media included a maintenance dose of Plasmocin (Invivogen ant-mpt) to prevent mycoplasma growth and antibiotics/antimycotics to prevent bacterial and fungal contamination. Cells were maintained at 37° C in 5% CO2 plus 20% O2 and regularly tested for mycoplasma using the MycoStrip Mycoplasma Detection Kit (Invivogen rep-mys-10). Primary cells were used within 2–8 passages and cell lines were used within 5–20 passages.
Breast tumor tissue microarray analysis
Antigen-retrieval was performed on a tissue microarray of paraffin-embedded breast tumor cores (US Biomax BR20829) at 95° C for 20 minutes. The microarray slide was rinsed in DI water then incubated with blocking buffer (5% BSA + 5% goat serum in TBS) for 1 hour at room temperature. The slide was then incubated with rabbit anti-periostin antibody (Abcam ab215199, 1:1000) overnight at 4° C. The next day, the slide was rinsed 3 times with TBS and incubated for 30 minutes at room temperature with Biocare’s MACH 3 Rabbit Probe (M3R533 G). Following another three TBS washes, the slide was incubated for 30 minutes at room temperature with Biocare’s MACH 3 Rabbit AP-Polymer (M3R533 G). The slide was then rinsed in TBS and developed using Biocare’s Warp Red Chromogen Kit (WR 806 H) for 8 minutes at room temperature. The slide was washed in TBS, counterstained for 3 seconds with hematoxylin QS, then rinsed in water and TBS. Finally, the microarray slide was dipped in 100% ethanol then xylene and mounted using Biocare’s Ecomount (EM897L). Fluorescent AP signal was imaged using a Nikon Eclipse Ti-E inverted microscope and NIS-Elements software and percent periostin positive area (by pixel) was quantified by thresholding in FIJI.
Immunoblotting
Collection of cell lysates and Western blotting were carried out as previously described using standard methods (30). For analysis of secreted proteins, conditioned media was collected and concentrated 10x using Microsep Advance Centrifugal Filters (Pall Laboratory MCP010C41). Samples probed for collagen I were prepared and run under non-reducing conditions in a 7.5% gel. Primary antibodies: rabbit anti-periostin (Abcam ab14041, 1:1000), rabbit anti-collagen I (Abcam ab34710, 1:1000). Secondary HRP-conjugated antibody: peroxidase goat anti-rabbit IgG (Vector Laboratories PI-1000, 1:10,000).
Real-time quantitative PCR (RT-qPCR)
Total RNA isolation was performed using a Quick-RNA Miniprep Kit (Zymo Research R1055) according to the manufacturer’s instructions. cDNA synthesis was completed using an iScript cDNA Synthesis Kit (Bio-Rad 1708891EDU), and qPCR reactions were carried out using a QuantStudio 12K Flex Real-time PCR System.
Immunofluorescence
Fixed frozen tissue sections were stained using the following antibodies and concentrations: Cy3-SMA (TCF C6198, 1:100), CD31 (BD Biosciences 550274, 1:50), F4/80 (BioRad MCA497, 1:50), CK14 (BioLegend 905301, 1:250), Pdpn (R&D Systems AF3244, 1:200), Prox1 (BioLegend 925202, 1:50), Lyve-1 (Abcam ab218535, 1:50), SMA (Sigma-Aldrich A5228, 1:50), Col1 (Abcam ab21286, 1:50), and Eng/CD105 (Thermo Fisher 14-1051-81). Antibodies were diluted in blocking buffer with 5% BSA, 5% goat serum, and 1% Triton X-100. Blocking buffer without goat serum was used for Pdpn staining. Primary incubations were performed overnight at 4° C, and secondary incubations were performed for 1 hour at room temperature. Nuclei were counterstained with DAPI and slides were mounted using Vectashield (Vector Labs H-1700-10). Slides were imaged using a Nikon Eclipse Ti-E inverted microscope and NIS-Elements software.
GeoMx Digital Spatial Profiling (DSP)
NanoString GeoMx DSP was used to quantify transcript numbers in spatially distinct populations of periostin-expressing cells in the naïve mammary glands of PostniZSGreen lineage tracing mice. Fixed frozen mammary glands were incubated with the NanoString mouse Whole Transcriptome Atlas (WTA) panel probes overnight, then stained with Cy3-SMA (TCF C6198), Pdpn (R&D Systems AF3244;Cy5 secondary), and the DNA stain Syto83 (Thermo S11364) as morphology markers to visualize tissue architecture. Stained slides were loaded onto the GeoMx instrument and scanned for region of interest (ROI) selection. Due to the filters in the optical system of the DSP platform, ZSGreen+ cells appear blue, SMA staining appears in green, and Pdpn staining appears in red in the ROIs. ROIs of ZSGreen+ cells near thin-walled Pdpn+ structures were selected to represent lymphatic-adjacent populations, and ROIs of ZSGreen+ cells near SMA+ structures were selected to represent duct- and blood vessel-adjacent populations. After UV illumination of the ROIs, the eluent was collected via microcapillary aspiration and transferred into individual wells of a microtiter plate. The collected aspirates in the microtiter plate were then transferred to a PCR plate for library prep with Seq Code primers. PCR products were pooled and purified, then subjected to Illumina NGS Sequencing (Next Seq 2000). FASTQ sequencing files obtained from the NGS run were processed into digital count conversion (DCC) files by NanoString’s GeoMx NGS Pipeline software. The DCC files were then uploaded onto the GeoMx DSP, and quality control checks and differential gene expression analysis were performed.
Second harmonic generation
Fixed frozen tumor sections were imaged using a Zeiss 710 Multiphoton Confocal microscope, and collagen fiber length measurements were performed in FIJI. The FIJI plug-ins TWOMBLI (31) and OrientationJ (http://bigwww.epfl.ch/demo/orientation/) (32) were used to analyze the collagen area (abundance), curvature (measured as mean change in angle along fibers), and directionality of the collagen fibers.
siRNA transfection
For in vitro periostin knockdown studies, primary human breast CAFs were transfected with 75 nm human periostin-targeting stealth siRNA (si-POSTN) (ThermoFisher siRNA ID: HSS116400) or 75 nm non-targeting control siRNA (si-Control) (DHARMACON D-001810-02-05) for 5 hours, then media was replaced. This siRNA transfection was repeated after 24 hours, and media was replaced with low-serum media. Cells were then used for functional assays.
Bulk RNAseq and analysis
Primary human breast CAFs were transfected with siRNA in quadruplicate as described above. RNA samples were harvested from cells and sent to Novogene for bulk RNA sequencing. Sequencing analysis was performed by the UVA Bioinformatics Core as previously described (33). On average we received 30 million paired ends for each of the replicates. RNAseq libraries were checked for their quality using the fastqc program (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). The results from fastqc were aggregated using multiqc software. In-house developed programs were used for adaptor identification, and any contamination of adaptor sequence was removed with cutadapt (https://cutadapt.readthedocs.io/en/stable/). Reads were mapped with the “splice aware” aligner ‘STAR’ to the transcriptome and genome of mm10 genome build. The HTseq software was used to count aligned reads that map onto each gene. The count table was imported into R to perform differential gene expression analysis using the DESeq2 package. Low expressed genes (genes expressed only in a few replicates and had low counts) were excluded from the analysis before identifying differentially expressed genes. Data normalization, dispersion estimates, and model fitting (negative binomial) were carried out with the DESeq function. The log-transformed, normalized gene expression of 500 most variable genes was used to perform an unsupervised principal component analysis. The differentially expressed genes were ranked based on the log2fold change and FDR corrected p-values. The ranked file was used to perform pathway analysis using GSEA software. The enriched pathways were selected based on enrichment scores as well as normalized enrichment scores.
ECM deposition assay
Primary human breast CAFs were seeded (30,000 cells per well) on a gelatin-coated chamber slide and transfected with siRNA as described above. 48 hours after the second siRNA treatment, cells were lysed using ammonium hydroxide-based extraction buffer. The deposited ECM was washed in PBS and fixed with 2% PFA for 20 minutes. The ECM was then rinsed in PBS and incubated in blocking buffer with 1% Triton for 30 minutes to reduce non-specific antibody binding. The ECM was then incubated with primary antibody (rabbit anti-collagen I ab34710, 1:200) for 1 hour at room temperature and secondary antibody (goat anti-rabbit IgG Alexa Fluor 594 1:100) for 1 hour at room temperature. Slides were mounted using Vectashield (Vector Labs H-1700-10) and imaged using a Nikon Eclipse Ti-E inverted microscope and NIS-Elements software. Collagen fiber length measurements were performed using FIJI.
Cell spreading assay
Primary human breast CAFs were seeded (1 x 105 cells/mL) in a 6-well gelatin coated dish and transfected with siRNA as described above. 24 hours following knockdown, CAFs were detached and re-seeded at 10,000 cells per well on a gelatin-coated chamber slide. The “rescue” wells were treated with 0.8 ug/mL recombinant human periostin (BioLegend 770506). After 24 hours, cells were fixed in 4% PFA and stained with Alexa Fluor 594 Phalloidin (ThermoFisher Scientific A12381). Cells were imaged using a Nikon Eclipse Ti-E inverted microscope and NIS-Elements software, and cell area measurements were performed in FIJI.
Wound closure scratch assay
Primary human breast CAFs were seeded (1 x 105 cells/mL) in a 6-well gelatin coated dish and transfected with siRNA as described above. The next day, a P1000 pipette tip was used to create a scratch down the middle of the cell monolayer in each well. Each well was washed once with PBS to removed detached cells and low serum media was then added. “Rescue” wells were treated with 0.8 ug/mL recombinant human periostin (BioLegend 770506). Scratches were imaged at 0 hours, 12 hours, and 24 hours using a Nikon Eclipse Ti-E inverted microscope and NIS-Elements software, and percent wound closure was calculated using area measurements in FIJI.
3D CAF/cancer cell spheroid invasion assay
Primary human breast CAFs were seeded (1 x 105 cells/mL) in a 6-well gelatin coated dish and transfected with siRNA as described above. The next day, CAFs were co-cultured 1:1 with MDA-MB-231mCherry human breast cancer cells in 20 uL hanging droplets containing 5% methylcellulose on the inverted lid of a 100 mm tissue culture dish containing PBS. The CAF/cancer cell co-cultures were incubated for 24 hours to allow aggregation of cells into a spheroid, as confirmed under light microscopy. The CAF/cancer cell spheroids were then embedded in either Matrigel (Corning 356237) or rat-tail collagen type I (Ibidi 50201) according to manufacturer’s instructions in a 96-well plate. Spheroids were imaged at 12 and 24 hours using a Nikon Eclipse Ti-E inverted microscope and NIS-Elements software, and invasive area was quantified using FIJI. Representative images of spheroids were taken using a Zeiss LSM 880 confocal microscope.
Trans-well migration assay
Primary human breast CAFs were seeded (1.5 x 105 cells/mL) in a 6-well gelatin coated dish and transfected with siRNA as described above. The next day, CAFs were co-cultured with MDA-MB-231mCherry human breast cancer cells into spheroids as described above, or MDA-MB-231mCherry cells were cultured into spheroids alone for mono-spheroid controls. Transwell membrane inserts (Fisher Scientific 07-200-150) were coated with 0.5% gelatin and seeded with 60,000 primary human lymphatic endothelial cells or 60,000 primary human umbilical vein endothelial cells (HUVECs). After 24 hours, CAF/cancer cell spheroids were embedded in rat-tail collagen type I (Ibidi 50201) in the transwell membrane inserts above the monolayer of endothelial cells. Inserts were cleared with a cotton swab and washed in PBS after 24 hours, so that only cells that had migrated across the lymphatic endothelial cell barrier and reached the bottom surface of the transwell insert remained. These cells were fixed in 4% PFA for 10 minutes at room temperature and washed in PBS. Transwell inserts were placed on slides and imaged using a Nikon Eclipse Ti-E inverted microscope and NIS-Elements software. Trans-migrated cancer cells were counted in FIJI.
Kaplan-Meier Analysis
Kaplan-Meier (KM) Plotter was used to generate survival curves for lymph node positive breast cancer patients from the Tang_2018 patient cohort that were stratified based on low versus high periostin protein expression.
Statistical analysis
All data points are shown, and horizontal lines on graphs represent median values. Descriptive numerical values in the text are expressed as mean value ± standard error of the mean (SEM). Sample sizes are listed in figure legends. All statistical analyses were performed using GraphPad Prism software, and P values less than 0.05 were considered significant.
Data and materials availability
The publicly available scRNAseq data generated by Wu et al. (34) are available at: https://zenodo.org/record/5031502#.Y_Og3i2cbUI. The GeoMx spatial profiling transcriptomic data generated in this study have been deposited in the NCBI GEO database under accession code GSE231806. The bulk RNAseq data generated in this study have been deposited in the NCBI GEO database under accession code GSE231594. The patient survival data analyzed in this study were obtained from the Tang_2018 dataset of the KM Plotter for breast protein at https://kmplot.com/analysis/index.php?p=service&cancer=breast_protein. All other raw data generated in this study are available upon request from the corresponding author.
Results
Periostin is enriched in CAFs and cycling perivascular-like cells in breast cancers and is associated with advanced disease stage and lymph node metastasis
As periostin has been shown to distinguish CAFs in triple-negative breast cancer samples (22), we turned to a comprehensive, spatially-resolved single-cell transcriptional atlas of molecularly diverse breast tumors to determine with greater resolution which cellular populations express periostin in human breast cancers (34). We found that periostin is not detected in breast cancer cells of any molecular subtype but is expressed by multiple stromal populations (Figure 1A). Periostin and associated matrix proteins are enriched in CAFs, including mesenchymal stem cell-like inflammatory CAFs (MSC iCAF-like) and myofibroblast-like CAFs (myCAF-like) as well as cycling perivascular-like (PVL) cells (Figure 1B). Despite being restricted to the tumor stroma, periostin may have important functional consequences during tumor progression as it is associated with disease recurrence in human breast cancer patients (27). Therefore, we hypothesized that its expression in human breast cancer tissues would correlate with disease stage and lymph node status. Indeed, when we stained a human breast tissue microarray of 200 tumor cores for periostin we found that periostin abundance positively associated with advanced disease stage (Figure 1C,D) and lymph node metastasis (Figure 1E). These data indicate that periostin is almost exclusively expressed by CAFs or CAF-like cells in the breast TME and its abundance associates with disease progression.
Figure 1. Periostin is enriched in CAFs and cycling perivascular-like cells in breast cancers and is associated with advanced disease stage and lymph node metastasis.
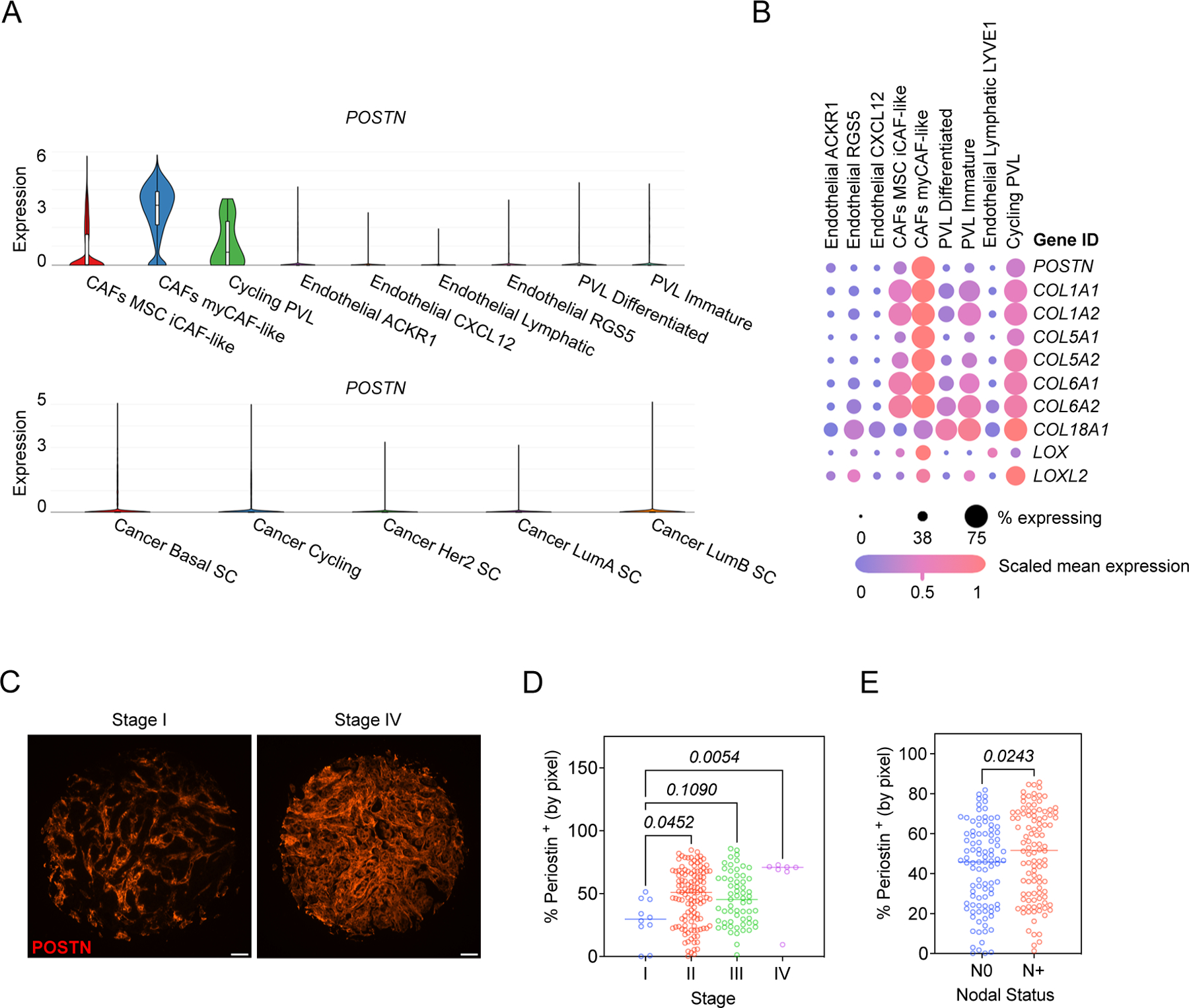
(A) Single-cell RNAseq data from the Wu et al dataset (34) were analyzed for periostin expression. Violin plots of differential expression of periostin (POSTN) in stromal populations (top) and breast cancer cells (bottom) are shown. MSC = mesenchymal stem cells, PVL = perivascular-like cells, and SC = subclass. (B) Expression of periostin and associated matrix proteins in stromal populations from the Wu et al. cohort. (C) Representative immunofluorescence images of human breast cancer tissues stained for periostin (in red). Scale bars: 100 μm. (D) Percentage of tissue area positive for periostin staining (by pixel), grouped by tumor stage (D) and patient’s lymph node status (N0 = no nodal involvement, N+ = nodal involvement) (E). Each data point represents an individual tumor core (N = 200). Statistics shown for Kruskal-Wallis test (D) and Mann-Whitney test (E).
Periostin-expressing cells surround tumor-naïve mammary ducts and blood vessels and are enriched at the lymphatic vessel periphery
Our human data supports existing murine studies linking periostin to metastasis (24–26). However, previous characterizations of periostin in primary and metastatic environments relied on immunostaining to characterize its abundance and distribution. Since periostin is secreted, immunostaining is unable to link periostin to a cellular source in the TME. While single-cell RNAseq data indicate that periostin expression is restricted to stromal populations in human breast cancers, these populations have not been studied in situ in murine tumor models. Thus, in vivo approaches to label periostin-expressing cells are required to characterize periostin’s source in the TME and to determine how these periostin-expressing populations function during tumor growth. We generated reporter mice (PostniZSGreen lineage tracing mice) to genetically mark periostin-expressing cells by crossing PostnMCM mice with ZSGreenl/s/l mice. Upon tamoxifen administration, periostin-expressing cells and their progeny are genetically labelled with ZSGreen and can be quantified and tracked across tissues using fluorescence microscopy. After generating PostniZSGreen mice, we first characterized the spatial distribution of ZSGreen+ cells in tumor-naïve mammary glands. We harvested mammary glands from tumor-naïve PostniZSGreen mice and stained for the following cell lineage markers: SMA (fibroblast and pericyte marker), CK14 (myoepithelial marker), F4/80 (macrophage marker), CD31 (endothelial marker), and Pdpn (lymphatic endothelial marker) (Figure 2A). While Pdpn is an established lymphatic endothelial cell marker (35–37), its expression has also been reported in fibroblasts (18,38,39). Therefore, we used Pdpn expression combined with tissue morphology to identify lymphatic vessels and confirmed the proximity of ZSGreen-labelled periostin+ cells and lymphatic vessels using two additional markers of lymphatic endothelium: Prox1 and Lyve-1 (Supplemental Figure 1). We found that periostin-expressing cells were relatively sparse in naïve tissues and surrounded mammary ducts and blood vessels as revealed by the CK14 and CD31 staining, respectively. Intriguingly, we also found that periostin-expressing cells localized to the lymphatic vessel periphery and were more enriched along thin-walled lymphatic vessels compared to large vessel endothelium marked by abundant SMA (Figure 2B). These data are consistent with the spatially resolved expression of periostin by cycling perivascular-like (PVL) stromal cells in human breast cancers as noted above using comprehensive single cell transcriptomics.
Figure 2. Periostin-expressing cells surround tumor-naïve mammary ducts and blood vessels and are enriched at the lymphatic vessel periphery.
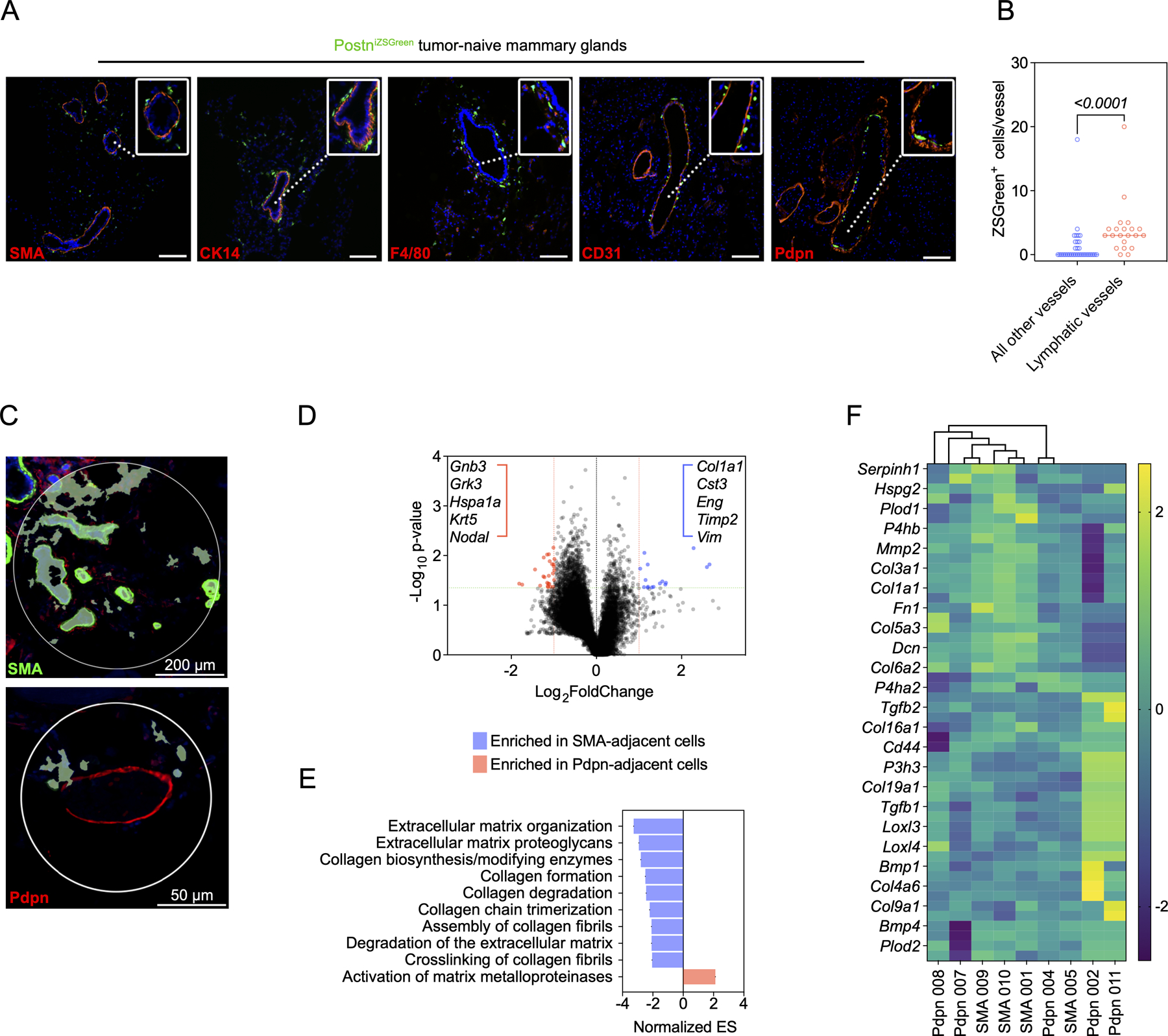
(A) Immunofluorescence images of tumor-naïve mammary glands from PostniZSGreen mice stained for the following markers (in red): SMA, CK14, F4/80, CD31, and Pdpn. Nuclei counterstained with DAPI. Scale bars: 200 μm, insets: 3x zoom. (B) Quantification of ZSGreen+ periostin-expressing cells per vessel in tissue sections of tumor-naïve mammary glands (n = 3 mammary glands per group). Each data point represents an individual vessel. Statistics shown for Mann-Whitney test. (C) Representative regions of interest (ROIs) used to select ZSGreen+ periostin-expressing cells for spatial RNA profiling (n = 4–5 ROIs per group, from the mammary glands of 3 mice). (D) Volcano plot of differential gene expression in ZSGreen+ cells located near ducts and blood vessels (SMA-adjacent) and lymphatic vessels (Pdpn-adjacent). Green line represents a p-value of 0.05 and red line represents the significance threshold. (E) Gene Set Enrichment Analysis (GSEA) showing upregulated pathways in spatially-defined ZSGreen+ populations. (F) Cluster analysis of ZSGreen+ cells from individual ROIs based on collagen-related genes.
Given the spatially distinct populations of periostin-expressing cells observed along the ducts, blood vessels, and lymphatic vessels of naïve mammary glands, we used GeoMx Digital Spatial Profiling (DSP) to molecularly characterize the subpopulations in situ. Using SMA and Pdpn as morphology markers to select regions of interest within the mammary glands, we performed whole transcriptome analysis on ZSGreen-labelled periostin-expressing cells within these regions (Figure 2C) and compared expression profiles of SMA-adjacent ZSGreen+ cells (near ducts and blood vessels) and Pdpn-adjacent ZSGreen+ cells (near lymphatic vessels). Spatial transcriptomics identified a number of differentially expressed genes between the periostin-expressing subpopulations (Figure 2D), and Gene Set Enrichment Analysis (GSEA) indicated that periostin-expressing cells located near ducts and blood vessels are enriched in genes involved in ECM organization as well as collagen synthesis and remodeling while lymphatic-adjacent periostin-expressing cells are enriched in genes related to the activation of matrix metalloproteinases which degrade the ECM (Figure 2E). However, cluster analysis of the individual subpopulations based on collagen-related genes revealed diversity among lymphatic-adjacent periostin-expressing cells, with multiple lymphatic-adjacent samples enriched in collagens and collagen cross-linking genes suggesting they also play a role in collagen synthesis and organization (Figure 2F). In sum, spatial transcriptomic analysis suggests that spatially distinct populations of periostin-expressing cells function together to remodel, synthesize, and organize the ECM within the mammary gland.
Highly-metastatic mammary tumors differentially activate periostin-expressing CAFs
Following characterization of periostin-expressing cells in the naïve mammary gland, we next used a mammary tumor model to assess the abundance, morphology, and spatial distribution of periostin-expressing cells in the primary TME and secondary sites of spontaneous metastasis. We used paired, differentially metastatic triple-negative mammary cancer cell lines EO771 and EO771.LMB to classify differences in periostin-expressing cell activation in low- versus highly-metastatic tumor contexts. These cell lines have been previously characterized to have differential capacities for spontaneous metastasis, with the poorly-metastatic parental EO771 line rarely reaching secondary sites and the highly-metastatic derivative EO771.LMB tropic to the lungs and lymph nodes (40). Importantly, the EO771 and EO771.LMB lines express negligible periostin compared to murine mammary CAFs (Figure 3A), so our lineage tracing strategy captures the predominant host-derived sources of periostin. Following tamoxifen administration to label periostin-expressing cells, PostniZSGreen mice were orthotopically injected with either EO771mCherry or EO771.LMBmCherry cells into the third mammary gland (Figure 3B). According to the previous characterization of the cancer cell lines, the difference in metastatic capacity is observed following primary tumor resection. Therefore, we incorporated this into our study design and harvested secondary tissues ~2 weeks after tumor resection, allowing more time for disseminated cells to reach and grow out at secondary sites. This experimental setup recapitulates human disease as human breast cancer patients often undergo a lumpectomy but may still progress to metastatic disease as a result of previously disseminated cancer cells emerging from dormancy and expanding in secondary sites. Since the periostin-expressing cells are genetically labelled with ZSGreen and the cancer cells are labelled with mCherry, we could quantify these populations using a thresholding strategy for positive fluorescence in histological sections (Supplemental Figure 2).
Figure 3. Highly-metastatic mammary tumors differentially activate periostin-expressing CAFs.
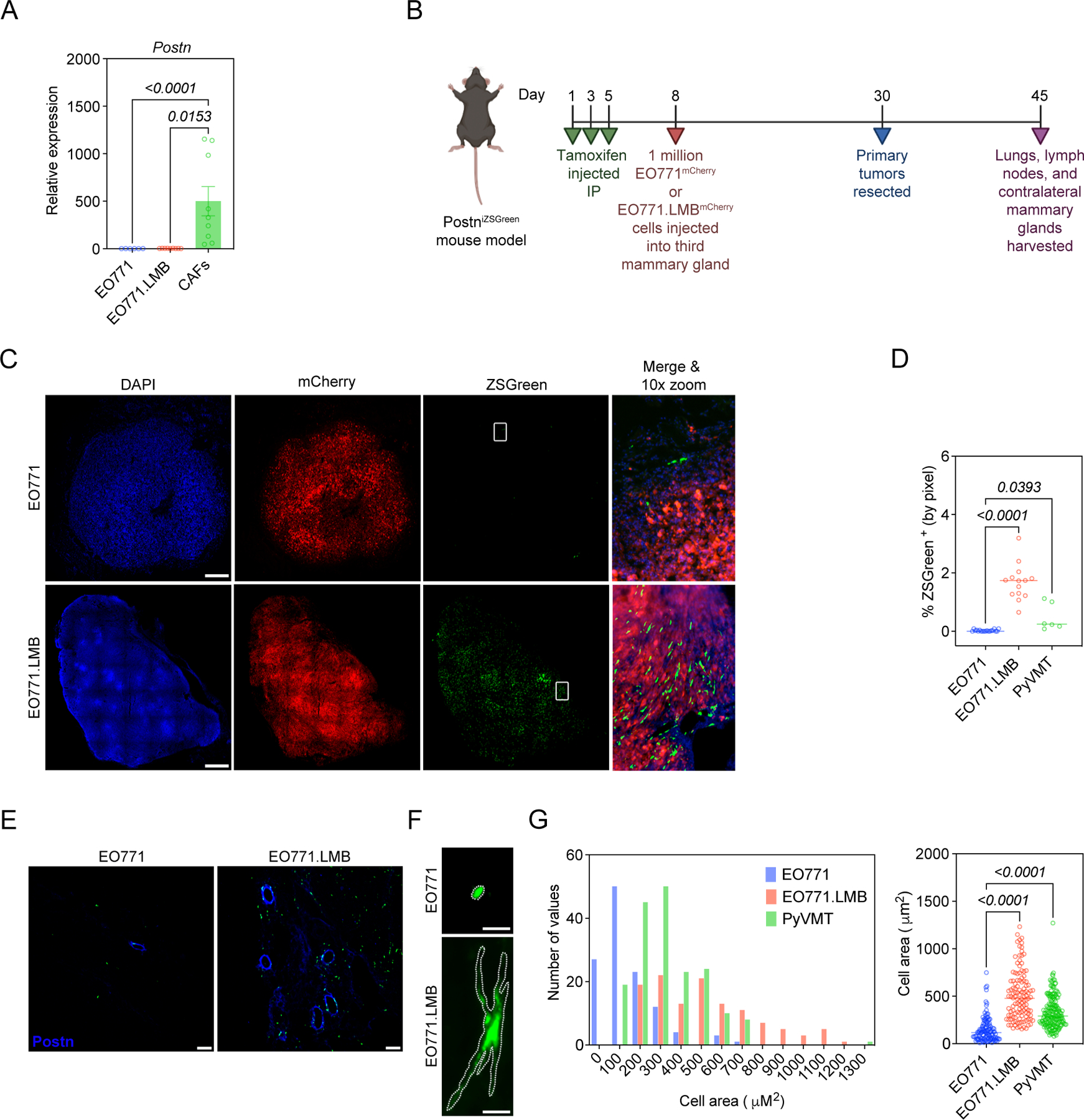
(A) qPCR analysis of periostin expression in mammary tumor cell lines and murine mammary CAFs. Data represent the mean ± SEM. Experiments were performed in triplicate and results compared using Kruskal-Wallis test. (B) Study design for EO771 vs EO771.LMB experiment in PostniZSGreen lineage tracing mice. (C) Tissue tilescans of primary mammary tumors from PostniZSGreen mice. Tumor cells labelled with mCherry and periostin-expressing cells genetically labelled with ZSGreen. Nuclei counterstained with DAPI. Scale bars: 500 μm. (D) Percentage of tissue area positive for ZSGreen in low-metastatic (EO771) tumors versus highly-metastatic (EO771.LMB and PyVMT) tumors (n = 3–7 mice per group). Each data point represents a histological section. Statistics shown for Kruskal-Wallis, Dunn’s multiple comparisons test. (E) Representative immunofluorescence images of mammary tumors from PostniZSGreen mice stained for periostin (in blue), with periostin-expressing cells genetically labelled with ZSGreen. Scale bars: 100 μm. (F) Representative images of individual ZSGreen-labelled periostin-expressing cells in EO771 and EO771.LMB tumors. Scale bars: 10 μm. (G) Area of individual ZSGreen-labelled cells in primary tumors represented as a histogram (left) and scatter plot (right). Each data point in the scatter plot represents an individual cell. 120–180 cells were measured from multiple tumor sections (n = 3–6 mice per group) and results compared using Kruskal-Wallis, Dunn’s multiple comparisons test. (Study design schematic created using BioRender.com).
When we examined the low-metastatic EO771mCherry tumors and highly-metastatic EO771.LMBmCherry tumors in the PostniZSGreen mice, we found that periostin-expressing cells were more abundant in highly-metastatic tumors with an ~ 84-fold increase in the percentage of tissue area positive for ZSGreen in EO771.LMB tumors compared to EO771 tumors (1.68% ± 0.2% versus 0.02% ± 0.01%) (Figure 3C,D). We also examined a second highly-metastatic variant of PyVMT in the PostniZSGreen mice and found a 24-fold increase in the percentage of tissue area positive for ZSGreen in these tumors compared to low-metastatic EO771 tumors (0.48% ± 0.2%) (Figure 3D). This difference in periostin-expressing cell abundance correlated with differences in secreted periostin in situ (Figure 3E). In addition to the difference in abundance of periostin-expressing cells, we also observed a difference in the morphology of the ZSGreen+ cells. Periostin-expressing cells in highly-metastatic EO771.LMB and PyVMT tumors exhibited the typical stellate shape of activated myofibroblasts and were 2–3-times larger on average compared to those found in low-metastatic EO771 tumors (509.7 ± 23.6 µm2 and 334.2 ± 12.6 µm2 versus 146.3 ± 12.0 µm2, respectively) (Figure 3F,G). In addition to their CAF-like morphology, a fraction of periostin-expressing cells in highly-metastatic EO771.LMB tumors co-localized with CAF markers including SMA, Collagen I, and Endoglin/CD105 (Supplemental Figure 3). These data suggest that highly-metastatic cancer cells differentially activate periostin-expressing CAFs in the primary tumor compared to their low-metastatic counterparts.
Periostin-expressing CAFs are more abundant in the metastatic niches of mice bearing highly-metastatic mammary tumors
Next, we examined the distribution of periostin-expressing CAFs in common metastatic sites of breast tumors, beginning with the draining axillary lymph nodes of the tumor-bearing mice as regional lymph nodes are often the first site of metastatic spread in breast cancers. Periostin-expressing cells were ~ 14-times more abundant in the axillary lymph nodes of mice bearing highly-metastatic EO771.LMB tumors, with 0.8% (± 0.1%) of the tissue area positive for ZSGreen compared to 0.06% (± 0.03%) in the lymph nodes of mice bearing low-metastatic EO771 tumors (Figure 4A,B). We then assessed the abundance of periostin-expressing cells in the lungs as this is another common site of breast cancer metastasis. Similar to the lymph nodes, we observed a significant increase in the percentage of lungs positive for ZSGreen in mice bearing highly-metastatic mammary tumors compared to those bearing low-metastatic tumors (0.2% ± 0.03% versus 0.08% ± 0.04%) (Figure 4C,D). Interestingly, we observed ZSGreen+ cells in a number of histological sections of lymph nodes and lungs without evidence of mCherry+ cancer cells in these same tissues, suggesting that periostin-expressing cells are present in the premetastatic niche. In support of this possibility, we also observed differential activation of periostin-expressing cells in the contralateral mammary glands of tumor-bearing mice, another common metastatic site for breast cancers (41,42). Though we did not detect mCherry+ cancer cells at these sites, there was an ~ 8-fold increase in the percentage of tissue area positive for ZSGreen in mice bearing highly-metastatic tumors compared to those bearing low-metastatic tumors (1.6% ± 0.2% versus 0.2% ± 0.1%) (Supplemental Figure 4).
Figure 4. Periostin-expressing CAFs are more abundant in the metastatic niches of mice bearing highly-metastatic mammary tumors.
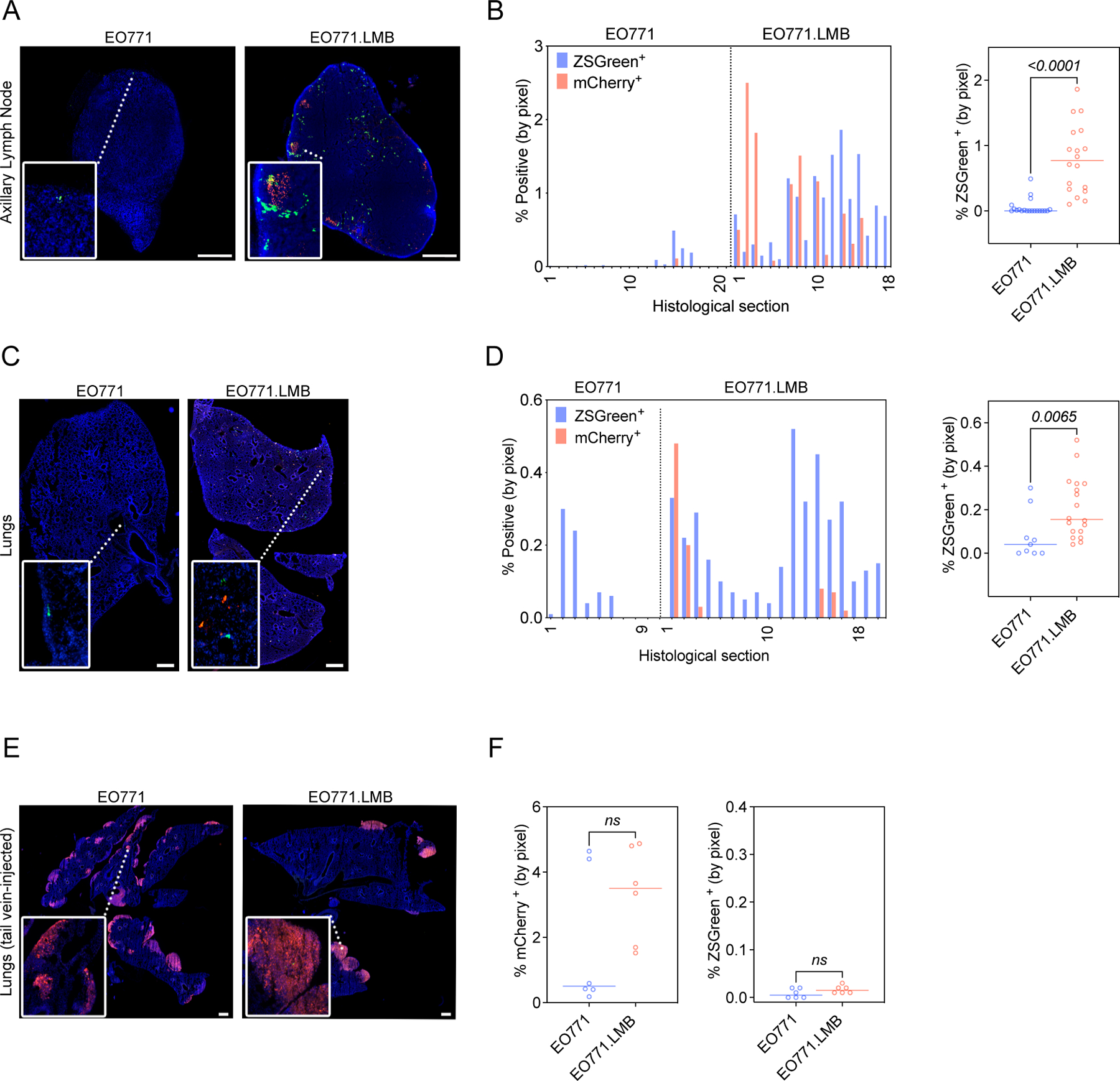
(A)Tissue tilescans of axillary lymph nodes from PostniZSGreen mice bearing low-metastatic (EO771) or highly-metastatic (EO771.LMB) mammary tumors. Tumor cells labelled with mCherry and periostin-expressing cells genetically labelled with ZSGreen. Nuclei counterstained with DAPI. Scale bars: 500 μm, insets: 3x zoom. (B) Percentage of tissue area positive for ZSGreen in serial sections of axillary lymph nodes from mice bearing EO771 or EO771.LMB mammary tumors represented as a histogram (left) and scatter plot (right). Each bar of the histogram represents a different histological section with matched mCherry and ZSGreen measurements. The scatter plot shows individual ZSGreen measurements with each point representing a different histological section (n = 6–7 lymph nodes per group). Statistics shown for Mann-Whitney test. (C) Tissue tilescans of lungs from PostniZSGreen mice bearing low-metastatic (EO771) or highly-metastatic (EO771.LMB) mammary tumors. Tumor cells labelled with mCherry and periostin-expressing cells genetically labelled with ZSGreen. Nuclei counterstained with DAPI. Scale bars: 500 μm, insets: 6x zoom. (D) Percentage of tissue area positive for ZSGreen in serial sections of lungs from mice bearing EO771 or EO771.LMB mammary tumors represented as a histogram (left) and scatter plot (right). Each bar of the histogram represents a different histological section with matched mCherry and ZSGreen measurements. The scatter plot shows individual ZSGreen measurements with each point representing a different histological section (n = 3–6 lungs per group). Statistics shown for Mann-Whitney test. (E) Tissue tilescans of lungs from PostniZSGreen mice injected via tail vein with EO771 or EO771.LMB tumor cells. Tumor cells labelled with mCherry and periostin-expressing cells genetically labelled with ZSGreen. Nuclei counterstained with DAPI. Scale bars: 500 μm, insets: 6x zoom. (F) Percentage of tissue area positive for mCherry (left) and ZSGreen (right) in serial sections of lungs from tail vein-injected mice. Each data point represents a different histological section (n = 3 mice per group). Statistics shown for Mann-Whitney test.
Since the spontaneous metastases we observed in the lungs resembled micrometastases, we hypothesized that a tail vein model of experimental metastasis would yield greater activation of periostin-expressing cells in the lungs, especially in the EO771.LMB-injected mice, as this would allow a greater number of tumor cells to colonize the lungs and activate periostin expression in tissue-resident cells via growth factor signaling. Surprisingly, though we found macrometastases of both the low-metastatic and highly-metastatic mammary cancer cells in the lungs following tail vein injection (Figure 4E), there was a lack of periostin activation in these tissues (Figure 4F), suggesting that the primary tumor must be established prior to activation of periostin at secondary sites.
Collagen fibers are longer and more aligned in highly-metastatic (periostinhigh) mammary tumors
Because periostin has been shown to promote collagen cross-linking through a mechanism dependent on BMP-1-mediated activation of lysyl oxidase (23,43), we next used second harmonic generation (SHG) imaging to visualize collagen fibers in low- versus highly-metastatic mammary tumors to determine whether differences in periostin+ cell abundance associated with intratumoral collagen structure. Coincident with an increase in periostin-expressing CAFs, we observed global changes to collagen matrix abundance and architecture in highly-metastatic tumors (Figure 5A). Total collagen fiber area was greater in highly-metastatic tumors (Figure 5B), and these fibers were straighter (Figure 5C) and more aligned (Figure 5D) when compared to low-metastatic counterparts whose collagen matrices were less dense and resembled the “curly” collagen fiber structures associated with normal mammary glands (44). Additionally, there was a 3.7-fold increase in collagen fiber length in highly-metastatic mammary tumors compared to low-metastatic mammary tumors (342.6 ± 14.9 µm versus 93.4 ± 5.4 µm) (Figure 5E). Taken together, these differences indicate that collagen matrices are more organized in highly-metastatic (periostinhigh) mammary tumors compared to low-metastatic (periostinlow) mammary tumors.
Figure 5. Collagen fibers are longer and more aligned in highly-metastatic (periostinhigh) breast tumors.
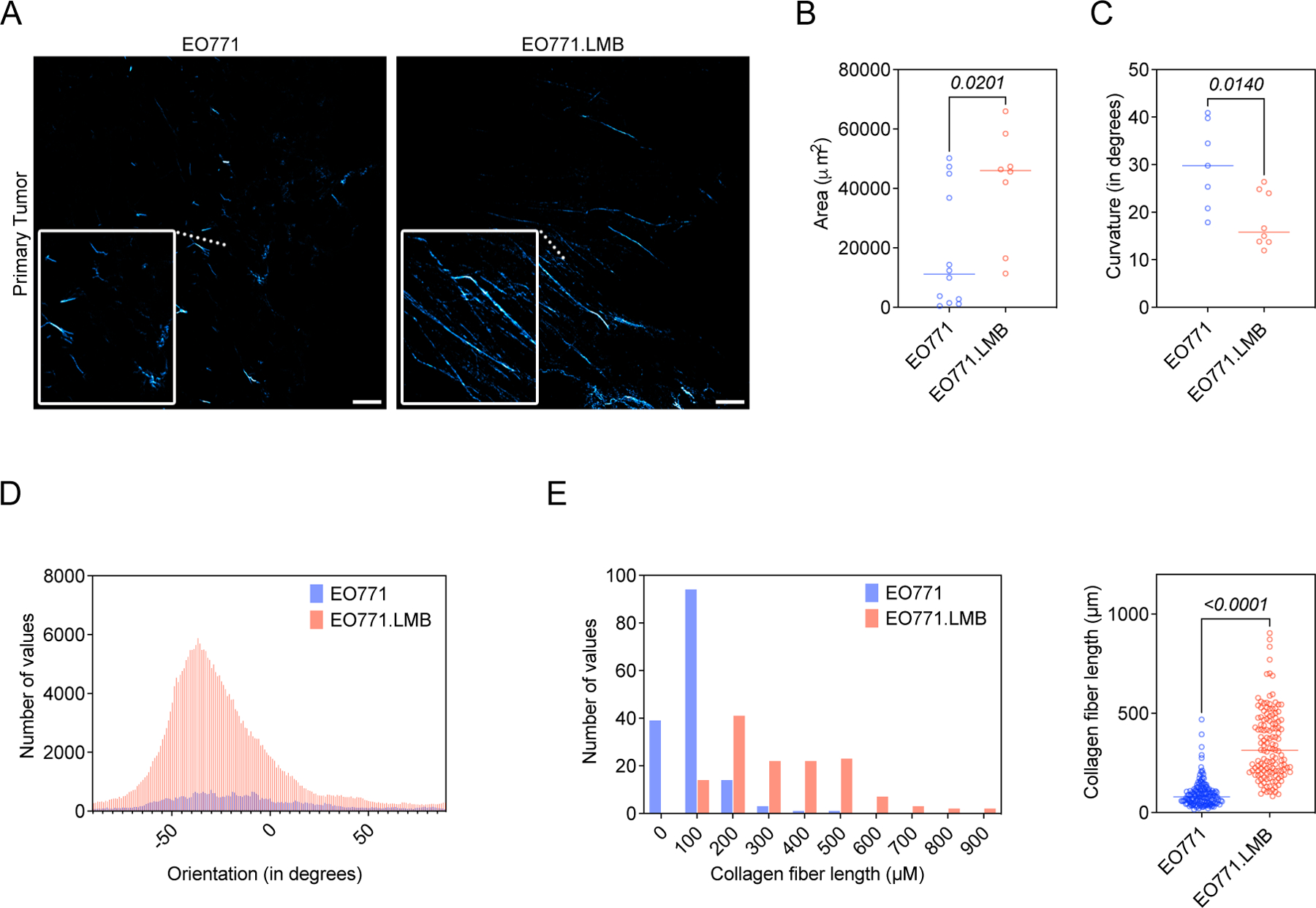
(A) Second harmonic generation (SHG) images of collagen fibers (pseudo-colored in cyan) in EO771 or EO771.LMB mammary tumors. Scale bars: 150 μm, insets: 3x zoom. (B) Total collagen area in primary tumors (n = 3–4 mice per group). Each data point represents a different histological section. Statistics shown for Mann-Whitney test. (C) Curvature of collagen fibers, measured as mean change in angle along fibers, in primary tumors (n = 3–4 mice per group). Each data point represents a histological section. Statistics shown for unpaired Student’s t test. (D) Representative histogram of collagen fiber orientation in primary tumors. A peaked histogram represents aligned fibers whereas a flat histogram represents random organization. (E) Quantification of collagen fiber length in primary tumors, represented as a histogram (left) and scatter plot (right). Each point represents an individual fiber. 130–160 fibers quantified from multiple histological sections (n = 3 mice per group) and results compared using a Mann-Whitney test.
Periostin knockdown in primary human breast CAFs alters collagen matrix architecture and inhibits collective cell invasion
Given that an aligned collagen matrix is a signature of more advanced breast tumors and can promote local invasion of cancer cells (45–49), we hypothesized that the in vivo differences in collagen matrix structure were related to differences in the pool of intratumoral periostin and could impact cancer cell migration. Thus, we knocked down periostin in primary human breast CAFs (Supplemental Figure 5A-C) to determine whether periostin expression confers functional properties to CAFs including their ability to deposit an organized collagen matrix. RNAseq of human breast CAFs showed that periostin knockdown secondarily reduced expression of matrisomal proteins, ECM regulators, and collagens among other matrix-related genes found in the NABA Gene Set Enrichment Analysis (GSEA) (Figure 6A, Supplemental Figure 5D). Secreted collagen I protein levels were reduced ~3-fold following periostin knockdown (Figure 6B), and the collagen fibers deposited by periostin-knockdown CAFs were significantly shorter than those deposited by CAFs treated with non-targeting control siRNA (201.6 ± 7.6 µm versus 120.0 ± 4.8 µm) (Figure 6C,D). Coupled with our in vivo observations that periostin-expressing cell area and matrix organization were associated with increased intratumoral periostin, these in vitro matrix alterations prompted us to test whether knocking down periostin would affect cell area in vitro, as the stellate-shaped morphology that is characteristic of CAFs is attributed to their ability to engage the ECM and form focal adhesions. Primary human breast CAF cell area was significantly reduced following periostin knockdown (2,199 ± 100.2 µm2 versus 1,643 ± 72.1 µm2), and this effect was rescued by addition of recombinant human periostin (2,534 ± 131.4 µm2) (Figure 6E, Supplemental Figure 5E). The ability of CAFs to spread is critical for their motility, so we next assessed whether periostin knockdown would also impede their ability to migrate. Indeed, periostin knockdown in human breast CAFs reduced migration, and addition of recombinant human periostin restored the migratory capacity of periostin-knockdown CAFs (Supplemental Figure 5F). Given this deficit in CAF migration following periostin knockdown, we hypothesized that ablating periostin in CAFs would also inhibit their ability to promote collective cell invasion. Therefore, we performed a 3D co-culture spheroid assay in which human breast CAFs were treated with either periostin-targeting siRNA (si-POSTN) or non-targeting control siRNA (si-Control) and co-cultured in spheroids with MDA-MB-231mCherry human breast cancer cells. Spheroids were then embedded in 3D matrices and invasive area was measured over time. We observed a selective impairment of collective cell invasion through a collagen matrix, as spheroids with periostin knockdown CAFs were significantly less invasive at 24 hours compared with control spheroids when embedded in type I collagen, but there was no difference in invasion when embedded in Matrigel which consists of laminins and other basement membrane proteins (Figure 6F,G). These data suggest that the ability of periostin-expressing CAFs to drive collective cell invasion is selectively dependent on collagen remodeling.
Figure 6. Periostin knockdown in primary human breast CAFs alters collagen matrix architecture and inhibits collective cell invasion.
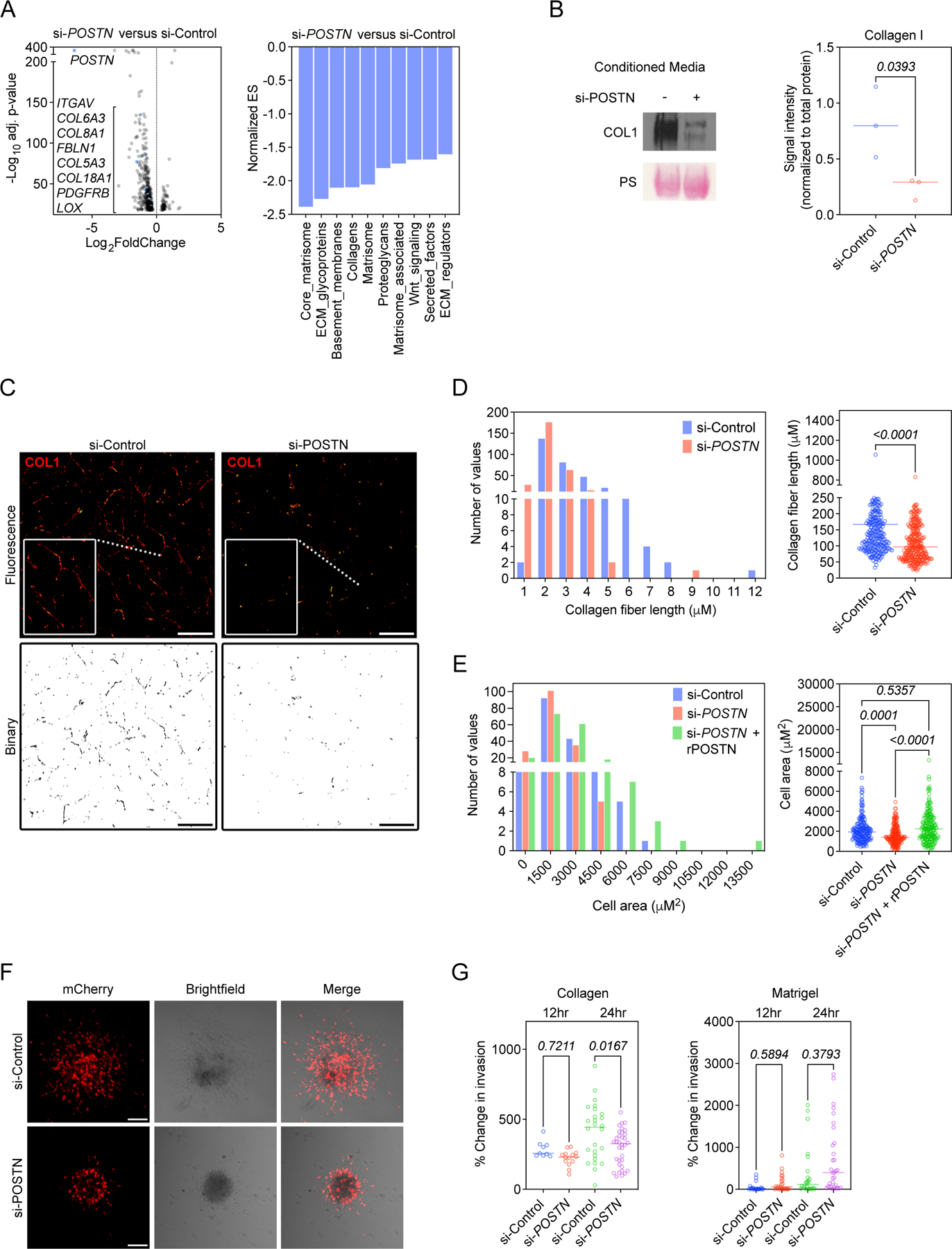
(A) Volcano plot of genes with significantly altered expression detected by bulk RNAseq following periostin knockdown in primary human breast CAFs (left) and NABA Gene Set Enrichment Analysis (GSEA) showing downregulated pathways in periostin knockdown cells (right). Knockdown performed in quadruplicate. (B) Western blot of secreted collagen I following periostin knockdown. Ponceau stain (PS) shown as loading control. Signal intensity quantified at right and compared using unpaired Student’s t test. Performed in triplicate. (C) Immunostaining of deposited collagen I by si-Control- vs. si-POSTN-treated human breast CAFs. Scale bars: 100 μm, insets: 2x zoom. Binary images of collagen matrices shown below. (D) Lengths of collagen fibers deposited by control versus periostin knockdown human breast CAFs represented as a histogram (left) and scatter plot (right). Each data point represents an individual fiber (n = 285–305 fibers per group). Experiments performed in triplicate and results compared using a Mann-Whitney test. (E) Cell area measurements of phalloidin-stained primary human breast CAFs treated with si-Control or si-POSTN ± recombinant human periostin (rPOSTN) represented as a histogram (left) and scatter plot (right). Experiment performed in triplicate with each data point representing an individual cell (n = 160–190 cells per group). Statistics shown for Kruskal-Wallis, Dunn’s multiple comparisons test. (F) Confocal images of spheroids consisting of MDA-MB-231mCherry human breast cancer cells and unlabeled primary human breast CAFs embedded in type I collagen (24 hr). Scale bars: 100 μm. (G) Percent change in invasive area of spheroids embedded in type I collagen (left) or Matrigel (right). Each data point represents an individual spheroid, carried out in biological triplicates. Statistics shown for ordinary one-way ANOVA, Tukey’s multiple comparisons test (left) and Kruskal-Wallis, Dunn’s multiple comparison’s test (right).
Periostin-expressing CAFs promote lymphatic metastasis by remodeling the extracellular matrix and directing lymphovascular invasion along organized collagen fibers
Taken together, our in vitro data implicated periostin-expressing CAFs in collagen-mediated collective invasion. To further explore the specific role of this population during tumor progression, we used a DTA depletion strategy to ablate periostin-expressing CAFs in vivo. We generated PostnDTA mice by crossing PostniZSGreen mice with Rosa-DTA mice. In this model, tamoxifen administration drives expression of diphtheria toxin in periostin-expressing cells resulting in their selective ablation. To confirm ablation of periostin-expressing cells, we quantified ZSGreen+ cells in the primary mammary tumors of control (PostniZSGreen) mice compared to periostin+ cell-depleted (PostnDTA) mice. There was a 75% reduction in the percentage of tissue area positive for ZSGreen in the mammary tumors of the PostnDTA mice, indicating successful ablation of the majority of periostin-expressing cells (Supplemental Figure 6). We then used these PostnDTA mice in our mammary tumor model to measure changes in matrix architecture and metastasis following ablation of periostin-expressing cells (Figure 7A). PostnDTA mice were treated with either tamoxifen or vehicle-only control, then injected with highly-metastatic EO771.LMBmCherry mammary cancer cells. Primary tumors were resected and a booster dose of tamoxifen (or vehicle-only control) was administered to ablate any periostin-expressing cells that may have been activated as a result of surgery as periostin is induced by tissue injury. Finally, secondary sites were harvested and evaluated for metastatic burden.
Figure 7. Periostin-expressing CAFs promote lymphatic metastasis by remodeling the extracellular matrix and directing lymphovascular invasion along organized collagen fibers.
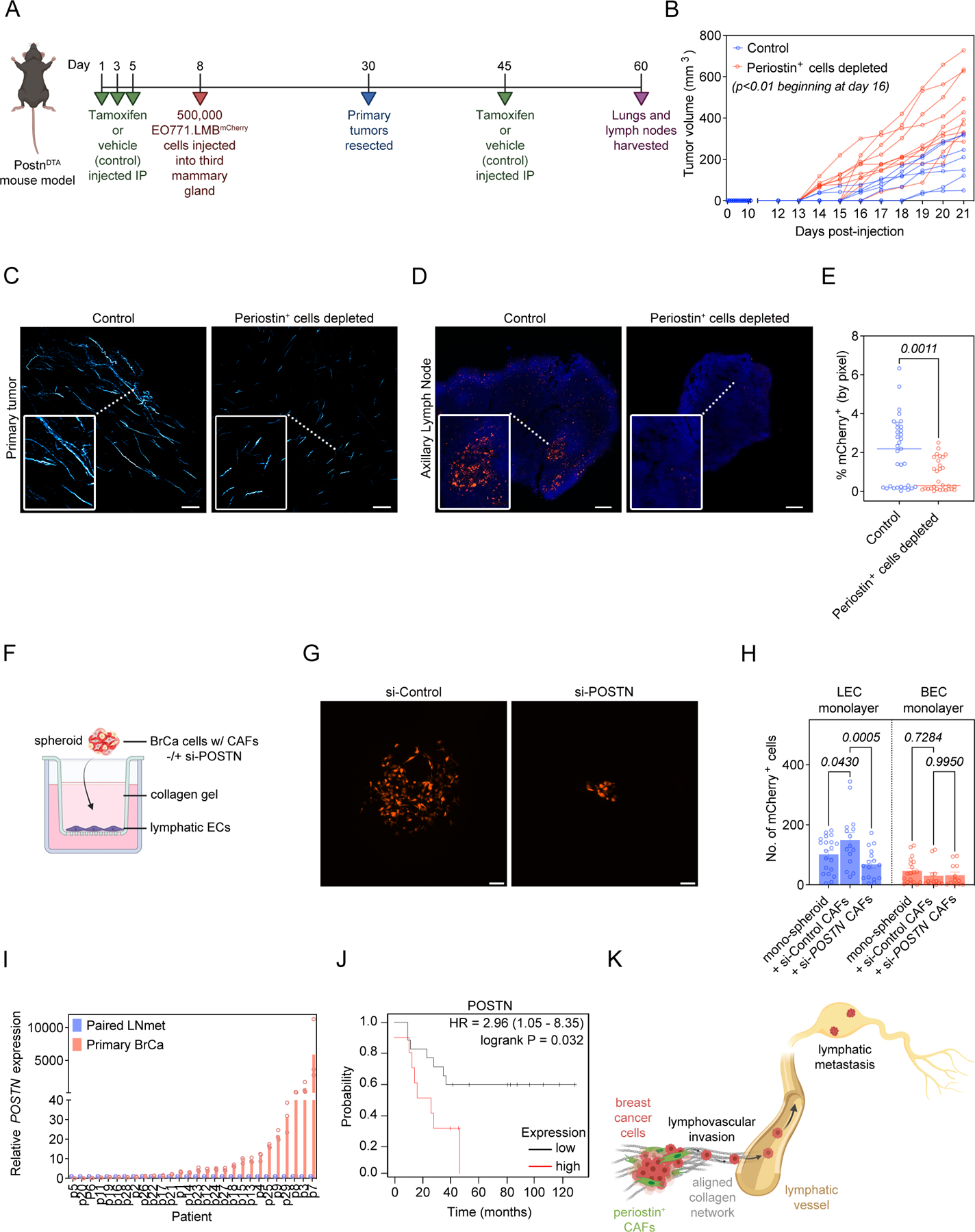
(A) Study design in PostnDTA mice. (B) Volume measurements of EO771.LMB tumors in control versus periostin+ cell-depleted mice. Each line represents an individual mouse (n = 8–10 mice per group). Statistics shown for multiple Mann-Whitney tests. (C) SHG images of intratumoral collagen fibers (pseudo-colored in cyan) in control versus periostin+ cell-depleted mice. Scale bars: 150 μm, insets: 3x zoom. (D) Tissue tilescans of axillary lymph nodes from control versus periostin+ cell-depleted mice bearing EO771.LMB mammary tumors. Tumor cells labelled with mCherry and nuclei counterstained with DAPI. Scale bars: 200 μm, insets: 3x zoom. (E) Percentage of tissue area positive for mCherry in sections of axillary lymph nodes from control versus periostin+ cell-depleted mice bearing highly metastatic EO771.LMB mammary tumors. Each data point represents an individual histological section (n = 8–10 mice per group). Statistics shown for Mann-Whitney test. (F) Experimental setup for in vitro lymphovascular invasion assay using MDA-MB-231mCherry human breast cancer cells, primary human breast CAFs, and primary human lymphatic endothelial cells. (G) Representative images of MDA-MB-231mCherry tumor cells that have invaded across the lymphatic endothelial cell barrier in the in vitro lymphovascular invasion assay. Scale bar: 100 μm. (H) Quantification of MDA-MB-231mCherry tumor cells that have invaded across the lymphatic endothelial cell (LEC) barrier or blood endothelial cell (BEC) barrier in the lymphovascular invasion assay. Spheroids consisted of either MDA-MB-231mCherry tumor cells alone (mono-spheroids) or co-cultures of MDA-MB-231mCherry tumor cells with primary human breast CAFs treated with either non-targeting control siRNA (si-Control) or periostin-targeting siRNA (si-POSTN). Each data point represents an individual spheroid. Experiment performed in triplicate. Statistics shown for 2-way ANOVA. (I) RT-qPCR analysis of periostin expression in paired primary breast cancer specimens (Primary BrCa) and lymph node metastases (Paired LNmet) from human breast cancer patients (n = 28 patients). qPCR performed in triplicate. (J) Kaplan-Meier plot of overall survival in lymph node positive breast cancer patients stratified by periostin protein expression (n = 27 patients). (K) Summary model. (Schematics created with BioRender.com).
Surprisingly, depleting periostin-expressing cells accelerated primary tumor growth which indicates a growth-restraining feature of periostin-expressing cells at sites of primary growth by breast cancer cells (Figure 7B). In line with previous observations, mammary tumors in which periostin-expressing cells had been depleted displayed impaired intratumoral collagen organization (Figure 7C), as collagen fibers were less aligned (Supplemental Figure 7A) and shorter in the tumors of periostin+ cell-depleted mice (235.0 ± 8.0 µm versus 154.9 ± 5.5 µm) (Supplemental Figure 7B). As expected, total collagen area was significantly reduced following periostin+ cell depletion (Supplemental Figure 7C), but we unexpectedly found that the curvature of collagen fibers in periostin+ cell-depleted tumors was reduced as well (Supplemental Figure 7D). This indicates that depleting periostin+ cells reduced collagen abundance, fiber length, and alignment, but did not revert the collagen fibers to the “curly” phenotype observed in the periostinlow EO771 primary tumors. Periostin+ cell depletion also dramatically reduced lymphatic metastasis of EO771.LMBmCherry mammary cancer cells. The metastatic burden in the draining axillary lymph nodes of periostin+ cell-depleted mice was reduced ~3-fold (2% ± 0.3% versus 0.8% ± 0.1%) (Figure 7D,E), suggesting that periostin-expressing cells enable lymphatic metastasis. Consistent with previous results, very few EO771.LMBmCherry cells reached the lungs in both the vehicle-control mice and periostin+ cell-depleted mice, and we did not observe a statistically significant reduction in pulmonary metastases following periostin+ cell depletion (Supplemental Figure 7E,F). In addition to changes in collagen organization, we also observed a shift in the immune landscape within primary tumors following periostin+ cell depletion with a significant increase in CD3+ T cells and CD68+ macrophages in both highly-metastatic tumor models as well as a significant increase in Foxp3+ regulatory T cells in the highly-metastatic PyVMT model (Supplemental Figure 8).
After confirming that periostin+ cell depletion reduced lymphatic metastasis but did not affect pulmonary metastasis in highly-metastatic PyVMT tumors (Supplemental Figure 9A-D), we next asked how attenuating periostin expression by blocking its upstream effector TGFβ would impact metastasis (Supplemental Figure 10A). We hypothesized that reducing TGFβ-mediated periostin expression by conditionally knocking out a receptor in the TGFβ pathway (Tgfbr2) in periostin-expressing cells would similarly reduce lymphatic metastasis. Using Postn-Cre:Tgfbr2fl/fl mice, we found a significant reduction in lymphatic metastasis of EO771.LMBmCherry cells following Tgfbr2 knockout (Supplemental Figure 10B,C). Given this reduction in lymphatic metastasis following periostin depletion in vivo, we then modelled lymphovascular invasion in vitro to determine whether periostin+ CAFs guide the intravasation of cancer cells across lymphatic endothelial cell (LEC) barriers. Using a modified spheroid/transwell migration assay (Figure 7F), we found that culturing MDA-MB-231mCherry human breast cancer cells into spheroids with periostin-expressing human breast CAFs enhanced their ability to invade across LEC monolayers compared to control mono-spheroids that consisted of MDA-MB-231mCherry cancer cells alone (149 ± 24 cells versus 101 ± 13 cells). However, periostin knockdown in CAFs attenuated their ability to promote cancer cell invasion across LEC monolayers (Figure 7G,H). This effect of periostin+ CAFs on cancer cell invasion appears to be specific to lymphatic vessels, as co-culturing MDA-MB-231mCherry cancer cells with either control periostin-expressing CAFs or si-POSTN CAFs did not affect cancer cell invasion across blood endothelial cell (BEC) barriers when we repeated the assay using human umbilical vein endothelial cells (HUVECs) (Figure 7H). These data indicate that periostin-expressing CAFs promote lymphovascular invasion of breast cancer cells.
Taken together, our data reveal a role for periostin-expressing CAFs in driving collagen-mediated lymphovascular invasion of cancer cells, resulting in lymphatic metastasis to the draining lymph node. To assess whether this pro-metastatic role for periostin within the primary tumor is reflected in clinical specimens, we used RT-qPCR analysis to measure relative periostin expression in paired primary breast tumors and lymph node metastases from 28 breast cancer patients obtained from the UNC Chapel Hill tissue biorepository. We found that periostin expression was higher in primary tumors compared to their paired lymph node samples in 68% of patients (Figure 7I), which is consistent with periostin functioning within the primary TME to promote breast cancer cell escape and eventual metastasis. To determine if periostin is associated with poor patient outcome, we used the Kaplan-Meier Plotter to perform survival analysis of lymph node positive breast cancer patients stratified by periostin protein expression (Figure 7J) (50). As predicted, high periostin expression significantly correlated with decreased overall survival probability in this patient cohort, further supporting a clinically-relevant role for periostin in driving disease progression (Figure 7K).
Discussion
Considerable progress has been made in the field of CAF biology using single-cell sequencing technologies to identify molecularly diverse CAF subpopulations. While expression profiles can indicate potential functions for subclasses of CAFs, the clustering algorithms used to analyze sequencing data can yield subpopulations that are not spatially or pathologically relevant. Therefore, there remains a need to track these cells in situ and validate the functions of CAF subpopulations to determine their biological importance during tumor progression. In the present study, we have used lineage tracing strategies to characterize a population of cells marked by expression of periostin, a matricellular protein associated with metastasis and expressed by the desmoplastic stroma of human breast cancers. We show that periostin is expressed by CAFs and perivascular-like cells and is enriched in advanced stage and lymph node positive human breast cancer samples. Our work also identifies a population of periostin-expressing CAFs that are enriched at the lymphatic vessel periphery and are differentially activated in highly-metastatic mammary tumors compared to their low-metastatic counterparts. In addition to quantitative differences, we observe phenotypic variation among periostin-expressing cells between tumor types. ZSGreen-labelled periostin+ cells in highly-metastatic tumors are larger and resemble activated myofibroblasts whereas periostin-expressing cells in low-metastatic tumors are smaller and rounded, reflecting the different activation states between the two populations. Together, this shows that the metastatic potential of cancer cells informs the activation of periostin-expressing cells in the primary TME. Interestingly, we find that the same is true at metastatic and premetastatic sites. We show that periostin-expressing cells are more abundant in the draining lymph nodes, lungs, and contralateral mammary glands of mice bearing highly-metastatic mammary tumors compared to those implanted with low-metastatic mammary tumors. A limitation of our spontaneous metastasis model is that relatively few mammary cancer cells reach the lungs before mice must be sacrificed. To address this, we used an experimental model of metastasis in which mammary cancer cells were injected into the tail vein of lineage tracing mice. Both low- and highly-metastatic mammary cancer lines established macrometastases in the lungs but marginally activated periostin-expressing cells despite a greater number of cancer cells present in these tissues. This indicates that periostin activation in a primary tumor is critical for robust activation of periostin at secondary sites. These findings support previous reports that periostin is activated in premetastatic (24) and metastatic niches (26) where it may promote cancer cell survival and outgrowth, though our study focuses on an earlier pro-metastatic role for periostin within the primary tumor.
We find that periostin-expressing cell abundance is associated with intratumoral collagen abundance and organization, with straighter, longer, and more aligned collagen fibers in highly-metastatic (periostinhigh) tumors. Our in vitro assays reveal that this difference in collagen matrix architecture is due to differences in intratumoral periostin abundance, as knocking down periostin expression in primary human breast CAFs reduces expression of a number of matrix-related proteins including collagens. This reduction in collagen secretion leads to a deficient and unorganized matrix, which impairs the ability of primary human breast CAFs to spread and migrate. These spreading and migratory defects are rescued by treatment with recombinant human periostin, suggesting that the differences in periostin-expressing cell morphology we observe in vivo in low- versus highly-metastatic tumors are directly related to differences in intratumoral periostin levels. We also show that periostin knockdown selectively inhibits collective cell invasion of primary human breast CAFs and breast cancer cells through collagen matrices. Though the timing of our in vitro assay is too short for CAFs to remodel collagen into the highly organized fibrillar networks we observe in periostinhigh tumors, our results reflect that the inhibitory effect of periostin knockdown on early steps of collagen remodeling is sufficient to reduce collective cell invasion. If CAF-derived periostin promotes collagen cross-linking as proposed, cancer cells may engage the resulting matrix differently in control versus periostin-depleted contexts. This could increase cancer cell proliferation in control conditions as stiffened ECM can drive tumor proliferation (46), though we observe the opposite in vivo with depletion of periostin-expressing cells increasing primary tumor growth despite impaired collagen organization. This accelerated tumor growth could be attributed to the loss of an organized collagen matrix, as fibrillar collagen can mechanically restrain tumor growth (51). Alternatively, periostin-expressing cell depletion may enhance primary tumor growth by shifting the immune landscape within tumors as has been previously observed upon CAF depletion in a model of pancreatic cancer (13). We observe an increase in T cells, including regulatory T cells, and macrophages in highly-metastatic tumors following periostin+ cell depletion, so it could be that immune populations are better able to access the primary tumor once periostin+ CAFs are depleted. However, the depletion process itself could invoke an inflammatory response as periostin-expressing cells undergo apoptosis and are cleared from the tissue, so the change in the immune milieu within the primary tumor may be unrelated to periostin+ CAFs and could instead be a feature of the mouse model. Therefore, an important future direction is to determine if periostin-expressing CAFs perform immune-modulatory functions in addition to their roles in matrix remodeling and to classify the specific immune populations present in periostin+ cell-depleted tumors to examine whether they support more aggressive tumor outgrowth.
Although our lineage tracing studies reveal that periostin-expressing cells make up a relatively small proportion of the total tumor tissue, they have a dramatic effect on the matrix architecture of the tumor microenvironment and lymphatic metastasis as evidenced by our periostin+ cell depletion tumor study. Our data reveal that periostin-expressing CAFs are instrumental in mediating lymphatic metastasis by depositing an organized collagen matrix and promoting lymphovascular invasion of breast cancer cells. CAFs and collagen alignment have been shown to play a role in hematogenous metastasis (52–63), but our work shows that periostin-expressing CAFs selectively promote lymphatic metastasis, as their depletion markedly reduces metastatic burden in the lymph node but does not significantly affect pulmonary metastasis. It could be that CAF-derived periostin generally promotes collagen remodeling and collective cell invasion towards both lymphatic and blood vessel interfaces within the primary tumor, but this enhanced invasion only leads to a difference in cancer cell invasion across lymphatic endothelial cell barriers and not blood endothelial cell barriers due to structural differences between the two types of vessels. Blood endothelial cells form tight junctions, making blood vessels inherently more restrictive, whereas lymphatic endothelial cells are characterized by discontinuous junctions that allow for the entry of fluid, macromolecules, and cells into lymphatic vessels during interstitial pressure regulation (64). This is reflected in our in vitro data that fewer cancer cells crossed the blood endothelial cell barrier versus the lymphatic endothelial cell barrier. These findings agree with and build upon recently published work showing that CAF-derived periostin can mediate metastasis by promoting lymphatic vessel permeability in an experimental model of popliteal lymph node metastasis (41), though this work used gain of function experiments to determine the effect of injected periostin on lymphatic metastasis of cervical squamous cell carcinoma whereas we have used genetic tools to determine the function of periostin+ cells in spontaneous breast cancer metastasis. Similarly, another study of periostin in experimental lymph node metastasis has shown an association between periostin deposition and lymphangiogenesis within lymph nodes prior to colonization by murine melanoma cells (65). This finding is consistent with our lineage tracing results showing periostin activation in lymph nodes as part of premetastatic niche formation, though our work emphasizes an ECM-remodeling role for periostin within the primary tumor site. Our mouse model recapitulates the desmoplastic stromal reaction linked to poor prognosis of breast cancer patients and allows us to deplete the periostin+ CAFs that contribute to this desmoplasia to reveal their critical role in collagen-mediated lymphovascular invasion. As our study is the first to lineage trace and deplete periostin-expressing cells in the tumor context, this strategy should be adapted to other orthotopic tumor models to reveal whether this CAF population shares a similar role in promoting the lymphatic metastasis of other cancers, especially those also characterized by desmoplasia such as pancreatic and lung cancer (19,66,67).
Densely aligned fibrillar collagen distinguishes progressive breast tumors from in situ lesions and is associated with increased periostin deposition in human patients (27), so early targeting or re-programming of the periostin+ CAF population could be an intervention to help prevent breast tumors from advancing to invasive disease. An important therapeutic consideration would be the timing of targeting periostin+ cells, as we observe increased primary tumor growth following periostin+ cell depletion similar to the paradoxical results of other CAF depletion studies (12,13). To avoid this effect and given that periostin is activated by tissue injury and inflammation, blocking periostin+ CAF activation may be most effective around the time of surgical resection of the tumor, though further preclinical studies are required to examine the effect of periostin blockade at different timepoints during tumor progression and whether this could be an effective strategy to normalize the ECM and attenuate lymphatic metastasis. The ability of desmoplasia to drive breast cancer metastasis by supporting cancer cell outgrowth at secondary sites has been established (52,68), but attempts to target mediators of this process have been primarily preclinical with limited and largely unsuccessful clinical trials (69–71). Our study identifies a population of CAFs, including PVL-CAFs, responsible for the collagen remodeling that drives desmoplasia and demonstrates their role in promoting lymphovascular invasion and lymphatic metastasis, revealing a potential new avenue for therapeutic intervention in the metastatic cascade.
Supplementary Material
Significance Statement.
Highly-metastatic breast cancer cells activate a population of periostin-expressing cancer-associated fibroblasts that remodel the extracellular matrix to promote escape of cancer cells into lymphatic vessels and drive colonization of proximal lymph nodes.
Acknowledgments
JLN previously received support from an NCI Ruth L. Kirschstein NRSA for Individual Predoctoral Fellows Award (F31CA247407-02) and the NIH/NCI training grant T32CA009109. ACD is supported by grants from the American Cancer Society (129755-RSG-16-176-DDC), the National Institutes of Health/National Cancer Institute (2RO1 CA177875 and RO1 CA2558451), the Melanoma Research Alliance (ID612638), and funds from the Emily Couric Cancer Center at the University of Virginia. Portions of this research were supported by the NCI Cancer Center Support Grant 5P30CA044579 and by the UVA Genome Analysis and Technology Core (RRID:SCR_018883). This work used the Zeiss 710 multiphoton confocal microscope and Zeiss LSM 880 confocal microscope in the UVA Advanced Microscopy Facility which is supported by the University of Virginia School of Medicine. We would like to thank Natalia Dworak, Dr. Stacey Criswell, and Dr. Adrian Halme for their assistance using this equipment. Illumina NGS sequencing for the spatial RNA profiling was performed by the University of Minnesota Genomics Center. We would also like to thank Marya Dunlap-Brown of the UVA Molecular Immunologic and Translational Sciences (MITS) Core for her help with mouse surgeries. All mouse tissues were sectioned using the Thermo CryoStar NX50 cryostat in the UVA Biorepository and Tissue Research Facility (BTRF), and we wish to thank Angela Miller for her training and technical assistance. We would also like to thank Barushi Amarasinghe of the UVA BTRF for his assistance with immunohistochemistry. BioRender was used to design all schematics.
Footnotes
Competing interests
There are no competing interests to declare.
There are no conflicts of interest to disclose
References
- 1.Redig AJ, McAllister SS. Breast cancer as a systemic disease: a view of metastasis. J Intern Med 2013;274:113–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mohammed RAA, Martin SG, Gill MS, Green AR, Paish EC, Ellis IO. Improved Methods of Detection of Lymphovascular Invasion Demonstrate That It is the Predominant Method of Vascular Invasion in Breast Cancer and has Important Clinical Consequences. The American Journal of Surgical Pathology 2007;31 [DOI] [PubMed] [Google Scholar]
- 3.Houvenaeghel G, Cohen M, Classe JM, Reyal F, Mazouni C, Chopin N, et al. Lymphovascular invasion has a significant prognostic impact in patients with early breast cancer, results from a large, national, multicenter, retrospective cohort study. ESMO Open 2021;6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rakha EA, Martin S, Lee AH, Morgan D, Pharoah PD, Hodi Z, et al. The prognostic significance of lymphovascular invasion in invasive breast carcinoma. Cancer 2012;118:3670–80 [DOI] [PubMed] [Google Scholar]
- 5.Nathanson SD, Kwon D, Kapke A, Alford SH, Chitale D. The Role of Lymph Node Metastasis in the Systemic Dissemination of Breast Cancer. Annals of Surgical Oncology 2009;16:3396–405 [DOI] [PubMed] [Google Scholar]
- 6.Sabatier R, Jacquemier J, Bertucci F, Esterni B, Finetti P, Azario F, et al. Peritumoural vascular invasion: A major determinant of triple-negative breast cancer outcome. European Journal of Cancer 2011;47:1537–45 [DOI] [PubMed] [Google Scholar]
- 7.Liu T, Zhou L, Li D, Andl T, Zhang Y. Cancer-Associated Fibroblasts Build and Secure the Tumor Microenvironment. Frontiers in Cell and Developmental Biology 2019;7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Labernadie A, Kato T, Brugués A, Serra-Picamal X, Derzsi S, Arwert E, et al. A mechanically active heterotypic E-cadherin/N-cadherin adhesion enables fibroblasts to drive cancer cell invasion. Nature Cell Biology 2017;19:224–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marusyk A, Tabassum DP, Janiszewska M, Place AE, Trinh A, Rozhok AI, et al. Spatial Proximity to Fibroblasts Impacts Molecular Features and Therapeutic Sensitivity of Breast Cancer Cells Influencing Clinical Outcomes. Cancer Res 2016;76:6495–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Erdogan B, Ao M, White LM, Means AL, Brewer BM, Yang L, et al. Cancer-associated fibroblasts promote directional cancer cell migration by aligning fibronectin. J Cell Biol 2017;216:3799–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hirata E, Girotti MR, Viros A, Hooper S, Spencer-Dene B, Matsuda M, et al. Intravital imaging reveals how BRAF inhibition generates drug-tolerant microenvironments with high integrin β1/FAK signaling. Cancer Cell 2015;27:574–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014;25:735–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014;25:719–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Helms EJ, Berry MW, Chaw RC, DuFort CC, Sun D, Onate MK, et al. Mesenchymal Lineage Heterogeneity Underlies Non-Redundant Functions of Pancreatic Cancer-Associated Fibroblasts. Cancer Discovery 2021:candisc.0601.2021 [DOI] [PMC free article] [PubMed]
- 15.Bartoschek M, Oskolkov N, Bocci M, Lövrot J, Larsson C, Sommarin M, et al. Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nature Communications 2018;9:5150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davidson S, Efremova M, Riedel A, Mahata B, Pramanik J, Huuhtanen J, et al. Single-Cell RNA Sequencing Reveals a Dynamic Stromal Niche That Supports Tumor Growth. Cell Reports 2020;31:107628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Puram SV, Tirosh I, Parikh AS, Patel AP, Yizhak K, Gillespie S, et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017;171:1611–24.e24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Friedman G, Levi-Galibov O, David E, Bornstein C, Giladi A, Dadiani M, et al. Cancer-associated fibroblast compositions change with breast cancer progression linking the ratio of S100A4+ and PDPN+ CAFs to clinical outcome. Nature Cancer 2020;1:692–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lambrechts D, Wauters E, Boeckx B, Aibar S, Nittner D, Burton O, et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat Med 2018;24:1277–89 [DOI] [PubMed] [Google Scholar]
- 20.Chen S, Zhu G, Yang Y, Wang F, Xiao Y-T, Zhang N, et al. Single-cell analysis reveals transcriptomic remodellings in distinct cell types that contribute to human prostate cancer progression. Nature Cell Biology 2021;23:87–98 [DOI] [PubMed] [Google Scholar]
- 21.Öhlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med 2017;214:579–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu SZ, Roden DL, Wang C, Holliday H, Harvey K, Cazet AS, et al. Stromal cell diversity associated with immune evasion in human triple-negative breast cancer. The EMBO Journal 2020;39:e104063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maruhashi T, Kii I, Saito M, Kudo A. Interaction between periostin and BMP-1 promotes proteolytic activation of lysyl oxidase. J Biol Chem 2010;285:13294–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Z, Xiong S, Mao Y, Chen M, Ma X, Zhou X, et al. Periostin promotes immunosuppressive premetastatic niche formation to facilitate breast tumour metastasis. J Pathol 2016;239:484–95 [DOI] [PubMed] [Google Scholar]
- 25.Soikkeli J, Podlasz P, Yin M, Nummela P, Jahkola T, Virolainen S, et al. Metastatic outgrowth encompasses COL-I, FN1, and POSTN up-regulation and assembly to fibrillar networks regulating cell adhesion, migration, and growth. Am J Pathol 2010;177:387–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Malanchi I, Santamaria-Martínez A, Susanto E, Peng H, Lehr H-A, Delaloye J-F, et al. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature 2012;481:85–9 [DOI] [PubMed] [Google Scholar]
- 27.Risom T, Glass DR, Averbukh I, Liu CC, Baranski A, Kagel A, et al. Transition to invasive breast cancer is associated with progressive changes in the structure and composition of tumor stroma. Cell 2022;185:299–310.e18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kanisicak O, Khalil H, Ivey MJ, Karch J, Maliken BD, Correll RN, et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nature Communications 2016;7:12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brauer HA, Makowski L, Hoadley KA, Casbas-Hernandez P, Lang LJ, Romàn-Pèrez E, et al. Impact of Tumor Microenvironment and Epithelial Phenotypes on Metabolism in Breast Cancer. Clinical Cancer Research 2013;19:571–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xiao L, Kim DJ, Davis CL, McCann JV, Dunleavey JM, Vanderlinden AK, et al. Tumor Endothelial Cells with Distinct Patterns of TGFβ-Driven Endothelial-to-Mesenchymal Transition. Cancer Research 2015;75:1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wershof E, Park D, Barry DJ, Jenkins RP, Rullan A, Wilkins A, et al. A FIJI macro for quantifying pattern in extracellular matrix. Life Sci Alliance 2021;4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Püspöki Z, Storath M, Sage D, Unser M. Transforms and Operators for Directional Bioimage Analysis: A Survey. Adv Anat Embryol Cell Biol 2016;219:69–93 [DOI] [PubMed] [Google Scholar]
- 33.Banerjee A, Li D, Guo Y, Mei Z, Lau C, Chen K, et al. A reengineered common chain cytokine augments CD8+ T cell-dependent immunotherapy. JCI Insight 2022;7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu SZ, Al-Eryani G, Roden DL, Junankar S, Harvey K, Andersson A, et al. A single-cell and spatially resolved atlas of human breast cancers. Nature Genetics 2021;53:1334–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lund AW, Wagner M, Fankhauser M, Steinskog ES, Broggi MA, Spranger S, et al. Lymphatic vessels regulate immune microenvironments in human and murine melanoma. The Journal of Clinical Investigation 2016;126:3389–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mezey É, Szalayova I, Hogden CT, Brady A, Dósa Á, Sótonyi P, et al. An immunohistochemical study of lymphatic elements in the human brain. Proceedings of the National Academy of Sciences 2021;118:e2002574118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015;523:337–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Neri S, Ishii G, Hashimoto H, Kuwata T, Nagai K, Date H, et al. Podoplanin-expressing cancer-associated fibroblasts lead and enhance the local invasion of cancer cells in lung adenocarcinoma. Int J Cancer 2015;137:784–96 [DOI] [PubMed] [Google Scholar]
- 39.Shindo K, Aishima S, Ohuchida K, Fujiwara K, Fujino M, Mizuuchi Y, et al. Podoplanin expression in cancer-associated fibroblasts enhances tumor progression of invasive ductal carcinoma of the pancreas. Mol Cancer 2013;12:168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Johnstone CN, Smith YE, Cao Y, Burrows AD, Cross RS, Ling X, et al. Functional and molecular characterisation of EO771.LMB tumours, a new C57BL/6-mouse-derived model of spontaneously metastatic mammary cancer. Dis Model Mech 2015;8:237–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alkner S, Tang M- HE, Brueffer C, Dahlgren M, Chen Y, Olsson E, et al. Contralateral breast cancer can represent a metastatic spread of the first primary tumor: determination of clonal relationship between contralateral breast cancers using next-generation whole genome sequencing. Breast Cancer Research 2015;17:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vichapat V, Garmo H, Holmberg L, Fentiman IS, Tutt A, Gillett C, et al. Patterns of metastasis in women with metachronous contralateral breast cancer. British Journal of Cancer 2012;107:221–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Norris RA, Damon B, Mironov V, Kasyanov V, Ramamurthi A, Moreno-Rodriguez R, et al. Periostin regulates collagen fibrillogenesis and the biomechanical properties of connective tissues. J Cell Biochem 2007;101:695–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Egeblad M, Rasch MG, Weaver VM. Dynamic interplay between the collagen scaffold and tumor evolution. Curr Opin Cell Biol 2010;22:697–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Provenzano PP, Eliceiri KW, Campbell JM, Inman DR, White JG, Keely PJ. Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Medicine 2006;4:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009;139:891–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Conklin MW, Eickhoff JC, Riching KM, Pehlke CA, Eliceiri KW, Provenzano PP, et al. Aligned collagen is a prognostic signature for survival in human breast carcinoma. Am J Pathol 2011;178:1221–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van Helvert S, Storm C, Friedl P. Mechanoreciprocity in cell migration. Nat Cell Biol 2018;20:8–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Friedl P, Alexander S. Cancer invasion and the microenvironment: plasticity and reciprocity. Cell 2011;147:992–1009 [DOI] [PubMed] [Google Scholar]
- 50.Ősz Á, Lánczky A, Győrffy B. Survival analysis in breast cancer using proteomic data from four independent datasets. Scientific Reports 2021;11:16787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bhattacharjee S, Hamberger F, Ravichandra A, Miller M, Nair A, Affo S, et al. Tumor restriction by type I collagen opposes tumor-promoting effects of cancer-associated fibroblasts. J Clin Invest 2021;131. [DOI] [PMC free article] [PubMed]
- 52.Dumont N, Liu B, Defilippis RA, Chang H, Rabban JT, Karnezis AN, et al. Breast fibroblasts modulate early dissemination, tumorigenesis, and metastasis through alteration of extracellular matrix characteristics. Neoplasia 2013;15:249–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Condeelis J, Segall JE. Intravital imaging of cell movement in tumours. Nature Reviews Cancer 2003;3:921–30 [DOI] [PubMed] [Google Scholar]
- 54.Wang W, Wyckoff JB, Frohlich VC, Oleynikov Y, Hüttelmaier S, Zavadil J, et al. Single cell behavior in metastatic primary mammary tumors correlated with gene expression patterns revealed by molecular profiling. Cancer Res 2002;62:6278–88 [PubMed] [Google Scholar]
- 55.Han W, Chen S, Yuan W, Fan Q, Tian J, Wang X, et al. Oriented collagen fibers direct tumor cell intravasation. Proceedings of the National Academy of Sciences 2016;113:11208–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goetz Jacky G, Minguet S, Navarro-Lérida I, Lazcano Juan J, Samaniego R, Calvo E, et al. Biomechanical Remodeling of the Microenvironment by Stromal Caveolin-1 Favors Tumor Invasion and Metastasis. Cell 2011;146:148–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wullkopf L, West AV, Leijnse N, Cox TR, Madsen CD, Oddershede LB, et al. Cancer cells’ ability to mechanically adjust to extracellular matrix stiffness correlates with their invasive potential. Mol Biol Cell 2018;29:2378–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Barker HE, Chang J, Cox TR, Lang G, Bird D, Nicolau M, et al. LOXL2-mediated matrix remodeling in metastasis and mammary gland involution. Cancer Res 2011;71:1561–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Alexander S, Weigelin B, Winkler F, Friedl P. Preclinical intravital microscopy of the tumour-stroma interface: invasion, metastasis, and therapy response. Curr Opin Cell Biol 2013;25:659–71 [DOI] [PubMed] [Google Scholar]
- 60.Wolf K, Alexander S, Schacht V, Coussens LM, von Andrian UH, van Rheenen J, et al. Collagen-based cell migration models in vitro and in vivo. Semin Cell Dev Biol 2009;20:931–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ilina O, Bakker GJ, Vasaturo A, Hofmann RM, Friedl P. Two-photon laser-generated microtracks in 3D collagen lattices: principles of MMP-dependent and -independent collective cancer cell invasion. Phys Biol 2011;8:015010. [DOI] [PubMed] [Google Scholar]
- 62.Lee HO, Mullins SR, Franco-Barraza J, Valianou M, Cukierman E, Cheng JD. FAP-overexpressing fibroblasts produce an extracellular matrix that enhances invasive velocity and directionality of pancreatic cancer cells. BMC Cancer 2011;11:245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Venning FA, Wullkopf L, Erler JT. Targeting ECM Disrupts Cancer Progression. Front Oncol 2015;5:224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang F, Zarkada G, Yi S, Eichmann A. Lymphatic Endothelial Cell Junctions: Molecular Regulation in Physiology and Diseases. Front Physiol 2020;11:509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gillot L, Lebeau A, Baudin L, Pottier C, Louis T, Durré T, et al. Periostin in lymph node pre-metastatic niches governs lymphatic endothelial cell functions and metastatic colonization. Cell Mol Life Sci 2022;79:295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Whatcott CJ, Diep CH, Jiang P, Watanabe A, LoBello J, Sima C, et al. Desmoplasia in Primary Tumors and Metastatic Lesions of Pancreatic Cancer. Clinical Cancer Research 2015;21:3561–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Altorki NK, Markowitz GJ, Gao D, Port JL, Saxena A, Stiles B, et al. The lung microenvironment: an important regulator of tumour growth and metastasis. Nature Reviews Cancer 2019;19:9–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cox TR, Bird D, Baker AM, Barker HE, Ho MW, Lang G, et al. LOX-mediated collagen crosslinking is responsible for fibrosis-enhanced metastasis. Cancer Res 2013;73:1721–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ferreira S, Saraiva N, Rijo P, Fernandes AS. LOXL2 Inhibitors and Breast Cancer Progression. Antioxidants (Basel) 2021;10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nandi T, Pradyuth S, Singh AK, Chitkara D, Mittal A. Therapeutic agents for targeting desmoplasia: current status and emerging trends. Drug Discovery Today 2020;25:2046–55 [DOI] [PubMed] [Google Scholar]
- 71.Huang J, Zhang L, Wan D, Zhou L, Zheng S, Lin S, et al. Extracellular matrix and its therapeutic potential for cancer treatment. Signal Transduction and Targeted Therapy 2021;6:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.