

Abstract
Neuropathic pain is a common, debilitating chronic pain condition caused by damage or disease affecting the somatosensory nervous system. Understanding the pathophysiological mechanisms underlying neuropathic pain is critical for developing new therapeutic strategies to treat chronic pain effectively. Tiam1 is a Rac1 guanine nucleotide exchange factor (GEF) that promotes dendritic and synaptic growth during hippocampal development by inducing actin cytoskeletal remodeling. Here, using multiple neuropathic pain animal models, we show that Tiam1 coordinates synaptic structural and functional plasticity in the spinal dorsal horn via actin cytoskeleton reorganization and synaptic NMDAR stabilization and that these actions are essential for the initiation, transition, and maintenance of neuropathic pain. Furthermore, antisense oligonucleotides (ASOs) targeting spinal Tiam1 persistently alleviate neuropathic pain sensitivity. Our findings suggest that Tiam1-coordinated synaptic functional and structural plasticity underlies the pathophysiology of neuropathic pain and that intervention of Tiam1-mediated maladaptive synaptic plasticity has long-lasting consequences in neuropathic pain management.
In Brief:
Current treatment options for neuropathic pain are limited. In this study, Li et al. identify Tiam1-coordinated maladaptive synaptic structural and functional plasticity in spinal excitatory neurons as the pathophysiological mechanism that initiates, transits, and sustains neuropathic pain, thus providing a promising therapeutic target to treat neuropathic pain effectively.
INTRODUCTION
Neuropathic pain is a type of chronic pain triggered by nerve injury or disease that results in pathological hyperexcitability of somatosensory nociceptive circuits, a condition that negatively impacts the quality of life1,2. Unfortunately, current treatment options for neuropathic pain primarily focus on symptom suppression and are limited in use due to their low efficacy and undesirable side effects3. Thus, a major challenge for pain research is to elucidate the pathophysiological mechanisms underlying neuropathic pain and harness this knowledge to develop effective therapeutic strategies for long-lasting pain management.
Despite diverse pathological triggers (e.g., nerve injury, diabetes, chemotherapy drugs), a common feature of neuropathic pain is central sensitization induced by functional and structural maladaptive synaptic plasticity in the spinal dorsal horn that results in pain hypersensitivity4,5. The functional plasticity underlying neuropathic pain involves diminished synaptic inhibition (disinhibition) and increased synaptic excitation of spinal dorsal horn neurons6–8. Structurally, increases in the size and density of dendritic spines, the primary postsynaptic sites of excitatory synapses, have been reported in the superficial spinal cord of diverse models of neuropathic pain and account for the long-term nature of neuropathic pain9–12. However, the mechanisms controlling how persistent nociceptive activity drives synaptic plasticity in the spinal dorsal horn and how synaptic structural and functional plasticity are coordinated during neuropathic pain development remain unclear.
In neurons, the Rho-family small GTPase Rac1 typically promotes actin polymerization that drives dendritic spine formation, growth, and stabilization, whereas RhoA induces actin-myosin contractility, resulting in spine shrinkage and loss13. Rho GTPases function by cycling between an active GTP-bound and inactive GDP-bound form, and their activation state and downstream signaling are tightly controlled by guanine nucleotide exchange factors (GEFs, activators) and GTPase-activating proteins (GAPs, inhibitors)13,14. We previously identified the multifunctional Rac1-GEF Tiam1 as a critical regulator of Rac1-dependent dendritic spine and excitatory synapse development in hippocampal neurons15–17. Tiam1 is recruited to activated N-methyl-D-aspartate-type glutamate receptors (NMDARs) and TrkB and EphB receptor tyrosine kinases where it induces local Rac1 activation and actin polymerization, resulting in dendritic spine remodeling15,16,18. Notably, all three of these synaptic receptors are expressed in the spinal dorsal horn and have been implicated in the development of neuropathic pain6,7,19. Furthermore, an epigenome-wide association study (EWAS) of 1708 monozygotic and dizygotic Caucasian twins found that Tiam1 was significantly associated with widespread chronic pain20. Here, we present evidence that Tiam1 coordinates persistent nociceptive activity-induced structural and functional plasticity via actin cytoskeletal reorganization and synaptic NMDAR stabilization in spinal dorsal horn neurons, which together underpins the pathophysiology of neuropathic pain. Moreover, we develop an antisense oligonucleotide (ASO) strategy targeting spinal Tiam1 for the potential treatment of neuropathic pain.
RESULTS
Tiam1 activity is required for the development of neuropathic pain.
To determine the role of Tiam1 in the development of neuropathic pain, we first investigated whether Tiam1 is activated in the spinal dorsal horn of mice subjected to neuropathic pain. We conducted an affinity-precipitation assay using GST-Rac1G15A, a guanine nucleotide-free form of Rac1 that preferentially binds to activated GEFs21. The active-GEF assay was performed on spinal dorsal horn homogenates from mice subjected to sham or spared nerve injury (SNI), a widely used animal model of neuropathic pain (Fig. 1A)22,23. We found that the levels of Tiam1 that precipitated with Rac1G15A from SNI mice were markedly higher than from sham controls 3 weeks after surgery (Fig. 1B), indicating that Tiam1 is activated in the dorsal spinal horn of mice with neuropathic pain.
Figure 1. Tiam1 is essential for the development of neuropathic pain.
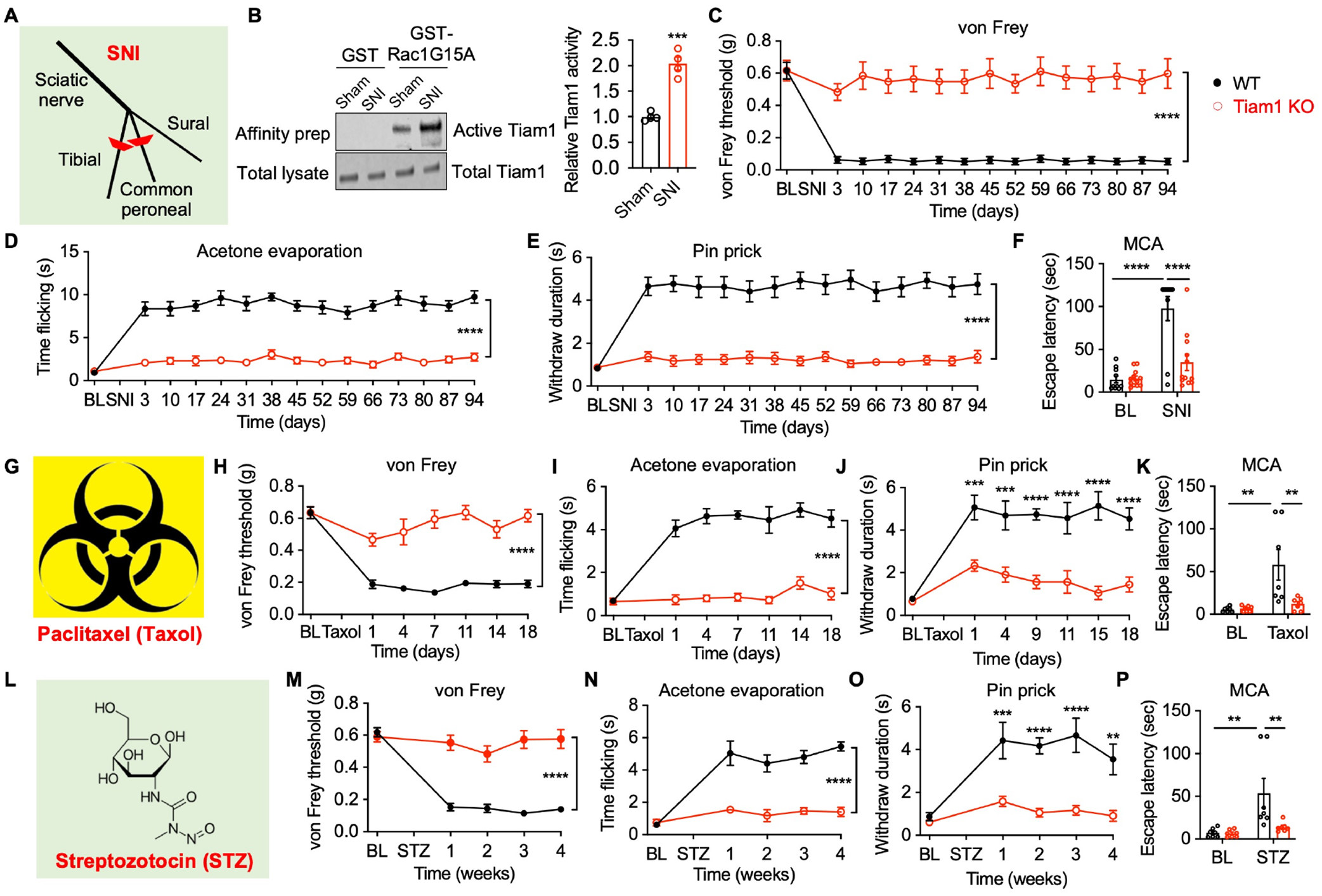
(A) Depiction of the spared nerve injury (SNI) neuropathic pain model.
(B) Representative blots and quantification of the active-GEF assay showing that Tiam1 is activated in the spinal dorsal horn of mice subjected to neuropathic pain (3 weeks after SNI, n = 4 mice). Total Tiam1 levels were used as the loading control. Mann-Whitney U-test (sham vs SNI).
(C-F) Tiam1 deletion prevents SNI-induced neuropathic pain. Time course for changes in von Frey withdrawal thresholds (C), time flicking with acetone evaporation (D), and withdrawal duration to the pinprick of global Tiam1 KO mice and WT littermates before and after SNI (E). (F) Escape latency in the mechanical conflict-avoidance (MCA) assay 3 weeks after SNI. Two-way ANOVA analysis followed by Dunnett’s post-hoc comparison between groups (n = 10–12 mice/group. von Frey: P < 0.0001, F14,328 = 4.106; acetone: P < 0.0001, F14,330 = 9.780; pinprick: P < 0.0001, F14,330 = 5.157; MCA: P = 0.0006, F1,40 = 13.89).
(G-K) Tiam1 deletion blocks paclitaxel (Taxol)-induced neuropathic pain. Two-way ANOVA analysis followed by Dunnett’s post-hoc comparison between groups (n = 7–10 mice/group. von Frey: P < 0.0001, F6,126 = 13.71; acetone: P < 0.0001, F6,84 = 14.52; pinprick: P < 0.0001, F6,98 = 9.063; MCA: P = 0.0039, F1,24 = 10.22).
(L-P) Tiam1 deletion prevents streptozotocin (STZ)-induced neuropathic pain. Two-way ANOVA analysis followed by Dunnett’s post-hoc comparison between groups (n = 6–10 mice/group. von Frey: P < 0.0001, F4,83 = 19.43; acetone: P < 0.0001, F4,55 = 19.72; pinprick: P < 0.0001, F4,70 = 7.469; MCA: P = 0.0058, F1,24 = 9.182).
Data are presented as means ± s.e.m. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
See also Figures S1 and S2.
To investigate whether activated Tiam1 in the spinal dorsal horn contributes to neuropathic pain, we generated Tiam1 global knockout (Tiam1 KO) mice by crossing Tiam1fl/fl mice with an Ella-Cre transgenic line. We confirmed Tiam1 loss in the spinal cord and dorsal root ganglion (DRG) via western blot analysis (Fig. S1A). Mice lacking Tiam1 are viable, fertile, and display no gross alterations in DRG, spinal cord, or brain structures17. Global Tiam1 KO mice also perform as well as wild-type (WT) littermates on an accelerating rotarod, suggesting that they do not have deficits in motor coordination, motor learning, or balance17. Importantly, no differences were observed between naïve global Tiam1 KO mice and their WT littermates in their responses to von Frey filaments (i.e., mechanical sensitivity) or a hotplate (i.e., heat sensitivity) (Fig. S1B, C), indicating that Tiam1 is not necessary for nociceptive pain. We thus subjected adult WT and global Tiam1 KO mice to SNI surgery and then tested the mice using behavioral assays over a 3-month period to assess the development of neuropathic pain under clinically relevant conditions. As expected, following SNI surgery, WT mice displayed a marked hypersensitivity to innocuous mechanical von Frey filament stimuli (Fig. 1C), increased flicking times in response to acetone evaporation (Fig. 1D), and increased withdrawal duration in response to pinprick stimulation (Fig. 1E) during the entire monitoring period, indicating that SNI induces prolonged tactile allodynia and hyperalgesia in WT mice. Strikingly, in global Tiam1 KO mice, SNI failed to cause neuropathic pain hypersensitivity, as shown by the absence of statistically significant differences in von Frey thresholds (Fig. 1C), flicking times (Fig. 1D), or withdrawal duration (Fig. 1E) compared to pre-surgery baselines. To further evaluate Tiam1’s role in neuropathic pain, we used an operant mechanical conflict-avoidance (MCA) test to measure non-reflexive pain behavior, which involves mice choosing either to remain in a brightly lit compartment or to escape to a dark compartment by crossing an array of height-adjustable nociceptive probes24,25. Three weeks after SNI surgery, WT mice, but not global Tiam1 KO mice, exhibited a significant increase in escape latency from the white-lit chamber at a probe height of 5 mm (Fig. 1F), suggesting that Tiam1 deletion also prevents the development of non-reflexive chronic pain behavior. Notably, pre-surgery reflexive and non-reflexive pain behaviors were not different between global Tiam1 KO mice and WT controls (Fig. 1C–F). A similar reduced mechanical and thermal hypersensitivity was observed when global Tiam1 KO mice were subjected to the chronic constriction injury (CCI) model of the neuropathic pain26 (Fig. S2A–D). These data indicate that Tiam1 plays an essential role in the development of peripheral nerve injury-induced neuropathic pain.
To explore Tiam1’s role in other types of neuropathic pain, we utilized the chemotherapy-induced peripheral neuropathy model27 (Fig. 1G). As expected, in WT mice, paclitaxel treatment (i.p. 2mg/kg/day for 5 days) induced a marked hypersensitivity to innocuous mechanical von Frey filament stimuli (Fig. 1H), increased flicking times with acetone evaporation (Fig. 1I), and increased withdrawal duration in response to pinprick stimulation (Fig. 1J) over a 2-week testing period. In contrast, in global Tiam1 KO mice paclitaxel treatment did not significantly change the tactile threshold, cold allodynia, or mechanical pinprick hyperalgesia relative to baseline values (Fig. 1H–J). Additionally, two weeks after paclitaxel treatment, WT mice, but not global Tiam1 KO mice, exhibited a significant increase in escape latency in the MCA assay (Fig. 1K). These data indicate that Tiam1 deletion prevents the development of chemotherapy-induced peripheral neuropathy.
We next used a streptozotocin-induced diabetic neuropathy model (i.p. 150mg/kg, single dose)28–30 to determine the role of Tiam1 in diabetes-induced neuropathic pain (Fig. 1L). Following streptozotocin injection, WT mice, but not global Tiam1 KO mice, displayed significant hypersensitivity to innocuous mechanical von Frey filament stimuli (Fig. 1M), increased flicking times with acetone evaporation (Fig. 1N), and increased withdrawal duration in response to pinprick stimulation (Fig. 1O) during a 4-week monitoring period, indicating that Tiam1 is required for the development of reflexive chronic pain behaviors. Likewise, the MCA assay showed that Tiam1 deletion prevents the development of non-reflexive pain (Fig. 1P). Together, these results reveal that Tiam1 is also required for the development of diabetes-induced neuropathic pain.
Tiam1 expressed in spinal dorsal horn neurons, not DRG or excitatory forebrain neurons, is essential for the development of neuropathic pain.
Persistent neuropathic pain is mediated by maladaptive changes in peripheral, spinal, and brain nociceptive circuits2,31. To test whether genetic deletion of Tiam1 from nociceptors affects the development of neuropathic pain, we crossed Tiam1fl/fl mice with an Advillin-Cre driver line (Fig. S3A). No differences were observed in responses to von Frey stimuli or acetone evaporation before or after SNI surgery between Advillin-Tiam1 cKO mice and their littermate controls throughout the 3 weeks of testing (Fig. S3B, C). Similarly, Tiam1 deletion from DRG neurons had no effect on paclitaxel- or streptozotocin-induced pain hypersensitivity (Fig. S3D–G). These data indicate that Tiam1 expression in DRG neurons is unnecessary for nociceptive or persistent neuropathic pain.
Since Tiam1 regulates excitatory synapse development and spine morphogenesis in the hippocampus17, we next crossed Tiam1fl/fl mice with CaMKIIα-Cre transgenic mice to delete Tiam1 specifically from postnatal forebrain excitatory neurons32,33 (Fig. S4A, B). Conditional deletion of Tiam1 from forebrain excitatory neurons did not alter basal mechanical or thermal sensitivity (Fig. S4C, D). Moreover, Tiam1 forebrain cKO mice exhibited similar mechanical and thermal hypersensitivity relative to littermate controls throughout the monitoring period following SNI surgery (Fig. S4C, D). These data indicate that, like DRG neurons, Tiam1 expression in forebrain excitatory neurons is not required for the development of neuropathic pain.
To investigate the role of spinal Tiam1 in neuropathic pain development, we specifically deleted Tiam1 in spinal dorsal horn neurons by performing intra-spinal dorsal horn microinjections of a rAAV8 vector expressing Cre-GFP under the control of the neuronal-specific promotor human synapsin (rAAV8-hSyn-Cre-GFP) in Tiam1fl/fl mice. Intra-spinal dorsal horn microinjections of a rAAV8 vector expressing GFP alone (rAAV8-hSyn-GFP) served as a control (Fig. 2A). rAAV8-hSyn-Cre-GFP injections resulted in a 60% reduction in Tiam1 levels in the spinal dorsal horn (Fig. 2B; Fig. S5E). Tiam1 deletion from spinal dorsal horn neurons did not affect basal nociceptive sensitivity (Fig. S5A, B). However, following SNI surgery, mice injected with the Cre-GFP-expressing virus developed significantly less pain hypersensitivity in response to mechanical stimuli (von Frey filaments) and cold (acetone evaporation) compared to mice injected with control GFP-expressing virus (Fig. 2C, D; Fig. S5C, D). Furthermore, Tiam1 deletion from spinal dorsal horn neurons prevented the development of chemotherapy- and diabetics-induced peripheral neuropathy (Fig. 2E–H). While intra-spinal dorsal microinjection of rAAV8-hSyn-Cre-GFP in Tiam1fl/fl mice might also disrupt Tiam1 expression in some motor neurons (Fig. 2A), this possibility is unlikely to confound our behavioral results since we recently showed that global Tiam1 KO mice perform as well as WT littermates on open field activity and accelerating rotarod, suggesting that they do not have deficits in locomotion, motor coordination, motor learning, or balance17. Thus, these results suggest that Tiam1 expressed in spinal cord dorsal horn neurons is required for the development of neuropathic pain.
Figure 2. Tiam1 deletion from spinal dorsal horn neurons prevents neuropathic pain development.
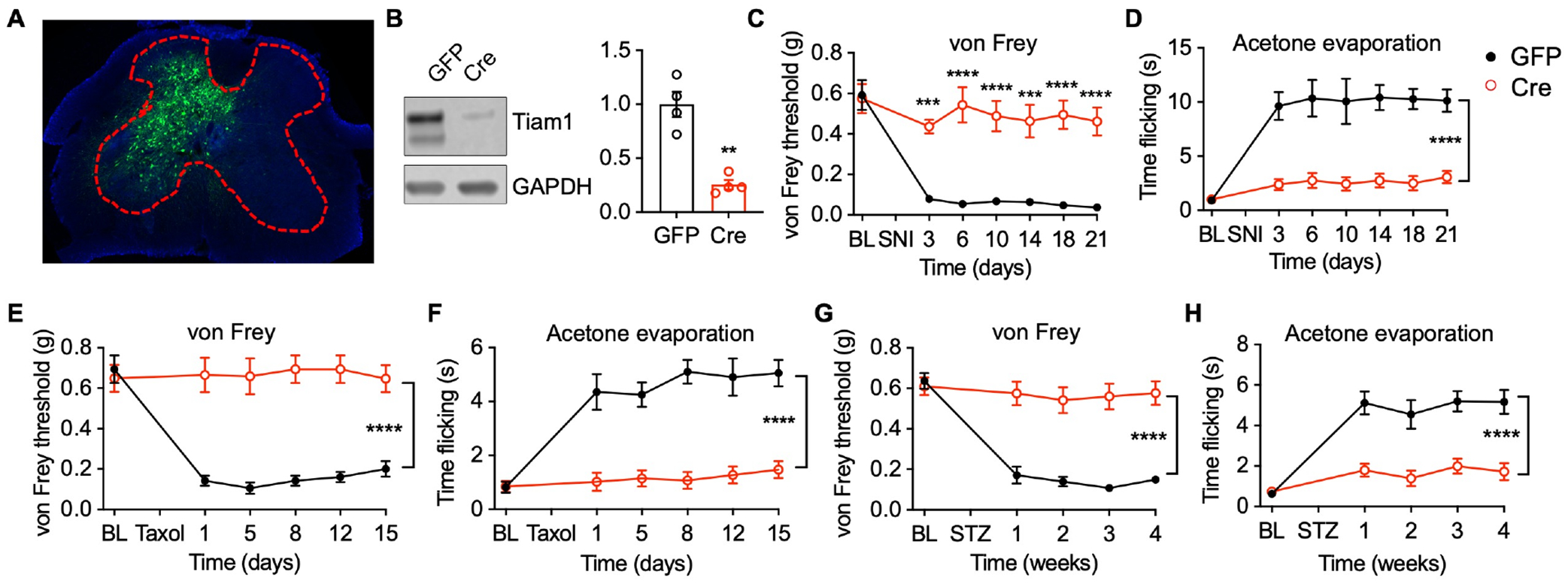
(A) Intra-spinal dorsal horn microinjection of rAAV8-hSyn-GFP (GFP).
(B) Representative blots and quantification show that injection of rAAV8-hSyn-Cre-GFP (Cre) downregulates Tiam1 protein levels in the spinal dorsal horn of Tiam1fl/fl mice. GAPDH was used as the loading control. Mann-Whitney U-test (n = 4).
(C and D) Tiam1 deletion from spinal dorsal horn neurons prevents SNI-induced neuropathic pain. Time course for changes in von Frey withdrawal thresholds and time flicking with acetone evaporation of Tiam1fl/fl mice infected with rAAV8-hSyn-GFP (GFP) or rAAV8-hSyn-Cre-GFP (Cre) before and after SNI. Two-way ANOVA analysis followed by Dunnett’s post-hoc test (n = 7–8 mice/group. von Frey: P < 0.0001, F6,84 = 7.477; acetone: P < 0.0001, F6,91 = 8.992).
(E and F) Tiam1 ablation from spinal dorsal horn neurons precludes paclitaxel (Taxol)-induced neuropathic pain. Two-way ANOVA analysis followed by Dunnett’s post-hoc test (n = 8 mice/group. von Frey: P < 0.0001, F5,84 = 6.408; acetone: P < 0.0001, F5,84 = 8.940).
(G and H) Tiam1 deletion from spinal dorsal horn neurons inhibits streptozotocin (STZ)-induced neuropathic pain. Two-way ANOVA analysis followed by Dunnett’s post-hoc test (n = 9–10 mice/group. von Frey: P < 0.0001, F4,84 = 14.69; acetone: P < 0.0001, F4,70 = 14.90).
Data are presented as means ± s.e.m. **P < 0.01, ***P < 0.001, ****P < 0.0001.
See also Figures S3, S4, and S5.
Tiam1 expressed in excitatory neurons, not inhibitory neurons, is necessary for neuropathic pain development.
Previous studies indicate that all spinal dorsal horn neurons express either Slc17a6 (vesicular glutamate transporter, vGluT2) or Slc32a1 (vesicular GABA transporter, vGat), representing glutamatergic excitatory interneurons or GABAergic inhibitory interneurons34,35. To investigate which of these cell types Tiam1 functions in to regulate the development of neuropathic pain, we deleted Tiam1 from either excitatory or inhibitory neurons by crossing Tiam1fl/fl mice with a vGluT2-Cre line or a vGat line, respectively (Fig. 3A and Fig. S6A). SNI failed to cause neuropathic pain hypersensitivity in vGluT2-Tiam1 cKO mice, but not in vGat-Tiam1 cKO mice, in both reflexive pain assays and the MCA non-reflexive pain assay (Fig. 3B–D and Fig. S6B–D). These data indicate that Tiam1 expressed in excitatory neurons, rather than inhibitory neurons, determines the development of neuropathic pain.
Figure 3. Tiam1 deletion from excitatory neurons prevents the development of neuropathic pain.
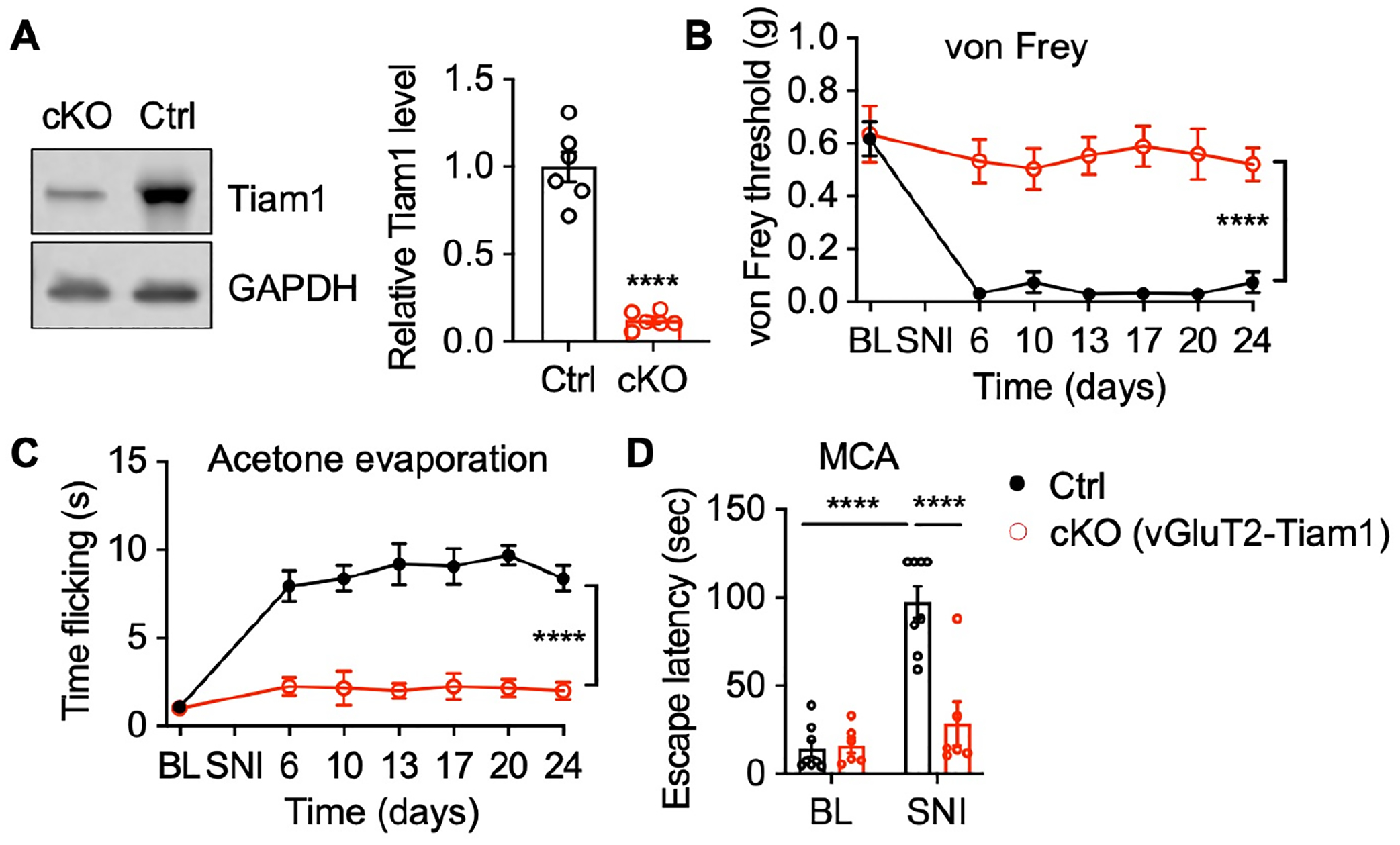
(A) Western blots and quantification showing Tiam1 deletion from vGluT2-expressing excitatory neurons in Tiam1 cKO mice (cKO, vGluT2-Cre∷Tiam1fl/fl) compared to littermate controls (Ctrl, vGluT2-Cre). Cre-dependent AAV vector expressing eGFP (AAV8-pCAG-DIO-eGFP) was injected into the spinal dorsal horn of control mice and vGluT2-Tiam1 cKO mice via intra-spinal dorsal horn microinjection. eGFP-expressing spinal dorsal horn neurons were isolated with flow cytometry for western blot analysis. GAPDH was used as the loading control. Two-tailed Student’s t-test (n = 6).
(B-D) Tiam1 deletion from vGluT2-expressing excitatory neurons prevents SNI-induced neuropathic pain. von Frey withdrawal thresholds (B), time flicking with acetone evaporation (C), and escape latency in the MCA assay (D) of vGluT2-Tiam1 cKO mice and littermate controls before and after SNI. Two-way ANOVA analysis followed by Dunnett’s post-hoc test (n = 6–8 mice/group. von Frey: P < 0.0001, F8,64 = 9.601; acetone: P < 0.0001, F8,64 = 9.957; MCA: P < 0.0001, F1,24 = 34.12).
Data are presented as means ± s.e.m. ****P < 0.0001.
See also Figure S6.
Tiam1 mediates synaptic structural plasticity in neuropathic pain.
Since our results indicate that Tiam1 signaling in excitatory spinal dorsal horn neurons is required to develop neuropathic pain, we investigated the underlying mechanisms by which Tiam1 promotes neuropathic pain. Tiam1 regulates dendritic spine remodeling during brain development by modulating actin dynamics15,16,18. Dendritic spines are generally found on spinal cord interneurons that receive convergent inputs, representing more than 90% of the excitatory synaptic connections11. Emerging evidence suggests that dorsal horn neuron dendritic spine dysgenesis contributes to nociceptive hyperexcitability associated with neuropathic pain resulting from spinal cord injury, peripheral nerve injury, diabetic neuropathy, and thermal burn injury4,5,9,36,37. We, therefore, investigated whether Tiam1 regulates dendritic spine remodeling during the development of neuropathic pain. To visualize neuron morphology, we sparsely labeled spinal dorsal horn neurons by performing intra-spinal dorsal horn microinjections of low titer GFP-expressing AAV virus (rAAV8-hSyn-GFP) on WT and global Tiam1 KO mice. Two weeks after virus injections, we performed sham or SNI surgeries on the mice. Then, three weeks after surgery, mice were perfused and sectioned (Fig. 4A). High-resolution confocal imaging was performed to analyze dendritic spines on GFP-expressing wide dynamic range (WDR) neurons from the spinal dorsal horn36,38 (Fig. 4B). Consistent with previous findings, nerve injury increased the density of dendritic spines on WDR neurons from WT mice36 (Fig. 4B, C). Actin polymerization is a driving force for dendritic spine remodeling39–41. Actin exists in two forms: monomeric globular actin (G-actin) and polymerized filamentous actin (F-actin). F-actin to G-actin ratio reflects the balance between actin polymerization and depolymerization42. In WT mice, SNI surgery increased the F-actin to G-actin ratio in the spinal dorsal horn (Fig. 4D), consistent with nerve injury inducing dendritic spine remodeling (Fig. 4B, C).
Figure 4. Tiam1 mediates synaptic structural plasticity in neuropathic pain.
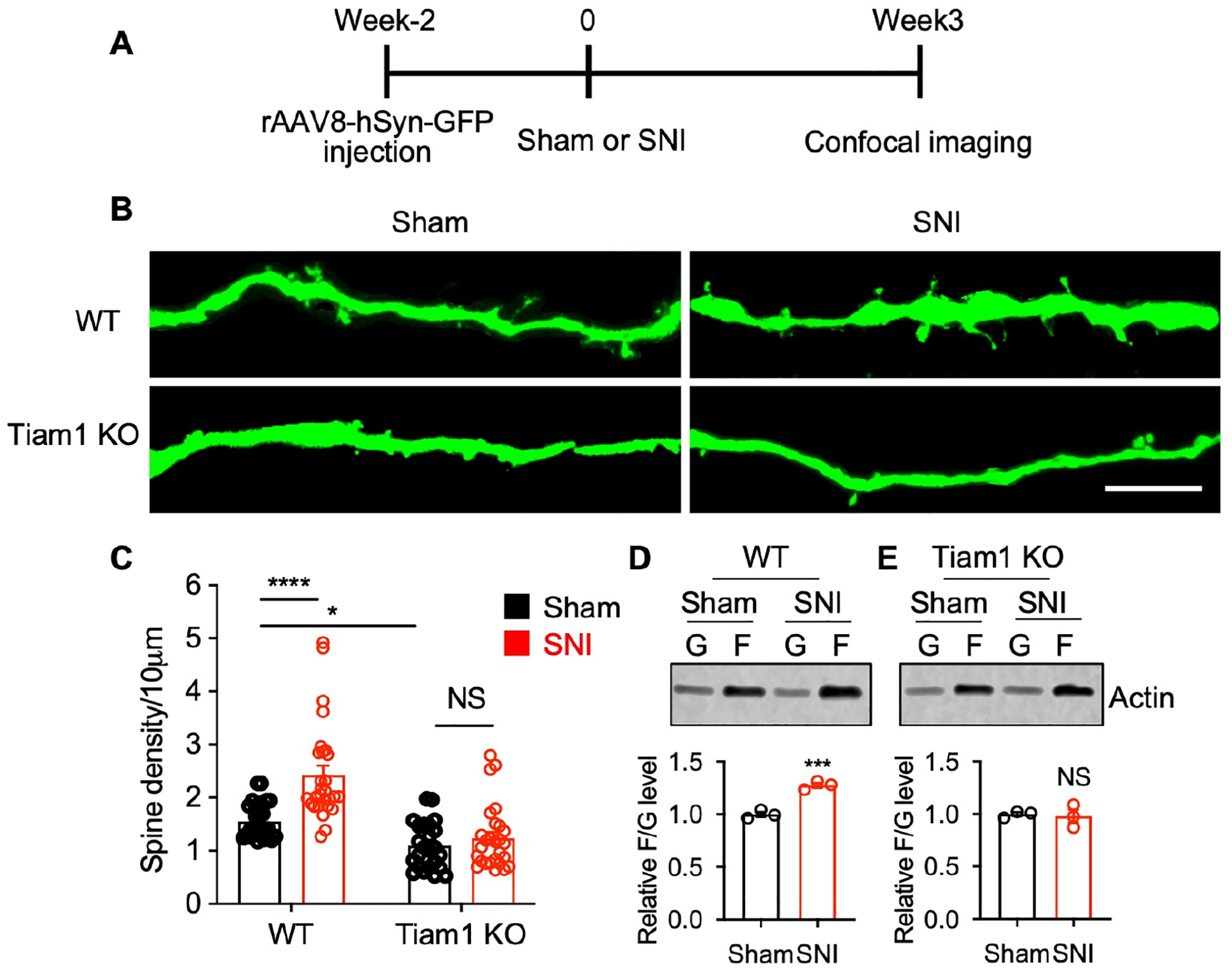
(A) Experimental paradigm for dendritic spine analysis.
(B and C) Representative confocal images and quantification of dendritic spine density showing the increase in dendritic spines in dorsal horn spinal neurons following SNI in WT mice, but not global Tiam1 KO mice (scale bar, 10 μm). Two-way ANOVA analysis followed by Dunnett’s post-hoc test (n = 24–28 neurons from 3 mice/group. P < 0.0001, F1,103 = 47.77).
(D and E) Tiam1 deletion attenuates nerve injury-stimulated actin polymerization in the spinal dorsal horn. Western blots and quantification revealed that SNI increased the F- to G-actin ratio in the spinal dorsal horn of WT mice (D), but not global Tiam1 KO mice (E). Mann-Whitney U-test (n = 3).
Data are presented as means ± s.e.m. *P < 0.05, ***P < 0.001, ****P < 0.0001, NS, no significant difference.
See also Figure S7.
In contrast to WT mice, nerve injury in Tiam1 global KO mice did not change the density of dendritic spines on GFP-expressing WDR neurons (Fig. 4B, C), nor did it alter the F-actin to G-actin ratio in the spinal dorsal horn (Fig. 4E). Similarly, Tiam1 deletion also attenuated paclitaxel (Taxol)- and streptozotocin (STZ)-stimulated actin polymerization in the spinal dorsal horn (Fig. S7). We also noticed a slight reduction in the spine density of spinal dorsal horn neurons in sham global Tiam1 KO mice compared to sham WT mice (Fig. 4B, C). Tiam1 has a well-established role in regulating dendritic spine/synapse development in the hippocampus and may play a similar role in the spinal cord. However, since we observe the same results in blocking neuropathic pain development in global Tiam1 KO mice as we do when we genetically ablate or inhibit Tiam1 in adult mice (Figs. 1 and 2 and NSC23766 results below), we do not believe that Tiam1’s developmental effects are involved in its role in neuropathic pain. These results suggest that Tiam1 regulates synaptic structural plasticity of spinal dorsal horn neurons in neuropathic pain.
Tiam1 regulates synaptic functional plasticity in neuropathic pain.
Synaptic NMDAR-mediated central sensitization in the spinal dorsal horn plays a critical role in the development of the neuropathic pain8,43–45. Since Tiam1 loss attenuates nerve injury-induced actin polymerization (Fig. 4D, E) and actin polymerization facilitates synaptic NMDAR activity46, we next investigated whether Tiam1 is required for these NMDAR changes. Consistent with previous reports, we found that in WT mice, nerve injury (3 weeks after SNI) significantly increased synaptic levels of NMDAR subunits GluN1 and GluN2B, with no effect on AMPAR subunit levels (Fig. 5A, B)8,45. In contrast, nerve injury did not alter synaptic NMDAR subunit levels in global Tiam1 KO mice (Fig. 5A, B). Electrophysiological recordings showed that SNI markedly increased the amplitude of puff NMDA currents of spinal dorsal horn neurons in WT mice (Fig. 5D). Importantly, the SNI-induced increase in synaptic NMDAR activity was abrogated in global Tiam1 KO mice (Fig. 5D). As expected, no difference was detected in synaptic AMPAR activity (Fig. S8). These findings indicate that Tiam1 is essential for the increase in synaptic NMDAR activity of spinal dorsal horn neurons during the development of neuropathic pain.
Figure 5. Tiam1 regulates synaptic functional plasticity in neuropathic pain.
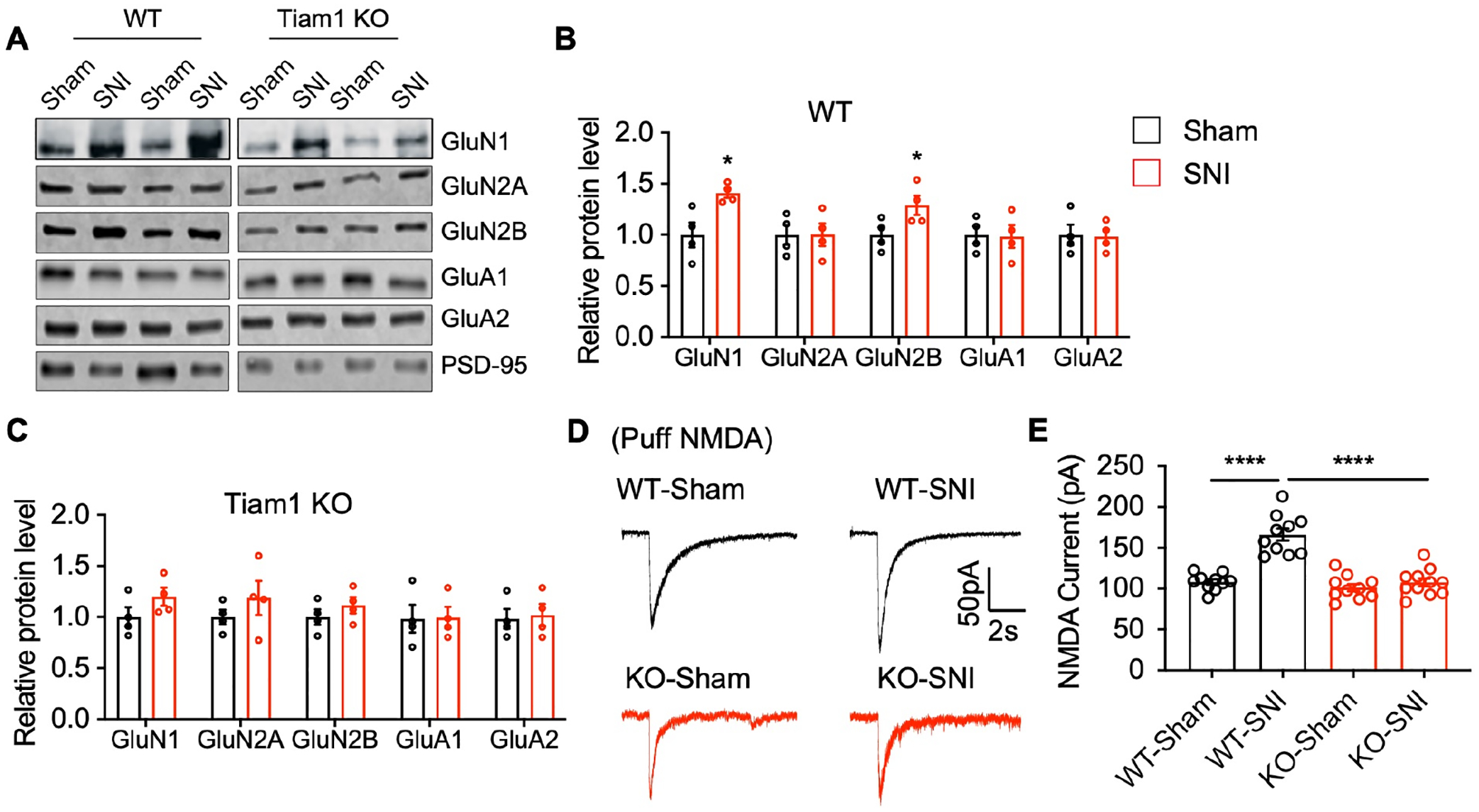
(A-C) Western blots and quantification show that nerve injury (3 weeks after SNI) significantly increases synaptic NMDAR subunit (GluN1 and GluN2B) levels in the spinal dorsal horn of WT mice, but not global Tiam1 KO mice. Synaptic AMPAR subunits show no significant difference between sham and SNI for either group. PSD-95 was used as the loading control. Mann-Whitney U-test (n = 4, sham vs SNI).
(D and E) Representative current traces and mean changes in NMDAR currents elicited by puff application of 100 mM NMDA to spinal dorsal horn neurons in WT and global Tiam1 KO mice 3 weeks after SNI or sham surgery. One-way ANOVA followed by Tukey’s post-hoc test (n = 10–11 neurons, P < 0.0001, F3,37 = 32.07).
Data are presented as means ± s.e.m. *P < 0.05, ****P < 0.0001.
See also Figures S8 and S9.
Tiam1-mediated neuropathic pain is Rac1-dependent.
Tiam1 functions as a GEF to activate the small GTPase Rac121. Notably, Rac1 plays an important role in neuropathic pain after injury9,36,37. To determine whether Tiam1-mediated neuropathic pain is dependent on Rac1 signaling, we performed intra-spinal dorsal horn microinjections of rAAV8-hSyn-Rac1Q61L-eGFP to express a constitutively-active Rac1 mutant in global Tiam1 KO mice (Fig. 6A). Microinjections of rAAV8-hSyn-eGFP served as a control. Two weeks after viral infections, we performed SNI surgery and measured mechanical and thermal sensitivity and non-reflexive pain behavior. The expression of constitutively-active Rac1Q61L-eGFP, but not control eGFP, in the spinal dorsal horn rescued neuropathic pain phenotypes in global Tiam1 KO mice subjected to SNI, as evidenced by marked hypersensitivity to von Frey filament stimuli, increased flicking time with acetone evaporation and increased escape latency from the white-lit chamber in the operant MCA assay (Fig. 6B–D). Electrophysiological recordings also showed that Rac1Q61L expression in the spinal dorsal horn rescued the abrogated synaptic NMDAR activity in global Tiam1 KO mice subjected to neuropathic pain (3 weeks after SNI) (Fig. 6E, F). These data indicate that Tiam1-mediated neuropathic pain phenotypic manifestations in the spinal dorsal horn are dependent on Rac1 GTPase signaling.
Figure 6. Tiam1 regulates neuropathic pain behaviors via Rac1 signaling.
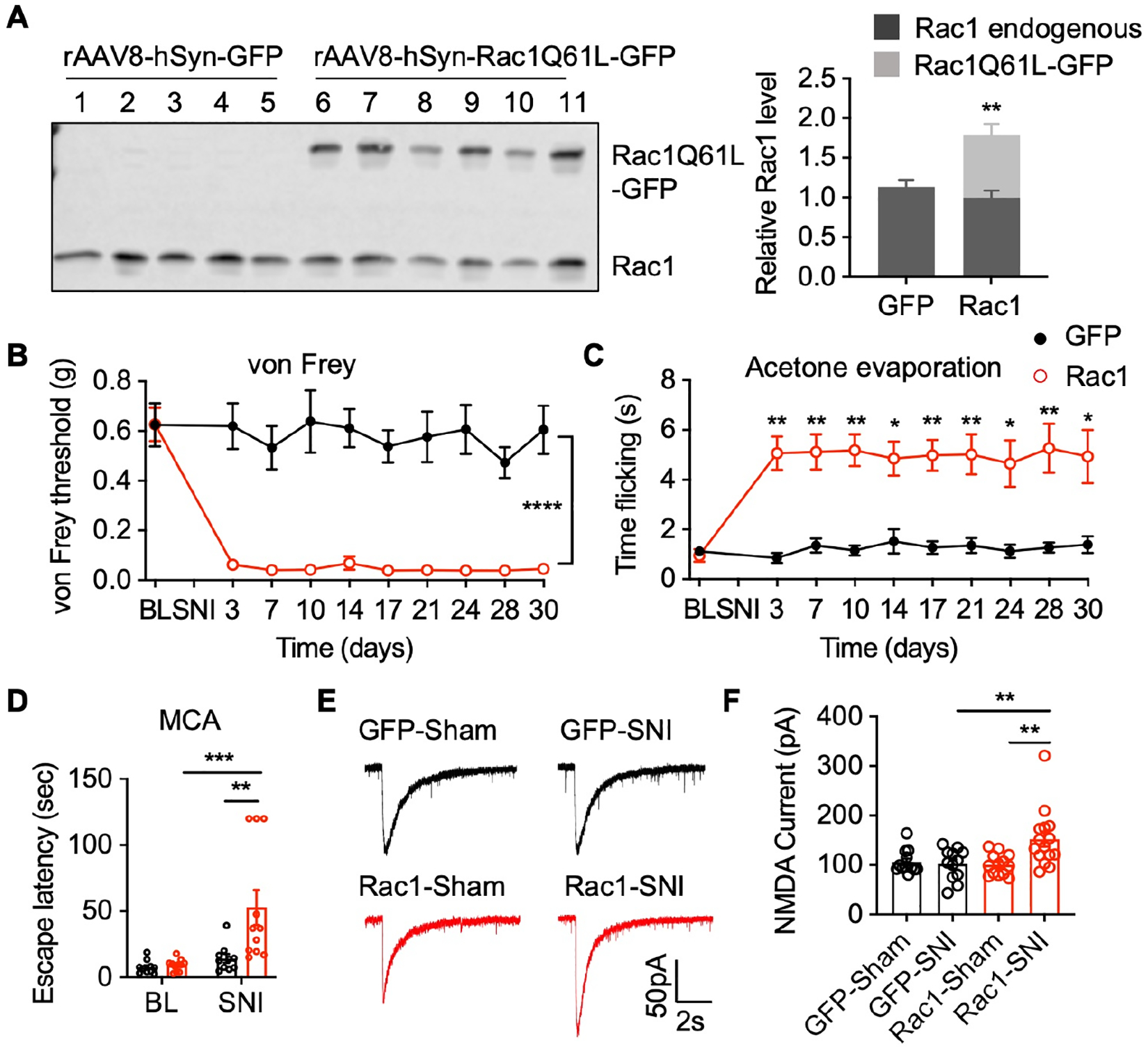
(A) Western blots and quantification of endogenous Rac1 and constitutively-active Rac1Q61L-GFP transgene expression in the spinal dorsal horn of global Tiam1 KO mice infected with control rAAV8-hSyn-GFP (GFP) or rAAV8-hSyn-Rac1Q61L-GFP (Rac1) via intra-spinal dorsal horn microinjection. Student t-test (n = 5–6). Endogenous Rac1 was used as the loading control. (B-D) Rac1Q61L expression in the spinal dorsal horn reverses the diminished neuropathic pain phenotype in global Tiam1 KO mice. von Frey withdrawal thresholds (B), time flicking with acetone evaporation (C), and escape latency in the MCA assay (D) of global Tiam1 KO mice infected with rAAV8-hSyn-GFP (GFP) or rAAV8-hSyn-Rac1Q61L-GFP (Rac1) before and after SNI. Two-way ANOVA analysis followed by Dunnett’s post-hoc test (n = 5–11 mice/group. von Frey: P < 0.0001, F9,130 = 6.812; acetone: P = 0.0308, F9,124 = 2.141; MCA: P = 0.0013, F1,38 = 12.07).
(E and F) Rac1Q61L expression in the spinal dorsal horn rescues the diminished NMDAR activity in global Tiam1 KO mice. Original current traces and mean changes in NMDAR currents elicited by puff application of 100 mM NMDA to spinal dorsal horn neurons in global Tiam1 KO mice infected rAAV8-hSyn-GFP (GFP) or rAAV8-hSyn-Rac1Q61L-GFP (Rac1) 3 weeks after SNI surgery. One-way ANOVA followed by Tukey’s post-hoc test (n = 12–15 neurons. P = 0.0015, F3,48 = 5.960).
Data are presented as means ± s.e.m. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Tiam1-Rac1 signaling is essential for the initiation, transition, and maintenance of neuropathic pain.
Our genetic strategy revealed that global Tiam1 loss or Tiam1 deletion from spinal dorsal horn excitatory neurons prevents the development of neuropathic pain (Figs. 1–3) and that Tiam1-mediated neuropathic pain is dependent on Rac1 GTPase signaling (Fig. 6). However, the precise role of Tiam1-Rac1 signaling at different stages of chronic neuropathic pain is unknown. To directly assess the role of Tiam1-Rac1 signaling in the initiation of neuropathic pain, we treated neuropathic pain mice with NSC23766 (5 mg/kg/day, i.p.), a widely used small molecule inhibitor that prevents Rac1 activation by the Rac1-specific GEFs Tiam1 and Trio47, at the time of SNI surgery and examined pain behaviors. As expected, a 4-day treatment with NSC23766 (given on the day of SNI surgery and the following 3 days) completely abolished SNI-induced persistent tactile allodynia and mechanical hyperalgesia for at least 3 weeks (Fig. 7A–C), indicating that Tiam1-Rac1 signaling is required for the initiation of neuropathic pain.
Figure 7. Inhibiting Tiam1-Rac1 signaling prevents the initiation and transition of neuropathic pain and reverses established neuropathic pain.
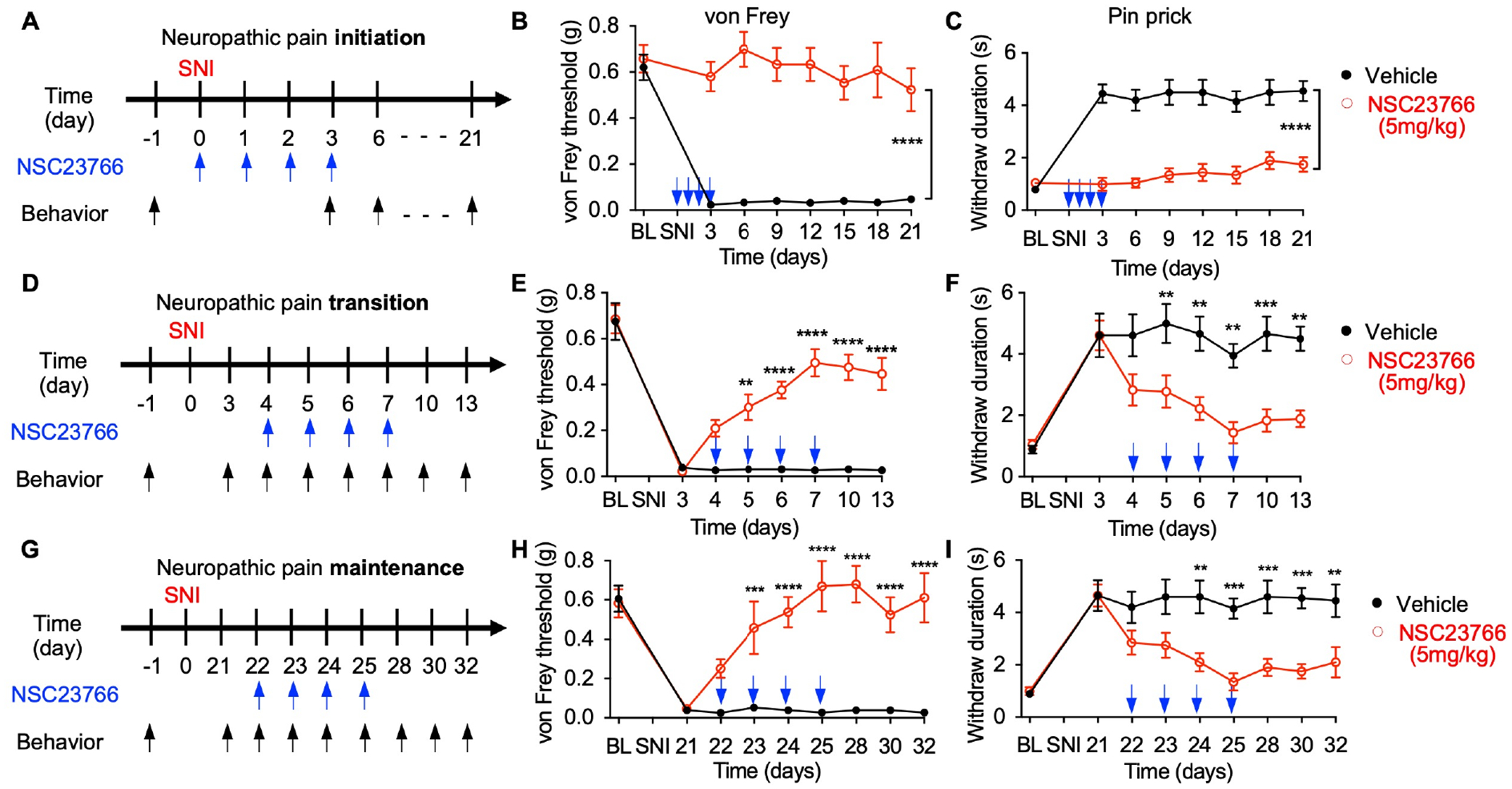
(A-C) NSC23766 treatment prevents the initiation of neuropathic pain. (A) Experimental paradigm. Time course for changes in von Frey thresholds (B) and withdrawal duration to pinprick (C) of SNI mice treated with 4 consecutive daily injections of NSC23766 (5 mg/kg) starting at the time of surgery. Blue arrows indicate NSC23766 injections. Two-way ANOVA analysis followed by Dunnett’s post-hoc test (n = 10 mice/group. von Frey: P < 0.0001, F7,144 = 7.725; pinprick: P < 0.0001, F7,144 = 8.966).
(D-F) NSC23766 prevents the transition of neuropathic pain. Vehicle or NSC23766 (5 mg/kg) treatment was started 4 days after SNI surgery for 4 consecutive days, and behaviors were tested 60 min after drug administration. Two-way ANOVA analysis followed by Dunnett’s post-hoc test (n = 6–9 mice/group. von Frey: P < 0.0001, F7,106 = 33.84; pinprick: P < 0.0001, F7,128 = 10.22).
(G-I) NSC23766 reverses established neuropathic pain. Vehicle or NSC23766 (5 mg/kg) treatment was started 3 weeks after SNI surgery for 4 consecutive days, and behaviors were tested 60 min after drug administration. Two-way ANOVA analysis followed by Dunnett’s post-hoc test (n = 10 mice/group. von Frey: P < 0.0001, F8,161 = 9.928; pinprick: P < 0.0001, F8,162 = 8.852).
Data are presented as means ± s.e.m. **P < 0.01, ***P < 0.001, ****P < 0.0001.
See also Figures S10, S11, and S12.
Neuropathic pain hypersensitivity gradually develops over the first 3 days following periphery nerve injury (SNI model) and is fully established as a chronic pain state after 3 weeks. We next asked whether Tiam1-Rac1 signaling is also required to transmit and/or maintain neuropathic pain. To this end, we treated neuropathic pain mice with NSC23766 (5 mg/kg/day, i.p.) at the transition and maintenance stages of neuropathic pain, i.e., around 3 days and 3 weeks after SNI surgery, respectively, and measured pain sensitivity (Fig. 7D, G). Four-day treatments with NSC23766 prevented the transition from acute to chronic neuropathic pain (starting 4 days after SNI) (Fig. 7D–F) and reversed established neuropathic pain (starting 22 days after SNI) (Fig. 7G–I). Significantly, NSC23766 treatment did not affect pain sensitivity in sham animals (Fig. S10A–C). Since NSC23766-mediated Rac1 inhibition was recently reported to contribute to motor neuron death in the amyotrophic lateral sclerosis (ALS) model48, we measured the activity of the apoptosis marker caspase-3 in the spinal ventral horn of NSC23766-treated mice but detected no difference in caspase-3 activity (Fig. S10D, E). Likewise, we have not detected any enhanced cell death or motor deficiency in Tiam1 global KO mice17. However, we did find that 4-day NSC23766 treatment attenuated SNI-induced actin polymerization and SNI-induced increases in the levels of synaptic NMDAR subunits GluN1 and GluN2B, as well as the amplitude of puff NMDA current in the spinal dorsal horn (Fig. S11 and Fig. S12). Taken together, these results suggest that Tiam1-Rac1 signaling is critical for the initiation, transition, and maintenance of neuropathic pain by driving maladaptive synaptic plasticity.
Antisense oligonucleotide (ASO) targeting of spinal Tiam1 alleviates neuropathic pain hypersensitivity.
Our results argue that Tiam1 is a promising therapeutic target for treating neuropathic pain. Antisense oligonucleotides (ASOs) are short, synthetic, single-stranded oligodeoxynucleotides that alter target protein expression by modulating mRNA processing or degradation49. To determine whether neuropathic pain could be treated by locally reducing Tiam1 levels in a potentially clinically relevant manner, we designed five 20 bp ASOs targeting different sites of the rat Tiam1 mRNA that could be used to reduce Tiam1 levels in rats (Fig. 8A and Table S1). We used rats, a different rodent animal model, to perform therapeutic studies because rats are larger (for intrathecal injections), have more complex cognitive abilities than mice, and their behavior better mimics behavior seen in humans50. After screening Tiam1 ASOs for their ability to reduce Tiam1 levels in cultured rat cortical neurons, ASO-1 was found to be the most effective at reducing Tiam1 expression, with an approximately 75% decrease in protein level (Fig. 8B). Since a single centrally-delivered ASO has been shown to target the spinal cord with a long duration of activity51, we administered ASO-1 or a control ASO to rats via intrathecal injection to validate the in vivo suppression efficacy of ASO-1. Two weeks after a single lumbar puncture injection of 100 μg Tiam1 ASO-1, we found that ASO-1 administration resulted in an approximately 50% reduction of Tiam1 protein levels in the spinal dorsal horn (Fig. 8C and Fig. S13A). To test whether Tiam1 ASO-1 treatment alleviates pain hypersensitivity in a rat neuropathic pain model, we intrathecally injected Tiam1 ASO-1 or control ASO 3 weeks after SNI surgery. Treatment with control ASO did not affect the mechanical or thermal withdrawal thresholds of SNI-treated rats (Fig. 8D, E). However, one week after lumbar puncture injection of Tiam1 ASO-1, SNI-induced pain hypersensitivity in rats was significantly reduced, and this effect lasted another 2 weeks (Fig. 8D, E). In contrast, neither Tiam1 ASO-1 nor control ASO treatment affected basal pain sensitivity in sham rats (Fig. S13B, C). These data indicate that ASO targeting of spinal Tiam1 alleviates neuropathic pain hypersensitivity.
Figure 8. ASO targeting of spinal Tiam1 alleviates neuropathic pain hypersensitivity.
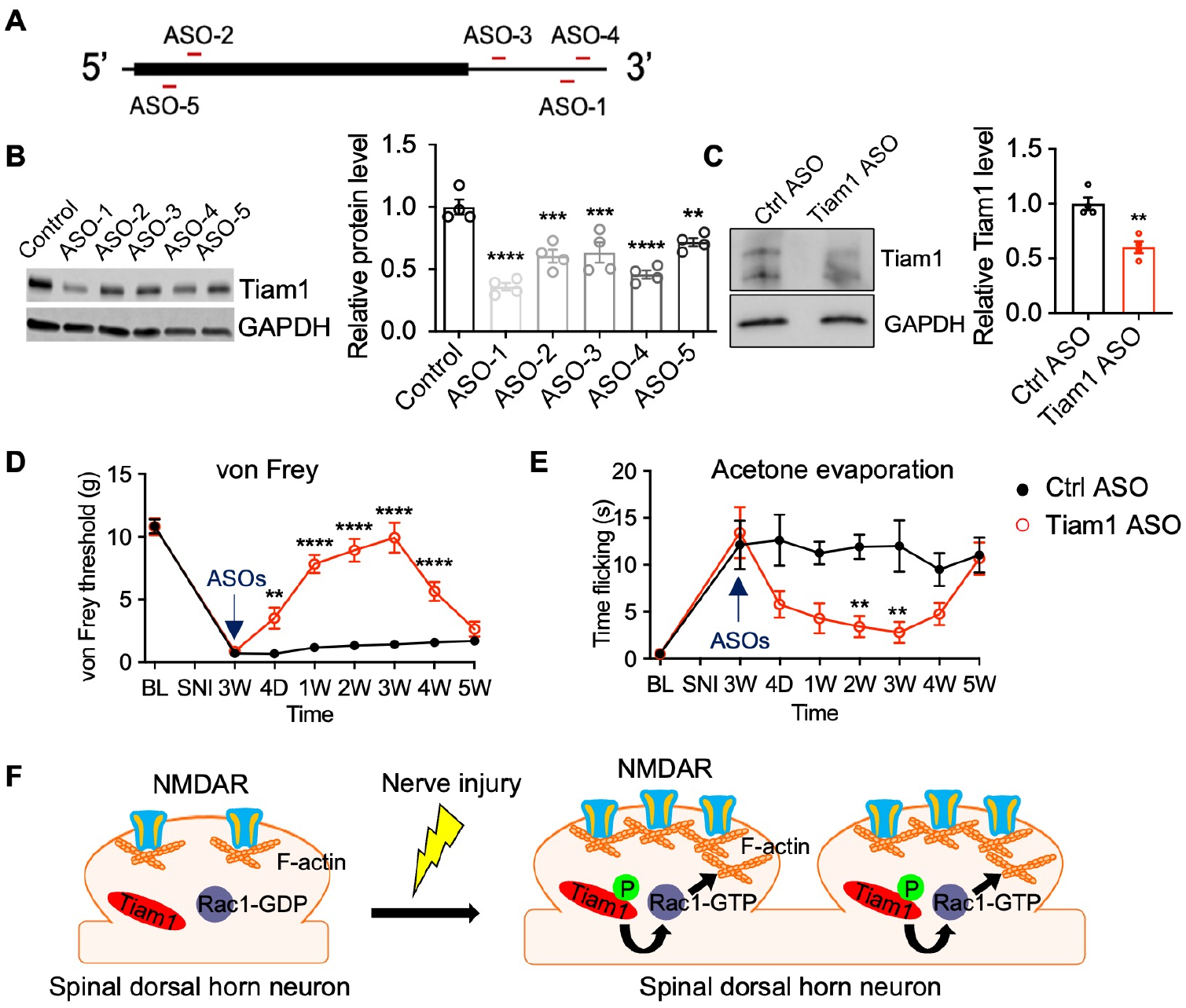
(A) Location of ASO targeting sequences in rat Tiam1 mRNA. The box represents an open reading frame.
(B) Western blots and quantification of Tiam1 levels from primary rat cortical neuron cultures treated with control ASO or different Tiam1 ASOs. GAPDH was used as the loading control. One-way ANOVA followed by Tukey’s post-hoc test (n = 4. P < 0.0001, F5,18 = 18.53).
(C) Western blots and quantification of Tiam1 protein levels in the spinal dorsal horn 2 weeks after lumbar puncture injection of control ASO or Tiam1 ASO-1. GAPDH was used as the loading control. Mann-Whitney U-test (n = 4).
(D and E) Time courses for changes in the von Frey threshold and flicking time of acetone evaporation after lumbar puncture injection of control ASO or Tiam1 ASO-1 in neuropathic pain rats (3 weeks after SNI). Two-way ANOVA analysis followed by Dunnett’s post-hoc test (n = 8 rats/group. von Frey: P < 0.0001, F7,112 = 54.07; acetone: P < 0.0001, F7,112 = 8.136).
(F) Model of Tiam1-coordinated synaptic structural and functional plasticity underlying the pathophysiology of neuropathic pain.
Data are presented as means ± s.e.m. **P < 0.01, ***P < 0.001, ****P < 0.0001.
See also Figures S13, S14, and Table S1.
DISCUSSION
The Rac1-GEF Tiam1 is an established regulator of the dendrite, spine, and synapse development, which couples synaptic NMDAR, TrkB, and EphB receptors to Rac1 activation and actin cytoskeletal remodeling in hippocampal neurons during brain development15–18,52. Here, we found that Tiam1-Rac1 signaling is also essential for the development and maintenance of neuropathic pain by orchestrating synaptic structural and functional plasticity via actin cytoskeleton reorganization and synaptic NMDAR stabilization in spinal dorsal horn excitatory neurons (Fig. 8H). Moreover, we show that targeting spinal Tiam1 with an ASO is an effective strategy to alleviate neuropathic pain hypersensitivity. Thus, our study has uncovered a pathophysiological mechanism that initiates, transits, and sustains neuropathic pain, and it has identified a promising therapeutic target for treating neuropathic pain with long-lasting consequences.
Maladaptive synaptic plasticity within nociceptive circuits in the spinal dorsal horn is a key mechanism underlying the development of chronic pain4,5. Functional alterations in nociceptive networks are accompanied by structural remodeling and reorganization of synapses, cells, and circuits, and this structural plasticity likely accounts for the long-term nature of chronic pain5,53,54. Dendritic spine remodeling, in particular, provides a structural-based mechanism for modifying and maintaining long-term changes in the synaptic function12. A diverse array of signaling pathways regulates the formation and architecture of dendritic spines, often by modulating the underlying actin cytoskeletal organization13,55. In several animal models of neuropathic pain, increases in the density of dendritic spines in the superficial spinal cord have been reported, which is mediated by Rac1 signaling9,36,37. Here, we show that in response to various forms of nerve injury, the Rac1-GEF Tiam1 induces actin polymerization in the spinal dorsal horn, as indicated by an increase in the F- to G-actin ratio, and drives dendritic spine growth. Unexpectedly, Tiam1 also controls synaptic NMDAR levels, but not TrkB or EphB receptors (Fig. S14), and thereby modulates NMDAR-mediated synaptic transmission. Given that actin serves as a scaffold for synaptic proteins in addition to regulating the spine structure46, it is possible that Tiam1-induced actin polymerization facilitates synaptic NMDAR activity and stabilization in addition to spine morphogenesis. Our study, therefore, identifies Tiam1 as an essential player in the pathogenesis of neuropathic pain that coordinates actin cytoskeletal dynamics, dendritic spine morphogenesis, and synaptic receptor function in spinal dorsal horn excitatory neurons in response to nerve damage.
Tiam1 is well placed to propagate the signaling from nociceptive activity to neuropathic pain hypersensitivity via modulation of spinal dorsal horn neuron synapses. As a Rac1-GEF that couples synaptic receptors to Rac1 signaling, Tiam1 is highly expressed in the spinal cord as well as the developing and adult brain. We and others have previously shown that Tiam1 is recruited to activated synaptic NMDAR, TrkB, and EphB receptors during brain development, resulting in its phosphorylation and activation, enabling Tiam1 to induce localized Rac1 signaling and actin remodeling that drives dendritic spine development15,16,18,52. In the context of neuropathic pain, nociceptive activity activates synaptic NMDAR, TrkB, and EphB receptors6–8. By functioning downstream of these activated receptors in the spinal dorsal horn, Tiam1 likely acts as a convergence point to integrate multiple signaling inputs that promote robust Rac1-dependent maladaptive synaptic structural and functional plasticity in spinal dorsal horn neurons, leading to pain hypersensitivity and the long-term nature of neuropathic pain. In terms of how Tiam1 is activated, in addition to being tyrosine phosphorylated following the activation of TrkB and EphB receptor tyrosine kinases16,18,52, Tiam1 is phosphorylated on serine/threonine residues and activated following Ca2+ influx through glutamate-stimulated NMDARs15. Notably, Tiam1 has recently been shown to form a reciprocally activating signaling complex (RAKEC) with CaMKIIα, which stably activates CaMKIIα and Rac1 and maintains an enlarged spine morphology56. Tiam1 may also provide an amplification step in the nociceptive activity-dependent regulation of synaptic plasticity. Since Tiam1 is an enzyme, only a few Tiam1 molecules may be required to activate multiple Rac1 molecules, which would result in enhanced actin cytoskeletal remodeling. Finally, because Tiam1 controls both dendritic spine density and synaptic function, it likely provides a critical link in the coordination of nociceptive activity-dependent synaptic structural and functional plasticity.
Our results indicate that Tiam1 mediates spinal dorsal horn synaptic plasticity that underlies the pathophysiology of neuropathic pain, suggesting that targeting Tiam1 may be an effective strategy to treat neuropathic pain. Interestingly, we noticed that earlier intervention produces a longer-lasting, more robust effect on reducing pain hypersensitivity compared to late intervention (Fig. 7). The reasons may be as below: In the early stages of neuropathic pain development, nociceptive activity activates spinal Tiam1, which induces downstream signaling pathways that initiate synaptic structural and functional plasticity, leading to pain hypersensitivity. In the later stages of neuropathic pain development, Tiam1-mediated maladaptive synaptic structural and functional plasticity already contribute to pain hypersensitivity and the persistent nature of pain. Therefore, for early treatment, reducing pain hypersensitivity only requires preventing Tiam1-induced maladaptive synaptic plasticity. However, for late treatment, reducing pain hypersensitivity not only involves preventing the ongoing nociceptive activity-induced maladaptive synaptic plasticity, but also requires a reversal of the established maladaptive synaptic plasticity. It is also possible that Tiam1 blocks inflammatory-induced pain, which can occur immediately following SNI surgery, but this needs to be further investigated. Taken together, our findings uncover a pathophysiological mechanism that underlies the development and maintenance of different types of neuropathic pain and identifies a novel therapeutic target that could be translated into the clinic to treat neuropathic pain with long-lasting consequences.
STAR * METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Lingyong Li (lingyongli@uabmc.edu).
Material Availability
This study did not generate new unique reagents.
Data and Code Availability
This study did not generate any unique datasets or code.
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Animals.
All animal protocols were approved by the Institutional Animal Care and Use Committee of Baylor College of Medicine and were conducted in accordance with the National Institutes of Health Guidelines. Global Tiam1 KO mice were generated as described17. For conditional knockout of Tiam1 from postnatal forebrain excitatory neurons, Tiam1flox/flox mice were crossed with CaMKIIα-Cre mice, and the resulting CaMKIIα-Cre∷Tiam1+/flox mice were then crossed with Tiam1flox/flox mice to obtain CaMKIIα-Cre∷Tiam1flox/flox (CaMKIIα-Tiam1 cKO mice) as well as Tiam1flox/flox littermates (control) for use in experiments. For conditional knockout of Tiam1 from DRG neurons, Tiam1flox/flox mice were crossed with Advillin-Cre mice, and the resulting Advillin-Cre∷Tiam1+/flox mice were then crossed with Tiam1flox/flox mice to obtain Advillin-Cre∷Tiam1flox/flox (Advillin-Tiam1 cKO mice) as well as Tiam1flox/flox littermates (control) for use in experiments. For conditional knockout of Tiam1 from excitatory neurons, Tiam1flox/flox mice were crossed with vGluT2-Cre mice, and the resulting vGluT2-Cre∷Tiam1+/flox mice were then crossed with Tiam1+/flox mice to obtain vGluT2-Cre∷Tiam1flox/flox (vGluT2-Tiam1 cKO mice) and as well as vGluT2-Cre littermates (control). For conditional knockout of Tiam1 from inhibitory neurons, Tiam1flox/flox mice were crossed with vGat-Cre mice, and the resulting vGat-Cre∷Tiam1+/flox mice were then crossed with Tiam1+/flox mice to obtain vGat-Cre∷Tiam1flox/flox (vGat-Tiam1 cKO mice) and as well as vGat-Cre littermates (control). Genotyping of Tiam1 mice was determined by PCR from tail DNA using the primers: P1: ACG TGT GTT AAT TAG CCA GGT TTG ATG G, P2: GAT CCA CTA GTT CTA GAG CGG CCG AA, P3: CTA CCC GGA GGA AGT GGA AGC ACT ACT. Genotyping of CaMKIIα-Cre mice was determined by PCR from tail DNA using the primers: P1: GCA TTA CCG GTC GAT GCA ACG AGT GAT GAG, P2: GAG TGA ACG AAC CTG GTC GAA ATC AGT GCG. Genotyping of Advillin-Cre mice was determined by PCR from tail DNA using the primers: P1: CTT TGT GAT GTT TCA GTT CCA G, P2: AGG ATC TGC ACA CAG ACA GGA. Genotyping of vGluT2-Cre mice was determined by PCR from tail DNA using the primers: P1: ACA CCG GCC TTA TTC CAA G, P2: AAG AAG GTG CGC AAG ACG, P3: CTG CCA CAG ATT GCA CTT GA. Genotyping of vGat-Cre mice was determined by PCR from tail DNA using the primers: P1: GCA TTT CTG GGG ATT GCT TA, P2: GTC ATC CTT AGC GCC GTA AA, P3: CTA GGC CAC AGA ATT GAA AGA TCT, P4: GTA GGT GGA AAT TCT AGC ATC ATC C. Tiam1flox/flox mice were maintained on a 129SvEv background, while CaMKIIα-Cre mice, Advillin-Cre mice, vGluT2-Cre mice, and vGat-Cre mice were maintained on a C57BL6J background. Mice were housed in two to four per cage, under a 12-hour light and 12-hour dark-light cycle (lights on at 06:00 and off at 18:00), with ad libitum access to food and water. All experiments used age-matched male and female mice.
Antisense oligonucleotides (ASOs) synthesis.
The ASOs were synthesized, modified, and purified by Thermal Fish Scientific as previously described51,57. Lyophilized ASOs were dissolved in sterile saline and diluted to the desired concentration for dosing. The ASO solution was sterilized through a 0.2-mm filter before use. The ASO sequences, start points, and modifications used in the study are shown in Supplementary Table 1.
Pain models.
Spared nerve injury (SNI) model.
SNI surgery was conducted according to the method described previously22. Briefly, animals were anesthetized with 2% isoflurane. A heating pad was used to maintain the core body temperature of the animals at 37 °C. An incision was made on the left lateral thigh to expose the sciatic nerve. We ligated and sectioned the common peroneal and tibial nerves (leaving the sural nerve intact) with a 5–0 silk suture under a surgical microscope. The sham procedure consisted of the same surgery without nerve ligation and section.
Chronic constriction injury (CCI) model.
Chronic constriction injury of the sciatic nerve (CCI) was conducted to the methods published previously58. Briefly, the mice were anesthetized with 2% isoflurane. A heating pad was used to maintain the core body temperature of the animals at 37 °C. The left lateral sciatic nerve was quickly exposed at mid-thigh level, 5 mm of that was carefully free of surrounding connective tissue. Then three knots were laced up loosely around the sciatic nerve with 5–0 suture from distal to proximal, 1 mm apart, with the first knot next to the trifurcation. The proper tightness depended on the slight tremors of the hind limb. The sham procedure consisted of the same surgery without a sciatic nerve tie.
Chemotherapy-induced peripheral neuropathy model.
Chemotherapy-induced peripheral neuropathy model was conducted as previously described27. Mice were treated with paclitaxel (IP, once daily for 5 days). The control mice were injected with the same volume of vehicle.
Streptozotocin-induced diabetic neuropathy model.
Streptozotocin-induced diabetic neuropathy model was based on previous studies28–30. Mice were administrated with streptozotocin STZ (IP, 150mg/kg, single dose). Diabetes was confirmed 6- and 12-day postinjection by measuring blood glucose levels (glucose > 12 mmol/l). Only animals with established diabetes were then tested for mechanical allodynia and thermal hyperalgesia. The control mice were injected with the same volume of vehicle.
Viral injection.
A new minimally-invasive method for microinjection of the virus into the mouse spinal dorsal horn without laminectomy was conducted to methods published previously59. Briefly, mice were anesthetized with 2% isoflurane and placed in a stereotaxic frame (Kopf). A heating pad was used to maintain the core body temperature of the animals at 37 °C. The skin was incised at Th12–L3 and custom-made clamps were attached to the rostral and caudal sites of the vertebral column. Paraspinal muscles around the left side of the interspace between Th13 and L1 vertebrae were removed, and the dura mater and the arachnoid membrane were carefully incised using the tip of a 27G needle to make a small window to allow the glass micropipette insert directly into the spinal dorsal horn (500 μm lateral from the midline and 250 μm in depth from the surface of the dorsal root entry zone) via interspace between Th13 and L1 vertebrae.
A volume of 0.5μl rAAV8-hSyn-GFP (2.6×10E12 gc/mL, UNC Vector Core), rAAV8-hSyn-Cre-GFP (2.4×10E12 gc/mL, UNC Vector Core), AAV8-pCAG-DIO-eGFP (1.2×10E12 gc/mL, UNC Vector Core), or rAAV8-hSyn-Rac1Q61L-GFP (1×10E12 gc/mL, BCM Vector Core) was injected using glass micropipette attached to a Hamilton microsyringe connected to an infusion pump at a rate of 200 nl/min. After injection, the glass micropipette remained in place for 5 min, and the skin was sutured.
Lumbar puncture injection.
ASO (100 μg/rat) was administrated via intermittent lumbar puncture as described before60. Briefly, rats were anesthetized with 2% isoflurane. The lumbar region was shaved and prepared with Betadine solution, and the intervertebral spaces were widened by placing the animal on a Plexiglas tube. Animals were then injected at the lumbar vertebral space between L5 and L6 using a 32-gauge needle connected to a syringe filled with 100 μg ASO dissolved in 20 μL of saline. Correct subarachnoid positioning of the tip of the needle was verified by a tail- or paw-flick test.
Behavioral assessments.
The experimenters performing the behavioral tests and quantitative analyses were blinded to mouse genotypes and treatments for all behavioral tests. Behavioral assessments were performed between 09:00 and 14:00.
von Frey filament assay.
The mechanical sensitivity of mouse hindpaws was assessed as described in numerous prior reports8,60. Animals were acclimated to the testing room and apparatus for 45 min each on the two days before the beginning of testing and on each subsequent testing day. Mechanical sensitivity was assessed using a series of calibrated von Frey filaments (Stoelting, Wood Dale, IL). These filaments were applied perpendicular to the plantar surface of the hind paw with sufficient force to bend the filament. Rapid withdrawal of the paw away from the stimulus within 4 s was characterized as a positive response. If there was no response, the filament of the next greater force was applied. After a response, the filament of the next lower force was applied. 0.04 −1.4 g von Frey filaments were employed in mouse assay, and 0.4 −15 g von Frey filaments were employed in rat assay. We calculated the tactile stimulus force that produced a 50% likelihood of a withdrawal response using the “up-down” method8,61.
Acetone evaporation assay.
To evaluate affective-motivational responses evoked by thermal stimulation, we applied a single, unilateral 50-μl drop of acetone (evaporative cooling) to the left hind paw, and the duration of reflexive withdrawal or attending behavior (lifting, shaking, licking of the paw) was recorded for up to 60 s after the stimulation. To prevent behavioral sensitization that can result from multiple noxious stimulations and then averaging those responses, only one drop of acetone was applied during a given testing session62,63.
Pinprick assay.
Briefly, animals were first placed in a square Plexiglas chamber on top of a wire mesh table. Animals were acclimated to this testing arena for 1 hour the day before testing and for an additional 30 minutes immediately before testing. A small insect pin (tip diameter = 0.03 mm, Fine Science Tools, USA) was applied with minimal pressure to the plantar surface of the left hind paw, taking care not to penetrate the skin. The duration of the reflexive withdrawal or attending behavior (lifting, shaking, licking of the paw) was recorded for up to 60 s after the stimulation.
Mechanical conflict-avoidance (MCA) assay.
Voluntary aversion to a noxious stimulus was assessed using a commercial three-chambered apparatus, the Mechanical Conflict-Avoidance (MCA) System (Noldus, USA). The MCA System contains an aversive lighted area, a walkway with mechanical probes, and an attractive dark area. The mice were placed in the aversive lighted area and allowed to escape from this area to the preferred dark area through the walkway. The walkway consists of mechanical probes that are painful but not sharp enough to cause any tissue damage when walking on. Longer latencies to escape the light chamber indicate increased motivation to avoid the probes, and this escape latency is the measure of pain-related behavior in this test. We performed the operant MCA test on mice with modifications recently described in detail24,25. The test was performed for 2 days. Before the testing day, mice were acclimated to the MCA unit for 5 min with the LEDs switched off, the barrier door open, and the mechanical probe height set to zero. For testing, mice were placed into the lighted chamber with the lid closed. The LEDs were switched on after 10 s, and the barrier was removed 20 s after that. Each mouse’s behavior started from the introduction into the lighted chamber until the mouse crossed the halfway point of the walkway with mechanical probes during the 120 s cutoff. After running all mice in a particular cohort at a probe height of 0 mm, the process was repeated with the probe height set to 5 mm. The duration between the barrier opening and the mouse crossing the midpoint of the walkway with mechanical probes was quantified and expressed as ‘escape latency’.
Biochemical assays.
Affinity-precipitation assay for Tiam1 GEF activity.
The Tiam1 activity was measured using a previously described affinity precipitation assay64. Briefly, the spinal dorsal horn from treated mice was isolated and homogenized in cold lysis buffer (25 mM HEPES, pH 7.4, 0.1 M NaCl, 1% NP40, 5 mM MgCl2, 10% glycerol, 1 mM DTT, 10 μg/ml leupeptin, 10 μg/ml aprotinin and 1 mM sodium orthovanadate), and centrifuged at 15,000g for 30 min. The supernatant was incubated with 30 μg of GST-Rac1G15A bound to GSH-agarose beads for 2 hr at 4 °C and mixed gently on a rocking shaker. After washing with lysis buffer 3 times, beads were resuspended in Laemmli buffer. Samples were resolved by SDS-PAGE, transferred to the nitrocellulose membrane, blocked with 5% fat-free milk in 0.1% Tween-PBS, and incubated with anti-Tiam1 antibody (1:1000). Active Tiam1 was determined by western blot analysis from the precipitated fraction and normalized to total protein (input).
F-actin to G-action ratio.
The F-actin to G-actin ratio was determined by western blot, as previously described65,66. Briefly, the two forms of actin differ in that F-actin is insoluble, whereas G-actin is soluble. The spinal dorsal horn from sham or SNI-treated mice was isolated and homogenized in cold lysis buffer (10 mM K2HPO4, 100 mM NaF, 50 mM KCl, 2 mM MgCl2, 1 mM EGTA, 0.2 mM DTT, 0.5% Triton X-100, 1 mM sucrose, pH 7.0) and centrifuged at 15,000g for 30 min. Soluble actin (G-actin) was measured in the supernatant. The insoluble F-actin in the pellet was resuspended in lysis buffer plus an equal volume of buffer 2 (1.5 mM guanidine hydrochloride, 1 mM sodium acetate, 1 mM CaCl2, 1 mM ATP, 20 mM Tris-HCl, pH 7.5) and incubated on ice for 1 h to convert F-actin into soluble G-actin, with gentle mixing every 15 min. The samples were centrifuged at 15,000g for 30 min, and F-actin was measured in this supernatant. Samples from the supernatant (G-actin) and pellet (F-actin) fractions were proportionally loaded and analyzed by western blotting.
Synaptosome preparation.
Synaptosome preparation was performed as in our previous publications8. The spinal dorsal horn (L4 and partial L5 spinal cord segment) from sham or SNI mice was homogenized using glass-Teflon homogenizer in 10 volumes of ice-cold HEPES-buffered sucrose (0.32 M sucrose, 1 mM EGTA, and 4 mM HEPES at pH 7.4) containing a protease inhibitor cocktail (Sigma-Aldrich). The homogenate was centrifuged at 1,000 g for 10 min at 4 °C to remove the nuclei and large debris. The supernatant was centrifuged at 10,000 g for 15 min to obtain the crude synaptosome fraction. The synaptosome pellet was lysed via hypoosmotic shock in 9 volumes of ice-cold HEPES buffer with the protease inhibitor cocktail for 30 min. The lysate was centrifuged at 25,000 g for 20 min at 4 °C to obtain the synaptosome membrane fraction for the following immunoblotting experiments.
Single-cell dissociation and flow cytometry.
The spinal dorsal horn neurons dissociation was performed with Multi Tissue Dissociation Kit (Miltenyi Biotec) with modification. Briefly, spinal dorsal horn tissue chunks were incubated in 1 ml of enzyme P solution for 1 h at 37 °C and 5% CO2. After 10 min of incubation, tissues were triturated briefly with a P1000 pipette tip and returned. Cells were triturated another four times (around 30 each) with a P200 pipette tip over the rest of the remaining incubation time. At room temperature, cell suspensions were centrifuged at 350g for 10 min, resuspended in 1 ml PBS with the proteinase inhibitor cocktail (Sigma-Aldrich), and centrifuged again. The supernatant was removed, and 1 ml PBS with a proteinase inhibitor cocktail was added to the cells. Cells were passed through a 70-μm cell strainer to remove debris. Samples were centrifuged (350 g for 8 min at 4 °C), resuspended in 0.5 ml of PBS with the proteinase inhibitor cocktail, and kept on ice for flow cytometry. Cells were sorted via the Sony SH800. GFP-positive cells were used for subsequent immunoblotting experiments.
Immunoblotting.
The protein samples were homogenized in RIPA buffer containing (in mM) 50 Tris-HCl (pH 7.4), 1% NP-40, 0.1%SDS, 150 NaCl, 1 EDTA, 1 Na3VO4, and 1 NaF in the presence of a proteinase inhibitor cocktail (Sigma-Aldrich). The lysates were centrifuged at 13,000 rpm for 30 min at 4 °C. The supernatant was carefully collected, and the protein concentration was measured using a DC Protein Assay Kit (Bio-Rad). 30 μg of the total proteins from each sample were loaded and separated using 4–15% Tris-HCl SDS-PAGE gels. The resolved proteins were transferred to an Immobilon-P membrane (Millipore). The membrane was treated with 5% nonfat dry milk in TBST at 25 °C for 1 hr and then incubated in TBST supplemented with 0.1% Triton X-100 and 1% BSA and primary antibodies overnight at 4 °C. The membrane was washed three times and then incubated with horseradish peroxidase-conjugated secondary antibodies for 1 h at 25 °C. The protein band was revealed using an ECL Plus Detection Kit (Thermo Fisher Scientific, Waltham, MA), and the protein band density was quantified with the Odyssey Fc Imager (LI-COR Biosciences, Lincoln, NE) and normalized to the control protein band on the same blot. Tiam1 was detected using rabbit anti-Tiam1 antibody (sc-872, 1:1,000; Santa Cruz) or sheep anti-Tiam1 antibody (AF5038, 1:1,000; R&D Systems); Actin was detected using mouse anti-Actin antibody (MAB1501, 1:10,000; Millipore); GluN1 was detected using rabbit anti-GluN1 antibody (G8913, 1:1,000; Sigma) or rabbit anti-GluN1 antibody (AB52177, 1:1,000; Abcam); GluN2A was detected using rabbit anti-GluN2A (PA5–35377, 1:1,000; Thermo Fisher Scientific); GluN2B was detected using anti-mouse GluN2B (75–002, 1:1,000; NeuroMab); GluA1 was detected using mouse anti-GluA1 antibody (75–327, 1:1,000; NeuroMab); GluA2 was detected by using rabbit anti-GluA2 antibody (ab10529, 1:1,000; Millipore); EphB was detected by using rabbit anti-EphB pan antibody (PA5–39736, 1:1,000; Thermo Fisher Scientific); TrkB was detected by using rabbit anti-TrkB antibody (Ab187041, 1:1,000; Abcam); Caspase 3 was detected by suing rabbit anti-Caspase 3 antibody (9662, 1:1,000; Cell Signaling); PSD-95 was detected by using rabbit anti-PSD-95 antibody (ab18258, 1:2,000; Abcam); GAPDH was detected by using mouse anti-GAPDH (sc-47724, 1:1,000; Santa Cruz).
Morphological analysis.
To observe the effects of Tiam1 on spine remodeling in the spinal dorsal horn, rAAV8-hSyn-eGFP (UNC vector core) was injected via intra-spinal dorsal horn microinjection and was used to specifically label the neurons. Spinal cord lumbar sections (40 μm thick) were collected from mice perfused with 4% PFA, and only dendritic spines on neurons labeled with eGFP were selected for the spine analysis in a blinded manner as previously described17. All dendritic spine images of wide dynamic range (WDR) neurons were captured using a Laser Scanning Confocal Microscope (LSCM, Zeiss LSM 880, Germany) with a 63x oil 210 immersion objective. Z series were taken at an interval of 0.37 μm for each dendrite. Spine morphometric analysis was done in a blinded manner using Imaris software (Bitplane Scientific Software) as previously described67. Five criteria allowed us to sample and analyze whole cells with morphology similar to those observed for WDR neurons identified by previous studies9,36,38: (1) neurons were located within lamina 4 and 5; (2) eGFP positive neurons must have had dendrites and spines that were completely impregnated, appearing as a continuous length; (3) at least one dendrite extended into an adjacent lamina relative to the origin of the cell body; (4) at least one-half of the primary dendritic branches remained within the thickness of the tissue section, such that their endings were not cut and instead appeared to taper into an ending; and (5) the cell body diameter fell between 20 and 50 μm.
Spinal cord slice preparation and electrophysiology.
The spinal cord slices were obtained following the previous protocol68 with some modifications. In brief, mice were anesthetized with 3% isoflurane and removed the lumbar spinal cords via laminectomy. The spinal cords at the L4–L5 level were placed in ice-cold sucrose artificial cerebrospinal fluid containing (in mM) 234 sucrose, 3.6 KCl, 1.2 MgCl2, 2.5 CaCl2, 1.2 NaH2PO4, 12.0 glucose, and 25.0 NaHCO3, pre-saturated with 95% O2 and 5% CO2. The spinal cord tissue was glued onto the stage of a vibratome, and transverse slices (400 μm) of spinal cords were cut in ice-cold sucrose artificial cerebrospinal fluid and then preincubated in Krebs solution oxygenated with 95% O2 and 5% CO2 at 34 °C for at least 1 h before being transferred to the recording chamber. The Krebs solution contained (in mM) 117.0 NaCl, 3.6 KCl, 1.2 MgCl2, 2.5 CaCl2, 1.2 NaH2PO4, 11.0 glucose, and 25.0 NaHCO3. The spinal cord slices were placed in a glass-bottom chamber and continuously perfused with Krebs solution at 5.0 ml/min at 34 °C maintained by an inline solution heater and a temperature controller. We filled a glass pipette (5–10 MΩ) with an internal solution containing (in mM) 135.0 potassium gluconate, 5.0 TEA, 2.0 MgCl2, 0.5 CaCl2, 5.0 HEPES, 5.0 EGTA, 5.0 Mg-ATP, 0.5 Na-GTP and 10 lidocaine (lignocaine) N-ethyl bromide (adjusted to pH 7.2–7.4 with 1 M KOH; 290–300 mOsmol/L). The WDR neurons were identified for recording as previously described9,36,38. Postsynaptic NMDAR currents were elicited by puff application of 100 μM NMDA to the recorded neuron using a positive pressure system (4 p.s.i., 15 ms; Toohey Company, Fairfield, NJ), and puff application of the vehicle produced no currents. The tip of the puff pipette was placed 150 μm away from the recorded neuron. To minimize the Mg2+ block of NMDARs, the puff NMDA currents were recorded in an extracellular solution containing no Mg2+, 10 μM glycine, and 1 μM tetrodotoxin at a holding potential of −60 mV8,60. The pipette internal solution contained (in mM) 110.0 Cs2SO4, 2.0 MgCl2, 0.1 CaCl2, 1.1 EGTA, 10.0 HEPES, 2.0 Mg-ATP and 0.3 Na2GTP (pH was adjusted to 7.25 with 1.0 M CsOH; 280–300 mOsmol/L).
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses were performed using Prism 9 software (GraphPad Software Inc., San Diego, CA). No statistical methods were used to predetermine sample sizes, but our sample sizes are similar to those reported in previous publications8,45. The normality test was performed by the Shapiro–Wilk test. We used a Mann-Whitney U-test or two-tailed Student’s t-test to compare two groups and a one-way or two-way analysis of variance (ANOVA, followed by Dunnett’s or Tukey’s post-hoc tests) to compare more than two groups. Data are presented as means ± s.e.m. All the behavioral, electrophysiological, biochemical, and morphological data were obtained by counterbalancing experimental conditions with controls and analyzed in a blinded fashion. The following analyses were not completed blinded to experimental conditions: Immunoblotting. Statistical significance was accepted when P < 0.05.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-Tiam1 | Santa Cruz | SC-872 |
| Sheep anti-Tiam1 | R&D Systems | AF5038 |
| Rabbit anti-Rac1 | Cell Signaling | 4651 |
| Mouse anti-Actin | Millipore | MAB1501 |
| Rabbit anti-GluN1 | Sigma | G8913 |
| Rabbit anti-GluN1 | Abcam | AB52177 |
| Rabbit anti-GluN2A | Thermo Fisher | PA5-35377 |
| Mouse anti-GluN2B | NeuroMab | 75-002 |
| Mouse anti-GluA1 | NeuroMab | 75-327 |
| Rabbit anti-GluA2 | Millipore | Ab10529 |
| Rabbit anti-PSD-95 | Abcam | Ab18258 |
| Rabbit anti-EphB | Thermo Fisher | PA5-39736 |
| Rabbit anti-TrkB | Abcam | Ab187041 |
| Rabbit anti-Caspase 3 | Cell Signaling | 9662 |
| Mouse anti-GAPDH | Santa Cruz | Sc-47724 |
| Peroxidase Donkey anti-rabbit IgG | Jackson Immuno Res | 711-035-152 |
| Peroxidase Goat anti-mouse IgG | Jackson Immuno Res | 115-035-003 |
| Chemicals, peptides, and recombinant proteins | ||
| NSC23766 | Tocris | 2161 |
| NMDA | Tocris | 0114 |
| AMPA | Tocris | 0254 |
| Streptozotocin | Sigma-Aldrich | S0130 |
| L-Glutamate | Sigma-Aldrich | 49621 |
| Bicuculline | Sigma-Aldrich | 14343 |
| (2R)-amino-5-phosphonopentanoate (AP5) | Abcam | ab120003 |
| Spermine | Sigma-Aldrich | 85590 |
| Protease and phosphatase inhibitors | Thermo Fisher | 78440 |
| 4–15% Mini-PROTEAN Precast Protein Gels | Bio-Rad | 4560000 |
| Pierce Fast Western Blot Kit, ECL Substrate | Thermo Fisher | 35050 |
| RIPA Lysis Buffer | Thermo Fisher | 89900 |
| Experimental models: Organisms/strains | ||
| Tiam1 floxed mice | Cheng et al., 2021 | PMID: 33328293 |
| CaMKIIα-Cre mice | Jackson Laboratory | 005359 |
| Advillin-Cre mice | Jackson Laboratory | 026516 |
| vGluT2-Cre mice | Jackson Laboratory | 016963 |
| vGat-Cre mice | Jackson Laboratory | 017535 |
| Recombinant DNA | ||
| rAAV8-hSynapsin-eGFP | UNC Vector core | N/A |
| rAAV8-hSynapsin-Cre-eGFP | UNC Vector core | N/A |
| rAAV8-hSynapsin-Rac1Q61L-GFP | BCM Vector core | N/A |
| rAAV8-pCAG-DIO-eGFP | UNC Vector core | N/A |
| Software and algorithms | ||
| Prism 9.0 | GraphPad Software | https://www.graphpad.com/ |
| ImageJ | ImageJ | https://imagej.nih.gov/ij/download.html |
| Clampfit 10.0 software | Axon Instruments | www.moleculardevices.com |
Highlights.
Tiam1 in spinal excitatory neurons determines the development of neuropathic pain
Tiam1 coordinates synaptic structural and functional plasticity in neuropathic pain
Tiam1-Rac1 signaling initiates, transits, and sustains neuropathic pain
ASOs targeting spinal Tiam1 alleviate neuropathic pain sensitivity
ACKNOWLEDGEMENTS
We thank Drs. Andreas Tolias, Federico Scala, and De-Pei Li for technical advice and support. We also received technical assistance and resource from the BCM IDDRC Neurobehavioral Core. This work was supported by grants from the Department of Defense W81XWH-20-10790 (L.L.), the National Institutes of Health NS124141 (L.L. and K.F.T.), the Mission Connect/TIRR Foundation (L.L. and K.F.T.), and the National Institutes of Health NS062829 (K.F.T.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST DISCLOSURE
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information including 14 figures and 1 table can be found in this article online.
REFERENCES
- 1.Costigan M, Scholz J, and Woolf CJ (2009). Neuropathic pain: a maladaptive response of the nervous system to damage. Annu. Rev. Neurosci 32, 1–32. 10.1146/annurev.neuro.051508.135531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Finnerup NB, Kuner R, and Jensen TS (2021). Neuropathic pain: from mechanisms to treatment. Physiol. Rev 101, 259–301. 10.1152/physrev.00045.2019. [DOI] [PubMed] [Google Scholar]
- 3.Jayakar S, Shim J, Jo S, Bean BP, Singeç I, and Woolf CJ (2021). Developing nociceptor-selective treatments for acute and chronic pain. Sci. Transl. Med 13, eabj9837. 10.1126/scitranslmed.abj9837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Woolf CJ, and Salter MW (2000). Neuronal plasticity: increasing the gain in pain. Science 288, 1765–1769. 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]
- 5.Kuner R (2010). Central mechanisms of pathological pain. Nat. Med 16, 1258–1266. 10.1038/nm.2231. [DOI] [PubMed] [Google Scholar]
- 6.Battaglia AA, Sehayek K, Grist J, McMahon SB, and Gavazzi I (2003). EphB receptors and ephrin-B ligands regulate spinal sensory connectivity and modulate pain processing. Nat. Neurosci 6, 339–340. 10.1038/nn1034. [DOI] [PubMed] [Google Scholar]
- 7.Coull JAM, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, and De Koninck Y (2005). BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature 438, 1017–1021. 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- 8.Chen J, Li L, Chen S-R, Chen H, Xie J-D, Sirrieh RE, MacLean DM, Zhang Y, Zhou M-H, Jayaraman V, et al. (2018). The α2δ−1-NMDA Receptor Complex Is Critically Involved in Neuropathic Pain Development and Gabapentin Therapeutic Actions. Cell Rep. 22, 2307–2321. 10.1016/j.celrep.2018.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tan AM, Stamboulian S, Chang Y-W, Zhao P, Hains AB, Waxman SG, and Hains BC (2008). Neuropathic pain memory is maintained by Rac1-regulated dendritic spine remodeling after spinal cord injury. J. Neurosci 28, 13173–13183. 10.1523/JNEUROSCI.3142-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim SK, and Nabekura J (2011). Rapid synaptic remodeling in the adult somatosensory cortex following peripheral nerve injury and its association with neuropathic pain. J. Neurosci 31, 5477–5482. 10.1523/JNEUROSCI.0328-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tan AM, and Waxman SG (2012). Spinal cord injury, dendritic spine remodeling, and spinal memory mechanisms. Exp. Neurol 235, 142–151. 10.1016/j.expneurol.2011.08.026. [DOI] [PubMed] [Google Scholar]
- 12.Tan AM, and Waxman SG (2015). Dendritic spine dysgenesis in neuropathic pain. Neurosci. Lett 601, 54–60. 10.1016/j.neulet.2014.11.024. [DOI] [PubMed] [Google Scholar]
- 13.Duman JG, Blanco FA, Cronkite CA, Ru Q, Erikson KC, Mulherkar S, Saifullah AB, Firozi K, and Tolias KF (2021). Rac-maninoff and Rho-vel: The symphony of Rho-GTPase signaling at excitatory synapses. Small GTPases, 1–34. 10.1080/21541248.2021.1885264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bos JL, Rehmann H, and Wittinghofer A (2007). GEFs and GAPs: critical elements in the control of small G proteins. Cell 129, 865–877. 10.1016/j.cell.2007.05.018. [DOI] [PubMed] [Google Scholar]
- 15.Tolias KF, Bikoff JB, Burette A, Paradis S, Harrar D, Tavazoie S, Weinberg RJ, and Greenberg ME (2005). The Rac1-GEF Tiam1 couples the NMDA receptor to the activity-dependent development of dendritic arbors and spines. Neuron 45, 525–538. 10.1016/j.neuron.2005.01.024. [DOI] [PubMed] [Google Scholar]
- 16.Tolias KF, Bikoff JB, Kane CG, Tolias CS, Hu L, and Greenberg ME (2007). The Rac1 guanine nucleotide exchange factor Tiam1 mediates EphB receptordependent dendritic spine development. Proc Natl Acad Sci USA 104, 7265–7270. 10.1073/pnas.0702044104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheng J, Scala F, Blanco FA, Niu S, Firozi K, Keehan L, Mulherkar S, Froudarakis E, Li L, Duman JG, et al. (2021). The Rac-GEF Tiam1 Promotes Dendrite and Synapse Stabilization of Dentate Granule Cells and Restricts Hippocampal-Dependent Memory Functions. J. Neurosci 41, 1191–1206. 10.1523/JNEUROSCI.3271-17.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lai K-O, Wong ASL, Cheung M-C, Xu P, Liang Z, Lok K-C, Xie H, Palko ME, Yung W-H, Tessarollo L, et al. (2012). TrkB phosphorylation by Cdk5 is required for activity-dependent structural plasticity and spatial memory. Nat. Neurosci 15, 1506–1515. 10.1038/nn.3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bourinet E, Altier C, Hildebrand ME, Trang T, Salter MW, and Zamponi GW (2014). Calcium-permeable ion channels in pain signaling. Physiol. Rev 94, 81–140. 10.1152/physrev.00023.2013. [DOI] [PubMed] [Google Scholar]
- 20.Livshits G, Malkin I, Freidin MB, Xia Y, Gao F, Wang J, Spector TD, MacGregor A, Bell JT, and Williams FMK (2018). Genome-wide methylation analysis of a large population sample shows neurological pathways involvement in chronic widespread musculoskeletal pain. BÓL 19, 11–22. 10.5604/01.3001.0012.5920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arthur WT, Ellerbroek SM, Der CJ, Burridge K, and Wennerberg K (2002). XPLN, a guanine nucleotide exchange factor for RhoA and RhoB, but not RhoC. J. Biol. Chem 277, 42964–42972. 10.1074/jbc.M207401200. [DOI] [PubMed] [Google Scholar]
- 22.Decosterd I, and Woolf CJ (2000). Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain 87, 149–158. 10.1016/S0304-3959(00)00276-1. [DOI] [PubMed] [Google Scholar]
- 23.Shields SD, Eckert WA, and Basbaum AI (2003). Spared nerve injury model of neuropathic pain in the mouse: a behavioral and anatomic analysis. J. Pain 4, 465–470. 10.1067/s1526-5900(03)00781-8. [DOI] [PubMed] [Google Scholar]
- 24.Harte SE, Meyers JB, Donahue RR, Taylor BK, and Morrow TJ (2016). Mechanical conflict system: A novel operant method for the assessment of nociceptive behavior. PLoS ONE 11, e0150164. 10.1371/journal.pone.0150164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shepherd AJ, and Mohapatra DP (2018). Pharmacological validation of voluntary gait and mechanical sensitivity assays associated with inflammatory and neuropathic pain in mice. Neuropharmacology 130, 18–29. 10.1016/j.neuropharm.2017.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bennett GJ, and Xie YK (1988). A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 33, 87–107. 10.1016/0304-3959(88)90209-6. [DOI] [PubMed] [Google Scholar]
- 27.Höke A, and Ray M (2014). Rodent models of chemotherapy-induced peripheral neuropathy. ILAR J. 54, 273–281. 10.1093/ilar/ilt053. [DOI] [PubMed] [Google Scholar]
- 28.Pittenger G, and Vinik A (2003). Nerve growth factor and diabetic neuropathy. Exp. Diabesity Res 4, 271–285. 10.1155/EDR.2003.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Colleoni M, and Sacerdote P (2010). Murine models of human neuropathic pain. Biochim. Biophys. Acta 1802, 924–933. 10.1016/j.bbadis.2009.10.012. [DOI] [PubMed] [Google Scholar]
- 30.Islam MS (2013). Animal models of diabetic neuropathy: progress since 1960s. J. Diabetes Res 2013, 149452. 10.1155/2013/149452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peirs C, and Seal RP (2016). Neural circuits for pain: Recent advances and current views. Science 354, 578–584. 10.1126/science.aaf8933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tsien JZ, Chen DF, Gerber D, Tom C, Mercer EH, Anderson DJ, Mayford M, Kandel ER, and Tonegawa S (1996). Subregion- and cell type-restricted gene knockout in mouse brain. Cell 87, 1317–1326. 10.1016/s0092-8674(00)81826-7. [DOI] [PubMed] [Google Scholar]
- 33.Dragatsis I, and Zeitlin S (2000). CaMKIIalpha-Cre transgene expression and recombination patterns in the mouse brain. Genesis 26, 133–135. 10.1002/(sici)1526–968x(200002)26:2<133∷aid-gene10>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 34.Abraira VE, Kuehn ED, Chirila AM, Springel MW, Toliver AA, Zimmerman AL, Orefice LL, Boyle KA, Bai L, Song BJ, et al. (2017). The cellular and synaptic architecture of the mechanosensory dorsal horn. Cell 168, 295–310.e19. 10.1016/j.cell.2016.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Häring M, Zeisel A, Hochgerner H, Rinwa P, Jakobsson JET, Lönnerberg P, La Manno G, Sharma N, Borgius L, Kiehn O, et al. (2018). Neuronal atlas of the dorsal horn defines its architecture and links sensory input to transcriptional cell types. Nat. Neurosci 21, 869–880. 10.1038/s41593-018-0141-1. [DOI] [PubMed] [Google Scholar]
- 36.Tan AM, Chang Y-W, Zhao P, Hains BC, and Waxman SG (2011). Rac1-regulated dendritic spine remodeling contributes to neuropathic pain after peripheral nerve injury. Exp. Neurol 232, 222–233. 10.1016/j.expneurol.2011.08.028. [DOI] [PubMed] [Google Scholar]
- 37.Tan AM, Samad OA, Fischer TZ, Zhao P, Persson A-K, and Waxman SG (2012). Maladaptive dendritic spine remodeling contributes to diabetic neuropathic pain. J. Neurosci 32, 6795–6807. 10.1523/JNEUROSCI.1017-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Woolf CJ, and King AE (1987). Physiology and morphology of multireceptive neurons with C-afferent fiber inputs in the deep dorsal horn of the rat lumbar spinal cord. J. Neurophysiol 58, 460–479. [DOI] [PubMed] [Google Scholar]
- 39.Matus A, Ackermann M, Pehling G, Byers HR, and Fujiwara K (1982). High actin concentrations in brain dendritic spines and postsynaptic densities. Proc Natl Acad Sci USA 79, 7590–7594. 10.1073/pnas.79.23.7590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Furuyashiki T, Arakawa Y, Takemoto-Kimura S, Bito H, and Narumiya S (2002). Multiple spatiotemporal modes of actin reorganization by NMDA receptors and voltage-gated Ca2+ channels. Proc Natl Acad Sci USA 99, 14458–14463. 10.1073/pnas.212148999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Racz B, and Weinberg RJ (2004). The subcellular organization of cortactin in hippocampus. J. Neurosci 24, 10310–10317. 10.1523/JNEUROSCI.2080-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Honkura N, Matsuzaki M, Noguchi J, Ellis-Davies GCR, and Kasai H (2008). The subspine organization of actin fibers regulates the structure and plasticity of dendritic spines. Neuron 57, 719–729. 10.1016/j.neuron.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 43.Chaplan SR, Malmberg AB, and Yaksh TL (1997). Efficacy of spinal NMDA receptor antagonism in formalin hyperalgesia and nerve injury evoked allodynia in the rat. J. Pharmacol. Exp. Ther 280, 829–838. [PubMed] [Google Scholar]
- 44.Obara I, Goulding SP, Hu J-H, Klugmann M, Worley PF, and Szumlinski KK (2013). Nerve injury-induced changes in Homer/glutamate receptor signaling contribute to the development and maintenance of neuropathic pain. Pain 154, 1932–1945. 10.1016/j.pain.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li L, Chen S-R, Chen H, Wen L, Hittelman WN, Xie J-D, and Pan H-L (2016). Chloride homeostasis critically regulates synaptic NMDA receptor activity in neuropathic pain. Cell Rep. 15, 1376–1383. 10.1016/j.celrep.2016.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Peng Y, Zhao J, Gu Q-H, Chen R-Q, Xu Z, Yan J-Z, Wang S-H, Liu S-Y, Chen Z, and Lu W (2010). Distinct trafficking and expression mechanisms underlie LTP and LTD of NMDA receptor-mediated synaptic responses. Hippocampus 20, 646–658. 10.1002/hipo.20654. [DOI] [PubMed] [Google Scholar]
- 47.Gao Y, Dickerson JB, Guo F, Zheng J, and Zheng Y (2004). Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc Natl Acad Sci USA 101, 7618–7623. 10.1073/pnas.0307512101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stankiewicz TR, Pena C, Bouchard RJ, and Linseman DA (2020). Dysregulation of Rac or Rho elicits death of motor neurons and activation of these GTPases is altered in the G93A mutant hSOD1 mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis 136, 104743. 10.1016/j.nbd.2020.104743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rinaldi C, and Wood MJA (2018). Antisense oligonucleotides: the next frontier for treatment of neurological disorders. Nat. Rev. Neurol 14, 9–21. 10.1038/nrneurol.2017.148. [DOI] [PubMed] [Google Scholar]
- 50.Bryda EC (2013). The Mighty Mouse: the impact of rodents on advances in biomedical research. Mo Med 110, 207–211. [PMC free article] [PubMed] [Google Scholar]
- 51.Mohan A, Fitzsimmons B, Zhao HT, Jiang Y, Mazur C, Swayze EE, and Kordasiewicz HB (2018). Antisense oligonucleotides selectively suppress target RNA in nociceptive neurons of the pain system and can ameliorate mechanical pain. Pain 159, 139–149. 10.1097/j.pain.0000000000001074. [DOI] [PubMed] [Google Scholar]
- 52.Miyamoto Y, Yamauchi J, Tanoue A, Wu C, and Mobley WC (2006). TrkB binds and tyrosine-phosphorylates Tiam1, leading to activation of Rac1 and induction of changes in cellular morphology. Proc Natl Acad Sci USA 103, 10444–10449. 10.1073/pnas.0603914103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bushnell MC, Ceko M, and Low LA (2013). Cognitive and emotional control of pain and its disruption in chronic pain. Nat. Rev. Neurosci 14, 502–511. 10.1038/nrn3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kuner R, and Flor H (2016). Structural plasticity and reorganisation in chronic pain. Nat. Rev. Neurosci 18, 20–30. 10.1038/nrn.2016.162. [DOI] [PubMed] [Google Scholar]
- 55.Fu AK, and Ip NY (2017). Regulation of postsynaptic signaling in structural synaptic plasticity. Curr. Opin. Neurobiol 45, 148–155. 10.1016/j.conb.2017.05.016. [DOI] [PubMed] [Google Scholar]
- 56.Saneyoshi T, Matsuno H, Suzuki A, Murakoshi H, Hedrick NG, Agnello E, O’Connell R, Stratton MM, Yasuda R, and Hayashi Y (2019). Reciprocal Activation within a Kinase-Effector Complex Underlying Persistence of Structural LTP. Neuron 102, 1199–1210.e6. 10.1016/j.neuron.2019.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Swayze EE, Siwkowski AM, Wancewicz EV, Migawa MT, Wyrzykiewicz TK, Hung G, Monia BP, and Bennett CF (2007). Antisense oligonucleotides containing locked nucleic acid improve potency but cause significant hepatotoxicity in animals. Nucleic Acids Res. 35, 687–700. 10.1093/nar/gkl1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bennett GJ, Chung JM, Honore M, and Seltzer Z (2003). Models of neuropathic pain in the rat. Curr. Protoc. Neurosci Chapter 9, Unit 9.14. 10.1002/0471142301.ns0914s22. [DOI] [PubMed] [Google Scholar]
- 59.Kohro Y, Sakaguchi E, Tashima R, Tozaki-Saitoh H, Okano H, Inoue K, and Tsuda M (2015). A new minimally-invasive method for microinjection into the mouse spinal dorsal horn. Sci. Rep 5, 14306. 10.1038/srep14306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li L, Chen S-R, Zhou M-H, Wang L, Li D-P, Chen H, Lee G, Jayaraman V, and Pan H-L (2021). α2δ−1 switches the phenotype of synaptic AMPA receptors by physically disrupting heteromeric subunit assembly. Cell Rep. 36, 109396. 10.1016/j.celrep.2021.109396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chaplan SR, Bach FW, Pogrel JW, Chung JM, and Yaksh TL (1994). Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Methods 53, 55–63. 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- 62.Corder G, Tawfik VL, Wang D, Sypek EI, Low SA, Dickinson JR, Sotoudeh C, Clark JD, Barres BA, Bohlen CJ, et al. (2017). Loss of μ opioid receptor signaling in nociceptors, but not microglia, abrogates morphine tolerance without disrupting analgesia. Nat. Med 23, 164–173. 10.1038/nm.4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Corder G, Ahanonu B, Grewe BF, Wang D, Schnitzer MJ, and Scherrer G (2019). An amygdalar neural ensemble that encodes the unpleasantness of pain. Science 363, 276–281. 10.1126/science.aap8586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ru Q, Lu Y, Saifullah AB, Blanco FA, Yao C, Cata JP, Li D-P, Tolias KF, and Li L (2022). TIAM1-mediated synaptic plasticity underlies comorbid depression-like and ketamine antidepressant-like actions in chronic pain. J. Clin. Invest 132. 10.1172/JCI158545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zeng L-H, Xu L, Rensing NR, Sinatra PM, Rothman SM, and Wong M (2007). Kainate seizures cause acute dendritic injury and actin depolymerization in vivo. J. Neurosci 27, 11604–11613. 10.1523/JNEUROSCI.0983-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Huang W, Zhu PJ, Zhang S, Zhou H, Stoica L, Galiano M, Krnjević K, Roman G, and Costa-Mattioli M (2013). mTORC2 controls actin polymerization required for consolidation of long-term memory. Nat. Neurosci 16, 441–448. 10.1038/nn.3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tu Y-K, Duman JG, and Tolias KF (2018). The Adhesion-GPCR BAI1 Promotes Excitatory Synaptogenesis by Coordinating Bidirectional Trans-synaptic Signaling. J. Neurosci 38, 8388–8406. 10.1523/JNEUROSCI.3461-17.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Scala F, Kobak D, Bernabucci M, Bernaerts Y, Cadwell CR, Castro JR, Hartmanis L, Jiang X, Laturnus S, Miranda E, et al. (2021). Phenotypic variation of transcriptomic cell types in mouse motor cortex. Nature 598, 144–150. 10.1038/s41586-020-2907-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code.