

Summary
Soluble oligomers of amyloid β-protein (Aβ) have been defined as aggregates in supernatants following ultracentrifugation of aqueous extracts from Alzheimer’s disease brains and are believed to be upstream initiators of synaptic dysfunction, but little is known about their structures. We now report the unexpected presence of Aβ fibrils in synaptotoxic high-speed supernatants from AD brains extracted by soaking in aqueous buffer. The fibrils did not appear to form during preparation, and their counts by EM correlated with ELISA quantification. Cryo-EM structures of aqueous Aβ fibrils were identical to those from sarkosyl-insoluble homogenates. The fibrils in aqueous extracts were labelled by lecanemab, an Aβ aggregate-directed antibody reported to improve AD cognitive outcomes. Lecanemab protected against fibril synaptotoxicity. We conclude that Aβ fibrils are abundant in aqueous extracts from AD brains and have the same structures as those from plaques. These findings have implications for AD pathogenesis and drug design.
Introduction
Amyloid β (Aβ) and tau monomers aggregate into fibrils found in amyloid plaques and neurofibrillary tangles in the brains of people with Alzheimer’s disease (AD). Fibrils can be extracted from postmortem brains by ultracentrifugation in the presence of detergents such as N-lauryl sarcosine (sarkosyl). Pelleted detergent-insoluble fibrillar Aβ, however, has been reported less toxic than Aβ from aqueous ultracentrifugal supernatants prepared without detergent.1 Aqueous supernatants contain Aβ aggregates that disrupt synaptic plasticity1–3 and neurite morphology,4,5 promote tau phosphorylation,4,6,7 decrease memory,1 and accelerate Aβ seeding.8–11 These aqueous supernatant Aβ aggregates have been assumed to be non-fibrillar and termed “soluble oligomers” or “protofibrils.” Their neurotoxicity makes them therapeutic targets.
Lecanemab is an FDA-approved monoclonal antibody that improved cognitive outcomes in a phase 3 trial.12 Its success has been attributed to selectivity for soluble Aβ protofibrils over insoluble fibrils.13,14 However, a structural definition of protofibrils (or oligomers) from human brains does not exist.15 Here, we use “aggregate” instead of “protofibril” or “oligomer,” because oligo implies few monomers, whereas aggregates in aqueous brain extracts may comprise thousands of monomers. We use “protofilament” (not “protofibril,” which has no strict definition) to mean a single stack of β-pleated sheet-folded monomers; for Aβ, two protofilaments form one fibril.16 Nomenclature notwithstanding, improving AD therapy requires a structural understanding of aqueous Aβ aggregates in the human brain.
The term “soluble” is used for aggregates found in aqueous supernatants, but ultracentrifugation protocols vary and are often poorly described. Most Aβ aggregates aqueously extracted from AD brains have been reported to be high molecular weight (HMW): they elute in or near the void volume of size exclusion chromatography (SEC) columns (≥500 kDa).17,18 Low MW aggregates from AD brain, including covalent dimers,19 are particularly toxic,1,18 but disassembly of HMW aggregates ex vivo is often necessary to observe LMW aggregates and their toxicity.1,4,18 The structure of any of these is unknown.
Dynamic non-covalent interactions render extraction of aqueous Aβ aggregates under non-denaturing conditions challenging. A recent method soaks minced bits of AD cerebral cortex instead of homogenizing tissue, to avoid shearing plaques and other large Aβ aggregates; these soaking extracts exhibited a similar toxicity to homogenates.20,21 We previously reported a non-denaturing method to enrich for Aβ aggregates from AD soaking extracts by immunoprecipitation with the calcium-sensitive antibody B24 and were surprised to observe Aβ fibrils in the B24 eluates of putatively soluble extracts.22 We now report that Aβ fibrils are present in aqueous soaking extracts even without immunoprecipitation, have the same structure as plaque fibrils, inhibit synaptic transmission, and are bound and blocked by lecanemab.
Results
Aβ fibrils can be pelleted from aqueous extracts of Alzheimer’s disease brain
For immunoprecipitation using B24,22 we started with aqueous extracts prepared by mincing and soaking multiple regions of AD cerebral cortex in TBS, followed by ultracentrifugation and collection of the supernatant (Table 1).20 We previously immunoprecipitated Aβ with B24 from these soaking extracts in the presence of calcium and eluted it using EGTA, to avoid conditions which might denature aggregates.22 We found that the eluted HMW Aβ aggregates could be re-pelleted in a benchtop centrifuge at 20,000 g in a 1.5-ml tube for 1 h (pelleting distance ~1 cm), with no detectable Aβ remaining in the supernatants by ELISA. Immunogold labelling showed short Aβ fibrils in the pellets.22
Table 1.
The soaking extract method and its modifications for control experiments in this report
| Step # | Steps in standard protocol | Modifications of standard protocol for the specified control experiments, and relevant figure number |
|---|---|---|
| 1 | Frozen tissue thawed, grey matter dissected | Fig S3I–L: Fresh tissue |
| 2 | Tissue chopped using McIlwain Tissue Chopper set to 0.5 mm width | |
| 3 | Tissue bits soaked in 1:5 w:v extraction buffer 30 min 4°C with nutation in 50-ml tube |
Fig S2B: Tissue bits first rinsed with ~15 volumes extraction buffer before soaking Fig S3E: 1:15 w:v |
| 4 | 2,000 g fixed angle (pelleting distance ~4 cm) 10 min 4°C, top 90% supernatant collected | |
| 5 | 200,000 g in SW41Ti (pelleting distance ~9 cm) 110 min 4°C, top 90% of supernatant collected |
Fig S2A: Top 90% of supernatant divided into top 2 ml and bottom ~7 ml Fig 1C–G: 20,000 – 475,000 g in TLA100.3 (pelleting distance ~2 cm) for 110 min 4°C; top 90% of supernatant collected |
| 6 | Aliquot 1 ml into 1.5-ml Eppendorf LoBind tubes; store −80°C |
Fig S2E: Aliquot into siliconized or BSA-coated tubes Fig S3C, H: Store at 4°C Fig S3D: Dilute serially before freezing Fig S3E–F: Add various Aβ aggregation inhibitors before freezing Fig S3G: Filter through aluminum oxide filters before freezing |
| 7 (re-centrifugation) | Thaw aliquot(s), 20,000 g fixed angle (pelleting distance ~1 cm) 60 min 4°C. Retain input, supernatant and pellet for ELISA and/or EM |
We now asked if Aβ fibrils could be pelleted from soaking extracts without immunoprecipitation. We spun 1 ml of TBS soaking extracts (Table 1, Step #7) at 20,000 g for 1 h. Soluble Aβ aggregates would not be expected to pellet. However, a substantial proportion of aggregated Aβ (i.e., that which was detectable by a monomer-preferring Aβ ELISA only after disassembly in 5 M GuHCl) was depleted from the supernatants and recovered in the pellets (Fig 1A, center). Monomeric Aβ (i.e., not requiring GuHCl denaturation for detection by the monomer-preferring ELISA) was not depleted by re-centrifugation (Fig 1A, right), as expected. Examining the re-centrifugation pellets by EM revealed amyloid fibrils that reacted with N-terminal anti-Aβ antibody D54D2 (Fig 1B). In aqueous extracts from the minced AD brains, we also noticed fibrils resembling PHFs that stained with anti-tau antibodies (Fig S1). In this report, we focus on the Aβ fibrils. None were observed in the pellets from aqueous extracts of two control brains (C1 and C2), and a single fibril was seen on an EM grid of a third control brain (C3). The Aβ fibrils’ average length across thirteen brains was ~100 nm (Fig S2A). However, a Q-Q plot suggested undercounting of both very short fibrils (due to an inability to see them) and very long fibrils (due to their exceeding the micrograph size) (Fig S2B). Thus, there was probably a wider distribution of lengths than depicted in Fig S2A.
Fig 1. Aqueous soaking extracts of AD brain contain insoluble Aβ fibrils.

(A) Aliquots of aqueous soaking extracts from ultracentrifugal supernatants of AD cortex (see Methods and Table 1) were thawed and spun at 20,000 g in a tabletop centrifuge, and Aβ was quantified by ELISA with (center) and without (right) 5 M GuHCl. Note different y-axis scales. GuHCl denatures Aβ aggregates and allows detection by a monomer-preferring ELISA. Aβ detectable without GuHCl denaturation is monomeric. ELISAs revealed that aggregated Aβ but not Aβ monomers could be re-pelleted. (B) Fibrils in the pellets of re-centrifuged aqueous soaking extracts of brain AD7 were labeled by anti-Aβ N-terminal antibody D54D2. Scale bar = 100 nm. (C-F) A TBS soaking extract of brain AD6 was prepared by mincing cortex, soaking in TBS for 30 min, and a 10 min spin at 2,000 g (Table 1, steps 1–4). This supernatant was divided and ultracentrifuged in a TLA100.3 rotor (pelleting distance ~2 cm) at indicated g forces, and Aβ quantified in the supernatant. Monomer-preferring ELISA performed with (C) or without (D) GuHCl denaturation showed that Aβ aggregates but not monomers were depleted as the g-force increased, as did the ratio of Aβ concentration with:without GuHCl (E). (F) Increasing g-force abolished signal from two aggregate-preferring Aβ ELISAs. (G) Superdex 200 Increase size exclusion chromatography of the 475,000 g supernatant revealed a low MW Aβ profile. (H) Unlike brain, CSF Aβ aggregates were not depleted as g-force increased. (I) The Aβ concentration in soaking extracts after GuHCl denaturation correlated with the number of fibrils in the pellet after centrifugation by manual blinded counting of immunogold EM. Line represents simple linear regression. Drawings made with Biorender.com.
Aβ fibrils were not contaminants from the original ultracentrifugation pellets because they were present when only the top ~20% of the supernatant was collected and re-pelleted (Fig S2C). To rule out that the fibrils had adhered to the surface of the minced tissue rather than diffused out of the tissue into the soaking buffer, we rinsed the minced brain bits with 15 volumes of extraction buffer through a 100-μm filter prior to soaking. We found no reduction in the amount of extracted Aβ aggregates measured biochemically (Fig S2D) or as fibrils seen by EM. Further arguing in favor of diffusion of fibrils from the minced tissue into the aqueous buffer rather than carryover from the cut surface was the observation (also in Fig S2 of Hong et al.20) that diffusion of Aβ into the buffer required 15–30 min to plateau, similar to the diffusion rate of control soluble proteins.
Sufficient centrifugal force can pellet HMW Aβ aggregates
When accounting for the pelleting distance, our initial and re-centrifugation protocols were approximately equal in efficiency: the distance in thin-wall tubes in the SW41Ti rotor (initial soaking extract) is ~9 cm when full (Table 1, Step #5), whereas it is ~1 cm for a 1 ml microcentrifuge tube at a 45-degree angle in the benchtop centrifuge (Table 1, Step #7). Thus, the benchtop centrifugation should accomplish the same pelleting efficiency at an ~9-fold lower g-force. It was therefore possible that if higher g-forces had been used initially in preparing the soaking extracts, the fibrils would not be recovered from the supernatant during re-centrifugation. To test this, we modified the initial ultracentrifugation, using the fixed-angle TLA100.3 rotor (pelleting distance ~2 cm) at increasing g-forces, instead of the SW41Ti rotor (pelleting distance ~9 cm). We found that g-forces ≥250,000 g across the shorter distance depleted nearly all biochemically detectable aggregated Aβ from the supernatants (Fig 1C) but retained native monomers (Fig 1D). In 475,000 g supernatants, GuHCl denaturation released only 1.8-fold more Aβ monomers than without GuHCl, whereas in 20,000 g supernatants, GuHCl addition released 54-fold more (Fig 1E). This difference suggests that most Aβ in the 475,000 g supernatants consisted of native monomers and covalent dimers.1,19 To verify that Aβ aggregates were depleted from supernatants spun ≥ 250,000 g, we used two high-sensitivity ELISAs with aggregate-preferring capture antibodies23,24 not requiring GuHCl denaturation (Fig 1F). Neither assay could detect aggregates in the ≥250,000 g supernatants. Residual Aβ in the 475,000 g supernatants exhibited a LMW SEC profile (Fig 1G).
We conclude that most Aβ, and all its HMW aggregates from aqueous brain extracts, is made of suspended particles, consistent with the presence of fibrils. We compared this finding to Aβ in cerebrospinal fluid (CSF). Detection of CSF Aβ by aggregate-specific ELISA was low and unaffected by centrifugal force (Fig 1H). This suggests that the few Aβ aggregates detected in CSF were soluble, in contrast to those from aqueous extracts of AD brain.
Aβ fibrils in aqueous soaking extracts do not appear to form during sample preparation
We investigated if the Aβ fibrils were absent during life but polymerized from smaller non-fibrillar aggregates during the postmortem interval or sample processing. We used the standard soaking extract protocol using the SW41Ti rotor at 200,000 g,20 followed by re-centrifugation in the tabletop centrifuge at 20,000 g (Table 1).20
One possibility was that Aβ polymerization occurred along the plastic tube surface during re-centrifugation of the original soaking extracts. We re-centrifuged the standard soaking extracts and moved the supernatants to new tubes at specific time intervals. The Aβ in the pellets left behind was quantified by ELISA following GuHCl denaturation. If new fibril nucleation were occurring, we would have expected a lag in the accumulation of Aβ in the pellets, whereas pelleting of pre-existing particulates would be linearly time-dependent until they had been depleted from the supernatants. We observed no lag, suggesting no nucleation (Fig S2E, F). Storage of soaking extracts in siliconized or bovine serum albumin-coated tubes had no effect on subsequent re-pelleting compared to standard Eppendorf Protein LoBind polypropylene tubes (Fig S2G).
We investigated if Aβ polymerization occurred during freezing and thawing of freshly prepared soaking extracts. When proceeding directly from the initial SW41Ti rotor (Table 1, step 5) to a second spin in the benchtop centrifuge (step 7) at 4°C, Aβ aggregates measured by ELISA could still be pelleted (Fig S3A), and fibrils were observed by immuno-EM (Fig S3B). Nevertheless, freezing and thawing of a freshly prepared soaking extract resulted in the re-pelleting of more Aβ compared to storage at 4°C (Fig S3C). We also found that the re-pelleting was concentration-dependent: when the original ultracentrifugation supernatants were diluted at least four-fold before freezing, less aggregated Aβ could subsequently be re-pelleted after thawing (Fig S3D). This concentration dependence only occurred when dilution was performed after soaking and initial centrifugation. When dilution was performed by increasing the tissue weight-per-volume (w:v) soaking ratio from the usual 1:5 to 1:15 g/ml, similar amounts of Aβ were extracted, and the freeze/thaw dependence of the amount pelleted remained (Fig S3E). These results suggest that at w:v ratios of 1:5 and 1:15, the extraction of Aβ saturated at similar concentrations.
To assess further whether polymerization occurred during freeze/thaw, we added Aβ aggregation inhibitors, including aggregate-binding antibodies, 0.45% CHAPS,25 and scyllo-inositol,26,27 immediately after ultracentrifugation of the soaking extracts and before freezing the supernatants. None of these agents reduced the amount of Aβ that could be re-pelleted after thawing (Fig S3F, G). We also expected that if fibrils had formed ex vivo from monomers and/or soluble LMW aggregates during freeze/thaw, filtering the soaking extracts immediately after ultracentrifugation should have had no effect on the recovery and pelleting of Aβ. We found that freshly prepared soaking extracts were depleted of Aβ aggregates by a 20-nm pore size alumina filter (Fig S3H), but that they passed through a 100-nm filter, implying that Aβ aggregates were at least 20 nm in size prior to freezing the original soaking extracts. SEC of a freshly prepared TBS extract immediately after ultracentrifugation (without freezing) showed that Aβ still eluted in the void volume of a Superdex 200 Increase column (~≥500 kDa) (Fig S3I). Together, these results suggest that Aβ aggregates were HMW prior to freezing and storage of the soaking extracts. Increased pelleting of Aβ fibrils after freeze/thaw may be due to clumping of existing fibrils, but probably not to new fibril formation.
Another possibility was that polymerization occurred after death but before tissue freezing. To best exclude this artifact, we prepared an AD soaking extract and applied it to the Superdex 200 Increase column within 8 h of death without freezing either the tissue or the extract. Virtually all the ELISA-detectable aggregated Aβ eluted in the void volume of a Superdex 200 Increase column (Fig S3J–L), and fibrils could be pelleted upon re-centrifugation (Fig S3M).
Fibrils account for a substantial proportion of high-molecular weight Aβ aggregates in soaking extracts
We counted the D54D2-immunoreactive fibrils in the re-centrifugation pellets from 13 AD brains, using 30 EM fields each. The average number of fibrils per field correlated with the Aβ levels by ELISA after GuHCl denaturation (Fig 1I). The 13 brains appeared to separate into two groups, but our limited sample size could not determine if this was chance separation or two true populations.
Fibrils from soaking extracts of AD brain have the same structures as those from sarkosyl-insoluble fractions
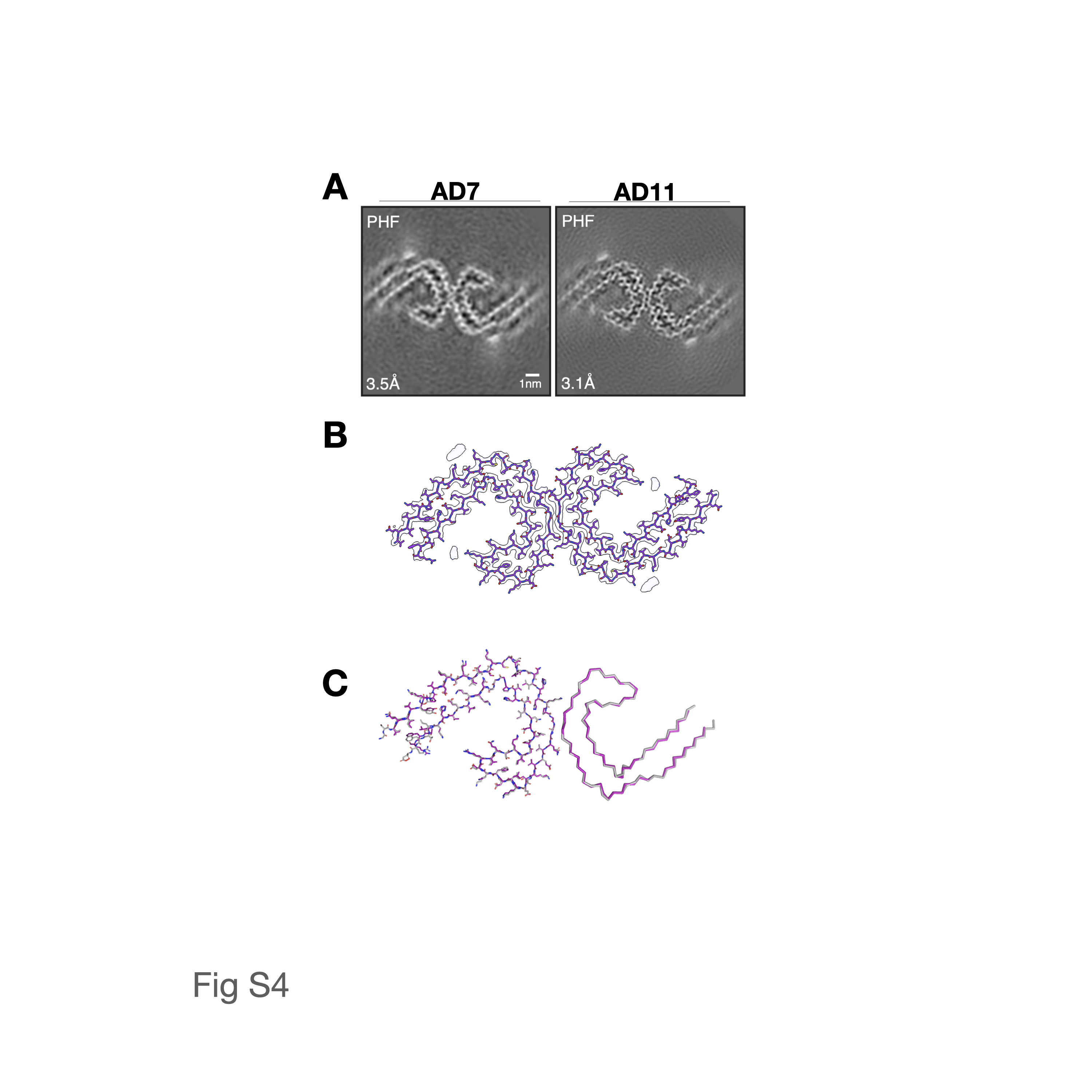
The cryo-EM structures of Aβ fibrils from sarkosyl-insoluble homogenates of AD brains were recently determined.16 In comparison, the aqueous soaking extract-derived pellets showed shorter fibrils that clumped less, consistent with their relative resistance to pelleting. Cryo-EM structure determination of soaking extract-derived fibrils from two cases of sporadic AD (AD7 and AD11) revealed identical structures to those of Aβ fibrils from sarkosyl-insoluble preparations18 (Fig 2). AD7 had only Type I fibrils, whereas AD11 had Type I and Type II fibrils.16 Tau fibrils from the re-centrifuged soaking extracts were structurally identical to PHFs from sarkosyl-insoluble fractions of AD brains (Fig S4).28
Fig 2. Aβ fibrils from aqueous extracts of AD brains have the same cryo-EM structures as fibrils from sarkosyl-insoluble homogenates.

(A) XY-cross-sections of cryo-EM maps of Aβ fibrils from soaking extracts of cases AD7 and AD11. For each map, a sum of the reconstructed densities for several XY-slices, approximating one β-rung, is shown. Aβ fibril types I and II16 are indicated at top left; the percentages of Type I and Type II fibrils relative to the total of imaged fibrils are shown at top right. Resolutions at bottom left. Scale bar = 1 nm. (B, C) Cryo-EM density maps (grey) and atomic models for Aβ Type I (orange), Aβ Type II (blue). (D, E), Comparison of the cryo-EM structures of Aβ Type I (B) fibrils and Aβ Type II (C) fibrils extracted from aqueous soaking extracts vs. sarkosyl-insoluble homogenates (in grey for Type I and Type II fibrils) from brains of AD patients.16 Structures are shown as sticks (above) for one protofilament and as ribbons (below) for the other.
Lecanemab labels Aβ fibrils from soaking extracts and protects from synaptotoxicity
Lecanemab is a humanized mouse monoclonal antibody raised against synthetic aggregates of Aβ bearing the AD-causing APP “Arctic” mutation E693G.13,29 Synthetic Aβ40[E693G] forms more soluble aggregates than wild-type Aβ40 in vitro; the mechanism of action of lecanemab is thought to relate to its preference for small, soluble Aβ aggregates over fibrils.30 Since lecanemab has been shown to immunoprecipitate Aβ from aqueous AD brain extracts,13 we asked whether it can bind to fibrils. Lecanemab decorated Aβ fibrils (Fig 3A, B) that had been re-pelleted from AD soaking extracts. We also examined antibody h1C22, which we previously reported binds better to synthetic Aβ oligomers than monomers or fibrils.5,22,23 We also observed specific h1C22 labelling of brain-derived Aβ fibrils (Fig 3C–E).
Fig 3. Lecanemab decorates Aβ fibrils from Alzheimer’s disease brains and protects from synaptotoxicity.

(A, B) Lecanemab labels pelleted Aβ fibrils from the re-centrifugation of AD7 TBS soaking extract with protein A gold (arrowheads). PHFs in the pellet did not react with lecanemab (arrows). (C, D) Immunogold labeling with Aβ aggregate-preferring h1C22 similarly decorates soaking extract fibrils. (E) Protein A gold alone does not label fibrils. (F) ELISA quantification of Aβ42 immunoprecipitated from AD6 aqueous extract followed by washing, elution and denaturation in 5 M GuHCl. Bapineuzumab, but not lecanemab, immunoprecipitated Aβ from the 250,000 g and 475,000 g (TLA100.3, pelleting distance ~2 cm) supernatants. (G-I) Immunogold of paraformaldehyde-fixed ultrathin cryosections from AD11 (G,H) and AD7 (I,J) brains reveals labeling of Aβ fibrils by lecanemab. (K) Protein A gold alone (brain AD7) shows minimal background labeling. Scale bar = 100 nm. (L, M) Soaking extracts were centrifuged for 2h, and the pellet resuspended in an equivalent volume of TBS. Supernatants or pellets from two AD soaking extracts were applied to mouse hippocampal slices at 5% v:v perfusate, and LTP was induced (arrow at time 0). (N) Lecanemab reversed the impairment of LTP due to the AD 12 soaking extract pellet. (O) The average of the last 10 minutes of recording in (N) showed a mean difference of 94% baseline fEPSP (95% CI, 61% - 127%, P<0.001). (P) The pelleted fraction of AD but not control brain soaking extracts impaired the hippocampal synaptic baseline without LTP induction. (Q) Lecanemab reversed impairment of baseline fEPSP slope by AD12 soaking extract pellet by a mean of 42% compared to isotype control (95% CI, 17 – 67, P = 0.002), quantified in (R). Error bars represent SD.
Since lecanemab labeled Aβ fibrils from aqueous brain extracts, we hypothesized that it would only immunoprecipitate Aβ from extracts prepared at g-forces that retain aggregates detectable by other means, such as aggregate-preferring ELISAs.24 We found that lecanemab could only immunoprecipitate Aβ from aqueous AD brain supernatants that had been ultracentrifuged at ≤100,000 g in the TLA100.3 rotor (Fig 3F), like 1C22 (Fig. 1F). Bapineuzumab, which binds both monomeric and aggregated Aβ, could still immunoprecipitate some Aβ from 250,000 and 475,000 g supernatants (Fig. 3F).
Lecanemab stains plaques and vascular amyloid by immunohistochemistry.31 To determine if it stains plaque fibrils, we prepared minimally fixed ultrathin cryosections from AD cortex to preserve antibody epitopes. Although cytoarchitecture was not well preserved, lecanemab labeled fibrils in situ (Fig 3G–J), with little background from protein A gold alone (Fig 3K).
We previously found that aqueous extracts of AD brain but not control brain can impair long-term potentiation (LTP) in wild-type mouse hippocampal slices.1,18,22,32 We asked if synaptotoxicity in this model was in the insoluble or soluble fraction of aqueously diffusible soaking extracts. Re-centrifugation demonstrated synaptotoxicity in pellets but not supernatants (Fig 3L, M). Lecanemab reversed synaptotoxicity by a mean of 93.7% fEPSP slope compared to isotype control (95% CI, 60.6% - 127%, P<0.0001) (Fig 3N, O). We observed that the resuspended fibril-rich pellets could impair the baseline field potential slope without LTP induction by high-frequency stimulation (HFS) (Fig 3P). Pellets from a re-centrifuged control brain soaking extract (C3) did not impair baseline synaptic transmission (Fig. 3P). Lecanemab prevented inhibition of the synaptic baseline compared to isotype control (mean difference 42% fEPSP slope, 95% CI 17% - 67%, P=0.002) (Fig 3Q, R).
Discussion
Our results indicate that at least some Aβ aggregates from aqueous extracts of AD brains are identical in structure to plaque-derived fibrils. While we cannot rule out the presence of some non-fibrillar HMW Aβ aggregates, a correlation between total Aβ levels and the number of Aβ fibrils suggests that fibrils are a substantial proportion of HMW Aβ aggregates (“oligomers,” “protofibrils”) that diffuse from AD cerebral cortex into aqueous extracts.
An unavoidable limitation of this work is the need to examine AD brains postmortem. Although we attempted to mitigate artifacts, we cannot exclude disassembly of some plaques into free-floating fibrils after death. We also cannot exclude some ex vivo aggregation of monomers or LMW aggregates (e.g. dimers) into fibrils. However, small-pore filters, inhibitors of Aβ aggregation, and the use of fresh tissue provided no evidence for these artifacts.
If short, dispersed Aβ fibrils can diffuse through the brain, they may be in equilibrium with plaques. APP transgenic mice show halos surrounding methoxy-XO4-labeled plaques that immunoreact with Aβ aggregate-preferring antibodies.33 These halos may consist of Aβ fibrils that are packed too loosely to shift the fluorescence spectra of thioflavin dyes and have more access to synaptic endings than fibrils from plaque cores. Supporting this model is our observation that diffusible Aβ fibrils have the same cryo-EM structures as those from sarkosyl-insoluble brain fractions. Experiments in mice in which microglia were depleted, or the AD risk gene TREM2 was knocked out, have shown that microglial plaque compaction may protect dendrites and synapses.34–36 Our observation of synaptotoxicity within the fibril-containing fractions of soaking extracts is compatible with this model.
Lecanemab prefers intermediate molecular weight synthetic Aβ aggregates over synthetic fibrils;13 however, in humans, lecanemab reduces amyloid PET signal, a marker of fibrillar Aβ,37 and labels amyloid plaques and blood vessel deposits by immunohistochemistry.31 Based on our finding that lecanemab labels Aβ fibrils in aqueous AD brain extracts, and that both lecanemab binding and lecanemab-dependent synaptotoxicity were restricted to the pelletable fractions, we conclude that lecanemab’s therapeutic target in the human brain includes diffusible fibrils with the same structures as those from plaques. It remains possible that lecanemab’s target includes non-fibrillar aggregates because we could not exclude their presence.
We also observed PHFs in soaking extracts. Others have observed that tau seeding activity in human aqueous extracts depends on the largest MW tau aggregates.10,11 Based on our results, we suspect that these seed-competent preparations contained PHFs with identical folds to those from sarkosyl-insoluble preparations.38 Supporting this model is the observation that short tau fibrils were the most seed-competent species in brain extracts from transgenic mice expressing human P301S tau.39 Future design of therapeutic agents against tau seeds should consider that they may be PHF.
STAR Methods
Resource Availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dennis Selkoe (dselkoe@bwh.harvard.edu).
Materials availability
Lecanemab and bapineuzumab were produced recombinantly from publicly available patent sequences. Human 1C22 (h1C22) was produced recombinantly after sequencing 1C22 and cloned with a human IgG1 backbone. 21F12, 2G3, and 3D6 were gifts from Elan Pharmaceuticals. 266 was produced from hybridoma cells purchased from ATCC. 71A1 was supplied by Abyssinia Biologics. No new unique reagents were generated in this study, but all antibodies are available for sharing upon request of the lead contact.
Data and code availability
All biochemical data reported in this paper will be shared by the lead contact upon request. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Cryo-EM maps have been deposited in the Electron Microscopy Data Bank (EMDB) under accession numbers EMD-15770, EMD-15771 and EMD-15772. Corresponding refined atomic models have been deposited in the Protein Data Bank (PDB) under accession numbers 8AZS, 8AZT and 8AZU.
Experimental Model and Study Participant Details
Brain donor demographic and pathologic information is described in Table S1. Data on gender, race, ancestry, ethnicity, and socioeconomic status were not available for the deidentified brain tissue samples. Low sample size precluded subgroup analysis by sex. Both subjects undergoing lumbar puncture were consented according to protocols approved by the Mass General Brigham Institutional Review Board.
All animal procedures were performed in accordance with the National Institutes of Health Policy on the Use of Animals in Research and were approved by the Harvard Medical School Standing Committee on Animals. Studies of mouse hippocampal slice recording used wild-type C57bl/6 mice aged 1–3 months purchased from Jackson Labs. The mice were group-housed 4 mice in each cage in a controlled room with 22–25 °C, 50% humidity and a 12 h light/dark circadian cycle with ad libitum access to food and water. Male and female mice were randomly assigned to treatment groups.
Method Details
Aqueous soaking extraction of fibrils
The soaking method for aqueous extraction of diffusible Aβ aggregates from AD brain has been described previously20,21 and is depicted in Table 1. Brain donor demographic and pathologic information is described in Table S1. Grey matter was dissected from fresh or frozen cortex (pooled tissue from frontal, parietal, and temporal lobes) of individuals with AD and minced using a McIlwain tissue chopper (razor blade) set at 0.05 mm. The resultant brain bits were soaked for 30 min in a 1:5 ratio (w:v) of TBS extraction buffer (25 mM Tris supplemented with 150 mM NaCl, as well as protease inhibitors 5 μg/ml leupeptin, 5 μg/ml aprotinin, 2 μg/ml pepstatin, 120 μg/ml 4-benzenesulfonyl fluoride hydrochloride, and phosphatase inhibitor 5 mM NaF, pH 7.2) in 50-ml Eppendorf Protein LoBind tubes, and then spun at 2,000 g in a Fiberlite F14–14 × 50cy rotor in a Sorvall Lynx 6000 centrifuge for 10 min at 4°C (pelleting distance ~4 cm depending on volume). TBS was chosen because its physiologic pH and salt concentration. We chose this over artificial CSF because the latter is not as well buffered. The top ~90% of the supernatants were transferred to thin-wall polypropylene tubes (Beckman catalog #331372) and spun using an SW41Ti rotor at 40,000 rpm for 110 minutes in an Optima L90K ultracentrifuge at 4°C (pelleting distance ~9 cm). The top ~90% supernatants were retained and frozen at −80°C in 1 ml aliquots. For re-centrifugation (see Results), the frozen aliquots were thawed and spun in an Eppendorf 5417R centrifuge equipped with an F-45–30-11 rotor or else a TOMY MX-307 centrifuge equipped with an AR015–24 rotor at 20,000 g for 2 h at 4°C (pelleting distance ~1 cm). Prior to this re-centrifugation, a 50 μl portion of each aliquot was retained for Aβ ELISA. The final supernatants, as well as the associated pellets, were collected and retained for ELISAs and electron microscopy (EM). For Aβ42 monomer ELISAs, pellets were resuspended and denatured in 50 μl TBS containing 5 M GuHCl and 5 mM EDTA. Similarly, for Aβ42 monomer ELISAs on the original soaking extracts (inputs) and their recentrifuged supernatants, each was denatured by mixing them at a 2:5 ratio (v:v) with TBS containing 7 M GuHCl and 5 mM EDTA, to reach a final GuHCl concentration of 5 M. For analysis by negative-staining EM, final pellets were resuspended in 15 μl cold water.
CSF Analysis
In order to obtain sufficient CSF for differential centrifugation experiments, we consented two subjects undergoing lumbar puncture for evaluation of normal pressure hydrocephalus (NPH) during which >30 cc of CSF is withdrawn. The CSF was frozen on dry ice within 15 minutes of lumbar puncture. It was later thawed and spun in 1-ml duplicates in a TLA120.2 rotor in thick-wall polycarbonate tubes (Beckman) for one hour at 4°C. The supernatants were then analyzed by Aβ aggregate-preferring ELISA.
ELISAs
Aβ monomer-preferring ELISAs were home-brew immunoassays run on the Meso Scale Discovery (MSD) platform, as described.22 Samples containing GuHCl were diluted before ELISA, such that the GuHCl concentration was <0.25 M, to avoid interference with the antibody reaction. All steps were at room temperature. Plates were coated with capture antibody in PBS overnight and blocked in 5% MSD Blocker A in TBS with 0.05% Tween-20 (TBS-T) for 1 h. Samples were then applied for 1.5 h after being diluted in 1% Blocker A in TBS-T. Plates were washed 3 times in TBS-T, then biotinylated detector antibody and MSD Strepavidin-Sulfotag (1:5000) were applied for 1.5 h. After three more washes in TBS-T, the plates were detected with 2x MSD read buffer. The lower limit of quantification (LLoQ) was defined as the lowest standard with a luminescence value at least twice the blank average, and the lower limit of detection (LLoD) was defined as the lowest standard with values greater than the blank average plus twice the blank standard deviation. Aβ aggregate-preferring ELISAs were home-brew assays run on the Millipore SMCxPRO platform, as described previously.24 Antibodies, sources, and concentrations are listed in Table S2.
Negative staining immunoelectron microscopy
Carbon-coated grids (EMS) were glow-discharged at 25 mA for 20 s. For liquid samples (resuspended final pellets from soaking extracts), 5 μl were adsorbed to the grid for 1–5 min. Excess liquid was blotted off with filter paper (Whatman #1). Grids were then floated on a drop of water, blotted again, and stained with 1% uranyl acetate for 15 s. Excess stain was blotted off with filter paper. For immunostaining after their adsorption to the grids, samples were blocked in 1% BSA for 10 min. Antibodies diluted in 1% BSA were then added for 20 min and washed 3 times in PBS. Protein A-gold (Cell Biology UMC Utrecht) in 1% BSA was applied for 30 min, followed by two washes in PBS and four washes in water. All grids were examined on a JEOL 1200EX transmission electron microscope equipped with an AMT 2k CCD camera. Except for lecanemab, primary antibodies were purchased from Cell Signaling Technologies and diluted as follows: Anti-Aβ N-terminus (D54D2) 1:10; anti-tau C-terminus (D1M9X) 1:20; anti-tau pS404 (D2Z4G) 1:50; anti-tau pT205 (E7D3E) 1:50. Lecanemab was obtained from Sanofi and used at a final concentration of 13.3 μg/ml. To quantify fibrils, thirteen 0.9-ml soaking extracts were centrifuged for 2h at 20,000 g in an Eppendorf FA-45–30-11 rotor, and the pellets were resuspended in 20 μl water. Samples were immunolabeled with D54D2 as above, and 30 micrographs were taken at random by one experimenter (AMS) from well-stained portions of each grid at 25,000x magnification. The image order was randomized by a second experimenter (ALM) and given to a third blinded experimenter (ME) who counted the number of immunolabeled fibrils. The second experimenter (ALM) measured length in ImageJ of all fibrils labeled by at least two immunogold particles.
For ultrathin cryosections, AD brain tissues were fixed (fresh or after freezing) in 4% paraformaldehyde and stored at 4°C. Prior to freezing in liquid nitrogen, the fixed tissue samples were infiltrated with 2.3 M sucrose in PBS for 15 min. Frozen samples were sectioned at a thickness of 60 nm at −120°C and the sections transferred to formvar/carbon coated copper grids. Grids were floated on PBS, and downstream immunolabeling was carried out as above. Contrasting/embedding of the labeled grids was carried out on ice in 0.3% uranyl acetate in 2% methyl cellulose for 10 min.
Size exclusion chromatography
Size exclusion chromatography of human brain soaking extracts was performed using ÄKTA FPLC and a Superdex 200 Increase 10/300 column (Cytiva). Extracts (350 μl) were injected into the column using a 0.5-ml loop. Running buffer was TBS (25 mM Tris, 150 mM NaCl, pH 7.4) at 0.5 ml/min. 0.5-ml fractions were collected and used directly for ELISA without lyophilization.
Electron cryo-microscopy
Four to five 1-ml aliquots of soaking extracts were thawed on ice and spun in 1.5 ml Eppendorf tubes in an Eppendorf FA-45–24-11 rotor at 20,000 g for 1 h. All the pellets were resuspended and combined into 1 ml cold water. Following a 30 min centrifugation at 20,000 g at 4°C, pellets were resuspended in 10 μl of 20 mM Tris-HCl, pH 7.4. Samples were centrifuged at 3,000 g for 3 min and applied to glow-discharged holey carbon gold grids (Quantifoil Au R1.2/1.3, 300 mesh), which were glow-discharged with an Edwards (S150B) sputter coater at 30 mA for 30 s. Aliquots (3 μl) of samples were applied to the grids and blotted for approximately 3–5 s with filter paper (Whatman) at 100% humidity and 4 °C, using a Vitrobot Mark IV (Thermo Fisher). Datasets were acquired on a Titan Krios G3 microscope (Thermo Fisher Scientific) operated at 300 kV. Images for case AD7 were acquired using a Falcon-4 detector without energy filter. Images for case AD11 were acquired on a Gatan K3 detector in super-resolution counting mode, using a Bio-quantum energy filter (Gatan) with a slit width of 20 eV. Images were recorded with a total dose of 40 electrons per Å2.
Movie frames were gain-corrected, aligned, dose-weighted and then summed into a single micrograph using RELION’s own motion correction program.40 Contrast transfer function (CTF) parameters were estimated using CTFFIND-4.1.41 All subsequent image-processing steps were performed using helical reconstruction methods in RELION.42,43 Fibrils were picked manually. Reference-free 2D classification was performed to select suitable segments for further processing. Deposited maps of Type I Aβ42 fibrils (EMD-13800), Type II Aβ42 fibrils (EMD-13809),16 and tau PHFs (EMD-3741 and EMD-0259)16 low-pass filtered to 10 Å were used as initial models. Combinations of 3D auto-refinement and 3D classifications were used to select the best segments for each structure. For all structures, Bayesian polishing40 and CTF refinement42 were performed to further increase the resolution of the reconstructions. Final reconstructions were sharpened using standard post-processing procedures in RELION, and overall resolutions were estimated from Fourier shell correlations at 0.143 between the independently refined half-maps, using phase randomisation to correct for convolution effects of a generous, soft-edged solvent mask.44
Atomic models comprising β-sheet rung were built in Coot,45 starting from available models for Type I Aβ42 fibrils (PDB:7Q4B), Type II Aβ42 fibrils (PDB:7Q4M), and PHFs (PDB:6HRE). Coordinate refinements were performed using Servalcat.46 For the Type II Aβ42 structure refinement, β-sheet hydrogen bond restraints via symmetry were used. Final models were obtained using refinement of the asymmetric unit against the unsharpened half-maps with helical symmetry constraints in Servalcat.
Immunoprecipitation
AD brain soaking extract supernatants were diluted 5-fold in TBS and divided into 0.5-ml aliquots in triplicate. Twenty μg bapineuzumab or lecanemab were added, along with 60 μl washed protein G beads (Bio-Rad) and incubated overnight nutating at 4°C. The beads were washed three times in cold TBS, then eluted in 0.1 ml 5 M GuHCl overnight at 4°C. Total Aβ42 was then quantified by MSD ELISA as above.
Hippocampal slice recording
Mice (1–3 month-old) were anesthetized with halothane and decapitated. Transverse acute hippocampal slices (350 μm) were cut in ice-cold oxygenated sucrose-enhanced artificial cerebrospinal fluid (aCSF) containing 206 mM sucrose, 2 mM KCl, 2 mM MgSO4, 1.25 mM NaH2PO4, 1 mM CaCl2, 1 mM MgCl2, 26 mM NaHCO3, 10 mM D-glucose, pH 7.4. Slices were incubated in aCSF that contained (in mM) 124 NaCl, 2 KCl, 2 MgSO4, 1.25 NaH2PO4, 2.5 CaCl2, 26 NaHCO3, 10 D-glucose saturated with 95% O2 and 5% CO2 (pH 7.4), in which they were allowed to recover for at least 90 min before recording. Recordings were performed in the same solution at RT in a chamber submerged in aCSF. To record field EPSPs (fEPSPs) in the CA1 region of the hippocampus, test stimuli were applied at low frequency (0.05 Hz) at a stimulus intensity that elicited a fEPSP amplitude that was 40–50% of maximum, and the test responses were recorded for 10 min before the experiment was begun to ensure stability of the response. Once a stable test response was attained, 0.5 ml supernatant or TBS-resuspended pellet from centrifuged AD brain TBS soaking extracts were added to 9.5 ml aCSF perfusate, and a baseline was recorded directly for synaptic transmission and an additional 30 min for LTP. Antibodies were added to the AD brain extract aliquots at 2.5 μg/mL, incubated with mixing for 30 min, then the mixture was added to the brain slice perfusion buffer. To induce LTP, we used two consecutive trains (1 s) of stimuli at 100 Hz separated by 20 s. Traces were obtained by pClamp 11 and analyzed using the Clampfit 11. The fEPSP magnitude was measured using the initial fEPSP slope and three consecutive slopes (1 min) were averaged and normalized to the mean value recorded 10 min before conditioning stimulus for synaptic transmission and LTP testing. All procedures involving mice were in accordance with the animal welfare guidelines of Harvard Medical School and Brigham Women’s Hospital.
Quantification and statistical analysis
We reserved null hypothesis significance testing of a lecanemab effect on electrophysiology for pre-specified confirmatory experiments derived from power analyses on preliminary data, using unpaired two-tailed student’s t-tests setting significance (α) to 0.05.47,48 Power analysis for effect of lecanemab on LTP using data in prior work comparing control buffer to AD brain extract suggested N=7 mice would allow 90% power to detect a difference between lecanemab and isotype control. The newly described phenomenon of synaptic baseline inhibition without LTP induction (Fig 3P) required a preliminary experiment for a power calculation to determine sample size for testing lecanemab effect. Brain AD8 with N=6 mice in the two groups (lecanemab vs isotype control) showed a mean absolute difference of 43% baseline fEPSP but compatible with a 95% confidence interval from 1.8% to 84%. Power analysis using these data suggested testing N=14 mice per group with a second brain extract (AD12) would give 90% power to detect a difference between lecanemab and isotype control.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank Matthew Frosch, Mel Feany and Michael Miller for brain tissue allocation. We thank Alexandra Golby and Mary-Beth Anketell for help acquiring CSF. Funded by NIH K08NS128329 (AMS), P01AG015379 (DJS), and RF1AG006173 (DJS); Alzheimer’s Association AACSF-21-849687 (AMS); the Davis Alzheimer’s Prevention Program (DJS); and Medical Research Council MC_UP_A025_1013 (SHWS) and MC_U105184291 (MG).
Footnotes
Declaration of Interests
DJS is a founding director and consultant of Prothena Biosciences. LL is a consultant for Korro Bio.
References
- 1.Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, et al. (2008). Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med 14, 837–842. 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hughes C, Choi ML, Yi J-H, Kim S-C, Drews A, George-Hyslop PS, Bryant C, Gandhi S, Cho K, and Klenerman D. (2020). Beta amyloid aggregates induce sensitised TLR4 signalling causing long-term potentiation deficit and rat neuronal cell death. Commun Biol 3, 79. 10.1038/s42003-020-0792-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, and Selkoe D. (2009). Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron 62, 788–801. 10.1016/j.neuron.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jin M, Shepardson N, Yang T, Chen G, Walsh D, and Selkoe DJ (2011). Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc Natl Acad Sci U S A 108, 5819–5824. 10.1073/pnas.1017033108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jin M, O’Nuallain B, Hong W, Boyd J, Lagomarsino VN, O’Malley TT, Liu W, Vanderburg CR, Frosch MP, Young-Pearse T, et al. (2018). An in vitro paradigm to assess potential anti-Aβ antibodies for Alzheimer’s disease. Nat Commun 9, 2676. 10.1038/s41467-018-05068-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De Felice FG, Wu D, Lambert MP, Fernandez SJ, Velasco PT, Lacor PN, Bigio EH, Jerecic J, Acton PJ, Shughrue PJ, et al. (2008). Alzheimer’s disease-type neuronal tau hyperphosphorylation induced by A beta oligomers. Neurobiol Aging 29, 1334–1347. 10.1016/j.neurobiolaging.2007.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Forny-Germano L, Lyra e Silva NM, Batista AF, Brito-Moreira J, Gralle M, Boehnke SE, Coe BC, Lablans A, Marques SA, Martinez AMB, et al. (2014). Alzheimer’s disease-like pathology induced by amyloid-β oligomers in nonhuman primates. J Neurosci 34, 13629–13643. 10.1523/JNEUROSCI.1353-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Langer F, Eisele YS, Fritschi SK, Staufenbiel M, Walker LC, and Jucker M. (2011). Soluble Aβ seeds are potent inducers of cerebral β-amyloid deposition. Journal of Neuroscience 31, 14488–14495. 10.1523/JNEUROSCI.3088-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Katzmarski N, Ziegler-Waldkirch S, Scheffler N, Witt C, Abou-Ajram C, Nuscher B, Prinz M, Haass C, and Meyer-Luehmann M. (2020). Aβ oligomers trigger and accelerate Aβ seeding. Brain Pathology 30, 36–45. 10.1111/BPA.12734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takeda S, Wegmann S, Cho H, DeVos SL, Commins C, Roe AD, Nicholls SB, Carlson GA, Pitstick R, Nobuhara CK, et al. (2015). Neuronal uptake and propagation of a rare phosphorylated high-molecular-weight tau derived from Alzheimer’s disease brain. Nat Commun 6, 8490. 10.1038/ncomms9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dujardin S, Commins C, Lathuiliere A, Beerepoot P, Fernandes AR, Kamath T. v, de Los Santos MB, Klickstein N, Corjuc DL, Corjuc BT, et al. (2020). Tau molecular diversity contributes to clinical heterogeneity in Alzheimer’s disease. Nat Med 26, 1256–1263. 10.1038/s41591-020-0938-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Dyck CH, Swanson CJ, Aisen P, Bateman RJ, Chen C, Gee M, Kanekiyo M, Li D, Reyderman L, Cohen S, et al. (2022). Lecanemab in early Alzheimer’s disease. N Engl J Med. 10.1056/NEJMOA2212948/SUPPL_FILE/NEJMOA2212948_APPENDIX.PDF. [DOI] [PubMed] [Google Scholar]
- 13.Tucker S, Möller C, Tegerstedt K, Lord A, Laudon H, Sjödahl J, Söderberg L, Spens E, Sahlin C, Waara ER, et al. (2015). The murine version of BAN2401 (mAb158) selectively reduces amyloid-β protofibrils in brain and cerebrospinal fluid of tg-ArcSwe mice. Journal of Alzheimer’s Disease 43, 575–588. 10.3233/JAD-140741. [DOI] [PubMed] [Google Scholar]
- 14.Lannfelt L, Möller C, Basun H, Osswald G, Sehlin D, Satlin A, Logovinsky V, and Gellerfors P. (2014). Perspectives on future Alzheimer therapies: amyloid-β protofibrils - a new target for immunotherapy with BAN2401 in Alzheimer’s disease. Alzheimers Res Ther 6, 16. 10.1186/alzrt246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Benilova I, Karran E, and de Strooper B. (2012). The toxic Aβ oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat Neurosci 15, 349–357. 10.1038/nn.3028. [DOI] [PubMed] [Google Scholar]
- 16.Yang Y, Diana A, Wenjuan Z, Melissa H, Sofia L, Manuel S, Abhay K, G. MA Y. P-CS, Jennifer M, et al. (2022). Cryo-EM structures of amyloid-β 42 filaments from human brains. Science (1979) 375, 167–172. 10.1126/science.abm7285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mc Donald JM, O’Malley TT, Liu W, Mably AJ, Brinkmalm G, Portelius E, Wittbold WM, Frosch MP, and Walsh DM (2015). The aqueous phase of Alzheimer’s disease brain contains assemblies built from ∼4 and ∼7 kDa Aβ species. Alzheimer’s & Dementia 11, 1286–1305. 10.1016/j.jalz.2015.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang T, Li S, Xu H, Walsh DM, and Selkoe DJ (2017). Large soluble oligomers of amyloid β-protein from Alzheimer brain are far less neuroactive than the smaller oligomers to which they dissociate. J Neurosci 37, 152–163. 10.1523/JNEUROSCI.1698-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brinkmalm G, Hong W, Wang Z, Liu W, O’Malley TT, Sun X, Frosch MP, Selkoe DJ, Portelius E, Zetterberg H, et al. (2019). Identification of neurotoxic cross-linked amyloid-β dimers in the Alzheimer’s brain. Brain 142, 1441–1457. 10.1093/brain/awz066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hong W, Wang Z, Liu W, O’Malley TT, Jin M, Willem M, Haass C, Frosch MP, and Walsh DM (2018). Diffusible, highly bioactive oligomers represent a critical minority of soluble Aβ in Alzheimer’s disease brain. Acta Neuropathol 136, 19–40. 10.1007/s00401-018-1846-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sideris DI, Danial JSH, Emin D, Ruggeri FS, Xia Z, Zhang YP, Lobanova E, Dakin H, De S, Miller A, et al. (2021). Soluble amyloid beta-containing aggregates are present throughout the brain at early stages of Alzheimer’s disease. Brain Commun 3, fcab147–fcab147. 10.1093/braincomms/fcab147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stern AM, Liu L, Jin S, Liu W, Meunier AL, Ericsson M, Miller MB, Batson M, Sun T, Kathuria S, et al. (2022). A calcium-sensitive antibody isolates soluble amyloid-β aggregates and fibrils from Alzheimer’s disease brain. Brain 147, 2528–2540. 10.1093/brain/awac023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang T, O’Malley TT, Kanmert D, Jerecic J, Zieske LR, Zetterberg H, Hyman BT, Walsh DM, and Selkoe DJ (2015). A highly sensitive novel immunoassay specifically detects low levels of soluble Aβ oligomers in human cerebrospinal fluid. Alzheimers Res Ther 7, 14. 10.1186/s13195-015-0100-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu L, Kwak H, Lawton TR, Jin S, Meunier AL, Dang Y, Ostaszewski B, Pietras A, Stern AM, and Selkoe DJ (2022). An ultra-sensitive immunoassay detects and quantifies soluble Aβ oligomers in human plasma. Alzheimer’s & Dementia 18, 1186–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Esparza TJ, Wildburger NC, Jiang H, Gangolli M, Cairns NJ, Bateman RJ, and Brody DL (2016). Soluble amyloid-beta aggregates from human Alzheimer’s disease brains. Sci Rep 6, 38187. 10.1038/srep38187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jin M, and Selkoe DJ (2015). Systematic analysis of time-dependent neural effects of soluble amyloid β oligomers in culture and in vivo: Prevention by scyllo-inositol. Neurobiol Dis 82, 152–163. 10.1016/j.nbd.2015.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McLaurin J, Golomb R, Jurewicz A, Antel JP, and Fraser PE (2000). Inositol Stereoisomers Stabilize an Oligomeric Aggregate of Alzheimer Amyloid β Peptide and Inhibit Aβ-induced Toxicity*. Journal of Biological Chemistry 275, 18495–18502. 10.1074/jbc.M906994199. [DOI] [PubMed] [Google Scholar]
- 28.Fitzpatrick AWP, Falcon B, He S, Murzin AG, Murshudov G, Garringer HJ, Crowther RA, Ghetti B, Goedert M, and Scheres SHW (2017). Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 547, 185–190. 10.1038/nature23002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nilsberth C, Westlind-Danielsson A, Eckman CB, Condron MM, Axelman K, Forsell C, Stenh C, Luthman J, Teplow DB, Younkin SG, et al. (2001). The “Arctic” APP mutation (E693G) causes Alzheimer’s disease by enhanced Aβ protofibril formation. Nat Neurosci 4, 887–893. 10.1038/nn0901-887. [DOI] [PubMed] [Google Scholar]
- 30.Lannfelt L, Möller C, Basun H, Osswald G, Sehlin D, Satlin A, Logovinsky V, and Gellerfors P. (2014). Perspectives on future Alzheimer therapies: amyloid-β protofibrils - a new target for immunotherapy with BAN2401 in Alzheimer’s disease. Alzheimers Res Ther 6, 16. 10.1186/alzrt246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johannesson M, Sahlin C, Söderberg L, Basun H, Fälting J, Möller C, Zachrisson O, Sunnemark D, Svensson A, Odergren T, et al. (2021). Elevated soluble amyloid beta protofibrils in Down syndrome and Alzheimer’s disease. Molecular and Cellular Neuroscience 114, 103641. 10.1016/J.MCN.2021.103641. [DOI] [PubMed] [Google Scholar]
- 32.Jin SX, Liu L, Li S, Meunier AL, and Selkoe DJ (2022). Aβ oligomers from human brain impair mossy fiber LTP in CA3 of hippocampus, but activating cAMP-PKA and cGMP-PKG prevents this. Neurobiol Dis 172, 105816. 10.1016/J.NBD.2022.105816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koffie RM, Meyer-Luehmann M, Hashimoto T, Adams KW, Mielke ML, Garcia-Alloza M, Micheva KD, Smith SJ, Kim ML, Lee VM, et al. (2009). Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci U S A 106, 4012–4017. 10.1073/pnas.0811698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meilandt WJ, Ngu H, Gogineni A, Lalehzadeh G, Lee S-H, Srinivasan K, Imperio J, Wu T, Weber M, Kruse AJ, et al. (2020). Trem2 deletion reduces late-stage amyloid plaque accumulation, elevates the Aβ42:Aβ40 ratio, and exacerbates axonal dystrophy and dendritic spine loss in the PS2APP Alzheimer's mouse model. The Journal of Neuroscience, 1819–1871. 10.1523/JNEUROSCI.1871-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Casali BT, MacPherson KP, Reed-Geaghan EG, and Landreth GE (2020). Microglia depletion rapidly and reversibly alters amyloid pathology by modification of plaque compaction and morphologies. Neurobiol Dis 142, 104956. 10.1016/j.nbd.2020.104956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Parhizkar S, Arzberger T, Brendel M, Kleinberger G, Deussing M, Focke C, Nuscher B, Xiong M, Ghasemigharagoz A, Katzmarski N, et al. (2019). Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat Neurosci 22, 191–204. 10.1038/s41593-018-0296-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Swanson CJ, Zhang Y, Dhadda S, Wang J, Kaplow J, Lai RYK, Lannfelt L, Bradley H, Rabe M, Koyama A, et al. (2021). A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Aβ protofibril antibody. Alzheimers Res Ther 13, 80. 10.1186/s13195-021-00813-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Falcon B, Zhang W, Schweighauser M, Murzin AG, Vidal R, Garringer HJ, Ghetti B, Scheres SHW, and Goedert M. (2018). Tau filaments from multiple cases of sporadic and inherited Alzheimer’s disease adopt a common fold. Acta Neuropathol 136, 699–708. 10.1007/S00401-018-1914-Z/FIGURES/5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jackson SJ, Kerridge C, Cooper J, Cavallini A, Falcon B, Cella CV, Landi A, Szekeres PG, Murray TK, Ahmed Z, et al. (2016). Short fibrils constitute the major species of seed-competent tau in the brains of mice transgenic for human P301S tau. Journal of Neuroscience 36, 762–772. 10.1523/JNEUROSCI.3542-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zivanov J, Nakane T, and Scheres SH (2019). A Bayesian approach to beam-induced motion correction in cryo-EM single-particle analysis. IUCrJ 6, 5–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rohou A, and Grigorieff N. (2015). CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J Struct Biol 192, 216–221. 10.1016/j.jsb.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zivanov J, Nakane T, and Scheres SHW (2020). Estimation of high-order aberrations and anisotropic magnification from cryo-EM data sets in RELION-3.1. IUCrJ2 7, 253–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.He S, and Scheres SHW (2017). Helical reconstruction in RELION. J Struct Biol 198, 163–176. 10.1016/j.jsb.2017.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen S, McMullan G, Faruqi AR, Murshudov GN, Short JM, Scheres SHW, and Henderson R. (2013). High-resolution noise substitution to measure overfitting and validate resolution in 3D structure determination by single particle electron cryomicroscopy. Ultramicroscopy 135, 24–35. 10.1016/j.ultramic.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Casañal A, Lohkamp B, and Emsley P. (2020). Current developments in Coot for macromolecular model building of Electron Cryo-microscopy and Crystallographic Data. Protein Science 29, 1055–1064. 10.1002/pro.3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yamashita K, Palmer CM, Burnley T, and Murshudov GN (2021). Cryo-EM single-particle structure refinement and map calculation using Servalcat. Acta Crystallographica Section D 77, 1282–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alger BE (2022). Neuroscience needs to test both statistical and scientific hypotheses. Journal of Neuroscience 42, 8432–8438. 10.1523/JNEUROSCI.1134-22.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Calin-Jageman RJ (2022). Better inference in neuroscience: test less, estimate more. Journal of Neuroscience 42, 8427–8431. 10.1523/JNEUROSCI.1133-22.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All biochemical data reported in this paper will be shared by the lead contact upon request. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Cryo-EM maps have been deposited in the Electron Microscopy Data Bank (EMDB) under accession numbers EMD-15770, EMD-15771 and EMD-15772. Corresponding refined atomic models have been deposited in the Protein Data Bank (PDB) under accession numbers 8AZS, 8AZT and 8AZU.