

Abstract
The RNA triphosphatase component (CaCet1p) of the mRNA capping apparatus of the pathogenic fungus Candida albicans differs mechanistically and structurally from the RNA triphosphatase of mammals. Hence, CaCet1p is an attractive antifungal target. Here we identify a C-terminal catalytic domain of CaCet1p from residue 257 to 520 and characterize a manganese-dependent and cobalt-dependent NTPase activity intrinsic to CaCet1p. The NTPase can be exploited to screen in vitro for inhibitors. The amino acids that comprise the active site of CaCet1p were identified by alanine-scanning mutagenesis, which was guided by the crystal structure of the homologous RNA triphosphatase from Saccharomyces cerevisiae (Cet1p). Thirteen residues required for the phosphohydrolase activity of CaCet1p (Glu287, Glu289, Asp363, Arg379, Lys396, Glu420, Arg441, Lys443, Arg445, Asp458, Glu472, Glu474 and Glu476) are located within the hydrophilic interior of an eight-strand β barrel of Cet1p. Each of the eight strands contributes at least one essential amino acid. The essential CaCet1p residues include all of the side chains that coordinate manganese and sulfate (i.e., γ phosphate) in the Cet1p product complex. These results suggest that the active site structure and catalytic mechanism are conserved among fungal RNA triphosphatases.
INTRODUCTION
RNA triphosphatase is an essential enzyme that catalyzes the first step of mRNA cap formation—the hydrolysis of the γ phosphate of triphosphate-terminated pre-mRNA to form a diphosphate end. Two distinct classes of eukaryotic RNA triphosphatases have been described. The RNA triphosphatases of metazoan species belong to a superfamily of phosphatases (which includes protein tyrosine phosphatases and dual specificity protein phosphatases) that act via formation and hydrolysis of a covalent enzyme–(cysteinyl-S)–phosphate intermediate (1–3). The metazoan RNA triphosphatase reaction requires no metal cofactor. In fact, metazoan RNA triphosphatases are inhibited by divalent cations. In contrast, the RNA triphosphatases of DNA viruses (poxviruses and baculoviruses) and the budding yeast Saccharomyces cerevisiae are strictly dependent on a divalent cation cofactor (4–10). The viral/fungal triphosphatase family is defined by three conserved collinear motifs (A, B and C) that include clusters of acidic and basic amino acids that are essential for catalytic activity (11,12) (Fig. 1). These motifs were initially uncovered by mutational studies of the vaccinia virus RNA triphosphatase (13). There is no amino acid sequence similarity whatsoever between the metazoan and viral/fungal triphosphatase families. Thus, RNA triphosphatase presents a remarkable case of complete divergence during the transition from fungal to metazoan species.
Figure 1.

Sequence conservation within the triphosphate tunnel of fungal RNA triphosphatase. The sequence of S.cerevisiae Cet1p from residue 304 to 539 is aligned to the homologous segments of C.albicans CaCet1p (286–520) and S.cerevisiae Cth1p (86–317). Gaps in the alignment are indicated by dashes (–). The β strands that comprise the triphosphate tunnel of Cet1p are shown above the amino acid sequence. Conserved motifs A (β1), B (β9) and C (β11) that define the metal-dependent RNA triphosphatase family are indicated below the sequence. The 14 charged amino acids of CaCet1p that were targeted for alanine substitution in the present study are highlighted in shaded boxes. The charged residues in motifs A, B and C of S.cerevisiae Cet1p that were shown previously to be essential for Cet1p function in vivo and in vitro are denoted by dots above the sequence.
RNA triphosphatase also presents an attractive target for antifungal drug development because the triphosphatase activity is essential for yeast cell growth (8,9), the enzyme is conserved among fungi (10,14) and metazoan species encode no obvious homologs of the fungal enzyme. Thus, an inhibitor of fungal RNA triphosphatase should have selectivity for the pathogen and minimal effect on the human host.
Large-scale screening for potential inhibitors of fungal RNA triphosphatase is simplified by the finding that the S.cerevisiae RNA triphosphatase Cet1p displays a robust ATPase activity in the presence of manganese (11,12). Cobalt is also an effective cofactor for ATP hydrolysis, but magnesium is not. (Note that magnesium is the preferred cofactor for the phosphohydrolase activity of Cet1p on RNA substrates.) Here we show that the RNA triphosphatase CaCet1p encoded by the pathogenic fungus Candida albicans also has an intrinsic manganese- or cobalt-dependent ATPase activity that resides within a C-terminal catalytic domain CaCet1(257–520)p.
To facilitate mechanistic and pharmacological studies of the metal-dependent RNA triphosphatases, we have pursued two parallel lines of investigation: (i) mutational analysis of yeast Cet1p, focusing on the identification of amino acids required for catalysis and (ii) crystallization of Cet1p and determination of its structure by X-ray diffraction. The mutational approach established the essentiality of the two glutamates in motif A and the three glutamates in motif C and engendered a prediction that motifs A and C comprise the metal-binding site on the enzyme (11,12).
The crystal structure of a biologically active fragment of Cet1p revealed surprising structural complexity for an apparently simple phosphohydrolase reaction (15). Cet1p adopts a novel enzyme fold whereby an antiparallel eight-strand β barrel forms a hydrophilic ‘triphosphate tunnel’ (Fig. 2). Multiple acidic side chains point into the tunnel cavity, including the essential glutamates of motifs A and C. The interior of the tunnel contains a single sulfate ion coordinated by basic side chains projecting into the tunnel. Insofar as sulfate is a structural analog of phosphate, it is likely that the side chain interactions of the sulfate reflect contacts made by the enzyme with the γ phosphate of the triphosphate-terminated RNA and nucleoside triphosphate substrates. A manganese ion within the tunnel cavity is coordinated with octahedral geometry to the sulfate, to the side chain carboxylates of the two essential glutamates in motif A and to a glutamate in motif C.
Figure 2.

The triphosphate tunnel and the metal-binding site. Stereo view of a cross section of the tunnel of S.cerevisiae Cet1p showing the 14 charged amino acids whose equivalents in CaCet1p were subjected to alanine mutagenesis in the present study. The figure highlights the network of side chain interactions that coordinate the sulfate and manganese ions. The manganese (blue sphere) interacts with octahedral geometry with the sulfate, three glutamates and two waters (red spheres). The image was prepared using SETOR (19). The amino acid numbers are those of Cet1p. The equivalent residue numbers in CaCe1tp are listed in Table 1.
The crystal structure of the S.cerevisiae triphosphatase provides a blueprint for further mutagenesis studies. Here we have initiated a structure-based mutational analysis of the C.albicans triphosphatase CaCet1p. We focus on the Candida enzyme for several reasons: (i) Candida CaCet1p is a genuine target for therapeutic drug discovery; (ii) the presumption that the structures and mechanisms of fungal triphosphatases are conserved can be tested by establishing a concordance of mutational effects on the Saccharomyces and Candida activities; (iii) targeting new residues for mutation based on the structure should ultimately pinpoint all of the functionally important side chains at the active site within the tunnel. We report the effects of alanine substitutions at 14 residues of CaCet1p that project into the triphosphate tunnel. We find that 13 of the side chains are important for triphosphatase activity. Mechanistic implications are discussed in light of the available structural data.
MATERIALS AND METHODS
Expression and purification of recombinant CaCet1p
The CaCET1, CaCET1(203–520), CaCET1(217–520), CaCET1(223–520), CaCET1(229–520), CaCET1(257–520) and CaCET1(267–520) genes were cloned into pET16-based bacterial expression vectors; the pET-CaCET1 plasmids were transformed into Escherichia coli BL21(DE3). Cultures (100 ml) of single ampicillin-resistant transformants were grown at 37°C in Luria-Bertani (LB) medium containing 0.1 mg/ml ampicillin until the A600 reached 0.5. The cultures were adjusted to 0.4 mM isopropyl-β-d-thiogalactopyranoside (IPTG) and incubation was continued at 37°C for 3 h. Cells were harvested by centrifugation and the pellet was stored at –80°C. All subsequent procedures were performed at 4°C. Thawed bacteria were resuspended in 5 ml of lysis buffer (50 mM Tris–HCl pH 7.5, 0.15 M NaCl, 10% sucrose). Cell lysis was achieved by addition of lysozyme and Triton X-100 to final concentrations of 50 µg/ml and 0.1%, respectively. The lysate was sonicated to reduce viscosity and insoluble material was removed by centrifugation in a Sorvall SS34 rotor at 18 000 r.p.m. for 45 min. The soluble lysates were applied to 1-ml columns of Ni-NTA agarose (Qiagen, Valencia, CA) that had been equilibrated with lysis buffer containing 0.1% Triton X-100. The columns were washed with 5 ml of lysis buffer containing 0.1% Triton X-100 and then eluted step-wise with a buffer solution (50 mM Tris–HCl pH 8.0, 100 mM NaCl, 2 mM DTT, 10% glycerol, 0.05% Triton X-100) containing 50, 100, 200, 500 and 1000 mM imidazole. SDS–PAGE analysis showed that the recombinant Candida proteins were recovered predominantly in the 200 mM imidazole eluate fractions. The peak fractions containing the recombinant proteins were pooled, adjusted to 5 mM EDTA and dialyzed against buffer C (50 mM Tris–HCl pH 8.0, 2 mM DTT, 5 mM EDTA, 10% glycerol, 0.05% Triton X-100) containing 50 mM NaCl. The dialysates were applied to 0.5-ml columns of phosphocellulose that had been equilibrated with 50 mM NaCl in buffer C. The columns were washed with the same buffer and then eluted step-wise with buffer C containing 0.1, 0.2, 0.5 and 1.0 M NaCl. The recombinant proteins were recovered predominantly in the 0.5 M NaCl eluate fractions. Protein concentrations were determined by the Bio-Rad dye-binding method with bovine serum albumin as the standard. The enzyme preparations were stored at –80°C.
Alanine-mutants of CaCet1p
Alanine-substitution mutations were introduced into the CaCET1(257–520) gene by the two-stage overlap extension method. The mutated genes were digested with NdeI and BamHI and then inserted into the bacterial expression vector pET16b. The pET-Δ256-Ala plasmids were transformed into E.coli BL21(DE3). Single transformants were inoculated into 100 ml of LB medium containing 0.1 mg/ml of ampicillin and grown at 37°C until the A600 reached 0.5. Recombinant protein expression was induced by placing the culture on ice for 30 min, followed by addition of IPTG to 0.4 mM and ethanol to 2% final concentration, and then continuing incubation for 24 h at 18°C with constant shaking. The cells were harvested by centrifugation and all subsequent procedures were performed at 4°C. The His-tagged CaCet1(257–520)-Ala proteins were purified from soluble bacterial lysates by nickel–NTA–agarose chromatography as described above. The 0.2 M imidazole eluate fractions containing CaCet1(257–520)p were dialyzed against buffer C containing 50 mM NaCl.
ATPase assay
Standard reaction mixtures (10 µl) containing 50 mM Tris–HCl pH 7.0 or 7.5, 5 mM DTT, 2 mM MnCl2 or CoCl2, 1 mM [γ-32P]ATP and enzyme were incubated for 15 min at 30°C. The reactions were quenched by adding 2.5 µl of 5 M formic acid. Aliquots of the mixtures were applied to a polyethyleneimine–cellulose TLC plate, which was developed with 1 M formic acid, 0.5 M LiCl. The extent of 32Pi release was quantitated by scanning the chromatogram with a FUJIX phosphorimager.
Km of CaCet1(257–520)p for ATP
Reaction mixtures (20 µl) containing 50 mM Tris–HCl pH 7.5, 5 mM DTT, 2 mM MnCl2, varying concentrations of [γ-32P]ATP and 0.1 ng of either wild-type CaCet1(257–520)p or the K291A mutant enzyme were incubated for 15 min at 30°C. The extent of 32Pi release at the lowest ATP concentrations tested was <15% of the input substrate. Km values were determined from double-reciprocal plots of the data.
RESULTS
Expression and purification of recombinant CaCet1p and N-terminal deletion mutants
The full-length 520-amino acid CaCet1p and the N-terminal deletion mutants CaCet1(203–520)p (Δ202), CaCet1(217–520)p (Δ216), CaCet1(223–520)p (Δ222), CaCet1(229–520)p (Δ228) and CaCet1(257–520)p (Δ256) were expressed in bacteria as His10-tagged fusions and purified from soluble bacterial lysates by Ni–agarose and phosphocellulose column chromatography. SDS–PAGE analysis of the polypeptide compositions of the Ni–agarose preparations of CaCet1p, Δ202 and Δ216 and their phosphocellulose elution profiles are shown in Figure 3. The Ni–agarose eluate fraction of CaCet1p contained, in addition to the full-length 66 kDa recombinant protein, many smaller polypeptides that are presumed to correspond to proteolytic fragments of CaCet1p that retain the N-terminal His-tag. Similar breakdown products were also noted during Ni–agarose purification of recombinant S.cerevisiae Cet1p (9). The recombinant Δ202 and Δ216 polypeptides were considerably more pure at the Ni–agarose step (Fig. 3, lane Ni) as were Δ222, Δ228 and Δ256 (not shown); the absence of smaller breakdown products in these preparations suggests that the N-terminal domain of CaCet1p is especially susceptible to proteolysis when expressed in bacteria.
Figure 3.
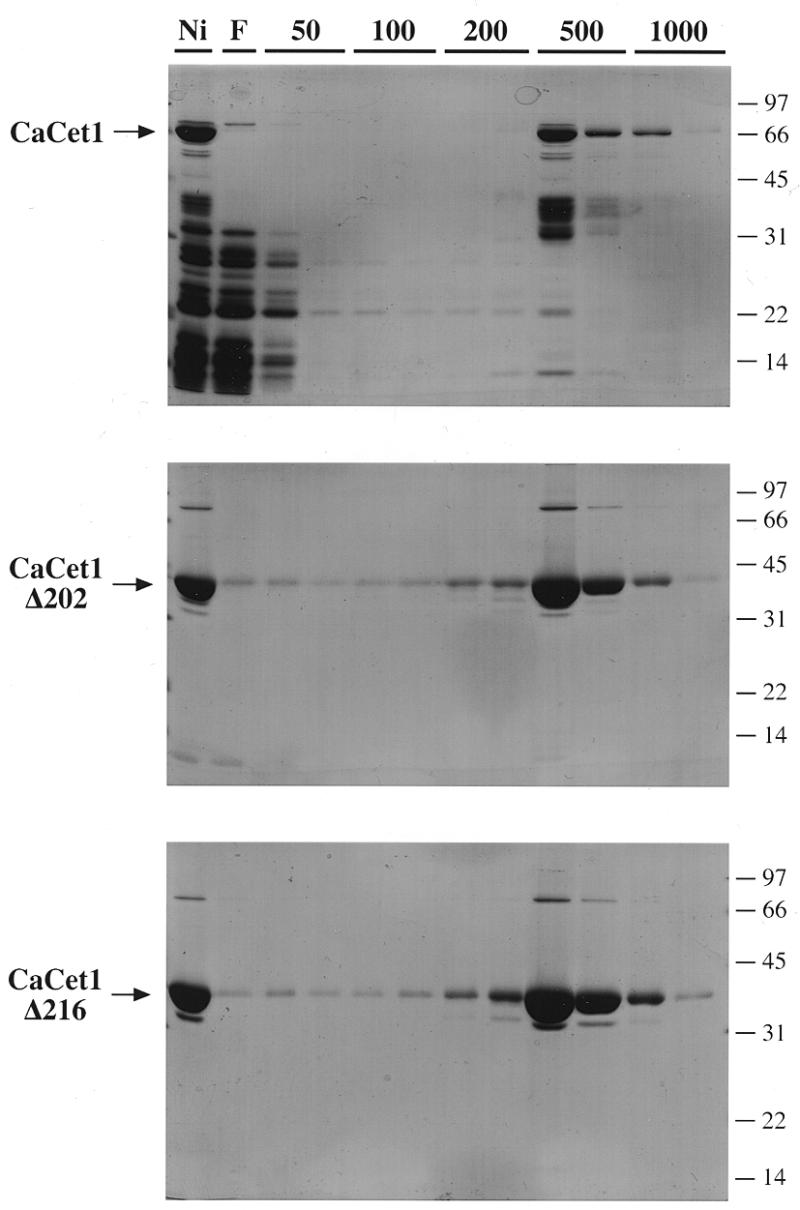
Purification of recombinant CaCet1p, CaCet1(203–520)p (Δ202) and CaCet1(217–520)p (Δ216). The phosphocellulose column elution profiles were analyzed by SDS–PAGE. Aliquots (20 µl) of the Ni–agarose fractions that were applied to the phosphocellulose columns (lane Ni); the phosphocellulose flow-through fraction (lane F), the phosphocellulose 50 mM NaCl wash fractions (lane 50), the 100 mM NaCl eluate fractions (lane 100), the 200 mM NaCl eluate fractions (lane 200), the 500 mM NaCl eluate fractions (lane 500) and the 1 M NaCl eluate fractions (lane 1000) were analyzed by electrophoresis through 12.5% polyacrylamide gels containing 0.1% SDS. The polypeptides were visualized by staining with Coomassie blue dye. The positions and sizes (kDa) of co-electrophoresed marker polypeptides are indicated at the right of each gel. The recombinant Candida proteins are indicated by arrows on the left.
Further purification of CaCet1p was achieved by phosphocellulose column chromatography. CaCet1p adsorbed to phosphocellulose and was step-eluted with buffer containing 0.5 M NaCl. Many, but not all, of the lower molecular weight contaminant polypeptides were removed at this step (Fig. 3). The Δ202 and Δ216 proteins adsorbed to phosphocellulose and were recovered in the 0.5 M NaCl eluate fractions (Fig. 3) as did the Δ222, Δ228 and Δ256 proteins (not shown). Figure 4A shows the polypeptide compositions of the phosphocellulose enzyme preparations analyzed by SDS–PAGE in the same gel. The apparent sizes of the predominant polypeptides corresponding to CaCet1p and serially truncated versions thereof are in good agreement with the calculated sizes of the His-tagged gene products.
Figure 4.
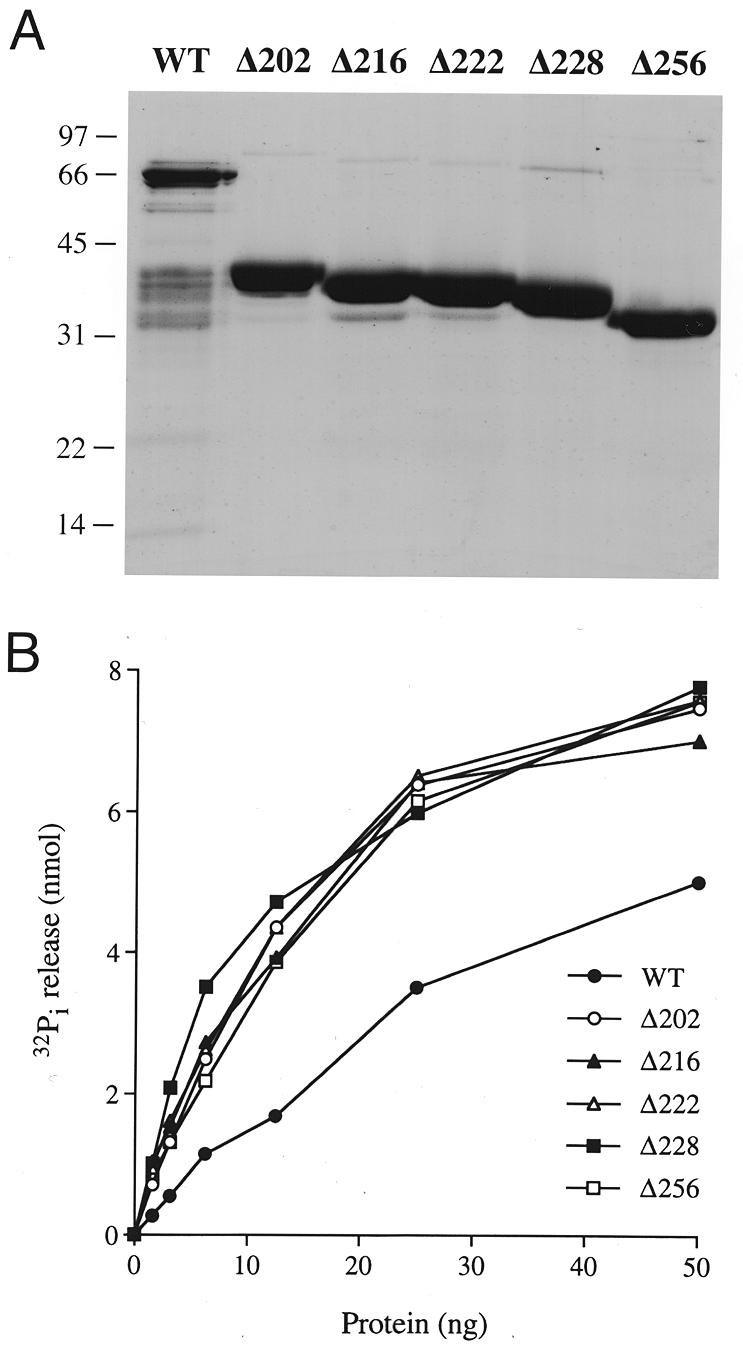
Manganese-dependent ATPase activity of CaCet1p and N-terminal deletion mutants of CaCet1p. (A) Polypeptide composition. Aliquots (4 µg) of the phosphocellulose preparations of recombinant CaCet1p, CaCet1(203–520)p (Δ202), CaCet1(217–520)p (Δ216), CaCet1(223–520)p (Δ222), CaCet1(229–520)p (Δ228) and CaCet1(257–520)p (Δ256) were analyzed by electrophoresis through a 12.5% polyacrylamide gel containing 0.1% SDS. Polypeptides were visualized by staining with Coomassie blue dye. The positions and sizes (in kDa) of marker proteins are indicated on the left. (B) ATPase activity. Reaction mixtures (10 µl) containing 50 mM Tris–HCl pH 7.5, 5 mM DTT, 2 mM MnCl2, 1 mM [γ-32P]ATP, and wild-type CaCet1p, Δ202, Δ216, Δ222, Δ228 or Δ256 as specified were incubated for 15 min at 30°C. 32Pi release is plotted as function of input protein.
Manganese-dependent and cobalt-dependent NTPase activity of CaCet1p
We showed previously that S.cerevisiae Cet1p has an intrinsic capacity to hydrolyze nucleoside triphosphates to nucleoside diphosphates and inorganic phosphate in the presence of a divalent cation cofactor—either manganese or cobalt (11). The divalent cation specificity of the NTPase is distinct from the RNA triphosphatase activity of Cet1p, which is supported by magnesium. The utility of an NTPase-based assay for in vitro screening of inhibitors of fungal RNA triphosphatases would be underscored by a demonstration that NTPase activity is a general property of fungal RNA triphosphatases (especially in pathogenic fungi like C.albicans) and not merely restricted to the enzyme from S.cerevisiae.
We found that the phosphocellulose preparations of recombinant CaCet1p, Δ202, Δ216, Δ222, Δ228 and Δ256 catalyzed the release of 32Pi from [γ-32P]ATP in the presence of manganese chloride and that the extent of ATP hydrolysis increased as a function of input enzyme (Fig. 4B). The specific activities of Δ202, Δ216, Δ222, Δ228 and Δ256 were almost identical. A specific activity for Δ202 of 390 pmol of ATP hydrolyzed per ng of protein in 15 min was calculated from the slope of the titration curve in the linear range. This corresponds to a turnover number of ~17 s–1 for Δ202. The specific activity of full-length CaCet1p (on a per ng basis) was one-half to one-third that of the truncated proteins (Fig. 4B); we attribute this to the presence of inactive contaminating polypeptides in the preparation. Further characterization of the biochemical properties of CaCet1p was performed with the Δ202 enzyme.
The metal specificity of CaCet1Δ202 was tested in reaction mixtures containing 1 mM ATP and 2 mM divalent cation (Fig. 5A). Cobalt was even more effective than manganese in activating the ATPase, whereas magnesium did not support ATP hydrolysis. Calcium and copper did not activate the ATPase; zinc was weakly active. The divalent cation specificity of the ATPase activity of recombinant full-length CaCet1p was the same as that of CaCet1Δ202 (data not shown). Cofactor titration experiments showed that hydrolysis of 1 mM ATP by CaCet1Δ202 was optimal at 1.25–2.5 mM manganese or cobalt and declined slightly as the divalent cation concentrations were increased to 5 and 10 mM (Fig. 5B). The titration curves were sharply sigmoidal at manganese or cobalt concentrations below the concentration of input ATP.
Figure 5.

Characterization of the triphosphatase activity of CaCet1(203–520)p. (A) Divalent cation requirement for ATP hydrolysis. Reaction mixtures (10 µl) containing 50 mM Tris–HCl (pH 7.5), 1 mM [γ-32P]ATP, 50 ng of Δ202 and 2 mM divalent cation as specified were incubated for 15 min at 30°C. Mg, Mn, Ca, Co and Zn were added as chloride salts; Cu was added as copper sulfate. (B) Dependence of ATPase activity on divalent cation concentration. Reaction mixtures (10 µl) containing 50 mM Tris–HCl pH 7.5, 50 ng of Δ202, 1 mM [γ-32P]ATP, and either MnCl2 or CoCl2 as specified were incubated for 15 min at 30°C. 32Pi release is plotted as a function of manganese or cobalt concentration. (C) RNA triphosphatase activity. Reaction mixtures (10 µl) containing 50 mM Tris–HCl (pH 7.5), 5 mM DTT, 1 mM MgCl2, 20 pmol (of triphosphate termini) of [γ-32P]poly(A) and Δ202 as specified were incubated for 15 min at 30°C. Reactions were quenched by adding 2.5 µl of 5 M formic acid. Aliquots (5 µl) of the mixtures were applied to a polyethyleneimine–cellulose TLC plate, which was developed with 0.75 M potassium phosphate (pH 4.3). 32Pi release is plotted as a function of input Δ202 protein.
RNA triphosphatase activity
The RNA triphosphatase activity of CaCet1Δ202 was gauged by the release of 32Pi from 2 µM [γ-32P]poly(A) in the presence of 1 mM magnesium (Fig. 5C). The specific activity, determined from the slope of the titration curve, was 32 pmol Pi released per ng of protein in 15 min. This corresponds to turnover number of ~1.4 s–1. This value is similar to the Vmax of 1 s–1 for the hydrolysis of [γ-32P]poly(A) by S.cerevisiae Cet1p (11).
Structure-based mutational analysis of CaCet1p
The catalytic domain of S.cerevisiae RNA triphosphatase Cet1p adopts a novel enzyme fold in which an eight-strand β barrel forms a topologically closed triphosphate tunnel (16). The β strands comprising the tunnel are displayed over the Cet1p protein sequence in Figure 1. Acidic and basic side chains that point into the tunnel are indicated in shaded boxes. All 14 of these charged residues are conserved in the C.albicans RNA triphosphatase CaCet1p and in S.cerevisiae Cth1p (Fig. 1). Cth1p is a non-essential protein with divalent cation-dependent RNA triphosphatase/NTPase activity that may participate in an RNA transaction unrelated to capping (12,16).
In order to elucidate structure–activity relationships for CaCet1p, we tested the effects of single alanine mutations at each of the 14 conserved side chains shown in Figure 1. These were: Glu287, Glu289 and Lys291 (β1), Asp363 (β5), Arg379 (β6), Lys396 (β7), Glu420 (β8), Arg441, Lys443 and Arg445 (β9), Asp458 (β10), and Glu472, Glu474 and Glu476 (β11). Note that at least one amino acid was mutated in each of the β strands that comprise the triphosphate tunnel. The Ala-mutations were introduced into the C-terminal catalytic domain, CaCet1(257–520)p, and the mutant polypeptides were expressed as His-tagged derivatives in E.coli in parallel with wild-type CaCet1(257–520)p. The recombinant proteins were purified from soluble bacterial extracts by Ni–agarose chromatography. SDS–PAGE analysis of the polypeptide compositions of the Ni–agarose protein preparations revealed similar extents of purification (Fig. 6). The 34 kDa CaCet1(257-520)p was the predominant polypeptide in every case.
Figure 6.

Expression and purification of mutated versions of CaCet1(257–520)p. Wild-type Cet1(257–520)p and the indicated single amino acid substitution mutants were expressed in bacteria at 18°C. Aliquots (5 µg) of the Ni–agarose preparations of the recombinant proteins were electrophoresed through a 12% polyacrylamide gel containing 0.1% SDS. Polypeptides were visualized by staining with Coomassie blue dye. The positions and sizes (in kDa) of marker proteins are indicated on the left.
Mutational effects on triphosphatase activity
The triphosphatase activities of the wild-type and mutant CaCet1(257–520)p proteins were assayed by the release of 32Pi from 1 mM [γ-32P]ATP in the presence of 2 mM manganese. Specific activity was determined from the slopes of the protein titration curves in the linear range of enzyme dependence. The specific activities of the 14 Ala-mutants in manganese-dependent ATP hydrolysis, normalized to the wild-type specific activity, are shown in Table 1. Alanine substitutions at 12 of the 14 positions examined elicited at least a 10-fold decrement in catalytic activity. The D363A mutation reduced activity to 12% of wild type. Only K291A was indifferent to side-chain removal.
Table 1. Effects of alanine mutations on the ATPase activity of CaCet1(257–520)p.
| Specific activity (% of wild type) | ||||
|---|---|---|---|---|
| Strand | Mutation | + Manganese | + Cobalt | Cet1p equivalent |
| β1 | E287A | <0.5 | <0.5 | E305 |
| E289A | <0.5 | <0.5 | E307 | |
| K291A | 67 | 74 | K309 | |
| β5 | D363A | 12 | 4 | D377 |
| β6 | R379A | <0.5 | <0.5 | R393 |
| β7 | K396A | <0.5 | <0.5 | K409 |
| β8 | E420A | <0.5 | <0.5 | E433 |
| β9 | R441A | 3 | <0.5 | R454 |
| K443A | 2 | <0.5 | K456 | |
| R445A | <0.5 | <0.5 | R458 | |
| β10 | D458A | 1 | <0.5 | D471 |
| β11 | E472A | 8 | <0.5 | E492 |
| E474A | <0.5 | <0.5 | E494 | |
| E476A | <0.5 | <0.5 | E496 | |
The Ni–agarose preparations of recombinant wild-type and mutant CaCet1(257–520)p were titrated for manganese- and cobalt-dependent ATPase activities as described under Materials and Methods. Two or three titration experiments were performed for each mutant protein and the specific activity was calculated from the average of the slopes of the titration curves. Each specific activity value was normalized to the specific activity of wild-type CaCet1(257–520)p with manganese or cobalt cofactors. The wild-type specific activity was the average of four titration experiments.
The proteins were also assayed for ATP hydrolysis with 2 mM cobalt included as the cofactor in lieu of manganese. The cobalt-dependent ATPase activities of the Ala-mutants were calculated from the slopes of the titration curves and then normalized to the wild-type specific activity (Table 1). The results were again quite stark. The specific activities of 12 of the mutants were <0.5% of the wild-type activity, and the D363A mutant displayed 4% of wild-type activity, whereas the K291A mutant was 78% as active as the wild-type protein.
Kinetic analysis
Kinetic parameters were determined for manganese-dependent ATP hydrolysis by wild-type CaCet1(257–520)p by measuring 32Pi release as a function of ATP concentration (Fig. 7). From a double-reciprocal plot of the data, we calculated a Km for ATP of 9 µM and a Vmax of 9 s–1. Potential effects of the K291A mutation on substrate binding might have been overlooked because the specific activity measurements in Table 1 were performed at 1 mM ATP, a substrate concentration more than two orders of magnitude above Km. Therefore, kinetic parameters were also determined for the K291A protein. The observed Km for ATP of 5 µM and a Vmax of 5 s–1 (data not shown) indicated that Lys291, though conserved among fungal RNA triphosphatases, is not important for either ATP binding or phosphohydrolase reaction chemistry.
Figure 7.

Dependence of ATP hydrolysis on ATP concentration. The extent of 32Pi release by wild-type Cet1(257–520)p (pmol/15 min) is plotted as a function of ATP concentration. The inset shows a double-reciprocal plot of the data.
DISCUSSION
Candida albicans RNA triphosphatase CaCet1p is a promising target for antifungal drug discovery. Initial genetic studies of the domain organization of CaCet1p entailed testing the ability of CaCET1 deletion alleles to complement S.cerevisiae cet1Δ strains (14; Schwer,B., Lehman,K., Saha,N. and Shuman,S., manuscript in preparation). Here we have analyzed the biochemical properties of recombinant CaCet1p and N-terminal deletion variants thereof. CaCet1p apparently consists of three domains: (i) an N-terminal segment from residue 1 to 202 that is dispensable for catalysis in vitro and for CaCet1p function in vivo; (ii) a segment from residue 203 to 228 that is dispensable for catalysis in vitro, but required for Cet1p function in vivo, said segment mediating the interaction of the triphosphatase with the guanylyltransferase component of the capping apparatus (17; Schwer,B., Lehman,K., Saha,N. and Shuman,S., manuscript in preparation); and (iii) a catalytic domain from residue 257 to 520.
We have shown that C.albicans RNA triphosphatase has nucleoside triphosphate phosphohydrolase activity in the presence of manganese or cobalt. An ATPase assay for CaCet1p function in vitro bypasses the costly and labor-intensive step of preparing triphosphate-terminated RNA substrates and is amenable to high-throughput screening for inhibitors using spectrophotometric assays of product formation. The failure of previous investigators to detect the ATPase activity of CaCet1p (10) can be attributed to their reliance on magnesium as the divalent cation cofactor. Manganese and cobalt-dependent NTPase activities have been described for Cet1p and Cth1p, and for vaccinia D1 and baculovirus LEF4. The turnover number of the Candida enzyme in ATP hydrolysis (9–17 s–1) is similar to the values for Cet1p (25 s–1), D1 (10 s–1) and LEF4 (30 s–1), and is slightly higher than that of Cth1p (2 s–1) (6,11,12,18). The Km of CaCet1p for ATP (9 µM) is slightly higher than that of Cet1p (3 µM), but lower than that of either LEF4 (43 µM), Cth1p (75 µM) or D1 (800 µM).
Here we have identified 13 residues within the catalytic domain of C.albicans CaCet1p that are important for manganese- dependent and cobalt-dependent nucleoside triphosphatase activity. Seven of the critical CaCet1p side chains (Glu287 and Glu289 in motif A; Arg441 and Lys443 in motif B; and Glu472, Glu474 and Glu476 in motif C) correspond to amino acids that are required for the activity of S.cerevisiae Cet1p in vivo and in vitro (these are denoted by dots in Fig. 1) (11,12). This group of amino acids is broadly conserved in the RNA triphosphatases encoded by poxviruses, African swine fever virus and baculoviruses. The glutamates in motif A and motif C comprise the metal binding site of Cet1p and their homologs are essential for catalysis by Cet1p (11,12), CaCet1p (this study), Cth1p (12), vaccinia D1 (13) and baculovirus LEF4 (5). Motifs A and C are located within strands β1 and β11 of Cet1p, which are widely separated in the primary structure, but closely approximated in the tertiary structure along the ‘floor’ of the tunnel. In motifs A and C of the fungal RNA triphosphatases, alternating charged side chains are interdigitated with alternating aliphatic/aromatic side chains (Fig. 1). This sequence pattern is reprised in motifs A and C of the poxvirus, ASFV and baculovirus RNA triphosphatases, suggesting that the metal-binding residues of the viral enzymes may also be located within β strands.
Six newly defined, important residues of CaCet1p (Asp363 in β5, Arg379 in β6, Lys396 in β7, Glu420 in β8, Arg445 in β9 and Asp458 in β10) are strictly conserved in S.cerevisiae Cet1p and Cth1p (Fig. 1). Our findings show that each of the eight strands of the β barrel contributes at least one essential component of the triphosphatase active site. This provides some rationale for the unusual tunnel structure of the fungal RNA triphosphatase. Note that Asp363, Arg379, Lys396, Glu420, Arg445 and Asp458 of CaCet1p have no recognizable counterparts in the poxvirus, African swine fever virus and baculovirus RNA triphosphatases, and that the sequences of these viral proteins display no persuasive similarities to the fungal protein sequences in the intervals between motifs A, B and C. Hence, the active sites of the poxvirus, African swine fever virus and baculovirus triphosphatases may adopt a different tertiary structure from the fungal triphosphatases (i.e., one without a fully closed tunnel), even though they share a common metal binding site. Note also that the viral proteins are bifunctional enzymes with RNA triphosphatase and RNA guanylyltransferase activities resident in a single polypeptide.
The molecular contacts of the essential CaCet1p residues are surmised from the crystal structure of Cet1p bound to manganese and sulfate (15). These side chains participate in a rich network of electrostatic and hydrogen bonds within the tunnel interior (Fig. 2). The interactions can be categorized as follows: (i) direct coordination of manganese (Glu305, Glu307 and Glu496 in Cet1p; Glu287, Glu289 and Glu476 in CaCet1p); (ii) coordination of solvent bound to manganese (Asp471 and Glu494 in Cet1p; Asp458 and Glu474 in CaCet1p); (iii) direct contact with sulfate (Arg393, Lys456 and Arg458 in Cet1p; Arg379, Lys443 and Arg445 in CaCet1p); (iv) interaction with solvent in the vicinity of sulfate (Asp377 and Glu433 in Cet1p; Asp363 and Glu420 in CaCet1p); and (v) side chain–side chain interactions (Arg454–Glu492 and Lys409–Glu305 in Cet1p; Arg441–Glu472 and Lys396–Glu287 in CaCet1p). How do these interactions instruct us about catalysis?
In the Cet1p crystal, which is construed to represent a product complex with Pi and manganese, the sulfate/phosphate oxygens are coordinated by three basic amino acids and by manganese as shown in Figure 2. Both arginines make bidentate contacts with the oxygens, while the lysine and manganese make monovalent contacts. The mutational analysis of CaCet1p shows that all three basic residues are essential and that virtually any perturbation of the coordination complex of manganese is deleterious. We infer a mechanism whereby cleavage of the β–γ phosphoanhydride bond occurs through a pentacoordinate phosphorane transition state in which the attacking water and the β phosphate leaving group are positioned apically. In this scheme, the basic side chains and the manganese would polarize the phosphate in the ground state and activate it for nucleophilic attack by water and also stabilize the negative charge on γ phosphate developed in the transition state. The essential basic residues are located in the upper part of the tunnel interior, the so-called walls and roof (15), whereas the manganese is bound by residues on the tunnel floor.
Manganese is octahedrally coordinated to the sulfate, to the side chain carboxylates of essential Cet1p residues Glu305, Glu307 and Glu496, and to two waters (Fig. 2). Removal of the equivalent acidic side chains of CaCet1p that contact the metal directly resulted in loss of activity, as one might expect. Yet CaCet1p was also inactivated by mutation of either Asp458 or Glu474 (Asp471 and Glu494 in Cet1p), which together coordinate one of the waters bound to manganese (Fig. 2). Glu305, a metal-binding glutamate, interacts with Nζ of Lys409, whose CaCet1p equivalent (Lys396) is essential for triphosphatase activity. Not all of the secondary interactions of the metal-binding glutamates seen in the crystal are functionally relevant, e.g., Cet1p Lys309 is within hydrogen bonding distance of the carboxylate of Glu307, yet mutation of the equivalent Lys291 side chain of CaCet1p had little impact on triphosphatase activity. These results indicate that the formation of a catalytic metal-binding site depends on a wide network of interactions of residues emanating from the floor and walls of the tunnel.
The essentiality of several of the CaCet1p residues analyzed here is not explained by interactions with either the metal or the γ phosphate in the structure of the product complex. There are likely to be additional interactions with the β and α phosphates in the substrate complex that contribute to substrate binding and catalysis. Essential CaCet1p residues Lys396 (Lys409 in Cet1p) and Arg441 (Arg454 in Cet1p) may play such a role. We speculate that an acidic residue might act as general base catalyst to promote attack of water on the γ phosphorus. A candidate for the role of general base is Glu420 of CaCet1p, an essential residue located in β5, the equivalent of which in the Cet1p structure (Glu433) is hydrogen-bonded to a water situated 3.7 Å from the sulfur center.
In summary, we have established structure–activity relationships for C.albicans RNA triphosphatase that confirm and extend the studies of the S.cerevisiae enzyme. We conclude that the catalytic mechanism and the structure of the active site are substantially conserved among fungal RNA triphosphatases. These findings inspire confidence that a mechanism-based inhibitor identified by screening against one fungal enzyme will display broad activity against RNA triphosphates from other fungal species.
REFERENCES
- 1.Takagi T., Moore,C.R., Diehn,F. and Buratowski,S. (1997) Cell, 89, 867–873. [DOI] [PubMed] [Google Scholar]
- 2.Ho C.K., Sriskanda,V., McCracken,S., Bentley,D., Schwer,B. and Shuman,S. (1998) J. Biol. Chem., 273, 9577–9585. [DOI] [PubMed] [Google Scholar]
- 3.Wen Y., Yue,Z. and Shatkin,A.J. (1998) Proc. Natl Acad. Sci. USA, 95, 12226–12231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shuman S. (1990) J. Biol. Chem., 265, 11960–11966. [PubMed] [Google Scholar]
- 5.Jin J., Dong,W. and Guarino,L.A. (1998) J. Virol., 72, 10011–10019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gross C.H. and Shuman,S. (1998) J. Virol., 72, 10020–10028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Itoh N., Mizumoto,K. and Kaziro,Y. (1984) J. Biol. Chem., 259, 13930–13936. [PubMed] [Google Scholar]
- 8.Tsukamoto T., Shibagaki,Y., Imajoh-Ohmi,S., Murakoshi,T., Suzuki,M., Nakamura,A., Gotoh,H. and Mizumoto,K. (1997) Biochem. Biophys. Res. Commun., 239, 116–122. [DOI] [PubMed] [Google Scholar]
- 9.Ho C.K., Schwer,B. and Shuman,S. (1998) Mol. Cell. Biol., 18, 5189–5198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamada-Okabe T., Mio,T., Matsui,M., Kashima,Y., Arisawa,M. and Yamada-Okabe,H. (1998) FEBS Lett., 435, 49–54. [DOI] [PubMed] [Google Scholar]
- 11.Ho C.K., Pei,Y. and Shuman,S. (1998) J. Biol. Chem., 273, 34151–34156. [DOI] [PubMed] [Google Scholar]
- 12.Pei Y., Ho,C.K., Schwer,B. and Shuman,S. (1999) J. Biol. Chem., 274, 28865–28874. [DOI] [PubMed] [Google Scholar]
- 13.Yu L., Martins,A., Deng,L. and Shuman,S. (1997) J. Virol., 71, 9837–9843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lehman K., Schwer,B., Ho,C.K., Rouzankina,I. and Shuman,S. (1999) J. Biol. Chem., 274, 22668–22678. [DOI] [PubMed] [Google Scholar]
- 15.Lima C., Wang,L.K. and Shuman,S. (1999) Cell, 99, 533–543. [DOI] [PubMed] [Google Scholar]
- 16.Rodriguez C.R., Takagi,T., Cho,E. and Buratowski,S. (1999) Nucleic Acids Res., 27, 2182–2188. [Google Scholar]
- 17.Ho C.K., Lehman,K. and Shuman,S. (1999) Nucleic Acids Res., 27, 4671–4678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Myette J.R. and Niles,E.G. (1996) J. Biol. Chem., 271, 11945–11952. [DOI] [PubMed] [Google Scholar]
- 19.Evans S.V. (1993) J. Mol. Graph., 11, 134–138. [DOI] [PubMed] [Google Scholar]