

Abstract
BACKGROUND:
Chronic kidney disease (CKD) accelerates the development of atherosclerosis, decreases muscle function, and increases the risk of amputation or death in patients with peripheral artery disease (PAD). However, the mechanisms underlying this pathobiology are ill-defined. Recent work has indicated that tryptophan-derived uremic solutes, which are ligands for the aryl hydrocarbon receptor (AHR), are associated with limb amputation in PAD. Herein, we examined the role of AHR activation in the myopathy of PAD and CKD.
METHODS:
AHR-related gene expression was evaluated in skeletal muscle obtained from mice and human PAD patients with and without CKD. Skeletal-muscle-specific AHR knockout mice (AHRmKO) with and without CKD were subjected to femoral artery ligation and a battery of assessments were performed to evaluate vascular, muscle, and mitochondrial health. Single nuclei RNA sequencing was performed to explore intercellular communication. Expression of the constitutively active AHR (CAAHR) was employed to isolate the role of the AHR in mice without CKD.
RESULTS:
PAD patients and mice with CKD displayed significantly higher mRNA expression of classical AHR-dependent genes (Cyp1a1, Cyp1b1, and Aldh3a1) when compared to either muscle from the PAD condition with normal renal function (P<0.05 for all three genes) or non-ischemic controls. AHRmKO significantly improved limb perfusion recovery and arteriogenesis, preserved vasculogenic paracrine signaling from myofibers, increased muscle mass and strength, as well as enhanced mitochondrial function in an experimental model of PAD/CKD. Moreover, viral-mediated skeletal muscle-specific expression of a CAAHR in mice with normal kidney function exacerbated the ischemic myopathy evidenced by smaller muscle masses, reduced contractile function, histopathology, altered vasculogenic signaling, and lower mitochondrial respiratory function.
CONCLUSIONS:
These findings establish AHR activation in muscle as a pivotal regulator of the ischemic limb pathology in CKD. Further, the totality of the results provide support for testing of clinical interventions that diminish AHR signaling in these conditions.
Keywords: peripheral artery disease, hindlimb ischemia, uremia, mitochondria
Graphical Abstract
INTRODUCTION
Peripheral arterial disease (PAD) affects 8–12 million Americans1 and is the third highest leading cause of cardiovascular mortality2. PAD is caused by atherosclerotic narrowing or occlusion in the lower extremities which leads to a spectrum of life-altering symptomatology including claudication, ischemic rest pain, as well as gangrene and/or non-healing ulcers that commonly result in major limb amputation especially when revascularization attempts fail or are not possible. Complicating the etiology of PAD, patients typically present with comorbid conditions or risk factors that accelerate disease evolution and are associated with poorer health outcomes. Among these, chronic kidney disease (CKD) is strongly associated with the development of atherosclerosis, decreased muscle function, and increased risk of amputation or death in PAD patients3–6. For example, mortality risk for PAD patients with CKD is ~2-to-4 times higher than PAD patients without CKD6–9. CKD has also been shown to be associated with increased failure rates of both endovascular and open surgical revascularization procedures in PAD management3,10. While the clinical evidence demonstrating that CKD exacerbates PAD pathobiology is undeniable, the cellular and physiological mechanisms underlying this pathology is ill-defined.
CKD leads to retention of waste products that can be harmful to other tissues/organs, including skeletal muscle11–15 and blood vessels16–18 where the combined effects presumably contribute to worsened PAD pathology. Unfortunately, many uremic metabolites are protein-bound and not adequately filtered by dialysis membranes and remain elevated despite treatment with renal replacement therapies19,20. A recent report demonstrated a strong correlation between tryptophan-derived uremic metabolites and adverse limb events in PAD patients21, highlighting the potential that specific biological pathways may contribute to the pathobiology of PAD in patients with CKD. Interestingly, many of these tryptophan-derived uremic metabolites have been shown to impair skeletal muscle mitochondrial function and drive atrophy22–25, potentially implicating skeletal muscle function and mitochondrial health as a site of coalescence between the pathobiology of CKD and PAD. Importantly, emerging evidence in PAD patients has demonstrated that muscle function/exercise capacity is a strong predictor of morbidity/mortality26–30.
Interestingly, several tryptophan-derived uremic metabolites (kynurenines and indoles) are ligands of the aryl hydrocarbon receptor (AHR), a ligand-activated transcription factor belonging to the basic helix-loop-helix/Per-ARNT-Sim (bHLH/PAS) family. Both mice and human patients with CKD display elevated expression of the Ahr31. The most widely studied of the uremic metabolites, indoxyl sulfate, causes muscle atrophy and mitochondrial dysfunction in mice with normal kidney function22,23,32, although the underlying mechanisms are unclear. Chronic activation of the AHR has been shown to reduce angiogenesis33,34 and increase atherosclerosis35–37, whereas chemical inhibition of the AHR has been shown to improve perfusion recovery in CKD mice following femoral artery ligation (FAL) and normalize post-ischemic angiogenesis21. Comparatively, little is known about the role of the AHR in mediating the toxic effects of uremic metabolite accumulation in skeletal muscle exposed to the CKD condition. However, a recent study reported that AHR activation in skeletal muscle could phenocopy the effects of tobacco smoking, a strong risk factor for PAD, on skeletal muscle pathology including atrophy and mitochondrial respiratory dysfunction38. Further, associations between the levels of CKD-associated AHR ligands and muscle mitochondrial energetics have been reported in rodents with CKD25. Based on the accumulation of evidence, we hypothesized that chronic AHR activation may mediate the myopathic condition in the presence of CKD and PAD.
METHODS.
Data Availability.
The data supporting the conclusions of this study are available from the corresponding author.
Detailed descriptions of the materials and experimental procedures can be found in the Expanded Material and Methods section of the Supplemental Material A Major Resources Table is available in the Supplemental Material. References related to methods employed included the following:25,39–47.
RESULTS
CKD and Uremic Toxin Exposure Result in Significant AHR Activation in Human and Mouse Skeletal Muscle.
To explore whether the uremic milieu caused by CKD resulted in activation of the AHR pathway in skeletal muscle, we obtained muscle biopsy specimens from the gastrocnemius of non-PAD adult controls, PAD patients without CKD, and PAD patients with CKD. Quantitative PCR analysis demonstrated significantly higher mRNA expression of classical AHR-dependent genes (Cyp1a1, Cyp1b1, and Aldh3a1) in PAD patients with CKD when compared to either PAD patients with normal renal function or non-PAD adult controls (Figure 1A). Patient characteristics are shown in Supplemental Table 1. Consistent with the clinical data, gastrocnemius skeletal muscle from C57BL6J mice with CKD and subjected to femoral artery ligation (FAL) displayed significantly increased mRNA expression of Ahr and Cyp1a1 (Figure 1B). Serum metabolomics of adenine-fed CKD mice confirmed a significant increase in tryptophan-derived uremic metabolites, including indoxyl sulfate, L-kynurenine, and kynurenic acid, that are known AHR ligands (Figure 1C). Given that the muscle specimens from human and mice contain other non-muscle cell types, we next explored the potential for AHR activation in isolated muscle cell culture systems. Acute (18h) exposure of murine (C2C12) myotubes or human primary myotubes to indoxyl sulfate, L-kynurenine, and kynurenic acid resulted in significant increases in Cyp1a1 mRNA levels (Figure 1D), a classical cytochrome P450 enzyme that is transcriptionally regulated by the AHR. Taken together, these data provide conclusive evidence of AHR activation in skeletal muscle exposed to the CKD condition.
Figure 1. Evidence of AHR activation by uremic metabolites in human and mouse skeletal muscle.
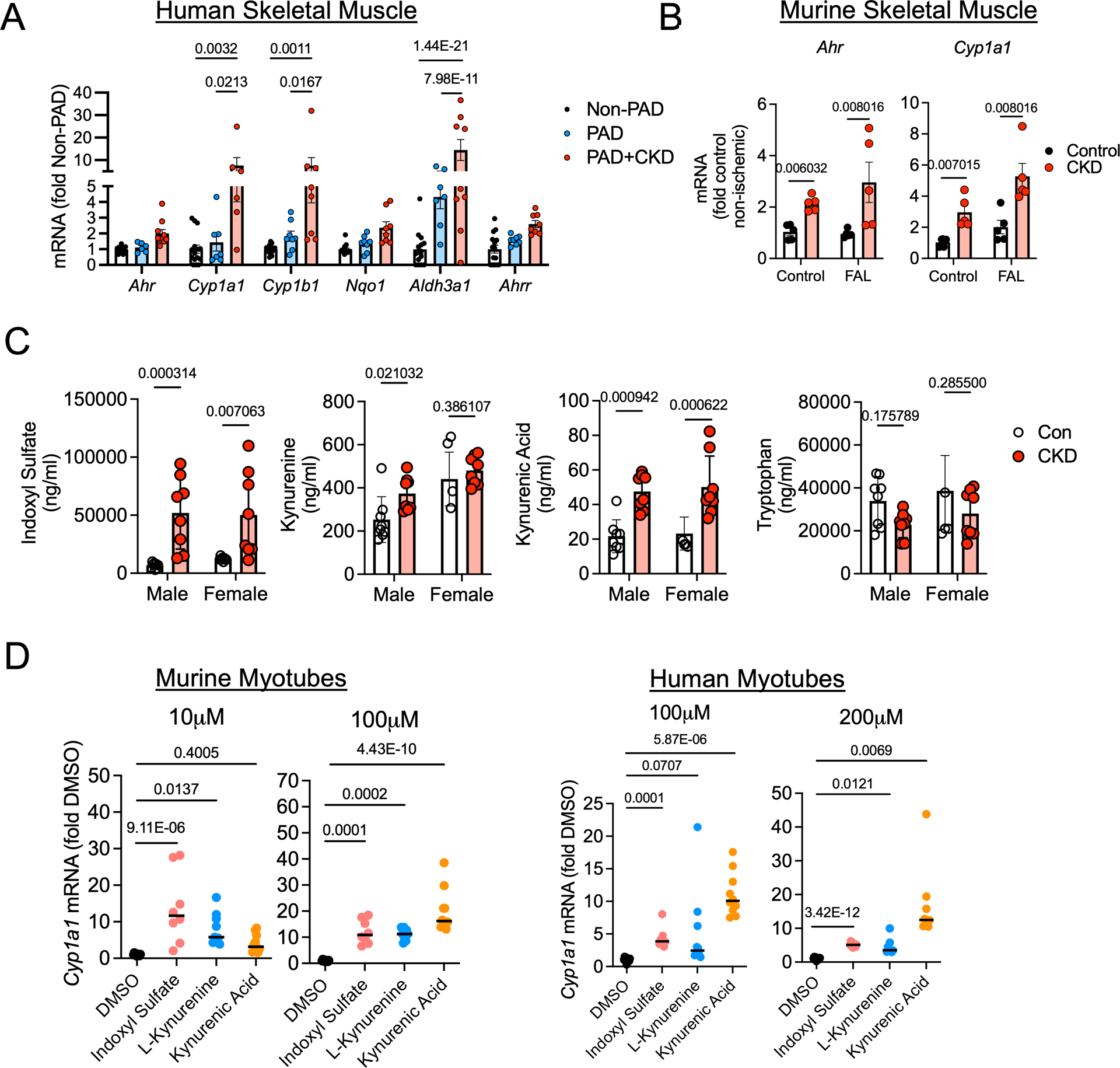
(A) Relative mRNA levels of AHR-related genes in gastrocnemius muscle specimens from non-PAD adults (n=15) and PAD patients with (n=10) and without CKD (n=8). (B) Relative expression of Ahr and Cyp1a1 in the gastrocnemius muscle of mice with and without CKD in both control and ischemic limbs (n=5/group). (C) Quantified levels of tryptophan-derived AHR ligands in the serum of mice with and without CKD (n=8 male Con, 4 female Con, 8 male CKD, 8 female CKD). (D) qPCR analysis of relative mRNA levels of Cyp1a1 following acute treatment with AHR ligands in both murine (n= 10 DMSO, 8 indoxyl sulfate, 9 L-kynurenine, and 10 kynurenic acid) and human cultured myotubes (n=10 DMSO, 9 indoxyl sulfate, 10 L-kynurenine, and 10 kynurenic acid). Panels A and D were analyzed using a one-way ANOVA with Sidak’s post hoc testing. Panels B and C were analyzed using Mann-Whitney tests. *P<0.05, Error bars represent the standard deviation.
Skeletal Muscle-Specific AHR Deletion Promotes Ischemic Muscle Perfusion Recovery in Mice with CKD.
Previous work has shown that chronic AHR activation reduces angiogenesis21,33,34,48 and promotes atherosclerosis35–37. Additionally, chronic activation of the AHR by dioxin leads to muscle weakness and atrophy38,49, symptoms commonly exhibited by PAD and CKD patients. The phenotype observed in models with chronic AHR activation is similar to the PAD patient with CKD, prompting us to hypothesize that the AHR might regulate limb pathology in this condition. To explore this, we generated an inducible skeletal muscle-specific AHR knockout mouse (AHRmKO, Figure 2A). Following administration of tamoxifen, deletion of the AHR in skeletal muscle was confirmed by immunoblotting (Figure 2B) and DNA recombination (Supplemental Figure 1A,B). Next, we randomized AHRmKO mice and their wildtype (AHRfl/fl) littermates to either a casein control (Con) or adenine-supplement diet to induce CKD for three weeks prior to subjecting the animals to FAL as an experimental model of PAD (Figure 2C). CKD mice, regardless of genotype, have significantly reduced glomerular filtration rate (GFR, Figure 2D), elevated blood urea nitrogen (BUN, Figure 2E), and reduced body weights (Figure 2F) compared to casein fed controls. Following FAL, CKD was found to significantly impair perfusion recovery in the paw in both male and female mice, as well as the gastrocnemius in female mice (Diet effect, P<0.035 in all cases; Figure 2G,H). Significant genotype effects were detected in both the gastrocnemius and paw of male and female mice, indicating that AHRmKO mice had improved gastrocnemius and paw perfusion recovery (Figure 2G,H). To assess the effects on the muscle vasculature, we labeled endothelial cells within the tibialis anterior muscle using fluorescently labeled Griffonia Simplicifolia lectin I isolectin B4. Fourteen days after FAL, there was a significant effect of diet in female (P=0.002), but not male mice, demonstrating that CKD reduced total capillary density in female mice (Figure 2I). Regardless of diet, there were no differences in total capillary density between AHRmKO and AHRfl/fl mice (Figure 2I). Analysis of pericyte abundance revealed a significant effect of diet in both male (P=0.0003) and female (P=0.019) (Figure 2J). A significant genotype effect was detected in male mice indicating the AHRmKO mice had greater pericyte abundance when compared to AHRfl/fl mice (P=0.016). Next, we examined the impact of CKD and muscle-specific AHR deletion on the abundance of arterioles by labeling muscle sections with alpha smooth muscle actin (αSMA). CKD was found to reduce arterioles abundance in both male and female AHRfl/fl mice (Figure 2K). Strikingly, AHRmKO mice with CKD, regardless of sex, display higher levels of arterioles in their ischemic limb (P=0.0007 and P=0.0006 for male and female respectively) compared to AHRfl/fl mice with CKD (Figure 2K). In the non-ischemic control limb, there were no significant adverse effects on capillary, arteriole, or pericyte density due to deletion of the AHR or the presence of CKD (Supplemental Figure 2).
Figure 2. Skeletal Muscle-Specific AHR deletion Promotes Ischemic Muscle Perfusion Recovery and Arteriogenesis in Mice with CKD.
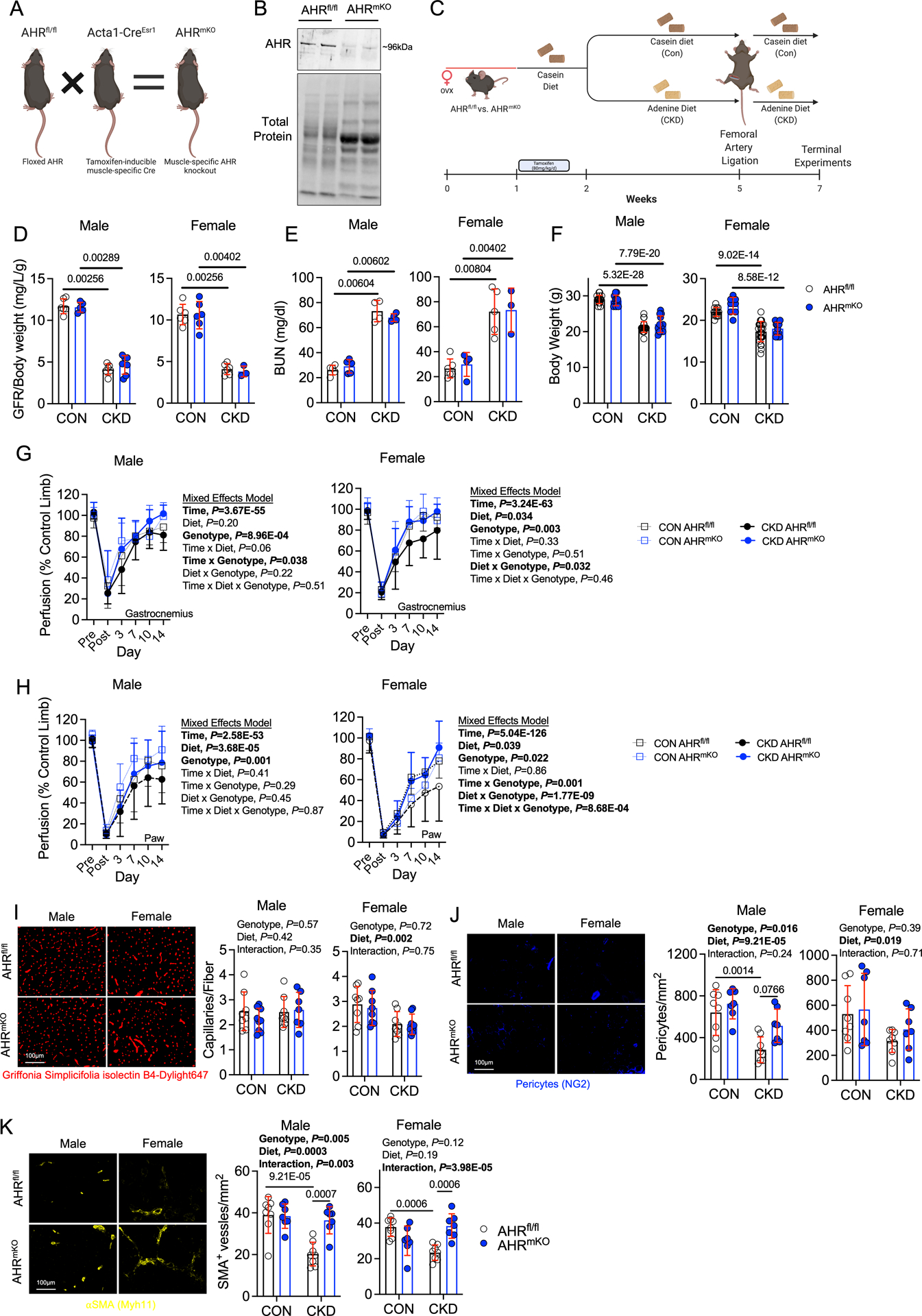
(A) Generation of inducible, muscle-specific AHR knockout mice and (B) immunoblotting confirmation of AHR deletion in muscle. (C) Graphical depiction of the experiment design. (D) Glomerular filtration rate (GFR) in mice with and without CKD (n=6 AHRfl/fl males/group, 5 AHRmKO male CON and 7 CKD, 6 AHRfl/fl female CON and 4 CKD, 6 AHRmKO female CON and 3 CKD). (E) Quantification of blood urea nitrogen (BUN) (n=5 AHRfl/fl male CON and 4 CKD, 5 AHRmKO male CON and 4 CKD, 6 AHRfl/fl female CON and 5 CKD, 4 AHRmKO female CON and 3 CKD). (F) Quantification of body weights (n=17 AHRfl/fl male CON and 21 CKD, 11 AHRmKO male CON and 12 CKD, 13 AHRfl/fl female CON and 25 CKD, 12 AHRmKO female CON and 14 CKD). (G) Quantification of perfusion recovery in the gastrocnemius muscle (n=14 AHRfl/fl male CON and 17 CKD, 13 AHRmKO male CON and 13 CKD, 10 AHRfl/fl female CON and 18 CKD, 9 AHRmKO female CON and 8 CKD). (H) Quantification of perfusion recovery in the paw (n=14 AHRfl/fl male CON and 17 CKD, 13 AHRmKO male CON and 13 CKD, 10 AHRfl/fl female CON and 18 CKD, 9 AHRmKO female CON and 8 CKD). Perfusion recovery was analyzed using mixed model analysis. (I) Quantification of capillary density in the tibialis anterior muscle (n=8 AHRfl/fl males/group, 9 AHRmKO male CON and 7 CKD, and n=8/genotype/diet in females). (J) Quantification of pericyte density in the tibialis anterior muscle (n=8 AHRfl/fl males/group, 7 AHRmKO males/group, and n=8 AHRfl/fl females/group, 7 AHRmKO females/group). (K) Quantification of arteriole density in the tibialis anterior muscle (n=8 AHRfl/fl males/group, 7 AHRmKO males/group, and n=8 AHRfl/fl females/group, 7 AHRmKO females/group). Statistical analyses with normally distributed data were performed using two-way ANOVA with Sidak’s post hoc testing for multiple comparisons when significant interactions were detected. Panels D-E were analyzed using Mann-Whitney tests. Error bars represent the standard deviation.
Skeletal Muscle-Specific Deletion of the AHR Preserves Muscle Mass, Contractile Function, and Mitochondrial Bioenergetics in Mice with CKD.
Because a progressive skeletal myopathy in CKD and PAD alone is associated with adverse long-term health outcomes in patients, we next examined the impact of AHR deletion on skeletal muscle health in mice with PAD and CKD. Consistent with a wealth of preclinical and clinical literature, mice with CKD displays markedly lower muscle weights in both the ischemic limb (Figure 3A) and non-ischemic limb (Supplemental Figure 3A) compared to their casein fed counterparts with normal kidney function. Despite the catabolic state of the CKD condition, the AHRmKO male mice had significantly larger masses of the ischemic tibialis anterior and extensor digitorum longus (EDL) muscles compared to AHRfl/fl male mice with CKD (Figure 3A), although muscle masses were still lower compared to casein fed mice (P<0.0001). Similar trends for increased muscle masses in AHRmKO mice were observed in the non-ischemic control limb muscles (Supplemental Figure 3A). In contrast to male mice, female AHRmKO mice with CKD did not exhibit increased muscle masses in the ischemic limb when compared to female AHRfl/fl mice with CKD (Figure 3A). Assessments of muscle force production revealed a significant diet effect in male mice (P=0.009), whereas the diet effect in female mice was trending but non-significant (P=0.08) (Figure 3B,C). Importantly, AHRmKO male mice had significantly higher levels of absolute force at both submaximal and maximal contractile states when compared to AHRfl/fl male mice with CKD (Figure 3B). Peak specific force levels (force normalized to area), an index of muscle quality, displayed a significant interaction (P=0.017) and post-hoc testing revealed a trending improvement in AHRmKO mice with CKD compared to AHRfl/fl mice with CKD (P=0.0573). In contrast to male mice, there were no detectable effect of genotype on muscle absolute or specific force levels (Figure 3C), indicating a sex-dependent effect of AHR deletion on muscle contractile function. Representative hematoxylin and eosin-stained and laminin-stained tibialis anterior muscles indicated a more preserved muscle architecture in AHRmKO mice with CKD (Figure 3D). Interestingly, the myofiber cross-sectional area (CSA) was not different between CKD mice with or without the AHR in the ischemic limb muscle (Figure 3E). However, in the non-ischemic control limb, CKD significantly decreased myofiber CSA in AHRfl/fl mice but not AHRmKO mice (Supplemental Figure 3B). Analysis of the total fiber number within the ischemic muscle uncovered a significant decrease in total fibers in CKD mice irrespective of sex (Figure 3F). Accordant with the improved muscle mass and absolute force levels, only AHRmKO male mice with CKD had significantly more myofibers compared to AHRfl/fl mice with CKD (Figure 3F). Male mice displayed a significant diet effect for the percentage of myofibers with centralized nuclei in the ischemic limb muscle, but no effects of AHR deletion were observed in either sex (Figure 3G). Additionally, analysis of the fiber type distribution within the non-ischemic control limb muscle indicated that neither CKD nor deletion of AHR altered the distribution of myosin fiber types (Supplemental Figure 3C). Moreover, skeletal muscle-specific AHR deletion had no effect on muscle size or contractile function in non-CKD mice that received a sham surgery (Supplemental Figure 4A-D).
Figure 3. Skeletal Muscle-Specific deletion of the AHR Preserves Ischemic Muscle Mass and Contractile Function in Mice with CKD.
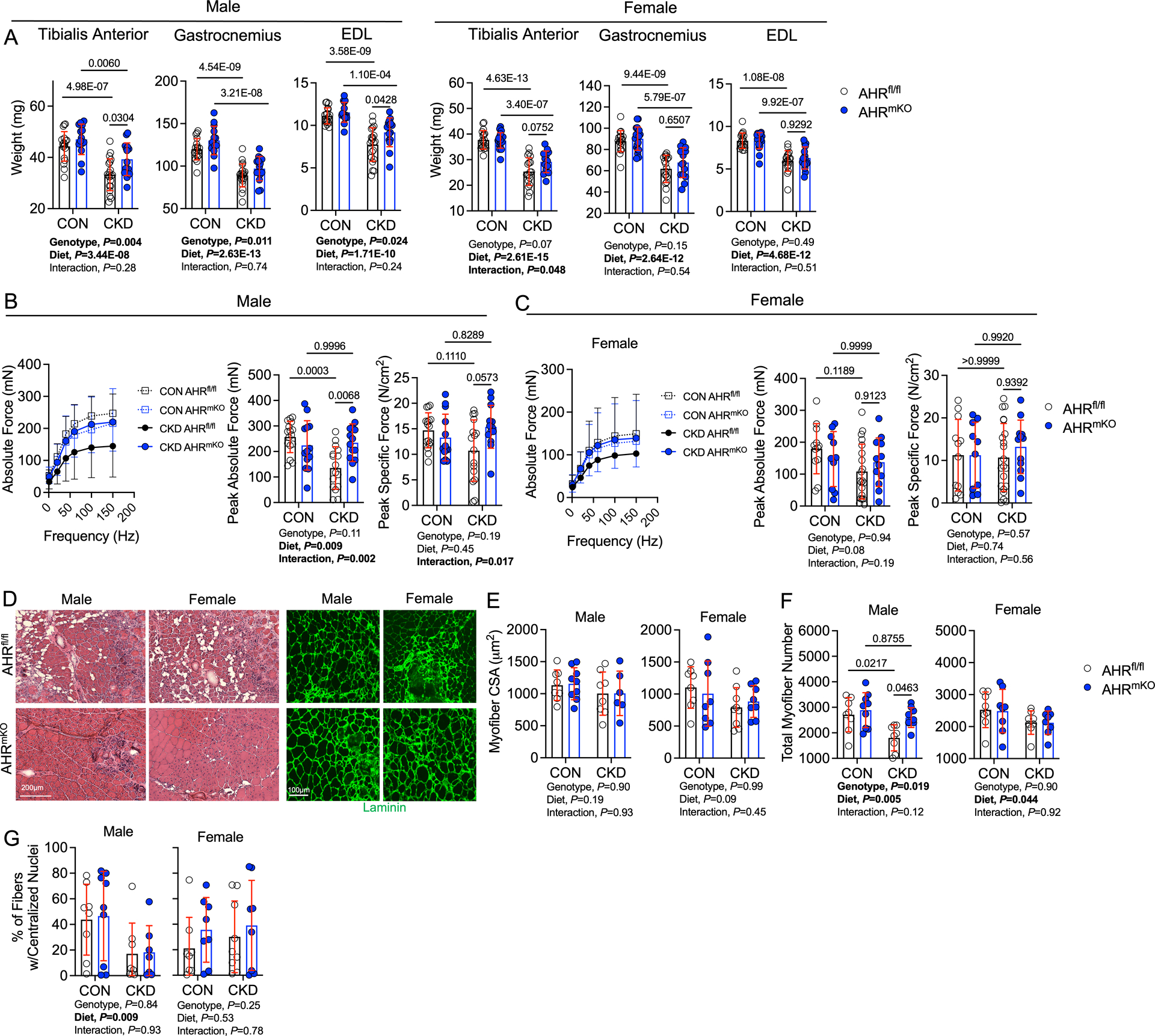
(A) Quantification of muscle weights in wildtype (AHRfl/fl) and AHRmKO mice (n=18 AHRfl/fl male CON and 20 CKD, 14 AHRmKO male CON and 15 CKD, 18 AHRfl/fl females/group, 20 AHRmKO female CON and 13 CKD). (B) Muscle contractile function quantification in male mice (n=16 AHRfl/fl male CON and 16 CKD, 13 AHRmKO male CON and 14 CKD). (C) Muscle contractile function quantification in female mice (n=12 AHRfl/fl female CON and 21 CKD, 10 AHRmKO female CON and 12 CKD). Specific force was calculated as the absolute force normalized to the cross-sectional area of the muscle. (D) Representative hematoxylin & eosin staining and laminin staining immunofluorescence from mice with CKD. (E) Quantification of the mean myofiber cross-sectional area of the tibialis anterior muscle (n=8 AHRfl/fl males/group, 9 AHRmKO male CON and 6 CKD, 8 AHRfl/fl female CON and 9 CKD, 8 AHRmKO females/group). (F) Quantification of the total fiber numbers in the tibialis anterior muscle (n=8 AHRfl/fl males/group, 9 AHRmKO males/group, 8 AHRfl/fl females/group, 8 AHRmKO females/group). (G) Quantification of the percentage of myofibers containing centralized nuclei within the tibialis anterior muscle (n=8 AHRfl/fl males/group, 9 AHRmKO male CON and 7 CKD, 8 AHRfl/fl female CON and 9 CKD, 8 AHRmKO females/group). Statistical analyses performed using two-way ANOVA with Sidak’s post hoc testing for multiple comparisons. Error bars represent the standard deviation.
Indoxyl sulfate, one of the most well characterized uremic metabolites, has been shown to impair mitochondrial respiratory function and increase reactive oxygen species (ROS) levels in mice22,23,32. Coincidently, mitochondrial function has been linked to PAD limb pathobiology50–55 and was identified as a potential site for the coalescence of PAD and CKD41. In light of these observations, we next asked whether the AHR may have a role in mediating mitochondrial dysfunction in mice with CKD and PAD. To assess mitochondrial function, mitochondria were isolated from the ischemic gastrocnemius muscles and respirometry was performed using a creatine kinase clamp system that measures mitochondrial respiratory function across a range of energy demands (akin to a stress test)56–58. When mitochondria were fueled by pyruvate and malate, AHRmKO male mice with CKD displayed higher rates of oxygen consumption at all levels of energy demand assessed when compared to AHRfl/fl male mice with CKD (Figure 4A). Quantification of conductance through the oxidative phosphorylation (OXPHOS) system (slope of oxygen consumption vs. ΔGATP) uncovered a significant interaction (P=0.003) and post-hoc testing revealed greater OXPHOS conductance in male AHRmKO mice compared to male AHRfl/fl mice (Figure 4A). Similarly, maximal uncoupled respiration (following the addition of FCCP) fueled by pyruvate/malate was significantly higher in male AHRmKO mice with CKD compared to AHRfl/fl male mice with CKD (P=0.0068, Figure 4A). Parallel experiments were also performed when fueling mitochondria with a medium chain fatty acid (octanoylcarnitine). These experiments revealed significant genotype effects, but no interactions between diet and genotype, indicating the AHRmKO male mice had improved mitochondrial fatty acid oxidation compared to AHRfl/fl male mice (Figure 4B). Interestingly, the effects of muscle-specific AHR deletion on mitochondrial function was absent in female mice, regardless of the kidney function or fuel source utilized (Figure 4C,D). Skeletal muscle-specific AHR deletion had no effect on mitochondrial function in non-CKD mice that received a sham surgery (Supplemental Figure 4E,F).
Figure 4. Skeletal Muscle-Specific deletion of the AHR Preserves Ischemic Muscle Mitochondrial Energetics in Mice with CKD.
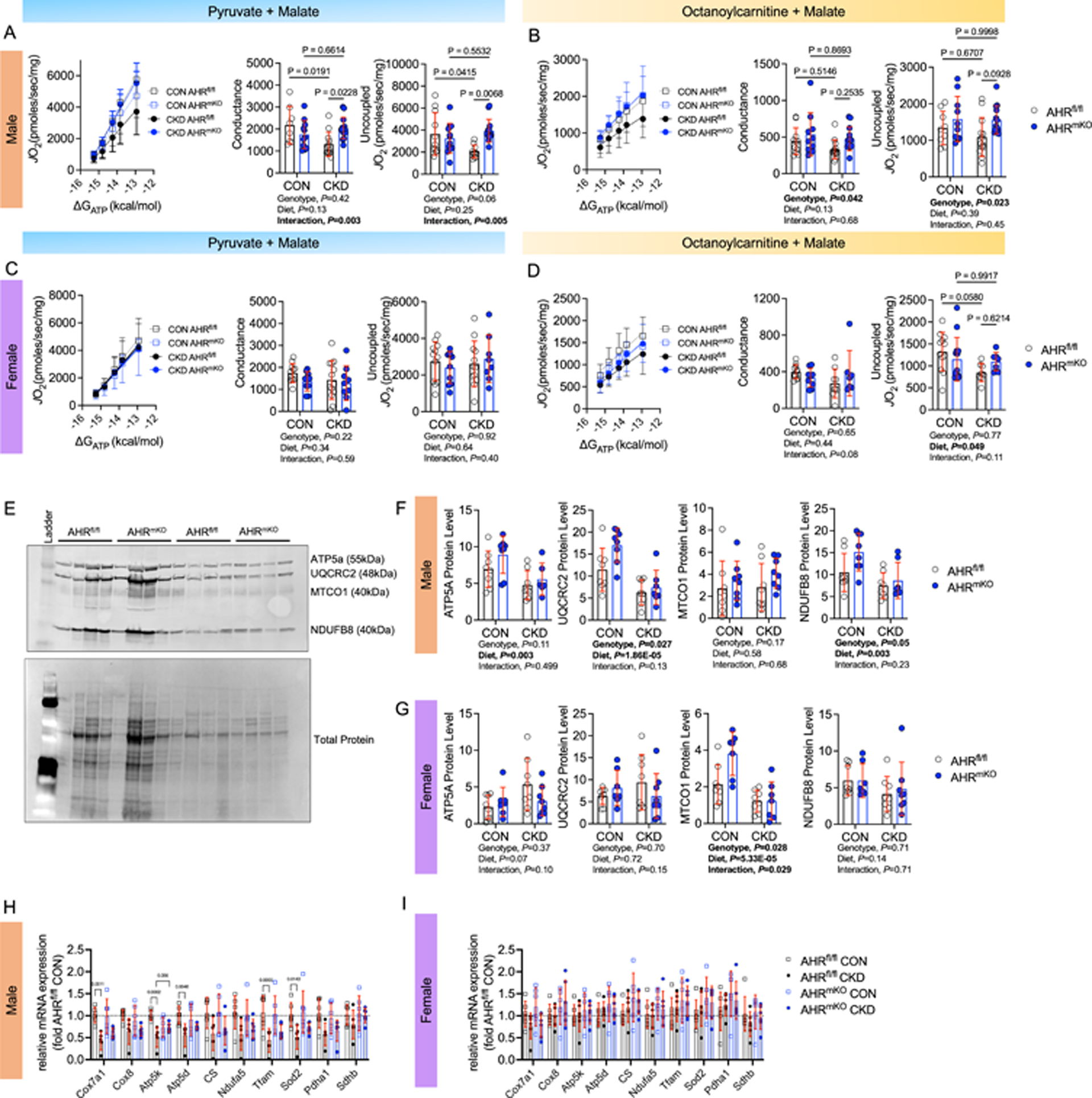
(A) Relationship between JO2 and ΔGATP when mitochondria were fueled with pyruvate and malate and quantification of the conductance (slope of JO2 and ΔGATP relationship) and uncoupled respiration rates in male mice (n=8 AHRfl/fl male CON and 12 CKD, 11 AHRmKO male CON and 14 CKD). (B) Relationship between JO2 and ΔGATP when mitochondria were fueled with octanoylcarnitine and malate and quantification of the conductance and uncoupled respiration rates in male mice (n=11 AHRfl/fl male CON and 13 CKD, 10 AHRmKO male CON and 13 CKD). (C) Relationship between JO2 and ΔGATP when mitochondria were fueled with pyruvate and malate and quantification of the conductance and uncoupled respiration rates in female mice (n=11 AHRfl/fl female CON and 13 CKD, 10 AHRmKO female CON and 9 CKD). (D) Relationship between JO2 and ΔGATP when mitochondria were fueled with octanoylcarnitine and malate and quantification of the conductance and uncoupled respiration rates in female mice (n=11 AHRfl/fl female CON and 9 CKD, 9 AHRmKO female CON and 7 CKD). (E) Western blotting membrane of mitochondrial electron transport system proteins (n=8/group/sex/genotype). (F) Quantification of mitochondrial protein abundance in male mice n=8/group/sex/genotype). (G) Quantification of mitochondrial protein abundance in female mice n=8/group/sex/genotype). (H) qPCR analysis of mitochondrial transcripts in male mice (n=6/group). (I) qPCR analysis of mitochondrial transcripts in female mice (n=6/group/sex/genotype). Statistical analyses performed using two-way ANOVA with Sidak’s post hoc testing for multiple comparisons. Error bars represent the standard deviation.
Next, mitochondrial content/density was explored using immunoblotting for select proteins in the electron transport system (Figure 4E). In male mice, a significant diet effect was found for ATP5A, UQCRC2, and NDUFB8 proteins indicating that CKD reduced the abundance of these proteins (Figure 4F, all blot images shown in Supplemental Figure 5). Further to this, AHRmKO mice were found to have significantly higher levels UQCRC2 and NDUFB8 protein levels (P=0.027 and P=0.05 respectively). In female mice, only MTCO1 protein levels were reduced in CKD mice (Figure 4G). Congruent with the findings from respirometry experiments, mitochondrial protein abundance was largely unaffected by the deletion of the AHR in female mice. At the level of the mRNA, the expression of several mitochondrial genes (Cox7a1, Atp5k, Atp5d, Tfam, Sod2) were significantly lower in male AHRfl/fl mice with CKD compared to AHRfl/fl mice without CKD (Figure 4H); however, these changes were attenuated in AHRmKO male mice with CKD. In contrast, mRNA levels for the examined mitochondrial genes were minimally impacted by either CKD or the deletion of the AHR in the ischemic muscle of female mice (Figure 4I). In the non-ischemic control limb, mitochondrial gene expression was minimally affected except for Ndufa5 and Tfam which were reduced in AHRfl/fl male mice with CKD but significantly greater in AHRmKO male mice with CKD (Supplemental Figure 6).
Single Nuclei RNA Sequencing (snRNA-seq) Reveals Differences in Cell Populations and Transcriptional Profiles in the Ischemic Muscle of AHRmKO mice.
To compare different nuclei populations in AHRfl/fl and AHRmKO skeletal muscles from CKD/PAD mice, we performed snRNA-seq on nuclei isolated from the tibialis anterior muscle of AHRfl/fl and AHRmKO harvested five days after FAL surgery (Figure 5A). Importantly, these muscle specimens were harvested at a timepoint when limb perfusion was not different between groups (Figure 5B). Nuclei were isolated using gentle homogenization, purified using the Chromium nuclei isolation kit (10x Genomics), and quality determined using fluorescence microscopy. From AHRfl/fl muscle, we captured 18,833 nuclei, and from AHRmKO muscle we captured 12,597 nuclei with high integrity. A median of 1,853 genes per nuclei were sequenced from AHRfl/fl muscle, whereas a median of 1,110 genes per nuclei were sequenced from AHRmKO muscle. The total number of genes detected was 25,964 and 25,009 in AHRfl/fl and AHRmKO mice respectively.
Figure 5. snRNA sequencing on ischemic muscle from AHRfl/fl and AHRmKO mice with CKD.
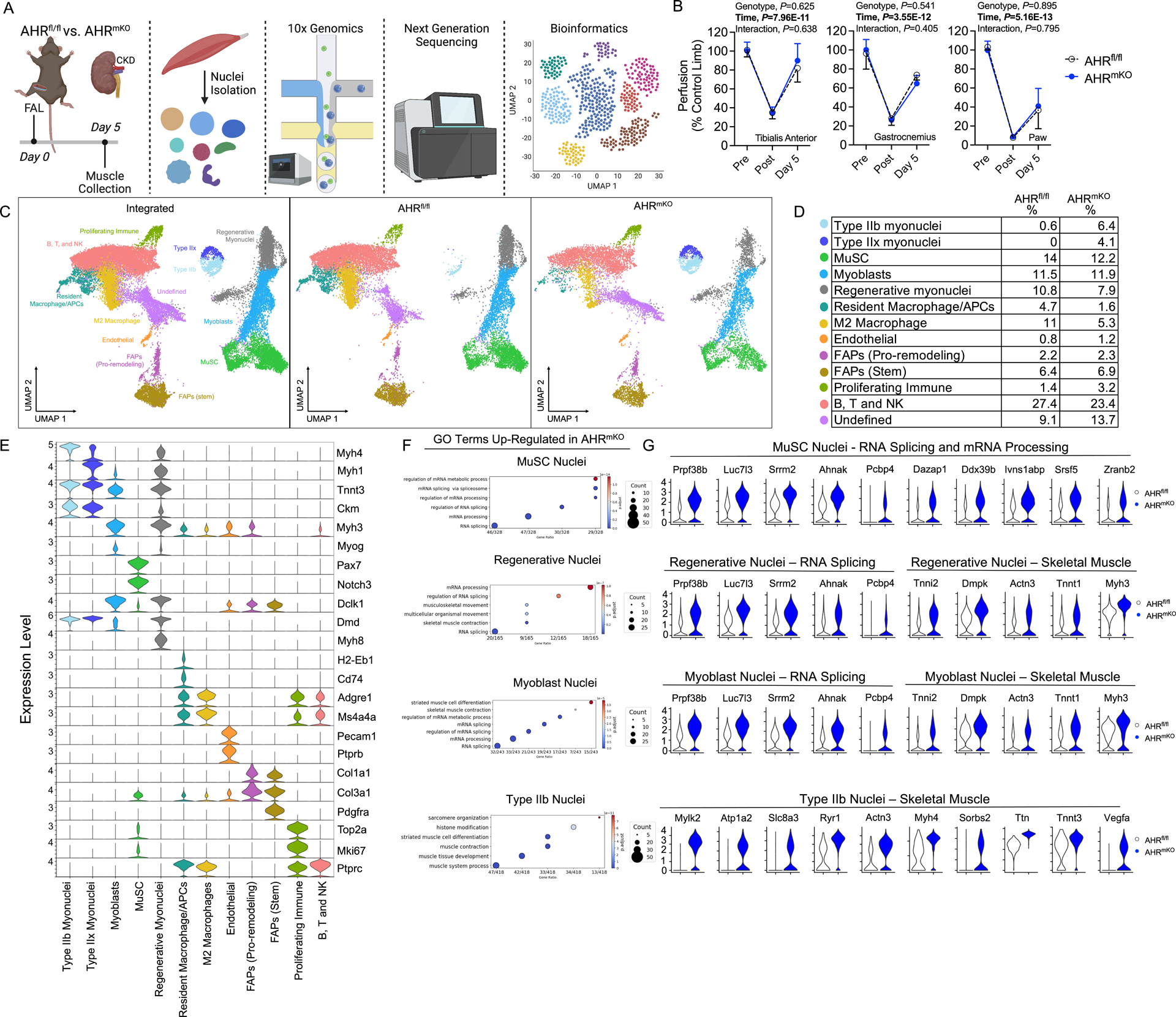
(A) Schematic of the experimental design. (B) Quantification of limb perfusion (n=3 males/group). (C) UMAPs visualization of the integrated datasets to identify clusters, as well as UMAPs for AHRfl/fl and AHRmKO muscle nuclei. (D) Percentage of nuclei within each cluster determined by group. (E) Violin plots showing z-score transformed expression of selected marker genes. (F) Dotplots of gene ontology (GO) analysis of significantly upregulated genes in AHRmKO myonuclei populations. (G) Violin plots showing normalized expression levels of select differentially expressed genes.
To compare AHRfl/fl and AHRmKO samples, we integrated the datasets resulting in a total of 31,430 nuclei for bioinformatic analysis including using uniform manifold approximation and projection (UMAP) to visualize and resolve different nuclear populations (Figure 5C). We identified 13 clusters of nuclei based upon their transcriptional profiles and assigned identities by examining normalized expression of values of top markers and known marker genes for each cluster (Figure 5C) and the relative percentages of each nuclei population are shown in Figure 5D. Violin plots for selected marker genes in each known nuclei population are depicted in Figure 5E. These analyses identified differences in the abundance of mature Type IIb and Type IIx myonuclei, with AHRmKO having more mature myonuclei compared to AHRfl/fl (Figure 5C,D). Considering the time course of muscle regeneration relative to the ischemic injury (day 5 post-FAL), this finding suggests that deletion of the AHR in skeletal muscle promotes the survival of mature myonuclei during severe limb ischemia in mice with CKD. Next, we explored differentially expressed genes and gene ontology (GO) pathways in myonuclei populations to uncover potential differences in transcriptional activity in AHRmKO mice. We identified that pathways related to RNA splicing, mRNA processing, and mRNA metabolic process, which are involved in muscle growth processes, were upregulated in AHRmKO muscle stem cell (MuSC), Regenerative, and Type IIx myonuclei populations (Figure 5F). In mature Type IIb and Type IIx myonuclei, upregulated pathways in AHRmKO mice included ‘muscle contraction’ and ‘generation of precursor metabolites and energy’, both of which are consistent with the observed preservation of muscle force production and mitochondrial function in AHRmKO mice with CKD following FAL surgery. Violin plots with expression levels of select differentially expressed genes involved in the top GO terms are shown in Figure 5G.
Skeletal Muscle-Specific Deletion of the AHR Preserves Paracrine Signaling Between the Muscle and Vasculature in Mice with CKD.
To investigate the potential intercellular communications that could explain the improved perfusion recovery in AHRmKO mice with CKD (Figure 2G,H), ligand-receptor interactions were calculated from snRNA sequencing data using CellChat59. Circle plots showing the overall intercellular communication occurring in AHRfl/fl and AHRmKO muscles are shown in Figure 6A. Circle sizes represent the number of cells, whereas the edge width represents the communication probability and edge color is set according to cell type. For the inferred angiogenic pathways, Vegf and Notch signaling pathways were present in AHRmKO muscles but were absent in AHRfl/fl muscles (Figure 6B). Moreover, the VEGF pathway communication was strong between the endothelial nuclei and mature Type IIb and IIx myonuclei populations of AHRmKO muscles. Circle plots for communications of each individual cell type are shown in Supplemental Figure 7. All significant signaling pathways were then ranked based upon their differences in relative overall information flow between AHRfl/fl and AHRmKO muscles (Figure 6C). Enriched angiogenic pathways in AHRmKO muscles included Vegf, Notch, and Pecam1. Enriched pathways in AHRfl/fl muscles included TGF-beta, collagen, granulin, and interestingly, angiopoietin. Analysis of the outgoing signaling patterns of secreting cells indicated that Type IIb and IIx myonuclei were the likely source of the angiogenic growth factor VEGF in AHRmKO muscles, whereas NOTCH ligands were most likely secreted from FAPs and myoblasts (Figure 6D). Analysis of the incoming communication pathways indicated that secreted VEGF was most likely interacting with VEGF receptor in endothelial cells of AHRmKO muscles (Figure 6D).
Figure 6. AHRmKO mice with CKD have preserved paracrine vasculogenic signaling between myofibers and vascular cells.
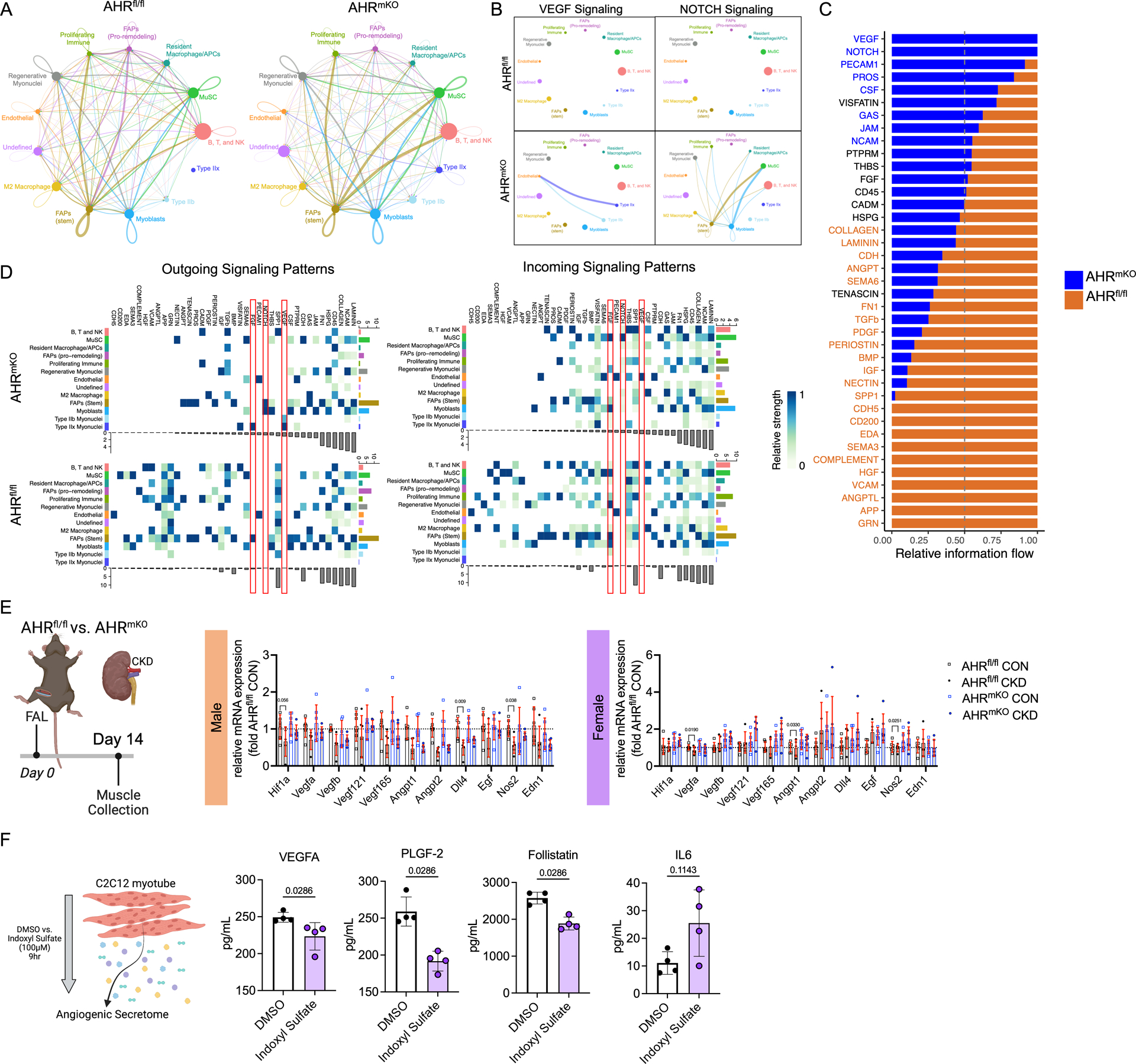
(A) Circle plots showing the overall intercellular communication occurring in AHRfl/fl and AHRmKO muscles. (B) Circle plots showing VEGF and NOTCH signaling communication in AHRfl/fl and AHRmKO muscles. (C) Ranked significant ligand-receptor communications for relative information flow between AHRfl/fl and AHRmKO muscles. (D) Enriched outgoing and incoming signaling patterns according to cell type based on signaling strength. (E) Targeted angiogenic and vascular reactivity qPCR analysis on total RNA from the ischemic hindlimb muscles (n=6/group/sex/genotype). (F) Analysis of the protein abundance of secreted angiogenic growth factors in conditioned media from C2C12 myotubes (n=4/group). Analyses in A-D were performed using CellChat. Panel E analyses involved a two-way ANOVA with Sidak’s post hoc testing for multiple comparisons. Panel F was analyzed with a Mann-Whitney test. Error bars represent the standard deviation.
Using traditional bulk qPCR in RNA isolated from ischemic muscle, the Hif1-alpha expression levels tended to be lower in AHRfl/fl male mice with CKD (P=0.056) but was not different in AHRmKO mice with CKD (Figure 6E). Considering that AHR forms a heterodimer with the aryl hydrocarbon receptor nuclear translocator (Arnt), also known as Hif1-beta, this finding suggests that chronic AHR activation may disrupt hypoxia signaling cascades through interactions of the AHR with hypoxia inducible transcription factors. Several angiogenic genes displayed a non-significant elevation of expression including Vegfa, Vegf121, Vegf165, Dll4, and Egf in male AHRmKO mice with CKD (Figure 6E). In female mice, disruptions in Hif1-alpha were not observed (Figure 6E). Female AHRfl/fl mice with CKD were found to have significantly lower expression angiogenic growth factors Vegfa and Angpt1, but these effects were abolished in AHRmKO females (Figure 6E). Similar to male mice, female AHRmKO also displayed non-significant increases in several other angiogenic genes including Vegf121 and Vegf165. Regarding vasomotor tone, both male and female AHRfl/fl mice with CKD, but not AHRmKO, displayed reduced expression of Nos2 which has a prominent role in regulating vasodilation (Figure 6E). Parallel analysis of these mRNAs in the non-ischemic limb demonstrated that AHRmKO mice with CKD has greater expression of several angiogenic transcripts in male (Vegf121, Angpt1, Angpt2, Egf) and female (Vegf121, Vegf165, Egf) compared to their AHRfl/fl correspondents with CKD (Supplemental Figure 8). Further to this, male AHRmKO mice with CKD had higher expression levels of Nos2 in their non-ischemic limb muscle compared to male AHRfl/fl mice with CKD (Supplemental Figure 8). These results suggest that angiogenic and vasodilatory gene expression was likely higher in the AHRmKO limb muscle prior to FAL surgery which likely contributed to the improved vascular recovery observed by laser Doppler flowmetry. To confirm a functional impact on paracrine signaling, we measured angiogenic growth factor levels in conditioned media taken from C2C12 myotubes treated with either vehicle of indoxyl sulfate to activate the AHR. Results from this experiment demonstrated that myotubes with AHR activation secreted significantly less VEGFa, placental growth factor 2 (PLGF-2), and follistatin, as well as a trending increase in IL6 (Figure 6F). The totality of these findings demonstrate that AHR activation in muscle diminishes paracrine vasculogenic signaling.
Expression of a Constitutively Active AHR Decreases Capillary Density and alters Angiogenic Gene Expression in Male Mice with Normal Kidney Function.
Because CKD results in the retention/accumulation of many uremic solutes that are not AHR ligands19,20 we sought to evaluate the role of AHR activation in mice with normal renal function. To accomplish this, we generated a constitutively active AHR (CAAHR) by deletion of the ligand binding domain (amino acids 277–418)60 and used adeno-associated virus (AAV) to express locally in the myofibers of the hindlimb driven by the human skeletal actin (HSA; ACTA1 gene) promoter (Figure 7A,B). In this experiment, two control groups were utilized that received either a green fluorescent protein (GFP) or the full coding sequence of the AHR which requires ligands for activation (AAV-HSA-AHR). qPCR analysis of muscle confirmed that both AAV-HSA-AHR and AAV-HSA-CAAHR treated mice had elevated expression of the Ahr, whereas Cyp1a1 mRNA levels were only increased in the AAV-HSA-CAAHR treated muscle, confirming constitutive AHR activation (Figure 7C). Interestingly, a significant interaction effect was detected for Cyp1a1 (P=0.001) indicating that females had greater expression than males treated with AAV-HSA-CAAHR. Following 12-weeks of AAV expression, FAL was performed and laser Doppler flowmetry measurements did not identify any significant alterations in perfusion recovery of the gastrocnemius muscle (Figure 7D). However, histological quantification of capillaries revealed that male mice treated with AAV-HSA-CAAHR had lower capillary density compared to GFP treated controls, whereas females treated with AAV-HSA-CAAHR had higher capillary density compared to GFP treated controls (Figure 7E). Quantification of pericyte abundance in the ischemic muscle revealed a similar density of pericytes in AAV-HSA-GFP and AAV-HSA-CAAHR treated mice (Figure 7F). A trending, but non-significant, decrease in arteriole abundance was observed in AAV-HSA-CAAHR mice (P=0.054, Figure 7G). RNA sequencing performed on the gastrocnemius muscle mice treated with either AAV-HSA-GFP or AAV-HSA-CAAHR showed that constitutive AHR activation decreased the expression of angiogenic growth factors (Vegfd, Fgf2, Egr3), genes involved in remodeling of the extracellular matrix (Mmp19, Mmp9, Timp3), vascular cell adhesion (Vcam1), and vasoreactivity (Ptgs2) (Figure 7H).
Figure 7. Expression of a Constitutively Active AHR Decreases Capillary Density and alters Angiogenic Signaling in Male Mice with Normal Kidney Function.
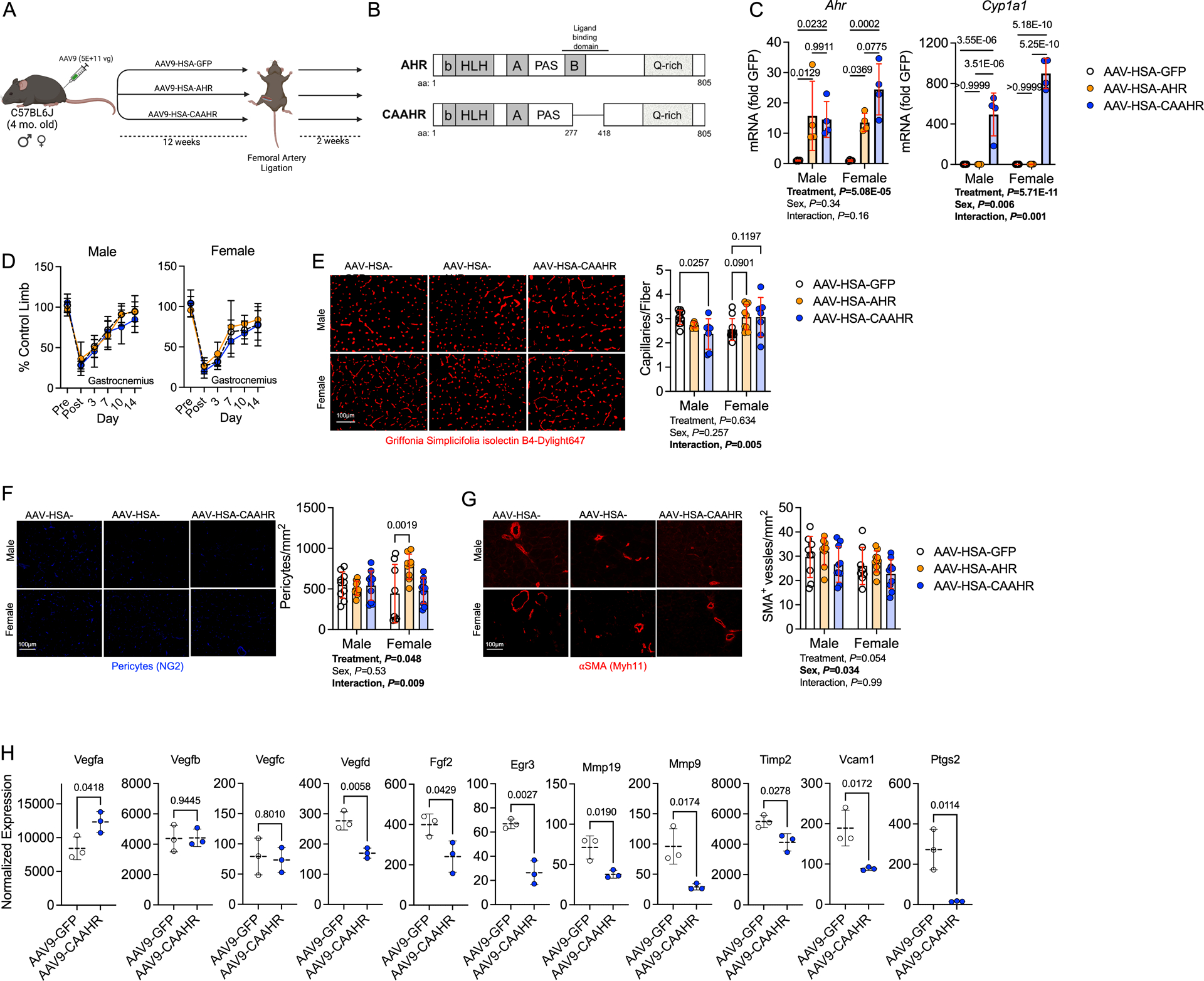
(A) Schematic of the experimental design. (B) Generation of a mutant constitutively active AHR (CAAHR). (C) Quantification of mRNA levels of Ahr and Cyp1a1 in mice (n=4/group/sex). (D) Perfusion recovery in the gastrocnemius muscle (n=10/group/sex). (E) Representative images of the tibialis anterior muscle labeled for endothelial cells and quantification of capillary density (n=10 AAV-HSA-GFP/sex, 6 male and 9 female AAV-HSA-AHR, and 7 AAV-HSA-CAAHR/sex). (F) Representative images of the tibialis anterior muscle labeled for pericytes and quantification of pericyte density (n=10 male and 8 female AAV-HSA-GFP, 10 male and 9 female AAV-HSA-AHR, and 10 AAV-HSA-CAAHR/sex). (G) Representative images of the tibialis anterior muscle labeled for arterioles and quantification of arteriole density (n=9 male and 8 female AAV-HSA-GFP, 9male and 10 female AAV-HSA-AHR, and 10 AAV-HSA-CAAHR/sex). (H) Vascular-associated gene expression in AAV-HSA-GFP and AAV-HSA-CAAHR muscle via bulk RNA sequencing analysis (n=3 males/group). Analysis of panels C-G was performed using two-way ANOVA with Sidak’s post hoc testing for multiple comparisons. Analysis in Panel H involved false-discovery rate corrected Wilcoxon test. Error bars represent the standard deviation.
Expression of a Constitutively Active AHR in Myofibers Exacerbates Ischemic Myopathy in Mice with Normal Kidney Function.
Next, we examined whether chronic AHR activation in skeletal myofibers, in the absence of renal dysfunction, was sufficient to drive myopathy in mice subjected to FAL. Consistent with the catabolic phenotype of CKD, the muscle weights of mice treated with AAV-HSA-CAAHR were significantly lower than those treated with either AAV-HSA-GFP or AAV-HSA-AHR (Figure 8A). Assessments of muscle contractile function demonstrated a further reduction in specific force levels, demonstrating that expression of the CAAHR severely diminishes muscle quality (force per area/size) (Figure 8B). Notably, the effect sizes of CAAHR expression on muscle function were larger in male mice compared to females (mean difference in peak specific force between GFP and CAAHR = 6.1 vs. 3.7 N/cm2 for males and females respectively). Histological analysis of ischemic tibialis anterior muscles uncovered pathological muscle remodeling in mice treated with CAAHR including increased numbers of centrally nucleated myofibers (males only), increased non-muscle areas (both sexes), and increased ischemic lesion areas (both sexes) (Figure 8D). Furthermore, AAV-HSA-CAAHR treated mice had a significantly smaller myofiber CSA compared to AAV-HSA-GFP controls (Figure 8E).
Figure 8. Expression of a Constitutively Active AHR in Myofibers Exacerbates Ischemic Myopathy in Mice with Normal Kidney Function.
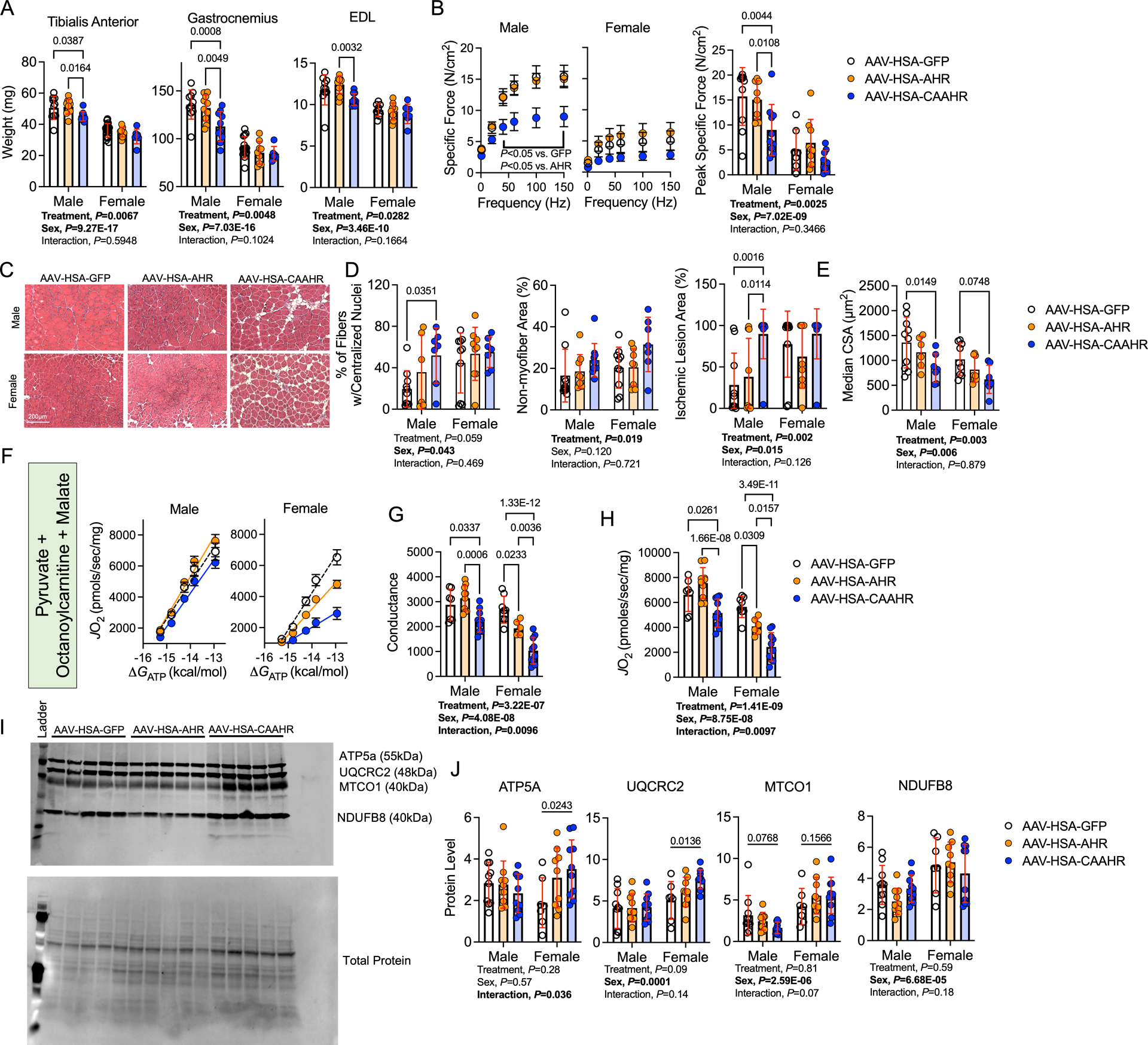
(A) Quantification of muscle weights from the ischemic limb (n=10 male and 14 female AAV-HSA-GFP, 10 male and 9 female AAV-HSA-AHR, and 10 male and 7 female AAV-HSA-CAAHR). (B) Force frequency curves in male and female mice. Specific force was calculated as the absolute force normalized to the cross-sectional area of the muscle (n=10 male and 7 female AAV-HSA-GFP, 10 male and 9 female AAV-HSA-AHR, and 10 male and 9 female AAV-HSA-CAAHR). (C) Representative hematoxylin & eosin staining of the ischemic tibialis anterior muscles. (D) Quantification of the histopathology of the ischemic tibialis anterior muscles (n=10 AAV-HSA-GFP/sex, 9 AAV-HSA-AHR/sex, and 10 AAV-HSA-CAAHR/sex). (E) Quantification of the mean myofiber CSA in the ischemic tibialis anterior muscles (n=10 male and 9 female AAV-HSA-GFP, 6 male and 8 female AAV-HSA-AHR, and 7 AAV-HSA-CAAHR/sex). (F) Relationship between JO2 and ΔGATP when mitochondria were fueled with pyruvate, octanoylcarnitine, and malate (n=7 male and 8 female AAV-HSA-GFP, 10 male and 6 female AAV-HSA-AHR, and 10 AAV-HSA-CAAHR/sex). (G) Quantification of the conductance (slope of JO2 and ΔGATP relationship) in the ischemic muscle mitochondria (n=7 male and 8 female AAV-HSA-GFP, 10 male and 6 female AAV-HSA-AHR, and 10 AAV-HSA-CAAHR/sex). (H) Maximal uncoupled respiration following the addition of a mitochondrial protonophore/uncoupler (FCCP) (n=7 male and 8 female AAV-HSA-GFP, 10 male and 6 female AAV-HSA-AHR, and 10 AAV-HSA-CAAHR/sex). (I) Western blotting membrane of mitochondrial electron transport system proteins. (J) Quantification of mitochondrial protein abundance in mice (n=10 male and 7 female AAV-HSA-GFP, 10 male and 9 female AAV-HSA-AHR, and 10 AAV-HSA-CAAHR/sex). Analysis performed using two-way ANOVA with Sidak’s post hoc testing for multiple comparisons. Error bars represent the standard deviation.
Expression of a Constitutively Active AHR in Myofibers Decreases OXPHOS Function in Ischemic Muscle of Mice with Normal Kidney Function.
To explore the impact of CAAHR expression on ischemic muscle mitochondrial OXPHOS function, we isolated mitochondria from the ischemic gastrocnemius muscle, energized them with a mixture of carbohydrates (pyruvate) and a medium chain fatty acid (octanoylcarnitine), and assessed the respiration rate across increasing levels of energy demand (Figure 8F). Quantification of OXPHOS conductance unveiled a significant defect in muscle mitochondrial OXPHOS in mice treated with AAV-HSA-CAAHR when compared to either GFP- or AHR-treated controls (Figure 8G). Following the addition of a mitochondrial uncoupler (the protonophore, FCCP), the maximal respiratory capacity (Figure 8H) was also significantly lower in AAV-HSA-CAAHR treated ischemic muscle from mice with normal function. These results are consistent with the preservation of muscle mitochondrial OXPHOS function in AHRmKO mice with CKD/PAD (Figure 4) and provide compelling evidence that chronic AHR activation alters skeletal muscle mitochondrial function.
Next, immunoblotting for electron transport system proteins was performed to assess if/how AAV-HSA-CAAHR treatment impacted mitochondrial content/density (Figure 8I, all blot images shown in Supplemental Figure 9). In this regard, no significant main effects for treatment were observed (Figure 8J). However, significant main effects of sex were observed for UQCRC2, MTCO1, and NDUFB8 demonstrating that female mice had increased mitochondrial protein abundance, a result that likely reflects compensatory mitochondrial biogenesis in response to poor OXPHOS function seen in Figure 8F–J. In some cases (ATP5A and UQCRC2), females treated with AAV-HSA-CAAHR had significant higher protein levels compared to AAV-HSA-GFP treated mice. Considering that OXPHOS function, determined using respirometric analyses of isolated mitochondria, was impaired in AAV-HSA-CAAHR treated mice, the increase protein abundance of the electron transport system proteins ATP5A and UQCRC2 may be indicative a compensatory response to the enzymatic impairments observed in these mitochondria. RNA sequencing performed on total RNA isolated from the gastrocnemius muscle of AAV-HSA-CAAHR treated mice also demonstrated altered mitochondrial gene expression when compared to AAV-HSA-GFP treated mice (Supplemental Figure 10).
DISCUSSION
In this study, we identified significant AHR activation in the limb muscle of mice and PAD patients with CKD and PAD and confirmed that tryptophan-derived uremic metabolites activate the AHR pathway in mouse and human muscle cells in vitro. Skeletal-muscle specific ablation of the AHR in mice with CKD improved limb muscle perfusion recovery, increased arteriogenesis and paracrine vasculogenic signaling, attenuated muscle atrophy, and improved muscle contractile function and mitochondrial OXPHOS in an experimental model of PAD. Furthermore, viral-mediated skeletal muscle-specific expression of a constitutively active AHR in mice with normal kidney function exacerbated the ischemic myopathy evidenced by smaller muscle masses, reduced contractile function, histopathology, and lower mitochondrial OXPHOS function and respiratory capacity. Importantly, deletion of the AHR in skeletal muscle had no effect on muscle health or function in mice subjected to sham surgery or those without CKD where ligand abundance is low. Coupled with the results of a recent study21, the findings herein suggest that future studies should explore the therapeutic potential of AHR inhibition as an adjuvant treatment option for PAD patients with CKD.
The pathogenesis of PAD unquestionably stems from the development of atherosclerosis in the peripheral vasculature resulting in compromised blood flow to the lower limbs. However, other factors beyond atherosclerosis have emerged as important contributors to limb symptoms and adverse limb events including microvascular disease61,62, endothelial dysfunction63, and skeletal muscle dysfunction64,65. Notably, CKD is recognized to be a strong, independent risk factor for PAD66–68 which has also been demonstrated to negatively impact both the vasculature69,70 and skeletal muscle independent of PAD71. The accumulation of several uremic metabolites including indoxyl sulfate, indole-3-acetic acid, L-kynurenine, and kynurenic acid (among others) have been directly linked to vascular and skeletal muscle pathophysiology in CKD23,24,32,70,72,73. Concerning for PAD patients, hazard ratios for the incidence of adverse limb events (after adjustment for age, sex, and eGFR) demonstrated a strong link to the levels of these specific uremic toxins21. Interestingly, each these tryptophan derived uremic toxins are established ligands of the AHR, suggesting that this ligand-activated cytosolic receptor could be a common link between uremic toxicity and the muscle and vascular pathology in CKD. Mechanistically, AHR activation has been shown to promote endothelial cell senescence74, promote atherosclerosis, inflammation36, and thrombosis73, as well as impair angiogenesis21,34,48. Importantly, AHR global knockout mice display improved limb perfusion recovery following treatment with the AHR ligand benzo[a]pyrene following hindlimb ischemia48,75. An intriguing observation from the current study is that deletion of the AHR in skeletal muscle cells of mice with CKD and PAD improved limb perfusion recovery within the gastrocnemius muscle (Figure 2G). Improved perfusion recovery corresponded with increased arteriole density in both male and female AHRmKO mice with CKD (Figure 2K), increased pericyte abundance in male AHRmKO mice, but normal capillary density following FAL. Gene expression analysis also revealed increase expression of Nos2, a vasodilatory gene, in AHRmKO mice with CKD. Considering that AHR deletion occurred only in myofibers, these findings suggest that AHR activation alters paracrine signaling events between the skeletal muscle and vasculature within the ischemic limb. To this end, interrogation of intercellular communication processes via snRNA sequencing demonstrated that AHRmKO mice with CKD had greater angiogenic signaling (Vegf, Notch, Fgf) between myonuclei and endothelial cells (Figure 6). qPCR confirmed higher expression levels of several vasculogenic genes in both the ischemic and non-ischemic limbs of AHRmKO mice with CKD. A functional deficit in angiogenic growth factor secretion was further confirmed in C2C12 myotubes, firmly establishing an important role of AHR activation in disrupting muscle-vascular communication. In contrast to CKD mice, AAV-HSA-CAAHR treated mice displayed relatively normal gastrocnemius muscle perfusion recovery (Figure 7D), as well as normal capillary and pericyte levels (Figure 7E,F). Contributing factors to this discrepancy likely stem from the heterogenous infection of AAV to the hindlimb muscles and the superficial penetration depth of laser Doppler which is incapable of reaching deeper portions of the gastrocnemius muscle. Moreover, laser Doppler flowmetry was performed using a small probe rather than a scanning imager which would have sampled more tissue area. However, it is worth highlighting that angiogenic signaling pathways and arteriole density were improved in AHRmKO with CKD and decreased in AAV-HSA-CAAHR treated mice. Nonetheless, of great concern for the PAD patient, the findings in the current study and others21 indicate that CKD negatively impacts both skeletal muscle and vasculature health, two critical components regulating walking performance and limb outcomes.
Skeletal muscle mitochondrial alterations have been documented in both PAD patients39,40,50–55,76,77 and CKD patients13,78–81, and thus could be a site for the coalescence of these conditions. In the current study, deletion of the AHR in skeletal muscle significantly improved mitochondrial OXPHOS in the ischemic muscle of CKD mice (Figure 4). However, this effect was primarily driven by the enhanced OXPHOS function in male mice rather than female mice, especially when mitochondria were fueled by carbohydrates. Conversely, AAV-driven AHR activation impaired mitochondrial OXPHOS in the ischemic limb of mice from both sexes when mitochondria were energized by a mixture of carbohydrates and fatty acids (Figure 8), suggesting that AHR mediates mitochondrial metabolism in a substrate-specific manner. Previous studies have identified sex-dependent transcriptional changes in the liver following treatment with the AHR ligand, TCDD82. While further work is needed to identify the specific mechanisms underlying AHR-dependent mitochondrial dysfunction in CKD/PAD, similar observations have been made in other cell/tissue types. For example, AHR activation has been suggested to decrease mitochondrial membrane potential, impair respiration, and/or increase ROS in spermatozoa83, hepatocytes84,85, pancreas86, cardiomyocytes87, and the brain88. Similarly, treatment of cultured myotubes with tobacco smoke condensate (which contains numerous AHR ligands) or expression of a CAAHR was reported to impair mitochondrial respiratory capacity38.
The AHR is most known for its role as a ligand activated transcription factor involved in xenobiotic toxicity. However, several reports have linked AHR activation to ischemic tissue damage. For example, cerebral ischemia (i.e., stroke) induces AHR activation via L-kynurenine and subsequent deletion of the AHR attenuated ischemic brain damage in mice89. This finding has been replicated using an AHR inhibitor in rats subjected to cerebral ischemia/reperfusion90. In cardiomyocytes, activation of the AHR by kynurenine has been shown to drive ROS production and promote apoptosis which could be prevented by AHR inhibition91. This study also demonstrated higher levels of cardiomyocyte apoptosis and larger infarct size following myocardial infarction in mice treated with exogenous kynurenine91. Paradoxically, it is worth noting that kynurenic acid, a known AHR ligand, has been reported to have cardioprotective effects in the setting of ischemia/reperfusion, although this cardioprotection does not appear to require the AHR92. Similarly, AHR activation in T-cells has also been reported to exert cardioprotective effects in mice with myocardial infarction93. In the current study, analysis of cell populations via snRNA sequencing five days following the induction of limb ischemia demonstrated that deletion of the AHR in skeletal muscle preserved mature myonuclei (Type IIb and IIx) (Figure 5). Given the acute phase of ischemic injury at the time of muscle collection, this finding may be consistent with the prevention of cell death pathways that has been reported in ischemic cardiomyocytes treated with AHR inhibitors91.
An unexpected finding of this study was the deletion of the AHR in skeletal muscle improved muscle size, muscle strength, and mitochondrial function in male, but not female mice with CKD. However, it is interesting to note that female AHRmKO mice with CKD displayed improved perfusion recovery compared to their AHRfl/fl counterparts with CKD (Figure 2), suggesting that the pathological effects of AHR activation in muscle may be disentangled from the limb hemodynamics. This assertion is support by previous work demonstrating that muscle-targeted therapies in mice subjected to FAL can improve muscle contractile function without corresponding improvements in limb perfusion recovery or angiogenesis94, as well as vascular-centric therapies improving angiogenesis without enhancing muscle contractile function46. While the exact mechanisms underlying the sex differences are unknown, several previous studies have reported sex-differences in the AHR reactivity in non-muscle tissues. For example, the hepatic transcriptional response to treatment with TCDD was muted in female mice when compared to males82. In rodents, CKD has been shown to alter AHR abundance in the kidney in a sex-dependent manner95. Additionally, the severity of gastric tumors induced by expression of a constitutively active AHR was worse in male mice compared to female mice60. Intriguingly, the AHR/ARNT heterodimer has been reported to be physically associated with the estrogen receptor and may regulate its transcriptional activity96, providing a potential explanation for the sex-dependent effects observed in this study. The AHR has also been implicated to promote proteasomal degradation of the estrogen receptor via the cullin 4B ubiquitin ligase97. Additional mechanistic experimentation is required to fully establish the sex-dependent AHR mechanisms in the context of CKD-PAD pathobiology.
There are some limitations in this study that are worthy of discussion. First, this study employed younger mice despite the well-known association between PAD prevalence and increasing age. However, it is important to note that mice were enrolled into studies between the ages of 4–6 months to ensure the skeletal muscle and vasculature were fully matured. Further to this, PAD/CLTI patients with CKD often suffer from numerous other co-morbid conditions such as hypertension, diabetes, and hyperlipidemia that may co-exist with CKD but were not present in this study. These comorbidities coupled with the genetic and environmental risk factors (i.e., smoking, poor diet, low physical activity) that are typically present are all contributing factors to patient outcomes. Additionally, PAD/CLTI is a progressive disease where atherosclerosis develops slowly over time and is hastened by the presence of CKD. In contrast, a rapid and severe decrease in limb blood was induced by FAL in the experiments herein. As such, the conditions of chronic ischemia are not well modeled by the FAL surgery employed in this study. Nevertheless, a benefit of the FAL surgery stems from the ability to use clear anatomical landmarks for surgical ligatures which results in a consistent post-operative outcome. Finally, it was not possible to perform temporal analyses within this study design which could have allowed exploration of the dynamic changes in arteriogenesis and angiogenesis in the ischemic hindlimb and the impact AHR activation had on these processes.
CONCLUSIONS.
These data demonstrate that chronic Ahr activation, such as that in chronic kidney disease, is an important regulator of murine ischemic myopathy. Ahr activation, which has been previously observed in blood from PAD patients with CKD and linked to adverse limb events21, was also observed in skeletal muscle specimens from PAD patients with CKD. Ablation of the AHR in skeletal muscle of mice with CKD significantly improved limb perfusion recovery and preserved paracrine vasculogenic signaling, improved muscle mass and contractile function, as well as enhanced mitochondrial energetics in an experimental model of PAD. Overall, these findings are an important step in elucidating the complex pathobiology of PAD in the context of renal insufficiency and provide support for testing of interventions that diminish Ahr signaling in these conditions.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What is known?
Peripheral artery disease (PAD) patients with chronic kidney disease (CKD) have worsen myopathic symptoms and increased risk for adverse limb events and mortality compared with patients with normal kidney function.
The accumulation of tryptophan-derived uremic solutes has been associated with adverse limb events in PAD patients with CKD.
What new information does this article contribute?
Analysis of limb muscle from both mice and PAD patients uncovered that the accumulation of tryptophan-derived uremic solutes in CKD causes activation of the aryl hydrocarbon receptor (AHR), a ligand-activated transcription factor.
Deletion of the AHR in skeletal muscle of mice with CKD improved vasculogenesis, muscle contractile function, and mitochondrial energetics following femoral artery ligation.
In mice with normal kidney function, muscle-specific expression of an active AHR mutant worsened the ischemic myopathy caused by femoral artery ligation.
There is clear evidence that PAD patients with CKD have significantly higher risk of major adverse limb events and death compared to PAD patients with normal renal function. Thus, it is important to understand the biological mechanisms exacerbating PAD outcomes in CKD to improve medical management in these patients. Using skeletal muscle biopsies from PAD patients and muscle cell culture systems, we discovered that the accumulation of tryptophan-derived uremic solutes results in activation of the AHR. Deletion of the AHR specifically in skeletal muscle of mice with CKD led to improved perfusion recovery, enhanced arteriogenesis, preservation of paracrine vasculogenic signaling from skeletal myocytes, as well as improved muscle contractile and mitochondrial function following femoral artery ligation. Conversely, expression of a mutant AHR with constitutive activation in mice with normal kidney function worsened ischemic vasculogenesis, muscle strength, and mitochondrial function in the ischemic limb. Unexpectedly, some sex-dependent effects of AHR activation were observed with male mice have greater improvements in limb outcomes. In summary, this study establishes AHR activation in muscle as a critical regulator of ischemic pathology in the CKD condition.
Sources of Funding:
This study was supported by National Institutes of Health (NIH) grant R01-HL149704 (T.E.R.). S.T.S was supported by NIH grant R01HL148597. S.A.B was supported by NIH grant R01DK119274. L.F.F. was supported by NIH grants R01HL130318 and R21AG873239. K.K. was support by the American Heart Association grant POST903198. T.T. was support by NIH grant F31-DK128920.
ABBREVIATIONS
- AAV
adeno-associated virus
- AHR
aryl hydrocarbon receptor
- AHRmKO
muscle-specific AHR knockout mouse
- CAAHR
constitutively active AHR
- CKD
chronic kidney disease
- FAL
femoral artery ligation
- GFP
green fluorescent protein
- GFR
glomerular filtration rate
- MuSC
muscle stem cell
- OXPHOS
oxidative phosphorylation
- PAD
peripheral arterial disease
- ROS
reactive oxygen species
Footnotes
Disclosures: None.
Supplemental Material:
References
- 1.Ostchega Y, Paulose-Ram R, Dillon CF, Gu Q, Hughes JP. Prevalence of peripheral arterial disease and risk factors in persons aged 60 and older: data from the National Health and Nutrition Examination Survey 1999–2004. J Am Geriatr Soc. 2007;55:583–589. doi: 10.1111/j.1532-5415.2007.01123.x [DOI] [PubMed] [Google Scholar]
- 2.Fowkes FG, Rudan D, Rudan I, Aboyans V, Denenberg JO, McDermott MM, Norman PE, Sampson UK, Williams LJ, Mensah GA, et al. Comparison of global estimates of prevalence and risk factors for peripheral artery disease in 2000 and 2010: a systematic review and analysis. Lancet. 2013;382:1329–1340. doi: 10.1016/S0140-6736(13)61249-0 [DOI] [PubMed] [Google Scholar]
- 3.Heideman PP, Rajebi MR, McKusick MA, Bjarnason H, Oderich GS, Friese JL, Fleming MD, Stockland AH, Harmsen WS, Mandrekar J, et al. Impact of Chronic Kidney Disease on Clinical Outcomes of Endovascular Treatment for Femoropopliteal Arterial Disease. J Vasc Interv Radiol. 2016;27:1204–1214. doi: 10.1016/j.jvir.2016.04.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaminski MR, Raspovic A, McMahon LP, Lambert KA, Erbas B, Mount PF, Kerr PG, Landorf KB. Factors associated with foot ulceration and amputation in adults on dialysis: a cross-sectional observational study. BMC Nephrol. 2017;18:293. doi: 10.1186/s12882-017-0711-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O’Hare AM, Sidawy AN, Feinglass J, Merine KM, Daley J, Khuri S, Henderson WG, Johansen KL. Influence of renal insufficiency on limb loss and mortality after initial lower extremity surgical revascularization. J Vasc Surg. 2004;39:709–716. doi: 10.1016/j.jvs.2003.11.038 [DOI] [PubMed] [Google Scholar]
- 6.Lacroix P, Aboyans V, Desormais I, Kowalsky T, Cambou JP, Constans J, Bura Riviere A, investigators C. Chronic kidney disease and the short-term risk of mortality and amputation in patients hospitalized for peripheral artery disease. J Vasc Surg. 2013;58:966–971. doi: 10.1016/j.jvs.2013.04.007 [DOI] [PubMed] [Google Scholar]
- 7.O’Hare AM, Bertenthal D, Shlipak MG, Sen S, Chren MM. Impact of renal insufficiency on mortality in advanced lower extremity peripheral arterial disease. Journal of the American Society of Nephrology : JASN. 2005;16:514–519. doi: 10.1681/ASN.2004050409 [DOI] [PubMed] [Google Scholar]
- 8.Pasqualini L, Schillaci G, Pirro M, Vaudo G, Siepi D, Innocente S, Ciuffetti G, Mannarino E. Renal dysfunction predicts long-term mortality in patients with lower extremity arterial disease. J Intern Med. 2007;262:668–677. doi: 10.1111/j.1365-2796.2007.01863.x [DOI] [PubMed] [Google Scholar]
- 9.Liew YP, Bartholomew JR, Demirjian S, Michaels J, Schreiber MJ Jr. Combined effect of chronic kidney disease and peripheral arterial disease on all-cause mortality in a high-risk population. Clin J Am Soc Nephrol. 2008;3:1084–1089. doi: 10.2215/CJN.04411007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Patel VI, Mukhopadhyay S, Guest JM, Conrad MF, Watkins MT, Kwolek CJ, LaMuraglia GM, Cambria RP. Impact of severe chronic kidney disease on outcomes of infrainguinal peripheral arterial intervention. J Vasc Surg. 2014;59:368–375. doi: 10.1016/j.jvs.2013.09.006 [DOI] [PubMed] [Google Scholar]
- 11.Avin KG, Chen NX, Organ JM, Zarse C, O’Neill K, Conway RG, Konrad RJ, Bacallao RL, Allen MR, Moe SM. Skeletal Muscle Regeneration and Oxidative Stress Are Altered in Chronic Kidney Disease. PLoS One. 2016;11:e0159411. doi: 10.1371/journal.pone.0159411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gamboa JL, Billings FTt, Bojanowski MT, Gilliam LA, Yu C, Roshanravan B, Roberts LJ 2nd, Himmelfarb J, Ikizler TA, Brown NJ. Mitochondrial dysfunction and oxidative stress in patients with chronic kidney disease. Physiological reports. 2016;4. doi: 10.14814/phy2.12780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roshanravan B, Kestenbaum B, Gamboa J, Jubrias SA, Ayers E, Curtin L, Himmelfarb J, de Boer IH, Conley KE. CKD and Muscle Mitochondrial Energetics. Am J Kidney Dis. 2016;68:658–659. doi: 10.1053/j.ajkd.2016.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang XH, Mitch WE. +. Nat Rev Nephrol. 2014;10:504–516. doi: 10.1038/nrneph.2014.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang L, Wang XH, Wang H, Du J, Mitch WE. Satellite cell dysfunction and impaired IGF-1 signaling cause CKD-induced muscle atrophy. Journal of the American Society of Nephrology : JASN. 2010;21:419–427. doi: 10.1681/ASN.2009060571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hung SC, Kuo KL, Huang HL, Lin CC, Tsai TH, Wang CH, Chen JW, Lin SJ, Huang PH, Tarng DC. Indoxyl sulfate suppresses endothelial progenitor cell-mediated neovascularization. Kidney Int. 2016;89:574–585. doi: 10.1016/j.kint.2015.11.020 [DOI] [PubMed] [Google Scholar]
- 17.Jacobi J, Porst M, Cordasic N, Namer B, Schmieder RE, Eckardt KU, Hilgers KF. Subtotal nephrectomy impairs ischemia-induced angiogenesis and hindlimb re-perfusion in rats. Kidney Int. 2006;69:2013–2021. doi: 10.1038/sj.ki.5000448 [DOI] [PubMed] [Google Scholar]
- 18.Schellinger IN, Cordasic N, Panesar J, Buchholz B, Jacobi J, Hartner A, Klanke B, Jakubiczka-Smorag J, Burzlaff N, Heinze E, et al. Hypoxia inducible factor stabilization improves defective ischemia-induced angiogenesis in a rodent model of chronic kidney disease. Kidney Int. 2017;91:616–627. doi: 10.1016/j.kint.2016.09.028 [DOI] [PubMed] [Google Scholar]
- 19.Lekawanvijit S, Kompa AR, Krum H. Protein-bound uremic toxins: a long overlooked culprit in cardiorenal syndrome. American journal of physiology Renal physiology. 2016;311:F52–62. doi: 10.1152/ajprenal.00348.2015 [DOI] [PubMed] [Google Scholar]
- 20.Piroddi M, Bartolini D, Ciffolilli S, Galli F. Nondialyzable uremic toxins. Blood Purif. 2013;35 Suppl 2:30–41. doi: 10.1159/000350846 [DOI] [PubMed] [Google Scholar]
- 21.Arinze NV, Yin WQ, Lotfollahzadeh S, Napoleon MA, Richards S, Walker JA, Belghasem M, Ravid JD, Kamel MH, Whelan SA, et al. Tryptophan metabolites suppress the Wnt pathway and promote adverse limb events in chronic kidney disease. J Clin Invest. 2022;132. doi: ARTN e142260 10.1172/JCI142260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Enoki Y, Watanabe H, Arake R, Sugimoto R, Imafuku T, Tominaga Y, Ishima Y, Kotani S, Nakajima M, Tanaka M, et al. Indoxyl sulfate potentiates skeletal muscle atrophy by inducing the oxidative stress-mediated expression of myostatin and atrogin-1. Sci Rep. 2016;6:32084. doi: 10.1038/srep32084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sato E, Mori T, Mishima E, Suzuki A, Sugawara S, Kurasawa N, Saigusa D, Miura D, Morikawa-Ichinose T, Saito R, et al. Metabolic alterations by indoxyl sulfate in skeletal muscle induce uremic sarcopenia in chronic kidney disease. Sci Rep. 2016;6:36618. doi: 10.1038/srep36618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thome T, Salyers ZR, Kumar RA, Hahn D, Berru FN, Ferreira LF, Scali ST, Ryan TE. Uremic metabolites impair skeletal muscle mitochondrial energetics through disruption of the electron transport system and matrix dehydrogenase activity. Am J Physiol-Cell Ph. 2019;317:C701–C713. doi: 10.1152/ajpcell.00098.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thome T, Kumar RA, Burke SK, Khattri RB, Salyers ZR, Kelley RC, Coleman MD, Christou DD, Hepple RT, Scali ST, et al. Impaired muscle mitochondrial energetics is associated with uremic metabolite accumulation in chronic kidney disease. Jci Insight. 2021;6. doi: ARTN e139826 10.1172/jci.insight.139826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Singh N, Liu K, Tian L, Criqui MH, Guralnik JM, Ferrucci L, Liao Y, McDermott MM. Leg strength predicts mortality in men but not in women with peripheral arterial disease. J Vasc Surg. 2010;52:624–631. doi: 10.1016/j.jvs.2010.03.066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McDermott MM, Liu K, Ferrucci L, Tian L, Guralnik JM, Liao Y, Criqui MH. Decline in functional performance predicts later increased mobility loss and mortality in peripheral arterial disease. J Am Coll Cardiol. 2011;57:962–970. doi: 10.1016/j.jacc.2010.09.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McDermott MM, Liu K, Tian L, Guralnik JM, Criqui MH, Liao Y, Ferrucci L. Calf muscle characteristics, strength measures, and mortality in peripheral arterial disease: a longitudinal study. J Am Coll Cardiol. 2012;59:1159–1167. doi: 10.1016/j.jacc.2011.12.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jain A, Liu K, Ferrucci L, Criqui MH, Tian L, Guralnik JM, Tao H, McDermott MM. Declining walking impairment questionnaire scores are associated with subsequent increased mortality in peripheral artery disease. J Am Coll Cardiol. 2013;61:1820–1829. doi: 10.1016/j.jacc.2013.01.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leeper NJ, Myers J, Zhou M, Nead KT, Syed A, Kojima Y, Caceres RD, Cooke JP. Exercise capacity is the strongest predictor of mortality in patients with peripheral arterial disease. J Vasc Surg. 2013;57:728–733. doi: 10.1016/j.jvs.2012.07.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dou L, Poitevin S, Sallee M, Addi T, Gondouin B, McKay N, Denison MS, Jourde-Chiche N, Duval-Sabatier A, Cerini C, et al. Aryl hydrocarbon receptor is activated in patients and mice with chronic kidney disease. Kidney Int. 2018;93:986–999. doi: 10.1016/j.kint.2017.11.010 [DOI] [PubMed] [Google Scholar]
- 32.Enoki Y, Watanabe H, Arake R, Fujimura R, Ishiodori K, Imafuku T, Nishida K, Sugimoto R, Nagao S, Miyamura S, et al. Potential therapeutic interventions for chronic kidney disease-associated sarcopenia via indoxyl sulfate-induced mitochondrial dysfunction. J Cachexia Sarcopenia Muscle. 2017;8:735–747. doi: 10.1002/jcsm.12202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ichihara S, Yamada Y, Ichihara G, Nakajima T, Li P, Kondo T, Gonzalez FJ, Murohara T. A role for the aryl hydrocarbon receptor in regulation of ischemia-induced angiogenesis. Arteriosclerosis, thrombosis, and vascular biology. 2007;27:1297–1304. doi: 10.1161/ATVBAHA.106.138701 [DOI] [PubMed] [Google Scholar]
- 34.Salyers ZR, Coleman M, Balestrieri NP, Ryan TE. Indoxyl sulfate impairs angiogenesis via chronic aryl hydrocarbon receptor activation. Am J Physiol-Cell Ph. 2021;320:C240–C249. doi: 10.1152/ajpcell.00262.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Iwano S, Asanuma F, Nukaya M, Saito T, Kamataki T. CYP1A1-mediated mechanism for atherosclerosis induced by polycyclic aromatic hydrocarbons. Biochem Biophys Res Commun. 2005;337:708–712. doi: 10.1016/j.bbrc.2005.09.109 [DOI] [PubMed] [Google Scholar]
- 36.Wu D, Nishimura N, Kuo V, Fiehn O, Shahbaz S, Van Winkle L, Matsumura F, Vogel CF. Activation of aryl hydrocarbon receptor induces vascular inflammation and promotes atherosclerosis in apolipoprotein E−/− mice. Arteriosclerosis, thrombosis, and vascular biology. 2011;31:1260–1267. doi: 10.1161/ATVBAHA.110.220202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reynolds LM, Wan M, Ding J, Taylor JR, Lohman K, Su D, Bennett BD, Porter DK, Gimple R, Pittman GS, et al. DNA Methylation of the Aryl Hydrocarbon Receptor Repressor Associations With Cigarette Smoking and Subclinical Atherosclerosis. Circulation Cardiovascular genetics. 2015;8:707–716. doi: 10.1161/CIRCGENETICS.115.001097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thome T, Miguez K, Willms A, Burke SK, Chandran V, de Souza AR, Fitzgerald LF, Baglole C, Anagnostou ME, Bourbeau J, et al. Chronic aryl hydrocarbon receptor activity phenocopies smoking-induced skeletal muscle impairment. J Cachexia Sarcopeni. 2021. doi: 10.1002/jcsm.12826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ryan TE, Yamaguchi DJ, Schmidt CA, Zeczycki TN, Shaikh SR, Brophy P, Green TD, Tarpey MD, Karnekar R, Goldberg EJ, et al. Extensive skeletal muscle cell mitochondriopathy distinguishes critical limb ischemia patients from claudicants. JCI Insight. 2018;3. doi: 10.1172/jci.insight.123235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ryan TE, Kim K, Scali ST, Berceli SA, Thome T, Salyers ZR, O’Malley KA, Green TD, Karnekar R, Fisher-Wellman KH, et al. Interventional- and amputation-stage muscle proteomes in the chronically threatened ischemic limb. Clinical and Translational Medicine. 2022;12:e658. doi: 10.1002/ctm2.658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Berru FN, Gray SE, Thome T, Kumar RA, Salyers ZR, Coleman M, Dennis L, O’Malley K, Ferreira LF, Berceli SA, et al. Chronic kidney disease exacerbates ischemic limb myopathy in mice via altered mitochondrial energetics. Sci Rep. 2019;9:15547. doi: 10.1038/s41598-019-52107-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim K, Anderson EM, Thome T, Lu GY, Salyers ZR, Cort TA, O’Malley KA, Scali ST, Ryan TE. Skeletal myopathy in CKD: a comparison of adenine-induced nephropathy and 5/6 nephrectomy models in mice. Am J Physiol-Renal. 2021;321:F106–F119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qi Z, Whitt I, Mehta A, Jin J, Zhao M, Harris RC, Fogo AB, Breyer MD. Serial determination of glomerular filtration rate in conscious mice using FITC-inulin clearance. American journal of physiology Renal physiology. 2004;286:F590–596. doi: 10.1152/ajprenal.00324.2003 [DOI] [PubMed] [Google Scholar]
- 44.Rieg T A High-throughput method for measurement of glomerular filtration rate in conscious mice. J Vis Exp. 2013:e50330. doi: 10.3791/50330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thome T, Coleman MD, Ryan TE. Mitochondrial Bioenergetic and Proteomic Phenotyping Reveals Organ-Specific Consequences of Chronic Kidney Disease in Mice. Cells. 2021;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Salyers ZR, Coleman M, Le D, Ryan TE. AAV-mediated expression of PFKFB3 in myofibers, but not endothelial cells, improves ischemic muscle function in mice with critical limb ischemia. Am J Physiol Heart Circ Physiol. 2022;323:H424–H436. doi: 10.1152/ajpheart.00121.2022 [DOI] [PubMed] [Google Scholar]
- 47.Salyers ZR, Mariani V, Balestrieri N, Kumar RA, Vugman NA, Thome T, Villani KR, Berceli SA, Scali ST, Vasilakos G, et al. S100A8 and S100A9 are elevated in chronically threatened ischemic limb muscle and induce ischemic mitochondrial pathology in mice. JVS Vasc Sci. 2022;3:232–245. doi: 10.1016/j.jvssci.2022.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ichihara S, Yamada Y, Gonzalez FJ, Nakajima T, Murohara T, Ichihara G. Inhibition of ischemia-induced angiogenesis by benzo[a]pyrene in a manner dependent on the aryl hydrocarbon receptor. Biochem Biophys Res Commun. 2009;381:44–49. doi: 10.1016/j.bbrc.2009.01.187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maltais F, Decramer M, Casaburi R, Barreiro E, Burelle Y, Debigare R, Dekhuijzen PN, Franssen F, Gayan-Ramirez G, Gea J, et al. An official American Thoracic Society/European Respiratory Society statement: update on limb muscle dysfunction in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2014;189:e15–62. doi: 10.1164/rccm.201402-0373ST [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bhat HK, Hiatt WR, Hoppel CL, Brass EP. Skeletal muscle mitochondrial DNA injury in patients with unilateral peripheral arterial disease. Circulation. 1999;99:807–812. [DOI] [PubMed] [Google Scholar]
- 51.Pipinos II, Sharov VG, Shepard AD, Anagnostopoulos PV, Katsamouris A, Todor A, Filis KA, Sabbah HN. Abnormal mitochondrial respiration in skeletal muscle in patients with peripheral arterial disease. J Vasc Surg. 2003;38:827–832. [DOI] [PubMed] [Google Scholar]
- 52.Pipinos II, Judge AR, Zhu Z, Selsby JT, Swanson SA, Johanning JM, Baxter BT, Lynch TG, Dodd SL. Mitochondrial defects and oxidative damage in patients with peripheral arterial disease. Free Radic Biol Med. 2006;41:262–269. doi: 10.1016/j.freeradbiomed.2006.04.003 [DOI] [PubMed] [Google Scholar]
- 53.Gonzalez-Freire M, Moore AZ, Peterson CA, Kosmac K, McDermott MM, Sufit RL, Guralnik JM, Polonsky T, Tian L, Kibbe MR, et al. Associations of Peripheral Artery Disease With Calf Skeletal Muscle Mitochondrial DNA Heteroplasmy. Journal of the American Heart Association. 2020;9:e015197. doi: 10.1161/JAHA.119.015197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Groennebaek T, Billeskov TB, Schytz CT, Jespersen NR, Botker HE, Olsen RKJ, Eldrup N, Nielsen J, Farup J, De Paoli FV, et al. Mitochondrial Structure and Function in the Metabolic Myopathy Accompanying Patients with Critical Limb Ischemia. Cells. 2020;9. doi: 10.3390/cells9030570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Park SY, Pekas EJ, Anderson CP, Kambis TN, Mishra PK, Schieber MN, Wooden TK, Thompson JR, Kim KS, Pipinos, II. Impaired microcirculatory function, mitochondrial respiration, and oxygen utilization in skeletal muscle of claudicating patients with peripheral artery disease. Am J Physiol Heart Circ Physiol. 2022;322:H867–H879. doi: 10.1152/ajpheart.00690.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fisher-Wellman KH, Davidson MT, Narowski TM, Lin CT, Koves TR, Muoio DM. Mitochondrial Diagnostics: A Multiplexed Assay Platform for Comprehensive Assessment of Mitochondrial Energy Fluxes. Cell reports. 2018;24:3593–3606 e3510. doi: 10.1016/j.celrep.2018.08.091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Glancy B, Willis WT, Chess DJ, Balaban RS. Effect of calcium on the oxidative phosphorylation cascade in skeletal muscle mitochondria. Biochemistry. 2013;52:2793–2809. doi: 10.1021/bi3015983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Messer JI, Jackman MR, Willis WT. Pyruvate and citric acid cycle carbon requirements in isolated skeletal muscle mitochondria. Am J Physiol Cell Physiol. 2004;286:C565–572. doi: 10.1152/ajpcell.00146.2003 [DOI] [PubMed] [Google Scholar]
- 59.Jin S, Guerrero-Juarez CF, Zhang L, Chang I, Ramos R, Kuan CH, Myung P, Plikus MV, Nie Q. Inference and analysis of cell-cell communication using CellChat. Nat Commun. 2021;12:1088. doi: 10.1038/s41467-021-21246-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Andersson P, McGuire J, Rubio C, Gradin K, Whitelaw ML, Pettersson S, Hanberg A, Poellinger L. A constitutively active dioxin/aryl hydrocarbon receptor induces stomach tumors. Proc Natl Acad Sci U S A. 2002;99:9990–9995. doi: 10.1073/pnas.152706299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Beckman JA, Duncan MS, Damrauer SM, Wells QS, Barnett JV, Wasserman DH, Bedimo RJ, Butt AA, Marconi VC, Sico JJ, et al. Microvascular Disease, Peripheral Artery Disease, and Amputation. Circulation. 2019;140:449–458. doi: 10.1161/Circulationaha.119.040672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Olesen KKW, Anand SS, Gyldenkerne C, Thim T, Maeng M. Microvascular disease, peripheral artery disease, and the risk of lower limb amputation. Eur Heart J. 2020;41:2354–2354. [Google Scholar]
- 63.Gardner AW, Parker DE, Montgomery PS, Sosnowska D, Casanegra AI, Ungvari Z, Csiszar A, Sonntag WE. Endothelial Cell Inflammation and Antioxidant Capacity are Associated With Exercise Performance and Microcirculation in Patients With Peripheral Artery Disease. Circulation. 2014;130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hiatt WR, Armstrong EJ, Larson CJ, Brass EP. Pathogenesis of the limb manifestations and exercise limitations in peripheral artery disease. Circulation research. 2015;116:1527–1539. doi: 10.1161/CIRCRESAHA.116.303566 [DOI] [PubMed] [Google Scholar]
- 65.McDermott MM, Ferrucci L, Gonzalez-Freire M, Kosmac K, Leeuwenburgh C, Peterson CA, Saini S, Sufit R. Skeletal Muscle Pathology in Peripheral Artery Disease A Brief Review. Arterioscl Throm Vas. 2020;40:2577–2585. doi: 10.1161/Atvbaha.120.313831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Arinze NV, Gregory A, Francis JM, Farber A, Chitalia VC. Unique aspects of peripheral artery disease in patients with chronic kidney disease. Vasc Med. 2019;24:251–260. doi: 10.1177/1358863 [DOI] [PubMed] [Google Scholar]
- 67.O’Hare AM, Glidden DV, Fox CS, Hsu CY. High prevalence of peripheral arterial disease in persons with renal insufficiency: results from the National Health and Nutrition Examination Survey 1999–2000. Circulation. 2004;109:320–323. doi: 10.1161/01.CIR.0000114519.75433.DD [DOI] [PubMed] [Google Scholar]
- 68.Wattanakit K, Folsom AR, Selvin E, Coresh J, Hirsch AT, Weatherley BD. Kidney function and risk of peripheral arterial disease: results from the Atherosclerosis Risk in Communities (ARIC) Study. Journal of the American Society of Nephrology : JASN. 2007;18:629–636. doi: 10.1681/ASN.2005111204 [DOI] [PubMed] [Google Scholar]
- 69.Schiffrin EL, Lipman ML, Mann JF. Chronic kidney disease: effects on the cardiovascular system. Circulation. 2007;116:85–97. doi: 10.1161/CIRCULATIONAHA.106.678342 [DOI] [PubMed] [Google Scholar]
- 70.Ravid JD, Kamel MH, Chitalia VC. Uraemic solutes as therapeutic targets in CKD-associated cardiovascular disease. Nature Reviews Nephrology. 2021;17:402–416. doi: 10.1038/s41581-021-00408-4 [DOI] [PubMed] [Google Scholar]
- 71.Wang XH, Mitch WE, Price SR. Pathophysiological mechanisms leading to muscle loss in chronic kidney disease. Nat Rev Nephrol. 2022;18:138–152. doi: 10.1038/s41581-021-00498-0 [DOI] [PubMed] [Google Scholar]
- 72.Dou L, Bertrand E, Cerini C, Faure V, Sampol J, Vanholder R, Berland Y, Brunet P. The uremic solutes p-cresol and indoxyl sulfate inhibit endothelial proliferation and wound repair. Kidney Int. 2004;65:442–451. doi: 10.1111/j.1523-1755.2004.00399.x [DOI] [PubMed] [Google Scholar]
- 73.Kolachalama VB, Shashar M, Alousi F, Shivanna S, Rijal K, Belghasem ME, Walker J, Matsuura S, Chang GH, Gibson CM, et al. Uremic Solute-Aryl Hydrocarbon Receptor-Tissue Factor Axis Associates with Thrombosis after Vascular Injury in Humans. Journal of the American Society of Nephrology. 2018;29:1063–1072. doi: 10.1681/Asn.2017080929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Koizumi M, Tatebe J, Watanabe I, Yamazaki J, Ikeda T, Morita T. Aryl hydrocarbon receptor mediates indoxyl sulfate-induced cellular senescence in human umbilical vein endothelial cells. J Atheroscler Thromb. 2014;21:904–916. doi: 10.5551/jat.23663 [DOI] [PubMed] [Google Scholar]
- 75.Ichihara S, Yamada Y, Ichihara G, Nakajima T, Li P, Kondo T, Gonzalez FJ, Murohara T. A role for the aryl hydrocarbon receptor in regulation of ischemia-induced angiogenesis. Arterioscl Throm Vas. 2007;27:1297–1304. doi: 10.1161/Atvbaha.106.138701 [DOI] [PubMed] [Google Scholar]
- 76.Hart CR, Layec G, Trinity JD, Kwon OS, Zhao J, Reese VR, Gifford JR, Richardson RS. Increased skeletal muscle mitochondrial free radical production in peripheral arterial disease despite preserved mitochondrial respiratory capacity. Exp Physiol. 2018. doi: 10.1113/EP086905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.McDermott MM, Peterson CA, Sufit R, Ferrucci L, Guralnik JM, Kibbe MR, Polonsky TS, Tian L, Criqui MH, Zhao L, et al. Peripheral artery disease, calf skeletal muscle mitochondrial DNA copy number, and functional performance. Vasc Med. 2018:1358863X18765667. doi: 10.1177/1358863X18765667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gamboa JL, Roshanravan B, Towse T, Keller CA, Falck AM, Yu C, Frontera WR, Brown NJ, Ikizler TA. Skeletal Muscle Mitochondrial Dysfunction Is Present in Patients with CKD before Initiation of Maintenance Hemodialysis. Clin J Am Soc Nephro. 2020;15:926–936. doi: 10.2215/Cjn.10320819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kestenbaum B, Gamboa J, Liu S, Ali AS, Shankland E, Jue T, Giulivi C, Smith LR, Himmelfarb J, de Boer IH, et al. Impaired skeletal muscle mitochondrial bioenergetics and physical performance in chronic kidney disease. JCI Insight. 2020;5. doi: 10.1172/jci.insight.133289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Watson EL, Baker LA, Wilkinson TJ, Gould DW, Graham-Brown MPM, Major RW, Ashford RU, Philp A, Smith AC. Reductions in skeletal muscle mitochondrial mass are not restored following exercise training in patients with chronic kidney disease. Faseb J. 2020;34:1755–1767. doi: 10.1096/fj.201901936RR [DOI] [PubMed] [Google Scholar]
- 81.Xu CQ, Kasimumali A, Guo XJ, Lu RH, Xie KW, Zhu ML, Qian YY, Chen XH, Pang HH, Wang Q, et al. Reduction of mitochondria and up regulation of pyruvate dehydrogenase kinase 4 of skeletal muscle in patients with chronic kidney disease. Nephrology. 2020;25:230–238. doi: 10.1111/nep.13606 [DOI] [PubMed] [Google Scholar]
- 82.Lee J, Prokopec SD, Watson JD, Sun RX, Pohjanvirta R, Boutros PC. Male and female mice show significant differences in hepatic transcriptomic response to 2,3,7,8-tetrachlorodibenzo-p-dioxin. BMC Genomics. 2015;16. doi: ARTN 625 10.1186/s12864-015-1840-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fisher MT, Nagarkatti M, Nagarkatti PS. Aryl hydrocarbon receptor-dependent induction of loss of mitochondrial membrane potential in epididydimal spermatozoa by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Toxicol Lett. 2005;157:99–107. doi: 10.1016/j.toxlet.2005.01.008 [DOI] [PubMed] [Google Scholar]
- 84.He J, Hu B, Shi X, Weidert ER, Lu P, Xu M, Huang M, Kelley EE, Xie W. Activation of the aryl hydrocarbon receptor sensitizes mice to nonalcoholic steatohepatitis by deactivating mitochondrial sirtuin deacetylase Sirt3. Molecular and cellular biology. 2013;33:2047–2055. doi: 10.1128/MCB.01658-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hwang HJ, Dornbos P, Steidemann M, Dunivin TK, Rizzo M, LaPres JJ. Mitochondrial-targeted aryl hydrocarbon receptor and the impact of 2,3,7,8-tetrachlorodibenzo-p-dioxin on cellular respiration and the mitochondrial proteome. Toxicology and applied pharmacology. 2016;304:121–132. doi: 10.1016/j.taap.2016.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ghosh J, Chowdhury AR, Srinivasan S, Chattopadhyay M, Bose M, Bhattacharya S, Raza H, Fuchs SY, Rustgi AK, Gonzalez FJ, et al. Cigarette Smoke Toxins-Induced Mitochondrial Dysfunction and Pancreatitis Involves Aryl Hydrocarbon Receptor Mediated Cyp1 Gene Expression: Protective Effects of Resveratrol. Toxicol Sci. 2018;166:428–440. doi: 10.1093/toxsci/kfy206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Huang Y, Zhang J, Tao Y, Ji C, Aniagu S, Jiang Y, Chen T. AHR/ROS-mediated mitochondria apoptosis contributes to benzo[a]pyrene-induced heart defects and the protective effects of resveratrol. Toxicology. 2021;462:152965. doi: 10.1016/j.tox.2021.152965 [DOI] [PubMed] [Google Scholar]
- 88.Ren R, Fang Y, Sherchan P, Lu Q, Lenahan C, Zhang JH, Zhang J, Tang J. Kynurenine/Aryl Hydrocarbon Receptor Modulates Mitochondria-Mediated Oxidative Stress and Neuronal Apoptosis in Experimental Intracerebral Hemorrhage. Antioxidants & redox signaling. 2022. doi: 10.1089/ars.2021.0215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cuartero MI, Ballesteros I, de la Parra J, Harkin AL, Abautret-Daly A, Sherwin E, Fernandez-Salguero P, Corbi AL, Lizasoain I, Moro MA. L-kynurenine/aryl hydrocarbon receptor pathway mediates brain damage after experimental stroke. Circulation. 2014;130:2040–2051. doi: 10.1161/CIRCULATIONAHA.114.011394 [DOI] [PubMed] [Google Scholar]
- 90.Kwon JI, Heo H, Ham SJ, Chae YJ, Lee DW, Kim ST, Min J, Sung YS, Kim KW, Choi Y, et al. Aryl hydrocarbon receptor antagonism before reperfusion attenuates cerebral ischaemia/reperfusion injury in rats. Sci Rep-Uk. 2020;10. doi: ARTN 14906 10.1038/s41598-020-72023-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Melhem NJ, Chajadine M, Gomez I, Howangyin KY, Bouvet M, Knosp C, Sun YY, Rouanet M, Laurans L, Cazorla O, et al. Endothelial Cell Indoleamine 2, 3-Dioxygenase 1 Alters Cardiac Function After Myocardial Infarction Through Kynurenine. Circulation. 2021;143:566–580. doi: 10.1161/Circulationaha.120.050301 [DOI] [PubMed] [Google Scholar]
- 92.Wyant GA, Yu WY, Doulamis IP, Nomoto RS, Saeed MY, Duignan T, McCully JD, Kaelin WG. Mitochondrial remodeling and ischemic protection by G protein-coupled receptor 35 agonists. Science. 2022;377:621–U190. doi: 10.1126/science.abm1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Seong E, Lee JH, Lim S, Park EH, Kim E, Kim CW, Lee E, Oh GC, Choo EH, Hwang BH, et al. Activation of Aryl Hydrocarbon Receptor by ITE Improves Cardiac Function in Mice After Myocardial Infarction. Journal of the American Heart Association. 2021;10. doi: ARTN e020502 10.1161/JAHA.120.020502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dong G, Moparthy C, Thome T, Kim K, Yue F, Ryan TE. IGF-1 Therapy Improves Muscle Size and Function in Experimental Peripheral Arterial Disease. JACC: Basic to Translational Science. 2023;0. doi: doi:10.1016/j.jacbts.2022.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lu H, Lei X, Klaassen C. Gender differences in renal nuclear receptors and aryl hydrocarbon receptor in 5/6 nephrectomized rats. Kidney Int. 2006;70:1920–1928. doi: 10.1038/sj.ki.5001880 [DOI] [PubMed] [Google Scholar]
- 96.Ohtake F, Takeyama K, Matsumoto T, Kitagawa H, Yamamoto Y, Nohara K, Tohyama C, Krust A, Mimura J, Chambon P, et al. Modulation of oestrogen receptor signalling by association with the activated dioxin receptor. Nature. 2003;423:545–550. doi: 10.1038/nature01606 [DOI] [PubMed] [Google Scholar]
- 97.Ohtake F, Baba A, Takada I, Okada M, Iwasaki K, Miki H, Takahashi S, Kouzmenko A, Nohara K, Chiba T, et al. Dioxin receptor is a ligand-dependent E3 ubiquitin ligase. Nature. 2007;446:562–566. doi: 10.1038/nature05683 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting the conclusions of this study are available from the corresponding author.
Detailed descriptions of the materials and experimental procedures can be found in the Expanded Material and Methods section of the Supplemental Material A Major Resources Table is available in the Supplemental Material. References related to methods employed included the following:25,39–47.