

Abstract
Myelinating oligodendrocytes are essential for neuronal communication and homeostasis of the central nervous system (CNS). One of the most abundant molecules in the mammalian CNS is N-acetylaspartate (NAA), which is catabolized into L-aspartate and acetate by the enzyme aspartoacylase (ASPA) in oligodendrocytes. The resulting acetate moiety is thought to contribute to myelin lipid synthesis. In addition, affected NAA metabolism has been implicated in several neurological disorders, including leukodystrophies and demyelinating diseases such as multiple sclerosis. Genetic disruption of ASPA function causes Canavan disease, which is hallmarked by increased NAA levels, myelin and neuronal loss, large vacuole formation in the CNS, and early death in childhood. Although NAA’s direct role in the CNS is inconclusive, in peripheral adipose tissue, NAA-derived acetate has been found to modify histones, a mechanism known to be involved in epigenetic regulation of cell differentiation. We hypothesize that a lack of cellular differentiation in the brain contributes to the disruption of myelination and neurodegeneration in diseases with altered NAA metabolism, such as Canavan disease. Our study demonstrates that loss of functional Aspa in mice disrupts myelination and shifts the transcriptional expression of neuronal and oligodendrocyte markers towards less differentiated stages in a spatiotemporal manner. Upon re-expression of ASPA, these myelination and neuronal lineage markers are either improved or normalized, suggesting that NAA breakdown by Aspa plays an essential role in the maturation of neurons and oligodendrocytes. Also, this effect of ASPA re-expression is blunted in old mice, potentially due to limited ability of neuronal, rather than oligodendrocyte, recovery.
Graphical Abstract
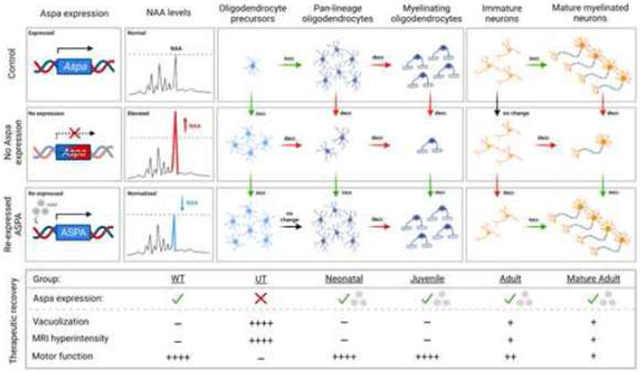
Introduction
Oligodendrocytes are the myelin-forming cells in the central nervous system (CNS) and play a vital role in efficient signal transduction, maintenance, and support of neuronal function (1, 2). Various injuries to oligodendrocytes can cause neurodegeneration, as reported in multiple sclerosis (MS) and leukodystrophies (3, 4). To correct injured or dysfunctional cells, CNS repair involves neuronal or glial transition from less-differentiated precursor states to mature cellular forms (5). During oligodendrocyte development, throughout which oligodendrocyte transcription factor 2 (Olig2) is continuously expressed, platelet-derived growth factor alpha (Pdgfrα+) positive oligodendrocyte precursor cells (OPCs) differentiate and progress from pre-myelinating to mature myelinating, myelin basic protein (Mbp+) positive oligodendrocytes (6, 10). Notably, altered Pdgfrα+ and Olig2+ expression levels have been reported in impaired oligodendrocyte lineage progression (6-9). For example, Pdgfrα inactivation drives OPC differentiation into mature oligodendrocytes and depletes existing OPC pools, suggesting that Pdgfrα+ OPCs play a critical role in oligodendrocyte maturation (9). Additionally, increased Olig2 expression promotes oligodendrocyte differentiation (10, 11).
In the CNS, aspartoacylase (ASPA) is expressed exclusively in oligodendrocytes, and loss-of-function mutations lead to failure of N-acetylaspartate (NAA) breakdown, thus causing Canavan disease (CD), a lethal leukodystrophy with progressive CNS spongy degeneration and dysmyelination (12-15). Furthermore, neuronal NAA, as well as its acetate residue, has been shown to epigenetically regulate gene expression in adipocytes and in oligodendrocytes through acetylation and methylation of histones, an important epigenetic mechanism in regulating cellular and oligodendrocyte differentiation (16, 40). Interestingly, in-vitro and in-vivo models have demonstrated that loss of Aspa delays differentiation of precursor to mature oligodendrocytes and increases proliferation of Olig2+ progenitors as a possible attempt of replenishing precursor pools for differentiation into mature myelinating oligodendrocytes (17).
Concomitantly, cortical neurons have been shown to decrease in Aspa−/− mice progressively, but it is unclear how this relates to oligodendrocyte changes (18). Neural and oligodendrocyte progenitor cells (NPCs and OPCs) are essential for CNS recovery (32) and OPCs are emerging as the rate-limiting cell-type for CNS remyelination (33). Nestin+ NPCs have been shown to generate OPCs and immature neurons in response to cellular signaling pathways and differential regulatory mechanisms (42, 43). These immature neurons are doublecortin (DCX) positive and eventually develop into mature neurons that are positive for RNA-binding fox-1 homolog-3 (RbFox3) transcripts. RbFox3 is the genetic encoder of the neuronal nuclei (NeuN) protein, which serves as a postmitotic neuronal marker (41). Growth associated protein 43 (GAP43) positive cells, which is a marker for neuronal growth cones during axonal regeneration (48) can be correlated with levels RbFox3+ cells to assess neuronal recovery otherwise unattainable in Aspa−/− mice. In this regard, we hypothesize that hindered CNS cellular differentiation disrupts myelination and contributes to neurodegeneration in diseases with affected NAA metabolism, such as Canavan disease.
Our study reveals that Aspa deficiency progressively disrupts myelin and neuronal architecture, and that this pathology is accompanied by an increased spatiotemporal expression of oligodendrocyte precursors and immature neuronal markers on in-situ single-cell analysis. Re-expressing ASPA in Aspa-deficient mice leads to normalized or overexpressed early-stage cellular markers and normalization of mature, differentiated oligodendrocytes over time (Figure S1). This is complemented by a time-dependent increase of mature neuronal cells and neuronal growth cones in a brain region-specific manner. These changes are paralleled by a phenotypical and biochemical normalization, suggesting that loss of Aspa function impedes the lineage progression of oligodendrocytes and neurons and that restoration of functional ASPA, with normalized NAA metabolism, enables the reversal of this cellular disruption in an age-dependent manner (Figure S1).
Results
ASPA re-expression reverses motor deficits and normalizes CNS pathology in Aspa-deficient mice
Aspa-deficient Nur7 mice develop progressive neurological symptoms, such as ataxia and tremors (19), which are attributed to affected neuronal viability resulting from dysmyelinating effects. To evaluate the correlation between CNS degeneration and recoverability, homozygous Nur7 mutant mice, which carry a nonsense mutation in the Aspa gene (Aspanur7/nur7), were injected with rAAV expressing ASPA (rAAVASPA) at pre-symptomatic neonatal (Neo) and symptomatic juvenile (Juv) age (Figure 1A). Injected animals were tested alongside age-matched, un-injected controls: wild-type (WT) and Aspanur7/nur7 (UT) mice.
Figure 1: Corrected motor function and normalized neuropathology after ASPA re-expression in neonatal and juvenile mice.

(A) Timeline of single rAAV administration per age group and their corresponding motor testing ages. Wildtype (WT), no injection Nur7 (UT), and injected Nur7 mice at either neonatal (Neo) or juvenile (Juv) ages were tested four weeks post the ASPA re-expression timepoint and at additional timepoints up to one year of age. (B) Accelerated rotarod and (C) balance beam motor testing of all experimental groups (n=9-10 each) up to one year of age (52 weeks). (D) MRI T2 sequences of coronal brain sections (hyper-intensive areas marked by dashed red line) and (E) hematoxylin and eosin (H&E) staining of brain regions at one year of age. CTX/STR: cortex/striatum; HIP/TH: hippocampus/thalamus; MB: midbrain; CB/PON: cerebellum/pons. One-way ANOVA with multi-comparison correction, mean ±SD, ns = not significant, * p<0.05; ** p<0.01, *** p<0.001, **** p<0.0001.
To assess overall motor ability, which serves as an indirect readout of affected CNS regions involved in motor coordination, all groups were motor tested at in four weeks post-injection and at additional time points up to one year of age (Figure 1A). Throughout the study, the Neo and Juv groups performed significantly better than the UT mice and equivalent to WT mice on the accelerating rotarod (Figure 1B) and balance beam (Figure 1C). In contrast, UT mice developed progressively worsening motor deficits, as evidenced by a decrease in time score performance on both assays from 4 to 52 weeks of age, suggesting a decline in motor function and progression of neurodegeneration.
MRI T2 imaging revealed that hyper-intensive signals (dashed-red outline) in affected brain regions such as the thalamus, midbrain, and cerebellum in UT mice were normalized in the Neo and Juv groups (Figure 1D). Paralleling the MRI findings, H&E staining depicted histopathological recovery in the Neo and Juv mice in the same brain regions (Figure 1E). These results suggest that expressing ASPA in pre-symptomatic and symptomatic mice can prevent and reverse CNS degeneration and associated phenotypes.
ASPA reconstitution promotes remyelination and increases axonal abundance in Aspa-deficient mice
Previously, it has been shown that Aspa deficiency negatively affects myelin (20). Given that the Juv group depicts pathological normalization, we hypothesized that a reversal of defective myelin might accompany recovery. First, to gauge the extent of neurodegeneration before ASPA reconstitution in symptomatic mice, sub-anatomical and global brain regions were examined at the six-week juvenile timepoint (Figure 2A, 2B); manifestation of vacuoles and brain matter loss were most prominent in the striatum, thalamus, cerebellum, and brainstem (Figure 2A).
Figure 2: Serial magnetic resonance spectroscopy (MRS) and imaging (MRI), electron microscopy (EM), and H&E demonstrate reversal of CNS pathology in juvenile group.

H&E images of (A) region-specific and (B) global differences at pre-injection timepoint (6 weeks old) between WT and UT groups. (C) Serial MRS of total N-acetylaspartate (NAA)/total creatine (tCr) ratio from 6 weeks of age (day of injection, week 0) and subsequently 1, 2, and 4 weeks post-injection; 7, 8, and 10 weeks of age respectively (n=3 per group and timepoint). (D) Midbrain T2 MRI sequence (hyper-intensive areas marked by dashed red line) and H&E images of thalamic regions and (E) EM at 2600x magnification depicting axons and myelination of anterior commissure corresponding to 1- and 4- weeks post-ASPA re-expression. Quantification of myelin and axon properties in the anterior commissure including (F) the G-ratio, (G) the number of myelin layers per axon, and (H) the number of myelinated axons per imaged section at 1- and 4- weeks post-ASPA re-expression. Two-way ANOVA with multi-comparison correction, mean ±SD, ns = not significant, * p<0.05; ** p<0.01, *** p<0.001, **** p<0.0001.
To test the hypothesized change in myelination, mice were injected with rAAVASPA at six weeks of age (Juv) and followed weekly for one month. Interestingly, a decrease in brain NAA levels was observed as early as one-week post-injection and was normalized by two weeks, suggesting NAA metabolization by ASPA (Figure 2C). Furthermore, this biochemical response to ASPA expression was paralleled by the gradual normalization in MRI signals and histopathology (Figure 2D).
In addition, the anterior commissure, a myelin-rich structure connecting both brain hemispheres, was subjected to electron microscopy (EM) to analyze myelin. Unlike WT mice which displayed a uniform pattern of numerous myelinated axons at both time points, a disorganized pattern of axons and variation in electron-dense structures, such as myelin (dark ring-like structures surrounding axons), was observed in the UT and Juv mice one-week post-injection (Figure 2E). However, by four weeks post-injection, myelin and axonal distribution in Juv mice began resembling the pattern of WT mice, indicating remyelination in the Juv group, whereas persisting vacuoles and defective myelination progressively increased in UT mice (Figure 2E). Differences in myelin characteristics and neuronal axons were reflected by the G-ratio (Figure 2F), the number of myelin layers per axon (Figure 2G), and the number of axons per section (Figure 2H), all of which were normalized in the ASPA-expressing Juv mice.
Restoration of ASPA activity increases Nestin expression in pre-dominantly affected brain-regions
Following the findings that neurodegeneration and myelin defects are reversible in the Juv group, we hypothesized that cellular recovery is underlying this regenerative process. Nestin+ cells are associated with stem cell properties and give rise to several cellular lineages in the brain (Figure 3A) (21, 22). Compared to WT and UT groups, the in-situ single-cell analysis revealed globally increased numbers of Nestin+ cells in Juv mice at one and four weeks post-ASPA reconstitution (Figure 3B). Notably, UT mice also exhibited an increase of Nestin+ cells from one to four weeks post-injection; however, these levels were significantly less than WT and Juv levels at the four-week mark (Figure 3B). To further understand how this change in Nestin+ cells is represented in the most affected brain regions, i.e., thalamus, cerebellum, brainstem, and corpus callosum, sub-anatomical imaging and analysis was performed (Figure 3C-D). Notably, in ASPA-expressing Juv mice, the thalamus, cerebellum, and brainstem (Figure 3C-D) and the hippocampus (Figure S2A) showed an overall increase of Nestin+ cells over time, with significant changes as early as one-week post-treatment. Interestingly, the corpus callosum did not depict significant differences in Nestin+ expression between the three groups at the four-week post-treatment timepoint (Figure 3C-D).
Figure 3: Nestin mRNA+ cells globally increase in the CNS in a region-specific manner following ASPA re-expression at juvenile age.

(A) Cell-lineage schematic of Nestin mRNA+ progenitor cells into oligodendrocyte or neuronal cell pools with their respective mRNA+ expressors. (B) Quantification of global Nestin mRNA+ cell counts for select brain regions: cortex (CTX), striatum (STR), corpus callosum (CC), hippocampus (HC), thalamus (TH), midbrain (MB), cerebellum (CB), and brainstem (BS). (C) Quantification of Nestin+ mRNA cell counts in the select brain regions: TH, CB, BS, and CC, and (D) representative images of selected positive cells (green-outline) for fluorescent Nestin+ mRNA puncta (white dots) in these regions at the four-week post-injection timepoint Two-way ANOVA with multi-comparison correction, confidence interval (CI) 99%, mean ±SD, ns = not significant, * p<0.0332; ** p<0.0021, *** p<0.0002, **** p<0.0001.
Oligodendrocyte differentiation increases post-ASPA expression in Aspa-deficient mice
Changes in Nestin+ cells may indicate active cellular repair, which expectedly could extend into oligodendrocyte lineage (Olig2+) to normalize myelin on EM in Juv mice. PDGFRα+ oligodendrocyte precursor cells (OPCs) possess the capacity to proliferate, giving rise to more differentiated pre-myelinating oligodendrocytes and ultimately, mature myelinating oligodendrocytes (MBP+) (23, 24).
Globally, OPCs in UT brains outnumbered WT mice and showed a near-consistent, static level of PDGFRα expression over time (Figure 4A). Interestingly, at both time points, ASPA-expressing Juv animals had significantly higher levels of PDGFRα+ OPCs than WT or UT on a global scale. In contrast to PDGFRa+ OPCs, UT mouse brains had fewer Olig2+ cells than WT or Juv mice (Figure 4B). Similarly, MBP expression was lower in UT brains than in Juv mice at either time point or less than in WT mice at the four weeks post-injection time point (Figure 4C). Additionally, analysis of brain regions showed that UT mice had significantly increased PDGFRα+ OPCs, in cerebellum and thalamus at one week (Figure 4D and Figure S3A) and cortex and thalamus at four weeks (Figure 4E and Figure S3A) post-injection, but significantly decreased levels of Olig2+ cells in most brain regions (Figure 4E and Figure S3A) at the four-week post-treatment time point when compared to the WT and Juv groups. Similarly, except for the thalamus and midbrain, MBP+ myelinating oligodendrocytes were decreased in UT brains compared to WT or Juv animals at the late-stage timepoint (Figure 4E and Figure S3A). Overall, these results suggest a disconnect between PDGFRα+ and Olig2+ or MBP+ cells, which could indicate a shift in oligodendrocyte lineage, either in the form of a disruption in the transition from OPCs to mature myelinating oligodendrocytes or a depletion of mature myelinating oligodendrocytes without sufficient compensation from OPCs.
Figure 4: Oligodendrocyte lineage marker mRNA+ cell counts increase in region-specific manner following ASPA re-expression at juvenile age.

(A-C) Total summed PDGFRa, Olig2, and MBP mRNA+ cell counts for CTX, STR, CC, HC, TH, MB, CB, and BS brain regions. (D) Quantification of mRNA+ cells at 1-week post-treatment timepoint (7 weeks of age) and (E) at 4-week post ASPA re-expression timepoint (10 weeks of age). Two-way ANOVA with multi-comparison correction, CI 99%, mean ±SD, ns = not significant, * p<0.0332; ** p<0.0021, *** p<0.0002, **** p<0.0001. Abbr.: platelet-derived growth factor receptor alpha (PDGFRa), oligodendrocyte transcription factor 2 (Olig2), myelin basic protein (MBP).
Functional ASPA activity recovers expression of neuronal cell markers
The close relationship between oligodendrocyte health and neuronal function is well documented, with oligodendrocyte injury leading to neuronal degeneration (18, 25). To explain the phenotypical and ultrastructural disease reversal after ASPA expression (Figures 1-2), we hypothesized that neuronal populations underwent a process of repair. Using doublecortin (DCX), a marker for neuronal precursor cells, pan CNS profiling showed that Juv mice consistently had lower DCX+ cells than UT and WT mice at both time points, with no difference between UT and WT groups, suggesting that ASPA re-expression leads to recruitment of neuronal precursor cells for neuronal recovery (Figure 5A). Upon further analysis, several brain regions demonstrated decreased DCX+ cells in the Juv group compared to the WT group, e.g., thalamus, cerebellum, and midbrain at one week (Figure 5D-E). Only the corpus callosum (Figure 5D) and striatum (Figure S3B) at one week and the cerebellum and brain stem at four weeks showed insignificant differences in DCX+ levels among the three groups (Figure 5D). To determine if this change in neuronal precursor cells was associated with changes in mature neurons, RbFox3+ cells were quantified. Consistently, UT mice had significantly fewer RbFox3+ cells than WT and Juv mice, predominately at the four-week post-injection timepoint, particularly in the cerebellum, brainstem, striatum, and hippocampus (Figure 5E and Figure S3B).
Figure 5: Neuronal mRNA+ cell counts increase in region-specific manner following ASPA re-expression at juvenile age.

(A-C) Total summed Doublecortin (DCX), RbFox3, and GAP43 mRNA+ cell counts for select brain regions (referenced in Fig 4). (D) Quantification of mRNA+ cells at the 1-week post-treatment timepoint (7 weeks of age) and (E) at the 4-week post-treatment timepoint (10 weeks of age). Two-way ANOVA with multi-comparison correction, CI 99%, mean ±SD, ns = not significant, * p<0.0332; ** p<0.0021, *** p<0.0002, **** p<0.0001. Abbr.: doublecortin (DCX), RNA binding fox-1 homolog-3 (RbFox3), growth associated protein 43 (GAP43).
Interestingly, the Juv group showed significantly improved numbers of RbFox3+ cells at both time points but could not achieve the same global levels as WT mice (Figure 5B). However, in several brain regions, RbFox3+ expression did achieve the same levels as WT, e.g., cerebellum, brain stem, and hippocampus, although not until the four weeks mark (Figure 5D-E and Figure S3B). To further confirm the degree of neuro-regeneration, Gap43, which marks neuronal growth cones and signifies a regeneration of synaptic relationships (26), was analyzed. Gap43+ cells globally increased in WT and Juv brains over time, whereas UT mice remained significantly below WT and Juv levels at both time points (Figure 5C). This trend persisted at the sub-anatomical level but was most pronounced four weeks after injection in all brain regions except in the midbrain (Figure 5D-E and Figure S3B).
Extent of CNS regeneration and motor recovery post-ASPA expression in Aspa-deficient mice is age-dependent
Based on the finding that symptomatic mice exhibit signs of oligodendrocyte and neuronal recovery post-ASPA expression, which seems to parallel the phenotypical and biochemical improvement, we hypothesized that efficacy of disease reversal depends on the extent of neurodegeneration at the time of ASPA reconstitution. Hence, Aspanur7/nur7 mice were injected with rAAVASPA at post-symptomatic time points of either adult (12 weeks) or mature adult (24 weeks) age (Figure 6A). On motor function testing, the adult group (Adu) showed some improvement until the end of the study at one year of age or 52 weeks. In contrast, the mature adult (Mat) group seemed non-responsive in motor function (Figures 6B-C). This was particularly interesting since MRI and histopathology suggested improvement of myelination and CNS pathology (Figure 6D-E), along with normalized NAA levels in the brain and urine, for both groups (Figure 6F-G).
Figure 6: Partial correction of motor function and neuropathology achieved in late-stage, post-symptomatic adult and mature adult injection timepoints.

(A) Timeline of single rAAV administration per age group and their corresponding motor testing ages. Wild-type (WT), non-injected Nur7 (UT), and rAAV injected Nur7 mice at either adult (Adu) or mature adult (Mat) ages were motor tested 4-weeks post the treatment timepoint and at additional timepoints up to one year of age (n=9-10 each). Motor tests included (B) accelerated rotarod and (C) balance beam performance assessments (WT: gray-outlined circle; UT: gray-outlined square; Adu: blue diamond; Mat: green hexagon). (D) MRI T2 sequences and (E) H&E staining of Adu and Mat groups at one year of age (52 weeks). (F) N-acetylaspartate (NAA) levels in the brain and (G) in urine, normalized to total creatine (tCr) and creatinine (Cr) respectively, after one year of age (52 weeks). Two-way ANOVA with multi-comparison correction, mean ±SD, ns = not significant, * p<0.05; ** p<0.01, *** p<0.001, **** p<0.0001.
To rule out limited rAAV transduction efficiency in progressively neurodegenerating cells as a limiting factor, which would lead to reduced ASPA expression, 11 CNS regions were analyzed for rAAV genomes and ASPA mRNA levels. Quantification per brain region suggested similar efficiencies in gene delivery (i.e., rAAV genomes) and transgene mRNA expression per vector genome across all age groups (Figure S4A-C).
Improvements were observed in some but not all measures of motor function for Adu and Mat groups. However, both rAAVASPA injected groups appeared healthier overall than UT mice, suggesting that either the rotarod and balance beam tests were not sensitive enough to capture motor function recovery or that ASPA restoration at such late stages may only induce efficient remyelination but not neuronal repair. To test the first hypothesis, we studied all six groups for gait analysis at one year of age. WT and all rAAVASPA-treated animals significantly outperformed UT mice. The Mat group performed better even when comparing the Mat group at one year of age to the six months old UT animals (Figure 7A-E), which suggested that Mat mice still retained the ability to improve their motor function above the level of symptomatic UT mice at six months of age.
Figure 7: Cat-walk gait analysis reveals extended therapeutic motor benefits throughout late-stage ASPA re-expression timepoints.

(A) Visualized gait pattern at one year of age (52 weeks); blue and purple indicate left hind and front leg and green and yellow indicate right hind and front leg, respectively. Gait analysis was performed at one year of age for each group (n=6-8 each) with five runs per mouse. (B) Regularity, (C) base support, (D) stride length of right and left front leg and (E) stride length of right and left hind leg at 26 weeks (6 months) for WT and Nur7 UT groups and at 52 weeks (12 months) for all groups. One-way ANOVA with Tukey’s multi-comparison correction, mean ±SD, ns = not significant, * p<0.0332; ** p<0.0021, *** p<0.0002, **** p<0.0001. (F) T-maze testing for spatial learning assessment at 10 weeks and 52 weeks of age. Nonparametric one-way ANOVA with Kruskal-Wallis multi-comparison test, mean ±SD, ns = not significant, * p<0.0332; ** p<0.0021, *** p<0.0002, **** p<0.0001.
Additionally, spatial memory testing was performed based on the finding that neuronal markers were improved in the hippocampus (Figure S3B), a region critical for memory function. All rAAVASPA injected groups, except for the Mat group, outperformed UT mice, even when comparing the one-year-old Adu group to 10-week-old UT mice (Figure 7F), suggesting recovery post-ASPA expression at a relatively late-stage of disease progression.
Discussion
In this study, in-situ profiling of CNS cellular lineage markers revealed a correlation between cell differentiation secondary to deficiency of functional Aspa and neurodegeneration, as reflected by increased neuronal and oligodendrocyte precursor cells (Figure 4 and Figure S3) and reduction of differentiated mature cells (Figure 4-5 and Figure S3A). This was paralleled by myelin loss and axonal degeneration, which was reversible to a pre-diseased state in an age-dependent manner post-functional ASPA expression.
Injuries to oligodendrocytes or neurons can compromise the function of one another (2, 27, 28). Correspondingly, neurons are affected in dysmyelinating disorders, including Canavan disease, which was previously reported in Aspa−/− mouse brains (17, 18); osmotic injury or direct NAA toxicity secondary to NAA accumulation has been proposed; however, several studies have since contested this hypothesis (29-31). Progressive injury to mature neurons may explain why replacing ASPA at late-disease stages, as represented by adult and mature adult groups, resulted in less symptomatic recovery despite normalized NAA levels in the brain and urine (Figures 6 and 7). This is also supported by normalized motor function and myelination in the juvenile group, despite RbFox3+ mature neuronal numbers remaining below WT levels (Figure 5B). Extrapolating this finding to mice with more advanced neurodegeneration (adult and mature groups) suggests that RbFox3+ mature neurons might have been reduced to a greater extent in these groups than in juvenile mice before and after re-expression of ASPA, thus limiting their symptomatic recovery. Additionally, quantifying rAAV genome and ASPA mRNA levels ruled out gene delivery as the responsible factor for the limited response in adult and mature adult mice (Figure S4).
Nestin+ NPCs give rise to PDGFRα+ OPCs, thus contributing to remyelination (21, 22, 34), which seems to align with the increase of Nestin+ cells above WT levels upon ASPA re-expression in Aspa−/− mice (Figure 3B and Figure S2). Interestingly, Nestin+ and PDGFRα+ cells in the UT mouse brains were on par or above WT (Figure 3B and Figure 4A), suggesting that the lineage transition at this stage might still have been intact. In addition to Nestin+ NPCs’ contribution, OPC repopulation depends on PDGFRα to prevent precocious OPC differentiation (9). This function might explain the persistence of PDGFRα elevation after ASPA reconstitution (Figure 4A, D-E, and Figure S3A), ultimately prompting the normalization of MBP+ mature, myelinating oligodendrocytes (Figure 4C-E) and myelin, as observed on EM imaging (Figure 2E). On the contrary, Olig2+ oligodendrocytes were significantly decreased in UT mice. Olig2 is critical for generating pre-myelinating and mature oligodendrocytes from OPCs (8), suggesting that the reduction of Olig2 in Aspa−/− mouse brains prevents this transition in the oligodendrocyte lineage.
Alternatively, Nestin expression has also been reported in reactive astrocytes (35) and since the presence of astrogliosis is well documented in Aspa−/− brains (36), this would expectedly increase Nestin+ cells in the UT group. However, levels of Nestin+ cells in UT mice were below WT and rAAV-injected groups, except in two brain regions: the cerebellum and midbrain (Figure 3D and Figure S2), thus indicating that increased Nestin+ cells in UT mice may be separate from disease-related astrogliosis.
ASPA expression in the brain is predominately found in oligodendrocytes and has been reported in other CNS cell-types such as astrocytes (44). rAAV9 transduction efficiency of oligodendrocytes is largely limited by selected promoter activity, age of treatment, and route of administration (46, 47), thus increasing the complexity of therapeutic strategy via rAAV9-oligodendrocyte targeting. On the other hand, previous research in Aspa−/− neonatal mice using rAAV9 has shown that both widespread ubiquitous or astrocyte-restricted ASPA expression can normalize CNS pathology (12). Relating these findings to our current study suggest that ubiquitous gene expression maintains the transcriptional capacity to alter oligodendrocyte and neuronal lineages towards more mature cellular states (Figures 3, 4, and 5, and Figure 2SA-B) in a therapeutic manner. In this regard, while it is possible that therapeutic effect in non-oligodendrocytes is driven by NAA metabolism in other CNS cell-types, as suggested by the NAA sink theory is astrocytes (12), it might also suggest substantial metabolic exchange between oligodendrocytes and neuronal and other glial cells that could compensate for NAA-derived L-aspartate and acetate. However, further studies are needed to identify individual cell-specific contributions of non-oligodendrocyte ASPA expression on OPC differentiation, myelination, and neuronal restoration.
The increase in Pdgfra+ OPCs and Mbp+ mature oligodendrocytes following ASPA re-expression is a brain region specific response. Myelinating oligodendrocytes were found to be transcriptionally increased in white matter regions, such as the corpus callosum, but more interestingly similar results were observed in gray matter regions such as the cortex and thalamus (Figure 4D-E and Figure S2A). These changes in oligodendrocyte composition in the gray matter may have therapeutic relevance for other demyelinating diseases such as multiple sclerosis which has historically been associated with myelin degradation in deep gray matter regions (45).
In summary, the present study suggests that loss of myelin and axonal injury in Aspa deficiency is attributable to changes in oligodendrocyte and neuronal differentiation secondary to interrupted NAA metabolism, which can be reversed upon ASPA re-expression in an age-dependent manner (Figure S1).
Methods
Animal procedures
The Nur7 mouse model, which carries a nonsense mutation in the Aspa gene (Aspanur7/nur7), was provided by Maria Traka (19). DNA from tail tips of 1-day-old pups were extracted using the QIAmp DNA mini kit or QIAcube robot (QIAgen, Hilden, Germany) and were genotyped using a TaqMan-based single-nucleotide-polymorphism (SNP) assay via ddPCR (BioRad, Hercules, CA, USA). Heterozygous Nur7 mice carrying an inverted balancer chromosome were bred, and homozygous Aspanur7/nur7 mutants, identified by dark pigmented ears and tails, were used in the experiments. All animal experiments were approved by the UMass Chan Medical School IACUC, Worcester, MA.
Age groups were defined as follows: neonatal (p1), juvenile (p42), adult (p84), and mature adult (p168). Corresponding single-administrative doses of rAAV9 delivering a codon-optimized human ASPA cDNA driven by the CB6 promoter with full Kozak element (3rd generation) (average weight in brackets (grams (g)): treated at p1 (1.5g), dose 4x1011GC; p42 (16g), p84 (23.6g), and p164 (23g) were injected intravenously with 4x1012GC.
Post-procedure, neonatal pups were cleaned with 70% ethanol and rubbed with bedding material before being returned to their cages. Mice treated at later time points were isoflurane-anesthetized and monitored until entirely awoken post-injection prior to returning to their cages.
Weights for the Neo group were recorded daily for the first 28 days as a general health assessment, after which weights were obtained every other week, for all mice, until the end of the study at one year of age.
Vector design and viral production
The vector used in this study was constructed and evaluated as previously described (12). Recombinant adeno-associated virus (rAAV) was produced by transient HEK 293 cell transfection and cesium-chloride (CsCl) sedimentation described previously (37). Vector preparations were titered by digital PCR (ddPCR), and purity was determined by 4-12% SDS-acrylamide gel electrophoresis and silver staining (Invitrogen, Carlsbad, CA). Transmission electron microscopy of negative stained rAAV virions at the UMass Chan Medical School Microscopy Core, Worcester, MA, was used to assess the morphology and integrity of the AAV virion.
Isolation of brain regions and DNA and RNA extraction
Brain regions were isolated as previously described (12). Mice were euthanized, brains were extracted, and midline was dissected into two halves. Brain regions were extracted from both hemispheres. Sample dissection was performed on an ice-cooled metal plate under a dissection microscope (39) and regions were immediately flash-frozen in liquid nitrogen. DNA and RNA were simultaneously extracted (Qiagen AllPrep Kit) and subjected to rAAV genome copy number quantification by ddPCR and rt-PCR/ddPCR to quantify hASPA mRNA as described before (12).
Motor function testing
Accelerating rotarod (Rotarod Series 8, IITC Life Science), balance beam (custom built by UMass Chan Medical School Machine Shop), T-maze (San Diego Instruments), and catwalk (Noldus) assays were used to test motor performance, endurance, coordination, spatial learning, and gait analysis.
Rotarod and balance beam testing were performed as described before (12). A maximum of 5 minutes per run was allotted, and latency until drop-off was recorded per run. Each mouse was tested three times, and the best run-time was used for analysis.
For T-maze assays, spontaneous and unrewarded testing was performed as described before (12). Mice were tested serially, and the T maze was cleaned between runs to avoid olfactory clues. Every mouse was tested 11 times, resulting in 10 alteration data points. Entry into one of the two arms was defined as complete crossing of the edge of the arm by all four paws.
For catwalk assays, mice were placed on the CatWalk walkway with the room lighting turned off. Mice were allowed to explore the walkway freely without interference or incentive to optimize the CatWalk camera system’s intensity and contrast. Data collection began post-optimization. The maximum time was 60 seconds to complete each run. A minimum of 5 complete runs was required per tested mouse. A complete run was defined as a full traverse from one end of the walkway to the other in one direction without turning or spinning around on the spot. Data collection by the CatWalk system was automatic, and data analysis of various parameters was conducted after each run.
Magnetic resonance imaging (MRI) and spectroscopy (MRS)
The imaging was done as described before (12). Briefly, animals were anesthetized with isoflurane in an induction chamber and transferred to the coil. Anesthesia was maintained at 2% of isoflurane using a nose cone, and mice were monitored continuously for vital signs. Imaging was performed using the Oxford 4.7-T/40-cm horizontal magnet equipped with a Bruker Biospec/Avance III HD console. A 1H radiofrequency mouse head coil (Bruker) was used. T2-weighted sequences were acquired using TurboRARE sequence with TR = 2,200 ms, TE = 36 ms, echo spacing = 12 ms, eight averages, and rare factor = 8. 1H MRS data were acquired using a single-voxel point-resolved spectroscopy sequence (PRESS) (repetition time = 2,500 ms, echo time = 16 ms, number of averages = 512, voxel size = 3 × 3 × 3 mm).
Mass spectrometry
The urine of n=3 mice for each group was collected and subjected to mass spectrometry to quantify N-acetylaspartate, normalized by creatinine. Creatinine and N-acetylaspartate were obtained from Cambridge Isotope Laboratories, Andover, MA, USA. The analysis was performed by the University of Massachusetts Amherst Mass spectrometry core facility.
Sample Preparation
N-acetylaspartic acid (NAA) concentration was determined using LC-MS/MS according to a method modified from Hušková et al. (38). Internal standard for NAA: 5 mg d3-NAA was dissolved in 1 mL 10% acetonitrile. 0.712 mL of this solution was made up to 100 mL with 10 % acetonitrile in water to give a final 1 x d3N stock solution of 200 uM d3-NAA. Calibrator solutions for NAA in the range 0.1 to 400 uM were used to produce a standard calibration curve for quantitation. To an aliquot of 40 uL of urine was added 160 uL of 1 x d3N in a 0.5 mL microcentrifuge tube. Samples were centrifuged at 14.5 krpm for 10 mins. The supernatant was drawn into a 1 mL disposable plastic syringe and filtered through a 0.45 um PTFE filter (Millex-LH 4mm syringe filter) into autosampler vials for LC-MS/MS analysis (see below). In cases where the measured NAA concentration was higher than the linear range, a 10-fold dilution was made, and the sample re-injected.
Creatinine concentration in urine and plasma was determined by modifying the method of Al-Dirbashi et al. (39). Internal standard for creatinine: 11.1 mg d3-creatinine was dissolved in 1 mL 10% acetonitrile. Then, 0.7 mL of this solution was made up to 100 mL with 10 % acetonitrile in water to give a final 1 x d3C stock solution of 669 uM d3-creatinine. Internal standard solutions were frozen in aliquots at −20°C for up to six months. Calibrator solutions for creatinine in the range 0.04 to 360 uM were used to produce a standard calibration curve for quantitation. Urine or plasma samples were thawed on ice. To an aliquot of 40 uL of urine was added 160 uL of 1 x d3C in a 0.5 mL microcentrifuge tube. Samples were centrifuged at 14.5 krpm for 10 mins. The supernatant was drawn into a 1 mL disposable plastic syringe and filtered through a 0.45 um PTFE filter (Millex-LH 4mm syringe filter) into autosampler vials for LC-MS/MS analysis. In cases where the measured creatinine concentration was higher than the linear range, a 10-fold dilution was made, and the sample re-injected.
LC-MS Analysis
A Waters H-Class UPLC system coupled to a Waters Xevo TQD triple quadrupole mass spectrometer was employed to analyze creatinine and NAA. Separation was performed on a Waters BEH C18 column (100 x 2.1 mm) preceded by a guard column and pre-column filter. Column temperature was 40°C. 2 uL samples were injected and separated by a gradient (A=0.1% formic acid in water, B=0.1% formic acid in acetonitrile) flow rate 0.2 mL/min consisting of 1 min hold at 5% B then stepping to 50% and holding for 3 min for a total run time of 4 min followed by re-equilibration for 4 min to starting conditions. All column effluent was directed into the ESI source of the triple quad mass spectrometer. Separate injections were used for creatinine and NAA determination. For creatinine determination, the system was operated in positive ion mode with desolvation temperature set to 300°C, source temperature 150°C, desolvation gas 300 L/hr, and source gas 20 L/hr. MRM transitions 114>73 and 114>86 were monitored for creatinine and 117>75, 117>89 for d3-creatinine with collision energy 10 eV and cone voltage 30V in each case. For NAA determination, the system was operated in negative ion mode at 3.8 kV capillary voltage, desolvation temperature 300°C, source gas 700 L/h, and cone gas 50 L/h MRM transitions for NAA 174>88 and 174>58 and 174>114 with collision energy 16, 18 and 10 eV respectively and constant cone voltage 30V. For d3-NAA, transitions were 177>91, 177>58, and 177>116. Dwell time was 25 ms, and argon was used for collision gas. Data analysis was performed using TargetLynx (Waters).
H&E Staining
Mice were euthanized and transcardially perfused with 4°C cold PBS and 4% paraformaldehyde (PFA). Tissues were excised and sliced using an Alto brain or spinal cord matrix (Roboz Surgical Instruments Inc., Gaithersburg, MD, USA). Mice tissues were stored in PFA at 4C overnight. UMass Chan Morphology Core did paraffin embedding and H&E staining. Sections were imaged using an Axioscope 50 (Zeiss) equipped with a DMC2900 camera (Leica Microsystems).
Transmission Electron Microscopy (EM)
Excised mouse brains were dissected along the interhemispheric cleft, and the anterior commissure was excised and immediately immersed in 2.5% glutaraldehyde in 0.1 M Sodium Cacodylate buffer, pH 7.2, and fixed overnight at 4 °C. Samples were processed at the UMass Chan Electron Microscopy Core following standard protocols. After fixation, samples were rinsed in the fixation buffer three times and post-fixed with 1% osmium tetroxide for 1h at room temperature. Samples were washed three times with DH2O, 20 minutes each, and were subsequently dehydrated through a graded ethanol series of 20% increments, followed by two changes in 100% ethanol. Next, samples were infiltrated with two changes of 100% Propylene Oxide and then with a 50%/50% propylene oxide / SPI-Pon 812 resin mix. Six fresh 100% SPI-Pon 812 resin changes were done the next day before polymerization at 68°C in flat embedding molds. Reorientation was done to obtain cross-sections of the anterior commissure. 1uM sections were stained with toluidine blue to confirm orientation. Approximately 70nm sections of the area were collected, positioned on copper support grids, and contrasted with Lead citrate and Uranyl acetate. Sections were studied using a CM10 TEM with 80Kv accelerating voltage, and images were taken using a Gatan TEM CCD camera. EM images were analyzed for the ratio of inner and outer diameter (g-ratio). On high magnification, myelin layers were counted per axon for each section, and the number of axons per section was quantified. At least four sections per mouse were analyzed.
In-situ single-cell mRNA staining and imaging
Mice were isoflurane-anesthetized and transcardially perfused with ice-cold 1X PBS followed by ice-cold 4% paraformaldehyde (PFA). Harvested brains were kept in 4% PFA at 4°C overnight, and then the following day, washed (x3) in 1X PBS and transferred into ice-cold, sterilely-filtered 30% sucrose. Once equilibrated, brains were embedded in a 1:2 OCT:30% sucrose mixture and frozen at −80°C until ready for sectioning. 10uM sagittal cryosections (Cryostar NX70, ThermoScientific, Waltham, MA) were collected on Superfrost Plus© microscope slides and kept at −80°C until ready for in-situ hybridization. Sections were pre-treated and stained according to the manufacturer’s RNAscope© Hiplex protocol (ACDBio, Newark, CA).
The workflow of the mRNA target detection is outlined in Figure S5A. In-situ hybridization of multiplexed Z-shaped probes was done on sagittal sections of either WT, UT, or Juv brains. Each probe contains an 18-25bp sequence complementary to the target mRNA, followed by a spacer sequence and a tail sequence complementary to the pre-amplifier. After probe binding, pre-amplifiers are added with sequences against their Z-probe tail sequence. To note, two Z-probes are required to be bound, in tandem, on the target mRNA to allow for recognition of pre-amplifier binding. Next, a series of additional amplifiers, each specific to their preceding amplifier strand sequence, is added, followed by sequence-specific fluorophores, which are also designed to bind to their preceding amplifier. This results in fluorescence detection of mRNA targets of interest, each hybridized to a pre-conjugated fluorophore, either FITC, Texas Red, or Cy5. Probes and probe reagents used for this experiment are listed in Table S1. Next, tissues were mounted using Prolong Gold Antifade Mountant with DAPI (Invitrogen P36931). Next, the whole brain sections were scanned at 20X magnification on a TissueFAXSiQ confocal slide scanner (TissueGnostics, Vienna, Austria) at the UMass Chan SCOPE Facility (RRID: SCR_022721). For multi-fluorescence detection of subsequent mRNA targets, the hybridized RNAscope© Hiplex fluorophores were irreversibly cleaved, and the next set of fluorophores was added to the tissue following the manufacturer’s protocol (ACDBio, Newark, CA).
Spatial quantification of single-cell RNAs
Slide-scanned images of whole-brain sagittal sections (n=3 per experimental group per timepoint) were imported into a spatial image analysis software (StrataQuest, TissueGnostics, Vienna, Austria). Scans from subsequent imaging rounds were cross-correlated through DAPI+ cell registration, and regions of interest: cortex, corpus callosum, hippocampus, striatum, thalamus, midbrain, cerebellum, and brainstem, were manually defined using an ROI generator. For analysis of single cells positive for fluorescent mRNA targets, nuclei masks of DAPI-stained tissues were identified using software detection. Next, a cellular mask (white outline) was generated around each nucleus to demark the perimeter of each cell body (Figure S5B). Then, to avoid false positive counts from tissue autofluorescence, detection parameters for maximum fluorescence intensity (x-axis) and total fluorescence intensity units (y-axis) were set for each channel/mRNA target, and the same settings per channel were applied to all experimental groups. Cells were then gated for the highest fluorescence intensity of mRNA puncta (Figure S5C). Finally, cell counts for all cells (white and green outlines), total mRNA+ cells (green outline only) (Figure S5D), and total DAPI+ cells were exported for quantification.
Software and statistics
Graphs were generated, and statistical calculations were performed using Prism 8 (GraphPad Software, Inc.). As stated in the legends, a 1-way or a 2-way ANOVA with multiple comparison corrections was used. P value < 0.05 was considered significant with confidence interval of 0.95, i.e. 95%. For the single-cell mRNA analysis, an alpha of 0.01 and confidence interval (CI) of 0.99, i.e. 99% was used. If not otherwise stated, a minimum of n = 3 mice per group was analyzed in each experiment.
Supplementary Material
Significance statement.
Homeostatic balance between oligodendrocytes and neurons is fundamental in maintaining neuronal conductivity and communication throughout the CNS. Functional impairments in either of these cell-types can result in neurodegenerative diseases, several of which have been linked to N-acetylaspartate metabolism between neurons and oligodendrocytes. This study demonstrates that disrupted N-acetylaspartate metabolism leads to progressive dysmyelination due to a deficiency of oligodendrocyte precursor turnover into mature myelinating cells. This in-turn augments neurodegeneration, in which a shortage of differentiated oligodendrocytes contributes to affected neuronal health. Normalizing N-acetylaspartate metabolism achieves therapeutic recovery of the oligodendrocyte lineage and promotes neuronal recovery in a time-dependent manner, ultimately leading to corrected CNS pathology.
Acknowledgment
Funding was provided by Jacob’s Cure, the National Tay-Sachs & Allied Diseases Association, Inc., Canavan’s Foundation, and NIH grants R01NS076991-01, 1P01AI100263-01, and 1P01HL131471-01.
Footnotes
Conflict of Interest Statement
G. Gao is a cofounder of biopharmaceutical companies, Voyager Therapeutics & ASPA Therapeutics and holds equities in the companies. G. Gao is an inventor on patents with potential royalties licensed to Voyager, ASPA, and other biopharmaceutical companies. D.J. Gessler is a cofounder of ASPA Therapeutics and holds equity in the company. G. Gao and D.J. Gessler are inventors on a pending patent (PCT/US2016/058197) that is relevant to the content of this manuscript, which may result in potential royalties if granted and licensed.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lappe-Siefke C, et al. , Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat Genet 33, 366–374 (2003). [DOI] [PubMed] [Google Scholar]
- 2.Fünfschilling U, et al. , Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 485, 517–521 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ciccarelli O, et al. , Pathogenesis of multiple sclerosis: insights from molecular and metabolic imaging. Lancet Neurology 13, 807–822 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Garcia L, Hacker JL, Sase S, Adang L, Almad A, Glial cells in the driver seat of leukodystrophy pathogenesis. Neurobiol Dis 146, 105087 (2020). [DOI] [PubMed] [Google Scholar]
- 5.Franklin RJM, Why does remyelination fail in multiple sclerosis? Nat Rev Neurosci 3, 705–714(2002). [DOI] [PubMed] [Google Scholar]
- 6.Mei F, et al. , Stage-Specific Deletion of Olig2 Conveys Opposing Functions on Differentiation and Maturation of Oligodendrocytes. J Neurosci 33, 8454–8462 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yu Y, et al. , Olig2 Targets Chromatin Remodelers to Enhancers to Initiate Oligodendrocyte Differentiation. Cell 152, 248–261 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang K, et al. , The Oligodendrocyte Transcription Factor 2 OLIG2 regulates transcriptional repression during myelinogenesis in rodents. Nat Commun 13, 1423 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Đặng TC, et al. , Powerful Homeostatic Control of Oligodendroglial Lineage by PDGFRα in Adult Brain. Cell Reports 27, 1073–1089.e5 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Copray S, et al. , Olig2 Overexpression Induces the In Vitro Differentiation of Neural Stem Cells into Mature Oligodendrocytes. Stem Cells 24, 1001–1010 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Wegener A, et al. , Gain of Olig2 function in oligodendrocyte progenitors promotes remyelination. Brain 138, 120–135 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gessler DJ, et al. , Redirecting N-acetylaspartate metabolism in the central nervous system normalizes myelination and rescues Canavan disease. Jci Insight 2, e90807 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lotun A, Gessler DJ, Gao G, Canavan Disease as a Model for Gene Therapy-Mediated Myelin Repair. Front Cell Neurosci 15, 661928 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adachi M, Schneck L, Cara J, Volk BW, Spongy degeneration of the central nervous system (Van Bogaert and Bertrand type; Canavan’s disease) A review. Hum Pathol 4, 331–347 (1973). [DOI] [PubMed] [Google Scholar]
- 15.Leone P, et al. , Long-Term Follow-Up After Gene Therapy for Canavan Disease. Sci Transl Med 4, 165ra163 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Prokesch A, et al. , N-acetylaspartate catabolism determines cytosolic acetyl-CoA levels and histone acetylation in brown adipocytes. Sci Rep-uk 6, 23723 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumar S, Biancotti JC, Matalon R, de Vellis J, Lack of aspartoacylase activity disrupts survival and differentiation of neural progenitors and oligodendrocytes in a mouse model of Canavan disease. J Neurosci Res 87, 3415–3427 (2009). [DOI] [PubMed] [Google Scholar]
- 18.Sohn J, et al. , Suppressing N-Acetyl-l-Aspartate Synthesis Prevents Loss of Neurons in a Murine Model of Canavan Leukodystrophy. J Neurosci 37, 413–421 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Traka M, et al. , Nur7 is a nonsense mutation in the mouse aspartoacylase gene that causes spongy degeneration of the CNS. J Neurosci 28, 11537–49 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maier H, Wang-Eckhardt L, Hartmann D, Gieselmann V, Eckhardt M, N-Acetylaspartate Synthase Deficiency Corrects the Myelin Phenotype in a Canavan Disease Mouse Model But Does Not Affect Survival Time. J Neurosci 35, 14501–14516 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park D, et al. , Nestin Is Required for the Proper Self-Renewal of Neural Stem Cells. Stem Cells 28, 2162–2171 (2010). [DOI] [PubMed] [Google Scholar]
- 22.Dahlstrand J, Lardelli M, Lendahl U, Nestin mRNA expression correlates with the central nervous system progenitor cell state in many, but not all, regions of developing central nervous system. Dev Brain Res 84, 109–129 (1995). [DOI] [PubMed] [Google Scholar]
- 23.Nogaroli L, et al. , Lysophosphatidic Acid can Support the Formation of Membranous Structures and an Increase in MBP mRNA Levels in Differentiating Oligodendrocytes. Neurochem Res 34, 182–193 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li H, Richardson WD, Evolution of the CNS myelin gene regulatory program. Brain Res 1641, 111–121 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Volkenhoff A, et al. , Glial Glycolysis Is Essential for Neuronal Survival in Drosophila. Cell Metab 22, 437–447 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Jin K, Wang X, Xie L, Mao XO, Greenberg DA, Transgenic ablation of doublecortin-expressing cells suppresses adult neurogenesis and worsens stroke outcome in mice. Proc National Acad Sci 107, 7993–7998 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Traka M, Podojil JR, McCarthy DP, Miller SD, Popko B, Oligodendrocyte death results in immune-mediated CNS demyelination. Nat Neurosci 19, 65–74 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee Y, et al. , Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 487, 443–8 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.von Jonquieres G, et al. , Uncoupling N-acetylaspartate from brain pathology: implications for Canavan disease gene therapy. Acta Neuropathol 135, 95–113 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baslow MH, Hypothesis: Molecular water pumps and the aetiology of Canavan disease: A case of the sorcerer’s apprentice. J Inherit Metab Dis 22, 99–101 (1999). [DOI] [PubMed] [Google Scholar]
- 31.Appu AP, et al. , Increasing N-acetylaspartate in the Brain during Postnatal Myelination Does Not Cause the CNS Pathologies of Canavan Disease. Front Mol Neurosci 10, 161 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Samanta J, et al. , Inhibition of Gli1 mobilizes endogenous neural stem cells for remyelination. Nature 526, 448–452 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kotter MR, Li W-W, Zhao C, Franklin RJM, Myelin Impairs CNS Remyelination by Inhibiting Oligodendrocyte Precursor Cell Differentiation. J Neurosci 26, 328–332 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xing YL, et al. , Adult Neural Precursor Cells from the Subventricular Zone Contribute Significantly to Oligodendrocyte Regeneration and Remyelination. J Neurosci 34, 14128–14146 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Duggal N, Schmidt-Kastner R, Hakim AM, Nestin expression in reactive astrocytes following focal cerebral ischemia in rats. Brain Res 768, 1–9 (1997). [DOI] [PubMed] [Google Scholar]
- 36.Mersmann N, et al. , Aspartoacylase-lacZ knockin mice: an engineered model of Canavan disease. Plos One 6, e20336 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun X, et al. , Molecular analysis of vector genome structures after liver transduction by conventional and self-complementary adeno-associated viral serotype vectors in murine and nonhuman primate models. Hum Gene Ther 21, 750–61 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huskova R, Chrastina P, Adam T, Schneiderka P, Determination of creatinine in urine by tandem mass spectrometry. Clin Chim Acta 350, 99–106 (2004). [DOI] [PubMed] [Google Scholar]
- 39.Al-Dirbashi OY, et al. , Stable isotope dilution analysis of N-acetylaspartic acid in urine by liquid chromatography electrospray ionization tandem mass spectrometry. Biomed Chromatogr 21, 898–902 (2007). [DOI] [PubMed] [Google Scholar]
- 40.Singhal NK, et al. , The neuronal metabolite NAA regulates histone H3 methylation in oligodendrocytes and myelin lipid composition. Experimental Brain Research 235, 279–292 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim KK, et al. , Identification of Neuronal Nuclei (NeuN) as Fox-3, a New Member of the Fox-1 Gene Family of Splicing Factors. The Journal of Biological Chemistry 45, 31052–31061 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Watson AES, et al. , Fractalkine signaling regulates oligodendroglial genesis from SVZ precursor cells. PNAS 16, 1968–1984 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Park TI, et al. , Adult Human Brain Neural Progenitor Cells (NPCs) and Fibroblast-Like Cells Have Similar Properties In Vitro but Only NPCs Differentiate into Neurons. PLoS One 7(6), e37742 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Y, et al. , Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 89, 37–53 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pontillo G, et al. , Unraveling Deep Gray Matter Atrophy and Iron and Myelin Changes in Multiple Sclerosis. American Journal of Neuroradiology 42(7), 1223–1230 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Eykens C, et al. , AAV9-mediated Gene Delivery of MCT1 to Oligodendrocytes Does Not Provide a Therapeutic Benefit in a Mouse Model of ALS. Molecular Therapy 20, 508–519 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ozgur-Gunes Y, et al. , Long-Term Disease Prevention with a Gene Therapy Targeting Oligodendrocytes in a Mouse Model of Adrenomyeloneuropathy. Human Gene Therapy 33, 17–18 (2022). [DOI] [PubMed] [Google Scholar]
- 48.Chung D, Shum A, Caraveo G, GAP-43 and BASP1 in Axon Regeneration: Implications for the Treatment of Neurodegenerative Diseases. Frontiers in Cell and Developmental Biology 8, e567537 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.