

Abstract
Environmental exposure to polycyclic aromatic hydrocarbons (PAH) has been shown to be associated with chronic disease outcomes through multiple mechanisms including altered regulation of the transcription factor peroxisome proliferator-activated receptor gamma (Ppar) γ. Because PAH exposure and Pparγ each have been associated with mammary cancer, we asked whether PAH would induce altered regulation of Pparγ in mammary tissue, and whether this association may underlie the association between PAH and mammary cancer. Pregnant mice were exposed to aerosolized PAH at proportions that mimic equivalent human exposures in New York City air. We hypothesized that prenatal PAH exposure would alter Pparγ DNA methylation and gene expression and induce the epithelial to mesenchymal transition (EMT) in mammary tissue of offspring (F1) and grandoffspring (F2) mice. We also hypothesized that altered regulation of Pparγ in mammary tissue would associate with biomarkers of EMT, and examined associations with whole body weight. We found that prenatal PAH exposure lowered Pparγ mammary tissue methylation among grandoffspring mice at postnatal day (PND) 28. However, PAH exposure did not associate with altered Pparγ gene expression or consistently with biomarkers of EMT. Finally, lower Pparγ methylation, but not gene expression, was associated with higher body weight among offspring and grandoffspring mice at PND28 and PND60. Findings suggest additional evidence of multi-generational adverse epigenetic effects of prenatal PAH exposure among grandoffspring mice.
Keywords: polycyclic aromatic hydrocarbons, prenatal exposure, mammary cancer, peroxisome proliferator-activated receptor gamma, epithelial to mesenchymal transition, multigenerational epigenetic effects
1. Introduction:
Exposure to polycyclic aromatic hydrocarbon (PAH) air pollutants has been associated with the development of both obesity(Rundle et al., 2012; Rundle et al., 2019; Yan et al., 2014) and breast cancer.(Gammon et al., 2002; Rundle et al., 2000; Sahay et al., 2019; Shen et al., 2017; White et al., 2014) While the mechanisms remain unclear, exposure to PAH has been shown to alter gene expression of the master regulator of adipogenesis peroxisome proliferator-activated receptor (Ppar) γ through direct ligand binding and through activation of the aryl hydrocarbon receptor (AhR).(Podechard et al., 2009; Yun et al., 2017) Previously, we have shown that the link between PAH exposure and obesity may occur through altered regulation of Pparγ in adipose tissue.(Yan et al., 2014) Whether this pathway may relate to breast cancer risk still needs to elucidated, as both the relationship between body weight and breast cancer risk, and between altered Pparγ regulation and cancer, remain complex.(Kotta-Loizou et al., 2012; Yun et al., 2018) For example, higher body weight has been shown to be either a protective factor or an adverse risk factor for breast cancer depending on the menopausal status and disease subtype.(Naik et al., 2019; Picon-Ruiz et al., 2017) Also, the presence of breast adiposity has been shown to lower the risk of developing breast cancer among pre- and post-menopausal women.(Pettersson et al., 2014; Soguel et al., 2017) Pparγ expression also has been found to be upregulated(Kotta-Loizou et al., 2012) and downregulated(Jiang et al., 2003) in breast tumors. Further, in vitro studies have demonstrated that Pparγ expression promotes(Ansari et al., 2022; Si et al., 2020) and inhibits(Xu et al., 2021) the epithelial to mesenchymal transition (EMT) in breast cancer cell lines.
Prenatal exposure to PAH may occur during a time period responsible for a higher risk of cancer subsequently in postnatal life. (Baur et al., 2021; Ekbom et al., 1992; Kehm et al., 2021; Sanderson et al., 1996; Weiss et al., 1997) The prenatal period, along with the pubertal, pregnancy, and menopause life stages, are times of heightened sensitivity to environmental exposures because they are characterized by breast tissue development and change.(Terry et al., 2019) Experimental and epidemiological studies have demonstrated the importance of environmental exposures specifically during the prenatal period to the development of breast cancer during subsequent adulthood.(Sahay et al., 2021; Terry et al., 2019) For example, in a population-based case-control study in western New York State, compared to postmenopausal women without breast cancer, postmenopausal women with breast cancer had 2.42 the odds of exposure to the highest concentrations of total suspended particles at birth, which were used as a proxy for PAH exposure.(Bonner et al., 2005) Also, newborn benzo[a]pyrene-DNA adducts levels have been found to be similar to that of the mothers, even though the transplacental dose of PAH to the fetus was 10 times lower than the delivered exposure to the mother.(Perera et al., 2005) This latter finding suggests that during the prenatal period, the susceptibility to PAH induced DNA damage could be much greater for the fetus than for the mother.
We asked whether a link between prenatal airborne PAH exposure and mammary cancer also may occur through altered regulation of Pparγ methylation and expression in mammary tissue. Our approach was to focus on the neoplastic biomarker of the EMT as a clinically relevant outcome.(Buyuk et al., 2022; Papadaki et al., 2019) EMT has been shown to precede tumor formation in mouse models of mammary cancer(Ye et al., 2015) and specifically in atypical ductal hyperplasia,(Hüsemann et al., 2008) a precancerous lesion. Here, the effects of prenatal PAH exposure on mammary Pparγ gene regulation and on EMT were measured following aerosolized exposures of PAH to mice at levels that mimic ambient conditions in New York City.(Rundle et al., 2012) We hypothesized that prenatal PAH exposure would lower Pparγ methylation and raise Pparγ gene expression as well as induce EMT in mammary tissue of offspring and grandoffspring mice during the prepubertal (postnatal day (PND) 28) and adult premenopausal (PND 60) age periods.(Silver, 1995) Secondarily, we hypothesized that altered Pparγ regulation in mammary tissue would associate with biomarkers of EMT in offspring and grandoffspring mice. We explored whether these latter relationships would strengthen following prenatal PAH exposure or differ by whole body weight.
2. Materials and Methods:
2.1. Animals
BALB/cByj female mice (Charles River Laboratories) were housed at Columbia University Irving Medical Center in a temperature, humidity and light controlled environment with ad libitum access to water and a phytoestrogen-free diet (D16020901 containing 22 kcal% fat and 23 kcal% protein, Research Diets, New Brunswick, NJ).(Sahay et al., 2021; Yan et al., 2014) Following at least 8–10 days of acclimatization, mice were mated. Offspring mice were allowed to wean until PND21. Males were sacrificed on PND21. Female offspring and grandoffspring mice were weighed individually at PND28 and PND60 as previously described.(Yan et al., 2014) Animal experiments were carried out in strict accordance with the principles and procedures of the National Research Council’s Guide for the Care and Use of Laboratory Animals and institutional guidelines.
2.2. PAH exposure
Exposure of the female dams to the PAH mixture was delivered through a customized chamber as developed by the Lovelace Respiratory Research Institute.(Chu et al., 2013; Miller et al., 2016; Sahay et al., 2021; Yan et al., 2014) The control aerosol consisted of 99.97% purified water, 0.02% Tween 80 and 0.01% antifoam (Sigma-Aldrich, St. Louis, MO). The mixed PAH solution had a concentration of 7.29 ng/m3 (3.69 ng/m3 pyrene, plus 3.60 ng/m3 from 8 other individual PAH (benz(a)anthracene, benzo(a)pyrene, benzo(b)fluoranthene, benzo(k)fluoranthene, benzo(g,h,i)perylene, chrysene, dibenz(a,h)anthracene and indeno(1,2,3,-CD)pyrene). The levels were intended to replicate the proportions of individual PAH that was measured among a cohort of NYC women participating in the Columbia Center for Children’s Environmental Health birth cohort using personal air sampling devices.(Perera et al., 2003; Yan et al., 2014) The aerosol was delivered beginning on gestational day (GD) 1–3 through GD 19–21 or until day of delivery for five hours a day, five days a week as described.(Chu et al., 2013; Yan et al., 2014)
2.3. Tissue dissection and immunohistochemistry
Immediately following euthanization via CO2 inhalation, mammary glands were removed using a dissection microscope, fixed in formalin, and embedded in paraffin. Immunohistochemistry (IHC) was performed using the Ventana Discovery Ultra instrument (Roche). This system allows for automated baking, deparaffinization and cell conditioning.(Basu et al., 2020) We focused on E-cadherin, a cell-cell adhesion molecule that is silenced by Snail, Slug, Twist, and ZEB1/2, the primary transcription factors involved in the regulation of epithelial and mesenchymal genes,(Wang, Zhou, 2013) as a biomarker of the epithelial state. We also focused on vimentin, a filament protein that promotes cell motility,(Battaglia et al., 2018) as a biomarker of the mesenchymal state. Staining was performed using anti-E-cadherin (1:500 dilution, Abcam, Cambridge, UK) and anti-vimentin (1:500 dilution, Abcam, Cambridge, UK) antibodies for 60 min. Secondary antibody Discovery OMNIMap anti-mouse-HRP (Roche) was used and the brown signal was obtained using the Discovery ChromoMap 3, 3’-diaminobenzidine (DAB) kit (Roche). Tissues were counterstained with hematoxylin in blue to visualize the nuclei. The percent of cells stained by DAB was assessed using the positive cell detection classifier function in Qupath(Baranova et al., 2021) in six random images per mouse. For 5 of the 33 mice (randomly selected) for which IHC was performed, a second masked reader annotated the mammary glands and used the positive cell detection classifier function. The intra-class correlation assessed agreement between readers as 0.99.
2.4. RNA extraction and quantitative RT PCR
RNA was extracted from mammary gland tissue using the RNeasy mini kit (Qiagen Sciences, Germantown, Maryland, USA). RNA concentration and purity were measured using a NanoDrop spectrophotometer (Thermo Scientific, Wilmington, DE, USA). 200 ng of RNA was transcribed using the iScript™ cDNA synthesis kit (Biorad, Hercules, CA, USA). Quantitative real-time PCR was performed using a 25-μl reaction volume containing 2 μl cDNA, 12.5 μl SYBR Green Mix (Biorad, Hercules, CA, USA), 9.5 μl H20, and 1 μl of forward and reverse primer for Pparγ, vimentin, and E-cadherin (Supplemental table 1). Amplifications were performed in duplicate with an initial incubation at 95 °C for 30 s, followed by 40 cycles of 95 °C for 10 s and 60 °C for 30 s, using a CFX Connect Real-Time PCR Detection System (Bio-Rad, Hercules, CA, USA). Duplicate samples were run together on the same plate. If the difference between duplicate samples exceeded 2 quantification cycle values, the sample was reanalyzed. Following amplification, a melt curve was used with 5 seconds each at increments of 0.5°C between 65°C and 95°C to confirm amplicon specificity. To minimize batch effects, samples from the control and exposure groups were run in equal proportions on cDNA synthesis and RT PCR plates.
2.5. DNA isolation and pyrosequencing
DNA was isolated from mammary gland tissue using the PureLink™ Genomic DNA Mini Kit (Invitrogen, Waltham, Massachusetts, USA). Genomic DNA concentration and purity were measured using the NanoDrop spectrophotometer. Pooled mammary tissue DNA from mice in the experiment that were not included in this study was incubated with M.Sssl CpG methyltransferase enzyme to create methylated control DNA (New England Biolabs, Ipswich, MA, USA). To create unmethylated control DNA, the REPLI-g Mini Kit (Qiagen Sciences, Germantown, Maryland, USA) was used to perform whole genome amplification of pooled mammary DNA, a process that causes the product to lose DNA methylation. 200 ng of DNA was bisulfite converted using the EZ DNA Methylation-Lightning Kit (Zymo Research, Orange, CA, USA). PCR and pyrosequencing primers (Supplemental Table 2) were designed using PyroMark Assay Design 2.0 software (Qiagen, Valencia, CA, USA). Three CpG sites (CpG-189, CpG-205, and CpG-241) in the promoter region of the Pparγ gene were analyzed based on previous reports.(Fujiki et al., 2009; Kamstra et al., 2014) PCR was performed with Qiagen Hot Star Taq DNA polymerase (Qiagen Sciences, Germantown, MD, USA) with the following concentrations: 1× PCR buffer, 1.5 μM MgCl2, 200 μM dNTP, 0.2 μM forward primer, and 0.2 μM reverse primer. The PCR was performed under the following conditions: 94 °C, 15 min; 45 cycles of 94 °C, 30 s; 56 °C, 30 s; 72 °C, 30 s; 72 °C, 5 min; and 4 °C hold. The PCR product was sequenced using the PyroMark Q96 MD. The PCR and pyrosequencing experiments were run with the methylated and unmethylated control DNA and a water blank. Samples from the control and exposure groups were run in equal proportions across bisulfite conversion, PCR, and pyrosequencing experiments to minimize batch effects.
2.6. Statistical analysis
Linear regression and t tests were used to assess for an association between PAH exposure and Pparγ promoter methylation and gene expression and vimentin and E-cadherin protein expression. Linear regression also was performed to assess whether Pparγ methylation or expression levels associated with vimentin and E-cadherin protein expression or with body weight. The latter analysis was repeated following stratification by PAH exposure group. Values of p<0.05 were considered statistically significant. All analyses were performed using IBM SPSS Statistics 24. Graphs were generated using the GraphPad Prism 9 software.
3. Results:
We asked whether prenatal PAH exposure would alter epigenetic regulation in the Pparγ promoter region in mammary tissue of female offspring and grandoffspring mice at both a mouse adolescent (PND28) and premenopausal (PND 60) time period. We found that PAH exposure lowered Pparγ methylation at CpG-189 and to a borderline extent at CpG-241 among grandoffspring mice at PND28 (Figure 1). However, PAH exposure did not alter methylation in the mammary glands of grandoffspring mice at PND60 nor among offspring mice at either time point (Figure 1 and Table 1). We also did not observe differences in mean Pparγ methylation between exposed and unexposed offspring mice at either timepoint and grandoffspring mice at PND60 in t test analyses. In contrast to our hypothesis, PAH exposure did not affect corresponding Pparγ relative gene expression in mammary tissue of grandoffspring mice at PND60 or offspring mice at either timepoint (Table 1). Pparγ methylation also did not correlate with Pparγ gene expression at any generation or timepoint (data not shown).
Figure 1.
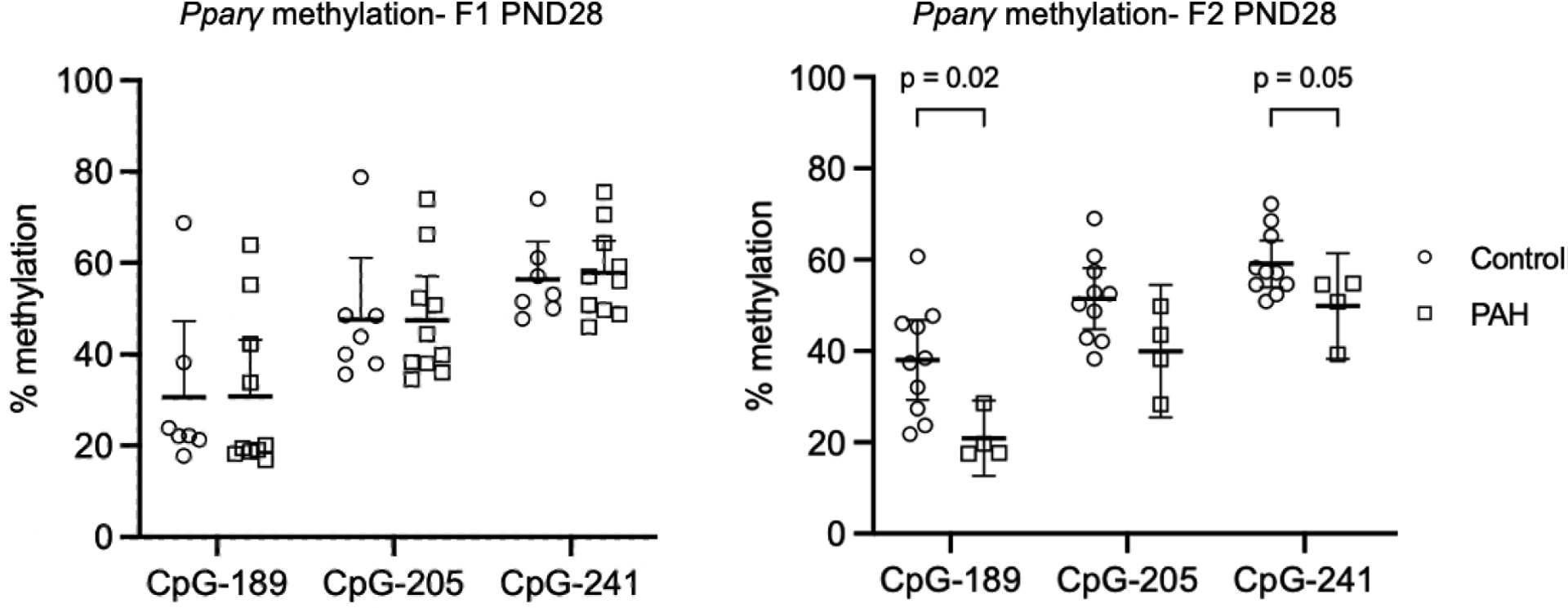
Prenatal PAH exposure lowered Pparγ methylation among grandoffspring (F2) mice at PND28, but not among offspring (F1) mice at PND28. Percent CpG methylation in mammary tissue of three sites (CpG-189, CpG-205, and CpG-241) in the Pparγ promoter region are shown following control (open circles) and PAH exposure (open squares) among F1 PND28 (control n = 7; PAH n = 10) and F2 mice at PND28 (control n = 10; PAH n = 10). Mean and 95% confidence intervals are marked. The p values for the association between prenatal PAH exposure and Pparγ methylation are shown for significant and borderline significant associations. Analyses were performed by t test. Abbreviations: PND: postnatal day, F1: first generation offspring, F2: second generation offspring.
Table 1.
Prenatal PAH is not associated with altered Pparγ relative gene expression among offspring (F1), grandoffspring (F2) mice, nor with Pparγ promoter methylation among offspring, grandoffspring mice at PND60. The B coefficient, 95% confidence intervals, and p values for the association between prenatal PAH exposure and Pparγ promoter methylation and relative gene expression are shown. Analyses were performed by linear regression. The linear regression analyses among mice at PND28 were not significant (data not shown).
| Generation + Timepoint | Sample Size | Ppary relative gene expression | Ppary methylation | ||||||
|---|---|---|---|---|---|---|---|---|---|
| CpG-189 | CpG-205 | CpG-241 | |||||||
| B Coefficient (95% CI) | P value | B Coefficient (95% CI) | P value | B Coefficient (95% CI) | P value | B Coefficient (95% CI) | P value | ||
| F1 PND60 | 21 | 0.01 (−0.53, 0.56) | 0.95 | 0.38 (−1.22, 14.26) | 0.09 | 0.36 (−1.40, 12.03) | 0.11 | 0.25 (−2.20, 7.44) | 0.27 |
| F2 PND60 | 16 | −0.27 (−1.12, 0.38) | 0.31 | 0.19 (−7.06, 14.02) | 0.49 | 0.29 (−4.15, 13.61) | 0.27 | 0.24 (−5.08, 12.72) | 0.37 |
We predicted that in the mammary tissue of mice exposed to PAH, the mesenchymal marker vimentin would be upregulated.(Shin et al., 2006) The epithelial marker E-cadherin could have been downregulated or upregulated, reflecting either a complete mesenchymal transition or a hybrid epithelial/mesenchymal state, that has been shown to occur in embryonic development and in circulating tumor cells of patients with breast cancer.(Armstrong et al., 2011; Nieto, 2013) However, we did not find an association between PAH exposure and vimentin or E-cadherin gene or protein expression (Tables 2,3).
Table 2.
Prenatal PAH exposure is not associated with vimentin or E-cadherin protein expression in offspring and grandoffspring mice at PND60. The B coefficient, 95% confidence intervals, and p values are shown for the association between prenatal PAH exposure and vimentin and e-cadherin protein expression among F1 PND60 and F2 PND60 mice.
| Generation + Time Point | Sample Size | Vimentin protein expression | E-cadherin protein expression | ||
|---|---|---|---|---|---|
| B Coefficient (95% CI) | P value | B Coefficient (95% CI) | P value | ||
| F1 PND60 | 21 | 0.28 (−15.70, 49.57) | 0.29 | 0.36 (−7.94, 45.03) | 0.16 |
| F2 PND60 | 16 | −0.12 (−40.12, 26.37) | 0.66 | 0.32 (−13.51, 50.59) | 0.24 |
Table 3.
Prenatal PAH exposure is not associated with vimentin or E-cadherin relative gene expression in offspring and grandoffspring mice. The B coefficient, 95% confidence intervals, and p values are shown for the association between prenatal PAH exposure and vimentin and e-cadherin relative gene expression among F1 PND28, F1 PND60, F2 PND28, and F2 PND60 mice.
| Generation + Time Point | Sample Size | Vimentin mRNA expression | E-cadherin mRNA expression | ||
|---|---|---|---|---|---|
| B Coefficient (95% CI) | P value | B Coefficient (95% CI) | P value | ||
| F1 PND28 | 17 | −0.31 (−3.12, 0.78) | 0.22 | 0.16 (−2.87, 5.17) | 0.55 |
| F1 PND60 | 21 | 0.1 (−1.08, 1.64) | 0.67 | −0.14 (−4.41, 2.39) | 0.54 |
| F2 PND28 | 14 | 0 (−3.39, 3.38) | 1 | 0.08 (−3.86, 5.00) | 0.78 |
| F2 PND60 | 16 | −0.16 (−2.46, 1.37) | 0.55 | 0.05 (−2.98, 3.57) | 0.85 |
Moreover, because Pparγ has been shown to be involved in tumorigenesis,(Lee et al., 2019) we next examined its association with biomarkers of EMT. Pparγ methylation at CpG-205 was associated with higher vimentin protein expression among offspring and grandoffspring mice at PND60 (B coefficient 0.36, 95% CI 0.04, 2.99, p = 0.05, n = 37) and among grandoffspring mice at PND60 at CpG-189 (B coefficient 0.53, 95% CI 0.06, 3.13, p = 0.04, n = 16) and CpG-241 (B coefficient 0.53, 95% CI 0.08, 3.50, p = 0.04, n = 16). Pparγ methylation was associated with higher E-cadherin gene expression among offspring mice at PND28 at CpG-241 (B coefficient 0.51, 95% CI 0.01, 0.40, p = 0.04, n = 17) and offspring and grandoffspring mice at PND28 and PND60 at CpG-205 (B coefficient 0.24, 95% CI 0.00, 0.17, p = 0.05, n = 68), and CpG-241 (B coefficient 0.29, 95% CI 0.02, 0.24, p = 0.02, n = 68). Despite the positive association between Pparγ methylation and vimentin protein expression, and in line with our hypothesis, Pparγ gene expression also was associated with higher vimentin gene expression among offspring mice at PND28 (B coefficient 0.54, 95% CI 0.22, 2.77, p = 0.02, n = 17) and among offspring and grandoffspring mice at PND28 and PND60 (B coefficient 0.27, 95% CI 0.10, 1.33, p = 0.02, n = 68).
Our group previously observed that lower Pparγ methylation in white and brown adipose tissue was associated with higher body weight among offspring and grandoffspring mice at PND60.(Yan et al., 2014) We sought to explore whether similar altered regulation would occur in mouse mammary tissue, also composed of brown and white adipose tissue, at PND28 and 60. (Colleluori et al., 2021) We found here that lower Pparγ mammary methylation at CpG-189, −205, and −241 was associated with higher body weight among offspring and grandoffspring mice at PND28 and 60 (CpG-189: B coefficient −0.29, 95% CI −0.16, −0.02, p = 0.02; CpG-205: B coefficient −0.29, 95% CI −0.19, −0.02, p = 0.02; CpG-241: B coefficient −0.28, 95% CI −0.25, −0.02, p = 0.02, n = 68). Contrary to our past findings of an association between PAH exposure and body weight,(Yan et al., 2014) comparable relationships were not detected in these models except that higher Pparγ methylation at CpG-205 was associated with higher body weight among grandoffspring mice at PND28 in the absence of PAH exposure (control exposure group B coefficient 0.65, 95% CI 0.00, 0.07, p = 0.04, n = 14). Also contrary to our hypothesis, the association between Pparγ gene expression and body weight became significant among offspring mice at both age points in the absence of PAH exposure (control mice: F1 PND28: B coefficient 0.82, 95% CI 0.05, 0.49, p = 0.03, n = 7; F1 PND60 B coefficient 0.72, 95% CI 0.29, 2.52, p = 0.02, n = 10).
4. Discussion:
We found that higher PAH exposure was associated with lower Pparγ methylation in mammary tissue among grandoffspring mice at PND28. This finding occurred in the absence of differences in Pparγ gene expression or consistent expression of biomarkers of EMT. Lower Pparγ methylation in the mammary tissue but not altered gene expression, was associated with higher body weight across two generations of mice. Prenatal exposure to PAH did not alter this relationship.
Epigenetic regulation of Pparγ, as for other genes, is known to be tissue specific.(Long et al., 2016) Accordingly, our finding of altered epigenetic regulation of Pparγ following prenatal PAH exposure in the mammary tissue among young offspring mice is novel. Interestingly, we observed altered epigenetic regulation of Pparγ among grandoffspring mice at the younger PND28 and not the older PND60 timepoint. PND28 represents the prepubescent adolescent life stage and PND60 represents the adult life stage prior to reproductive senescence.(Silver, 1995) Previous studies have demonstrated altered Pparγ promoter methylation in mice and rats in both the prepubertal(Gong et al., 2015) and adult(Yan et al., 2014) life stages in response to perinatal and prenatal environmental exposures. While methylation can induce stable alterations,(Ladd-Acosta, Fallin, 2019) our study design allowed us to ascertain that the effects of PAH on mammary methylation of Pparγ may not endure during adult mouse aging.(Mulder et al., 2021)
Interestingly, while the offspring mice were exposed to PAH in utero and the grandoffspring mice as germ cells of the offspring mice,(Xin et al., 2015) we observed altered epigenetic regulation of Pparγ in the grandoffspring generation only. Rodent studies have demonstrated that exposure to a variety of environmental chemicals can induce changes in DNA methylation in germ cells of prenatally exposed animals.(Pacchierotti, Spanò, 2015) While some timing and routes of exposure and some environmental toxicants have shown stronger effects on DNA methylation in the somatic cells of prenatally exposed animals,(Stouder et al., 2011) others have shown stronger effects in germ cells.(Somm et al., 2013) The periods of epigenomic reprogramming have been proposed as windows of susceptibility for environmental exposures and may affect the somatic and germ cells of offspring differently. (Pilsner et al., 2017) The first period of epigenomic reprogramming occurs in all cells of the embryo prior to implantation and the second occurs in primordial germ cells only.(Pilsner et al., 2017) As a result, exposure during the first reprogramming period may affect DNA methylation in somatic and germ cells equally, whereas exposure during the second period may only affect DNA methylation in germ cells.
The finding of lowered Pparγ methylation among grandoffspring mice at PND28 in the absence of a change in gene expression is contrary to findings from our group(Yan et al., 2014) and others(Chiu et al., 2021; Motawi et al., 2017) that have demonstrated that methylation is an important regulator of Pparγ gene transcription. In our previous mouse model of prenatal PAH exposure, we used the same model of exposure but looked at Pparγ methylation and expression in adipose tissue, as opposed to in mammary tissue.(Yan et al., 2014) It is possible that prenatal PAH exposure may affect Pparγ expression in adipose tissue differently than in mammary tissue, in line with the primary role of Pparγ in regulating adipogenesis.(Lee et al., 2019) Additionally, other CpG sites in the Pparγ promoter region may be more important in determining Pparγ expression than the CpG sites measured here. For instance, in our previous study, we observed lower Pparγ methylation at CpG-303 and CpG-195, but not CpG-189, at PND60 in adipose tissue of offspring and grandoffspring mice that were prenatally exposed to PAH. (Yan et al., 2014) In both studies, we did not observe a change in Pparγ methylation at CpG-189 among offspring or grandoffspring mice at PND60, although here we found lower methylation at CpG-189 among grandoffspring mice at PND28, a timepoint that was not measured in the previous study. Alternately, the discordance may be explained by pre- and post-transcriptional regulation of Pparγ gene expression in mammary tissue not measured here.(Lee et al., 2019) For instance, DNA hydroxymethylation was not measured but may have exerted oppositional effects on transcription.(Paluch et al., 2016) Another possibility is that silencing microRNAs (miRNAs), such as miRNAs 27b, 130b, and 138 that previously have been shown to lower Pparγ gene and protein expression, may have exerted unmeasured effects.(Motawi et al., 2017)
The absence of an association between prenatal PAH exposure and markers of EMT in mammary tissue may indicate that the altered methylation measured here was insufficient to drive the expression of EMT genes and proteins. Alternately, the prenatal PAH exposure, possibly as a result of the timing or insufficient dose of exposure, may not induce EMT in offspring and grandoffspring mice. Future studies that use a higher dose of exposure and that examine DNA methylation changes at more of the CpG sites in the Pparγ promoter region may be able to fill in that research gap. Although an association between PAH exposure and markers of EMT was absent, we observed select associations between Pparγ and biomarkers of EMT. To our knowledge, the regulation of EMT by Pparγ in mammary cells has only been studied previously in vitro. These studies demonstrated that Pparγ gene expression was necessary both to promote(Ansari et al., 2022; Si et al., 2020) and inhibit(Xu et al., 2021) EMT in breast cancer cell lines. Our novel in vivo findings were similarly contrasting. Pparγ has been shown to have pro-tumorigenic(Knower et al., 2013) and antineoplastic effects,(Apostoli et al., 2014) possibly based on the cell compartment in which Pparγ is expressed.(Avena et al., 2013) In a xenograft model, Pparγ expression in breast cancer cells inhibited tumor growth, whereas Pparγ expression in stromal cells promoted tumor growth.(Avena et al., 2013) More studies are needed to understand how Pparγ expression affects premalignant tissue.
Our finding of a relationship between lower Pparγ methylation and higher body weight is in line with studies that have found that the action of Pparγ in promoting adipogenesis is strongly regulated by epigenetic modification.(Motawi et al., 2017; Volberg et al., 2017) The explanation for the absence of an effect on mammary Pparγ gene transcription still needs to be elucidated, and similarly may indicate other forms of transcriptional regulation (Motawi et al., 2017; Paluch et al., 2016) or small effect sizes with low statistical power. An additional limitation of the study is that we had low statistical power to determine whether associations between Pparγ methylation and gene expression and body weight was strengthened following PAH exposure. It is possible that our relatively small sample sizes did not allow us to detect PAH induced small changes in Pparγ methylation and gene expression, and resulting changes in biomarkers of EMT, that we may have been able to detect with larger sample sizes. While our findings may indicate a lack of association, they also may indicate that our study lacked power to detect a true effect of PAH exposure. We also only examined female offspring and grandoffspring mice, and previous research suggests that the PAH effect on body weight may be more pronounced in males.(Kehm et al., 2021) We also acknowledge that EMT was not measured directly, and we relied on biomarkers of unproven robustness in this mouse model. Another limitation of the study is that we only examined 3 CpG sites in the Pparγ promoter region.
A strength of our study is that we investigated PAH exposure during the prenatal period when the risk induced by environmental exposures on subsequent offspring breast cancer may be heightened.(Terry et al., 2019) A second strength of our study was the physiological and airborne delivery of PAH at concentrations measured in urban air,(Chu et al., 2013; Rundle et al., 2012; Sahay et al., 2021; Yan et al., 2014) in attempt to replicate real world conditions.
5. Conclusions:
In conclusion, this study provides evidence that PAH exposure lowers Pparγ methylation in mammary tissue among grandoffspring of mice exposed to PAH during pregnancy. However, there was no evidence of an association between PAH exposure and Pparγ gene expression. Pparγ did associate inconsistently with markers of EMT and with higher body weight suggesting other potential clinically relevant outcomes. Overall, these results suggest some multigenerational epigenetic effects of PAH exposure. Further studies may be warranted to understand the full clinical consequences and underlying mechanisms of PAH exposure.
Supplementary Material
Prenatal PAH exposure lowered Pparγ methylation among grandoffspring mice
Lower Pparγ promoter methylation associated with higher body weight
PAH exposure did not associate with altered Pparγ gene expression
Evidence of multi-generational adverse epigenetic effects of prenatal PAH exposure
Acknowledgements:
The Biorepository and Pathology Core at the Icahn School of Medicine at Mount Sinai performed immunohistochemistry.
This work was supported by Breast Cancer and Environment Research Program (BCERP) initiative of NIEHS, grant number: 1U01ES026122.
Abbreviations:
- PAH
Polycyclic aromatic hydrocarbon
- Pparγ
Peroxisome proliferator activated receptor gamma
- EMT
Epithelial to mesenchymal transition
- AhR
Aryl hydrocarbon receptor
- PND
Postnatal day
- GD
Gestational day
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The protocol was approved by the Institutional Animal Care and Use Committee, Columbia University Irving Medical Center
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References:
- Ansari MI, Bano N, Kainat KM, Singh VK, Sharma PK, 2022. Bisphenol A exposure induces metastatic aggression in low metastatic MCF-7 cells via PGC-1α mediated mitochondrial biogenesis and epithelial-mesenchymal plasticity. Life Sci. 302, 1206–1249. 10.1016/j.lfs.2022.120649. [DOI] [PubMed] [Google Scholar]
- Apostoli AJ, Skelhorne-Gross GE, Rubino RE, Peterson NT, Di Lena MA, Schneider MM, SenGupta SK, Nicol CJ, 2014. Loss of PPARγ expression in mammary secretory epithelial cells creates a pro-breast tumorigenic environment. Int J Cancer. 134 (5), 1055–1066. 10.1002/ijc.28432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong AJ, Marengo MS, Oltean S, Kemeny G, Bitting RL, Turnbull JD, Herold CI, Marcom PK, George DJ, Garcia-Blanco MA, 2011. Circulating tumor cells from patients with advanced prostate and breast cancer display both epithelial and mesenchymal markers. Mol Cancer Res. 9 (8), 997–1007. 10.1158/1541-7786.Mcr-10-0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avena P, Anselmo W, Whitaker-Menezes D, Wang C, Pestell RG, Lamb RS, Hulit J, Casaburi I, Andò S, Martinez-Outschoorn UE, Lisanti MP, Sotgia F, 2013. Compartment-specific activation of PPARγ governs breast cancer tumor growth, via metabolic reprogramming and symbiosis. Cell Cycle. 12 (9), 1360–1370. 10.4161/cc.24289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranova K, Tran C, Plantinga P, Sangle N, 2021. Evaluation of an open-source machine-learning tool to quantify bone marrow plasma cells. J Clin Pathol. 74 (7), 462–468. 10.1136/jclinpath-2021-207524. [DOI] [PubMed] [Google Scholar]
- Basu A, Chiriboga L, Narula N, Zhou F, Moreira AL, 2020. Validation of PD-L1 clone 22C3 immunohistochemical stain on two Ventana DISCOVERY autostainer models: detailed protocols, test performance characteristics, and interobserver reliability analyses. J Histotechnol. 43 (4), 174–181. 10.1080/01478885.2020.1823105. [DOI] [PubMed] [Google Scholar]
- Battaglia RA, Delic S, Herrmann H, Snider NT, 2018. Vimentin on the move: new developments in cell migration. F1000Res. 7 (F1000 Faculty Rev), 1796. 10.12688/f1000research.15967.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baur B, Lee DI, Haag J, Chasman D, Gould M, Roy S, 2021. Deciphering the Role of 3D Genome Organization in Breast Cancer Susceptibility. Front Genet. 12, 788318. 10.3389/fgene.2021.788318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonner MR, Han D, Nie J, Rogerson P, Vena JE, Muti P, Trevisan M, Edge SB, Freudenheim JL, 2005. Breast cancer risk and exposure in early life to polycyclic aromatic hydrocarbons using total suspended particulates as a proxy measure. Cancer Epidemiol Biomarkers Prev. 14 (1), 53–60. Published 2005/01/26. [PubMed] [Google Scholar]
- Buyuk B, Jin S, Ye K, 2022. Epithelial-to-mesenchymal transition signaling pathways responsible for breast cancer metastasis. Cell Mol Bioeng. 15 (1), 1–13. 10.1007/s12195-021-00694-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu KC, Sisca F, Ying JH, Tsai WJ, Hsieh WS, Chen PC, Liu CY, 2021. Prenatal chlorpyrifos exposure in association with PPARγ H3K4me3 and DNA methylation levels and child development. Environ Pollut. 274, 116511. 10.1016/j.envpol.2021.116511. [DOI] [PubMed] [Google Scholar]
- Chu S, Zhang H, Maher C, McDonald JD, Zhang X, Ho SM, Yan B, Chillrud S, Perera F, Factor P, Miller RL, 2013. Prenatal and postnatal polycyclic aromatic hydrocarbon exposure, airway hyperreactivity, and Beta-2 adrenergic receptor function in sensitized mouse offspring. J Toxicol. 2013, 603581. 10.1155/2013/603581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colleluori G, Perugini J, Barbatelli G, Cinti S, 2021. Mammary gland adipocytes in lactation cycle, obesity and breast cancer. Rev Endocr Metab Disord. 22 (2), 241–255. 10.1007/s11154-021-09633-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekbom A, Trichopoulos D, Adami HO, Hsieh CC, Lan SJ, 1992. Evidence of prenatal influences on breast cancer risk. Lancet. 340 (8826), 1015–1018. 10.1016/0140-6736(92)93019-j. [DOI] [PubMed] [Google Scholar]
- Fujiki K, Kano F, Shiota K, Murata M, 2009. Expression of the peroxisome proliferator activated receptor gamma gene is repressed by DNA methylation in visceral adipose tissue of mouse models of diabetes. BMC Biol. 7, 38. 10.1186/1741-7007-7-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gammon MD, Santella RM, Neugut AI, Eng SM, Teitelbaum SL, Paykin A, Levin B,Terry MB, Young TL, Wang LW, Wang Q, Britton JA, Wolff MS, Stellman SD, Hatch M, Kabat GC, Senie R, Garbowski G, Maffeo C, Montalvan P,Berkowitz G, Kemeny M, Citron M, Schnabel F, Schuss A, Hajdu S, Vinceguerra V, 2002. Environmental toxins and breast cancer on Long Island. I. Polycyclic aromatic hydrocarbon DNA adducts. Cancer Epidemiol Biomarkers Prev. 11 (8), 677–685. [PubMed] [Google Scholar]
- Gong M, Liu J, Sakurai R, Corre A, Anthony S, Rehan VK, 2015. Perinatal nicotine exposure suppresses PPARγ epigenetically in lung alveolar interstitial fibroblasts. Mol Genet Metab. 114 (4), 604–612. 10.1016/j.ymgme.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hüsemann Y, Geigl JB, Schubert F, Musiani P, Meyer M, Burghart E, Forni G, Eils R, Fehm T, Riethmüller G, Klein CA, 2008. Systemic spread is an early step in breast cancer. Cancer Cell. 13 (1), 58–68. 10.1016/j.ccr.2007.12.003. [DOI] [PubMed] [Google Scholar]
- Jiang WG, Douglas-Jones A, Mansel RE, 2003. Expression of peroxisome-proliferator activated receptor-gamma (PPARgamma) and the PPARgamma co-activator, PGC-1, in human breast cancer correlates with clinical outcomes. Int J Cancer. 106 (5), 752–757. 10.1002/ijc.11302. [DOI] [PubMed] [Google Scholar]
- Kamstra JH, Hruba E, Blumberg B, Janesick A, Mandrup S, Hamers T, Legler J, 2014. Transcriptional and epigenetic mechanisms underlying enhanced in vitro adipocyte differentiation by the brominated flame retardant BDE-47. Environ Sci Technol. 48 (7), 4110–4119. 10.1021/es405524b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehm RD, Oskar S, Tehranifar P, Zeinomar N, Rundle AG, Herbstman JB, Perera F, Miller RL, Terry MB, 2021. Associations of prenatal exposure to polycyclic aromatic hydrocarbons with pubertal timing and body composition in adolescent girls: Implications for breast cancer risk. Environ Res. 196, 110369. 10.1016/j.envres.2020.110369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knower KC, Chand AL, Eriksson N, Takagi K, Miki Y, Sasano H, Visvader JE,Lindeman GJ, Funder JW, Fuller PJ, Simpson ER, Tilley WD, Leedman PJ, Graham J, Muscat GE, Clarke CL, Clyne CD, 2013. Distinct nuclear receptor expression in stroma adjacent to breast tumors. Breast Cancer Res Treat. 142 (1), 211–223. 10.1007/s10549-013-2716-6. [DOI] [PubMed] [Google Scholar]
- Kotta-Loizou I, Giaginis C, Theocharis S, 2012. The role of peroxisome proliferator-activated receptor-γ in breast cancer. Anticancer Agents Med Chem. 12 (9), 1025–1044. 10.2174/187152012803529664. [DOI] [PubMed] [Google Scholar]
- Ladd-Acosta C, Fallin MD, 2019. DNA methylation signatures as biomarkers of prior environmental exposures. Curr Epidemiol Rep. 6 (1), 1–13. 10.1007/s40471-019-0178-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JE, Schmidt H, Lai B, Ge K, 2019. Transcriptional and epigenomic regulation of adipogenesis. Mol Cell Biol. 39 (11), e00601–00618. 10.1128/mcb.00601-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long AS, Lemieux CL, Arlt VM, White PA, 2016. Tissue-specific in vivo genetic toxicity of nine polycyclic aromatic hydrocarbons assessed using the Muta™Mouse transgenic rodent assay. Toxicol Appl Pharmacol. 290, 31–42. 10.1016/j.taap.2015.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RL, Yan Z, Maher C, Zhang H, Gudsnuk K, McDonald J, Champagne FA, 2016. Impact of prenatal polycyclic aromatic hydrocarbon exposure on behavior, cortical gene expression and DNA methylation of the BDNF gene. Neuroepigenetics. 5, 11–18. 10.1016/j.nepig.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motawi TK, Shaker OG, Ismail MF, Sayed NH, 2017. Peroxisome proliferator-activated receptor gamma in obesity and colorectal cancer: the role of epigenetics. Sci Rep. 7 (1), 11180–11186. 10.1038/s41598-017-11180-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder RH, Neumann A, Cecil CAM, Walton E, Houtepen LC, Simpkin AJ,Rijlaarsdam J, Heijmans BT, Gaunt TR, Felix JF, Jaddoe VWV, Bakermans-Kranenburg MJ, Tiemeier H, Relton CL, van IMH, Suderman M, 2021. Epigenome-wide change and variation in DNA methylation in childhood: trajectories from birth to late adolescence. Hum Mol Genet. 30 (1), 119–134. 10.1093/hmg/ddaa280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik A, Monjazeb AM, Decock J, 2019. The obesity paradox in cancer, tumor immunology, and immunotherapy: potential therapeutic implications in triple negative breast cancer. Front Immunol. 10, 1940. 10.3389/fimmu.2019.01940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto MA, 2013. Epithelial plasticity: a common theme in embryonic and cancer cells. Science. 342 (6159), 1234850. 10.1126/science.1234850. [DOI] [PubMed] [Google Scholar]
- Pacchierotti F, Spanò M, 2015. Environmental impact on DNA methylation in the germline: state of the art and gaps of knowledge. Biomed Res Int. 2015, 123484. 10.1155/2015/123484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paluch BE, Naqash AR, Brumberger Z, Nemeth MJ, Griffiths EA, 2016. Epigenetics: A primer for clinicians. Blood Rev. 30 (4), 285–295. 10.1016/j.blre.2016.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadaki MA, Stoupis G, Theodoropoulos PA, Mavroudis D, Georgoulias V, Agelaki S, 2019. Circulating tumor cells with stemness and epithelial-to-mesenchymal transition features are chemoresistant and predictive of poor outcome in metastatic breast cancer. Mol Cancer Ther. 18 (2), 437–447. 10.1158/1535-7163.Mct-18-0584. [DOI] [PubMed] [Google Scholar]
- Perera F, Tang D, Whyatt R, Lederman SA, Jedrychowski W, 2005. DNA damage from polycyclic aromatic hydrocarbons measured by benzo[a]pyrene-DNA adducts in mothers and newborns from Northern Manhattan, the World Trade Center Area, Poland, and China. Cancer Epidemiol Biomarkers Prev. 14 (3), 709–714. 10.1158/1055-9965.Epi-04-0457. [DOI] [PubMed] [Google Scholar]
- Perera FP, Rauh V, Tsai WY, Kinney P, Camann D, Barr D, Bernert T, Garfinkel R, Tu YH, Diaz D, Dietrich J, Whyatt RM, 2003. Effects of transplacental exposure to environmental pollutants on birth outcomes in a multiethnic population. Environ Health Perspect. 111 (2), 201–205. 10.1289/ehp.5742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersson A, Graff RE, Ursin G, Santos Silva ID, McCormack V, Baglietto L, Vachon C, Bakker MF, Giles GG, Chia KS, Czene K, Eriksson L, Hall P, Hartman M, Warren RM, Hislop G, Chiarelli AM, Hopper JL, Krishnan K, Li J, Li Q, Pagano I, Rosner BA, Wong CS, Scott C, Stone J, Maskarinec G, Boyd NF, van Gils CH, Tamimi RM, 2014. Mammographic density phenotypes and risk of breast cancer: a meta-analysis. J Natl Cancer Inst. 106 (5). 10.1093/jnci/dju078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picon-Ruiz M, Morata-Tarifa C, Valle-Goffin JJ, Friedman ER, Slingerland JM, 2017. Obesity and adverse breast cancer risk and outcome: mechanistic insights and strategies for intervention. CA Cancer J Clin. 67 (5), 378–397. 10.3322/caac.21405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilsner JR, Parker M, Sergeyev O, Suvorov A, 2017. Spermatogenesis disruption by dioxins: epigenetic reprograming and windows of susceptibility. Reprod Toxicol. 69, 221–229. 10.1016/j.reprotox.2017.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podechard N, Fardel O, Corolleur M, Bernard M, Lecureur V, 2009. Inhibition of human mesenchymal stem cell-derived adipogenesis by the environmental contaminant benzo(a)pyrene. Toxicol In Vitro. 23 (6), 1139–1144. 10.1016/j.tiv.2009.05.011. [DOI] [PubMed] [Google Scholar]
- Rundle A, Hoepner L, Hassoun A, Oberfield S, Freyer G, Holmes D, Reyes M, Quinn J, Camann D, Perera F, Whyatt R, 2012. Association of childhood obesity with maternal exposure to ambient air polycyclic aromatic hydrocarbons during pregnancy. Am J Epidemiol. 175 (11), 1163–1172. 10.1093/aje/kwr455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rundle A, Tang D, Hibshoosh H, Estabrook A, Schnabel F, Cao W, Grumet S, Perera FP, 2000. The relationship between genetic damage from polycyclic aromatic hydrocarbons in breast tissue and breast cancer. Carcinogenesis. 21 (7), 1281–1289. [PubMed] [Google Scholar]
- Rundle AG, Gallagher D, Herbstman JB, Goldsmith J, Holmes D, Hassoun A, Oberfield S, Miller RL, Andrews H, Widen EM, Hoepner LA, Perera F, 2019. Prenatal exposure to airborne polycyclic aromatic hydrocarbons and childhood growth trajectories from age 5–14 years. Environ Res. 177, 108595. 10.1016/j.envres.2019.108595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahay D, Lloyd SE, Rivera JA, Jezioro J, McDonald JD, Pitiranggon M, Yan B, Szabolcs M, Terry MB, Miller RL, 2021. Prenatal polycyclic aromatic hydrocarbons, altered ERα pathway-related methylation and expression, and mammary epithelial cell proliferation in offspring and grandoffspring adult mice. Environ Res. 196, 110961. 10.1016/j.envres.2021.110961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahay D, Terry MB, Miller R, 2019. Is breast cancer a result of epigenetic responses to traffic-related air pollution? A review of the latest evidence. Epigenomics. 11 (6), 701–714. 10.2217/epi-2018-0158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson M, Williams MA, Malone KE, Stanford JL, Emanuel I, White E, Daling JR, 1996. Perinatal factors and risk of breast cancer. Epidemiology. 7 (1), 34–37. 10.1097/00001648-199601000-00007. [DOI] [PubMed] [Google Scholar]
- Shen J, Liao Y, Hopper JL, Goldberg M, Santella RM, Terry MB, 2017. Dependence of cancer risk from environmental exposures on underlying genetic susceptibility: an illustration with polycyclic aromatic hydrocarbons and breast cancer. Br J Cancer. 116 (9), 1229–1233. 10.1038/bjc.2017.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin SR, Sánchez-Velar N, Sherr DH, Sonenshein GE, 2006. 7,12-dimethylbenz(a)anthracene treatment of a c-rel mouse mammary tumor cell line induces epithelial to mesenchymal transition via activation of nuclear factor-kappaB. Cancer Res. 66 (5), 2570–2575. 10.1158/0008-5472.Can-05-3056. [DOI] [PubMed] [Google Scholar]
- Si L, Fu J, Liu W, Hayashi T, Nie Y, Mizuno K, Hattori S, Fujisaki H, Onodera S, Ikejima T, 2020. Silibinin inhibits migration and invasion of breast cancer MDA-MB-231 cells through induction of mitochondrial fusion. Mol Cell Biochem. 463 (1–2), 189–201. 10.1007/s11010-019-03640-6. [DOI] [PubMed] [Google Scholar]
- Silver LM Mouse genetics: concepts and applications. In: Oxford University Press; 1995: http://www.informatics.jax.org/silver/index.shtml. [Google Scholar]
- Soguel L, Durocher F, Tchernof A, Diorio C, 2017. Adiposity, breast density, and breast cancer risk: epidemiological and biological considerations. Eur J Cancer Prev. 26 (6), 511–520. 10.1097/cej.0000000000000310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somm E, Stouder C, Paoloni-Giacobino A, 2013. Effect of developmental dioxin exposure on methylation and expression of specific imprinted genes in mice. Reprod Toxicol. 35, 150–155. 10.1016/j.reprotox.2012.10.011. [DOI] [PubMed] [Google Scholar]
- Stouder C, Somm E, Paoloni-Giacobino A, 2011. Prenatal exposure to ethanol: a specific effect on the H19 gene in sperm. Reprod Toxicol. 31 (4), 507–512. 10.1016/j.reprotox.2011.02.009. [DOI] [PubMed] [Google Scholar]
- Terry MB, Michels KB, Brody JG, Byrne C, Chen S, Jerry DJ, Malecki KMC, Martin MB, Miller RL, Neuhausen SL, Silk K, Trentham-Dietz A, 2019. Environmental exposures during windows of susceptibility for breast cancer: a framework for prevention research. Breast Cancer Res. 21 (1), 96. 10.1186/s13058-019-1168-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volberg V, Yousefi P, Huen K, Harley K, Eskenazi B, Holland N, 2017. CpG methylation across the adipogenic PPARγ gene and its relationship with birthweight and child BMI at 9 years. BMC Med Genet. 18 (1), 7. 10.1186/s12881-016-0365-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhou BP, 2013. Epithelial-mesenchymal transition—a hallmark of breast cancer metastasis. Cancer Hallm. 1 (1), 38–49. 10.1166/ch.2013.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss HA, Potischman NA, Brinton LA, Brogan D, Coates RJ, Gammon MD, Malone KE, Schoenberg JB, 1997. Prenatal and perinatal risk factors for breast cancer in young women. Epidemiology. 8 (2), 181–187. 10.1097/00001648-199703000-00010. [DOI] [PubMed] [Google Scholar]
- White AJ, Teitelbaum SL, Stellman SD, Beyea J, Steck SE, Mordukhovich I, McCarty KM, Ahn J, Rossner P Jr., Santella RM, Gammon MD, 2014. Indoor air pollution exposure from use of indoor stoves and fireplaces in association with breast cancer: a case-control study. Environ Health. 13, 108. 10.1186/1476-069x-13-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin F, Susiarjo M, Bartolomei MS, 2015. Multigenerational and transgenerational effects of endocrine disrupting chemicals: A role for altered epigenetic regulation? Semin Cell Dev Biol. 43, 66–75. 10.1016/j.semcdb.2015.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Liu M, Yang Y, Wei C, Zhang X, Song H, Wang Y, Duan X, 2021. VSP-17 suppresses the migration and invasion of triple-negative breast cancer cells through inhibition of the EMT process via the PPARγ/AMPK signaling pathway. Oncol Rep. 45 (3), 975–986. 10.3892/or.2020.7916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Zhang H, Maher C, Arteaga-Solis E, Champagne FA, Wu L, McDonald JD, Yan B, Schwartz GJ, Miller RL, 2014. Prenatal polycyclic aromatic hydrocarbon, adiposity, peroxisome proliferator-activated receptor (PPAR) γ methylation in offspring, grand-offspring mice. PLoS One. 9 (10), e110706. 10.1371/journal.pone.0110706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X, Tam WL, Shibue T, Kaygusuz Y, Reinhardt F, Ng Eaton E, Weinberg RA, 2015. Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature. 525 (7568), 256–260. 10.1038/nature14897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun SH, Han SH, Park JI, 2018. Peroxisome proliferator-activated receptor γ and PGC-1α in cancer: dual actions as tumor promoter and suppressor. PPAR Res. 2018, 6727421. 10.1155/2018/6727421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun Y, Gao R, Yue H, Liu X, Li G, Sang N, 2017. Polycyclic aromatic hydrocarbon (PAH)-containing soils from coal gangue stacking areas contribute to epithelial to mesenchymal transition (EMT) modulation on cancer cell metastasis. Sci Total Environ. 580, 632–640. 10.1016/j.scitotenv.2016.12.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.