

Abstract
Background:
Myocardial infarction (MI) is a major risk factor for the development of heart failure with reduce ejection fraction (HFrEF). While previous studies have focused on HFrEF, the cardiovascular effects of ketone bodies in acute MI are unclear. We examined the effects of oral ketone supplementation as a potential treatment strategy in a swine acute MI model.
Methods:
Farm pigs underwent percutaneous balloon occlusion of the LAD for 80 minutes followed by 72 hours reperfusion period. Oral ketone ester or vehicle was administered during reperfusion and continued during the follow-up period.
Results:
Oral KE supplementation induced ketonemia 2–3 mmol/l within 30 minutes after ingestion. KE increased ketone(βHB) extraction in healthy hearts without affecting glucose and fatty acid (FA) consumption. During reperfusion, the MI hearts consumed less FA with no change in glucose consumption, whereas hearts from MI-KE-fed animals consumed more βHB and FA, as well as improved myocardial ATP production. A significant elevation of infarct T2 values indicative of inflammation was found only in untreated MI group compared to sham. Concordantly, cardiac expression of inflammatory markers, oxidative stress, and apoptosis were reduced by KE. RNA-seq analysis identified differentially expressed genes related to mitochondrial energy metabolism and inflammation.
Conclusions:
Oral KE supplementation induced ketosis and enhanced myocardial βHB extraction in both healthy and infarcted hearts. Acute oral supplementation with KE favorably altered cardiac substrate uptake and utilization, improved cardiac ATP levels, and reduced cardiac inflammation following MI.
Keywords: ketone ester, ketone, inflammation, metabolism, myocardial infarction, ischemia reperfusion injury
Graphical abstract
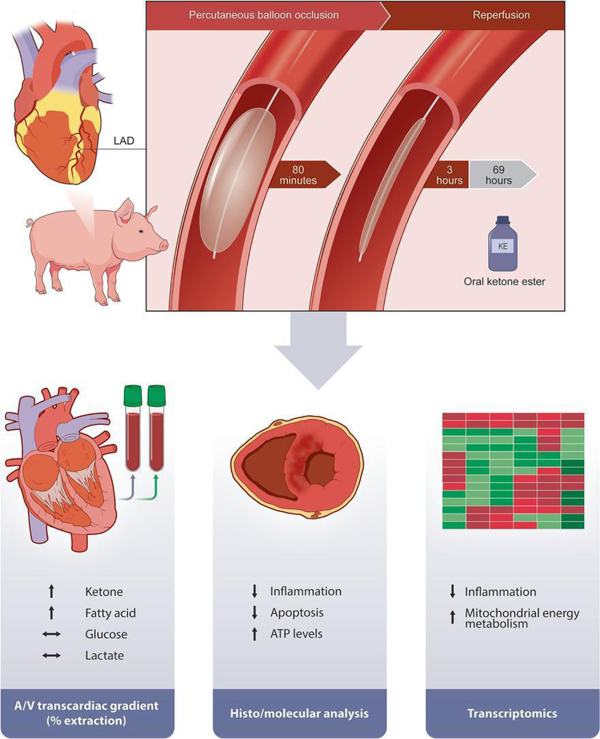
INTRODUCTION
Myocardial infarction (MI) refers to acute myocardial injury due to insufficient perfusion.[1] In the United States alone, it is estimated that someone suffers a MI every 40 seconds.[2] Despite the tremendous advances in the care of MI patients over the past two decades with reperfusion (either by primary percutaneous coronary intervention, fibrinolytic/ thrombolytic therapy or surgery) being the standard of care, MI remains the leading cause of mortality and morbidity worldwide as well as the main cause of heart failure with reduced ejection fraction (HFrEF).[3, 4]
Paradoxically, reperfusion may cause further damage to the myocardium. Reperfusion injury is another form of damage to the heart tissue that occurs following an MI caused by the sudden return of blood flow through the myocardium after an ischemic episode.[5] Rapid loss of cardiomyocytes following MI activates an intense inflammatory response. Of note, the inflammatory response to acute MI likely facilitates further injury as well as promoting adverse cardiac remodeling and eventual heart failure (HF).[5, 6] As such, efforts to develop new therapeutic strategies to promote timely resolution of the inflammatory response and prevent the subsequent post-MI HF are warranted.
Ketone bodies are endogenous metabolites that are linked to multiple biological processes,[7] including inflammation. Previous studies conducted in vitro, in animal models and in humans have revealed that ketones may exert anti-inflammatory properties in different tissues.[8–12] Some evidence in animals and humans also suggests that ketone supplementation improves cardiac function and ameliorates cardiac remodeling in chronic, post-MI HF,[13, 14] but the acute effects of ketone bodies in MI have not been established. As accumulating evidence indicates that timely resolution of inflammation may prevent adverse cardiac remodeling and post-MI HF,[6] we tested the hypothesis that oral ketone ester (KE) supplementation has early beneficial effects on the early phase of post-MI remodeling in swine.
METHODS
Animals
Male Yorkshire pigs were purchased from Tufts Cummings School of Veterinary Medicine (Boston, MA). The experimental protocol was approved by the IACUC at Massachusetts General Hospital (protocol number: 2019000253). The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication no. 85–23, revised 1996). We followed ARRIVE guidelines when reporting this study.
Ketone administration
Ketone ester (KE, βHB-butanediol monoester, ΔG®) was purchased from TDeltaS Global, Inc. (Oxfordshire, UK). KE was provided via drinking water by mixing the KE (550 mg/kgBW, twice daily) with non-caloric artificial sweetener sucralose (Splenda®) to mask the bitterness. This dose was previously tested and shown to stimulate ketonemia in humans.[15] KE is available on the market, it has been used in animals[13] and been shown to be safe and well tolerated in humans.[16]
Animal studies
Pigs were anesthetized and instrumented as described previously.[17] First, animals were sedated using tiletamine/zolazepam. Animals were then intubated, and anesthesia was maintained with 2% isoflurane. For the first cohort (dose determination study in healthy swine), simultaneous blood sampling from arterial and coronary sinus (CS) through cardiac catheterization was performed ~1 hour after KE ingestion to assess cardiac extraction of major substrates (i.e., FA, glucose and βHB). The percentage of cardiac extraction (uptake) of each substrate is calculated as the difference between the concentration in the coronary sinus and that in the artery (arterial-coronary sinus/ arterial concentration*100).[18] In the second cohort, to create a reproducible model of MI, a percutaneous transluminal coronary angioplasty dilation catheter was inflated to occlude flow distal to the second diagonal branch of the left anterior descending coronary artery (LAD). Coronary angiography was performed to visualize localization of the occlusion and patency of the second diagonal branch was ensured by contrast agent injection. The balloon was deflated after 80 minutes of ischemia. The sham animals underwent the same procedures, but without inflating the balloon. At the onset of reperfusion (~2.5 hours post-MI), after all animals were fully awake, animals were randomized to receive oral KE (MI-KE group) or vehicle (MI group) and continued for 72 hours. Simultaneous blood sampling from arterial and CS and cardiac magnetic resonance imaging (CMR) studies were performed on day 3 post-MI. Pigs were then subsequently sacrificed, and the hearts were collected for further analyses. Molecular analyses were performed in border zone region. All measurements were analyzed by a blinded investigator and were validated by external observer, when appropriate.
Cardiac magnetic resonance imaging (CMR)
Seventy-two hours post-MI, CMR imaging studies were performed on a 3 Tesla clinical scanner (Magnetom Prisma, Siemens Healthineers, Erlangen, Germany) Throughout the imaging procedures, anesthesia was maintained with 2% isoflurane. The CMR protocols included balanced steady-state free precession (bSSFP) cine sequences in CINE sequences in long axis view (4 and 2 chambers), as well as short axis views to cover the entire LV (1.4 × 1.4 × 6 mm3; electrocardiogram-gated; 35 cardiac phases; TR/TE = 3.2/1.6 ms; flip angle 50°). Commercially available T1 and T2 mapping sequences were included in the examination using standard parameters from Society of Cardiovascular Magnetic Resonance guidelines.[19] Late gadolinium enhancement (LGE) imaging was acquired 15 minutes after the gadolinium contrast agent injection (0.1 mmol/kg, Gadovist, Bayer Healthcare, Berlin, German) using phase sensitive inversion recovery FLASH (1.3 × 1.3 × 6 mm3; TR/TE/TI = 362/1.5/335 ms; flip angle 20°). The segmentation of left ventricle to quantify the LV function and infarct size was performed on Segment software (Medviso, Sweden), which has been 510 k FDA approved.
Blood measurements
βHB monitoring was performed daily, pen-side, using an Abbott Precision Xtra® handheld ketone meter (Abbott, Columbus, OH). Plasma βHB levels were quantified using Autokit 3-HB (Wako Chemicals, Germany). FA levels were analyzed by Animal Health Diagnostic Center at Cornell University. Glucose levels and other blood biochemistry panels were analyzed by Mass General Hospital Center for Comparative Research.
Quantitative real-time PCR
RNA was extracted from the border zone using TRIzol reagent (Invitrogen Corp., Carlsbad, CA, USA), as previously described[20] and the NanoDrop device (Thermo Scientific, Waltham, MA, USA) was used to measure RNA concentration. Random primer mix was used to prepare first-stranded DNA and thereafter used as a template for quantitative real-time reverse-transcriptase-PCR (qRT-PCR) (25 ng/reaction). mRNA levels obtained by a qRT-PCR using QuantStudio™ 5 Real-Time PCR System (Applied Biosystems™, Waltham, MA, USA). 36B4 reference gene were used to correct all measured mRNA expression. Primer sequences can be found in supplementary table S1.
ATP measurements
The ATP analysis was performed in the LV (border zone) using the ATP Assay Kit (colorimetric/fluorometric) from Abcam (#ab83355, Cambridge, UK) according to the manufacturer’s instructions. Results were then normalized by protein concentrations of each test, as previously described.
Histology
Pig hearts were fixed in 10% formalin solution and prepared for paraffin-embedding. After deparaffinization and rehydration, prior to TUNEL staining, heat induced antigen retrieval was performed (Retrievagen A (pH6.0) 550524, BD Biosciences) and paraffin-embedded tissue sections were incubated with TUNEL reaction mixture (In Situ Cell Death Detection Kit, TMR Red, 12156792910, MillipreSigma) for 1 hour at 37°C. For macrophage immunofluorescent staining (CD68), the sections blocked with 4% normal horse serum in PBS and a mouse anti-pig macrophages antibody (1:50, clone BA4D5, Cat# MCA2317GA, Bio-Rad) was incubated at 4°C overnight. A biotinylated horse anti-mouse IgG secondary antibody and streptavidin DyLight 594 (1:100, BA-2000 and 1:600, SA-5594, Vector Laboratories) were applied to detect pig macrophages. The tissue sections were counterstained with DAPI (1:3000, D21490, Thermo Fisher Scientific) and all the slides were scanned by a digital scanner NanoZoomer 2.0RS (Hamamatsu, Japan). The extent of TUNEL is normalized to total nuclear content using DAPI staining.
RNA sequencing (RNAseq)
RNA sequencing of the LV tissue (border zone) was performed by Novogene (Novogene Corporation Inc 8801 Folsom BLVD, Suite 290, Sacramento, CA) with Hiseq-4000 and libraries were sequenced using Illumina NovaSeq platforms (n=3 per group, MI-KE vs. MI). Differential expression analysis was performed using DESeq2. Raw p values were adjusted for multiple testing using the Benjamini-Hochberg procedure. Pathway analyses was performed using the gene ontology resource. Enriched biological processes and molecular functions classified according to gene ontology (GO) terms, and KEGG pathways were examined.
Statistical analyses
Data are presented as means ± standard errors of the mean (SEM) with all individual data points displayed. To compare normally distributed parameters, 1-way ANOVA followed by Tukey’s Post Hoc test was used. When skew distributed, a non-parametric Kruskal-Wallis test followed by a Dunn test with correction for multiple comparisons was used. Unpaired T-test or Mann-Whitney U test were applied to test the difference between 2 groups. Differences were considered significant at p < 0.05. GraphPad Prism for Mac (San Diego, CA, USA) was used to perform all statistical analysis. RNAseq and GSEA data analyses were performed with R and DESeq2 software, as described in Section RNA-seq. The implemented data analyzes methods had been discussed and consulted with an independent statistics consulting service.
RESULTS
We performed percutaneous balloon occlusion to induce MI or sham surgery on a total of 22 male Yorkshire pigs (40–50 kg). Seven pigs (31%) died on the table during MI procedure due to ventricular fibrillation and could not be resuscitated. The final group sizes were 4 for sham group, 6 for MI-veh group (MI group) and 5 for MI-KE group.
Oral KE supplementation induces ketosis and increases myocardial ketone extraction in healthy swine
First, we sought to develop a KE supplementation protocol that induces ketonemia in healthy pigs. The pigs received either vehicle drink or drink that was supplemented with the KE (550 mg/kg BW). 30 minutes after ingesting oral KE supplementation, circulating βHB were significantly elevated in pigs treated with KE compared with vehicle group (2.8 vs. 0.1 mmol/L, Figure 1A). We next sought to determine whether oral KE supplementation would change myocardial substate extraction. As expected, simultaneous blood samples taken from the coronary sinus and artery revealed a significant transcardiac extraction of βHB, without affecting the extraction of glucose or FA (Figure 1B–D).
Figure 1. Ketone ester increases circulating β-hydroxybutyrate (βHB) and myocardial βHB extraction in pigs.
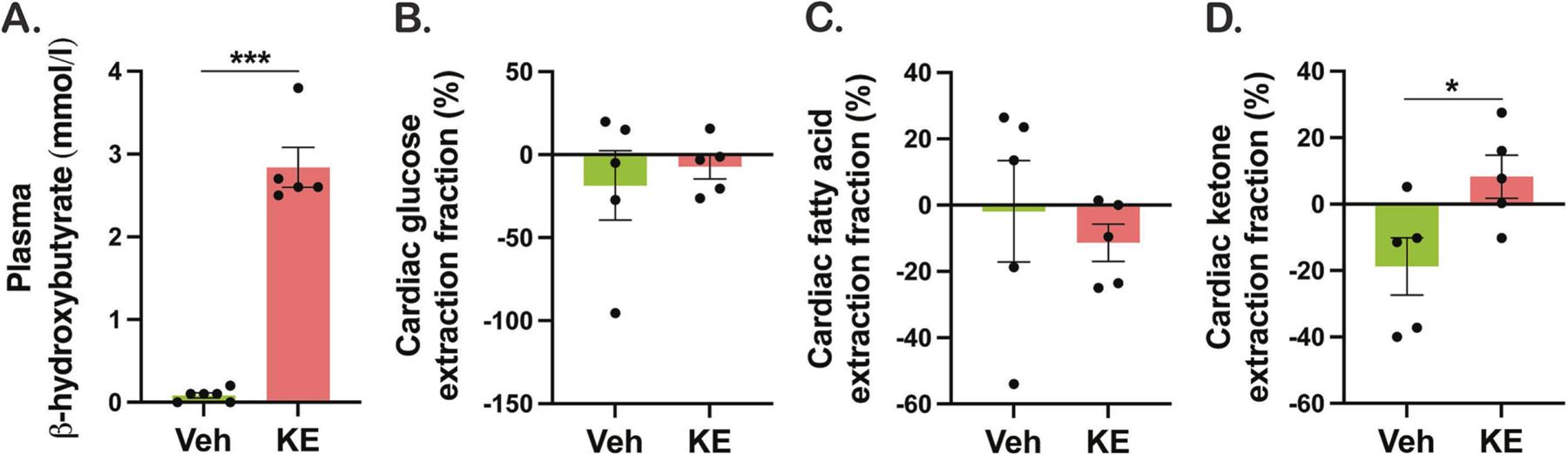
A. Plasma βHB concentrations measured 30 minutes after oral ketone ester ingestion. Cardiac extraction of (B) glucose, (C) fatty acid, and (D) βHB, calculated as the difference between the concentration in the coronary sinus and that in the artery. Veh indicates vehicle and KE, ketone ester. *p < 0.05, **p < 0.01, ***p < 0.001
CMR measures of myocardial tissue characteristics in post-MI swine
We next sought to determine the effects or oral KE supplementation in pigs after myocardial infarction. In our study, treatment with KE did not affect the clinical biochemistry parameters obtained from blood analysis (Table 1). A schematic representation of the treatment strategy with KE is depicted in Figure 2A. CMR analysis indicated that KE did not affect infarct size and cardiac function 72 hours post-MI. The MI size visualized by LGE-CMR was comparable between MI and MI-KE group (Figure 2B, Table 2). MI surgery resulted in a trend reduction in LVEF, but this trend did not reach statistical significance and did not differ between MI groups. T1 and T2 mapping were also performed to quantify the myocardial tissue characterization. No significant difference was found in focal T1 value in infarct and scar area among the groups (data not shown). Interestingly, the mean infarct zone T2 value was higher in MI-KE group, this was less than the increase seen in MI alone, and not significantly higher than sham (Figure 2C, p<0.05), whereas remote zone T2 remained comparable among the groups (Table 2). Other CMR parameters were comparable between MI and MI-KE treated animals (p>0.05 for all). All other relevant CMR parameters were depicted in Table 2.
Table 1. Clinical biochemistry parameters of swine blood samples.
| Parameters | Sham | MI | MI-KE |
|---|---|---|---|
| Sodium (mEq/l) | 141 ± 0.8 | 139 ± 0.9 | 140 ± 0.3 |
| Potassium (mEq/l) | 3.8 ± 0 | 3.7 ± 0.4 | 3.7 ± 0.1 |
| Chloride (mEq/l) | 100 ± 0.6 | 97 ± 1.2 | 97 ± 0.8 |
| Bicarbonate (mEq/l) | 28 ± 0.9 | 30 ± 0.8 | 29 ± 0.8 |
| Anion gap (mEq/l) | 18 ± 0.7 | 17 ± 0.3 | 18 ± 0.6 |
| Creatinine (mg/dl) | 1.1 ± 0 | 1.0 ± 0 | 0.0 ± 0.0 |
| Calcium (mg/dl) | 10.9 ± 0 | 10.6 ± 0.3 | 10.9 ± 0.1 |
| Phosphate (mg/dl) | 7.9 ± 0.5 | 8.1 ± 0.4 | 8.0 ± 0.5 |
N=4–5/group. Data are presented as means ± SEM.
Figure 2. CMR measures of myocardial tissue characteristics in post-MI swine.
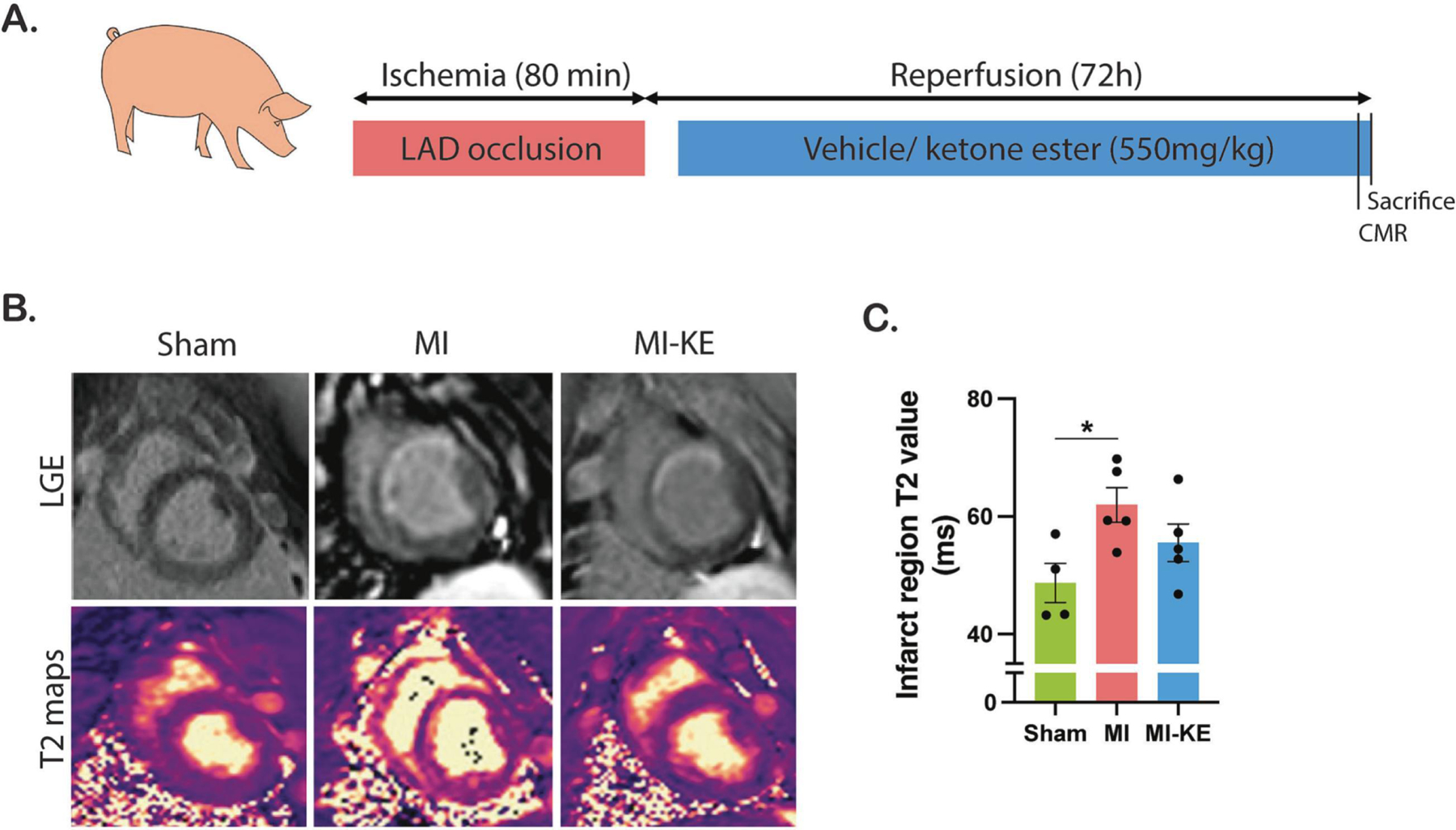
A. Experimental design of oral ketone ester supplementation in swine with MI. B. Representative late-gadolinium enhancement (LGE) images (top) and T2 maps (bottom). C. T2 value measured in the region of the infarct in MI pigs and its corresponding region in sham animals. Veh indicates vehicle and KE, ketone ester. *p < 0.05, **p < 0.01, ***p < 0.001.
Table 2. Cardiac magnetic resonance imaging (CMR) parameters for sham-operated and MI-operated pigs.
| CMR parameters | Sham | MI | MI-KE |
|---|---|---|---|
| Infarct size (%) | 0 | 19,3 ± 7 | 16 ± 5,3 |
| LVEF (%) | 54.8 ± 2.3 | 45.8 ± 2.6 | 45.2 ± 3 |
| EDV (ml) | 35,3 ± 12,3 | 58,8 ± 3 | 60,5 ± 6,8 |
| ESV (ml) | 22,3 ± 3,4 | 31,4 ± 2,6 | 31,8 ± 3,1 |
| Cardiac output (l/min) | 2.7 ± 0.5 | 2.6 ± 0.2 | 2.9 ± 0.3 |
| Stroke volume (ml) | 27.5 ± 3 | 27.8 ± 1.5 | 31 ± 2.4 |
| T1 remote (ms) | 1047,6 ± 51,8 | 1158,5 ± 134,6 | 1292,1 ± 93,9 |
| T1 infarct (ms) | 1066,6 ± 138,3 | 1283,9 ± 94,9 | 1246,1 ± 160,2 |
| T2 remote (ms) | 47,4 ± 3,9 | 47,7 ± 3,2 | 51,6 ± 3,4 |
| T2 infarct (ms) | 48,7 ± 3,3 | 62 ± 2,9 # | 56,2 ± 3,6 |
N=4–5/group. Data are presented as means ± SEM. MI indicates myocardial infarction; CMR, cardiac MRI; LVEF, left ventricular ejection fraction; EDV, end-diastolic volume; ESV, end-systolic volume. Veh indicates vehicle and KE, ketone ester. #P<0.05 vs. sham.
Treatment with KE alters myocardial substrate utilization and enhances myocardial energetics in swine following MI
We next evaluated the impact of KE treatment on major myocardial substrates (i.e., glucose, FA, lactate, and βHB). The difference in transcardiac glucose extraction did not reach statistical significance among the groups (Figure 3A), however, expression of the gene encoding the insulin-responsive glucose transporter 4 (GLUT4), Slc2a4, the transporter for glucose to enter cardiomyocytes, was decreased in MI group compared to sham and KE treatment normalized the GLUT4 expression to sham levels (Figure 3F). Myocardial uptake of FA was significantly reduced in MI group and the uptake was restored in MI-KE group (Figure 3B). Similarly, the decrease in expression of FA transporters CD36 and carnitine palmitoyl transferase-1β (CPT1-β) in MI group was recovered after KE treatment (Figure 3G–I). Ketone treatment had no significant effect on the lactate extraction.
Figure 3. Ketone ester treatment increases myocardial extraction of fatty acid and ketone and enhances myocardial energetics in pigs following ischemia reperfusion injury.
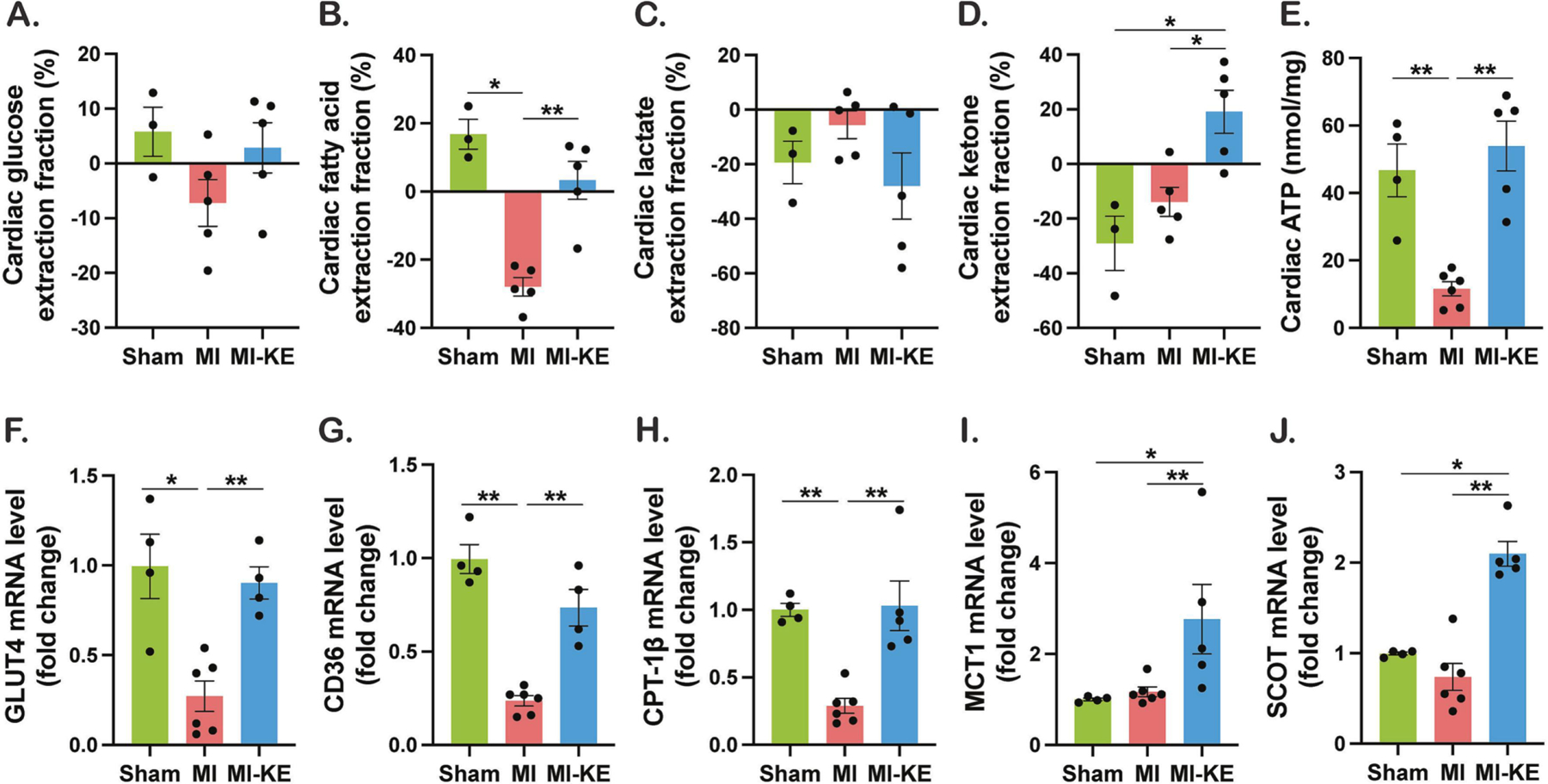
Cardiac extraction of glucose (A), fatty acid (B), lactate (C), and ketone (D). E. ATP levels in the LV tissue. Levels of mRNA encoding GLUT4 (glucose transporter 4, F), CD36 (G), CPT-1β (Carnitine palmitoyl transferase 1β, H), MCT1 (monocarboxylate transporter 1, I), and SCOT (succinyl CoA:3-oxo acid CoA transferase, J). Veh indicates vehicle and KE, ketone ester. *p < 0.05, **p < 0.01, ***p < 0.001.
Cardiac uptake of βHB was increased after KE treatment (Figure 3D) and accordingly, KE treatment increased both expression of the gene encoding MCT1 (monocarboxylate transporter 1), Slc16a1, the main transporter for ketone bodies to enter cardiomyocytes, and the enzyme catalyzing the second step of ketone oxidation, succinyl CoA:3-oxo acid CoA transferase (SCOT) (Figure 3I–J). These findings suggest that ketone bioavailability and cardiac ketone utilization are increased by KE. To determine whether KE supplementation affected myocardial energetics, cardiac ATP was measured. As expected, MI resulted in significant reduction of cardiac ATP. Remarkably, consistent with increased use of βHB as a fuel, cardiac ATP levels after IRI dramatically increased by KE feeding, restoring them to the normal levels seen in sham-operated controls (Figure 3E).
Treatment with KE suppressed cardiac inflammation, oxidative stress, and apoptosis in swine following MI
We next evaluated the effects of KE supplementation on cellular and molecular changes in the myocardium following MI. The transcript levels of proinflammatory cytokines IL (interleukin)-1, IL-6, IL-18, tumor necrosis factor alpha (TNF-α) and NLRP3 (NOD-, LRR- and pyrin domain-containing protein 3) were significantly elevated in MI hearts (Figure 4A–E). Furthermore, the transcript expression of markers for macrophage infiltration into cardiac tissue galectin-3 and adgre1 (F4/80) were significantly increased in MI hearts (Figure 4F–G). Importantly, KE treatment normalized of all these markers, suggesting that KE suppressed cardiac inflammation following MI (Figure 4A–G). These findings were further supported by the observation the number of CD68+ macrophages in the border zone (Figure 4I–J) were higher in the untreated group compared to the ketone group.
Figure 4. Ketone ester treatment attenuates cardiac injury, inflammation, oxidative stress, and apoptosis in pigs following ischemia reperfusion injury.
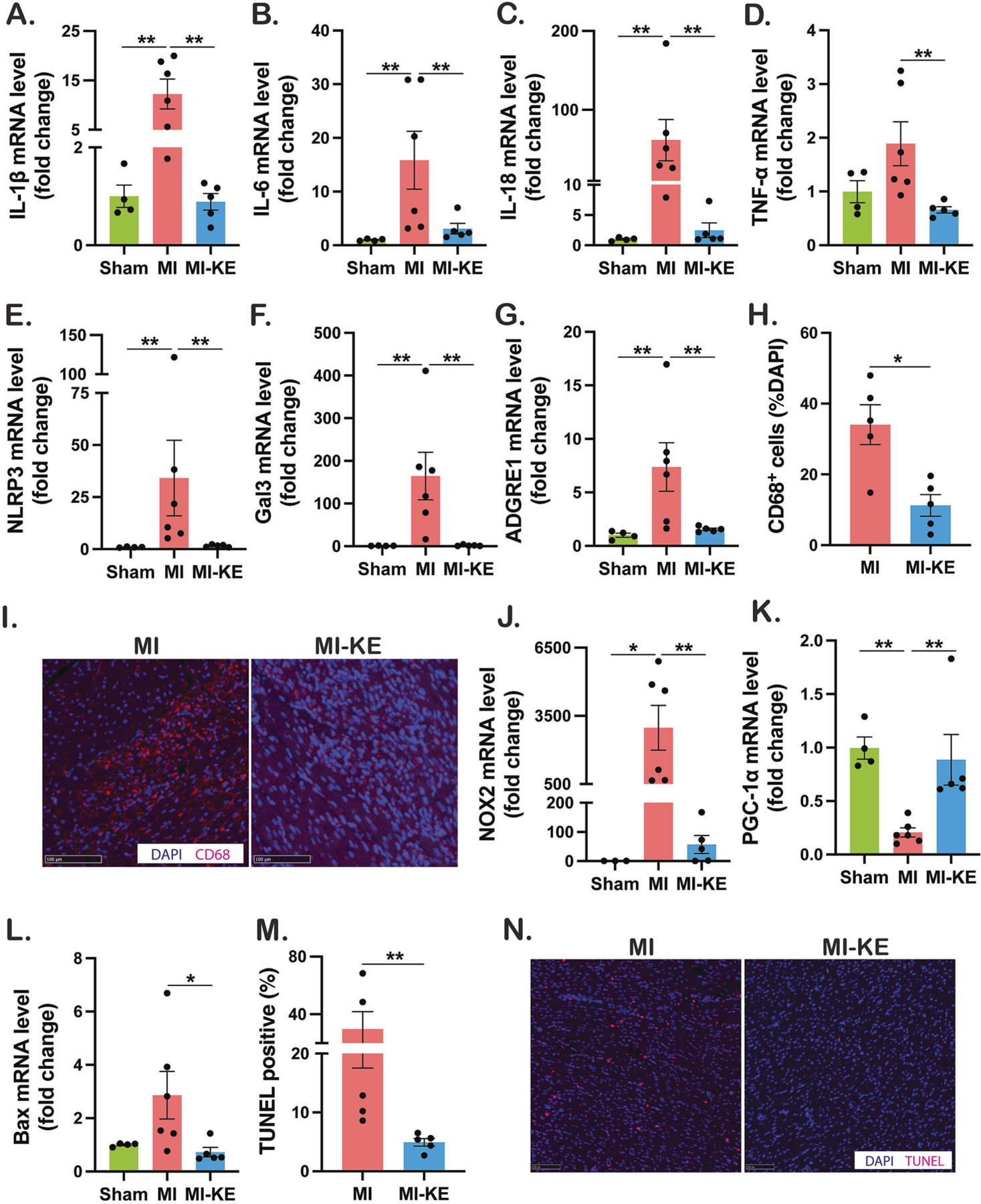
Measurement of mRNA levels to assess molecular markers for cardiac inflammation (A-G), and oxidative stress (J-K), and apoptosis (L), respectively, normalized to GAPDH. H-I. CD68 immunostaining and quantification in untreated MI and MI-KE hearts. M. Quantification of TUNEL positive cells in border zone. N. Apoptotic cells determined by TUNEL assay and DAPI staining. Veh indicates vehicle and KE, ketone ester. *p < 0.05, **p < 0.01, ***p < 0.001
To investigate the effect of KE on myocardial oxidative stress and mitochondrial biogenesis, we determined the transcript levels of the superoxide generating enzyme NOX2 and transcription factor for mitochondrial biogenesis PGC-1α. As expected, the transcript level of NOX2 was up-regulated and accompanied by diminished expression of the PGC-1α, and KE attenuated the NOX2 expression and normalized the PGC-1α expression (Figure 4J–K). Furthermore, reduction of transcript levels of Bax (Figure 4L), a trend for a lower Caspase 3 expression (data not shown), and fewer TUNEL-positive cell (Figure 4M–N) were observed after KE treatment, suggesting that cardiac apoptosis may be reduced by KE. Taken together, these data suggest that cardiac inflammation, oxidative stress, and apoptosis following MI are attenuated by KE treatment.
KE treatment induces transcriptome changes in the myocardium
Next, we performed RNA sequencing (RNA-seq) of left ventricular tissue to identify transcript profiles between MI and MI-KE group. Analyses of gene expression revealed 3560 genes that were differentially expressed between in MI-KE hearts compared to MI hearts. Of those, gene set enrichment identified 2116 significantly upregulated and 1444 downregulated genes (Figure 5A–B). Bioinformatic analysis of the genes revealed a role for KE in regulating multiple metabolism-related pathways in MI. Comparison of the most significantly enriched gene ontology (GO) demonstrated features that are universally affected in MI23, including terms for biological processes (cellular respiration, aerobic respiration, ATP metabolic process, energy pathways) and cellular components (mitochondrion). Interestingly, processes that were decreased including terms for biological processes innate immune response and inflammatory response (Figure 5C and supplementary data). Enrichment pathway analysis identified differences in KEGG pathways after KE treatment in MI hearts. Metabolism-related pathways, including FA degradation, pyruvate metabolism, BCAAs degradation, TCA cycle, carbon metabolism, cardiac muscle contraction, and oxidative phosphorylation were highly enriched after KE treatment (Supplementary Figure S1). In addition, inflammation-related pathways, including leukocyte trans endothelial migration, chemokine metabolism pathway, TLR signaling pathway, and B cell receptor signaling pathway were down-regulated (Supplementary Figure S2). Importantly, the expression of representative genes related to cardiac metabolism (GLUT4, CD36, CPT1-β, MCT1, BDH1, SCOT), inflammation (IL18, NLRP3, Gal3, ADGRE1), mitochondrial biogenesis (PGC-1α), and apoptosis (Bax, CASP3) (Figure 5D–P) corroborated the findings from the qPCR results. Taken together, these data suggest that KE induces distinct gene expression in MI hearts and that the beneficial effects of KE supplementation in MI may be driven by changes in biological pathways including cardiac energy metabolism and inflammation.
Figure 5. Ketone ester induces transcriptional changes in the myocardium.
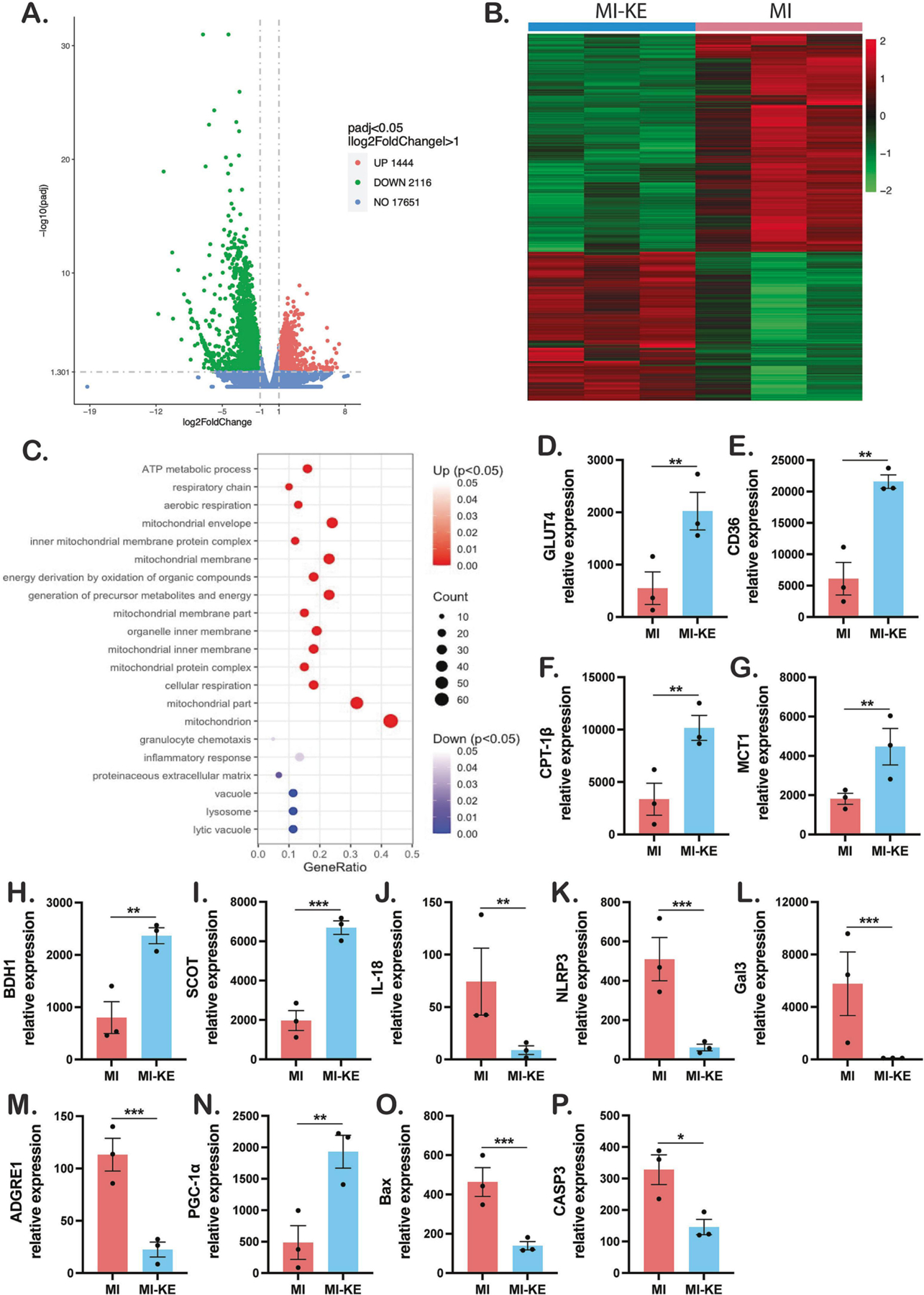
A. Differentially expressed genes in the ventricles from MI and MI-ketone pigs. B. Heat map showing the differentially expressed (up- or down- regulated) genes in individual hearts from MI-KE and MI pigs. Shaded color bar represents low (green) to high (red) expression level changes in MI-KE vs. MI hearts. C. GO analysis of upregulated (top 15) and downregulated genes in MI-KE vs. MI hearts. Comparison of the expression levels of the representative genes related to cardiac metabolism (glucose, fatty acid and ketone, D-I, respectively), inflammation (J-M), mitochondrial biogenesis (N), and apoptosis (O-P). Gene ratio indicates ratio of differentially expressed genes to all genes for this GO term. Veh indicates vehicle and KE, ketone ester. *p < 0.05, **p < 0.01, ***p < 0.001
DISCUSSION
In the present study, we tested the hypothesis that nutritional ketosis achieved by oral KE supplementation would provide early benefits against during the early phase of post-MI remodeling in swine. To achieve this goal, we first formulated a KE supplement that was able to induce ketonemia in pigs. Next, we determined the cardiac effects of KE supplementation following MI. Our results indicated that (1) nutritional ketosis can be achieved within 20–30 minutes after ingestion of KE in pigs; (2) Oral KE treatment altered cardiac substrate utilization post-MI; (3) KE treatment was effective to suppress cardiac inflammation, oxidative stress, apoptosis, and improve mitochondrial biogenesis; (4) KE treatment increased cardiac βHB uptake as well as induced genes involved in cardiac uptake and oxidation, and improved cardiac ATP levels post-MI; and (5) data from RNA-seq of LV tissue identify that KE significantly modulates biological pathway related mitochondria energy metabolism and reduces inflammation.
Our study is consistent with previous research demonstrating the cardioprotective effects of ketones in various models of CVD. Santos Gallego et al. investigated the effects of beta-hydroxybutyrate (BOHB) infusion during MI occlusion in a porcine model.[21] In their study, BOHB infusion started 30 minutes before coronary occlusion and continued until the procedure’s completion, while we administered oral KE approximately 3 hours after occlusion during the reperfusion period. The key difference between the two studies is the timing of MI induction and ketone body (KB) infusion. The authors evaluated the effects of KB infusion prior to and during MI and evaluated the outcomes four hours post-MI. In contrast, we focused on targeting the inflammatory pathway during the reperfusion period (72 hours) following 80 minutes of LAD occlusion. Their study showed less microvascular obstruction, improved LVEF and strain, while our study provides evidence that oral ketone ester administration during reperfusion can suppress the inflammatory response post-MI. It is unclear what the clinical implications of these differences are, but they could be critical in optimizing KB therapy for MI treatment. Nevertheless, this difference in timing reflects a potential clinical scenario where KB infusion may be used as a post-ischemic intervention, particularly in cases where reperfusion is delayed or ineffective. The comparison between the two studies highlights the potential clinical implications of ketone therapy for reducing myocardial injury post-MI and improving cardiac outcomes. Future studies are needed to compare the efficacy of different types of ketones and determine the optimal timing of administration.
Reperfusion is the top priority in patients with ST segment elevation MI (STEMI) and is essential to salvage viable myocardium, limit MI size, and improve clinical outcome. However, following the reperfusion and subsequent reperfusion injury, it is common for altered states in the tissue to occur with the onset of mitochondrial membrane damage, inflammatory response, chemoattractant release, formation of reactive oxygen species (ROS), altered metabolism, apoptosis, and various other effects.[6, 22] Moreover, the dysregulation of the immune cascade, unresolved inflammation, and overreactive fibrosis in the myocardium following MI may cause adverse remodeling and eventual post-MI HF.[23] A greater understanding of the mechanisms of phases of post-MI remodeling allows for the translation of potential clinical therapeutic targets. Of particular interest is the possibility of targeting notable inflammation pathways following MI.
Besides ketone bodies provide an efficient alternative energy substrate for metabolism in vital organs, accumulating evidence indicates that ketones may exert anti-inflammatory properties in different tissues in vitro, in animals and in human.[24] For instance, a previous study reported that βHB suppressed the NLRP3 inflammasome mediated IL-1β and IL-18 production in human monocytes.[8] Furthermore, circulating inflammatory markers were reduced after ketogenic diet consumption in humans.[25] In patients with diabetes and high cardiovascular risk, sodium-glucose co-transporter inhibitors (SGLT2i) attenuated NLRP3 activity, partially mediated by increased circulating βHB levels.[11] Ketosis induced by SGLT2i or KE also suppressed the activity of NLRP inflammasome and reversed the phenotypes in preclinical animal model of HFpEF.[9] Furthermore, numerous studies have analyzed the impacts of targeting inflammation for MI treatment. For instance, recent randomized trial of single dose IL-6 receptor inhibitor tocilizumab was associated with an increase in the myocardial salvage index in patients with acute STEMI.[26]
The results described herein provide additional rationale for the assessment of KE serving as potential treatment for MI. First, we demonstrated KE significantly suppressed cardiac inflammation following MI, alongside with substantial reduction in markers of cardiac oxidative stress, apoptosis, and improved mitochondrial biogenesis. This was concordant with CMR tissue level measurements with the non-significant elevation in infarct T2 values, indicative of inflammation. Increased T2 signal in CMR is thought to be caused by an increase in tissue water content, and can be a sensitive and specific marker for the detection of edema, a hallmark of tissue inflammation and injury.
In the course of our study, the observed increase in T2 signal in the MI group compared to the sham group is consistent with previous studies that have reported edema in the acute phase following MI. The finding of a non-significant increase in T2 signal in the MI-KE group compared to the sham group is an important result that should be carefully interpreted. Although this difference did not reach statistical significance, it is possible that the use of KE may have a subtle effect on T2 signal that was not detected in this study. Alternatively, the lack of a significant difference in T2 signal between the post-MI-KE and sham groups could be due to the protective effect of KE on the myocardium, as other biomarkers in the study also supported this observation. As noted previously, KE has been reported to have anti-inflammatory and antioxidant properties[7], which may have prevented or reduced myocardial injury and inflammation following MI. This could have resulted in a lower or more transient increase in T2 signal in the MI-KE group, which was not statistically distinguishable from the sham group. Further studies with larger sample sizes and more sensitive imaging techniques may be needed to fully elucidate the potential effects of KE on T2 signal following MI. Additionally, it would be important to investigate whether any potential changes in T2 signal are correlated with long-term functional outcomes, such as LVEF or incidence of heart failure. Interestingly, previous study reported that higher T2 values were correlated with worse prognosis in reperfused STEMI patients.[27] Taken together, KE treatment may result in less ongoing injury, which could similarly protect the heart over time and is consistent with the reduction in apoptosis.
Second, we validated KE supplementation altered cardiac substrate metabolism. It has been previously reported that in myocardial ischemia, glycolysis occurs during the loss of glucose and FA oxidation due to the lack of oxygen, thus becomes detrimental to conserving metabolic efficiency and flexibility.[28] KE did not influence cardiac lactate or glucose uptake after MI, but it did normalize the reduction of cardiac expression of main glucose transporter GLUT4 in MI group. Cardiac FA extraction and cardiac expression of transporters of FA CPT-1β and CD36 were reduced in MI pigs, and interestingly, KE treatment restored the ability of the MI hearts to utilize FA and correspondingly, it normalized the expression of all FA transporters. Our data extend previous observation[29] as we demonstrate that ketones do not influence the oxidation rates of other substrates. It is likely that the changes in FA extraction relate, at least in part, to the vasodilatory effects on the myocardium.[30]
Third, we revealed cardiac ketone uptake as well as the expression of cardiac ketone transporter MCT1 and the rate limiting enzyme in myocardial ketone oxidation SCOT were increased. Furthermore, nutritional ketosis achieved by KE supplementation was associated with the improvement of cardiac ATP levels. These findings suggest that nutritional ketosis results in further reprogramming of myocardial metabolism towards enhanced ketone utilization.
Finally, fourth, our RNA-seq data corroborate the findings above, in which KE supplementation may affect biological pathways such as improving mitochondrial energy metabolism and reducing cardiac inflammation. Taken together, we potentially established that nutritional ketosis represents an effective method against reperfusion injury in the acute setting of MI and suggest that KE supplementation may offer a therapeutic opportunity for patients with MI.
STUDY LIMITATIONS
Beyond issues inherent to the use of animal model to study human disease, our study has several limitations worth noting. First, ketone ester was administered after all animals were fully awake (~2.5 hours post-MI) due to KE only being able to be administered orally and should never be injected; thus, our study does not exclude the possibility that ketone could be more beneficial when given through infusion or intramyocardial injection.
Second, while the strength of this study included measurement of cardiac extraction of major substrates through direct blood sampling from arterial and coronary sinus, we did not measure myocardial blood flow. Thus, the flux (i.e absolute changes) in substrate extraction could not be determined. However, this method has been widely used and accepted to estimate cardiac extraction of substrates in previous studies.[29, 31] Furthermore, direct measurements of cardiac expression of markers related to cardiac metabolism by qPCR and unbiased GO analyses showed similar pattern and could overcome this limitation.
Third, while we describe multiple molecular effects of KE in the myocardium, we did not establish the mechanisms underlying these molecular changes. More mechanistic studies would be required including specific gain- and loss-of-function experiments, which are challenging in large animal models. Nevertheless, our study provides unique insight into the cardioprotective effects of KE in the context of acute MI and supports the exploration the chronic benefits of KE in post-MI HF setting.
Finally, the ketone levels achieved with oral KE supplementation were supraphysiological and not representative of normal physiological levels. Although KE supplementation has been shown to be safe and well-tolerated in both animals and humans, including healthy subjects and patients with HFrEF, the supraphysiological levels achieved in our study may not be directly applicable to clinical practice. Therefore, caution should be exercised when extrapolating the findings of our study to clinical scenarios. Additionally, given the small sample size in our study, further research with larger sample sizes may be beneficial to strengthen the generalizability and robustness of the findings.
CONCLUSION
Oral KE supplementation induced ketosis and enhanced myocardial βHB extraction in both healthy and infarcted hearts. Acute oral supplementation with KE altered myocardial substrate uptake and utilization, improved ATP levels, as well as mitigating markers of inflammation following MI. Evaluating the potential chronic benefits of these changes on long-term remodeling after MI will be an exciting direction for future studies.
Supplementary Material
Figure 6. Summary of relevant findings of the study.
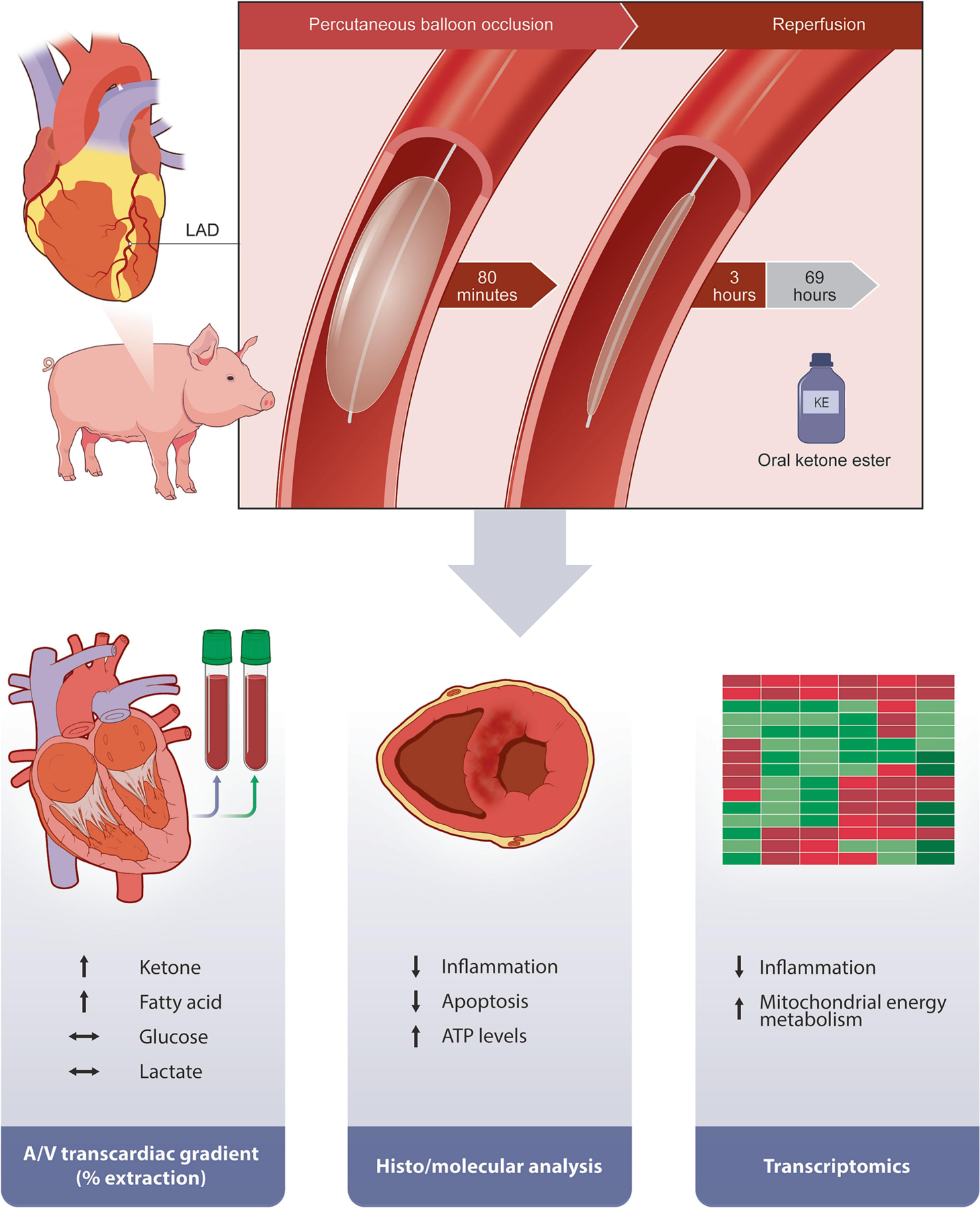
LAD indicates left anterior descending coronary artery; A/V indicates arterio-venosus.
Acknowledgments
We acknowledge Professor Kieran Clarke for support and expert advice throughout the project. We thank Yoshiko Imamoto, Benjamin Bonner and Danielle Kara for expert technical assistance.
Funding support and author disclosures
C.T.N is supported by grants from National Institute of Health (R01 HL151704, R01 HL159010, R01 HL135242).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
All authors have reported that they have no relationships relevant to the contents of this paper to disclose.
Declarations of interest: none
References
- [1].Thygesen K, Alpert JS, Jaffe AS, Chaitman BR, Bax JJ, Morrow DA, et al. Fourth Universal Definition of Myocardial Infarction (2018). J Am Coll Cardiol 2018;72:2231–64. [DOI] [PubMed] [Google Scholar]
- [2].Tsao CW, Aday AW, Almarzooq ZI, Alonso A, Beaton AZ, Bittencourt MS, et al. Heart Disease and Stroke Statistics-2022 Update: A Report From the American Heart Association. Circulation 2022;145:e153–e639. [DOI] [PubMed] [Google Scholar]
- [3].Heidenreich PA, Bozkurt B, Aguilar D, Allen LA, Byun JJ, Colvin MM, et al. 2022 AHA/ACC/HFSA Guideline for the Management of Heart Failure: Executive Summary: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. J Am Coll Cardiol 2022;79:1757–80. [DOI] [PubMed] [Google Scholar]
- [4].Faridi KF, Bhalla N, Atreja N, Venditto J, Khan ND, Wilson T, et al. New Heart Failure After Myocardial Infarction (From the National Cardiovascular Data Registries [NCDR] Linked With All-Payer Claims). Am J Cardiol 2021;151:70–7. [DOI] [PubMed] [Google Scholar]
- [5].Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med 2007;357:1121–35. [DOI] [PubMed] [Google Scholar]
- [6].Prabhu SD, Frangogiannis NG. The Biological Basis for Cardiac Repair After Myocardial Infarction: From Inflammation to Fibrosis. Circ Res 2016;119:91–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Yurista SR, Chong CR, Badimon JJ, Kelly DP, de Boer RA, Westenbrink BD. Therapeutic Potential of Ketone Bodies for Patients With Cardiovascular Disease: JACC State-of-the-Art Review. J Am Coll Cardiol 2021;77:1660–9. [DOI] [PubMed] [Google Scholar]
- [8].Youm YH, Nguyen KY, Grant RW, Goldberg EL, Bodogai M, Kim D, et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat Med 2015;21:263–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Deng Y, Xie M, Li Q, Xu X, Ou W, Zhang Y, et al. Targeting Mitochondria-Inflammation Circuit by β-Hydroxybutyrate Mitigates HFpEF. Circ Res 2021;128:232–45. [DOI] [PubMed] [Google Scholar]
- [10].Shimazu T, Hirschey MD, Newman J, He W, Shirakawa K, Le Moan N, et al. Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 2013;339:211–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kim SR, Lee SG, Kim SH, Kim JH, Choi E, Cho W, et al. SGLT2 inhibition modulates NLRP3 inflammasome activity via ketones and insulin in diabetes with cardiovascular disease. Nat Commun 2020;11:2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Fu SP, Li SN, Wang JF, Li Y, Xie SS, Xue WJ, et al. BHBA suppresses LPS-induced inflammation in BV-2 cells by inhibiting NF-κB activation. Mediators Inflamm 2014;2014:983401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Yurista SR, Matsuura TR, Silljé HHW, Nijholt KT, McDaid KS, Shewale SV, et al. Ketone Ester Treatment Improves Cardiac Function and Reduces Pathologic Remodeling in Preclinical Models of Heart Failure. Circ Heart Fail 2021;14:e007684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Nielsen R, Møller N, Gormsen LC, Tolbod LP, Hansson NH, Sorensen J, et al. Cardiovascular Effects of Treatment With the Ketone Body 3-Hydroxybutyrate in Chronic Heart Failure Patients. Circulation 2019;139:2129–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Cox PJ, Kirk T, Ashmore T, Willerton K, Evans R, Smith A, et al. Nutritional Ketosis Alters Fuel Preference and Thereby Endurance Performance in Athletes. Cell Metab 2016;24:256–68. [DOI] [PubMed] [Google Scholar]
- [16].Soto-Mota A, Vansant H, Evans RD, Clarke K. Safety and tolerability of sustained exogenous ketosis using ketone monoester drinks for 28 days in healthy adults. Regul Toxicol Pharmacol 2019;109:104506. [DOI] [PubMed] [Google Scholar]
- [17].Zhou Z, Nguyen C, Chen Y, Shaw JL, Deng Z, Xie Y, et al. Optimized CEST cardiovascular magnetic resonance for assessment of metabolic activity in the heart. J Cardiovasc Magn Reson 2017;19:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Arima Y, Izumiya Y, Ishida T, Takashio S, Ishii M, Sueta D, et al. Myocardial Ischemia Suppresses Ketone Body Utilization. J Am Coll Cardiol 2019;73:246–7. [DOI] [PubMed] [Google Scholar]
- [19].Messroghli DR, Moon JC, Ferreira VM, Grosse-Wortmann L, He T, Kellman P, et al. Clinical recommendations for cardiovascular magnetic resonance mapping of T1, T2, T2* and extracellular volume: A consensus statement by the Society for Cardiovascular Magnetic Resonance (SCMR) endorsed by the European Association for Cardiovascular Imaging (EACVI). J Cardiovasc Magn Reson 2017;19:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Yurista SR, Silljé HHW, Oberdorf-Maass SU, Schouten EM, Pavez Giani MG, Hillebrands JL, et al. Sodium-glucose co-transporter 2 inhibition with empagliflozin improves cardiac function in non-diabetic rats with left ventricular dysfunction after myocardial infarction. Eur J Heart Fail 2019;21:862–73. [DOI] [PubMed] [Google Scholar]
- [21].Santos-Gallego CG, Requena-Ibáñez JA, Picatoste B, Fardman B, Ishikawa K, Mazurek R, et al. Cardioprotective Effect of Empagliflozin and Circulating Ketone Bodies During Acute Myocardial Infarction. Circ Cardiovasc Imaging 2023;16:e015298. [DOI] [PubMed] [Google Scholar]
- [22].Gunata M, Parlakpinar H. A review of myocardial ischaemia/reperfusion injury: Pathophysiology, experimental models, biomarkers, genetics and pharmacological treatment. Cell Biochem Funct 2021;39:190–217. [DOI] [PubMed] [Google Scholar]
- [23].Davidson SM, Ferdinandy P, Andreadou I, Bøtker HE, Heusch G, Ibáñez B, et al. Multitarget Strategies to Reduce Myocardial Ischemia/Reperfusion Injury: JACC Review Topic of the Week. J Am Coll Cardiol 2019;73:89–99. [DOI] [PubMed] [Google Scholar]
- [24].Yurista SR, Nguyen CT, Rosenzweig A, de Boer RA, Westenbrink BD. Ketone bodies for the failing heart: fuels that can fix the engine? Trends Endocrinol Metab 2021;32:814–26. [DOI] [PubMed] [Google Scholar]
- [25].Forsythe CE, Phinney SD, Fernandez ML, Quann EE, Wood RJ, Bibus DM, et al. Comparison of low fat and low carbohydrate diets on circulating fatty acid composition and markers of inflammation. Lipids 2008;43:65–77. [DOI] [PubMed] [Google Scholar]
- [26].Broch K, Anstensrud AK, Woxholt S, Sharma K, Tøllefsen IM, Bendz B, et al. Randomized Trial of Interleukin-6 Receptor Inhibition in Patients With Acute ST-Segment Elevation Myocardial Infarction. J Am Coll Cardiol 2021;77:1845–55. [DOI] [PubMed] [Google Scholar]
- [27].Zia MI, Roifman I, Ghugre NR, Ignatius AJ, Strauss BH, Dick A, et al. Prognostic value of myocardial T2 mapping post reperfused acute myocardial infarction
- [28].Carley AN, Taegtmeyer H, Lewandowski ED. Matrix revisited: mechanisms linking energy substrate metabolism to the function of the heart. Circ Res 2014;114:717–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Monzo L, Sedlacek K, Hromanikova K, Tomanova L, Borlaug BA, Jabor A, et al. Myocardial ketone body utilization in patients with heart failure: The impact of oral ketone ester. Metabolism 2021;115:154452. [DOI] [PubMed] [Google Scholar]
- [30].Gormsen LC, Svart M, Thomsen HH, Søndergaard E, Vendelbo MH, Christensen N, et al. Ketone Body Infusion With 3-Hydroxybutyrate Reduces Myocardial Glucose Uptake and Increases Blood Flow in Humans: A Positron Emission Tomography Study. J Am Heart Assoc 2017;6. [DOI] [PMC free article] [PubMed]
- [31].Voros G, Ector J, Garweg C, Droogne W, Van Cleemput J, Peersman N, et al. Increased Cardiac Uptake of Ketone Bodies and Free Fatty Acids in Human Heart Failure and Hypertrophic Left Ventricular Remodeling. Circ Heart Fail 2018;11:e004953. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.