

Abstract
Glioblastoma (GB) is an astrocytic brain tumour with a low survival rate, partly because of its highly invasive nature. The GB tumour microenvironment (TME) includes its extracellular matrix (ECM), a variety of brain cell types, unique anatomical structures, and local mechanical cues. As such, researchers have attempted to create biomaterials and culture models that mimic features of TME complexity. Hydrogel materials have been particularly popular because they enable 3D cell culture and mimic TME mechanical properites and chemical composition. Here, we used a 3D collagen I-hyaluronic acid hydrogel material to explore interactions between GB cells and astrocytes, the normal cell type from which GB likely derives. We demonstrate three different spheroid culture configurations, including GB multi-spheres (i.e., GB and astrocyte cells in spheroid co-culture), GB-only mono-spheres cultured with astrocyte-conditioned media, and GB-only mono-spheres cultured with dispersed live or fixed astrocytes. Using U87 and LN229 GB cell lines and primary human astrocytes, we investigated material and experiment variability. We then used time-lapse fluorescence microscopy to measure invasive potential by characterizing the sphere size, migration capacity, and weight-averaged migration distance in these hydrogels. Finally, we developed methods to extract RNA for gene expression analysis from cells cultured in hydrogels. U87 and LN229 cells displayed different migration behaviors. U87 migration occurred primarily as single cells and was reduced with higher numbers of astrocytes in both multi-sphere and mono-sphere plus dispersed astrocyte cultures. In contrast, LN229 migration exhibited features of collective migration and was increased in monosphere plus dispersed astrocyte cultures. Gene expression studies indicated that the most differentially expressed genes in these co-cultures were CA9, HLA-DQA1, TMPRSS2, FPR1, OAS2, and KLRD1. Most differentially expressed genes were related to immune response, inflammation, and cytokine signalling, with greater influence on U87 than LN229. These data show that 3D in vitro hydrogel co-culture models can be used to reveal cell line specific differences in migration and to study differential GB-astrocyte crosstalk.
Graphical Abstract
Introduction
Glioblastoma (GB) is the most common primary malignant brain tumour in adults with a median five-year survival rate of 5.4%.1 Unlike other malignant tumours which metastasize through the circulatory system, GB rarely metastasizes, instead invading within the brain. GB cell migration mainly occurs in the perivascular space surrounding the blood vessels, extracellular spaces in the brain parenchyma, and along white matter tracts.2 The difficulty in eradicating these invading GB cells is a primary cause of tumour recurrence after surgery.3 Increased understanding of GB invasion could lead to new therapeutic options for this disease. However, GB migration is influenced by many complex factors in the tumour microenvironment (TME) including mechanical stresses4, extracellular matrix (ECM) composition and properties5, and behaviours of adjacent cells6, that are difficult to study in traditional 2D cell cultures or Boyden chamber cultures.
The GB TME comprises many cell types including astrocytes, oligodendrocytes, neurons in the brain parenchyma, native immune cells known as microglia and invading macrophages, along with pericytes and endothelial cells comprising the blood brain barrier.7 Emerging evidence suggests healthy astrocytes, potential cellular precursors of GB that compose 50% of native brain cells, are important mediators of GB invasion in the brain TME. Astrocytes directly contact and communicate with GB cells through gap junctions mediated by connexin43 (Cx43).8 Signalling molecules, such as microRNAs, can be transferred through these gap junctions to regulate GB migration,9 and Cx43 inhibition has been shown to decrease GB migration and invasion in vivo.10, 11 Astrocytes also form interconnected processes in the brain cortex and aligned tracts in white matter, both of which may serve as migration routes for GB cells.12 In the perivascular space, GB cells can take control of the vessels to facilitate tumour growth and invasion by causing astrocytes to retrieve their endfeet, which in normal brain cover ~99% of the vasculature.13, 14 Additionally, astrocytes support the production and activation of matrix metalloproteinases (MMPs) that increase tissue permeability, and reactive astrocytes in the brain TME secrete interleukin-6 (IL-6) which enhances MMP activity15. Reactive astrocytes observed in response to injury or disease pathology are characterized by unique genomic and phenotypic signatures.16 In GB, reactive astrocytes are likely associated with the activation of the nuclear factor kappa-B (NFκB) pathway by the NFκB ligand RANKL produced by GB cells.17
Most previous GB modelling studies employ costly animal models, brain slices with significant complexity, or traditional 2D cell cultures that fail to capture many aspects of the TME18. In particular, the reactive astrocyte phenotype is constitutively expressed in 2D monolayer culture on polystyrene, making studies of phenotype transition difficult.19 As such, several groups,20 including ours,21, 22 have developed biomaterials to more closely mimic features of brain TME that also allow study of the reactive astrocyte phenotype transition in 3D dispersed cell or spheroid culture.19 These materials are often composed of hyaluronic acid (HA) glycosaminoglycans combined with collagen or gelatine proteins to form hydrogels.23 HA comprises > 30% of brain tissue in normal and tumorous brain,24 whereas collagen I is produced by GB cells and is associated with GB invasiveness.25 Whereas hydrogel models have been well established for studies of dispersed cells and spheroids,26 the translation of these models to co-cultures employing GB cells and one or more additional cell types of interest is relatively underexplored.6, 27–31 Such models have been used to study GB interactions with endothelial cell networks6, 27, 28, microglia30, 31, astrocytes,29 and to approximate the perivascular niche.32 These 3D models are particularly useful for investigating astrocyte-GB interactions because they more closely mimic physiological astrocyte behaviours, such as the ability to induce a reactive astrocyte phenotype.
Here, we describe a 3D collagen I-hyaluronic acid (Col-HA) hydrogel system for GB-astrocyte co-culture studies. These Col-HA hydrogels have previously been shown to closely mimic the composition and physical properties of the brain.22 This model is compatible with a variety of configurations, including spheroids and mixed spheroid-dispersed cell cultures. Specifically, we demonstrate three GB-astrocyte configurations: (1) multi-spheres formed of GB cells and astrocytes, mimicking the process of tumorigenesis, (2) mono-spheres composed of only GB cells cultured with astrocyte-conditioned media to test the effect of soluble factors on GB spheroids, and (3) GB mono-spheres cultured with dispersed astrocytes to mimic the physiological abundance of astrocytes in the brain parenchyma in established tumours. This third GB mono-sphere plus dispersed astrocyte model permits GB induction of astrocyte behaviours through direct contact or soluble factors. To separate these effects, we also show a variation of this model in which astrocytes were fixed (i.e., dead) prior to encapsulation, preserving their surface ligands but not intracellular signalling response. Using these models, we explored GB cell migration response of two different GB lines: U87 cells associated with nodular behaviours and LN229 cells with infiltrating phenotypes.17 Results were collected and analysed through time lapse video microscopy, measuring total spheroid size, the area percent covered by migrating cells (migration capacity), and the weighted average migration distance of cell clusters from the spheroid surface (ad-distance). We then demonstrate methods for collecting and analysing RNA from selected co-culture systems for gene expression analysis of tumour signalling (i.e., Nanostring® nCounter Tumour Signalling 360™ panel). These hydrogel materials and methods for co-culture and analysis provide important tools for biologists and biomedical engineers to study complex interactions in the brain TME that will add to the growing body of work in biomaterial TME models.
Materials and methods
Cell culture
U87 (derived from a male patient) and LN229 (derived from a female patient) cell lines were obtained from the American Type Culture Association (ATCC), and primary normal human astrocytes (NHA) were obtained from Lonza. U87 cells were cultured in Dulbecco’s modified Eagle medium: nutrient mixture F-12 (DMEM/F-12, Gibco) supplemented with 10% foetal bovine serum (FBS, VWR), 1% penicillin-streptomycin (Corning), and 1x MycoZap (Lonza). LN229 cells were cultured in DMEM/F-12 supplemented with 5% FBS, 1% penicillin-streptomycin, and 1x MycoZap. NHAs were cultured in AGM™ Astrocyte Growth Medium BulletKit™ (Lonza CC3186). Cells were grown at 37 °C in an atmosphere of 5% CO2 and passaged at ~80% confluence.
Cell visualization
For visualization in 3D culture models, U87 and LN229 cells at low passage numbers were transfected with LentiBrite™ GFP Control Lentiviral Biosensor (Millipore Sigma 17–10387) at multiplicity of infection (MOI) = 40 following the manufacturer’s instructions. NHAs were stained with CellTracker Red CMPTX dye (Invitrogen ™) for 40 mins.
Generation of glioblastoma mono-spheres and glioblastoma-astrocyte multi-spheres
This study employed tumour spheroids composed of only cancer cells (i.e., mono-spheres) and spheroids comprised of tumour cells and astrocytes (i.e., multi-spheres). In addition, some mono-spheres were cultured in the presence of dispersed NHAs. GB mono-spheres for mono-sphere only and mono-sphere plus dispersed astrocyte studies were generated by seeding 1000 U87 or LN229 cells in 200 μl U87 growth media into interior wells of a Nunclon™ Sphera™ 96U-well microplate (Thermo Scientific™). U87 media was used because its increased FBS content was more compatible with astrocyte cultures. Multi-spheres of GB cells and astrocytes were generated by seeding interior wells at two different cell counts, 1000 or 2000 cells. A larger cell number was used in NHA rich conditions because U87 cells proliferate much faster than NHAs. Thus, more overall cells were needed to achieve a sphere size comparable to the conditions with a high U87 ratio. For 1000 cell conditions, NHA and U87 cells were seeded at ratios (N:U) = 1:5 or 1:10, whereas a total of 2000 cells was used for ratios = 10:1 or 5:1. In addition, we performed a control experiment with a fixed number of 1000 GB cells with astrocytes added at different ratios of N:U= 10:1, 5:1 1:5, and 1:10. All cells were seeded in 200 μl U87 growth media. A control of only astrocytes was also seeded at 2000 cells/well to compare to multi-sphere configurations. The outermost wells were filled with 250 μl phosphate buffered saline (PBS, Sigma Aldrich) to prevent evaporation. All spheres were cultured for 3 days prior to encapsulation in Col-HA biomaterials, and only wells with single, intact spheres were used for further experiments.
Hydrogel preparation and encapsulation
Collagen-hyaluronic acid (Col-HA) hydrogels were prepared by mixing collagen I solution (PureCol EZ Gel, Advanced Biomatrix, 5 mg/ml), thiolated HA (Glycosil, Advanced Biomatrix, 240 ± 30 kDa, 10 mg/ml in DMEM/F-12 or DI water), polyethylene glycol diacrylate (PEGDA) (Extralink, Advanced Biomatrix, 10 mg/ml in DI water), and DMEM/F-12. To form Col-HA prepolymer-solution, collagen/HA were added in equal volume and PEGDA/DMEM were both added at a 1:4 volume ratio compared to HA. PEGDA was used as a thiol-reactive crosslinker for enhanced gel stability. The final collagen concentration in this gel was 0.1 wt.%, and the final HA concentration was 0.2 wt.%. These concentrations were selected based on our previous work22, 33, 34 showing that this formulation closely mimics brain mechanical properties while promoting physiological cell behaviours.
After 3 days of spheroid only culture with no hydrogel, 150 μl of media in each well was carefully removed to avoid disturbing the sphere, and 50 μl of Col-HA gel with (NHA-loaded) or without (mono-spheres and multi-spheres) NHAs was added with a multichannel pipette. NHA-loaded gels (GB mono-spheres with dispersed NHAs) were prepared by resuspending NHAs in Col-HA gels at 5 × 105 cells/ml, 1 × 106 cells/ml, or 2 × 106 cells/ml before addition to the wells. The microplate was gently tapped to mix the gel solution with the residual media in the wells. The final concentrations of collagen and HA in each well were 1 mg/ml and 2 mg/ml, respectively, similar to our previous work.22, 34 Gels were incubated at 37 ºC in an atmosphere of 5% CO2 for 1 hour to form the hydrogel and then covered with 100 μl NHA (multi-spheres and NHA-loaded) or U87 (mono-spheres) growth media. For conditions in which fixed NHAs were used, NHAs were stained, fixed in 4% paraformaldehyde (PFA, Sigma Aldrich) and 4% sucrose (Sigma Aldrich) at room temperature for 10 min and washed with PBS before loading into the gels.
NHA-conditioned media
Some experiments were performed using NHA-conditioned media, which was generated by culturing NHAs as a sphere at 2000 cells/well seeding density for three days. Separate GB mono-spheres were cultured concurrently to reduce experimental variation. After encapsulation of GB mono-spheres in gels on day 3, NHA-conditioned media was added instead of U87 growth media.
Microscopy
Samples were imaged using a confocal inverted fluorescent microscope (Nikon, IX81) with a 4X objective for migration studies and a 10X objective to observe cell-cell interactions. GFP-transfected U87 and LN229 cells were captured in the FITC channel, CellTracker Red™-labelled NHAs were captured in the TRITC channel. Images were taken as z stacks through the depth of each sphere to capture the migration of cells from spheres completely.
Image processing
To quantify the migration of single cells and cell clusters from the spheres, images were intentionally saturated in the sphere region. This led to an uneven background with higher brightness in the sphere region and lower brightness away from the sphere. To compensate, images were batch processed in RStudio (R version 4.1.0) using the ‘SPE’ function in the ‘imager’ package to generate a homogenous background, and then thresholded using the ‘thresh’ function to generate black and white images (Supplementary Figure 1). For conditions in which z stacks were collected, thresholded images were manually patched and combined in ImageJ (NIH) to generate a single image capturing all cells (Supplementary Figure 2).
Sphere growth and migration measurements
Sphere growth was captured by measuring sphere Feret diameter daily after encapsulation on Day 3. Briefly, the sphere region was outlined using the ‘Multi Cell Outliner’ plugin in ImageJ from raw images in which the sphere was in focus. Regions outside of the sphere region of interest (ROI) were then cleared, and the outlined sphere was analysed for sphere diameter , area and location ( and ) using the ‘Analyze Particles’ function in ImageJ.
Sphere migration was captured by two factors: migration capacity, which describes the total area acquired by cells migrating out of the sphere; and adjusted migration distance (ad-distance), which describes the average distance travelled by cells or cell clusters migrating from the surface of the sphere. To measure the migration of cells from the sphere surface, the outlined sphere region was first subtracted from the thresholded images, and each cell cluster remaining on the frame was analysed for area and location ( and ) using ‘Analyze Particles’ function in ImageJ. Migration capacity was calculated as the percentage of the accumulated area of all migrating cell clusters to the total viable area of the frame (512 pixels by 512 pixels):
| Eqn. 1 |
To calculate the ad-distance, the migration distance of each cell cluster relative to the centre of the sphere was first calculated as:
| Eqn. 2 |
Ad-distance was then calculated as the difference between the average migration distance of cell clusters and the radius of the sphere:
| Eqn. 3 |
The same calculation was repeated for each sphere at all days of culture following encapsulation (i.e., post day 3).
Principal component analysis (PCA)
We observed an inevitable variation in the control groups at different experiment days. To characterize this, PCA was performed to examine the variation in sphere diameter, migration capacity, and ad-distance of three independent experiment groups performed on different days (E1-E3) and using two different batches of HA (B1 and B2) hydrogels. In RStudio (R version 4.1.0), PCA was performed with the “prcomp” function and results were visualized using the “factoextra” package.
RNA extraction
After completion of migration studies, hydrogels were harvested from 96-well plates for gene expression analysis. For RNA extraction of GB-NHA co-culture, astrocytes purchased from ScienCell were employed as Lonza discontinued their astrocyte cell line. For sufficient retrieval of RNA, multiple hydrogel replicates (N ~ 15–30) of the same experimental condition were pooled, digested in TRIzol (ThermoFisher 15596026), and mechanically lysed using an 18-gauge needle and syringe. Then, RNA extraction was performed following the manufacturer’s instructions for RNeasy Mini Kit (Qiagen 74104). Extracted RNA was analysed for yield and quality with a TapeStation RNA Bioanalyzer.
Tumour signalling assay
A tumour signalling assay was performed using NanoString nCounter™ technology based on direct detection of target molecules using color-coded molecular barcodes that provide digital quantification of the number of target molecules. Total RNA (85 ng) was hybridized overnight with nCounter™ Reporter (8 μl) probes in hybridization buffer and in an excess of nCounter™ Capture probes (2 μl) at 65 °C for 18 h. After overnight hybridization, excess probes were removed using a two-step magnetic bead-based purification procedure performed with an automated fluidic handling system (nCounter™ Prep Station). Biotinylated capture probe-bound samples were immobilized and recovered on a streptavidin-coated cartridge. The abundance of specific target molecules was then quantified using the nCounter™ digital analyser. Individual fluorescent barcodes and target molecules present in each sample were recorded with a charge-coupled device (CCD) camera by performing a high-density scan (325 fields of view). Images were processed internally into a digital format and exported as Reporter Code Count (RCC). The NanoString assay was run by the Genomics Shared Resource center at The Ohio State University.
Data plotting and statistics
All data were presented as average ± standard error and all plots were generated in SigmaPlot. Sphere diameter, migration capacity, and ad-distance of different conditions at each day of culture were compared using the Tukey HSD method in JMP Pro 12 at α = 0.05.
Gene expression analysis
For gene expression analysis, the following packages in R version 4.2.235 were utilized to import, normalize, analyse, and visualize Nanostring nCounter gene expression data: dplyr 1.0.10,36 ggplot2 3.4.0,35 BioCManager 1.30.19,37 NanoStringDiff 1.28.0,38 and EnhancedVolcano 1.16.0.39 The NanoStringDiff package utilizes both size factors and background level to normalize Nanostring nCounter data, a generalized linear model to characterize count data, and a likelihood ratio test to determine differentially expressed genes. Significantly differentially expressed genes (p < 0.05), as determined via NanoStringDiff, with log FC > 2 or < −2, or reduced gene lists (Table 1–2) were subsequently entered into Metascape40, a functional enrichment and gene annotation bioinformatics tool, to produce gene ontology plots.
Table 1:
Differentially expressed genes in LN229 vs. U87 co-cultures compared to mono-sphere cultures without astrocytes. Higher Log FC = more expression in LN229.
| LN229 vs. U87 Co-cultures | LN229 vs. U87 Mono-Spheres | Δlog FC (Co vs. Mono) | Notes | Ref. | |||
|---|---|---|---|---|---|---|---|
| Gene | Log FC | p value | Log FC | p value | |||
| CA9 | −3.9 | 0* | −35.9 | 0* | ↑ | -metabolic, increased in hypoxia; regulated by EGFR, STAT3 -associated w/increased GB invasion |
44 |
| FPR1 | −9.1 | 0* | −30.2 | 0* | ↑ | -inflammatory signaling; regulates STAT3, HIF-1a -increases GB invasion and cell survival |
45 |
| HGF | −2.3 | 0* | −36.8 | 0* | ↑ | -cytokine; only ligand for MET -enhanced GB cell survival, invasion, motility |
46 |
| IL7R | −26.6 | 2.6E-07 | −4.1 | 0* | - | -cytokine receptor; adaptive immune system -reduces % of infiltrating cancer cells |
47 |
| KLRG1 | −2.0 | 0.0038 | −28.5 | 9.5E-06 | ↑ | -immune checkpoint receptor; activated by E and N Cadherin ligands -inhibits immune response to cancer |
48 |
| TMPRSS2 | −30.2 | 0* | 4.2 | 0.023 | - | -transmembrane serine protease -associated with viral entry, prostate cancer -expressed by neurovascular astrocytes -regulates invasion and metastasis via HGF/MET activation |
49, 50 |
| CCR1 | −29.8 | 5.6E-12 | NS | - | -chemokine receptor for CCL5; regulates immune cell recruitment -upregulated at infiltrating GB tumor margin |
51 | |
| EDN1 | −2.5 | 5.6E-06 | NS | - | -vasoconstrictor; regulated by VEGF and ID1 -angiogenesis in GB |
52 | |
| HLA-DQA1 | −2.2 | 0* | NS | - | -immunoregulatory, part of MHC-II complex -high expression associated w/positive response to VEGF inhibitors |
53 | |
| KLRD1 | 2.6 | 0.034 | NS | ↑ | -immune response; NK-cell receptor -immunosuppressive via HLA-E binding |
54 | |
| KRT17 | −33.7 | 0* | NS | - | -intermediate filament; inhibits tumor suppressors -low in GB |
55 | |
| LAMC2 | −2.2 | 3.6E-09 | NS | - | -component of ECM protein laminin; associated with vasculogenic mimicry -highly expressed in U87/U251 cells |
56 | |
| NKX3–1 | −2.1 | 7.4E-05 | NS | - | -transcription factor -regulates the PI3K-AKT pathway; tumor suppressor -no known GB studies |
57 | |
| OAS2 | −24.7 | 8.6E-09 | NS | - | -regulates immunosuppression -increased in recurrent GB; drives stem-like behaviors |
58 | |
p value was <1E-12. NS = not significant.
Table 2:
Differentially expressed genes in LN229 and U87 co-cultures versus separate mono-sphere and astrocyte cultures. Higher Log FC values = more expression in co-culture.
| LN229 Co-Cultures vs. Separate | U87 Co-Cultures vs. Separate | Δlog FC (LN229 vs. U87) | Notes | Ref. | |||
|---|---|---|---|---|---|---|---|
| Gene | Log FC | p value | Log FC | p value | |||
| CA9 | 28.1 | 6.1E-07 | 5.4 | 6.1E-07 | ↑ | -metabolic, increased in hypoxia -regulated by EGFR, STAT3 -associated with increased invasion |
44 |
| EGLN3 | 26.1 | 0.014 | 3.4 | 0.014 | ↑ | -degrades HIFs; normalizes glioma vasculature -attenuates progression |
59 |
| FGF7 | 28.2 | 4.9E-08 | 2.5 | 4.9E-08 | ↑ | -fibroblast growth factor family -increases cell proliferation and survival |
60 |
| HLA-DQA1 | 27.7 | 2.0E-08 | 6.0 | 2.0E-08 | ↑ | -immunoregulatory, part of MHC-II complex -high expression associated with positive response to VEGF inhibitors |
53 |
| ITPR3 | 28.1 | 2.4E-08 | 2.4 | 2.4E-08 | ↑ | -calcium signaling -regulates cell death and survival -associated with GB migration and survival |
61, 62 |
| TMPRSS2 | −28.9 | 0.033 | 3.0 | 0.033 | - | -transmembrane serine protease -associated w/viral entry, prostate cancer -expressed by neurovascular astrocytes -regulates invasion and metastasis via HGF/MET activation |
49, 50 |
| ADH1A | NS | 23.6 | 0.037 | - | -catabolism -increased in brain cancers |
63 | |
| CD33 | NS | 28.5 | 0.00061 | - | -immune response; expressed on myeloid cells -possibly GB immunosuppressive |
64 | |
| CD38 | NS | 26.1 | 5.1E-05 | - | -regulates microglial activation; cell survival -MMP-12 expression |
65 | |
| FLT1 | NS | -30.1 | 0* | ↑ | -VEGF receptor -angiogenesis/tumorigenesis |
66 | |
| FPR1 | NS | 18.3 | 0* | - | -inflammatory signaling -regulates STAT3, HIF-1a -increases invasion and cell survival |
45 | |
| HAVCR2 | NS | 24.89 | 0.0036 | - | -immune checkpoint -poor prognostic indicator |
67 | |
| KLRK1 | NS | 28.5 | 0.036 | - | -immune surveillance, NK regulator -actin reorganization, cytokine release |
68 | |
| MAGEC2 | NS | 27.0 | 0.0053 | - | -cancer/testes gene -promotes amoeboid cell invasion via STAT3 |
69 | |
| MMP9 | NS | 27.3 | 0.0072 | - | -type IV collagenase, gelatinase -cell proliferation; poor GB prognostic indicator |
70 | |
| OAS2 | NS | 31.0 | 1.9E-05 | - | -regulates immunosuppression -increased in recurrent GB -drives stem-like behaviors |
58 | |
| TNFRSF18 | NS | 13.6 | 0.019 | - | -TNFα family -expressed on regulatory T cells |
71 | |
| KLRD1 | 28.4 | 0.0044 | NS | ↑ | -immune response, NK-cell receptor -immunosuppressive via HLA-E binding |
54 | |
| VEGFD | 26.4 | 0.017 | NS | ↑ | -VEGF family; regulate lymphatic vessels -prognostic for malignant transformation |
72 | |
p value was < 1E-12. NS = not significant.
Results
Characterization of batch-to-batch variation for GB mono-sphere encapsulation in Col-HA gels
A well-recognized problem with biomaterial models of tumour microenvironment is batch-to-batch (and experiment to experiment) variation.41 Variation in our culture model likely results from two factors: i) the heterogeneity of the tumour cells that is inevitable given that the spheres originate from a small number of cells (~1000 cells) and ii) batch variation in the naturally derived hydrogels comprising the culture matrix. To identify variability within our culture model, we first grew mono-spheres comprised of two GB cell lines, U87 and LN229, in Col-HA gels using two different batches of HA (B1 and B2) on three different days (E1, E2, E3). HA in particular exhibited variation because it was a single use material. It is provided by the manufacturer in units sufficient for one experiment.
U87 and LN229 GB mono-spheres were formed in liquid culture over 3 days to reach a sphere diameter >200 μm. Then, spheres were encapsulated in Col-HA gels (Supplementary Figure 3, Figure 1 A, B). Next, we compared the sphere diameter, migration capacity, and ad-distance data from each experiment and HA batch for both cell lines (Figure 1 C, D). Images were taken on day 3 to capture the sphere diameter at the end of liquid culture prior to encapsulation, and then daily to the end of culture (day 7 for U87 and day 9 for LN229). U87 mono-spheres were imaged over a shorter time duration because the migration front reached the limit of the visible frame (~1016 μm from the centre of the sphere) at day 7, preventing further meaningful analysis. Variance in sphere diameter at day 3 (prior to encapsulation) was detected for both cell lines. This could result from differences in proliferation rate or counting errors of the cells seeded on day 0, especially considering the low numbers of cells employed. Spheres cultured in batch 2 (B2) hydrogels had larger sphere diameters, but lower migration capacities and ad-distance, compared to spheres cultured in B1 hydrogels. This suggests that B2 gels supported cell proliferation over migration (Figure 1 C, D). Variation in sphere diameter, migration capacity, and ad-distance was also observed from experiment to experiment (E1, E2), but to a lesser degree.
Figure 1.
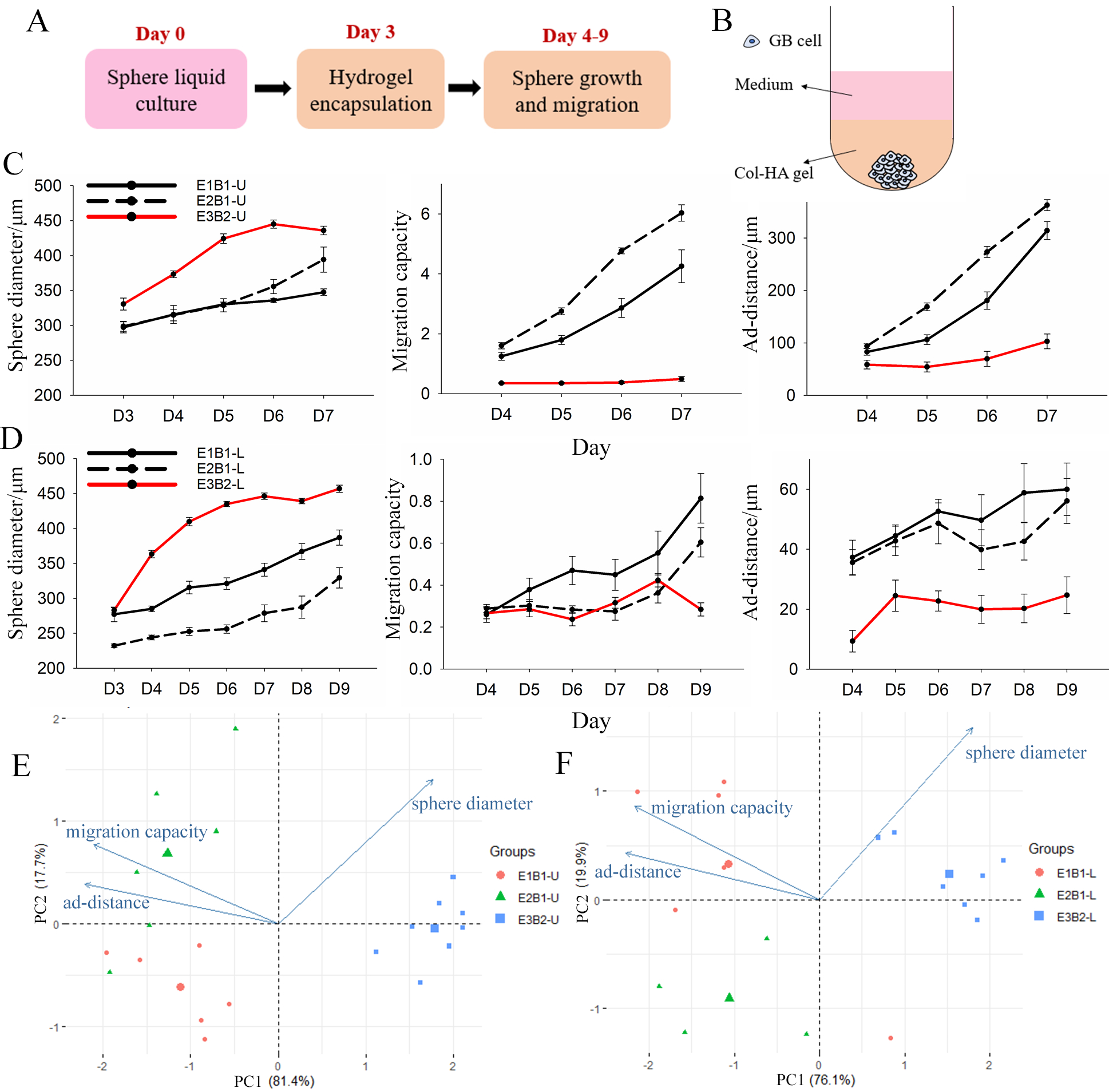
Three independent experiments (E1, E2, and E3) were performed with two batches of HA (B1 and B2) using U87 and LN229 mono-spheres encapsulated in Col-HA gels: A) experiment design and B) model configuration. (C, D) Sphere diameter, migration capacity, and adjusted migration distance (ad-distance) of C) U87 and D) LN229 mono-spheres are presented as average ± standard error. N = 4–8. (E, F) Principal component analysis (PCA) plots of E) U87 and F) LN229 mono-spheres shows separation of individual spheres in three experiment groups at the last day of culture (day 7 for U87 and day 9 for LN229).
To quantify these differences, principal component analysis (PCA) was performed on data from the last day of culture (day 7 for U87 and day 9 for LN229). PCA successfully separated the spheres from the three experiments into three groups by PC1 and PC2. E3B2 is separated from E1B1 and E2B1 mainly by PC1, which accounts for >75% of the total variation for both U87 and LN229 mono-spheres. Moreover, PC1 is contributed to by an even mix of sphere diameter (25.12%), migration capacity (35.70%), and ad-distance (39.18%) (Figure 1 E, F, Supplementary Table 1). This suggests that the spheres cultured in B2 gels are distinctly different from those cultured in B1 gels in all three dimensions of sphere growth and migration, with migration distance being the most affected. The E1B1 and E2B1 spheres are mainly separated by PC2, which is contributed to mostly by sphere diameter (74.28%), followed by migration capacity (22.07%), and the least of ad-distance (5.45%). Thus, day-to-day experimental variation appears to mainly influence sphere growth versus migration, in contrast to batch-to-batch variation caused by HA gels. Based on these results, we conclude that batch variation caused by the HA gels is the main factor affecting sphere migration behaviours, and therefore recommend that experiments be compared within the same batch of HA where possible.
Growth and migration of U87 mono-spheres and U87-NHA multi-spheres in Col-HA gels
To examine the interaction between GB cells and astrocytes, we first tested the effect of incorporating astrocytes during GB sphere formation by generating U87-NHA multi-spheres in liquid culture that were subsequently encapsulated in Col-HA gels (Figure 2A). U87 cells were studied because of their higher migration response compared to LN229 cells in mono-sphere culture without NHAs, which makes it easier to elucidate changes in migration response. U87-NHA multi-spheres at NHA:U87 (N:U) ratios of 10:1, 5:1, 1:5, 1:10 were formed and compared to control NHA only and U87 only mono-spheres. NHAs showed a much lower proliferation rate in culture than U87s, and NHA spheres were initiated with a greater number of cells. Despite this, the NHA-dominated spheres were still significantly smaller than the U87-dominated spheres at day 3 in liquid culture and throughout culture in Col-HA gels (Supplementary Figure 3, Figure 2B–C). After encapsulation, cells in NHA mono-spheres migrated in a ‘starfish’ form with long cell processes observed. U87s in all multi-spheres and U87 mono-spheres migrated as cell strands in regions close to the sphere, and more distant from the sphere surface, spread out as single cells. In N:U = 10:1 and 5:1 multi-spheres rich in NHAs, NHAs were found to mix evenly with U87s in the sphere region and migrate highly in alignment with the U87 cells migrating from the sphere. But for N:U = 1:5 and 1:10 multi-spheres rich in GB cells, NHAs were observed to be concentrated in the sphere centre and demonstrated little migration (Figure 2B, red). Interestingly, U87-NHA multi-spheres showed no statistical difference in U87 migration capacity in comparison to each other or to the U87 mono-spheres up to day 6 of culture. At day 7, NHA-dominated multi-spheres (N:U = 10:1 and 5:1) showed significantly lower migration capacity compared to other spheres (Figure 2B, green; Figure 2D). This could be the result of differences in the number of GB cells (i.e., total cell number was kept constant). Thus, we also performed a further experiment in which the number of GB cells was kept constant, and astrocytes were added at the same ratios as above (i.e., GB cell number kept constant) (Supplementary Figure 4). As expected, sphere sizes were larger for high N:U ratios, most likely reflecting the larger total number of cells. Despite this similar migration trends were observed. At day 7, NHA-dominated multi-spheres (N:U = 10:1 and 5:1) showed significant decreases in migration capacity, indicating that astrocytes reduce GB migration capacity. Migration distances of U87 mono- and multi-spheres were statistically indistinguishable through the 7 days of culture for both experiments (Figure 2E, Supplementary Figure 4). This suggests that astrocytes in the multi-sphere configuration reduce the total number of migrating cells (migration capacity), but not their distance travelled (ad-distance).
Figure 2.
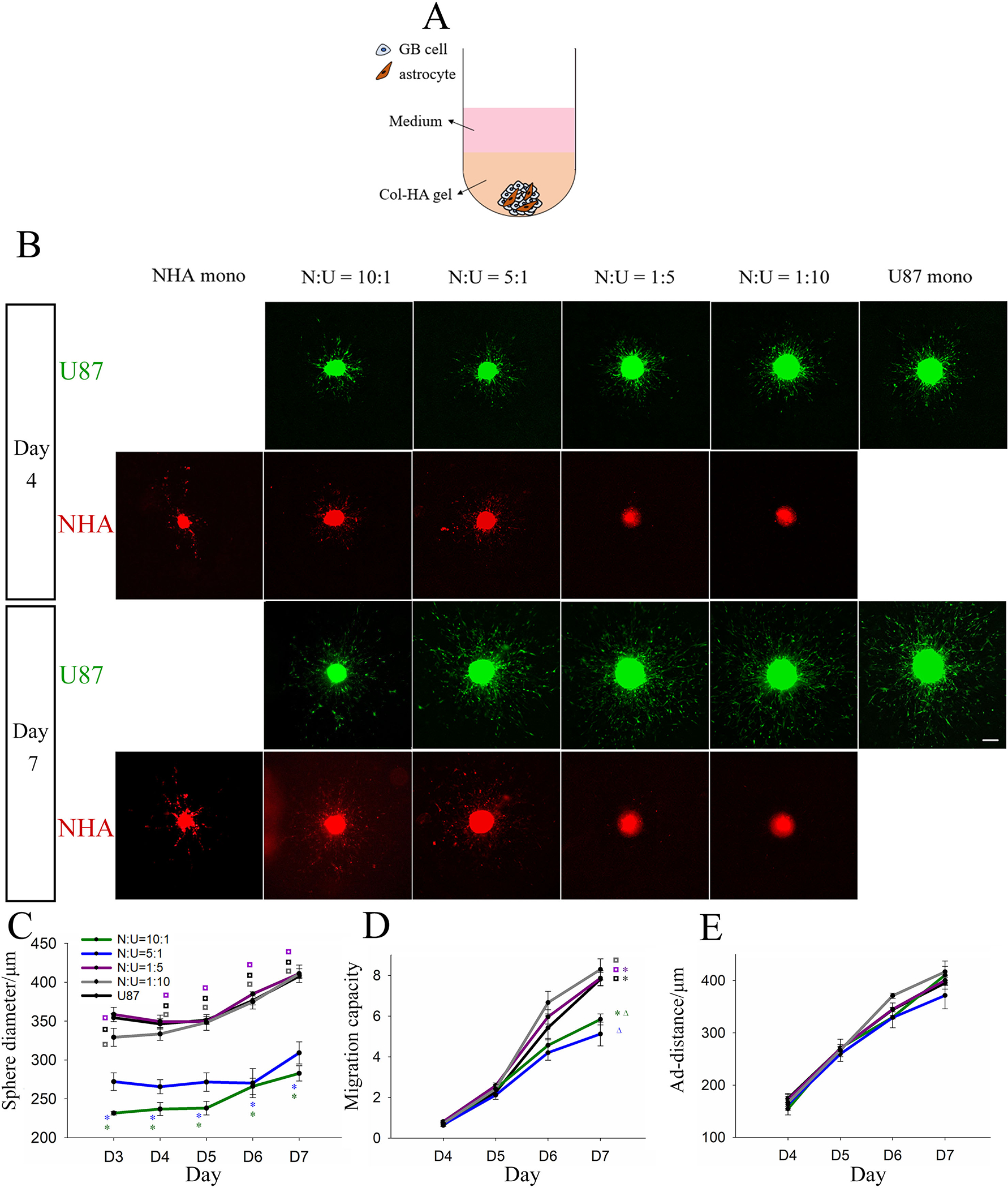
NHA mono-, U87 mono- and U87-NHA multi-spheres encapsulated in collagen-HA gels. A) Model configuration. B) Representative images of NHA (red) mono-, U87 (green) mono-, or U87-NHA multi-spheres at day 4 and day 7. Bar = 200 μm. GB C) sphere diameter, D) migration capacity, and E) adjusted migration distance (ad-distance) are presented as average ± standard error. N =4 or 5. *, ◘, and Δ are colour coded to the respective condition. Different symbol groups suggest groupings that are statistically different at that day. P < 0.05.
Growth and migration of U87 mono-spheres encapsulated in Col-HA gels with NHA-conditioned media
One possible reason for differences in GB migration in the presence of astrocytes is the secretion of soluble factors. To investigate this possibility, we formed U87 and NHA mono-spheres separately, and then encapsulated GB mono-spheres in Col-HA gels coated with conditioned media taken from NHA mono-sphere cultures (Figure 3A). Conditioned media failed to induce any difference in sphere size or migration as compared to the control with U87 growth media (Figure 3B–D), suggesting that differences in migration resulted from either GB-induced change in the astrocyte that would not be captured in separate culture or from direct contact with GB cells.
Figure 3.
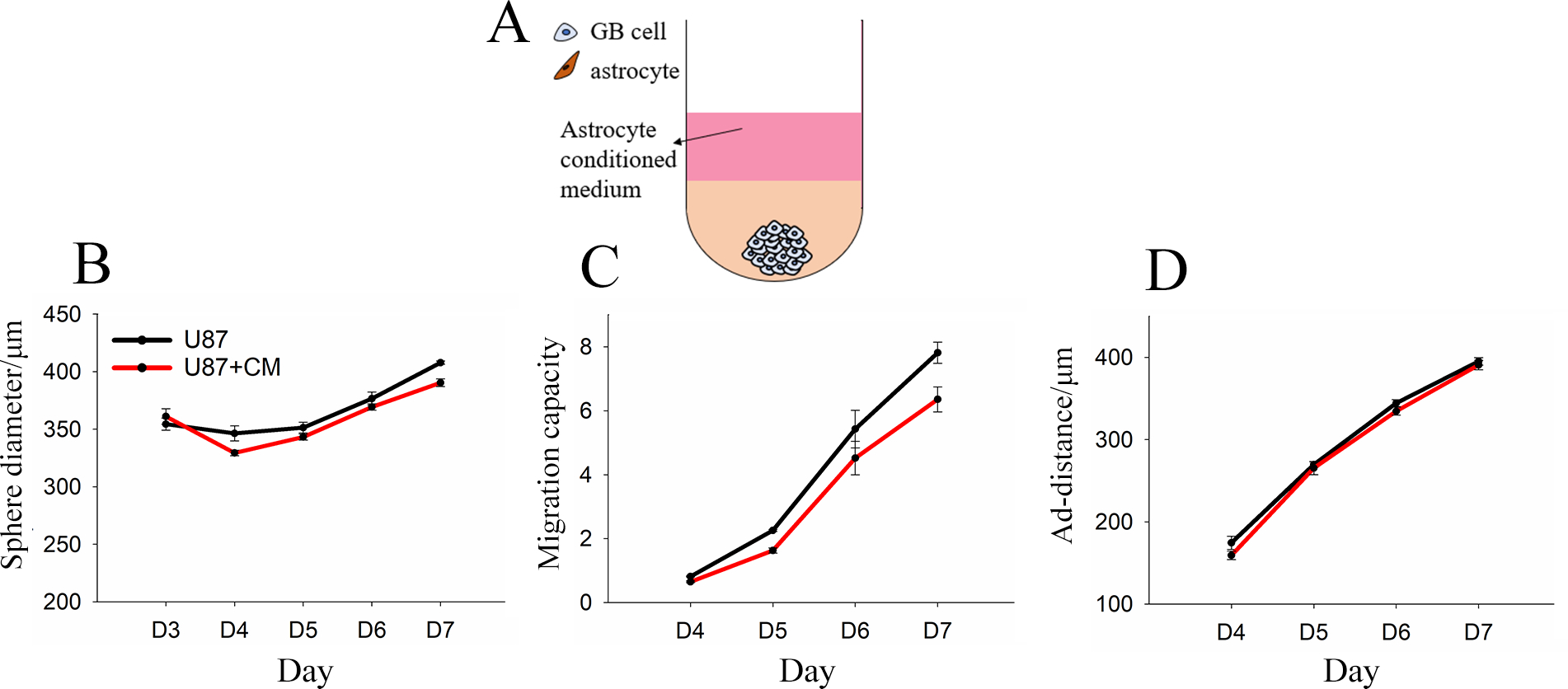
U87 mono-spheres encapsulated in collagen-HA gels with or without NHA-conditioned media (CM): A) Model configuration. B) Sphere diameter, C) migration capacity, and D) adjusted migration distance (ad-distance) are presented as average ± standard error. N = 4 or 5. No significant differences were observed on any culture day. P > 0.05.
Growth and migration of U87 and LN229 mono-spheres encapsulated in Col-HA gels loaded with live NHAs
The final model configuration that we investigated consisted of GB mono-spheres cultured with dispersed astrocytes. This configuration most closely resembles the in vivo morphology in which a tumour mass is in contact with astrocytes in the TME-adjacent brain parenchyma. In these experiments, both U87 and LN229 cells were employed to evaluate potential gene expression differences driving the differential response of these cell lines. GB mono-spheres (U87 or LN229) were encapsulated in NHA-loaded Col-HA gels at high cell density (5 × 105 or 1 × 106 NHAs/ml, Supplementary Figure 5A). LN229 mono-spheres appeared less compact in liquid culture compared to U87 mono-spheres, potentially reflecting their more infiltrative phenotype.17 During gel encapsulation, a small number of loose cells were inevitably dislodged from the LN229 spheres that could be detected distant from the sphere region (Supplementary Figure 5B). Those cells were manually excluded in image processing and were not included in the migration calculations. Consistent with our prior observations (Figure 1), U87 and LN229 mono-spheres (no NHAs) showed a similar trend in sphere growth (~17.7% increase in sphere diameter from day 4 to 7), but distinct differences in migration patterns. U87 mono-spheres migrated mainly in the form of single cells or small clusters, keeping a clear boundary of the sphere mass. LN229 mono-spheres showed little single cell migration until day 9 and mainly migrated by extending dense cell strands from the spheres. As a result, LN229 mono-spheres demonstrated a more diffuse boundary, with reduced migration capacity and a smaller ad-distance, compared to the U87 mono-spheres (Figure 4).
Figure 4.
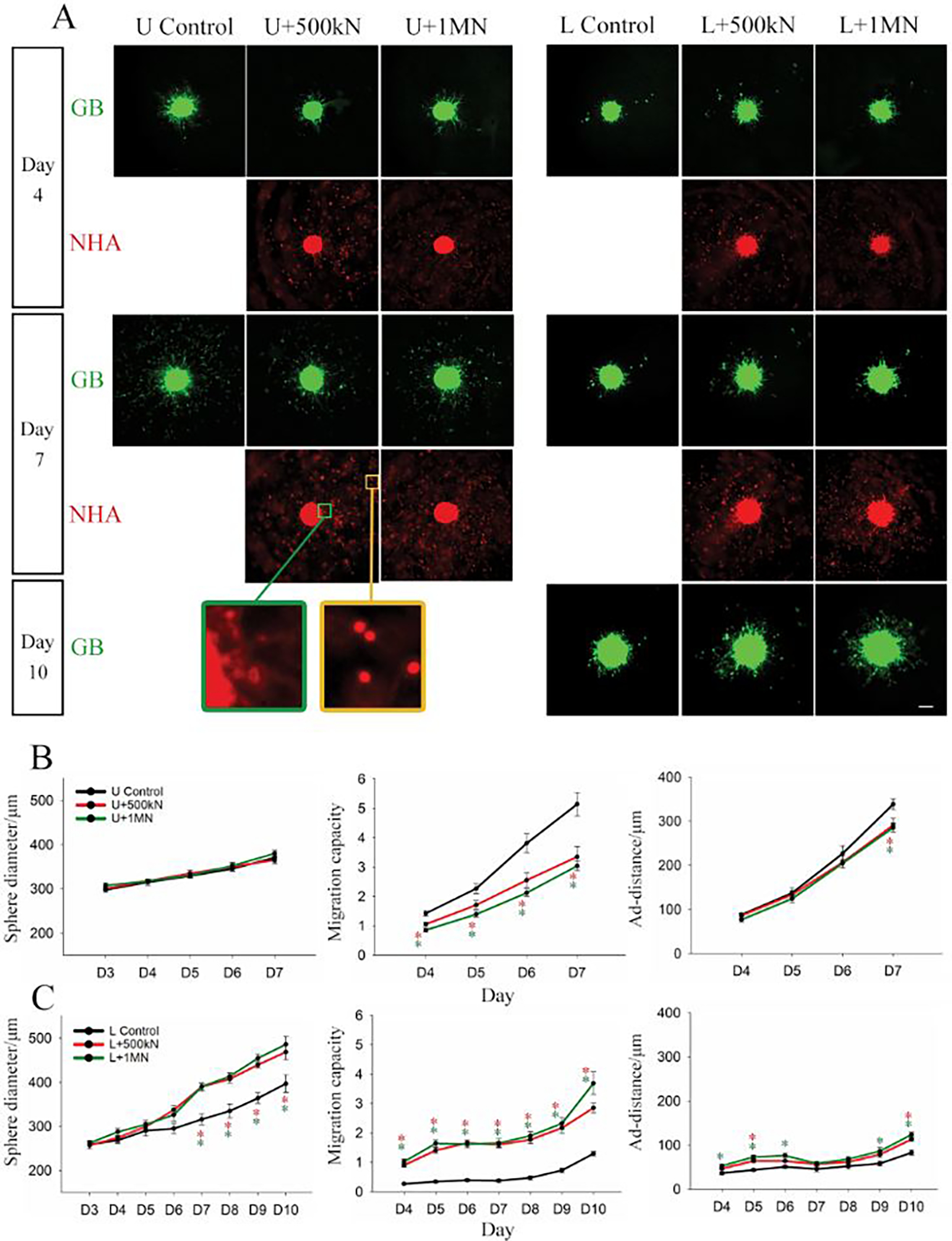
U87 (U) and LN229 (L) mono-sphere growth and migration in collagen-HA gels loaded with NHA at concentrations of 500k (500kN) or 1M (1MN) cells/ml. A) Representative images of U87 or LN229 (green) mono-spheres and NHA (red) at day 4, day 7, and day 10 (LN229 only). NHA staining became weak with culture and therefore was not captured after day 7. Inset shows change in NHA morphology based on distance from the sphere. Bar = 200 μm. Sphere diameter, migration capacity, and adjusted migration distance (ad-distance) of B) U87 and C) LN229 mono-spheres are presented as average ± standard error. N = 8~12. * is colour coded to the respective condition and suggests the group is statistically different from the control at that day. P < 0.05. The two concentrations of NHAs are not significantly different from each other in both U87 and LN229 at all experiment days. P>0.05.
Interestingly, immediately after gel solidification (within 4 hours after encapsulation), we observed co-localization of NHAs on the surface of both U87 and LN229 mono-spheres, which was al established in extended culture (Figure 4A, Supplementary Figure 5B, Supplementary Figure 6). Time lapse images of U87 mono-spheres after gel encapsulation show dynamic reorganization of GB cells on the sphere surface (Supplementary Video 1). Like the multi-sphere configuration, NHAs were highly aligned to GB cells migrating from the spheres. Particularly, NHAs adjacent to the spheres adopted an elongated morphology, whereas NHAs located further from the sphere surface were mostly rounded (Figure 4A, inset). For U87 spheres, co-culture with NHAs did not statistically change sphere size, but did induce a constant decrease in migration capacity (~35% lower than the control) throughout culture. Ad-distance was only reduced at day 7, the last day of observation. For LN229 spheres, co-culture with NHA significantly increased sphere size, migration capacity, and ad-distance with the migration capacity being most affected. The migration capacity of LN229 spheres co-cultured with 1 × 106 NHAs/ml was ~337% higher than the control at day 7, and ~184% higher than control at day 10. The LN229 data is in direct opposition to results observed for U87 cells, indicating that this culture model can detect differences in GB behaviors based on cell heterogeneity. There were no statistical differences observed for the two different NHA loading densities for both U87 and LN229 spheres (Figure 4B, C).
Growth and migration of U87 and LN229 mono-spheres encapsulated in Col-HA gels loaded with fixed NHAs
Given the observation of NHA co-localization with GB cells on the sphere surface (Figure 4A), we hypothesized that surface ligands may mediate these interactions. To examine if astrocytes regulate GB sphere migration through surface ligands (Figure 4A), we next encapsulated U87 mono-spheres with live or fixed NHAs at a density of 2 × 106 NHAs/ml (Supplementary Figure 7). Fixed NHAs were treated with formaldehyde to prevent secreted signaling, while maintaining surface receptors as intact as possible. High NHA loading density was chosen to compensate for the loss of proliferation resulting from fixation. Immediately after gel solidification, we observed prominent attachment of live NHAs to U87 and LN229 mono-spheres (Supplementary Figure 7A), whereas the fixed NHAs showed lower, but still observable, attachment (Supplementary Figure 7B). During extended culture, the intensity of the signal from fixed NHAs increased for LN229 spheres, but remained constant with low intensity for U87 spheres (Figure 5A).
Figure 5.
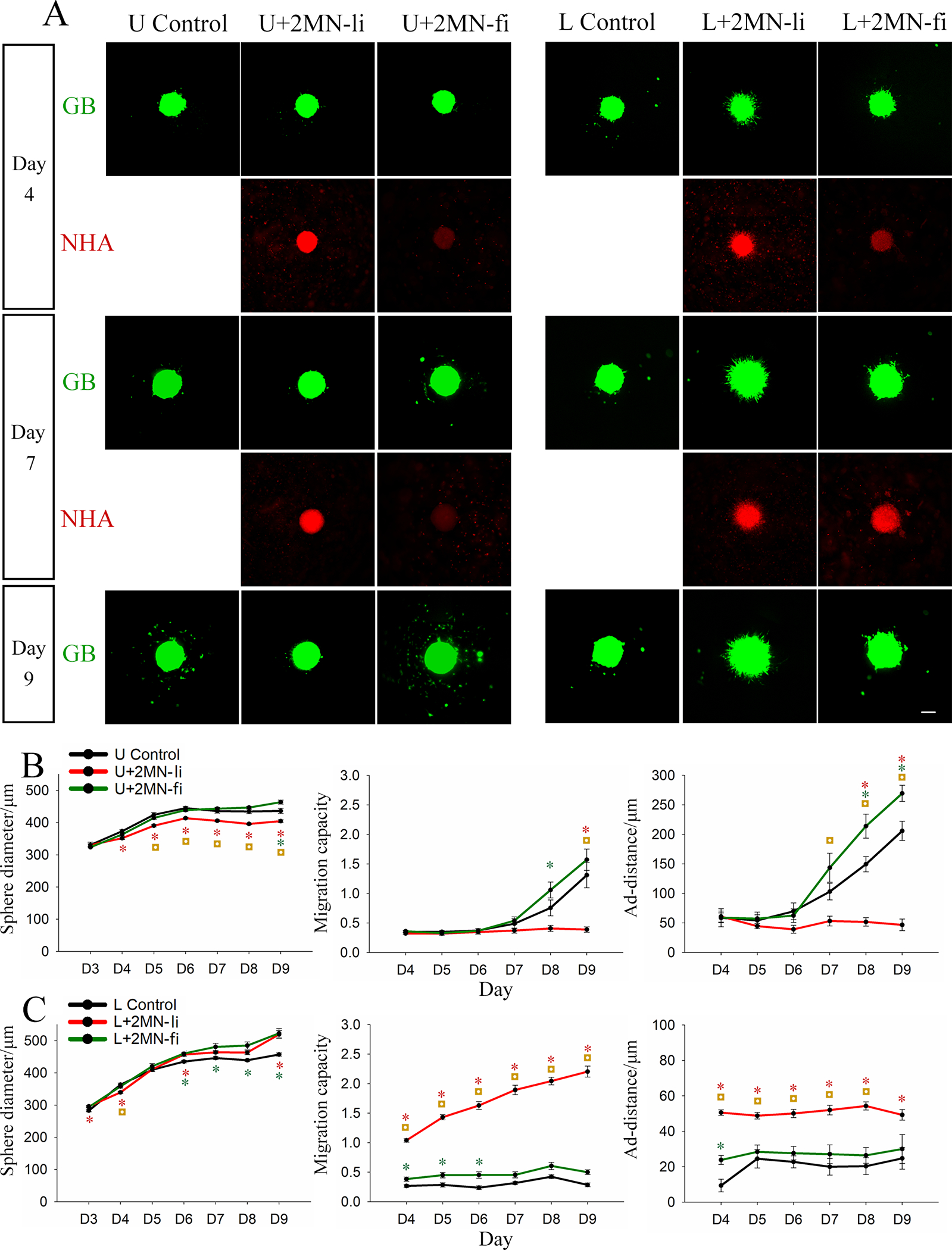
U87 (U) and LN229 (L) mono-sphere growth and migration in collagen-HA gels loaded with live (2MN-li) or fixed NHA (2MN-fi) at 2M cells/ml. A) Representative images of U87 or LN229 (green) mono-spheres and NHAs (red) at day 4, day 7, and day 9. NHA staining became weak over the culture duration, and therefore, was not captured after day 7. Scale bar = 200 μm. (B, C) Sphere diameter, migration capacity, and adjusted migration distance (ad-distance) of B) U87 and C) LN229 mono-spheres are presented as average ± standard error. N = 6~8. * is colour coded to the respective condition and suggests the group is statistically different from the control on that day. ◘ suggests that the live NHA and fixed NHA groups are significantly different from each other on that day. P < 0.05.
For U87 spheres, fixed NHAs induced changes in sphere growth and migration that contrasted with those of live NHAs. Whereas live NHAs decreased U87 sphere size with time, fixed NHAs did not affect sphere size until day 9, when a slightly increased sphere size was observed. U87 sphere migration capacity and ad-distance were reduced by live NHAs throughout culture; however, fixed NHAs did not affect either until day 7 at which point both migration capacity and ad-distance were enhanced compared to the control (Figure 5B). For LN229 spheres, fixed NHAs induced similar, but less pronounced, changes in sphere growth and migration compared to live NHAs. Sphere size was slightly enhanced by both live and fixed NHA, but only live NHAs increased migration capacity and ad-distance from day 4 to day 9. Fixed NHAs yielded statistically significant differences in migration capacity from day 4 to day 6, and in ad-distance at day 4, but these were not observed in extended culture (Figure 5C). For extended cultures (day 7 onwards), both U87 and LN229 cells exposed to fixed astrocytes exhibited opposite migration trends compared to their respective live-astrocyte conditions.
This disparity was also observed in our previous work examining GB and astrocyte co-cultures on white matter tract-mimetic electrospun fibers42. Migration of patient-derived GB cells increased in the presence of live astrocytes and astrocyte-conditioned media, but decreased in the presence of fixed astrocytes. This was attributed to possible competing signaling pathways involving soluble signaling vs. signaling via cell surface receptors. It is also possible that cell surface receptor damage induced by fixation43 altered results, or alternatively, that different cell receptors are expressed by NHAs in the presence of GB (fixed cells were cultured separately from GB cells before fixation and addition). Nonetheless, these results are consistent with our prior findings and suggest that cell surface molecules may play a role in GB migration, although this role is complicated and likely influenced by a host of other factors. Further work is needed to discern the exact mechanisms that govern astrocyte-GB interactions via cell surface receptors.
Analysis of gene expression changes for U87 and LN229 mono-spheres in the presence of astrocytes
RNA extraction from hydrogels was performed using similar methods for tissue processing, including mechanical homogenization and chemical lysis. The number of cells in each gel culture was small, and thus multiple replicates (N ~ 15–30) for each gene expression sample were pooled to obtain sufficient RNA (>300 ng) for analysis. RINe scores ≥ 9.3 were achieved for all conditions, indicating high levels of RNA integrity. Thus, we recommend this approach for processing hydrogel TME mimetic samples. Samples were then evaluated using the Nanostring™ nCounter™ Tumour Signalling 360 gene expression analysis chip for 760 tumour associated genes. We examined gene expression of pooled samples from U87 and LN229 because of their differing migration behaviours (Figures 1, 4–5). Three configurations were employed. Mono-spheres cultured in the presence of dispersed astrocytes (i.e., GB-mono-sphere plus NHA co-culture) (N=2) comprised the experimental condition. Two control conditions included mono-spheres cultured without astrocytes (i.e., GB mono-sphere only) (N=2) and isolated mono-sphere RNA mixed with RNA isolated from dispersed astrocytes cultured in hydrogels separately (i.e., separate cultures) (N=1) to preclude cross-talk.
We first compared LN229 to U87 samples in mono-culture without astrocyte and co-culture with astrocytes to identify differences between cell lines (Table 144–58). In co-culture, 31 genes were differentially expressed genes (DEGs) with a log fold change (FC) > 2 and a p value < 0.05 (Supplementary Table 2, Supplementary Figure 8). In contrast, control LN229 vs. U87 mono-sphere-only cultures displayed 138 DEGs that met the same significance criteria (Supplementary Table 3, Supplementary Figure 9). This suggests that the addition of astrocytes to the cultures may have mitigated some of the differences in gene expression between these two cell lines. This finding is potentially important for studies using this panel to represent GB heterogeneity, as our findings show that differences between these cell lines are reduced in the presence of astrocytes.
Of the 31 DEGs in co-culture, 17 were differentially expressed with similar log FC values in control mono-sphere cultures lacking astrocytes, and thus likely reflect genotypic differences between these two cell lines (Supplementary Tables 2–3, grey). However, there were 5 genes differentially expressed in both culture conditions that differed by more than 10 in log FC and 1 DEG that had a change in log FC sign. There were also 8 DEGs unique to co-culture conditions (Table 1). These genes could reflect cell-line specific, differential reprogramming of either GB cells or astrocytes that could impact migration. However, they could also reflect contributions from the astrocytes in culture.
To identify differences resulting specifically from cross-talk as opposed to the presence of astrocytes alone, we also compared each GB co-culture to a sample comprised of mixed RNA from GB cells and astrocytes grown separately (Table 244, 45, 49, 50, 53, 54, 58–72). Co-cultures vs. mixed separate culture comparisons for LN229 and U87 cells yielded 55 and 58 DEGs, respectively (Supplementary Tables 4–5, Supplementary Figures 10–11), reflecting differences in GB-astrocyte crosstalk as opposed to differences arising from the presence of astrocyte RNA. Of these, 19 were differentially expressed in both LN229 and U87 cultures at similar levels and likely reflect crosstalk changes occurring in both lines (Supplementary Tables 4–5, gray). There were also 5 DEGs varying by a log FC of more than 10 and one with a different sign (Table 2). In addition, there were 28 and 31 DEGs unique to LN229 and U87 cultures, respectively (Supplementary Tables 4–5). These genes reflect cell-line specific differences in crosstalk between GB cells and astrocytes. Of these, 2 vs. 11 DEGs had |log FC| values > 10, for LN229 and U87, respectively (Table 2), indicating that U87 cells had a stronger response to co-culture with astrocytes than LN229 cells. Six of these genes appeared in both comparisons (Tables 1–2): CA9, HLA-DQA1, TMPRSS2, FPR1, OAS2, and KLRD1, and represent the greatest differences in GB-astrocyte crosstalk between U87 and LN229 GB cells.
To identify DEGs potentially associated with migration, we also analysed expression of genes associated with changes in the ECM and cell-binding to ECM. Cell migration proceeds by binding to the ECM through cell surface receptors, such as integrins. There were no differences detected between cell lines for integrin gene expression in co-culture, except for lower LN229 expression of ITGA5, which encodes the α5 integrin protein that associates with β1 to form a fibronectin receptor. However, this difference was also observed in mono-sphere culture and is therefore not related to GB-astrocyte crosstalk. In our co-culture model, astrocytes are expected to modify the hydrogel composition by depositing their own ECM molecules, such as laminin, fibronectin, and vitronectin.73 There were no differences observed between cell lines in co-culture for collagen IV or XI, and the Tumor360 panel does not test for hyaluronan synthase, fibronectin, or vitronectin. However, there was a decrease in LAMC2 for LN229 vs. U87 co-cultures. LAMC2 encodes the laminin γ2 chain, a part of laminin 5, a molecule that promotes cell adhesion and migration and that is associated with tumour invasion.74 Additionally, MMP9 was strongly differentially expressed in U87 co-culture vs. mono-sphere only cultures, but there was no significant difference for LN229 co-cultures vs. mono-spheres only cultures. MMP9 is a type IV collagenase and gelatinase that may alter local ECM density. Thus, in addition to migratory signalling, the TME ECM is also likely altered to influence migration. Interestingly, U87 cells showed highest change in ECM gene regulation in co-culture with dispersed astrocytes, but reduced migration compared to LN229 cells.
From gene expression analysis, we identified a reduced gene set most closely associated with differential GB-astrocyte crosstalk (i.e., those listed in Tables 1–2 and the 28 and 31 DEGs unique to LN229 and U87 cultures in co-culture vs. separate culture analysis). Most of these genes are associated with cytokine signalling or immune regulation, suggesting that exposure of GB cells to astrocytes reprograms immune signalling. Metascape Gene Ontology (GO) biological processes enrichment analysis was performed on this reduced gene set (Figure 6, Supplementary Figures 12–14). All the processes identified in LN229 vs. U87 co-culture comparisons are also present in the other comparisons (LN229 vs. U87 mono-spheres without astrocytes and LN229 or U87 co-cultures vs. LN229 or U87 separate cultures). Relevant to the cell migration changes we observed locomotion (GO:0040011) and localization (GO:0051179) processes were both altered in co-culture. Immune regulation (GO:0002376) was also strongly implicated. These data suggest a potential link between immune signalling and GB migration.
Figure 6.

Metascape Gene Ontology (GO) biological processes enrichment analysis of differentially expressed genes in LN229 vs. U87 co-cultures that are not also differentially expressed in mono-culture without astrocytes.
Discussion
Astrocytes comprise the main cell type of the brain stroma, forming networks of processes throughout the brain that support neuronal migration.15 Astrocyte endfeet also comprise part of the blood-brain barrier (BBB).15 In GB, astrocyte processes may provide paths for tumour cell migration, similar to Scherer’s structures.2, 12 In their activated morphology, which is induced by exposure to GB, reactive astrocytes may facilitate tumour cell migration by altering the brain ECM through release of MMPs, altered ECM deposition75, and release of regulatory cytokines (e.g., IL-6).15 The crosstalk between astrocytes and GB cells is likely complex because GB is a tumour of astrocytic origin, and GB tumours can be induced from astrocytes by mutating core pathways such as Akt, RB, Kras, and PTEN in mouse models.76, 77
This study explored and validated 3D in vitro brain TME mimetic hydrogels models to study GB migration in GB-astrocyte co-culture (Table 3). Previous GB-astrocyte co-culture models include in vitro 2D and Boyden chamber assays, brain slices, and animal models.11, 78 In vitro models are especially helpful in elucidating the particular role of astrocytes, separate from other brain cells present in brain slices and in vivo models. In addition, they are amenable to migration studies permitting analysis of collective and single cell migration phenomena. Previously, we constructed Col-HA hydrogels and used them to study single cell migration of dispersed GB cells.34 This model was also used by Herrera-Perez et al. for the co-culture of dispersed patient-derived GB cells and primary astrocytes (ScienCell).79 In that study, they found that astrocytes enhanced the migration speed and directionality of GB cells.79 Our model builds upon this previous work by investigating interactions of astrocytes with GB spheroids in three configurations: multi-cellular spheroids comprised of GB cells and astrocytes, mono-spheres comprised of GB cells cultured with astrocyte-conditioned media, and mono-spheres comprised of GB cells cultured with dispersed astrocytes. Our study employed 2 different cell lines, U87 and LN229, reflecting nodular versus infiltrative tumours, respectively.17 Spheroid cultures more closely mimic the presence of an established tumour mass in contact with adjacent normal tissue than dispersed cell cultures that mimic earlier stages of tumorigenesis.
Table 3:
Summary of findings for different co-culture configurations. Arrow direction indicates change relative to the control (no astrocytes or no conditioned media).
| Culture Configuration | Sphere size | Migration capacity | Ad-distance | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GB-astrocyte multi-spheres Figure 2 |
U ↓ |
U ↓* |
U NS |
|||||||||
| GB mono-spheres in astrocyte-conditioned media Figure 3 |
U NS |
U NS |
U NS |
|||||||||
| GB mono-spheres with dispersed astrocytes Figure 4 |
U ↓ |
L ↑ |
U ↓ |
L ↑ |
U ↓* |
L ↑ |
||||||
| GB mono-spheres cultured with live vs. fixed dispersed astrocytes Figure 5 |
U/Li ↓ |
U/Fi ↑* |
L/Li ↑ |
L/Fi NS |
U/Li ↓* |
U/Fi ↑* |
L/Li ↑ |
L/Fi NS |
U/Li ↓* |
U/Fi ↑* |
L/Li ↑ |
L/Fi NS |
Indicates no statistically significant change until day 7 and/or after. NS = no significant change. U = U87 and L=LN229. Li = live astrocytes and F = fixed astrocytes.
In the absence of astrocytes, the Col-HA model supported the growth and migration of cells from both U87 and LN229 mono-spheres (Figures 1–2). Whereas both cell lines were seeded at 1000 cells/well for sphere formation, U87 mono-spheres had a higher rate of sphere growth (i.e., larger sphere diameter) in liquid culture and after gel encapsulation. U87 mono-spheres also showed a larger migrating population (migration capacity) and longer migration distance (ad-distance) than LN229 spheroids. U87 cells have been previously reported to proliferate and migrate faster than LN229 cells in dispersed 3D cell culture models, consistent with our observations for spheroid culture and validating our results.80
Next, we explored GB-astrocyte interactions in Col-HA hydrogels. Initially, only U87 GB cells were tested because of their high degree of sphere growth and migration observed in mono-culture. We also compared multi-spheres containing U87 cells and astrocytes to astrocyte-only spheroids and GB mono-spheres (Figure 2). Astrocyte-only mono-spheres formed a loose structure during liquid culture and grew into starfish-like networks with long processes extending from the sphere into the hydrogel. However, the astrocytes in multi-spheres were tightly packed in the sphere centre by U87 cells, and when encapsulated, migrated from the sphere mainly as single cells aligned with migrating U87 cells. In the NHA-rich multi-spheres (N:U = 10:1 and 5:1), we observed evidence of collective migration, as both U87 and NHA extended fine cell strands highly co-localizing with each other at regions close to the sphere. This structure closely resembles ‘leader-follower’ behaviour in which non-invasive cells follow the migration of leader, invading cells, which has been observed in multi-cellular spheroid co-cultures in other cancer types.81 This could potentially result from astrocyte reprogramming by the GB cells to migrate in a GB-like pattern. Previous co-culture studies employing Boyden chambers have shown that astrocytes can be activated by GB cells, enhancing GB cell migration through secretion of signalling molecules.82, 83 In this study, we explored this possibility using a configuration in which conditioned media from astrocytes cultured independently was added to GB mono-sphere cultures (Figure 3). However, this configuration did not elicit changes in migration. This supports the hypothesis that astrocyte reprogramming by GB cells is required for migration changes and validates the multi-sphere model as a potential means to study astrocyte reprogramming. However, in this model, astrocytes are in direct contact with GB cells.
To examine interactions of GB spheres with astrocytes more distant from the sphere surface, we also studied a configuration consisting of GB mono-spheres embedded in Col-HA gels containing dispersed astrocytes (Figure 4). This configuration more closely resembles the physiology of an established tumour, and we tested both U87 and LN229 cells. In both cell lines, we observed co-localization of astrocytes on GB mono-spheres within 1 hour of encapsulation and throughout culture. Interestingly, co-localization was reduced but not abrogated by astrocyte fixation prior to encapsulation (Figure 5), suggesting that astrocyte co-localization is at least partially mediated directly by GB cells. Time lapse imaging revealed the GB sphere surface as dynamic, with continuous reorganization that may facilitate astrocyte transport to the sphere surface (Supplementary Video 1). However, since recruitment of live astrocytes was much more robust than with fixed astrocytes, astrocyte interactions likely contribute to this behaviour as well. This process may be comparable to astrocyte response to traumatic brain injury (TBI). In TBI, astrocytes proliferate and rearrange themselves to form astroglial scars surrounding the lesion site, which helps limit inflammatory response.84 Interestingly, LN229 xenograft models have shown a greater ability to induce GFAP+ phenotypes, suggestive of reactive astrocytes, than U87 xenografts,17 and here LN229 cells showed greater recruitment of fixed astrocytes to the tumour sphere surface than U87 spheres (Figure 5).
In contrast to Herrera-Perez et al.,79 which employed three different patient-derived GB cell lines and observed similar results for each of them in dispersed cell cultures, we observed mixed migration results based on cell line in this model (Figure 4). Addition of dispersed astrocytes (Lonza) to U87 “nodular” mono-sphere cultures decreased GB migration, whereas addition of astrocytes to LN229 “infiltrative” mono-sphere cultures increased GB migration. In addition to these changes in migration capacity and migration distance, we also observed differences in migration behaviours between the two cell types. U87 cells and astrocytes displayed primarily single cell migration behaviours, whereas LN229 cultures evidenced collective migration of GB cells and astrocytes in the form of dense cell strands with clear leading edges penetrating the surrounding gel. The strands formed by astrocytes in the sphere region were highly aligned to the strands formed by LN229 cells but shorter in length, suggesting that astrocytes at the sphere region potentially migrated away from the sphere by following the LN229 cells. In the mode of collective migration, astrocytes likely supported the migration of LN229 cells instead of acting as a physical barrier.
The contrasting observations in our study from those of Herrera-Perez et al.79may result from culture configuration (i.e., spheroids vs. dispersed cells), but it is more likely that GB cell genotype and phenotype yield differential response to astrocyte co-culture. Previously, spheroid culture of LN229 cells was shown to induce increased expression of stem-cell associated markers, such as Nestin, SOX2, Musashi-1, and CD44, an HA binding protein, whereas these were not observed in U87 spheroids.85 We detected an upregulation of SOX2 in LN229 mono-spheres compared to U87 mono-spheres (log FC = 29.8, P value = 0, Supplementary Table 3), but not in co-cultures with dispersed astrocytes. LN229 cells also secrete RANKL protein, a known driver of astrocyte activation, at a level 3 times higher than U87 cells.17 When injected into mice, LN229 xenografts were found to be infiltrative with high GFAP expression in the tumour periphery, suggestive of astrocyte activation, whereas U87 xenografts were nodular with low astrocyte GFAP expression in the tumour periphery.17 Thus, LN229 cells are more stem-like, more infiltrative, and more likely to generate reactive astrocyte phenotypes than U87 cells.
To examine the possibility of cell line-specific astrocyte-GB crosstalk, we performed a Nanostring™ nCounter™ Tumor360 gene expression analysis (Tables 1–2, Figure 6). We found significant differences in gene expression between LN229 and U87 cultures (Table 1) and between co-cultures of GB mono-spheres plus dispersed NHAs, mono-spheres (no NHAs), and separate cultures of GB cells and NHAs whose RNA was mixed after extraction (Table 2). The majority of genes implicated were associated with immune response, inflammation, or cytokine signalling (Tables 1–2), locomotion (GO:0040011), localization (GO:0051179, or immune regulation (GO:0002376) processes (Figure 6).
Interestingly, U87 cells that displayed reduced migration in the presence of astrocytes also showed the greatest sensitivity to GB-astrocyte crosstalk. The top three processes identified between U87 co-cultures and separate cultures were response to stimulus (GO:0050896), immune regulation, and locomotion, whereas the same comparison for LN229 cells yielded positive regulation of biological processes (GO:0048518), metabolic process (GO: 0008152), and developmental processes (GO:0032502) (Supplementary Figures 12–14). U87 cells also showed lower recruitment of fixed astrocytes to the tumour-sphere surface, and in xenografts,17 fewer GFAP+ cells near the tumour periphery. These data suggest that astrocytes may mount a more robust immune response to U87 tumours, limiting their migration, and implying that the reactive phenotype observed in association with LN229 xenografts may be less capable of mitigating tumour invasion. However, it is also possible that differences in other signalling pathways yield increased migration of LN229 cells in the presence of astrocytes.
Our gene expression analyses implicated six genes that appeared across all comparisons (Tables 1–2): CA9, FPR1, TMPRSS2, HLA-DQA1, KLRD1, and OAS2. We did not observe any differences between cell lines in co-culture for SOX2, CD44,85 or RANKL (TNFSF11)17 that were implicated in previous studies, although we did observed changes in SOX2 in mono-culture. Of the six genes highlighted by our study, CA9 and FPR1 are associated with increased invasion through STAT3 signalling44, 45, and TMRPSS2 is associated with invasion through the HGF/MET nexus.46, 49, 50 The remaining genes, HLA-DQA1, KLRD1, and OAS2, are associated with immune response.53, 54, 58 Thus STAT3 and HGF/MET signalling likely mediate observed differences in cell migration.
Conclusions
Here, we described a co-culture spheroid model for examining astrocyte-GB interactions in 3D hydrogels mimicking the brain tumour microenvironment. We characterized GB and astrocyte migration behaviours and quantified GB sphere growth and migration in three model configurations recapitulating different aspects of tumour formation and invasion. The two model cell lines chosen in this study, U87 and LN229, demonstrated distinct migration levels different from each other, but consistent with previous xenograft studies.17 The presence of astrocytes reduced U87 spheroid migration, but promoted migration from LN229 spheres. Specifically, we observed a scar-like structure formed by the astrocytes on the surface of GB spheres, which potentially acted as a physical barrier to cell migration. However, LN229 spheres overcame this barrier through collective migration behaviours that promoted astrocyte migration from the sphere surface. These observations are supported by differential gene expression studies between U87 and LN229 co-cultures that implicate immune signalling genes as the most altered, with greater impact on U87 cultures than LN229 cultures. Changes in migration may occur through STAT3 or TMPRSS2-mediated MET/HGF signalling and are probably promoted by local alteration of the ECM, such as changes in laminin 5 and MMP9. These data show that 3D in vitro hydrogel co-culture models can be used to reveal cell line specific differences in migration and to study differential GB-astrocyte crosstalk.
Supplementary Material
Acknowledgements
We gratefully acknowledge the Alumni Grants for Graduate Research and Scholarship at The Ohio State University for supporting this work. This work was funded in part by a generous donation from the Paul Bigley family, and is dedicated to his memory. The authors acknowledge Paolo Fadda the Genomics Shared Resource center at The Ohio State University Comprehensive Cancer Center for assistance with gene expression processing. Research reported in this publication was supported by The Ohio State University Comprehensive Cancer Center and the National Institutes of Health under grant number P30 CA016058. We acknowledge the NanoString™ for partial support of gene expression materials.
Footnotes
Conflicts of interest
The authors would like to disclose that the Nanostring™ NCounter™ Tumor360 chip was obtained at a 50% discount from Nanostring as part of a promotion and award won by YC. This contribution is unrelated to the experiments or its findings.
References
‡ Genomics data and analysis for this paper are available from DataDryad at doi:10.5061/dryad.fxpnvx0wv.
- 1.Tan AC, Ashley DM, Lopez GY, Malinzak M, Friedman HS and Khasraw M, Ca-a Cancer Journal for Clinicians, 2020, 70, 299–312. [DOI] [PubMed] [Google Scholar]
- 2.Scherer HJ, Am. J. Cancer, 1938, 34, 333–351. [Google Scholar]
- 3.Giese A, Bjerkvig R, Berens ME and Westphal M, J. Clin. Oncol, 2003, 21, 1624–1636. [DOI] [PubMed] [Google Scholar]
- 4.Calhoun MA, Cui Y, Elliott EE, Mo X, Otero JJ and Winter JO, Sci. Rep, 2020, 10, 311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ulrich TA, de Juan Pardo EM and Kumar S, Cancer Res, 2009, 69, 4167–4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ngo MT and Harley BA, Advanced Healthcare Materials, 2017, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prager BC, Xie Q, Bao S and Rich JN, Cell Stem Cell, 2019, 24, 41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang W, Couldwell WT, Simard MF, Song H, Lin JHC and Nedergaard M, Cancer Res, 1999, 59, 1994–2003. [PubMed] [Google Scholar]
- 9.Hong XT, Sin WC, Harris AL and Naus CC, Oncotarget, 2015, 6, 15566–15577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oliveira R, Christov C, Guillamo JS, de Bouard S, Palfi S, Venance L, Tardy M and Peschanski M, BMC Cell Biol, 2005, 6, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sin WC, Aftab Q, Bechberger JF, Leung JH, Chen H and Naus CC, Oncogene, 2016, 35, 1504–1516. [DOI] [PubMed] [Google Scholar]
- 12.Gritsenko PG, Ilina O and Friedl P, J. Pathol, 2012, 226, 185–199. [DOI] [PubMed] [Google Scholar]
- 13.Mathiisen TM, Lehre KP, Danbolt NC and Ottersen OP, Glia, 2010, 58, 1094–1103. [DOI] [PubMed] [Google Scholar]
- 14.Watkins S, Robel S, Kimbrough IF, Robert SM, Ellis-Davies G and Sontheimer H, Nat. Commun, 2014, 5, 4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brandao M, Simon T, Critchley G and Giamas G, Glia, 2019, 67, 779–790. [DOI] [PubMed] [Google Scholar]
- 16.Escartin C, Galea E, Lakatos A, O’Callaghan JP, Petzold GC, Serrano-Pozo A, Steinhäuser C, Volterra A, Carmignoto G, Agarwal A, Allen NJ, Araque A, Barbeito L, Barzilai A, Bergles DE, Bonvento G, Butt AM, Chen W-T, Cohen-Salmon M, Cunningham C, Deneen B, De Strooper B, Díaz-Castro B, Farina C, Freeman M, Gallo V, Goldman JE, Goldman SA, Götz M, Gutiérrez A, Haydon PG, Heiland DH, Hol EM, Holt MG, Iino M, Kastanenka KV, Kettenmann H, Khakh BS, Koizumi S, Lee CJ, Liddelow SA, MacVicar BA, Magistretti P, Messing A, Mishra A, Molofsky AV, Murai KK, Norris CM, Okada S, Oliet SHR, Oliveira JF, Panatier A, Parpura V, Pekna M, Pekny M, Pellerin L, Perea G, Pérez-Nievas BG, Pfrieger FW, Poskanzer KE, Quintana FJ, Ransohoff RM, Riquelme-Perez M, Robel S, Rose CR, Rothstein JD, Rouach N, Rowitch DH, Semyanov A, Sirko S, Sontheimer H, Swanson RA, Vitorica J, Wanner I-B, Wood LB, Wu J, Zheng B, Zimmer ER, Zorec R, Sofroniew MV and Verkhratsky A, Nat. Neurosci, 2021, 24, 312–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim JK, Jin X, Sohn YW, Jin X, Jeon HY, Kim EJ, Ham SW, Jeon HM, Chang SY, Oh SY, Yin J, Kim SH, Park JB, Nakano I and Kim H, Cancer Lett, 2014, 353, 194–200. [DOI] [PubMed] [Google Scholar]
- 18.Hoarau-Véchot J, Rafii A, Touboul C and Pasquier J, Int. J. Mol. Sci, 2018, 19, 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zuidema JM, Gilbert RJ and Gottipati MK, Cells Tissues Organs, 2018, 205, 372–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dundar B, Markwell SM, Sharma NV, Olson CL, Mukherjee S and Brat DJ, Glia, 2020, 68, 2173–2191. [DOI] [PubMed] [Google Scholar]
- 21.Rao SS, Nelson MT, Xue R, Dejesus JK, Viapiano MS, Lannutti JJ, Sarkar A and Winter JO, Biomaterials, 2013, 34, 5181–5190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rao SS, DeJesus J, Short AR, Otero JJ, Sarkar A and Winter JO, ACS Appl. Mater. Interfaces, 2013, 5, 9276–9284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rao SS, Lannutti JJ, Viapiano MS, Sarkar A and Winter JO, Tissue engineering. Part B, Reviews, 2014, 20, 314–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Delpech B, Maingonnat C, Girard N, Chauzy C, Maunoury R, Olivier A, Tayot J and Creissard P, Eur. J. Cancer, 1993, 29A, 1012–1017. [DOI] [PubMed] [Google Scholar]
- 25.Payne LS and Huang PH, Mol. Cancer Res, 2013, 11, 1129–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heffernan JM, Overstreet DJ, Le LD, Vernon BL and Sirianni RW, Ann. Biomed. Eng, 2015, 43, 1965–1977. [DOI] [PubMed] [Google Scholar]
- 27.Avci NG, Fan Y, Dragomir A, Akay YM and Akay M, IEEE Trans. Nanobioscience, 2015, 14, 790–796. [DOI] [PubMed] [Google Scholar]
- 28.Duong Thanh N, Fan Y, Akay YM and Akay M, IEEE Trans. Nanobioscience, 2016, 15, 289–293. [DOI] [PubMed] [Google Scholar]
- 29.Civita P, Leite DM and Pilkington GJ, Int. J. Mol. Sci, 2019, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen J-WE, Jan L, Leary S, Sarkaria JN, Steelman AJ, Gaskins HR and Harley BAC, J. Neuroinflammation, 2020, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leite DM, Zvar Baskovic B, Civita P, Neto C, Gumbleton M and Pilkington GJ, FASEB J, 2020, 34, 1710–1727. [DOI] [PubMed] [Google Scholar]
- 32.Ngo MT, Sarkaria JN and Harley BAC, Advanced Science, 2022, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Calhoun MA, Bentil SA, Elliott E, Otero JJ, Winter JO and Dupaix RB, ACS Biomaterials Science & Engineering, 2019, 5, 3964–3973. [DOI] [PubMed] [Google Scholar]
- 34.Cui Y, Cole S, Pepper J, Otero JJ and Winter JO, Biomaterials Science, 2020, 8, 4821–4831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wickham H, ggplot2: Elegant Graphics for Data Analysis, Springer-Verlag, New York, NY, 2016. [Google Scholar]
- 36.Wickham H, François R, Henry L and Müller K, dplyr: A Grammar of Data Manipulation R packageversion 1.0.10,, https://CRAN.R-project.org/package=dplyr). [Google Scholar]
- 37.Morgan M, BiocManager: Access the Bioconductor Project Package Repository R package version 1.30.19, https://CRAN.Rproject.org/package=BiocManager). [Google Scholar]
- 38.Wang H, Zhai T and Wang C, NanoStringDiff: Differential Expression Analysis of NanoString nCounterData R package version 1.28.0, https://bioconductor.org/packages/release/bioc/html/NanoStringDiff.html). [Google Scholar]
- 39.Blighe K, Rana S and Lewis M, EnhancedVolcano: Publication-ready volcano plots with enhanced colouring and labeling R package version 1.16.0, https://github.com/kevinblighe/EnhancedVolcano). [Google Scholar]
- 40.Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C and Chanda SK, Nat. Commun, 2019, 10, 1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schuessler TK, Chan XY, Chen HJ, Ji K, Park KM, Roshan-Ghias A, Sethi P, Thakur A, Tian X, Villasante A, Zervantonakis IK, Moore NM, Nagahara LA and Kuhn NZ, Cancer Res, 2014, 74, 5359–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grodecki J, Short AR, Winter JO, Rao SS, Otero JJ, Lannutti JJ and Sarkar A, Biotechnol. Prog, 2015, 31, 1406–1415. [DOI] [PubMed] [Google Scholar]
- 43.Short AR, Czeisler C, Stocker B, Cole S, Otero JJ and Winter JO, Macromol. Biosci, 2017, 17, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reuss AM, Groos D, Buchfelder M and Savaskan N, Int. J. Mol. Sci, 2021, 22, 5518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Torrisi F, Alberghina C, D’Aprile S, Pavone AM, Longhitano L, Giallongo S, Tibullo D, Di Rosa M, Zappalà A, Cammarata FP, Russo G, Ippolito M, Cuttone G, Li Volti G, Vicario N and Parenti R, Biomedicines, 2022, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cruickshanks N, Zhang Y, Yuan F, Pahuski M, Gibert M and Abounader R, Cancers, 2017, 9, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang C, Kong L, Kim S, Lee S, Oh S, Jo S, Jang I and Kim T-D, Int. J. Mol. Sci, 2022, 23, 10412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hu QP, Kuang JY, Yang QK, Bian XW and Yu SC, Int. J. Cancer, 2016, 138, 2804–2812. [DOI] [PubMed] [Google Scholar]
- 49.Chen A, Zhao W, Li X, Sun G, Ma Z, Peng L, Shi Z, Li X and Yan J, Frontiers in Immunology, 2022, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lucas JM, Heinlein C, Kim T, Hernandez SA, Malik MS, True LD, Morrissey C, Corey E, Montgomery B, Mostaghel E, Clegg N, Coleman I, Brown CM, Schneider EL, Craik C, Simon JA, Bedalov A and Nelson PS, Cancer Discovery, 2014, 4, 1310–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Isci D, D’Uonnolo G, Wantz M, Rogister B, Lombard A, Chevigne A, Szpakowska M and Neirinckx V, Cancers, 2022, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Choi S-H, Park MJ and Kim H, Oncology Letters, 2022, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Urup T, Michaelsen SR, Olsen LR, Toft A, Christensen IJ, Grunnet K, Winther O, Broholm H, Kosteljanetz M, Issazadeh-Navikas S, Poulsen HS and Lassen U, Mol. Oncol, 2016, 10, 1160–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cheray M, Bessette B, Lacroix A, Melin C, Jawhari S, Pinet S, Deluche E, Clavere P, Durand K, Sanchez-Prieto R, Jauberteau M-O, Battu S and Lalloue F, J. Cell. Mol. Med, 2017, 21, 244–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Baraks G, Tseng R, Pan C-H, Kasliwal S, Leiton CV, Shroyer KR and Escobar-Hoyos LF, Cancer Res, 2022, 82, 1159–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ling G, Wang S, Song Z, Sun X, Liu Y, Jiang X, Cai Y, Du M and Ke Y, Cancer Biology & Therapy, 2011, 12, 978–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shergalis A, Bankhead A, Luesakul U, Muangsin N and Neamati N, Pharmacol. Rev, 2018, 70, 412–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tatari N, Khan S, Livingstone J, Zhai K, McKenna D, Ignatchenko V, Chokshi C, Gwynne WD, Singh M, Revill S, Mikolajewicz N, Zhu C, Chan J, Hawkins C, Lu J-Q, Provias JP, Ask K, Morrissy S, Brown S, Weiss T, Weller M, Han H, Greenspoon JN, Moffat J, Venugopal C, Boutros PC, Singh SK and Kislinger T, Acta Neuropathologica, 2022, 144, 1127–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sciorra VA, Sanchez MA, Kunibe A and Wurmser AE, PLoS One, 2012, 7, e40053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu ZQ, Ren JJ, Zhao JL, Zang J, Long QF, Du JJ, Jia XT, Gu NB, Di ZL, Qian YH and Li SZ, Sci. Rep, 2020, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Parys JB, Bultynck G and Vervliet T, in Cellular Biology of the Endoplasmic Reticulum, eds. Agellon LB and Michalak M, Springer International Publishing, Cham, 2021, DOI: 10.1007/978-3-030-67696-4_11, pp. 215–237. [DOI] [Google Scholar]
- 62.Kang SS, Han K-S, Ku BM, Lee YK, Hong J, Shin HY, Almonte AG, Woo DH, Brat DJ, Hwang EM, Yoo SH, Chung CK, Park S-H, Paek SH, Roh EJ, Lee S. j, Park J-Y, Traynelis SF and Lee CJ, Cancer Res, 2010, 70, 1173–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jelski W, Laniewska-Dunaj M, Orywal K, Kochanowicz J, Rutkowski R and Szmitkowski M, Neurochem. Res, 2014, 39, 2313–2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Raychaudhuri B, Rayman P, Ireland J, Ko J, Rini B, Borden EC, Garcia J, Vogelbaum MA and Finke J, Neuro-oncol, 2011, 13, 591–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Levy A, Blacher E, Vaknine H, Lund FE, Stein R and Mayo L, Neuro-oncol, 2012, 14, 1037–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang Q, Xu B, Du J, Xu X, Shang C, Wang X and Wang J, Int. J. Mol. Med, 2018, 41, 2139–2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ding M, Li Y.-a., Lu Z and Hou G, Biomed Research International, 2022, 2022. [Google Scholar]
- 68.Chitadze G and Kabelitz D, Scand. J. Immunol, 2022, 96, e13201. [DOI] [PubMed] [Google Scholar]
- 69.Song X, Hao J, Wang J, Guo C, Wang Y, He Q, Tang H, Qin X, Li Y, Zhang Y and Yin Y, Oncogene, 2017, 36, 1476–1486. [DOI] [PubMed] [Google Scholar]
- 70.Xue Q, Cao L, Chen XY, Zhao J, Gao L, Li SZ and Fei Z, Oncol Lett, 2017, 13, 1325–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Miska J, Rashidi A, Chang AL, Muroski ME, Han Y, Zhang L and Lesniak MS, Cancer Immunol. Immunother, 2016, 65, 1555–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jiang J, Wang S, Chen Y, Wang C, Qu C and Liu Y, Medicine (Baltimore), 2018, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Oh LY and Yong VW, Glia, 1996, 17, 237–253. [DOI] [PubMed] [Google Scholar]
- 74.Miyazaki K, Cancer Sci, 2006, 97, 91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kawataki T, Yamane T, Naganuma H, Rousselle P, Anduren I, Tryggvason K and Patarroyo M, Exp. Cell Res, 2007, 313, 3819–3831. [DOI] [PubMed] [Google Scholar]
- 76.Sonoda Y, Ozawa T, Aldape KD, Deen DF, Berger MS and Pieper RO, Cancer Res, 2001, 61, 6674–6678. [PubMed] [Google Scholar]
- 77.Schmid RS, Simon JM, Vitucci M, McNeill RS, Bash RE, Werneke AM, Huey L, White KK, Ewend MG, Wu J and Miller CR, Neuro-oncol, 2016, 18, 962–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chekhonin IV, Chistiakov DA, Grinenko NF and Gurina OI, Cell. Mol. Neurobiol, 2018, 38, 1179–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Herrera-Perez RM, Voytik-Harbin SL, Sarkaria JN, Pollok KE, Fishel ML and Rickus JL, PLoS One, 2018, 13, e0194183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Diao WW, Tong XZ, Yang C, Zhang FR, Bao C, Chen H, Liu LY, Li M, Ye FF, Fan QH, Wang JF and Ou-Yang ZC, Sci. Rep, 2019, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Carey SP, Starchenko A, McGregor AL and Reinhart-King CA, Clin. Exp. Metastasis, 2013, 30, 615–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gagliano N, Costa F, Cossetti C, Pettinari L, Bassi R, Chiriva-Internati M, Cobos E, Gioia M and Pluchino S, Oncol. Rep, 2009, 22, 1349–1356. [DOI] [PubMed] [Google Scholar]
- 83.Chen WL, Xia TL, Wang DH, Huang B, Zhao P, Wang J, Qu X and Li XG, Oncotarget, 2016, 7, 62425–62438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Burda JE, Bernstein AM and Sofroniew MV, Exp. Neurol, 2016, 275 Pt 3, 305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hong X, Chedid K and Kalkanis SN, Int. J. Oncol, 2012, 41, 1693–1700. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.