

Abstract
Alport syndrome, a type IV collagen disorder, leads to glomerular disease and, in some patients, hearing loss. AS is treated with inhibitors of the renin-angiotensin system; however, a need exists for novel therapies, especially those addressing both major pathologies. Sparsentan is a single-molecule dual endothelin type-A and angiotensin II type 1 receptor antagonist (DEARA) under clinical development for focal segmental glomerulosclerosis and IgA nephropathy. We report the ability of sparsentan to ameliorate both renal and inner ear pathologies in an autosomal-recessive Alport mouse model. Sparsentan significantly delayed onset of glomerulosclerosis, interstitial fibrosis, proteinuria, and glomerular filtration rate decline. Sparsentan attenuated glomerular basement membrane defects, blunted mesangial filopodial invasion into the glomerular capillaries, increased lifespan more than losartan, and lessened changes in profibrotic/proinflammatory genes pathways in both the glomerular and renal cortical compartments. Notably, treatment with sparsentan, but not losartan, prevented accumulation of extracellular matrix in the strial capillary basement membranes in the inner ear and reduced susceptibility to hearing loss. Improvements in lifespan and in renal and strial pathology were observed even when sparsentan was initiated after development of renal pathologies. These findings suggest that sparsentan may address both renal and hearing pathologies in Alport syndrome patients.
Keywords: sparsentan, chronic kidney disease, genetic disorders, extracellular matrix, Alport syndrome, stria vascularis, basement membrane
Introduction
Alport syndrome (AS) is a congenital basement membrane collagen disorder associated with progressive failure of the glomerular filtration barrier, hearing loss, and anterior lenticonus. AS culminates in end-stage renal disease (ESRD) in some patients as early as the third decade of life [1]. The vast majority of AS cases are due to COL4A3/4 haploinsufficiency, while autosomal recessive inheritance is rare (approximately 1%) [2]. Approximately 5% of the cases are X-linked caused by pathogenic variants in COL4A5 and are at much higher risk for CKD [3]. In AS, the glomerular basement membrane (GBM) of the kidney and strial capillary basement membrane (SCBM) in the inner ear, lacks the physiological association of three type IV collagen chains (α3, α4, and α5). Instead, only the type IV collagen (α1)2α2 GBM network remains, which is thinner and contains fewer interchain crosslinks [4]. In the kidney, this abnormal GBM composition results in a compromised ability to withstand biomechanical stresses on the glomerular filtration barrier leading to glomerular disease [5]. For similar reasons, AS is also frequently associated with progressive hearing loss.
Current standard-of-care (SOC) renin-angiotensin-aldosterone system (RAAS) inhibitors, including angiotensin-converting enzyme inhibitors (ACEIs) and angiotensin receptor blockers (ARBs), have beneficial renal effects in AS patients that could be mediated by pleiotropic nephroprotective effects, including a reduction in intraglomerular pressure resulting in alleviation of biomechanical stresses on the GBM [6–9]. However, AS patients still progress to premature kidney failure at an unacceptable rate, underscoring the need to develop new treatments [11,12].
Endothelin 1 (ET-1), a vasoactive peptide with pleiotropic effects in renal pathophysiology, has emerged as an important factor in the progression of AS. Endothelin-1 (ET-1), a vasoactive peptide, is an agonist for two G-protein-coupled receptors, endothelin type A receptor (ETAR), and endothelin type B receptor (ETBR). Activation of ETAR results in vasoconstriction, proliferation, inflammation, extracellular matrix production, and fibrosis.
We showed that ET-1 is induced in the endothelium of Alport glomeruli, where it binds to ETARs on mesangial cells. The mechanism downstream of ETAR remains only partially elucidated. Activation of CDC42/RAC1 leads to mesangial filopodia invasion of glomerular capillaries, resulting in the deposition of mesangial proteins, including laminin α2, in the GBM [13]. Laminin α2 directly injures podocytes by activating focal adhesion kinase and NF-κB [14]. ETAR blockade has been shown to prevent mesangial filopodial invasion and to ameliorate glomerular disease in Alport mice [13].
Inner ear pathology in AS is associated with progressive thickening of the SCBM [15]. The stria vascularis (see supplementary material, Figure S1) plays an important role in maintaining the endocochlear potential and, thus, hearing [16]. In mouse models of AS, SCBM thickens due to accumulation of basement membrane proteins leading to hypoxia, metabolic stress, and elevated susceptibility to noise-induced hearing loss [17]. In parallel with the beneficial effects of ETAR antagonism in the AS kidney, amelioration of pathologies in the stria vascularis were also observed [17], indicating a role of ET-1 in the pathophysiology of AS-associated hearing loss. While the molecular mechanisms downstream of ETAR activation in the stria are not clearly understood, it is likely that similar pathways to those in the mesangial cells may play a role. However, whether ETAR antagonism lessens susceptibility to functional hearing loss was not determined.
Sparsentan is a single molecule that antagonizes both ETAR and angiotensin II subtype 1 receptor (AT1R). It is currently undergoing phase 3 clinical trials for the treatment of focal segmental glomerulosclerosis (FSGS) and IgA nephropathy (IgAN) [18,19]. Studies in experimental models support a wide spectrum of beneficial effects of dual inhibition of the RAAS and ET system in the treatment of kidney diseases [20]. We hypothesized that dual inhibition of ET and AngII receptors with sparsentan could serve as a potentially efficacious treatment for glomerular and renal tubulointerstitial disease in the autosomal recessive COL4A3−/− mouse model of AS, while also delaying the onset of inner ear pathology and susceptibility to functional hearing loss.
Materials and methods
Mice.
Male and female COL4A3 knockout (KO) mice, on the 129/Sv background, were used [21]. All experiments were done in accordance with approved IACUC protocols at Boystown National Research Hospital and Washington University, with full attention and compliance with USDA recommendations for the care and use of animals in research (https://www.fs.usda.gov/research/about/animalcare last accessed March 31st, 2023). Animals were housed in rooms with 70 °F temperature, 55% relative humidity and a 14/10-h light/dark cycle. Animals had free access to rodent chow (Envigo, Indianapolis, IN, USA) and water. The genetic status of Col4a3 KO mice was confirmed by a genomic PCR test on DNA isolated from tail clippings. Mice were caged together based on sex. Experimental design was focused to minimize the number of animals employed and to minimize pain and distress.
Animal studies.
Male and female Col4a3 KO mice were randomized and stratified by weight into treatment groups. Mice were given vehicle (0.5% methylcellulose 4000 cps, Sigma #64625; 0.25% Tween-80 in distilled water, Sigma #P1754) or sparsentan by oral gavage once daily. Losartan was given in the drinking water (DW) to deliver 10 mg/kg daily. In studies where losartan was initiated at 3 wk of age, losartan (10 mg/kg) was administered by oral gavage between 3 and 4 wk of age. In life-span studies, AS mice were dosed until they lost more than 15% of peak body weight. For the hearing loss studies, additional control groups were included using AS mice that were given water by oral gavage between 3–4 wk of age (VW). A schematic of the studies is depicted in Figure 1.
Figure 1. Schematic overview of studies in AS mice.
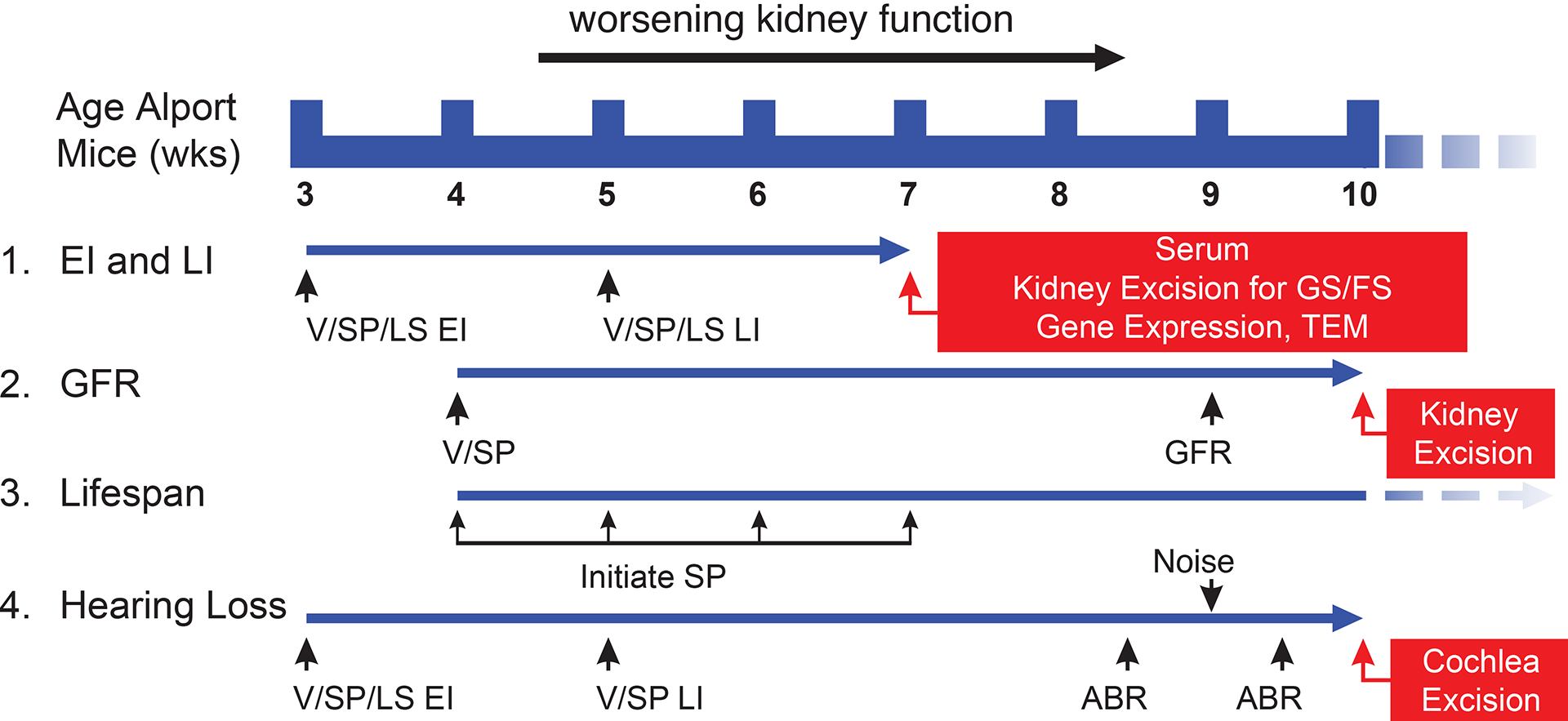
Studies examining the ability of sparsentan (SP) or losartan (LS) to delay the progression of the renal and inner ear pathology and dysfunction in AS mice were run in several different paradigms. (1) EI and LI: Treatment in AS mice was started at 3 wk of age (EI) or 5 wk of age (LI). Studies were terminated at 7 wk of age for collection of serum and removal and processing of kidneys for assessment of pathology. RNA from isolated glomeruli and cortex harvested from mice in EI studies were used to analyze gene expression. (2) GFR: In AS mice treated from 4 wk of age, assessment of GFR was conducted at 9 wk of age, and GS and FS were determined at 10 wk of age. (3) Lifespan: Treatment of AS mice was initiated at 4, 5, 6, or 7 wk of age, and UP/C was determined on a weekly basis. (4) Hearing loss: AS mice were treated from 3 wk of age (EI) or 5 wk of age (LI). Pre-noise ABR was assessed at 8.5 wk of age, and a second ABR test was performed 5 days following noise exposure. In EI studies, cochleae were excised following the second ABR test. Striae were isolated and processed for TEM to histology and measure SCBM width.
Blood pressure (BP) measurements.
BP was measured using a tail cuff and Non-Invasive Blood Pressure System (CODA2 High Throughput System, Kent Scientific, Torrington, CT, USA).
Measurements of urine protein to creatinine ratio (UP/C).
Urine was collected weekly. UP was measured using a standard Bradford assay (BioRad Inc, Hercules, CA, USA). UP was normalized to creatinine (UP/C) (BioAssay Systems Creatinine Assay kit, #DICT-500.
Immunofluorescence (IF) microscopy.
Fresh frozen OCT-embedded kidneys were sectioned at 8-μm and fixed in acetone for 10 min at −20 °C. Slides were incubated overnight at 4 °C in the appropriate primary antibody and blocking solution. Dual staining was performed by incubating slides using antibodies for CD45 at 1:100 (catalog 14–0451–85; Thermo Fisher Scientific, Waltham, MA, USA), collagen type I (COLI) at 1:200 (catalog CL50151AP; Cedarlane Labs, Burlington, Canada), and fibronectin at 1:500 (catalog F3648; Sigma-Aldrich, St. Louis, MO, USA) in 7% milk blocking solution. Slides were rinsed with PBS and incubated for 1 h at room temperature with Alexa Fluor secondary antibodies at 1:500 in blocking solution. Slides were rinsed in PBS and mounted using Vectashield Mounting Medium with DAPI (catalog H-1200; Vector Laboratories, Inc., Newark, CA, USA). Images of the CD45, COLI and fibronectin stains were captured using a Zeiss Axio Zoom V16 with 80× magnification (Zeiss Group, Oberkochen, Germany). Confocal images were captured using a Zeiss LSM 800 with a 63× NA: 1.4 oil objective (Zeiss Group).
Tubulointerstitial fibrosis (TIF) and glomerulosclerosis (GS) scoring.
TIF and GS scoring was performed blinded by two individual scorers. For GS, both fibronectin and Masson’s trichrome were employed. Masson’s trichrome staining was performed by the University of Nebraska Medical Center’s Tissue Science Facility. The total number of sclerotic (>50% glomerular area) and healthy glomeruli were recorded and scored for each cryosection and the sclerotic proportion was expressed as a percentage. The Fibrosis score was determined by expressing the COLI-immunopositive area as a % of the total cortical area and scoring as follows: 0, <5%; 1, 5% to ≤10%; 2, >10% to ≤25%; 3, >25% to ≤50%; 4, >50% to ≤75%; and 5, >75% to 100%. Since increased immune cell infiltration frequently accompanies fibrosis, IF with anti-CD45 antibodies was used to visualize the presence of inflammatory cells.
Transmission electron microscopy.
The GBM and SCBM were prepared and examined as described previously [13,14,22].
Gene expression analysis.
Kidney cortical glomeruli were isolated following transcardiac perfusion with 4-μm Dynabeads (Life Technologies, Carlsbad, CA, USA) using a magnet, as described previously [23]. Gene expression levels were determined using the RT² Profiler PCR Array (Array: PAMM-120ZA, Qiagen, Hilden, Germany), which assesses 84 genes involved in dysregulated tissue remodeling during the repair and healing of wounds. Raw counts were normalized using the average value across biological replicates of five reference genes (Actb, B2m, Gapdh, Hprt1 and Rplp0). Fold-changes (FC) were calculated as the ratio of the normalized gene expression in the test sample to the normalized gene expression in the control sample (2-ΔCt). P values were calculated using Student’s t test and the replicate 2-ΔCt values for each gene in the control group and test groups, and unadjusted P values <0.05 were considered for downstream analyses. The heatmap visualization was generated via R software (version 4.2.0, The R Foundation, Vienna, Austria) using the package “ComplexHeatmap” (https://bioconductor.org/packages/release/bioc/html/ComplexHeatmap.html last accessed March 31, 2023) with default clustering parameters. For graphical visualization and interpretation, FC were converted to FC-regulation (−1/FC) and color-coded with the linear interpolation function “colorRamp2” (https://cran.r-project.org/web/packages/colorRamp2/index.html last accessed March 31, 2023) with a scale from −10 to 10. FC-regulation values below −10 and above 10 were color-coded as −10 and 10.
Transdermal glomerular filtration rate (GFR) method.
GFR was determined using a transdermal device (MediBeacon, Creve Coeur, MO, USA), as previously described [24]. For device placement and injection of FITC-labeled sinistrin, mice were anesthetized with isoflurane. An imager device was mounted onto each animal’s back. Background signal was recorded for 2 min prior to the retro-orbital injection of 150 mg/kg FITC-sinistrin (in PBS) (Fisher Scientific, #NC1570801). Data were analyzed using Mannheim Pharma and Diagnostics Lab software (MediBeacon, Creve Coeur, MO, USA) [25]. GFR (ml/min) was calculated from the decrease in fluorescence intensity over time. T½ was calculated in minutes from the injection start to the 50% overall luminance point [26].
Inner ear studies.
All procedures for hearing studies were conducted at Washington University (St. Louis, MO, USA). The colony was established using mice from the Cosgrove lab to ensure consistency with the renal studies. Dosing was performed as described for the renal studies.
Hearing evaluation with the auditory brainstem response (ABR).
Threshold evaluations were conducted in mice at 8.5 and 9.5 wk of age (following exposure to noise stress) that had been treated with vehicle, vehicle-water, sparsentan, or losartan from 3 or 5 wk of age (vehicle or sparsentan only). Mice were sedated using avertin (2,2,2-tribromoethanol, 250 mg/kg; Sigma-Aldrich) and underwent ABR testing as described previously [17]. The magnitude of the hearing loss post-noise was calculated by subtracting the ABR hearing threshold for pre-noise from that of post-noise hearing testing.
Metabolic stress via exposure to noise.
Following and initial hearing assessment, mice were exposed to a metabolically stressful noise (106 dB SPL OB centered at 10 kHz for 10 h); this noise has been previously shown not to cause damage to the organ of Corti and hair cells [17,27]. Following the noise exposure, the mouse was removed and returned to vivarium housing. Five days after the noise exposure, the mouse underwent a post-noise hearing assessment followed by euthanasia.
Basement membrane width.
Cochleae were removed for analysis following the post-noise ABR measurement. Preparation of the cochlea for transmission electron microscopy (TEM) was conducted as described previously [28]. SCBM thickness measurements were calculated as described previously [17].
Statistical analyses.
Data are shown as mean ±SD, mean or median (CI) as indicated. The number of replicates for each experiment is shown in the figure legends. Significance was set at a probability level of <0.05. The statistical method used for comparison of groups is indicated in the figure or table legend and was performed with GraphPad Prism 9 (GraphPad Software, San Diego, CA, USA) software or SAS version 9.4 (SAS Institute, Cary, NC, USA) as indicated.
Results
Studies comparing the effects of sparsentan with those of losartan on renal and auditory function were conducted in several experiments with different designs as illustrated in Figure 1. The dose of sparsentan was selected following a pilot study (supplementary material, Table S1).
Early-intervention (EI) studies compared the effects of sparsentan (AS-SP; 120 mg/kg) in AS mice to those in losartan-treated AS mice (AS-LS; 10 mg/kg) in mice treated from ages 3 to 7 wk. Sparsentan and losartan-treated AS mice showed significant attenuation of the development of proteinuria (P<0.05), TIF and GS (P<0.001, P<0.01 respectively; Figure 2A and supplementary material, Figure S2). In late-intervention (LI) studies sparsentan attenuated UP/C at 7 wk (P<0.05), whereas the antiproteinuric effect in losartan-treated mice was not significant (supplementary material, Table S2). Sparsentan also significantly ameliorated GS and TIF in 7-wk-old mice to levels comparable to those in untreated 5-wk-old AS mice (AS-UT) (P<0.01 and P<0.05, respectively (Figure 2), whereas the effect of losartan on GS (Figure 2D) or the TIF score (Figure 2E) was not significant compared to 7-wk-old AS-V mice.
Figure 2. Sparsentan delays development of GS, fibrosis, and GBM morphology more than losartan in AS mice.
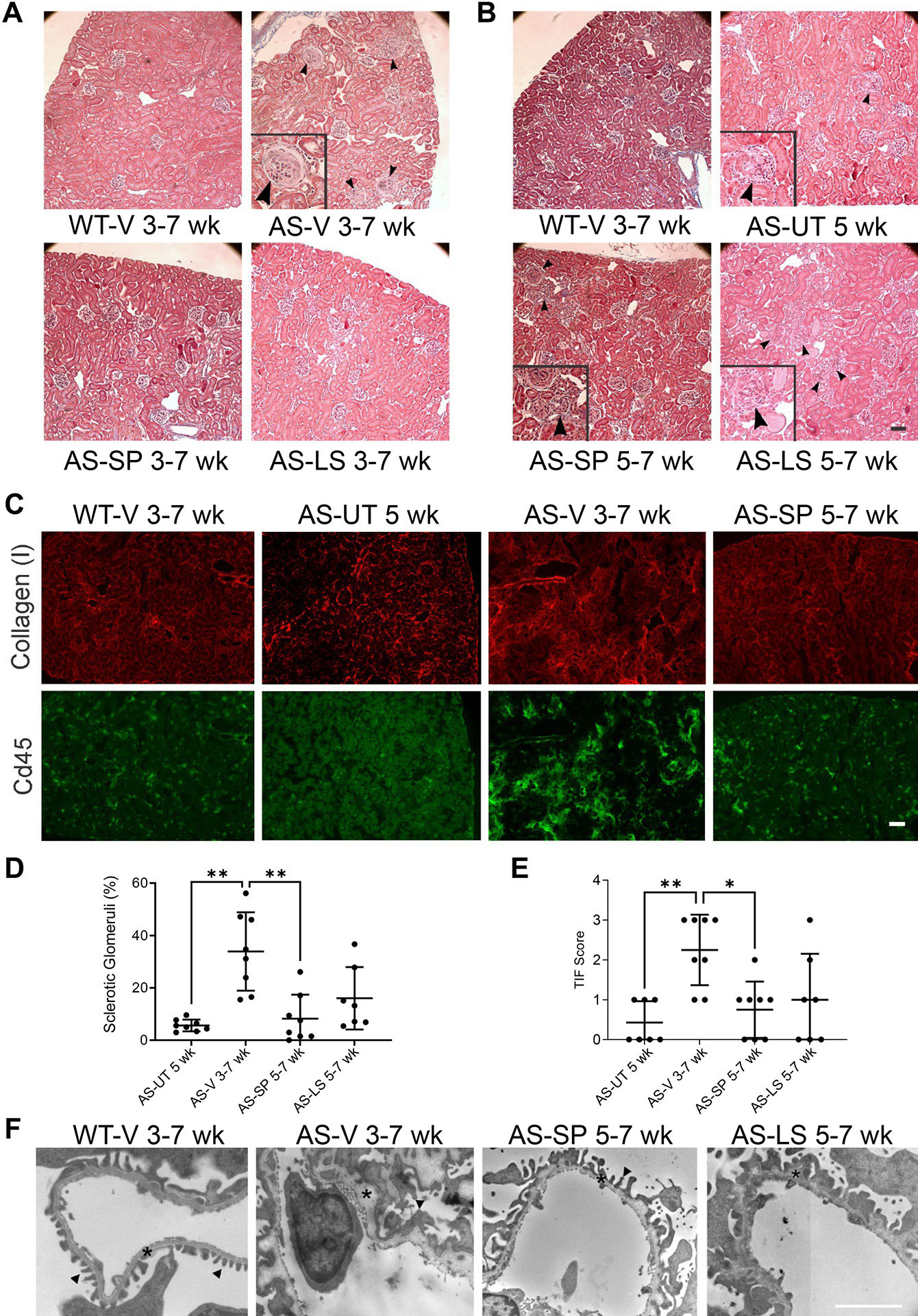
(A, B) Trichrome staining of representative kidney sections from EI and LI studies (n=8, except for AS-LS 5–7 wk of age, n=7). Compared to the kidney section from a WT-V mouse treated from 3–7 wk of age, the representative kidney section from a 3–7 wk of age AS-V mouse contained an increased number of sclerotic glomeruli, as indicated by the enlarged glomerulus denoted by black arrows in the inset. In kidney sections from AS-SP or AS-LS EI mice, sclerotic glomeruli were largely absent such that the pathology was comparable to that of the WT-V mice. The representative section from an AS-UT 5-wk-old mouse depicts the increase in GS at baseline before treatment in LI, as indicated by the enlarged glomerulus (black arrows in the inset). The GS in AS-SP 5–7-wk-old mice tended to be less than that in AS-LS 5–7-wk-old mice, as indicated by the difference in the number of black arrows between the images. (C) Representative cortical sections from AS mice in LI studies showing that fluorescence was attenuated in AS-SP 5–7-wk-old mice compared to those from AS-V 3–7-wk-old mice following staining with anti-COLI antibodies (red) or anti-CD45 antibodies (green), illustrating the antifibrotic and anti-inflammatory effects of sparsentan. (D) Quantification of GS following IF with anti-fibronectin antibodies showed that in AS-SP 5–7-wk-old mice, the percentage of sclerotic glomeruli in kidney sections was significantly ameliorated (**P<0.01) compared to that in AS-V 3–7-wk-old mice. GS in AS-UT 5-wk-old mice was also significantly lower than that in AS-V 3–7-wk-old mice (**P<0.01). (E) Quantification of TIF in LI studies via anti-COLI IF depicting significant prevention of TIF in AS-SP 5–7-wk-old mice (*P<0.05) compared to AS-V 3–7-wk-old mice. TIF in AS-UT 5-wk-old mice was also significantly lower than that in AS-V 3–7-wk-old mice (**P<0.01). (F) Glomerular ultrastructural images from WT-V 3–7-wk-old mice, AS-V 3–7-wk-old mice, AS-SP 5–7-wk-old mice, and AS-LS 5–7-wk-old mice illustrate the GBM thickening and dysmorphology (asterisks) and podocyte effacement (black arrows) and preservation of GBM morphology in AS-SP 5–7-wk-old mice compared to AS-V 3–7-wk-old mice. AS-LS 5–7-wk-old mice had little alterations in GBM compared to AS-V 3–7-wk-old mice. Panel D data were statistically analyzed using Brown-Forsythe and Welch 1-way ANOVA tests with Dunnett’s T3 multiple comparison test. Panel E data were statistically analyzed using the Kruskal–Wallis test with Dunn’s multiple comparison test.
Ultrastructural analysis of glomeruli in mice from EI studies demonstrated that sparsentan largely prevented GBM dysmorphology and ameliorated podocyte effacement (supplementary material, Figure S2B), to a greater extent than observed in in AS-LS mice. In LI studies, AS-SP mice exhibited an intermediate GBM dysmorphology and foot process effacement, but these abnormalities were qualitatively less than those observed in AS-V 7-wk-old mice (Figure 2F). Losartan had no appreciable effect on GBM dysmorphology when administered in LI studies (Figure 2F).
Sparsentan delayed the decline in kidney function.
The beneficial effects of sparsentan on renal function were also assessed by measuring GFR. Figure 3A shows that while GFR was markedly reduced in the 9-wk-old AS-V mice (P<0.001), the GFR in 9-wk-old AS-SP mice was, after 5 weeks of treatment, significantly higher than that in AS-V mice (P<0.0001) and was maintained at a level comparable to that of WT-V mice. Moreover, examination of kidney sections from 10-wk-old mice (after 6 wk treatment) revealed that preservation of kidney function in AS-SP mice coincided with a significant attenuation of the development of GS (P<0.01; Figure 3B) and TIF (P<0.01) compared to AS-V mice (Figure 3C,D).
Figure 3. Sparsentan delayed the decline in GFR and renal pathology in aged AS mice.
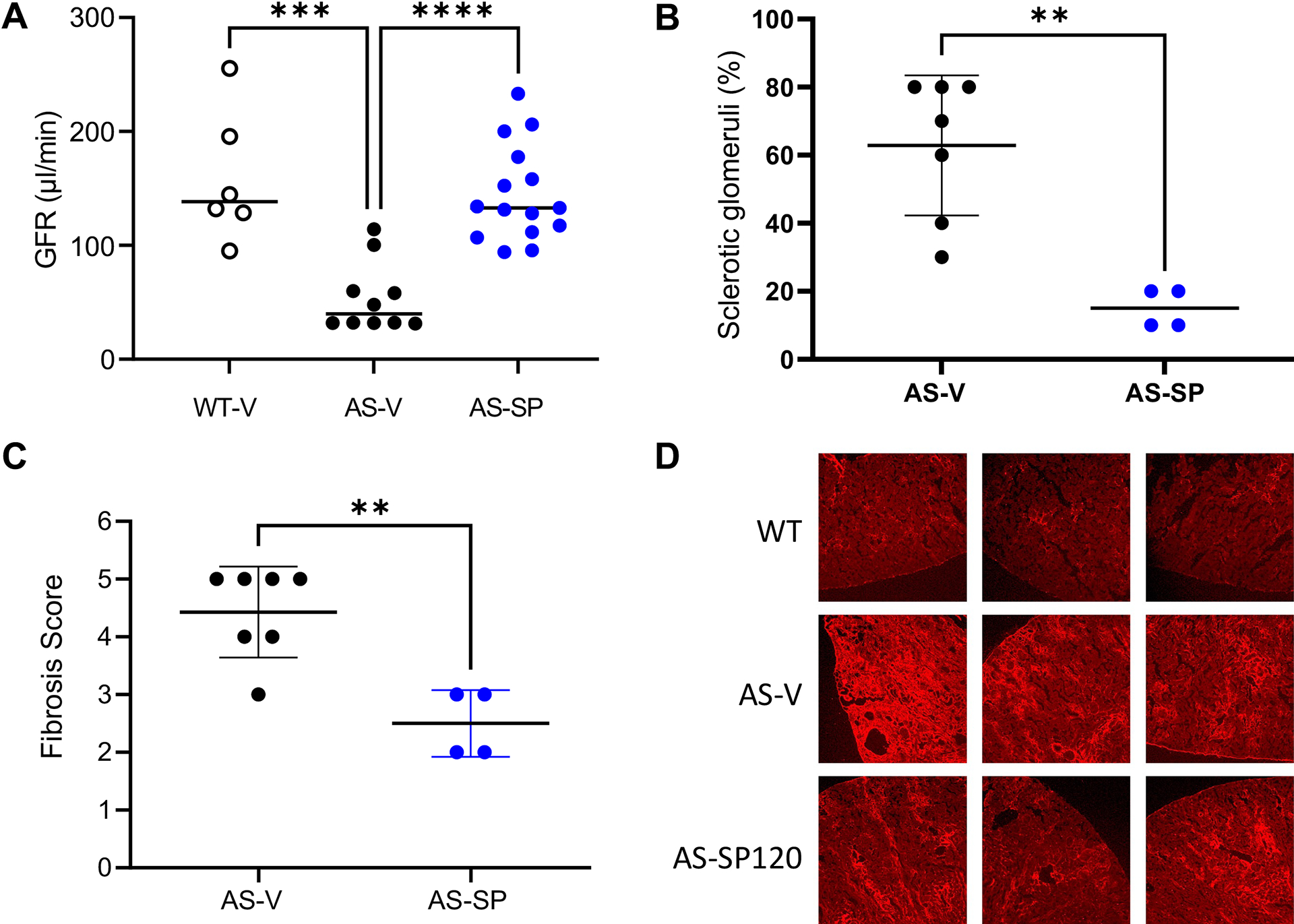
GFR and renal pathology in WT and AS mice treated with vehicle or sparsentan from 4 wk of age. (A) Comparison of GFR at 9 wk of age. ***P<0.001, ****P<0.0001, WT-V (n=6) or AS-SP (n=15) compared to AS-V mice (n=10). Preservation of GFR in AS-SP mice compared to AS-V mice was associated with amelioration at 10 wk of age of (B) GS and (C) TIF. **P<0.01 AS-SP (n=7) compared to AS-V (n=4). (D) Representative sections from three WT-V, AS-V, and AS-SP mice following staining with an anti-COLI antibody, indicating the attenuation of fibrosis in 10-wk-old AS-SP mice. Data in panel A are shown as their median; data in B and C are shown as their mean±SD. Analysis of panel A data used 1-way ANOVA followed by Tukey’s multiple comparison test. Analysis of panels B and C data was conducted using a 2-tailed Student’s t test. All analyses were performed using GraphPad Prism.
Sparsentan treatment attenuated profibrotic/proinflammatory gene expression in both the glomerular and cortical compartments of the kidney.
Of the 84 genes in the mouse fibrosis array panel, 32 and 17 genes were found differentially expressed (DE; significance level P<0.05) in the renal cortex and isolated glomeruli, respectively, of AS mice relative to WT mice in EI studies where mice were treated from 3 to 7 wk of age (Figure 4). By contrast, expression levels of multiple DE genes in the cortex and glomeruli, such as the proinflammatory and profibrotic mediators Ccl2 and Ccr2, Il1b, and other signal transduction genes, such as serpin family E member 1 (Serpine1), Tissue inhibitor of metalloproteinase-1 (Timp1) thrombospondin 2 (Thbs2), and Myc in sparsentan-treated AS mice tended to be closer to those observed in the WT mice (Figure 4A,B).
Figure 4. Sparsentan treatment attenuated expression of proinflammatory/profibrotic genes in both the cortical and glomerular compartments of the kidney.
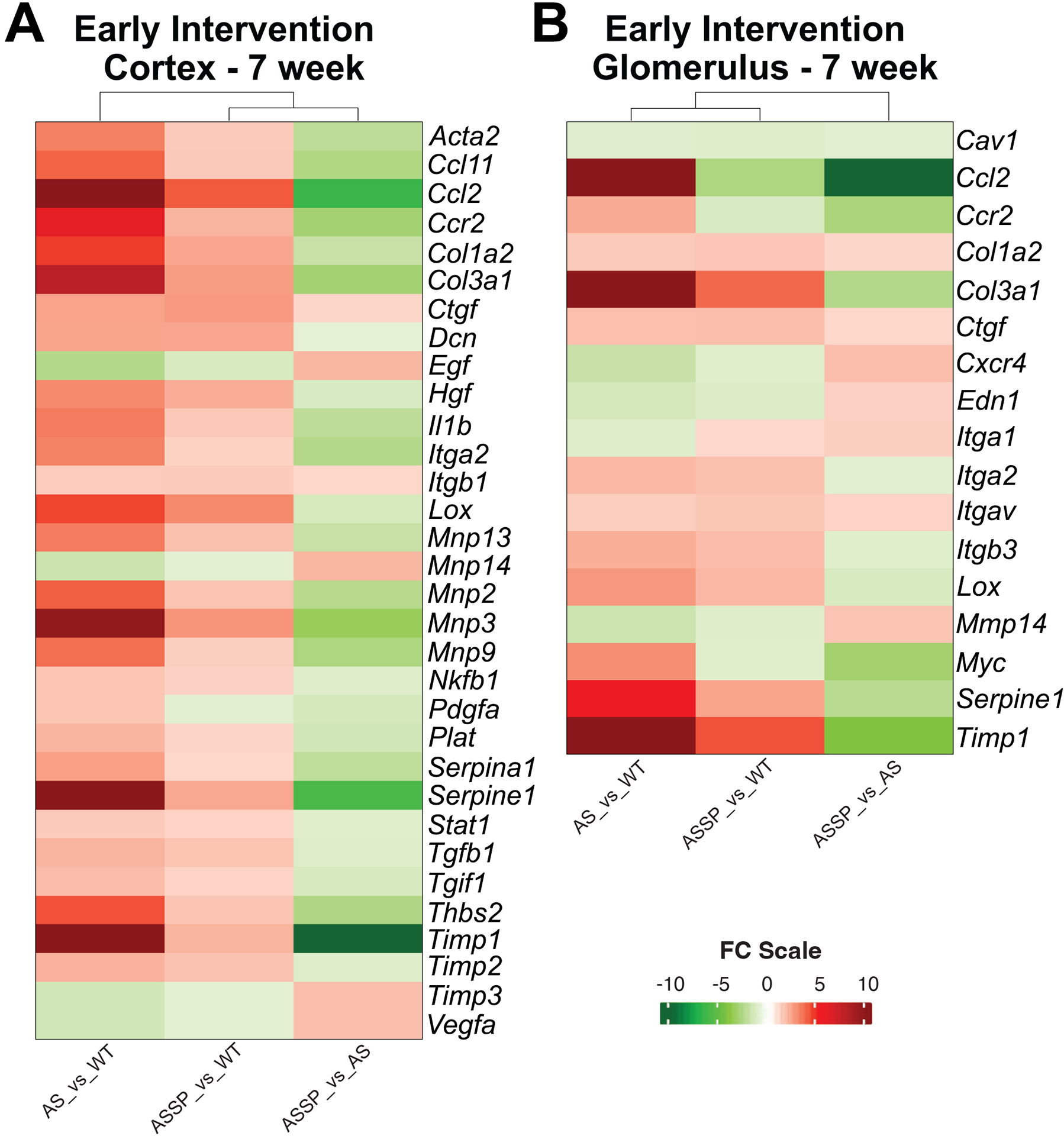
Heat map of DE genes from the mouse fibrosis microarray in the (A) cortex and (B) glomeruli of AS mice treated in EI studies from 3–7 wk old with vehicle or sparsentan (120 mg/kg) and compared to tissue from WT-V mice. Of the 84 genes in the panel investigated, 32 and 17 genes were found differentially expressed (P<0.05) in the renal cortex and glomeruli of AS mice, respectively, relative to WT mice. Changes in expression levels of several of the DE genes were ameliorated or trended toward normalization when AS mice were treated with sparsentan. Statistical analysis is described in the Methods section under Gene expression analysis.
Sparsentan treatment extended lifespan even when initiated in mice with renal pathology.
Sparsentan treatment started at 4 wk significantly extended lifespan compared to vehicle (Figure 5A median lifespan, AS-V mice, 67.5 days; AS-SP mice, 118.0 days; P<0.01; supplementary material, Table S3). Losartan treatment started at 4 wk was less effective than sparsentan in extending lifespan (median lifespan, AS-LS mice, 84.0 days; P<0.05; supplementary material, Table S3). These beneficial effects were not associated with differences in BP between the groups (supplementary material, Table S4). However, these effects paralleled the delay in the development of proteinuria in AS-SP mice. For AS mice started on treatment at 4 wk, the increase in UP/C was delayed in animals receiving sparsentan compared to those treated with vehicle (Figure 5B). The median UP/C in AS-SP mice was significantly lower than in AS-V mice at 7 (P<0.05), 8 (P<0.01), 9 (P<0.01), and 10 (P<0.05) wk of age (supplementary material, Table S5). The median UP/C of AS-LS mice, however, was significantly lower than that of AS-V mice only at 8 wk of age (P<0.05) and was significantly higher than that of AS-SP mice from 7–11 wk of age. Moreover, the median lifespan of AS-SP mice was significantly increased compared to that of AS-V mice even when sparsentan treatment was started at 5, 6, or 7 wk of age (P<0.01; supplementary material, Table S3), despite progressive increases in GS and TIF in untreated mice at these ages (Figure 5C,D).
Figure 5. Sparsentan extended lifespan and delayed proteinuria even when treatment initiated in mice with developed renal structural changes.
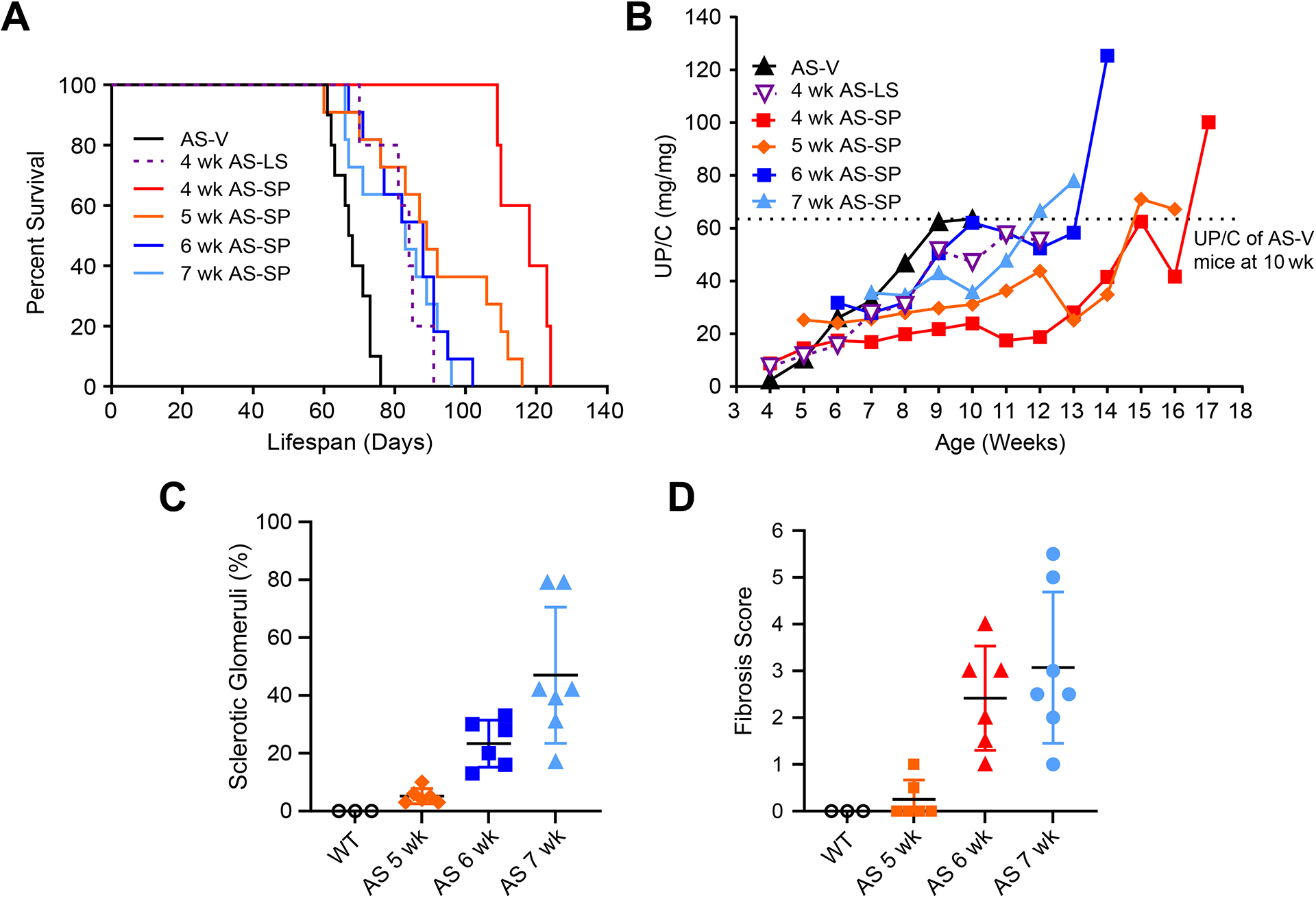
Lifespan and UP/C of AS-V, AS-LS, or AS-SP mice treated from 4, 5, 6, or 7 wk of age, where renal structural changes and proteinuria were evident from 5 wk of age. (A) Kaplan–Meier plot showing the percent survival of AS-V, AS-LS, or AS-SP mice with treatment initiated at different ages. (B) Mean UP/C over the course of the lifespan study in mice shown in panel A. Dashed line indicates the mean UP/C of AS-V mice at 10 wk of age. (C) Sclerotic glomeruli fraction in untreated AS mice at 5, 6, and 7 wk of age (n=6, n=6, and n=7, respectively). (D) TIF in untreated AS mice at 5, 6, and 7 wk of age (n=6, n=6, and n=7, respectively). Dosing with study treatment AS-LS, 10 mg/kg; AS-SP, 120 mg/kg. The number of mice surviving at each week during the life-span studies is presented in supplementary material, Table S7.
Sparsentan lessened susceptibility to hearing loss in AS mice.
To determine whether sparsentan impacted susceptibility to hearing loss, we exposed mice to a metabolically stressful noise to which the WT mice are resistant. This noise enhances the hypoxic stress on the inner ear of the AS mice [16,17]. AS mice and their WT littermates were treated from 3–9.5 wk of age with sparsentan, losartan, or their vehicles before examining hearing function using the ABR technique. Hearing was assessed before (at 8.5 wk of age) and 5 days after exposure to the noise. AS-V and AS-VW mice were more susceptible to hearing loss than their WT counterparts (Figure 6A and 6B, respectively), particularly in the low-to-mid test frequencies (supplementary material, Table S6). AS-SP and WT-SP mice showed equal ability to recover from the noise stress (Figure 6C). AS-LS mice exhibited no recovery of hearing loss; in fact, hearing loss was significantly greater at 16 kHz (P<0.05) in AS-LS mice than in AS-VW (Figure 6D).
Figure 6. Sparsentan but not losartan protected against hearing loss in AS mice.
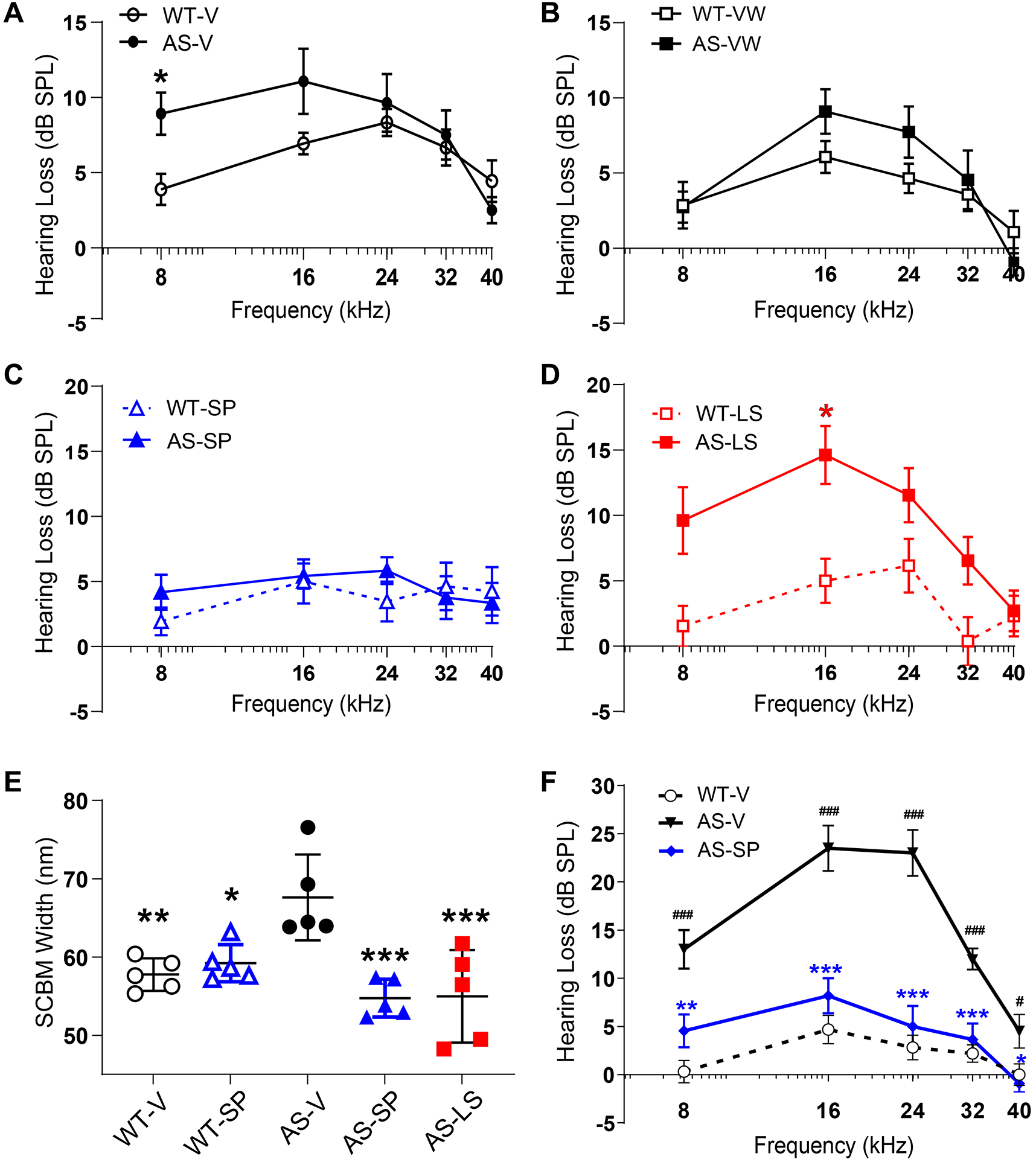
Hearing loss from a metabolic noise stress in AS or WT mice following treatment with vehicle, losartan, or sparsentan from 3–9 wk of age (EI) or 5–9 wk of age (LI; sparsentan only) was assessed by ABR measurements at frequencies of 8–40 kHz. AS-V (A) and AS-VW (B) were susceptible to the metabolically stressful noise as evidenced by hearing loss 5 days following exposure versus the recovery by WT-V or WT-VW mice, respectively (*P<0.05 for WT-V versus AS-V), (C) Recovery of hearing loss in AS-SP mice was equivalent to that of WT-SP mice, (D) Hearing loss in AS-LS mice was worse compared to AS-VW mice (*P<0.05 WT-LS versus AS-LS), (E) SCBM width in WT and AS mice treated with vehicle, SP, or LS from 3–9.5 wk of age in EI studies (n=5), determined from ultrastructural images obtained following excision of the cochlea after the post-noise ABR measurement. Both SP and LS significantly prevented the increase in SCBM width observed in AS-V mice. *P<0.05 for AS-V versus WT-SP; **P<0.01 for AS-V versus WT-V; ***P<0.001 for AS-V versus AS-SP or AS-LS. Notably, the average SCBM in the AS-LS mice had thinned to less than the normal basement membrane width of 50–60 nm, while the remaining three mice showed average SCBM widths consistent with those from WT-V mice. (F) Late treatment at 5–9 wk of age with sparsentan significantly attenuated susceptibility to hearing loss in AS mice compared to AS-V mice at all frequencies tested: WT-V, n=16; AS-V, n=10; AS-SP, n=11. ###P<0.001, #P<0.05 for AS-V versus WT-V; ***P<0.001, **P<0.01, *P<0.05 for AS-V versus AS-SP.
Data in (A–E) are shown as mean dB SPL±SEM. Data in (E) are shown for each stria as well as the group mean±SD (nm). Analysis (A–D and F) was performed using 2-way ANOVA followed by the Holm-Šídák multiple comparison test (GraphPad Prism). Analysis in (E) was performed using 1-way ANOVA and Tukey’s post-hoc multiple comparison test (GraphPad Prism).
Preliminary studies showed equivalent SCBM thickness in AS-V and AS-VW mice, therefore SCBM comparisons used only AS-V mice (supplementary material, Figure S3). SCBM thickness in Figure 6E revealed, as previously described [17], that AS-V mice had a significantly wider SCBM than did WT-V mice (P<0.01). By contrast, the SCBM thickness in AS mice following treatment with sparsentan or losartan was equivalent to that in WT-V mice and significantly thinner than that in AS-V mice (P<0.001). Interestingly, several AS-LS mice had very thin SCBMs. Examination of the strial ultrastructure (supplementary material, Figure S4) showed markedly fewer occurrences of lucent vacuoles and intracellular edema in AS-SP compared to AS-V mice, whereas AS-LS mice had pathology reminiscent of that observed in the AS-V mice. Importantly, although losartan treatment in the AS mice prevented SCBM thickening, this did not translate into a functional hearing improvement. The SCBM differences between sparsentan and losartan were further underscored in pilot EI studies during which AS mice were treated with vehicle, sparsentan (200 mg/kg), or losartan (10 mg/kg). SCBMs were examined by IF for expression of laminin α2, laminin α5, and collagen (IV) α1 chain [17]. Results (supplementary material, Figure S5) show that these ECM molecules were equally present in WT-V and AS-SP mice SCBMs but were more abundant in SCBMs of AS-V mice. Losartan did not prevent the ECM protein accumulation seen in AS mice SCBMs. Importantly, sparsentan intervention begun in 5-wk-old mice with renal pathology still protected against the noise susceptibility of the AS-V mouse (Figure 6F). Compared to WT-V mice, AS-V mice had a significantly elevated hearing threshold across test frequencies (8–32 kHz P<0.001, 40 kHz P<0.05). Moreover, in comparison to the AS-V mice, AS-SP mice exhibited significant reduction of hearing loss at all test frequencies (8 kHz, P<0.01; 16–32 kHz, P<0.001; 40 kHz, P<0.05) (Figure 6F).
Discussion
In this study, dual inhibition of ETAR and AT1R by sparsentan in AS mice prevented the development of proteinuria, renal molecular and structural changes, ameliorated the development of the hearing loss, and extended lifespan when administered either early in disease development or late post-development of glomerular structural changes. Lifespan improvement in AS mice has been previously linked to preservation of kidney function [29–33]. This study also demonstrated preservation of GFR with sparsentan treatment, consistent with the treatment-associated extension of lifespan in the AS mice.
Previous studies exploring the effects of ET and RAS inhibition have suggested the involvement of both systems in the pathophysiology of renal complications of AS. In fact, AT1R antagonists or ACE inhibitors (ACEi) are considered to be SOC for AS patients. In this study, comparisons were made with losartan as an AT1R antagonist, a relevant comparator to the ARB moiety of sparsentan. Sparsentan, offered greater renal protection and lifespan extension than losartan, particularly when treatment was initiated in mice later in their disease, suggesting additive nephroprotective effects of ETAR to SOC treatment approaches. While ACEi show good renoprotection in both mice and man when administered early (preemptive therapy), renoprotection is lost when administered after renal disease is established [9,30].
Previous studies [13] provided strong evidence for the involvement of ET-1 acting via ETAR in the pathophysiology of AS renal dysfunction. The ETAR antagonist sitaxentan improved GS, TIF, proteinuria, and BUN, and normalized GBM ultrastructure and profibrotic gene expression [13]. The effects of RAAS inhibition on the development of kidney injury and extension of lifespan in AS models have been more extensively studied. Gross, et al [30] found that ramipril ameliorated the development of proteinuria and structural changes in AS mice (129/Sv Col4a3−/− mice as used in this study). Ramipril significantly prolonged lifespan when administered from 4 wk of age to a greater extent than observed with sparsentan in the present studies. However, in AS mice, when ramipril treatment was started at 7 wk of age, there was no significant effect of lifespan [30]. This markedly contrasts with the ability of sparsentan in this study with treatment initiated at 7 wk of age to extend median lifespan by 15 days, suggesting potential benefit of dual ETAR/AT1R inhibition over current SOC therapies.
In the microarray study, DEGs between AS and WT mice align well with a previous study where whole-transcriptome analysis was performed on AS mouse kidney tissue [34]. In the renal cortical and glomerular compartment, sparsentan treatment of AS mice attenuated changes in the expression of a number of proinflammatory, profibrotic, proliferative, and adhesion pathway genes previously implicated in the AS pathogenesis. Some DE genes were attenuated by ETAR antagonism based on previous studies, such as several Mmp, Timp1, Ccl2 and Ccr2, Il1b, and Thbs2 [13,29,35,36]. Notably upregulation of Serpine1, which plays several roles, including regulation of MMPs, and impacting podocyte loss in a mouse FSGS model was ameliorated by sparsentan in both the cortex and glomeruli [37]. Also of interest is Myc [38], important in the developing mouse kidney [39] and a key player in renal fibrosis [40]. Thus, at the molecular level, sparsentan treatment impacts a wide range of pathways associated with development of renal disease in AS mice. Further support for sparsentan attenuating changes in genes regulating matrix deposition, dual immunostaining in glomeruli demonstrated an absence of laminin α2 in the GBM and of integrin α8 in the glomerulus of sparsentan-treated mice compared to AS-V mice (supplementary material, Figure S6).
Hearing loss frequently accompanies AS, particularly in patients with COL4A5 mutations [41]. Thus far, no therapeutic intervention ameliorated the inner ear pathology and/or function in AS mice. The connection between ETAR-mediated dysregulation of SCBM proteins and SCBM thickening is supported by the fact that the strial and glomerular pathologies are functionally similar [13,17,42]. Here, we showed that sparsentan indeed does preserve stria vascularis morphology and SCBM width. Moreover, sparsentan, most likely through its ETAR antagonism, allowed mice to recover from a metabolically stressful noise even when administered after proteinuria and renal disease were established. Notably, losartan was ineffective in preventing stress-related hearing loss in AS mice.
Sparsentan has recently been shown to ameliorate a variety of functional, structural, and molecular characteristics of renal injury in other models of glomerular diseases [43–45] and, even more importantly, to have a profound antiproteinuric effect in patients with FSGS [46]. Whether the renoprotection offered by dual antagonism at the ETAR and AT1R in the AS mouse studies will translate to the clinic awaits results from a recently initiated trial with sparsentan in pediatric patients with selected glomerular diseases (EPPIK; https://clinicaltrials.gov/ct2/show/NCT05003986), also open to AS patients, and by an ongoing phase 2 clinical trial with the ETAR antagonist atrasentan in AS patients (https://clinicaltrials.gov/ct2/show/NCT04573920).
In summary, our study demonstrates that dual ETAR/AT1R antagonism with sparsentan in the AS mouse model shows two significant advantages over treatment with an ARB alone. First, it significantly improved renal function and lifespan even when treatment was started in animals with established proteinuria, TIF, and GS. Second, sparsentan treatment prevented sensitivity to hearing loss, again, even when administered after significant renal damage was established. If translated to the clinic, sparsentan might have the potential to preserve both hearing and renal function in patients with Alport syndrome.
Supplementary Material
SUPPLEMENTARY MATERIAL ONLINE
Supplementary materials and methods
Figure S1. Structural pathology of the stria vascularis
Figure S2. In early intervention (EI) studies sparsentan delays development of GS, fibrosis, and GBM morphology more than losartan in AS mice
Figure S3. Sparsentan prevented thickening of the SCBM in AS mice
Figure S4. Sparsentan attenuated strial pathology in Alport mice
Figure S5. Sparsentan but not losartan prevents ECM accumulation in the SCBM of AS mice
Figure S6. Sparsentan inhibits mesangial filopodial invasion of glomerular capillary loops in AS mice
Table S1. Dose-dependent effects of sparsentan in AS mice treated from 3–7 wk of age in pilot EI studies
Table S2. UP/C in 7-wk-old mice in EI and LI studies and TIF score in EI
Table S3. Median life span of AS-V, AS-SP or AS-LS mice starting treatment at different ages
Table S4. BP during life span studies with treatment started at 4 wk of age
Table S5. Median UP/C of AS-V, AS-SP, or AS-LS at different ages of treatment initiation
Table S6. Hearing loss following metabolic noise stress in early intervention studies
Table S7. The number of mice surviving at each week during the life-span studies (relates to Figure 5)
Acknowledgements
The authors would like to thank James Hasson, Ping He, and Heeral Nandola for their help with statistical analysis. The authors would also like to acknowledge Patricia Bedard for her critical discussions and review of the data and manuscript. Editorial support was provided by Anthony DiLauro, ELS, and Stephen Bublitz, ELS, of MedVal Scientific Information Services, LLC (Princeton, NJ, USA) and was funded by Travere Therapeutics.
This research was partially conducted at the Integrated Biomedical Imaging Facility at Creighton University, Omaha, NE. This facility is supported by the Creighton University School of Medicine and grants GM103427 and GM 139762 from the National Institute of General Medical Science (NIGMS), a component of the National Institutes of Health (NIH). The facility was constructed with support from grants from the National Center for Research Resources (RR016469) and the NIGMS (GM103427).
Funding statement
The mouse Alport studies were conducted in the laboratories of DC at Boys Town National Hospital and of MAG at Washington University and were funded by Travere Therapeutics, Inc.
Footnotes
Conflicts of interest: CJ, RK, MN, and TP are employees of Travere Therapeutics, Inc., and may have an equity or other financial interest in Travere Therapeutics. All other authors have declared no conflicts of interest.
Data availability statement
The PCR microarray data is available in GEO with accession number GSE228756 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE228756). Other data used for the analyses in this manuscript are available on request from the corresponding author.
References
Reference 47 is cited only in the supplementary material.
- 1.Kashtan CE. Alport syndrome: achieving early diagnosis and treatment. Am J Kidney Dis. 2021;77(2):272–279. [DOI] [PubMed] [Google Scholar]
- 2.Gibson J, Fieldhouse R, Chan MMY, et al. Prevalence estimates of predicted pathogenic COL4A3-COL4A5 variants in a population sequencing database and their Implications for alport syndrome. J Am Soc Nephrol. 2021;32(9):2273–2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Groopman EE, Povysil G, Goldstein DB, Gharavi AG. Rare genetic causes of complex kidney and urological diseases. Nat Rev Nephrol. 2020;16(11):641–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gunwar S, Ballester F, Noelken ME, Sado Y, Ninomiya Y, Hudson BG. Glomerular basement membrane. Identification of a novel disulfide-cross-linked network of α3, α4, and α5 chains of type IV collagen and its implications for the pathogenesis of Alport syndrome. J Biol Chem. 1998;273(15):8767–8775. [DOI] [PubMed] [Google Scholar]
- 5.Meehan DT, Delimont D, Cheung L, et al. Biomechanical strain causes maladaptive gene regulation, contributing to Alport glomerular disease. Kidney Int. 2009;76(9):968–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Webb NJ, Shahinfar S, Wells TG, et al. Losartan and enalapril are comparable in reducing proteinuria in children with Alport syndrome. Pediatr Nephrol. 2013;28(5):737–743. [DOI] [PubMed] [Google Scholar]
- 7.Webb NJ, Shahinfar S, Wells TG, et al. Losartan and enalapril are comparable in reducing proteinuria in children. Kidney Int. 2012;82(7):819–826. [DOI] [PubMed] [Google Scholar]
- 8.Savva I, Pierides A, Deltas C. RAAS inhibition and the course of Alport syndrome. Pharmacol Res. 2016;107:205–210. [DOI] [PubMed] [Google Scholar]
- 9.Gross O, Tönshoff B, Weber LT, et al. A multicenter, randomized, placebo-controlled, double-blind phase 3 trial with open-arm comparison indicates safety and efficacy of nephroprotective therapy with ramipril in children with Alport’s syndrome. Kidney Int. 2020;97(6):1275–1286. [DOI] [PubMed] [Google Scholar]
- 10.Boeckhaus J, Hoefele J, Riedhammer KM, et al. Lifelong effect of therapy in young patients with the COL4A5 Alport missense variant p.(Gly624Asp): a prospective cohort study. Nephrol Dial Transplant. 2022. [DOI] [PubMed] [Google Scholar]
- 11.Gross O, Licht C, Anders HJ, et al. Early angiotensin-converting enzyme inhibition in Alport syndrome delays renal failure and improves life expectancy. Kidney Int. 2012;81(5):494–501. [DOI] [PubMed] [Google Scholar]
- 12.Temme J, Peters F, Lange K, et al. Incidence of renal failure and nephroprotection by RAAS inhibition in heterozygous carriers of X-chromosomal and autosomal recessive Alport mutations. Kidney Int. 2012;81(8):779–783. [DOI] [PubMed] [Google Scholar]
- 13.Dufek B, Meehan DT, Delimont D, et al. Endothelin A receptor activation on mesangial cells initiates Alport glomerular disease. Kidney Int. 2016;90(2):300–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Delimont D, Dufek BM, Meehan DT, et al. Laminin α2-mediated focal adhesion kinase activation triggers Alport glomerular pathogenesis. PLoS One. 2014;9(6):e99083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cosgrove D, Samuelson G, Meehan DT, et al. Ultrastructural, physiological, and molecular defects in the inner ear of a gene-knockout mouse model for autosomal Alport syndrome. Hear Res. 1998;121(1–2):84–98. [DOI] [PubMed] [Google Scholar]
- 16.Thulasiram MR, Ogier JM, Dabdoub A. Hearing function, degeneration, and disease: spotlight on the stria vascularis. Front Cell Dev Biol. 2022;10:841708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meehan DT, Delimont D, Dufek B, et al. Endothelin-1 mediated induction of extracellular matrix genes in strial marginal cells underlies strial pathology in Alport mice. Hear Res. 2016;341:100–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Komers R, Diva U, Inrig JK, Loewen A, Trachtman H, Rote WE. Study design of the phase 3 sparsentan versus irbesartan (DUPLEX) study in patients with focal segmental glomerulosclerosis. Kidney Int Rep. 2020;5(4):494–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barratt J, Rovin B, Carroll K, et al. PROTECT in immunoglobulin A nephropathy (IgAN): study design of a phase 3, randomized, double-blind, international, active-controlled study of the efficacy and safety of sparsentan [abstract]. Kidney Dis. 2018;4(3):142. [Google Scholar]
- 20.Komers R, Plotkin H. Dual inhibition of renin-angiotensin-aldosterone system and endothelin-1 in treatment of chronic kidney disease. Am J Physiol Regul Integr Comp Physiol. 2016;310(10):R877–R884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cosgrove D, Samuelson G, Pinnt J. Immunohistochemical localization of basement membrane collagens and associated proteins in the murine cochlea. Hear Res. 1996;97(1–2):54–65. [PubMed] [Google Scholar]
- 22.Zallocchi M, Johnson BM, Meehan DT, Delimont D, Cosgrove D. α1β1 integrin/Rac1-dependent mesangial invasion of glomerular capillaries in Alport syndrome. Am J Pathol. 2013;183(4):1269–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takemoto M, Asker N, Gerhardt H, et al. A new method for large scale isolation of kidney glomeruli from mice. Am J Pathol. 2002;161(3):799–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scarfe L, Schock-Kusch D, Ressel L, et al. Transdermal measurement of glomerular filtration rate in mice. J Vis Exp. 2018;(140):58520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schreiber A, Shulhevich Y, Geraci S, et al. Transcutaneous measurement of renal function in conscious mice. Am J Physiol Renal Physiol. 2012;303(5):F783–F788. [DOI] [PubMed] [Google Scholar]
- 26.Schock-Kusch D, Xie Q, Shulhevich Y, et al. Transcutaneous assessment of renal function in conscious rats with a device for measuring FITC-sinistrin disappearance curves. Kidney Int. 2011;79(11):1254–1258. [DOI] [PubMed] [Google Scholar]
- 27.Yoshida N, Hequembourg SJ, Atencio CA, Rosowski JJ, Liberman MC. Acoustic injury in mice: 129/SvEv is exceptionally resistant to noise-induced hearing loss. Hear Res. 2000;141(1–2):97–106. [DOI] [PubMed] [Google Scholar]
- 28.Gratton MA, Rao VH, Meehan DT, Askew C, Cosgrove D. Matrix metalloproteinase dysregulation in the stria vascularis of mice with Alport syndrome: implications for capillary basement membrane pathology. Am J Pathol. 2005;166(5):1465–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rubel D, Frese J, Martin M, et al. Collagen receptors integrin alpha2beta1 and discoidin domain receptor 1 regulate maturation of the glomerular basement membrane and loss of integrin alpha2beta1 delays kidney fibrosis in COL4A3 knockout mice. Matrix Biol. 2014;34:13–21. [DOI] [PubMed] [Google Scholar]
- 30.Gross O, Beirowski B, Koepke ML, et al. Preemptive ramipril therapy delays renal failure and reduces renal fibrosis in COL4A3-knockout mice with Alport syndrome. Kidney Int. 2003;63(2):438–446. [DOI] [PubMed] [Google Scholar]
- 31.Gross O, Girgert R, Beirowski B, et al. Loss of collagen-receptor DDR1 delays renal fibrosis in hereditary type IV collagen disease. Matrix Biol. 2010;29(5):346–356. [DOI] [PubMed] [Google Scholar]
- 32.Rubel D, Stock J, Ciner A, et al. Antifibrotic, nephroprotective effects of paricalcitol versus calcitriol on top of ACE-inhibitor therapy in the COL4A3 knockout mouse model for progressive renal fibrosis. Nephrol Dial Transplant. 2014;29(5):1012–1019. [DOI] [PubMed] [Google Scholar]
- 33.Rubel D, Boulanger J, Craciun F, et al. Anti-microRNA-21 therapy on top of ACE inhibition delays renal failure in Alport syndrome mouse models. Cells. 2022;11(4):594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sampson NS, Ryan ST, Enke DA, Cosgrove D, Koteliansky V, Gotwals P. Global gene expression analysis reveals a role for the alpha 1 integrin in renal pathogenesis. J Biol Chem. 2001;276(36):34182–34188. [DOI] [PubMed] [Google Scholar]
- 35.Adams JC, Lawler J. The thrombospondins. Cold Spring Harb Perspect Biol. 2011;3(10):a009712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cosgrove D, Meehan DT, Delimont D, et al. Integrin α1β1 regulates matrix metalloproteinases via P38 mitogen-activated protein kinase in mesangial cells: implications for Alport syndrome. Am J Pathol. 2008;172(3):761–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kobayashi N, Ueno T, Ohashi K, et al. Podocyte injury-driven intracapillary plasminogen activator inhibitor type 1 accelerates podocyte loss via uPAR-mediated β1-integrin endocytosis. Am J Physiol Renal Physiol. 2015;308(6):F614–F626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kurbegovic A, Trudel M. The master regulators Myc and p53 cellular signaling and functions in polycystic kidney disease. Cell Signal. 2020;71:109594. [DOI] [PubMed] [Google Scholar]
- 39.Bates CM, Kharzai S, Erwin T, Rossant J, Parada LF. Role of N-myc in the developing mouse kidney. Dev Biol. 2000;222(2):317–325. [DOI] [PubMed] [Google Scholar]
- 40.Shen Y, Miao N, Wang B, et al. c-Myc promotes renal fibrosis by inducing integrin αv-mediated transforming growth factor-β signaling. Kidney Int. 2017;92(4):888–899. [DOI] [PubMed] [Google Scholar]
- 41.Boeckhaus J, Strenzke N, Storz C, Gross O, On Behalf Of The Gpn Study G, Early Pro-Tect Alport I. Characterization of sensorineural hearing loss in children with Alport Syndrome. Life (Basel). 2020;10(12):360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cosgrove D, Meehan DT, Grunkemeyer JA, et al. Collagen COL4A3 knockout: a mouse model for autosomal Alport syndrome. Genes Dev. 1996;10(23):2981–2992. [DOI] [PubMed] [Google Scholar]
- 43.Jenkinson C, Moldoveanu Z, Komers R, et al. Protective effects of sparsentan from proliferative glomerular injury induced by administration of human immune complexes in a murine model of experimental iga nephropathy [poster]. Presented at International Society of Nephrology World Congress of Nephrology, April 12–15, 2019, Melbourne, Australia. [Google Scholar]
- 44.Nagasawa H, Suzuki H, Jenkinson C, et al. The dual endothelin type A receptor (ETAR) and angiotensin II type 1 receptor (AT1R) antagonist, sparsentan, protects against the development of albuminuria and glomerulosclerosis in the gddY mouse model of IgA nephropathy [poster]. Presented at American Society of Nephrology Kidney Week, October 22–25, 2020, Virtual. [Google Scholar]
- 45.Gyarmati G, Shroff U, Izuhara A, Komers R, Bedard PW, Peti-Peterdi J. Sparsentan improves glomerular blood flow and augments protective tissue remodeling in mouse models of focal segmental glomerulosclerosis (FSGS) [poster]. Presented at European Renal Association - European Dialysis and Transplant Association (ERA-EDTA) Congress, June 5–8, 2021, Berlin, Germany/Virtual. [Google Scholar]
- 46.Trachtman H, Nelson P, Adler S, et al. DUET: a phase 2 study evaluating the efficacy and safety of sparsentan in patients with FSGS. J Am Soc Nephrol. 2018;29(11):2745–2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rodgers KD, et al. The laminins in the murine inner ear: developmental transitions and expression in cochlear basement membranes. Hear Res 2001; 158: 39–50. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPLEMENTARY MATERIAL ONLINE
Supplementary materials and methods
Figure S1. Structural pathology of the stria vascularis
Figure S2. In early intervention (EI) studies sparsentan delays development of GS, fibrosis, and GBM morphology more than losartan in AS mice
Figure S3. Sparsentan prevented thickening of the SCBM in AS mice
Figure S4. Sparsentan attenuated strial pathology in Alport mice
Figure S5. Sparsentan but not losartan prevents ECM accumulation in the SCBM of AS mice
Figure S6. Sparsentan inhibits mesangial filopodial invasion of glomerular capillary loops in AS mice
Table S1. Dose-dependent effects of sparsentan in AS mice treated from 3–7 wk of age in pilot EI studies
Table S2. UP/C in 7-wk-old mice in EI and LI studies and TIF score in EI
Table S3. Median life span of AS-V, AS-SP or AS-LS mice starting treatment at different ages
Table S4. BP during life span studies with treatment started at 4 wk of age
Table S5. Median UP/C of AS-V, AS-SP, or AS-LS at different ages of treatment initiation
Table S6. Hearing loss following metabolic noise stress in early intervention studies
Table S7. The number of mice surviving at each week during the life-span studies (relates to Figure 5)
Data Availability Statement
The PCR microarray data is available in GEO with accession number GSE228756 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE228756). Other data used for the analyses in this manuscript are available on request from the corresponding author.