

Abstract
Dysfunctional signaling in midbrain reward circuits perpetuates diseases characterized by compulsive overconsumption of rewarding substances such as substance abuse, binge eating disorder, and obesity. Ventral tegmental area (VTA) dopaminergic activity serves as an index for how rewarding stimuli are perceived and triggers behaviors necessary to obtain future rewards. The evolutionary linking of reward with seeking and consuming palatable foods ensured an organism’s survival, and hormone systems that regulate appetite concomitantly developed to regulate motivated behaviors. Today, these same mechanisms serve to regulate reward-directed behavior around food, drugs, alcohol, and social interactions. Understanding how hormonal regulation of VTA dopaminergic output alters motivated behaviors is essential to leveraging therapeutics that target these hormone systems to treat addiction and disordered eating. This review will outline our current understanding of the mechanisms underlying VTA action of the metabolic hormones ghrelin, glucagon-like peptide-1, amylin, leptin, and insulin to regulate behavior around food and drugs of abuse, highlighting commonalities and differences in how these five hormones ultimately modulate VTA dopamine signaling.
Introduction
Public health epidemics including obesity and substance abuse are characterized by dysregulation of the brains reward processing centers [1–3]. The inherently rewarding properties of palatable foods and drugs drive cravings and increase motivation to repeat the experience and chronic overconsumption of highly rewarding substances leads to fundamental alterations to the brain’s perception of reward [4–6]. The ventral tegmental area (VTA) dopamine neurons regulate many aspects of reward saliency including reinforcement, decision making, attention, and impulsivity [7–9]. The VTA is a heterogeneous nucleus receiving a wide range of inputs and in turn innervating many different nuclei. Three prominent targets of VTA projections are the nucleus accumbens (NAc), medial prefrontal cortex (mPFC) and central nucleus of the amygdala (CeA) [10, 11]. Tremendous work has been done to understand the connectivity of VTA circuits, mapping which nuclei provide input to which projection targets, and how these VTA input-output circuits are anatomically organized and discriminately activated to regulate specific aspects of reward-directed behavior [7, 8, 10–12].
VTA dopamine neurons are activated by rewarding stimuli and changes in dopaminergic output reveal how VTA activity is modulated by external and internal factors [13–16]. However, the paradigm that higher levels of dopamine signaling equate to a higher value reward is a vast and incomplete simplification. There is much debate within the field how different aspects of reward (saliency and valance) are communicated, on what timescale, and how dopamine can signal both motivation and learning [17]. Dopamine plays a primary function in motivation to stimulate movement and an increase in dopamine firing can directly trigger behavior [18]. In seeking rewards, an animal learns to differentiate between actual and anticipated outcomes, and dopamine neurons respond to unexpected stimuli to convey this reward prediction error [19, 20]. When deciding whether to pursue a reward, past experiences, the value of the reward, and the work (time and physical effort) required to obtain the reward are considered, all of which and more are mediated by dopamine [17, 21, 22].
Dopamine release in the NAc has been most extensively studied in midbrain dopamine circuity and is the parameter measured in much of the work discussed herein. NAc dopamine release has been identified to encode the perceived saliency (importance) of a reward independent of its valance [23], reinforce learning based on feedback from tangible actions rather than the value of an expected reward [21, 24], and also signals the prediction and duration of aversive stimuli which are additive to concurrent rewarding stimuli [25]. Another factor to consider is that dopamine nerve firing activity does not necessarily translate with terminal dopamine release [17]. Dopamine terminals are heavily innervated with projections from outside nuclei which modulate dopamine release via glutamatergic, cholinergic, and opioid signaling [26, 27]. Changes in extracellular dopamine concentrations and dopamine neuron firing rates are common metrics used to assess the hormonal regulation of mesolimbic signaling in the studies covered in this review, which are ultimately a simplification of the full physiologic picture.
Metabolic hormones control not only the intake of palatable foods but also regulate behaviors around addictive substances through action in the VTA [28]. Ghrelin, leptin, insulin, glucagon-like peptide-1 (GLP-1) and amylin are metabolic hormones that prominently regulate nutrient utilization, energy expenditure, and appetite and all act in the VTA through interacting mechanisms to affect motivated behavior [29–33]. This review provides a comprehensive update of the regulation of midbrain dopamine circuits by peripheral feeding hormones [28, 34], cataloguing what is currently known about the mechanisms by which these hormones influence VTA activity and how these actions regulate food intake and behavior around drugs of abuse. The review will first explore how the peripheral orexigenic signal ghrelin stimulates dopamine-driven motivation for food and addictive substances before turning to address how the satiation hormones GLP-1, amylin, leptin, and insulin act to suppress dopamine output and mitigate the consumption of rewarding substances. Better understanding of the reward-modulating effects of these hormone systems will help develop more effective treatments for obesity and addictive disorders.
Ghrelin
Enteroendocrine P/D1 cells of the gastrointestinal tract, chiefly the stomach, produce the hormone ghrelin that acts centrally at growth hormone secretagogue receptor type 1a (GHSR) to stimulate appetite and enhance motivation for foods and rewarding stimuli [35]. The GHSR is expressed in a distributed network of central nuclei that include but are not limited to the hippocampus, many hypothalamic subnuclei, the dorsal vagal complex (DVC) of the hindbrain, the facial motor nucleus, the laterodorsal tegmental area (LDTg), the parabrachial nucleus (PBN), the substantia nigra, and the VTA, that each contribute to the physiological actions of ghrelin [36, 37]. While the stomach is the dominant source of ghrelin in the body, the hypothalamus has also been identified as a central ghrelin source [38], although reports have been inconsistent and the physiological relevance of hypothalamic produced ghrelin is unresolved [39]. Although no study to date has tracked peripherally labeled ghrelin appearance in the VTA, peripheral ghrelin increases VTA dopamine activity [40]. Consistent with a direct VTA-site of action for ghrelin, fluorescently labeled ghrelin injected intracerebroventricularly (ICV) is detected in all subnucleus of the VTA and is proposed to be transported into brain parenchyma via ependymal cells through a receptor independent mechanism [41, 42]. Indeed, extensive work has demonstrated how ghrelin action in the VTA drives motivation for rewarding substances.
Mechanism
ICV and intra-VTA ghrelin induce cFos in both dopamine and GABA VTA neurons [42], although the majority of ghrelin dependent VTA manipulations explore changes in dopaminergic activity. Ghrelin binding and GHSR expression was observed in VTA dopamine neurons, and the ghrelin stimulated increase in VTA dopamine neuron firing frequency was absent in GHSR knockout mice [43]. Additionally, peripheral ghrelin increased and GHSR antagonist injection decreased dopamine turnover in the VTA, further supporting that ghrelin increases VTA dopaminergic activity [40].
The dominant projection target of VTA dopamine neurons is the NAc [7], and NAc dopamine levels are often used to indicate VTA dopaminergic activity. Intra-VTA, ICV, and peripheral ghrelin administration alone was sufficient to elevate NAc dopamine turnover and extracellular dopamine concentrations in the NAc shell, but not core [44–46]. Systemic administration of a GHSR antagonist blocked the ability of ICV ghrelin to increase dopamine release in the VTA and the NAc shell, and systemic ghrelin-induced NAc dopamine release was blocked by VTA GHSR antagonism [47, 48], effectively pinpointing the VTA as the site of action for ghrelin to stimulate NAc dopamine release. VTA ghrelin signaling not only increased dopamine release in the NAc, but also the mPFC and CeA, two other significant VTA projection sites [49].
Food stimulates NAc dopamine release, and intake of both regular and palatable foods increase NAc dopamine levels, with a stronger effect following palatable food intake [50, 51]. Ghrelin modulates food-evoked NAc dopamine levels as GHSR knockout mice do not increase NAc dopamine release after peanut butter consumption [52]. Furthermore, ICV ghrelin administration enhanced NAc dopamine spikes in response to sucrose pellet retrieval and this was attenuated by ICV GHSR antagonist delivery [50]. Surprisingly, this study did not see an enhancement of sucrose retrieval evoked NAc dopamine spikes with intra-VTA ghrelin injection [50]. Perhaps ghrelin action on a circuit that provides input to the VTA rather than within the VTA augments sucrose stimulated NAc dopamine activity.
Ghrelin’s regulation of NAc dopamine levels is dependent on food availability. With food available, peripheral ghrelin increased NAc dopamine levels, but in the absence of food ghrelin decreased NAc dopamine levels [53]. The effect in both directions was blocked by VTA GHSR antagonism [53]. This could highlight a role of ghrelin to only induce positive reinforcement if an anticipated reward it received. Direct VTA ghrelin injection increased NAc dopamine release with or without food present, suggesting that ghrelin action at a separate brain region regulates the food consumption dependent modulation of VTA-to-NAc dopaminergic transmission [53]. Further complicating the interpretation, while systemic ghrelin treatment prior to chow intake increased NAc dopamine levels, this was not observed when a ghrelin injection preceded palatable food intake [51]. This suggests that palatable foods and ghrelin increase NAc dopamine levels through partially independent mechanisms that when combined may cancel one another out as negative feedback to discontinue palatable food intake.
Although untreated food deprived rats consumed more chow than sated rats injected peripherally with ghrelin, fasting-induced chow intake only minimally increased NAc dopamine levels compared to ghrelin treatment [53]. This implies that the magnitude of NAc dopamine signaling is not correlated with the amount of food consumed. In fact, VTA-to-NAc dopamine signaling more prominently regulates food reward directed motivated behaviors like progressive ratio (PR) than basal food intake. While both fasting and intra-VTA ghrelin increased chow intake and PR responding for sucrose, pretreatment of the NAc with a dopamine 1 receptor (D1R) or dopamine 2 receptor (D2R) antagonist impaired only PR performance without diminishing chow intake [54]. Thus, mesolimbic dopamine transmission is an essential component of ghrelin-regulated food-reward behaviors while sites outside the VTA mediate ghrelin-stimulated chow intake. One study suggested that VTA NPY1 receptors were involved in mediating the effect of VTA ghrelin to stimulate chow intake but not PR performance [55], but more work is required to understand how NPY and ghrelin signaling may interact in the VTA.
Like food, drugs stimulate NAc dopamine release, and the increase in NAc dopamine release induced by alcohol, amphetamines, cocaine, morphine, and nicotine were all attenuated by peripheral GHSR antagonist administration [47, 56–59]. Specifically, administration of ICV and an intra-VTA GHSR antagonist or the lack of GHSR expression also decreased alcohol stimulated NAc dopamine release [60, 61].
Cholinergic input from the LDTg to the VTA regulates midbrain ghrelin action. Ghrelin action in the LDTg increased NAc dopamine levels and locomotor activity [45, 62]. LDTg ghrelin signaling stimulated VTA acetylcholine levels and VTA antagonism of nicotinic acetylcholine receptors (nACh) prevented the increase in NAc dopamine levels following LDTg ghrelin injection [62], implying that acetylcholine released from LDTg terminals in the VTA drives NAc dopamine output (Figure 1; 1). Further supporting this idea, peripheral administration of nACh receptor antagonists blocked the effect of ICV ghrelin to increase locomotion and NAc dopamine release [63], and of ICV or intra-VTA ghrelin to increase food intake [64]. Peripheral and central nACh antagonism similarly attenuated fasting-induced refeeding and blocked the conditioned place preference to palatable food [64]. In the LDTg, the GHSR is co-expressed with choline acetyltransferase, verifying that LDTg ghrelin signaling stimulates cholinergic neurons [64]. Signaling at VTA nACh receptor subtypes alpha 3, beta 2, and beta 3 partially mediated the increased locomotion and NAc dopamine levels following VTA ghrelin delivery [65], and chronic ICV ghrelin treatment increased VTA nACh receptor beta 2 mRNA [66]. Together, these findings identify LDTg-to-VTA-to-NAc as an important pathway underlying ghrelin regulation of motivated behaviors.
Figure 1.
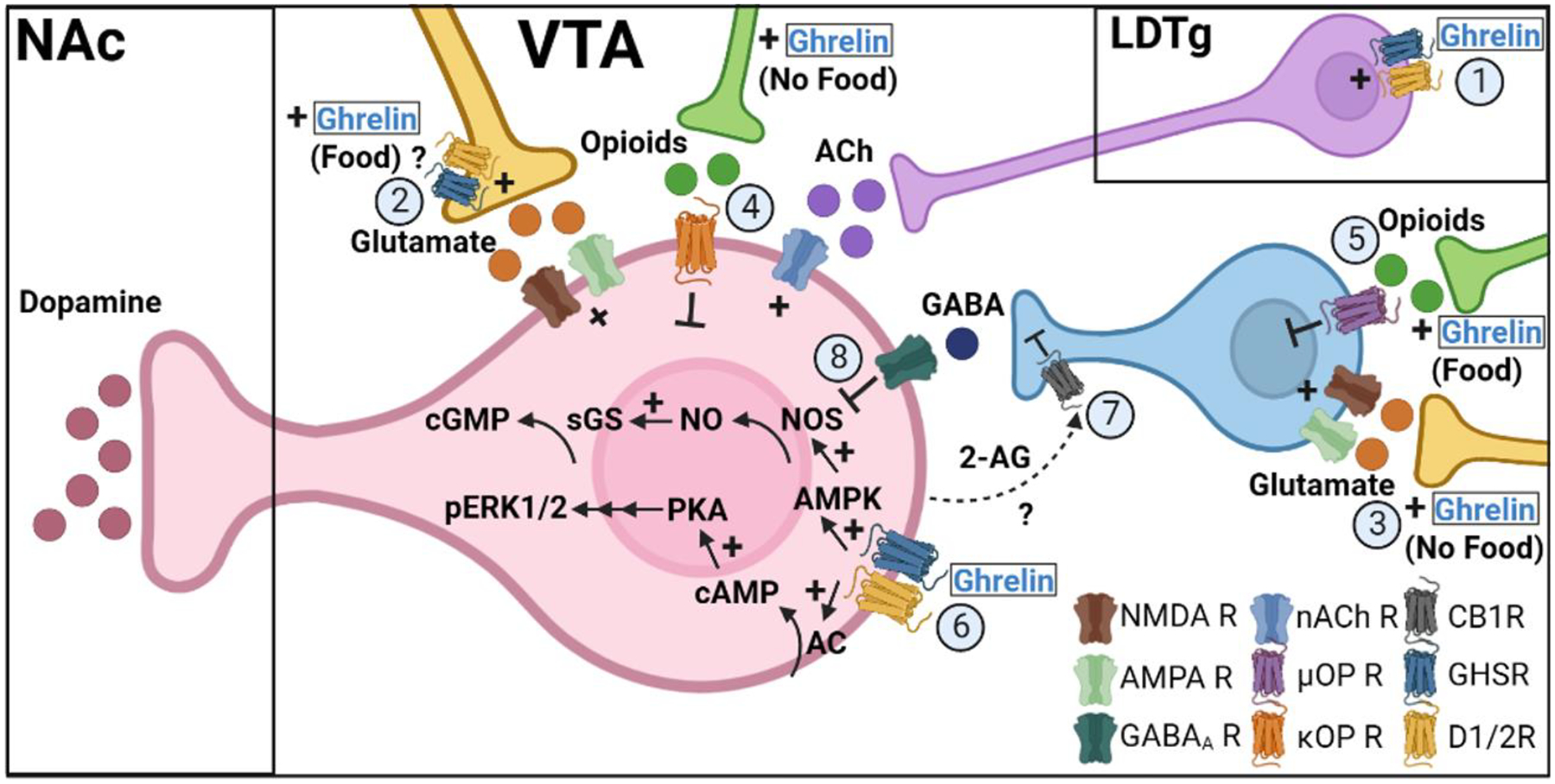
Ghrelin signaling mechanisms in the VTA. Ghrelin increases VTA dopamine neurons activity through multiple mechanisms. Ghrelin stimulates excitatory LDTg-to-VTA cholinergic signaling at nACh receptors on VTA dopamine neurons (1). In the presence of food, ghrelin increases glutamate-mediated excitation of VTA dopamine neurons through a possible presynaptic mechanism (2), yet in the absence of food, ghrelin stimulates glutamatergic inputs to GABA interneurons to inhibit VTA dopamine neurons (3). Ghrelin also stimulates ĸ-opioid receptor inhibition of VTA dopamine neurons to decrease dopamine output in the absence of food (4) but stimulates μ-opioid receptor inhibition of GABA interneurons to disinhibit VTA dopamine signaling in the presence of food (5). Ghrelin acts directly on VTA dopamine neurons to stimulate downstream ERK1/2 phosphorylation and NO production (6) and this possibly reduces local GABA signaling at dopamine neurons by stimulating inhibitory endocannabinoid signaling at presynaptic CB1 receptors on GABA interneurons (7). Mechanisms which lessen GABA interneuron inhibition of dopamine neurons disinhibit NO synthase, a downstream mediator of GHSR signaling (8).
Abbreviations: VTA (ventral tegmental area), NAc (nucleus accumbens), LDTg (laterodorsal tegmental nueclus), NMDA R (N-methyl-D-aspartate receptor), AMPA R (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor), GABAA R (GABA-A receptor), nACh R (nicotinic acetylcholine receptor), μOP R (μ-opioid receptor), ĸOP R (ĸ-opioid receptor), CB1R (cannabinoid receptor type 1), GHSR (growth hormone secretagogue receptor), D1/2R (dopamine receptor 1 or 2), 2-AG (2-arachidonoylglycerol), AC (adenylyl cyclase), cAMP (cyclic adenosine monophosphate), PKA (protein kinase A), pERK1/2 (phosphorylated extracellular signal-regulated kinase 1/2), AMPK (AMP-activated protein kinase), NOS (nitric oxide synthase), NO (nitric oxide), sGS (soluble guanylyl cyclase), cGMP (cyclic guanosine monophosphate). Created in BioRender.com.
Glutamatergic inputs to VTA dopamine neurons are also involved in ghrelin’s regulation of VTA dopaminergic activity. Ghrelin increased the action potential frequency in VTA dopamine neurons, and this was blocked by combined AMPA and NMDA receptor antagonists but was not affected by GABA-A receptor blockade [43] (Figure 1; 2). Additionally, peripheral ghrelin-stimulated locomotion and NAc dopamine release were both attenuated by VTA NMDA receptor antagonism [48]. The state-dependent effect of ghrelin to increase NAc dopamine levels in the presence of food and decrease NAc dopamine levels in the absence of food was blocked in both directions by NMDA receptor antagonism [53]. Again, the blockade of NAc dopamine release was uncoupled from ghrelin-induced hyperphagia [53]. Interestingly, GABA-A receptor antagonism only blocked ghrelin-increased NAc dopamine release in the presence of food, implying food-dependent role of ghrelin signaling on VTA GABA neurons [53] (Figure 1; 3). These data corroborate that ghrelin, presumably through a presynaptic mechanism, increases glutamatergic signaling at NMDA receptors to depolarize VTA dopamine neurons.
VTA opioid receptor signaling has also been implicated in the food state dependent ghrelin regulation of VTA-to-NAc dopamine signaling. The ghrelin mediated decrease in NAc dopamine levels in the absence of food was blocked by VTA kappa opioid receptor antagonism (Figure 1; 4), while the effect of ghrelin to increase NAc dopamine levels in the presence of food was blocked by VTA mu opioid receptor antagonism [51] (Figure 1; 5). Surprisingly, the absence of NAc dopamine release with ghrelin treatment in the presence of palatable food was stimulated after kappa opioid receptor or GHSR antagonism [51], suggesting that kappa opioid receptor signaling downstream of ghrelin prevents the release of dopamine in the NAc normally stimulated by palatable food. Intra-VTA naltrexone, which primarily antagonizes mu opioid receptors but has some activity at kappa opioid receptors, blocked VTA ghrelin stimulated PR responding for sucrose without altering the increase in chow intake [55]. Peripheral ghrelin-induced locomotor activity was not affected by peripheral naltrexone or intra-VTA orexin-A receptor antagonism [48]. However, intra-VTA orexin-A potentiated NAc dopamine release evoked by sucrose pellet retrieval and VTA orexin antagonism blocked the effect of ICV ghrelin to increase sucrose consumption [50].
It was recently discovered that within the VTA, ghrelin signaling is mediated by an oligomeric complex that includes the functional GHSR (GHSR1a), a truncated nonfunctional isoform GHSR1b, and D1R [67]. Activation of this receptor complex stimulated cAMP and ERK1/2 phosphorylation and ghrelin induced activation of VTA dopamine neurons was blocked by GHSR antagonism, D1R antagonism, or PKA inhibition [67] (Figure 1; 6). VTA D1R antagonism also blocked the central ghrelin induced increase in locomotion [47]. These results suggest that both dopamine and ghrelin can activate the same receptor complex to mediate VTA ghrelin action. Another mechanism by which ghrelin promotes VTA dopamine activity is through increasing excitatory and decreasing inhibitory input to VTA dopamine neurons. In wildtype but not GHSR knockout mice, peripheral ghrelin increased the number of glutamatergic synapses and decreases the number of GABAergic synapses connecting with VTA dopamine neurons [43]. The functional relevance of these anatomical changes is that ghrelin treatment increased the frequency of miniature excitatory post-synaptic currents (mEPSCs) and decreased the frequency of miniature inhibitory post-synaptic currents (mIPSCs) recorded from VTA dopamine neurons [43]. The decreased inhibitory input may result from an endocannabinoid-mediated retrograde presynaptic mechanism. Presynaptic CB1Rs regulate GABA release onto VTA dopamine neurons, and VTA CB1R antagonism blocked the ability of VTA ghrelin to stimulate locomotor activity but not chow intake [68] (Figure 1; 7).
GABA interneurons in the VTA signal locally to suppress activation of neighboring dopamine neurons and disrupt motivated behaviors [69, 70]. Limiting GABA signaling at VTA dopamine neurons, through either direct GABA interneuron ghrelin signaling or a cannabinoid retrograde presynaptic mechanism, may stimulate dopaminergic activity through a nitric oxide (NO) mediated mechanism. VTA dopamine neurons express NO synthase [71] and agonism of GABA-A and -B receptors inhibit NO synthesis [72, 73]. Recent work has implicated NO as a mediator of VTA ghrelin reward driven behaviors. Systemic ghrelin increased NO levels in the VTA, which through recruitment of soluble guanylyl cyclase stimulates cGMP production [74]. Inhibition NO or cGMP synthesis prevented systemic ghrelin from increasing locomotion, NAc dopamine release, or inducing condition place preference [74]. NO has been identified as a critical mediator of ghrelin signaling in other brain areas [75–78]. Selective ablation of VTA GABA neurons increased locomotor activity [79], and the locomotion stimulating effect of ghrelin was attenuated by systemic pretreatment with a GABA-B receptor agonist [74]. Together these findings propose that tonic signaling from VTA GABA interneurons suppress NO synthesis and dopaminergic transmission. In turn, ghrelin signaling at VTA GABA interneurons disinhibits NO production and dopamine neuron firing to stimulate motivated behaviors (Figure 1; 8). In fact, in other brain regions AMPA and NMDA glutamate signaling has been shown to increase NO production [73, 80, 81], suggesting that in addition to GABA, ghrelin-mediated enhancement of presynaptic glutamatergic signaling onto VTA dopamine neurons may also converge to stimulate NO signaling.
There are many receptor systems and intracellular signaling molecules implicated in VTA ghrelin signaling to enhance the motivation for a variety of rewarding stimuli. These mechanisms potentiated by ghrelin that promote addictive and compulsive behaviors to seek and consume rewarding substances all converge to stimulate VTA dopamine neuron activity and dopamine transmission at projection targets such as the NAc.
Behavior
Food
Intraparenchymal delivery of ghrelin to the VTA consistently increased the intake of chow diet and the degree of hyperphagia correlated positively with body weight in lean mice [42, 54, 82, 83]. Peripheral ghrelin-induced chow intake was blocked by antagonizing VTA GHSRs [43], highlighting the VTA as an important site of ghrelin action. Intra-VTA ghrelin also increased HFD intake in sated mice, but this no longer correlated with body weight, suggesting a diet-induced loss of appropriate central integration of metabolic information [83]. In fact, diet-induced obesity decreased VTA GHSR mRNA expression [84].
Ghrelin’s regulation of food intake is influenced by nutrient state, and hyperphagia following a fast is partially mediated by elevated ghrelin levels. Heightened ghrelin action in the fasted state is driven by nutritional need, as peripheral glucose attenuates intra-VTA ghrelin-induced hyperphagia and glucoprivation enhances VTA ghrelin-stimulated feeding [85]. Both ghrelin and GHSR knockout mice ate less following a fast and GHSR knockout mice showed diminished fasting-induced VTA neuronal activation [43, 86]. Interestingly, 6 days of chronic infusion but not acute injection of a GHSR antagonist in the VTA reduced fasting-stimulated chow intake [43, 87]. However, in the later study rats underwent 2h of operant testing for sucrose pellets after the GHSR antagonist injection before chow intake was recorded, which likely altered the response to chow. HFD intake after food deprivation was increased following VTA ghrelin injection and was blocked by VTA GHSR antagonist pretreatment [88], suggesting that exogenous ghrelin can further enhance fasting-induced refeeding of palatable foods.
Macronutrient selection is a determining factor in the nutritional and rewarding qualities of a meal and numerous studies have explored the role of VTA ghrelin signaling on food preference in the face of palatable and non-palatable choices. Some studies report that VTA ghrelin action increases intake of the less palatable option. A study examining how VTA ghrelin signaling regulates intake of different macronutrients found that VTA ghrelin injection acutely increased intake of chow but not lard or sucrose [89]. Moreover, in mice with simultaneous access to both chow and peanut butter, ICV and intra-VTA ghrelin delivery selectively increased chow intake [68].
However, more often it has been reported that VTA ghrelin signaling promotes palatable food intake and especially lipid-rich foods. When offered only chow or peanut butter, ICV ghrelin increased the intake of either while intra-VTA ghrelin stimulated intake of peanut butter but not chow [52]. GHSR knockout mice had reduced intake of peanut butter but not chow and peripheral GHSR antagonist administration over 7 days decreased daily ensure intake without altering chow intake [52]. Additionally, ghrelin knockout mice showed reduced intake of sucrose and sucralose but not HFD [83], and food deprived GHSR knockout mice had diminished intake of chow but not lard or sugar [89]. Looking specifically at the VTA in mediating these effects, bilateral VTA lesion attenuated the effect of ICV ghrelin to stimulate intake of peanut butter without affecting chow intake [52]. In rats simultaneously offered carbohydrate, protein, and fat rich foods, intra-VTA ghrelin did not alter intake of any diet but VTA antagonism of GHSRs selectively reduced intake of the fat rich diet [82]. Thus, although the data on VTA ghrelin’s regulation of macronutrient food choice are not unanimous, and differences in experimental design, species, and data interpretation likely contribute to these reported differences, most findings support a role for ghrelin to more potently stimulate intake of palatable fat-rich foods. Beyond stimulating food intake, ghrelin also influences food liking through regulating taste perception. Whole-body ghrelin knockout and female GHSR knockout mice displayed reduced taste responsivity to lipids [90, 91], although whether the VTA has any role in this effect or if ghrelin-enhanced taste responsiveness is mediated solely by gustatory GHSRs remains to be determined.
In a model that isolated VTA ghrelin signaling by restoring GHSR expression only in the VTA of GHSR knockout mice, body weight, ad libitum chow intake, and sucrose intake during a 1h access period under basal conditions or after peripheral ghrelin administration were not affected [92]. Nonetheless, placement in a novel stressful environment increased food intake, respiratory exchange ratio, and oxygen consumption, suggesting an increase in energy expenditure [92]. In fact, the only parameter that remained elevated after mice were acclimated to the environment was the increase in oxygen consumption [92]. Overall, these studies suggest that blockade of global and VTA ghrelin action suppresses palatable food intake, and isolating VTA ghrelin action signaling is not necessarily sufficient to increase consumption of rewarding food.
Ghrelin signaling in the VTA also increased the motivation to receive a sucrose reward during a PR task [54, 66, 82, 87, 93, 94], and this was blocked by VTA dopamine depletion [94]. Furthermore, peripheral, ICV, and intra-VTA injection of a GHSR antagonist alone was sufficient to decrease PR performance under food deprived conditions [66, 82, 87]. Peripheral treatment with ghrelin increased conditioned place preference for food in lean mice, but not in HFD-maintained mice, suggesting a diet-induced ghrelin resistance to induce condition a place preference [83]. Interestingly, in the absence of food, ghrelin induced an aversion for the conditioned side in both chow and HFD fed mice [83]. The presence or absence of food after ghrelin treatment elicits opposite behaviors, suggesting that ghrelin can regulate both positive and negative valance around food reward. Hunger can induce a negative affect and ghrelin’s action to promote food acquisition may be viewed as serving to both remove the negative feelings of hunger in addition to engaging a food-dependent reward response. Thus, ghrelin stimulated appetite that does not result in acquiring food likely heightens the negative affect of hunger and may explain the conditioned place aversion response.
While VTA ghrelin signaling increases motivation to receive a food reward, it was found that intra-VTA ghrelin failed to increase lever pressing or VTA dopamine levels stimulated by a food-reward paired cue [44, 93, 95]. However, in rats previously trained to lever press for chocolate, following an extinction period VTA ghrelin enhanced cue-induced reinstatement of lever pressing in both sated and food deprived conditions [96]. This highlights a role of ghrelin to increase food cravings, which are associated with high levels of impulsivity around food choices and impulsivity is a driving factor in overeating and obesity [97–100]. Not surprisingly, ICV and VTA ghrelin signaling increased food-reward directed impulsive behavior [44]. Disinhibition of cautious behaviors in an unfamiliar environment is one aspect of impulsivity that is stimulated by hunger and ghrelin. In fact, removing ghrelin signaling by GHSR knockout or VTA infusion of a GHSR antagonist increased the latency to approach a novel palatable food item [101] and VTA lesion decreased ICV ghrelin-stimulated food exploratory behavior [52]. Fasting ghrelin levels also correlated positively with food-cue reactivity in visual, taste, and reward related brain areas and reported appetite in humans [102]. Furthermore, in a meta-analysis of fMRI studies to evaluate how ghrelin modulates reward responses in humans, it was found that ghrelin unambiguously increased reward responses in the motivational neurocircuitry including the mesolimbic pathway [103]. Thus, ghrelin signaling can strongly motivate reward seeking in humans.
Impulsivity is also a factor in the development of binge eating disorders [104–106]. A binge eating model of 2h restricted HFD access each day induces hyperphagia during this period and robustly activates the VTA [107]. In this model, ICV and intra-VTA ghrelin surprisingly increase chow and decrease HFD intake [108], suggesting that ghrelin may differentially modulate food choice in an established binge eating model. Overnight fasting to elevate endogenous ghrelin also stimulated chow intake but did not suppress HFD intake [108]. In rats with restricted HFD access, intra-VTA ghrelin increased HFD feeding and this was blocked by antagonizing VTA GHSRs [88]. Four weeks of chronic ICV ghrelin increased binge HFD intake but GHSR knockout mice exhibited binging behavior similar to controls [108]. However, during the first 4 days of establishing binging behavior, GHSR knockout mice failed to escalate HFD intake and showed blunted diet-induced VTA neuronal activation [107]. Together, these results supporting that VTA ghrelin signaling exacerbates binging of palatable foods and is involved in initiating but not critical to maintain binge eating.
Drugs of Abuse
Similar to ghrelin’s actions on food rewards, VTA ghrelin signaling enhances the reward saliency of drugs with high abuse potential. The VTA mediates heightened locomotion in response to rewarding substances to further increase reward seeking actions, and VTA ghrelin signaling has a role in mediating this behavior. Mice with GHSR expression only in the VTA showed elevated cocaine-stimulated locomotion compared to full body GHSR knockouts [92], while peripheral administration of a GHSR antagonist decreased cocaine and amphetamine-induced locomotor activity [57, 109]. Cocaine induced a strong conditioned place preference and this was enhanced by intra-VTA or peripheral ghrelin administration [83, 110, 111], and conversely attenuated by intra-VTA or peripheral GHSR antagonism [57, 110]. Although diet-induced obese mice lost ghrelin-stimulated conditioned place preference for food, their ghrelin-enhanced place preference for cocaine was intact [83]. VTA ghrelin signaling alone is sufficient to induce conditioned place preference, and subthreshold dose of VTA ghrelin potentiates cocaine-induced conditioned place preference [111]. In fact, cocaine’s rewarding properties in part are mediated by ghrelin as cocaine increased systemic ghrelin concentrations through an adrenergic beta-1 receptor mediated mechanism and cocaine increased VTA sensitivity to ghrelin by increasing GHSR expression on VTA dopamine but not GABA or glutamate neurons [112]. Peripheral GHSR antagonist administration decreased cocaine seeking behavior and cue-dependent reinstatement and reduced direct electrical self-stimulation of VTA dopamine neurons in the presence and absence of cocaine [112]. This corroborates that VTA dopamine signaling is inherently rewarding and is the site of ghrelin’s modulation of the rewarding properties of cocaine.
Ghrelin’s actions on addictive substances are not restricted to cocaine. Indeed, opioids elevate systemic ghrelin levels and increase GHSR expression on VTA dopamine but not GABA neurons [113], and blockade of VTA GHSRs decreased heroin seeking behavior [114]. Peripheral injection of a GHSR antagonist decreased oxycodone self-administration and PR performance, and morphine- and nicotine-stimulated locomotor activity and conditioned place preference [56, 58, 59, 113]. Ghrelin may also participate in the reinforcing effects of cannabis as systemic GHSR antagonism mitigated VTA cannabinoid receptor 1 (CB1R) agonist induced NAc dopamine release and turnover [115]. Thus, ghrelin stimulates the drive to seek and consume many drugs of abuse.
Alcohol intake has been linked with VTA ghrelin signaling. In fact, plasma ghrelin levels correlated with the severity of alcohol cravings in newly abstinent alcoholics and polymorphisms in the GHSR and pro-ghrelin gene were associated with heavy alcohol use [116, 117]. Furthermore, ghrelin infusion increased alcohol self-administration and neuroactivity in the NAc of alcohol dependent individuals [118], supporting that ghrelin promotes alcohol consumption. Corroborating this notion, rats genetically predisposed to consume a high amount of alcohol had higher VTA GHSR mRNA expression [119]. Interestingly, alcohol intake decreased circulating ghrelin levels [119], and 10 months of voluntary alcohol consumption downregulated VTA GHSR mRNA [120]. Peripheral and intra-VTA ghrelin stimulated alcohol intake, preference for alcohol, alcohol-induced locomotor activity, and potentiated cocaine-induced alcohol intake [60, 109, 120–122], while peripheral GHSR antagonist injection reduced alcohol-dependent conditioned place preference, locomotion, and alcohol binging behavior [60, 120]. Furthermore, mice lacking either the GHSR or ghrelin hormone showed decreased alcohol-induced conditioned place preference and locomotion [60, 61]. These studies demonstrate a clear regulation of VTA ghrelin signaling on alcohol intake.
Interestingly, VTA ghrelin has been shown to improve cognitive behavior and physical activity on the forced swim, open field, and rotarod tasks, suggesting ghrelin may help to mitigate depressive like behaviors [123]. But VTA ghrelin may also be implicated in depression-mediated hyperphagia and high doses of ghrelin can induce anxiety-like behaviors [121, 124]. Thus, the role of VTA ghrelin action in mood regulation at current understanding is inconclusive.
Activity and Arousal
VTA-to-NAc dopaminergic activity stimulates both goal-directed locomotion and more generalized wakefulness and arousal [125]. Ghrelin signaling was found to stimulate both generalized and reward-directed locomotor activity [45, 126]. In fact, peripheral, ICV, and intra-VTA ghrelin all increased locomotion [45, 47, 52, 63, 65, 74], and ICV and peripheral ghrelin-induced locomotion was blocked by VTA GHSR antagonism [47, 48]. This supports that activation of mesolimbic circuits is required for ghrelin-induced locomotion. Indeed, in one study where peripheral ghrelin administration failed to alter locomotion, ghrelin likely did not reach the VTA as no cFos was detected in the VTA [42]. Lastly, although GHSR knockout mice with selective reintroduction of GHSR in the VTA did not alter baseline locomotion, ghrelin-stimulated locomotion was not tested [92]. Thus, VTA GHSR signaling may not regulate movement under basal conditions with low ghrelin levels. Together, these data suggest that through activating VTA-to-NAc dopamine signaling, ghrelin stimulates generalized arousal and goal-directed locomotor activity to seek a food or drug reward. Under high ghrelin fasting conditions, these dual functions of wakefulness and movement are hypothesized to work in parallel to keep an organism receptive to opportunities to obtain food.
Social Interactions
VTA dopamine signaling reinforces addictive substances and natural rewards such as sexual behavior and positive social interactions. VTA ghrelin signaling increased while VTA GHSR antagonism decreased female directed male sexual behaviors assessed by the latency to initiate sex, number and duration of mounts [40]. Ghrelin deficient mice showed decreased male active seeking behavior for a female sexual partner [127]. Interestingly, fasted rats with elevated endogenous ghrelin decreased anticipatory sex behavior and this was further attenuated by blocking VTA ghrelin signaling in the food deprived state [127]. However, VTA ghrelin did not enhance anticipatory sex behavior in sated rats that already displayed a high level of sexual motivation [127]. These results suggest that ghrelin action in the VTA enhances motivation for sex, however under food deprived conditions ghrelin action at sites other than the VTA prioritizes nutrient seeking.
Ghrelin further regulates non-sexual social interactions, and the absence of GHSRs or peripheral GHSR antagonism increased the latency to approach a new animal and decreased the time spent interacting with the unknown animal [101]. This is mediated by VTA ghrelin signaling as VTA GHSR antagonism increased while selective expression of the GHSR in the VTA decreased the latency to begin a novel social interaction [101]. Additionally, VTA GHSR blockade decreased and VTA ghrelin increased locomotion to explore novel food and non-food objects and environments and two polymorphisms in the GHSR gene are associated with novelty seeking personalities [128]. Thus, VTA ghrelin signaling motivated behavior for a wide range of pleasurable stimuli.
Cumulatively, these studies conclude that VTA ghrelin signaling incentivizes the consumption of food, cocaine, opioids, nicotine, cannabis, alcohol, sex, and novel experiences, and increasing locomotion to participate in these activities is a primary mechanism to seek and indulge in rewarding experiences. Now we will explore how the satiation hormones GLP-1, amylin, leptin, and insulin act in the VTA to mitigate reward-directed motivated behaviors.
Glucagon-like Peptide-1
Glucagon-like peptide-1 (GLP-1) is synthesized principally from two sources within the body: the intestinal L-cells in response to nutrient ingestion and centrally as a neuropeptide from preproglucagon (PPG) expressing neurons in the nucleus tractus solitarius (NTS) and reticular formation of the caudal brainstem [29]. To a much lesser extent, GLP-1 is also synthesized in the olfactory bulbs with little to no projection output [129]. PPG neurons in the brainstem project to and release endogenous GLP-1 in the VTA [130]. Thus, this central source of GLP-1 is thought to be the predominant endogenous GLP-1 ligand acting on mesolimbic reward circuits. In contrast, exogenously administered GLP-1 to either the periphery or ICV acts in a very distributed fashion on GLP-1 receptors (GLP-1R) expressed throughout the brain in nuclei of relevance to energy balance, including but not limited to the CeA, hypothalamic subnuclei, ventrolateral medulla, hippocampus, cortex, bed nucleus of the stria terminalis (BNST), olfactory bulb, preoptic area, lateral habenula, substantia nigra, PBN, hindbrain DVC, locus ceruleus, LDTg, NAc, and VTA [131–133]. Peripherally administered GLP-1 readily crosses the blood brain barrier and acts in the VTA to affected motivated aspects of ingestive behavior. Indeed, systemically injected fluorescently labeled exendin-4 (Ex-4), a GLP-1 receptor (GLP-1R) agonist, is detected in neurons and glia of the VTA [134, 135]. As discussed herein, a growing body of evidence supports the role of VTA GLP-1R signaling in regulating motivated behaviors for rewarding stimuli.
Mechanism
NAc dopamine release is commonly used as an indicator of VTA-to-NAc dopamine transmission [7], and as ghrelin stimulated this pathway, much evidence supports that satiation hormones mute NAc dopamine signaling. Peripheral GLP-1R agonists attenuated NAc dopamine release evoked by alcohol, amphetamine, cocaine, and nicotine [136–140]. Although systemic cocaine elevated dopamine release in both the NAc core and shell, the mitigating effect of ICV Ex-4 delivery was found to be selective for dopamine signaling in the NAc core [139]. ICV Ex-4 did not attenuate the magnitude of NAc dopamine release evoked by electrical stimulation of VTA neurons or rates of NAc dopamine reuptake [139]. These results support a model in which VTA GLP-1 action suppresses NAc dopamine release likely by dampening the excitability of dopamine neurons.
Indeed, the decrease in VTA dopamine activity induced by systemic lithium chloride was mediated by central GLP-1R signaling [141]. In the context of food reward, ICV Ex-4 decreased the spike in VTA dopamine neuron firing activity in response to a sucrose predictive cue, but surprisingly did not alter spontaneous VTA dopamine firing [142]. The amplitude of cue-evoked dopamine activity was correlated with sucrose approach and licking behaviors [142], highlighting VTA-to-NAc dopamine signaling in reward context associations. While the effect of Ex-4 was of equal magnitude in males and females, female rats had a lower baseline dopamine response to the sucrose paired cue [142]. Surprisingly, GLP-1 signaling was found to increase VTA mRNA and protein expression of tyrosine hydroxylase (TH) [143–145], the rate limiting enzyme in dopamine synthesis, which implies an increase in dopamine production in contrast to the well document effect of GLP-1 to decrease VTA dopamine output.
The role of VTA glutamatergic signaling on the anorectic effects of VTA GLP-1 and how this modulates dopaminergic activity has also been investigated. It was found that intra-VTA AMPA but not NMDA receptor antagonism attenuated intra-VTA Ex-4-induced hypophagia, and Ex-4 increased the frequency of spontaneous mEPSCs in VTA dopamine neurons [143]. This study also found that Ex-4 decreased the paired-pulse ratio in VTA dopamine neurons but did not alter the action potential frequency in response to injected current [143], suggesting that GLP-1s effects are presynaptically mediated. We hypothesize that these dopamine neurons are unlikely to innervate the NAc in the face of multiple lines of evidence that VTA GLP-1R signaling decreases NAc dopamine release. In turn, we propose that VTA dopamine neurons stimulated by GLP-1 on presynaptic glutamatergic terminals project to a different target such as the amygdala (Figure 2; G1).
Figure 2.

Satiation hormone signaling mechanisms in the VTA. The distinct mechanisms outlined for each hormone are indicated in the figure, legend, and manuscript text by the first letter of that hormone and the sequential number. A question mark is placed near any mechanisms which are hypothesized.
GLP-1: GLP-1 is proposed to excite presynaptic glutamate terminals that innervate a subset of VTA dopamine neurons which project to the CeA (G1). GLP-1Rs also may be expressed postsynaptically on some VTA-to-NAc projecting dopamine neurons (G2). GLP-1 decreases VTA dopaminergic activity by increasing GABAergic input to VTA dopamine neurons through a possible presynaptic mechanism to increase glutamatergic excitation of VTA GABA interneurons (G3), stimulating long-range GABA projections from the LDTg (G4), and possibly direct activation of VTA GABA interneurons (G5).
Amylin: CTR is expressed on a subset of VTA dopamine neurons which may innervate the CeA (A1) but likely do not innervate the NAc. Amylin is proposed to decrease VTA dopamine neuron activity by increasing local (A2) and LDTg-to-VTA (A3) projecting GABA-mediated inhibition of VTA dopamine neurons. Dimerization of CTR with a RAMP is required to form the amylin receptor complex, while sCT binds strongly to CTR in the absence of a RAMP. Many studies investigating VTA amylin signaling used sCT and the profile of RAMP co-expression with CTR in different VTA populations is unknown, so CTR alone is depicted here.
Leptin: Leptin decreases VTA dopamine neuron activity by increasing inhibitory GABA signaling at VTA dopamine neurons by directly activating VTA GABA interneurons through a pERK1/2 dependent mechanism (L1) and inhibiting long-range LH-to-VTA GABA projections that inhibition VTA GABA interneurons (L2). Leptin signaling on LH-to-VTA GABA neurons may regulate VTA dopamine neurons TH expression (L3). Leptin also acts directly on VTA dopamine neurons that project to the CeA through a pSTAT3 and cAMP dependent mechanism (L4). Lastly, leptin may act through a presynaptic mechanism to inhibit release of glutamate onto VTA dopamine neurons (L5).
Insulin: Insulin decreases VTA dopamine neuron activity through an mTOR dependent mechanism by increasing the membrane transport (I1) and expression (I2) of DAT to enhance synaptic dopamine clearance and stimulating the synthesis and retrograde release of inhibitory endocannabinoid signaling at glutamate neuron presynaptic CB1 receptors to decrease glutamatergic excitation of VTA dopamine neurons (I3).
Abbreviations: VTA (ventral tegmental area), NAc (nucleus accumbens), LDTg (laterodorsal tegmental nueclus), LH (lateral hypothalamus), CeA (central nucleus of the amygdala), NMDA R (N-methyl-D-aspartate receptor), AMPA R (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor), GABAA R (GABA-A receptor), GABAB R (GABA-B receptor), CTR (calcitonin receptor subunit of amylin receptor), RAMP (receptor activity modifying protein), DAT (dopamine transporter), CB1R (cannabinoid receptor type 1), GLP-1R (glucagon-like peptide-1 receptor), leptin R (LepR), insulin R (InsR), TH (tyrosine hydroxylase), 2-AG (2-arachidonoylglycerol), pSTAT3 (phosphorylated signal transducer and activator of transcription 3), cAMP (cyclic adenosine monophosphate), pERK1/2 (phosphorylated extracellular signal-regulated kinase 1/2), PI3K (phosphoinositide 3-kinase), PIP3 (phosphatidylinositol 3,4,5-triphosphate), pAKT (phosphorylated protein kinase B), mTOR (mammalian target of rapamycin). Created in BioRender.com.
These findings in contrast to those described in a latter report [146] which suggested that postsynaptic GLP-1R signaling on VTA dopamine neurons decreased AMPA but not NMDA evoked EPSCs. This report argued in support of a postsynaptic GLP-1R mechanism to reduce dopamine excitatory synaptic strength, as bath applied Ex-4 decreased the frequency and amplitude of spontaneous mEPSCs in VTA-to-NAc dopamine neurons but did not affect the paired-pulse ratio of electrically stimulated EPSCs [146]. Thus, GLP-1Rs may also be expressed postsynaptically on VTA-to-NAc dopamine neurons (Figure 2; G2), but the mechanism for how GLP-1 diminishes glutamatergic input to these neurons is unknown.
The discrepancies between these two studies might be explained by several factors. AMPA receptor blockade in vivo is not isolated to VTA-to-NAc dopamine neurons and would alter glutamatergic signaling in all VTA circuits. For instance, inhibiting glutamatergic input to VTA GABAergic interneurons that depress VTA-to-NAc dopamine projections would enhance dopamine output and could explain the mitigated anorectic effect of Ex-4 (Figure 2; G3). Additionally, the two papers identified VTA dopamine neurons using different criteria and the first report recorded from dopamine neurons of an unidentified projection target. VTA dopamine neurons are extremely heterogeneous and their properties are in part dictated by their projection pathway [9, 147], making it difficult to compare the electrophysiological results in these two papers. Species differences between mouse and rat may also be a factor. More studies are needed to unravel the role of glutamatergic signaling in VTA GLP-1 action, as well as the role for postsynaptic GLP-1R on non-dopaminergic neurons and glia of the VTA and how these actions may also influence dopaminergic transmission to affect reward behaviors.
Many nuclei that innervate the VTA express GLP-1Rs and through action on these projections, GLP-1R signaling may also modulate reward-directed behaviors. One area in which the effects of GLP-1 signaling has begun to be characterized is the LDTg. GLP-1Rs are expressed, at least in part, on LDTg GABA neurons that project to the VTA and silencing of this pathway blocked the effect of Ex-4 to decrease cocaine seeking behavior [148], suggesting that GLP-1 action in the LDTg increases inhibitory GABAergic input to the VTA (Figure 2; G4). LDTg Ex-4 also decreased alcohol intake and sexual interaction behaviors [149, 150], lending support that LDTg-to-VTA GLP-1R expressing GABAergic neurons likely innervate and inhibit the activity of VTA dopamine neurons. GLP-1R signaling in the VTA may also increase VTA GABA interneuron inhibition of VTA dopamine neurons (Figure 2; G5) as bath applied Ex-4 increased the amplitude of spontaneous mIPSCs onto VTA-to-NAc dopamine neurons from midbrain slices [146]. Thus, GLP-1R signaling may not only decrease excitatory inputs but also increase inhibitory inputs to VTA dopamine neurons. Peripheral and central sources of GLP-1 decrease the motivation for and consumption of rewarding substances by diminishing VTA dopamine output.
No study to date has identified the phenotypes of GLP-1R expressing VTA neurons on specified input-output curcuits, but mechanistic studies provide some clues and suggests GLP-1Rs may be expressed on VTA-to-NAc dopamine neurons, GABA interneurons within the VTA, on LDTg-to-VTA GABA projections, and presynaptically on glutamatergic inputs to VTA dopamine neurons and local VTA GABA neurons (Figure 2.) Further work into the mechanisms and intracellular molecules that mediate VTA GLP-1 action are necessary to optimize the use of GLP-1 based therapeutics to target VTA regulated motivated behaviors.
Behavior
Food
It is not surprising that in reducing the dopamine stimulating effects of rewarding substances, GLP-1 signaling also mitigates reward-directed behavior. Peripheral, ICV, and intra-VTA administration of Ex-4 decreased operant responding for a sucrose pellet reward, conditioned place preference for a chocolate reward, and ad libitum chow and HFD intake and body weight [130, 143, 151–154]. Intra-VTA Ex-4 reduced HFD intake by decreasing both the number of meals and meal size over 24h [143]. Injection of a GLP-1R antagonist into the VTA blocked peripheral Ex-4-mediated suppression of HFD intake [143], confirming that the VTA is directly accessible as a central target for peripherally delivered GLP-1R agonists that regulates palatable food intake. The central source of GLP-1 also regulates palatable food intake as chemogenetic activation of hindbrain PPG neurons and VTA GLP-1 nerve terminals decreased HFD intake [146].
VTA GLP-1R signaling also diminishes motivation for food rewards. Even in overnight fasted rats, VTA Ex-4 suppressed PR responding and chow intake and ICV pretreatment with a GLP-1R antagonist prevented Ex-4-mediated suppression of PR performance [153]. Furthermore, the stimulatory effect of VTA ghrelin and NPY signaling to increase PR responding for sucrose was blocked by intra-VTA Ex-4 [151, 154].
While systemic Ex-4 decreases intake of either chow or HFD [145], GLP-1R agonism in the VTA appears to preferentially suppress intake of palatable high-energy dense foods without robustly affecting intake of standard chow in rodents. Indeed, in rats with 1h access to a sucrose solution prior to normal ad libitum chow access, intra-VTA Ex-4 robustly decreased sucrose intake, while only mildly diminishing the intake of chow at the last timepoint [130]. In rats with ad libitum choice access to both HFD and chow, intra-VTA Ex-4 actually increased early chow intake, while robustly suppressing the intake of HFD throughout the day [130]. These data support that when offered two food choices, VTA GLP-1 signaling more potently suppresses intake of the palatable option. These effects are not limited to a pharmacological action of GLP-1R agonism, as VTA GLP-1R blockade alone increased intake of HFD [130], confirming that endogenous signaling at VTA GLP-1Rs physiologically regulates intake of palatable foods.
Two studies have investigated sex-differences in VTA GLP-1 signaling with inconsistent results. The first found that VTA Ex-4 more potently suppressed motivation for a sucrose reward in female than male mice, but equally suppressed ad libitum chow intake in both sexes [155]. Meanwhile another study found no difference in the effect of ICV Ex-4 to inhibit cue-evoked sucrose intake behavior between males and females [142]. Further investigation is required to determine any influence of sex on VTA GLP-1 signal.
Lastly and importantly, GLP-1 signaling in the VTA does not elicit the most common side effects reported with peripheral GLP-1R agonism, i.e. nausea and vomiting. Indeed, there are no reports noting illness-like behaviors by GLP-1R activation specific to the VTA [130, 153].
Evidence supports that midbrain GLP-1 signaling also dampens the rewarding value of food in humans, as Ex-4 decreased food-induced neuroactivity in reward related brain areas in obese subjects compared to lean subjects [156]. When looking at different reward phases, Ex-4 decreased brain reward-system activity to anticipation of a food-reward but increased activity in these areas to consumption of a food-reward in participants with obesity, which may serve to reduce cravings and prevent overeating [157]. However, in a meta-analysis of fMRI studies investigating the effects of GLP-1 on central reward responses there was insufficient convergence of data from a limited number of studies to conclude that GLP-1 reduces activity of reward circuits [103].
Drugs of Abuse
VTA GLP-1 signaling mitigates the rewarding properties of substances with high addictive potential. Intra-VTA Ex-4 decreased and VTA GLP-1R knockdown increased cocaine self-administration on a PR task [158]. Furthermore, systemic and intra-VTA Ex-4 decreased cocaine-priming induced reinstatement of drug seeking behavior [134]. Peripheral Ex-4 also reduced cocaine and amphetamine stimulated locomotor activity and conditioned place preference [137]. Surprisingly, cocaine stimulates central GLP-1 neurotransmission. This mechanism is thought to involve a cocaine-mediated increase in corticosterone release, which in turn increases activation of NTS PPG neurons and release of GLP-1 in the VTA [158]. This hypothesis was supported by data showing that brainstem administration of corticosterone decreased cocaine self-administration and this effect was blocked by VTA GLP-1R antagonism [158], corroborating that through engaging a stress response, cocaine increases endogenous VTA GLP-1 signaling as a negative feedback to inhibit further cocaine use. In fact, extinction from cocaine decreased NTS PPG mRNA expression [134].
Alcohol intake is similarly attenuated by VTA GLP-1 action. Peripheral and VTA Ex-4 and GLP-1 decreased operant responding for alcohol self-administration, alcohol conditioned place preference, 2-bottle choice alcohol intake, and alcohol stimulated locomotion [138, 149, 152, 159, 160]. The posterior VTA was found to mediate GLP-1’s effects on alcohol intake, as injection of Ex-4 into the posterior but not anterior VTA decreased alcohol-stimulated locomotion [140]. In contrast to other reports, this aforementioned study did not see an effect of VTA Ex-4 to decrease conditioned place preference dependent alcohol memory or alcohol intake [140]. Systemic GLP-1R antagonism was sufficient to increase alcohol intake [160], supporting that endogenous GLP-1R signaling acts to suppress alcohol consumptive behaviors. Peripheral GLP-1R agonist treatment also prevented alcohol deprivation in alcohol trained rats from inducing hyper consumption of alcohol [149], suggesting that GLP-1 dampens alcohol cravings. However, VTA Ex-4 was not found to decrease the motivation for alcohol reacquisition after an extinction period [149, 159]. In addition to regulating alcohol consumption, GLP-1R activation regulates fluid intake more broadly [161, 162], and intra-VTA Ex-4, as well as peripheral administration of a high dose of Ex-4 reduces water intake [149]. Yet, there seems to be a lower threshold for VTA GLP-1Rs to affect alcohol than water intake as intra-VTA GLP-1, and treatment with lower doses of Ex-4 in the VTA and periphery were sufficient to decrease alcohol but not water or food intake [149, 160]. Thus, VTA GLP-1R activation appears to have a higher sensitivity to mitigate alcohol intake behaviors compared to food and water.
Intra-VTA Ex-4 attenuated the stimulatory effect of cocaine and amphetamine to promote alcohol intake and blocked the ability of VTA ghrelin to further potentiate alcohol intake [163]. VTA Ex-4 also decreased VTA ghrelin and NPY action from increasing alcohol intake in a 2-bottle access test [151]. The rewarding effects of nicotine were also blunted by GLP-1 action and peripheral Ex-4 attenuated nicotine-induced locomotion and conditioned place preference [137]. The effects of GLP-1 extend beyond drugs of abuse, indicating a broader involvement of VTA GLP-1 in motivated behavior. For example, Ex-4 delivery to the posterior but not anterior VTA decreased male anticipatory sexual interactions and mounting behaviors during sex [150]. Thus, GLP-1 signaling in the VTA has been identified to attenuate motivation for not only food, but also other rewarding stimuli including cocaine, amphetamine, alcohol, nicotine, and sex.
Activity and Arousal
VTA dopamine and GABA neurons regulate activity and arousal [164, 165], yet intra-VTA Ex-4 injection did not alter general locomotor activity or exploratory (rearing) behavior in the absence of food [153]. Peripheral Ex-4 also did not affect exploratory locomotor activity while assessing food-induced conditioned place preference [153]. Furthermore, spontaneous locomotor activity was not influenced by ICV or peripheral Ex-4 or viral ablation of NTS PPG neurons, which innervate the VTA [166, 167]. Still, some evidence suggests that GLP-1 regulates activity as peripheral treatment with a dual incretin receptor agonist (GLP-1 and glucose-dependent insulinotropic polypeptide) stimulated exploratory and locomotor activity in a mouse model of Parkinson’s disease with significantly damaged VTA function and motor activity. However, whether this is mediated by VTA GLP-1R signaling is unknown and GLP-1 enhancing locomotion is a surprising finding because GLP-1 typically decreases activity. Indeed, global knockout of GLP-1R elevated locomotion [168] and as mentioned in the above section, peripheral and VTA treatment with GLP-1 agonists suppressed drug-stimulated locomotion. Although further work is needed to determine if and how VTA GLP-1R signaling regulates activity and arousal, these data suggest that alone GLP-1 action in the VTA does little to affect activity but can attenuate heightened locomotion induced by a stimulant. Of note, injection of Ex-4 into the NAc transiently reduced both locomotor and rearing activity [153].
Amylin
The satiation hormone amylin is co-released with insulin from pancreatic beta cells following a meal and acts centrally to regulate energy homeostasis [169, 170]. Amylin signaling in the VTA has received less attention compared to the other peripheral metabolic hormones examined in this review, but interest in amylin’s control of feeding behaviors and motivated behavior largely through action in the VTA is growing [171]. The amylin receptor is composed of a primary calcitonin receptor (CTR) subunit that heterodimerizes with 1 of 3 receptor activity modifying protein (RAMP1, 2 or 3) [172, 173]. The VTA expresses CTR and equal levels of RAMP1–3 [174], but it is unknown which RAMPs co-express with CTR in which populations of VTA neurons. Peripheral administration of a fluorescently labeled long-lasting CTR agonist salmon calcitonin (sCT) is detected in VTA neurons [175], supporting that peripherally released amylin penetrates the blood brain barrier to signal in the VTA. However, sCT is a ligand for both the amylin receptor and CTR when it is not paired with a RAMP [172, 176], and binds more widely than amylin [177]. This difference in receptor activation between amylin and sCT should be acknowledged in the interpretation of these studies. In addition to the VTA, expression of CTR and binding of radiolabeled sCT and amylin are detected in many nuclei across the brain including but not limited to hypothalamic nuclei, BNST, substantia nigra, NAc, locus coeruleus, LDTg, DVC of the hindbrain, the reticular formation, preoptic area, CeA, and dorsal raphe [177–180], all of which contribute to amylin’s regulation of the energy balance [31, 181].
Mechanism
Activation of CTR in the VTA has been shown to decrease NAc dopamine release in response to multiple rewarding stimuli. Intra-VTA sCT reduced phasic dopamine in the NAc core in response to retrieval of a sucrose pellet in rats [182] and abolished alcohol-stimulated dopamine release in the NAc shell in mice [175]. Furthermore, peripheral sCT reduced cocaine-stimulated NAc shell dopamine release in mice [183]. In anesthetized rats, peripheral sCT but not amylin decreased NAc phasic dopamine release evoked by electrical stimulation of the VTA, and interestingly this effect was greatly attenuated in area postrema (AP) and parabrachial nucleus (PBN) lesioned rats [184]. These results imply that the AP and PBN are important downstream areas that mediate peripheral sCT’s ability to suppress VTA stimulated phasic dopamine responses. The lack of an effect for peripheral amylin to modulate NAc dopamine release could result from differences in the pharmacokinetic and receptor activating profiles between amylin and sCT [172, 185].
These studies all support that VTA amylin receptor signaling decreases VTA dopaminergic output. Indeed, 5 days of peripheral sCT treatment decreased VTA dopamine turnover [186], indicating reduced VTA dopamine production. Moreover, pretreating the NAc with dopamine 1 and 2 receptor agonists blocked the anorectic effects of intra-VTA sCT [182], demonstrating that a reduction in NAc dopamine receptor signaling is necessary for VTA amylin receptor induced hypophagia. It is estimated that over 60% of VTA CTR is expressed on dopamine neurons, while 12% of all VTA dopamine neurons express CTR [182]. The intracellular signaling cascades downstream of VTA amylin receptor activation have not been established. Amylin receptor signaling has been shown to stimulate cAMP formation, calcium mobilization, ERK phosphorylation, and STAT-3 phosphorylation, although which intracellular signaling pathways are activated is highly dependent on the CTR:RAMP phenotype and location of the receptor complex [172, 173, 176, 187, 188]. Interestingly, intra-VTA coadministration of subthreshold doses of amylin and leptin produced hypophagia and weight loss [189]. One explanation for this is that amylin and leptin signaling in the VTA converge on a common intracellular pathway.
Although a significant portion of VTA CTR is expressed on dopamine neurons, it is unlikely that activation of these amylin receptors, which is stimulatory and predicted to increase neuron firing, is responsible for decreasing VTA dopamine output in the NAc. Accordingly, we hypothesize that the 60% of CTR expression found on dopaminergic neurons do not innervate the NAc but instead project to other VTA targets like the CeA (Figure 2; A1). Recently it was discovered that intra-VTA leptin reduces NAc dopamine release through an indirect mechanism to increase local GABA-mediated inhibition of neighboring VTA-to-NAc projecting dopamine neurons [190]. If leptin and amylin induce satiation through a common mechanism in the VTA, non-dopaminergic CTR signaling may activate VTA GABA interneurons to inhibit dopamine projections (Figure 2; A2). Additionally, it has been hypothesized that amylin inhibits VTA dopaminergic activity through long-range GABAergic projections coming from the LDTg [191] (Figure 2; A3). More work is needed to establish which intracellular signaling cascades on which population of VTA cells mediate the amylin-dependent reduction in the motivation for and consumption of rewarding substances.
Behavior
Food
sCT administered in the VTA decreased chow intake and body weight over 24h primarily by decreasing the meal size and acutely decreasing the number of meals eaten [174]. This reduction in food intake did not induce behaviors indicative of nausea, changes in locomotion, or anxiety-like behaviors [174, 192], supporting the hypothesis that neither feeling of malaise nor reduced physical activity contribute to the anorectic effect of sCT action in the VTA. Amylin and leptin signaling in the VTA cooperatively suppress chow intake also through a reduction in meal size [189]. VTA amylin receptor signaling is physiologically relevant for the control of food intake as injection of an amylin receptor antagonist in the VTA alone increased 24h chow intake and reduced the effect of peripheral sCT to suppress chow intake [174]. Furthermore, viral mediated knockdown of CTR expression selectively in the VTA increased ad libitum food intake and body weight gain in rats maintained on a HFD but did not change caloric intake in rats maintained on a chow diet [182]. The diet specific impact suggests that while VTA CTR activation can suppress chow intake, the endogenous role of CTR in the VTA is more evident in the control of palatable food intake.
Supporting the hypothesis that amylin receptor signaling in the VTA modulates motivated aspects of ingestive behavior, sCT injected into the VTA reduced PR lever presses for a sucrose pellet reward and the intake of a sucrose solution and intralipid during a 1h access period [174, 192]. Interestingly, when both sucrose and intralipid were presented as a choice to rats, VTA sCT preferentially suppressed intralipid intake [192]. In fat-preferring rats, peripheral sCT reduced the intake of shortening, chow, and sucrose and only the effect on shortening intake were partially reversed by VTA blockade of amylin receptors [192]. While in sucrose-preferring rats peripheral sCT also reduced the intake of shortening, chow, and sucrose but pre-treatment with an amylin receptor antagonist in the VTA partially reversed the suppression of all 3 food options [192]. Together, these results indicate that VTA amylin receptor signaling more prominently suppresses fat intake but does also attenuate sugar intake, taking into account that individual macronutrient preference can influence these effects. The regulation of VTA amylin receptor signaling extends beyond palatable foods as VTA sCT also suppress intake of the non-nutritive sweetener sucralose and angiotensin II stimulated-water intake but not ad libitum water intake [192]. Thus, VTA amylin action regulates a range of motivated ingestive behaviors. From a translational perspective, there are multiple amylin/CTR-agonist compounds in various stages of clinical trials for obesity/diabetes treatment. However, at present, the sole FDA-approved amylin analogue pramlintide, is reported to reduce body weight and appetite in obese patients [193, 194]. However, it is not clear whether pramlintide or native amylin modulates the activity of reward-circuits in humans.
Drugs of Abuse
A few recent studies have investigated if VTA amylin receptor signaling modulates intake of high-abuse potential substances. Intra-VTA sCT abolished alcohol-induced locomotor activity but did not modulate alcohol reward dependent memory in a conditioned place preference in mice [175]. In rats with 12 weeks of intermittent alcohol exposure, sCT injected into the VTA did not affect chow intake or body weight but diminished 24h alcohol intake [175]. Interestingly, intra-VTA sCT only mildly and non-significantly reduced cocaine-stimulated locomotion in mice [183]. More detailed investigations are warranted to understand the role of VTA amylin signaling in the motivation for drugs of abuse.
Leptin
The adipose tissue derived hormone leptin functions as a long-term adiposity signal to regulate food intake and energy balance [195]. Circulating leptin levels are proportional to the amount of body adipose stores [196] and leptin crosses the blood brain barrier through a receptor-dependent transport mechanism [197, 198] to act centrally at the long-form leptin receptor (LepR) [33, 199]. An abundance of evidence discussed below supports that VTA leptin signaling is physiologically relevant in the control of motivated behaviors, but no study to date has explicitly detected peripheral leptin in the VTA. Through this section, phosphorylation of the transcription factor STAT3, an intracellular pathway classically engaged by leptin signaling, is often used to assess leptin signaling activity.
Mechanism
Peripheral administration of leptin decreased the firing frequency of VTA dopamine neurons in vivo by 40% and bath application of leptin in a slice preparation decreased the action potential frequency of VTA dopamine neurons by 20% [200]. In another slice preparation study, leptin decreased the firing frequency in 71% of dopamine neurons [201]. Firing of VTA dopamine neurons that project to the NAc is inhibited by D2R auto-activation [202, 203]. In 41% of leptin-responsive VTA dopamine neurons, leptin attenuated the D2R agonist-induced decrease in firing rates [201]. This finding suggests that VTA leptin signaling primarily decreases dopaminergic activity but also has a mechanism to indirectly increase dopamine neuron excitability by inhibiting D2R auto-inhibition. One study reported that leptin did not alter firing activity of VTA dopamine or GABA neurons, but the authors of this study only recorded from 5–7 neurons and theoretically could have selected leptin-unresponsive neurons [204].
Evidence also supports that leptin dampens VTA dopamine activity in response to reward cues. Peripheral leptin attenuated the VTA dopamine spike induced by presentation of a food reward cue under food deprived conditions [95]. Whether VTA leptin modulates dopamine neuron activity in response to reward retrieval in addition to the reward cue has not been investigated. LepR knockout from dopamine neurons increased baseline VTA dopamine burst firing without affecting the average firing rate [205], suggesting that even in the absence of a reward leptin action in the VTA regulates basal dopaminergic tone. Leptin regulation of VTA dopamine nerve activity translates into altered NAc dopamine release. Intra-VTA leptin attenuated cocaine-stimulated NAc dopamine levels [206] and, in turn, the antagonism or knockout of VTA LepRs enhanced cocaine-induced NAc dopamine levels [207].
While 66.7% of VTA LepRs are expressed on dopaminergic neurons, they do not project directly to the NAc or mPFC [190]. In fact, only 5.3% of VTA-to-NAc and essentially no VTA-to-mPFC projecting neurons express the LepR, suggesting that VTA leptin signaling indirectly decreases dopamine output to these nuclei [190, 208]. Leptin increased the frequency without changing the amplitude of mIPSCs in VTA-to-NAc dopamine neurons, showing a presynaptic increase in inhibitory inputs to this pathway. Leptin depolarized and increased the frequency of spontaneous action potentials in VTA GABAergic interneurons that innervate VTA-to-NAc dopamine neurons [190], verifying that VTA leptin signaling increases local inhibition of dopaminergic transmission to the NAc (Figure 2; L1).
The lateral hypothalamus (LH) is another prominent source of GABAergic input to the VTA and a subset of this population expresses the LepR (LHLepRb) [209, 210]. Surprisingly, LHLepRb-to-VTA neurons made monosynaptic connections with VTA GABA but not VTA dopamine neurons and leptin decreased spontaneous action potentials in these neurons [190] (Figure 2; L2). These findings suggest that LHLepRb-to-VTA neurons exert a tonic inhibition of VTA GABA interneurons that is suppressed by leptin, resulting in further inhibition of dopaminergic VTA-to-NAc projections. Substantiating this idea, photostimulation of LHLepRb-to-VTA projections increased neuronal activity in VTA dopamine neurons and increased motivation for both sucrose and chow pellets in a PR task in ad libitum fed but not fasted mice, while inhibition of this pathway reduced PR task performance [190, 210]. Activation of this pathway was also found to abolish discrimination of food reward-associated cues during training and extinction phases, whereas leptin-mediated inhibition of LHLepRb-to-VTA neurons increases cue discrimination [211]. Thus, this pathway specifically regulates acquisition of learned appetitive behavior likely through modulating VTA dopamine error signals.
The LH is not the only hypothalamic nucleus connected with VTA leptin signaling. Short-term overexpression of exogenous leptin in the VTA in chow rats increased pSTAT3 not only in the VTA but also in the LH, ARC, DMH, and VMH [212]. On the other hand, overexpression of a leptin antagonist in the VTA prevented leptin-induced pSTAT3 in the MBH and blockade of MBH leptin signaling weakened the hypophagia caused by VTA leptin overexpression [213]. These results suggest that VTA leptin action trans-activates the hypothalamus and the full-strength response to VTA leptin signaling requires leptin action in the MBH.
Leptin also affects VTA dopaminergic activity through regulating VTA TH expression. In lean rats, ICV leptin decreased VTA TH mRNA, in line with the action of leptin to downregulate VTA dopaminergic transmission [214]. However, high-fat/high-sugar feeding downregulates VTA TH mRNA and in rats on this obesogenic diet, ICV leptin increased VTA TH mRNA [214]. Ob/ob mice deficient in leptin have reduced VTA TH mRNA and protein compared to wildtype mice and VTA TH protein levels were restored by 3-day subcutaneous leptin infusions but a single intra-VTA leptin injection did not elevate VTA TH mRNA [209, 215]. The opposing results in lean versus models of obesity indicate a state-dependent regulation of leptin on VTA TH expression that may involve an interaction between leptin and neurotensin-1, which is released from a subset of LH-to-VTA projecting neurons and also regulates VTA TH expression [216]. While diet-induced obese and ob/ob mouse models are both obese, diet-induced obesity confers a degree of insulin and leptin resistance while ob/ob mice are in fact highly sensitive to exogenous leptin administration. These physiological differences, along with differences in the site of leptin administration between these models, could contribute to the different timescale in the aforementioned observed responses.
Interestingly, injection of leptin in the LH increased VTA TH mRNA in ob/ob mice [209], suggesting that leptin signaling in LHLepRb-to-VTA neurons, and not leptin action in the VTA itself, regulates VTA TH expression (Figure 2; L3). Changes in VTA TH expression following peripheral and ICV, but not intra-VTA leptin administration, support the hypothesis that the LH is the site of action for leptin regulation of VTA TH expression. Although a decrease in VTA TH protein expression was observed following overexpression of leptin in the VTA, this manipulation trans-activates the hypothalamus, and thus could result from VTA-LH-VTA signaling [212]. Accompanying the increase in VTA TH mRNA, LH leptin injection also increased NAc dopamine content [209], confirming that increases in VTA TH expression translate into higher levels of dopamine output. Further supporting this notion, ob/ob mice with diminished VTA TH expression show a blunted release of dopamine in the NAc in response to electrical stimulation of VTA afferents that is due to lower presynaptic dopamine levels and not increased dopamine reuptake [215].
While no LepR expressing VTA dopamine neurons project to the NAc or mPFC, they do robustly innervate the CeA [190, 208] (Figure 2; L4). In ob/ob mice, acute peripheral leptin induces phosphorylation of the cAMP dependent response element (CREB) in the VTA and CeA but not NAc [208]. Within the CeA, cocaine- and amphetamine-regulated transcript (CART) neurons are one population targeted by VTA LepR expressing neurons, and the leptin administration downregulates the elevated CeA CART expression in ob/ob mice [208]. The CeA regulates anxiety [217] and evidence supports that VTA leptin signaling on dopamine neurons innervating the CeA mitigate anxiety. LepRs knockout selective in dopamine neurons increased basal VTA dopaminergic tone and produced an anxiogenic phenotype which is attenuated by CeA dopamine 1 receptor (D1R) antagonism [205]. Furthermore, intra-VTA and systemic leptin decreased anxiety-like behaviors, and this was attenuated by VTA JAK2/STAT3 inhibition [218]. VTA LepR knockout eliminated leptin-induced pSTAT3 and promoted anxiogenic behaviors [218], suggesting that VTA pSTAT3 is involved in mediating leptins regulation of anxiety (Figure 2; L4). However, other evidence suggests this effect may be more prominently driven by CeA than VTA leptin action. LepR knockdown in the CeA but not VTA increased anxiety-like behaviors [207], and chemogenetic activation of VTA LepR expressing neurons was not anxiogenic [219]. Finally, STAT3 knockout from dopamine neurons did not alter baseline anxiety-like behaviors, but this was not investigated in the context of a leptin challenge [220].
The LepR is a tyrosine kinase receptor that upon ligand binding phosphorylates JAK2 to initiate multiple internal signal transduction pathways, the classic marker of LepR activation being phosphorylation of the transcription factor STAT3 [221]. Intra-VTA and peripheral leptin injection and overexpression of leptin in the VTA induces VTA pSTAT3 in dopaminergic and GABAergic neurons [200, 212, 215, 222]. More specifically, VTA leptin phosphorylates STAT3-Tyr705 and ERK1/2-Thr202/Tyr204 but does not induce phosphorylation of ATK-Ser473/Thr308 or S6K1-Thr389 [222, 223]. Investigating the mechanism by which leptin inhibits dopamine transmission, one study found that leptin depressed glutamatergic AMPA and NMDA mediated EPSCs in VTA dopamine neurons and decreased the frequency but not amplitude of mEPSCs [224] (Figure 2. L5). Application of a PI3K or JAK2, but not MEK1/2 inhibitor disrupted the leptin-dependent reduction in AMPA-mediated EPSCs [224], demonstrating that JAK2 and PI3K are necessary for leptin-induced synaptic depression of glutamatergic inputs to VTA dopamine neurons. This presynaptic reduction in EPSCs represents a separate mechanism from the increase in IPSCs by which leptin inhibits VTA dopamine transmission.
Others have investigated the role of leptin-recruited signaling molecules in mediating leptins reductions in food intake and body weight. VTA inhibition of PI3K or mTOR did not affect the anorectic response to intra-VTA leptin, yet inhibiting the first step in the leptin signaling cascade with a subthreshold JAK2 inhibitor did block the negative energy balance effects of intra-VTA leptin [223]. As expected, central MEK1/2 inhibition prevented the intra-VTA leptin-induced downstream phosphorylation of VTA ERK1/2 but did not disrupt leptin-dependent pSTAT3 recruitment [222]. Despite leaving STAT3 phosphorylation intact, central MEK1/2 inhibition prevented the leptin-induced decrease in VTA dopamine neuron firing frequency (Figure 2; L1) and intra-VTA leptin-induced hypophagia [222], suggesting that pSTAT3 does not mediate the anorectic effect of VTA leptin signaling. In fact, the finding that STAT3 knockout in dopamine neurons did not alter the anorectic response to ICV or VTA leptin injection or ad libitum HFD intake or compulsive sucrose consumption supports that VTA STAT3 does not regulate leptin-induced hypophagia [220]. However, dopamine neuron STAT3 knockout decreased operant responding for sucrose pellets, amphetamine-stimulated locomotion, and NAc dopamine release [220], underscoring the importance of STAT3 in mesolimbic regulated motivation for rewards.
Overall, while VTA LepRs do reduce ad libitum food intake through a MEK1/2 dependent mechanism, leptin more potently decrease the motivation for than consumption of a reward (sucrose, cocaine). Leptin signaling in the VTA inhibits dopamine neuron firing through multiple mechanisms including increasing local and long-rang inhibitory GABA input, decreasing excitatory glutamatergic input through a PI3K dependent mechanism, and downregulating dopamine neuron TH expression through a LH-to-VTA signaling mechanism. At present understanding, VTA leptin-stimulated pSTAT3 regulates the motivation aspect of reward seeking and likely the anxiolytic and physical activity reducing properties of leptin. As recent work has begun to highlight the importance of leptin signaling in VTA GABA neurons, elucidating the mechanisms underlying GABA mediated inhibition of VTA dopamine output will be essential to understanding leptin regulation of motivated behaviors.
Behavior
Food
As expected, the leptin-induced reduction in VTA dopamine output corresponds with diminished motivation for a variety of rewarding stimuli. Delivery of leptin in the VTA dose-dependently decreased 24h intake of chow in rats [200], and chemogenetic activation of VTA LepR expressing neurons decreased chow intake only in food restricted mice [219]. Intra-VTA leptin also decreased food intake and body weight equally in rats maintained on a high-fat or low-fat/high-carbohydrate diet for 16 weeks [225]. However, the heaviest quartile of HFD fed rats had a reduced anorectic response to leptin, indicating the development of VTA leptin resistance in obesity. Diet-induced central leptin resistance contributes to the hyperphagia associated with obesity and is thought to be attributed to chronically elevated leptin levels [226]. In fact, central overexpression of exogenous leptin in lean rats induced leptin resistance in the VTA, assessed by a normalization of the initial reduction in chow intake within 37 days and body weight after 280 days and reduced VTA STAT3 phosphorylation (pSTAT3) in response to exogenous ICV leptin, that resembled the HFD-induced decrease in VTA leptin responsiveness [227]. Exogenous leptin overexpression specifically in the VTA at the onset of HFD feeding initially decreased food intake but this normalized within 14 days, while body weight remained lower for 50 days before returning to the level of controls [212]. Chronic VTA leptin overexpression also greatly dampened the VTA pSTAT3 response to ICV leptin injection [212].
Conversely, overexpression of a dominant negative leptin mutant in the VTA, effectively antagonizing LepRs, increased ad libitum chow intake and body weight over 20 days [213]. Knockdown of LepRs in the VTA similarly increased daily chow intake and dark cycle locomotion without inducing weight gain [200]. These studies demonstrate that chronic increases in VTA leptin signaling initially create a negative energy balance before leading to the development of leptin resistance, while a sustained absence of LepR signaling in the VTA promotes hyperphagia. Thus, endogenous leptin action in the VTA serves to regulate normal energy homeostasis. Interestingly, peripheral leptin pretreatment did not attenuate intra-VTA ghrelin-induced hyperphagia [85].
VTA leptin signaling also modulates motivated behaviors around a food reward. In fact, while higher doses of intra-VTA leptin were necessary to suppress chow intake, a low dose was required to decrease brain reward function [228]. Chemogenetic activation of VTA LepR expressing neurons reduced the number of active lever presses during a PR test for a sucrose solution reward in food restricted but not ad libitum fed mice [190, 219]. The absence of a leptin-suppressed motivation for food reward in satiated mice suggests that higher leptin levels in the fed state of healthy animals may already maximally engage VTA leptin signaling. Interestingly, the number of licks for a freely available 20% sucrose solution was not affected by stimulation of VTA LepR expressing neurons under either sated or food deprived conditions [219], indicating that at least for sucrose, VTA leptin signaling reduces the motivation to work for a food reward but not affect consumption of the rewarding food [190, 219]. However, chronic knockdown of VTA LepRs increases the sensitivity to palatable foods as a 2-bottle access test between water and a low sucrose concentration solution (0.2%) that was not favored by controls was preferred 2:1 in LepR-knockdown rats [200]. Additionally, VTA LepR knockdown increased the hyperphagia observed during the first 3 days of HFD exposure [200]. These results conclude that VTA leptin signaling attenuates the motivation to work for a food reward in a nutrition-state dependent manner and may tamper the rewarding properties of taste-subthreshold or novel palatable foods.
Drugs of Abuse
Beyond ingestion of food rewards, VTA leptin action mitigates the rewarding properties of drugs of abuse. VTA administration of leptin decreased cocaine-induced conditioned place preference and sensitivity to cocaine-predictive cues during extinction testing yet did not reduce self-administration of cocaine [206]. Thus, like with sucrose, VTA leptin action may downregulate some rewarding property of cocaine but does not reduce cocaine taking. Reciprocally, knockdown of VTA LepRs increased cocaine-dependent place preference [207]. Interestingly, previous exposure to a saccharin reward increased VTA LepR mRNA expression and decreased cocaine-dependent conditioned place preference [207]. More evidence that VTA leptin signaling regulates reward salience from drugs of abuse comes from findings that intra-VTA administration of leptin dose-dependently decreases active lever presses for heroin infusion in food deprived but not sated rats [114], and ob/ob mice deficient in leptin show a diminished locomotor response to amphetamines which is reversed following leptin infusion [215]. Surprisingly, no work has investigated VTA leptin signaling in regulating alcohol consumption, but preclinical and clinical work suggests a relationship between leptin and alcohol addiction [229].
Activity and Arousal
Running wheels can be used to assess physical activity in rodents, yet rodents often voluntarily engage in wheel running to excess and this activity is classified as an addictive-like behavior [230]. In fact, rats with ad libitum choice access to chow and HFD and a running wheel don’t develop a preference for HFD and as little as 2 days of wheel running increases the maximal pSTAT3 response to centrally administered leptin selectively in the VTA [231]. In aged rats maintained on a HFD, wheel running elevated basal and leptin-stimulated pSTAT3 only in the VTA and reduced HFD intake and body weight [232]. These studies provide evidence that wheel running may substitute for the reward of HFD and through heightened VTA sensitivity to leptin, blunt the drive to consume palatable foods. Interestingly, mice lacking STAT3 in dopamine neurons exhibited increased voluntary wheel running and heightened rewarding effect of running as exhibited in a conditioned place preference task, which is abolished by VTA leptin injection [220]. The finding that VTA leptin decreased the reward associated with running in dopamine neuron STAT3 knockout mice implies that while STAT3 regulates running reward it is either not necessary for leptins effects on physical activity or leptin action is mediated by non-dopamine neurons [220]. Intra-VTA leptin also suppresses wheel running activity in a rat model of anorexia and human patients with anorexia nervosa treated with exogenous leptin show reductions in hyperactivity [233, 234]. Surprisingly, chemogenetic activation of LepR expressing VTA neurons and acute intra-VTA leptin injection did not change locomotor activity, yet chronic knockdown of VTA LepRs increased dark cycle locomotion [190, 200, 218, 219]
Insulin
Insulin secretion from pancreatic beta cells is stimulated by hyperglycemia and insulin is best characterized for its role in glucoregulation through peripheral and central mechanisms, but it also may act centrally as an anorectic signal [30, 235]. Insulin is transported across the blood brain barrier through an insulin receptor (InsR) independent saturable mechanism [236, 237]. However, the kinetics of postprandial insulin transport to the VTA have not been explored. Nonetheless, the InsR is expressed on dopamine neurons in the VTA [238] and insulin signaling in the VTA has been postulated to be part of its coordinated effect to communicate peripheral energy status and decrease the saliency of energy-rich palatable foods and other rewarding stimuli. The VTA rapidly develops insulin resistance in response to HFD feeding [239] and is a confounding variable to interpret the effects of VTA insulin signaling in diabetic and obese states.
Mechanism
Insulin blunts the drive for rewarding substances by reducing VTA dopaminergic transmission. Intra-VTA insulin decreased release of dopamine in the NAc in response to cocaine and electrical stimulation of dopamine neuron burst firing [240]. VTA dopamine neurons can release dopamine not only at projection sites but locally within the VTA from the soma and dendrites or from its own axon collaterals [241]. This somatodendritic release of dopamine can regulate the activity of dopamine projections by both acting at autoinhibitory D2Rs and presynaptic D1Rs on GABAergic and glutamatergic inputs to the VTA to modify release of those neurotransmitters [242]. Thus, the ultimate effect of local VTA dopamine signaling on dopamine output and behavior is hard to anticipate, and the role of satiation hormones on this process virtually unexplored. One study investigating how insulin regulates somatodendritic dopamine release found that insulin dose dependently attenuated electrically evoked dopamine concentrations within the VTA which was blocked by pretreatment with an InsR antagonist, and PI3K and mTOR inhibitors [243], indicating the suppressive effect of insulin on VTA dopamine levels requires PI3K and mTOR activation downstream of the InsR. These effects were independent of protein synthesis and were not due to alterations in dopamine release as insulin still suppressed VTA dopamine concentrations when dopamine release was reduced in the presence of low calcium or elevated by increasing the voltage of electrical stimulation [243]. However, the insulin-mediated reduction in VTA dopamine levels was absent in the presence of a dopamine transporter (DAT) inhibitor or in DAT knockout mice [243], concluding that insulin decreases the level of dopamine in the VTA by stimulating DAT-mediated dopamine reuptake (Figure 2; I1).
Indeed, bath application of insulin to NAc slices enhanced DAT-mediated dopamine clearance and this effect was blocked by InsR antagonism [244]. However, diet-induced obese mice had diminished basal rates of dopamine reuptake and it was not enhanced by insulin [244]. In fact, lower dopamine reuptake in the NAc correlated with worsened peripheral glucose tolerance, suggesting the development of peripheral and central insulin resistance, and application of an insulin sensitizing molecule restored the ability of insulin to increase NAc dopamine reuptake [244]. NAc InsR expression was not different between lean and obese mice [244], pointing to diet-induced differences in InsR function and regulation of DAT activity. Insulin increases translocation of central DAT to the plasma membrane [245] and within the VTA hypoinsulinemic states like fasting and type 1 diabetes decreased VTA DAT mRNA expression and activity, while hyperinsulinemic states and chronic ICV insulin infusion increased VTA DAT mRNA expression [246–249] (Figure 2; I2). Thus, insulin acutely decreases dopamine signaling at VTA projection targets by increasing synaptic dopamine clearance and this is disrupted with HFD feeding.
Insulin also reduces VTA dopamine output by causing long term depression of excitatory inputs onto VTA dopamine neurons. AMPA and NMDA mediated EPSCs were decreased by insulin and this effect blocked by InsR antagonist pretreatment but once initiated were not reversed by InsR antagonism [250]. Insulin did not alter GABA-induced IPSCs in VTA dopamine neurons [250]. Insulin induced long term depression of VTA dopamine neurons required Akt and mTOR but not PKA and was not mediated by postsynaptic internalization of AMPA receptors [250]. This suggested insulin may be acting through a presynaptic mechanism and insulin decreased the paired pulse ratio of stimulated EPSCs [250], indicating a decrease in the probability of neurotransmitter release onto VTA dopamine neurons.
Endocannabinoids act in a retrograde fashion to modulate presynaptic signaling [251], and blockade of CB1R in the VTA prevented insulin’s ability to decrease the frequency of incoming mEPSCs [250] (Figure 2; I3). Endocannabinoid synthesis and presynaptic CB1R signaling were required for the initiation but not maintenance of insulin-induced long-term depression, and agonizing CB1R decreased AMPA EPSCs to a similar degree as insulin, suggesting a common mechanism [250]. Stimulating endogenous insulin release with a sweetened high fat meal immediately prior to recordings eliminated the effect of applied insulin to decrease AMPA-mediated EPSCs and the frequency of mEPSCs, but these responses were restored 1h after meal termination [250], supporting that endogenous postprandial insulin signaling is sufficient to decrease glutamatergic input to VTA dopamine neurons.
Short term (24h) exposure to HFD increased the frequency of mEPSCs onto VTA dopamine neurons 2 days later but were still sensitive to inhibition by insulin [14]. Immediately but not 2 days post high fat diet exposure, the paired pulse ratio in dopamine neurons was elevated [14], suggesting an increase in presynaptic neurotransmitter release probability. Antagonizing CB1Rs decreased the paired pulse ratio, increased AMPA evoked EPSCs, and increased spontaneous mEPSCs [14], suggesting that acute sweetened high fat diet exposure transiently increased presynaptic endocannabinoid tone as a compensatory mechanism to reduce the release probability of glutamate onto VTA dopamine neurons. This increase in glutamatergic input was reflected by an increase in the number of excitatory synapses onto VTA dopamine neurons, while excitatory synapses onto non-dopamine neurons or inhibitory synapses onto either were not changed [14]. However, this VTA adaptation to offset increased dopaminergic excitation after short term HFD exposure is not sustained under chronic conditions. In a hyperinsulinemic mouse model, insulin-induced synaptic depression of VTA dopamine neurons was also impaired, suggesting that chronic pathological hyperinsulinemia disrupts normal insulin action in the VTA [252].
Surprisingly, it has also been reported that insulin increased the spike frequency in 50% of VTA dopamine neurons [253]. This effect still occurred under conditions of blocked synaptic inputs [253], implying this effect was mediated by InsR signaling on dopamine neurons. Knockout of the InsR from dopamine neurons did not affect basal spike frequency but prevented the insulin-induced increase in firing [253]. However, the frequency of glutamatergic EPSCs was diminished in dopamine neurons lacking the InsR, while IPSC frequency was unaltered [253]. Why these results differ from the findings described above is, however, uncertain.
Intranasal insulin administration enables effective delivery to the brain with minimal systemic spillover [254] and labeling of fluorescently tagged insulin delivered intranasally was detected in the VTA in rats [255]. Intranasal insulin also reduced dopaminergic activity and strengthened functional connectivity within dopaminergic networks in humans [256]. During a glucose challenge, insulin sensitive but not insulin resistant patients showed reduced VTA activation in response to high calorie food images, suggesting that insulin resistance disrupts the normal inhibition of reward responding to food cues in the sated state [257, 258]. Intranasal insulin in overnight fasted insulin sensitive, but not insulin resistant patients reduced ratings of food picture palatability and this correlated with diminished activity of VTA-to-NAc projections [259], and in overweight nondiabetic individuals the success of dietary intervention could be predicted from baseline responsiveness to intranasal insulin-induced inhibition of VTA-to-NAc activity [260]. Thus, VTA insulin signaling is critical in mitigating cravings for palatable foods and highlights the need to address reward dysfunction in individuals at risk for metabolic disorders. Mechanistic work supports that the development of insulin resistance is an important factor in diet-induced disruptions to normal VTA regulation of reward-directed behaviors.
Behaviors
Food
In mice trained to consume chow during a 4h restricted period each day, intra-VTA insulin did not alter food consumption during this time [243]. However, intake of HFD presented following the normal feeding window was robustly suppressed by intra-VTA insulin delivery [243], suggesting that VTA insulin action inhibits palatable food intake in sated mice and is particularly relevant to decrease intake of palatable foods in excess to the body’s nutritional need. In line with these results, VTA injection of insulin only mildly suppressed food intake in chow fed rats when given at supraphysiologic, but not physiologic concentrations [228]. However, intra-VTA insulin given at a low dose decreased the sensitivity to rewarding intracranial electrical self-stimulation, indicating a decrease in reward function [228].
Knockout of the InsR from dopamine neurons increased daily chow intake and adiposity in males and females and increased the preference for low concentrations of a sucrose (1–2%) solution in a 2-bottle choice test [253], indicating that endogenous insulin signaling on dopamine neurons suppresses the sensitivity to the rewarding properties of sucrose. Interestingly, VTA insulin administration dose dependently decreased conditioned place preference for a food reward (Froot loops) but did not reduce PR performance for either sucrose or the more powerful reinforcer, sweetened condensed milk [250]. Another study found that while intra-VTA insulin blocked opioid-induced sucrose intake, it did not reduce operant responding for sucrose self-administration [261]. These findings indicate that VTA insulin action reduces the salience of food reward associated cues, but not the motivation to obtain a palatable reinforcer. Many studies did not examine the effect of intra-VTA insulin action on glycemia which itself regulates appetite and should be considered as a potential secondary mediator of insulin’s anorectic effects.
Drugs of Abuse
A few studies have investigated VTA insulin signaling in the rewarding properties of addictive substances. Insulin delivery in the VTA blocked, while intranasal insulin attenuated, cocaine-stimulated locomotor activity [240, 253]. These effects were reversed by intra-VTA delivery of an InsR antagonist [240].
Surprisingly, the InsR substrate 2 (IRS2), a protein phosphorylated downstream of InsR activation and other neurotrophic factors, seems to enhance the rewarding properties of drugs of abuse. Overexpression of IRS2 in the VTA increased conditioned place preference to morphine and cocaine and drug-stimulated locomotion, while VTA expression of a dominant negative IRS2 caused conditioned place avoidance to morphine and cocaine and decreased drug-induced locomotor activity [262, 263]. IRS2 phosphorylates Akt downstream and VTA expression of a dominant negative Akt similarly reduced conditioned place preference to morphine [263]. Chronic morphine reduced place preference for morphine, inhibited pAkt levels, and decreased VTA dopamine neuron cell body size [263]. These results are opposite what would be expected for stimulation of IRS2-Akt downstream of InsRs signaling. However, the manipulations in these studies may not mimic the effects of insulin and VTA insulin signaling in these models was not investigated. Nonetheless, these studies support that changes in the IRS2-Akt pathway are important in long term actions of drugs of abuse.
Activity and Arousal
Intra-VTA insulin decreased food anticipatory behaviors including food-seeking locomotion, rearing, and digging for both chow and HFD [14, 250], while in mice sated on HFD, VTA injection of an InsR antagonist increased food anticipatory behaviors to HFD. Importantly, intra-VTA insulin did not alter basal locomotion [250, 253]. Thus, VTA insulin signaling may reduce motivation to seek a food-reward. Overall, more work is needed to clarify the role of VTA insulin in regulating food intake and drug use.
Remaining Questions
The neuroscience, obesity, addiction, and feeding fields have contributed extensively to our knowledge of how metabolic hormones influence the behavioral responses to rewarding stimuli. The most is currently understood about mechanisms mediating ghrelin and leptin action in the VTA and the least about amylin and GLP-1. Information as fundamental as which neuron phenotypes in which input-output VTA circuits express the receptors for these hormone systems is largely unknown. The VTA is an incredibly heterogeneous nucleus made up of multiple sub-nuclei that are differentially composed of dopaminergic, GABAergic, glutamatergic neurons, as well as neurons that co-release glutamate or GABA with dopamine and glutamate/GABA co-releasing neurons [9, 147]. Beyond this, 16 distinct cell type clusters have been genetically identified for the rat VTA [264], providing a new level of data resolution. What contribution these different pathways have on specific aspects of motivated behaviors and how these are affected by ingestive processes is just beginning to be unraveled [265]. The majority of the studies discussed in this review either assessed generalized VTA dopamine signaling, or if they did investigate a projection target it was almost always the NAc, in which the VTA or NAc subregions involved were not known. Dopamine release in different areas of the NAc control specific aspects of motivated behavior [266], the nuanced nature of which has not been appreciated until recently. A better understanding of the organization of VTA connections with technological advancements will undoubtedly help explain inconsistencies in the existing literature and greatly expand our appreciation for hormonal regulation of VTA-influenced behaviors.
Conclusion
Ghrelin, GLP-1, amylin, leptin, and insulin signaling in the VTA regulate a wide range of motivated behaviors. Ghrelin action on its own is rewarding and potentiates the motivation for and reinforcement of addictive substances through stimulating VTA dopaminergic activity. In opposition, the satiation hormones act through discrete and overlapping mechanisms to decrease VTA dopamine output, reduce the rewarding value of stimuli, and decrease behaviors directed to obtain these rewards. Given that the GHSR forms complexes with both D1 and D2 receptors and dopamine signaling modulates receptor activity [267, 268], the action of satiation hormones to diminish VTA dopamine levels could reduce activity at the GHSR complex [163]. Thus, GLP-1, amylin, leptin, and insulin action diminish the positive valance of a reward by directly decreasing VTA dopamine levels which in turn may help terminate signaling that encourages reward seeking behavior.
In healthy individuals, the balance between these orexigenic and anorexigenic systems helps increase the incentive salience of food reward under energy deficit conditions and dampens this desire once energy levels are replenished [269]. However, the palatability of the modern food environment evokes reward signaling despite sufficient energy storage, contribute to hyperphagia and obesity [270, 271]. In turn, chronic consumption of palatable foods dysregulates VTA function and reward perception, compounding the difficulty to limit consumption of high-energy palatable foods. As little as 3 days of HFD feeding increases VTA pro-inflammatory markers and weeks of HFD intake cause VTA insulin resistance, oxidative stress, inflammation, and microglia infiltration [239, 272, 273]. While transient exposure to palatable foods increases VTA TH expression and dopamine signaling [14, 274], as little as 2 weeks to 25 weeks of HFD feeding or dysregulated eating in a model of binge eating disorder decreases VTA TH expression, midbrain dopamine network activity, and VTA-to-NAc dopamine turnover [272, 275–279]. This depression of baseline dopamine reward circuits could result in chronic feelings of being under-stimulated. Moreover, HFD feeding decreases the ability of non-palatable foods to alleviate the negative valance of hunger and blunts the rewarding properties of amphetamine and sucrose [277, 280], suggesting that the value of any rewarding stimuli is diminished in obesity. In turn, this may cause individuals to consume higher levels of rewarding substances in attempts to feel pleasure. This highlights an interaction between obesity and addictive disorders and necessitates an even greater urgency to understand the molecular basis of reward and how therapeutic interventions targeting endogenous hormone systems can be leveraged to treat reward dysfunction.
Highlights.
The VTA is a key modulator of reward-directed behaviors.
Ghrelin induces motivation and amplifies the drive for rewarding stimuli.
GLP-1, amylin, leptin, and insulin dampen food and drug-induced reward.
These hormones affect VTA activity through converging and divergent mechanisms.
VTA reward processing is a target to treat diseases of overconsumption.
Financial Support and Conflicts of Interest
This work was supported by NIH-DK105155 (M.R.H) and NIH-DK127591 (C.E.G). M.R.H. receives research funding from Boehringer Ingelheim and Eli Lilly & Co. that was not used in support of these studies. M.R.H. is a chief executive officer of Cantius Therapeutics, LLC, that pursues biological work unrelated to the current study. No other potential conflicts of interest relevant to this article were reported.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Berridge KC, et al. , The tempted brain eats: pleasure and desire circuits in obesity and eating disorders. Brain Res, 2010. 1350: p. 43–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morales I and Berridge KC, ‘Liking’ and ‘wanting’ in eating and food reward: Brain mechanisms and clinical implications. Physiol Behav, 2020. 227: p. 113152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gardner EL, Addiction and brain reward and antireward pathways. Adv Psychosom Med, 2011. 30: p. 22–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leigh SJ and Morris MJ, The role of reward circuitry and food addiction in the obesity epidemic: An update. Biol Psychol, 2018. 131: p. 31–42. [DOI] [PubMed] [Google Scholar]
- 5.Winder DG, et al. , Synaptic plasticity in drug reward circuitry. Curr Mol Med, 2002. 2(7): p. 667–76. [DOI] [PubMed] [Google Scholar]
- 6.Cooper S, Robison AJ, and Mazei-Robison MS, Reward Circuitry in Addiction. Neurotherapeutics, 2017. 14(3): p. 687–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fields HL, et al. , Ventral tegmental area neurons in learned appetitive behavior and positive reinforcement. Annu Rev Neurosci, 2007. 30: p. 289–316. [DOI] [PubMed] [Google Scholar]
- 8.Flores-Dourojeanni JP, et al. , Temporally Specific Roles of Ventral Tegmental Area Projections to the Nucleus Accumbens and Prefrontal Cortex in Attention and Impulse Control. J Neurosci, 2021. 41(19): p. 4293–4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morales M and Margolis EB, Ventral tegmental area: cellular heterogeneity, connectivity and behaviour. Nat Rev Neurosci, 2017. 18(2): p. 73–85. [DOI] [PubMed] [Google Scholar]
- 10.Beier KT, et al. , Topological Organization of Ventral Tegmental Area Connectivity Revealed by Viral-Genetic Dissection of Input-Output Relations. Cell Rep, 2019. 26(1): p. 159–167 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Breton JM, et al. , Relative contributions and mapping of ventral tegmental area dopamine and GABA neurons by projection target in the rat. J Comp Neurol, 2019. 527(5): p. 916–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Derdeyn P, et al. , Uncovering the Connectivity Logic of the Ventral Tegmental Area. Front Neural Circuits, 2021. 15: p. 799688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dela Cruz JA, Coke T, and Bodnar RJ, Simultaneous Detection of c-Fos Activation from Mesolimbic and Mesocortical Dopamine Reward Sites Following Naive Sugar and Fat Ingestion in Rats. J Vis Exp, 2016(114). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu S, et al. , Consumption of palatable food primes food approach behavior by rapidly increasing synaptic density in the VTA. Proc Natl Acad Sci U S A, 2016. 113(9): p. 2520–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pascoli V, et al. , Sufficiency of Mesolimbic Dopamine Neuron Stimulation for the Progression to Addiction. Neuron, 2015. 88(5): p. 1054–1066. [DOI] [PubMed] [Google Scholar]
- 16.Di Chiara G and Imperato A, Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci U S A, 1988. 85(14): p. 5274–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berke JD, What does dopamine mean? Nat Neurosci, 2018. 21(6): p. 787–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schultz W and Romo R, Dopamine neurons of the monkey midbrain: contingencies of responses to stimuli eliciting immediate behavioral reactions. J Neurophysiol, 1990. 63(3): p. 607–24. [DOI] [PubMed] [Google Scholar]
- 19.Schultz W, Apicella P, and Ljungberg T, Responses of monkey dopamine neurons to reward and conditioned stimuli during successive steps of learning a delayed response task. J Neurosci, 1993. 13(3): p. 900–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eshel N, et al. , Dopamine neurons share common response function for reward prediction error. Nat Neurosci, 2016. 19(3): p. 479–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Syed EC, et al. , Action initiation shapes mesolimbic dopamine encoding of future rewards. Nat Neurosci, 2016. 19(1): p. 34–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hamid AA, et al. , Mesolimbic dopamine signals the value of work. Nat Neurosci, 2016. 19(1): p. 117–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kutlu MG, et al. , Dopamine release in the nucleus accumbens core signals perceived saliency. Curr Biol, 2021. 31(21): p. 4748–4761 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Coddington LT, Lindo SE, and Dudman JT, Mesolimbic dopamine adapts the rate of learning from action. Nature, 2023. 614(7947): p. 294–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goedhoop JN, et al. , Nucleus accumbens dopamine tracks aversive stimulus duration and prediction but not value or prediction error. Elife, 2022. 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jones JL, et al. , Basolateral amygdala modulates terminal dopamine release in the nucleus accumbens and conditioned responding. Biol Psychiatry, 2010. 67(8): p. 737–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cachope R, et al. , Selective activation of cholinergic interneurons enhances accumbal phasic dopamine release: setting the tone for reward processing. Cell Rep, 2012. 2(1): p. 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Narayanan NS, Guarnieri DJ, and DiLeone RJ, Metabolic hormones, dopamine circuits, and feeding. Front Neuroendocrinol, 2010. 31(1): p. 104–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kanoski SE, Hayes MR, and Skibicka KP, GLP-1 and weight loss: unraveling the diverse neural circuitry. Am J Physiol Regul Integr Comp Physiol, 2016. 310(10): p. R885–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu S and Borgland SL, Insulin actions in the mesolimbic dopamine system. Exp Neurol, 2019. 320: p. 113006. [DOI] [PubMed] [Google Scholar]
- 31.Mietlicki-Baase EG and Hayes MR, Amylin activates distributed CNS nuclei to control energy balance. Physiol Behav, 2014. 136: p. 39–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Revitsky AR and Klein LC, Role of ghrelin in drug abuse and reward-relevant behaviors: a burgeoning field and gaps in the literature. Curr Drug Abuse Rev, 2013. 6(3): p. 231–44. [DOI] [PubMed] [Google Scholar]
- 33.Xu L, Leptin action in the midbrain: From reward to stress. J Chem Neuroanat, 2014. 61–62: p. 256–65. [DOI] [PubMed] [Google Scholar]
- 34.Liu S and Borgland SL, Regulation of the mesolimbic dopamine circuit by feeding peptides. Neuroscience, 2015. 289: p. 19–42. [DOI] [PubMed] [Google Scholar]
- 35.Yanagi S, et al. , The Homeostatic Force of Ghrelin. Cell Metab, 2018. 27(4): p. 786–804. [DOI] [PubMed] [Google Scholar]
- 36.Zigman JM, et al. , Expression of ghrelin receptor mRNA in the rat and the mouse brain. J Comp Neurol, 2006. 494(3): p. 528–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mani BK, et al. , Neuroanatomical characterization of a growth hormone secretagogue receptor-green fluorescent protein reporter mouse. J Comp Neurol, 2014. 522(16): p. 3644–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ferrini F, et al. , Ghrelin in central neurons. Curr Neuropharmacol, 2009. 7(1): p. 37–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cabral A, et al. , Is Ghrelin Synthesized in the Central Nervous System? Int J Mol Sci, 2017. 18(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Prieto-Garcia L, et al. , Ghrelin and GHS-R1A signaling within the ventral and laterodorsal tegmental area regulate sexual behavior in sexually naive male mice. Psychoneuroendocrinology, 2015. 62: p. 392–402. [DOI] [PubMed] [Google Scholar]
- 41.Cabral A, Fernandez G, and Perello M, Analysis of brain nuclei accessible to ghrelin present in the cerebrospinal fluid. Neuroscience, 2013. 253: p. 406–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cornejo MP, et al. , Ghrelin Recruits Specific Subsets of Dopamine and GABA Neurons of Different Ventral Tegmental Area Sub-nuclei. Neuroscience, 2018. 392: p. 107–120. [DOI] [PubMed] [Google Scholar]
- 43.Abizaid A, et al. , Ghrelin modulates the activity and synaptic input organization of midbrain dopamine neurons while promoting appetite. J Clin Invest, 2006. 116(12): p. 3229–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Anderberg RH, et al. , The Stomach-Derived Hormone Ghrelin Increases Impulsive Behavior. Neuropsychopharmacology, 2016. 41(5): p. 1199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jerlhag E, et al. , Ghrelin administration into tegmental areas stimulates locomotor activity and increases extracellular concentration of dopamine in the nucleus accumbens. Addict Biol, 2007. 12(1): p. 6–16. [DOI] [PubMed] [Google Scholar]
- 46.Quarta D, et al. , Systemic administration of ghrelin increases extracellular dopamine in the shell but not the core subdivision of the nucleus accumbens. Neurochem Int, 2009. 54(2): p. 89–94. [DOI] [PubMed] [Google Scholar]
- 47.Edvardsson CE, Vestlund J, and Jerlhag E, A ghrelin receptor antagonist reduces the ability of ghrelin, alcohol or amphetamine to induce a dopamine release in the ventral tegmental area and in nucleus accumbens shell in rats. Eur J Pharmacol, 2021. 899: p. 174039. [DOI] [PubMed] [Google Scholar]
- 48.Jerlhag E, et al. , Glutamatergic regulation of ghrelin-induced activation of the mesolimbic dopamine system. Addict Biol, 2011. 16(1): p. 82–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pierre A, et al. , Effects of ghrelin receptor activation on forebrain dopamine release, conditioned fear and fear extinction in C57BL/6J mice. J Neurochem, 2020. 154(4): p. 389–403. [DOI] [PubMed] [Google Scholar]
- 50.Cone JJ, McCutcheon JE, and Roitman MF, Ghrelin acts as an interface between physiological state and phasic dopamine signaling. J Neurosci, 2014. 34(14): p. 4905–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kawahara Y, et al. , Food reward-sensitive interaction of ghrelin and opioid receptor pathways in mesolimbic dopamine system. Neuropharmacology, 2013. 67: p. 395–402. [DOI] [PubMed] [Google Scholar]
- 52.Egecioglu E, et al. , Ghrelin increases intake of rewarding food in rodents. Addict Biol, 2010. 15(3): p. 304–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kawahara Y, et al. , Peripherally administered ghrelin induces bimodal effects on the mesolimbic dopamine system depending on food-consumptive states. Neuroscience, 2009. 161(3): p. 855–64. [DOI] [PubMed] [Google Scholar]
- 54.Skibicka KP, et al. , Divergent circuitry underlying food reward and intake effects of ghrelin: dopaminergic VTA-accumbens projection mediates ghrelin’s effect on food reward but not food intake. Neuropharmacology, 2013. 73: p. 274–83. [DOI] [PubMed] [Google Scholar]
- 55.Skibicka KP, et al. , Ghrelin interacts with neuropeptide Y Y1 and opioid receptors to increase food reward. Endocrinology, 2012. 153(3): p. 1194–205. [DOI] [PubMed] [Google Scholar]
- 56.Engel JA, Nylander I, and Jerlhag E, A ghrelin receptor (GHS-R1A) antagonist attenuates the rewarding properties of morphine and increases opioid peptide levels in reward areas in mice. Eur Neuropsychopharmacol, 2015. 25(12): p. 2364–71. [DOI] [PubMed] [Google Scholar]
- 57.Jerlhag E, et al. , Ghrelin receptor antagonism attenuates cocaine- and amphetamine-induced locomotor stimulation, accumbal dopamine release, and conditioned place preference. Psychopharmacology (Berl), 2010. 211(4): p. 415–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jerlhag E and Engel JA, Ghrelin receptor antagonism attenuates nicotine-induced locomotor stimulation, accumbal dopamine release and conditioned place preference in mice. Drug Alcohol Depend, 2011. 117(2–3): p. 126–31. [DOI] [PubMed] [Google Scholar]
- 59.Sustkova-Fiserova M, et al. , Ghrelin receptor antagonism of morphine-induced accumbens dopamine release and behavioral stimulation in rats. Psychopharmacology (Berl), 2014. 231(14): p. 2899–908. [DOI] [PubMed] [Google Scholar]
- 60.Jerlhag E, et al. , Requirement of central ghrelin signaling for alcohol reward. Proc Natl Acad Sci U S A, 2009. 106(27): p. 11318–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jerlhag E, et al. , The alcohol-induced locomotor stimulation and accumbal dopamine release is suppressed in ghrelin knockout mice. Alcohol, 2011. 45(4): p. 341–7. [DOI] [PubMed] [Google Scholar]
- 62.Jerlhag E, et al. , Concomitant release of ventral tegmental acetylcholine and accumbal dopamine by ghrelin in rats. PLoS One, 2012. 7(11): p. e49557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jerlhag E, et al. , Ghrelin stimulates locomotor activity and accumbal dopamine-overflow via central cholinergic systems in mice: implications for its involvement in brain reward. Addict Biol, 2006. 11(1): p. 45–54. [DOI] [PubMed] [Google Scholar]
- 64.Dickson SL, et al. , Blockade of central nicotine acetylcholine receptor signaling attenuate ghrelin-induced food intake in rodents. Neuroscience, 2010. 171(4): p. 1180–6. [DOI] [PubMed] [Google Scholar]
- 65.Jerlhag E, et al. , Alpha-conotoxin MII-sensitive nicotinic acetylcholine receptors are involved in mediating the ghrelin-induced locomotor stimulation and dopamine overflow in nucleus accumbens. Eur Neuropsychopharmacol, 2008. 18(7): p. 508–18. [DOI] [PubMed] [Google Scholar]
- 66.Skibicka KP, et al. , Role of ghrelin in food reward: impact of ghrelin on sucrose self-administration and mesolimbic dopamine and acetylcholine receptor gene expression. Addict Biol, 2012. 17(1): p. 95–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Navarro G, et al. , Complexes of Ghrelin GHS-R1a, GHS-R1b, and Dopamine D(1) Receptors Localized in the Ventral Tegmental Area as Main Mediators of the Dopaminergic Effects of Ghrelin. J Neurosci, 2022. 42(6): p. 940–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kalafateli AL, et al. , A cannabinoid receptor antagonist attenuates ghrelin-induced activation of the mesolimbic dopamine system in mice. Physiol Behav, 2018. 184: p. 211–219. [DOI] [PubMed] [Google Scholar]
- 69.Tan KR, et al. , GABA neurons of the VTA drive conditioned place aversion. Neuron, 2012. 73(6): p. 1173–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.van Zessen R, et al. , Activation of VTA GABA neurons disrupts reward consumption. Neuron, 2012. 73(6): p. 1184–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Klejbor I, et al. , The relationships between neurons containing dopamine and nitric oxide synthase in the ventral tegmental area. Folia Histochem Cytobiol, 2004. 42(2): p. 83–7. [PubMed] [Google Scholar]
- 72.Wirtshafter D and Sheppard AC, Localization of GABA(B) receptors in midbrain monoamine containing neurons in the rat. Brain Res Bull, 2001. 56(1): p. 1–5. [DOI] [PubMed] [Google Scholar]
- 73.Fedele E, Varnier G, and Raiteri M, In vivo microdialysis study of GABA(A) and GABA(B) receptors modulating the glutamate receptor/NO/cyclic GMP pathway in the rat hippocampus. Neuropharmacology, 1997. 36(10): p. 1405–15. [DOI] [PubMed] [Google Scholar]
- 74.Engel JA, et al. , Ghrelin activates the mesolimbic dopamine system via nitric oxide associated mechanisms in the ventral tegmental area. Nitric Oxide, 2022. 131: p. 1–7. [DOI] [PubMed] [Google Scholar]
- 75.Borner T, et al. , Lipopolysaccharide inhibits ghrelin-excited neurons of the arcuate nucleus and reduces food intake via central nitric oxide signaling. Brain Behav Immun, 2012. 26(6): p. 867–79. [DOI] [PubMed] [Google Scholar]
- 76.Abtahi S, et al. , Ghrelin enhances food intake and carbohydrate oxidation in a nitric oxide dependent manner. Gen Comp Endocrinol, 2017. 250: p. 9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Morley JE, et al. , Nitric oxide is a central component in neuropeptide regulation of appetite. Peptides, 2011. 32(4): p. 776–80. [DOI] [PubMed] [Google Scholar]
- 78.Aguggia JP, et al. , Growth hormone secretagogue receptor signaling in the supramammillary nucleus targets nitric oxide-producing neurons and controls recognition memory in mice. Psychoneuroendocrinology, 2022. 139: p. 105716. [DOI] [PubMed] [Google Scholar]
- 79.Shank EJ, et al. , Selective ablation of GABA neurons in the ventral tegmental area increases spontaneous locomotor activity. Behav Neurosci, 2007. 121(6): p. 1224–33. [DOI] [PubMed] [Google Scholar]
- 80.Zhou Y, et al. , An AMPA glutamatergic receptor activation-nitric oxide synthesis step signals transsynaptic apoptosis in limbic cortex. Neuropharmacology, 2006. 51(1): p. 67–76. [DOI] [PubMed] [Google Scholar]
- 81.Shim I, et al. , Role of nitric oxide synthase inhibitors and NMDA receptor antagonist in nicotine-induced behavioral sensitization in the rat. Eur J Pharmacol, 2002. 443(1–3): p. 119–24. [DOI] [PubMed] [Google Scholar]
- 82.King SJ, et al. , Motivation to obtain preferred foods is enhanced by ghrelin in the ventral tegmental area. Horm Behav, 2011. 60(5): p. 572–80. [DOI] [PubMed] [Google Scholar]
- 83.Lockie SH, et al. , Diet-induced obesity causes ghrelin resistance in reward processing tasks. Psychoneuroendocrinology, 2015. 62: p. 114–20. [DOI] [PubMed] [Google Scholar]
- 84.Blanco-Gandia MC, et al. , Changes in gene expression and sensitivity of cocaine reward produced by a continuous fat diet. Psychopharmacology (Berl), 2017. 234(15): p. 2337–2352. [DOI] [PubMed] [Google Scholar]
- 85.Lockie SH, et al. , Glucose availability regulates ghrelin-induced food intake in the ventral tegmental area. J Neuroendocrinol, 2019. 31(7): p. e12696. [DOI] [PubMed] [Google Scholar]
- 86.Lamont EW, et al. , Ghrelin-deficient mice have fewer orexin cells and reduced cFOS expression in the mesolimbic dopamine pathway under a restricted feeding paradigm. Neuroscience, 2012. 218: p. 12–9. [DOI] [PubMed] [Google Scholar]
- 87.Skibicka KP, et al. , Ghrelin directly targets the ventral tegmental area to increase food motivation. Neuroscience, 2011. 180: p. 129–37. [DOI] [PubMed] [Google Scholar]
- 88.Wei XJ, et al. , Ghrelin signaling in the ventral tegmental area mediates both reward-based feeding and fasting-induced hyperphagia on high-fat diet. Neuroscience, 2015. 300: p. 53–62. [DOI] [PubMed] [Google Scholar]
- 89.Schele E, et al. , Centrally Administered Ghrelin Acutely Influences Food Choice in Rodents. PLoS One, 2016. 11(2): p. e0149456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cai H, et al. , Altered lipid and salt taste responsivity in ghrelin and GOAT null mice. PLoS One, 2013. 8(10): p. e76553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Calder AN, et al. , Ghrelin Receptors Enhance Fat Taste Responsiveness in Female Mice. Nutrients, 2021. 13(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Skov LJ, et al. , Exploring the Behavioral and Metabolic Phenotype Generated by Re-Introduction of the Ghrelin Receptor in the Ventral Tegmental Area. Int J Mol Sci, 2017. 18(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sommer S and Hauber W, Ghrelin receptor activation in the ventral tegmental area amplified instrumental responding but not the excitatory influence of Pavlovian stimuli on instrumental responding. Neurobiol Learn Mem, 2016. 134 Pt B: p. 210–5. [DOI] [PubMed] [Google Scholar]
- 94.Weinberg ZY, Nicholson ML, and Currie PJ, 6-Hydroxydopamine lesions of the ventral tegmental area suppress ghrelin’s ability to elicit food-reinforced behavior. Neurosci Lett, 2011. 499(2): p. 70–3. [DOI] [PubMed] [Google Scholar]
- 95.van der Plasse G, et al. , Modulation of cue-induced firing of ventral tegmental area dopamine neurons by leptin and ghrelin. Int J Obes (Lond), 2015. 39(12): p. 1742–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.St-Onge V, Watts A, and Abizaid A, Ghrelin enhances cue-induced bar pressing for high fat food. Horm Behav, 2016. 78: p. 141–9. [DOI] [PubMed] [Google Scholar]
- 97.Meule A and Kubler A, Double trouble. Trait food craving and impulsivity interactively predict food-cue affected behavioral inhibition. Appetite, 2014. 79: p. 174–82. [DOI] [PubMed] [Google Scholar]
- 98.Georgii C, et al. , Food craving, food choice and consumption: The role of impulsivity and sham-controlled tDCS stimulation of the right dlPFC. Physiol Behav, 2017. 177: p. 20–26. [DOI] [PubMed] [Google Scholar]
- 99.VanderBroek-Stice L, et al. , Multidimensional assessment of impulsivity in relation to obesity and food addiction. Appetite, 2017. 112: p. 59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Murphy CM, Stojek MK, and MacKillop J, Interrelationships among impulsive personality traits, food addiction, and Body Mass Index. Appetite, 2014. 73: p. 45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Park SB, et al. , Contribution of growth hormone secretagogue receptor (GHSR) signaling in the ventral tegmental area (VTA) to the regulation of social motivation in male mice. Transl Psychiatry, 2021. 11(1): p. 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kroemer NB, et al. , Fasting levels of ghrelin covary with the brain response to food pictures. Addict Biol, 2013. 18(5): p. 855–62. [DOI] [PubMed] [Google Scholar]
- 103.Schulz C, Vezzani C, and Kroemer NB, How gut hormones shape reward: A systematic review of the role of ghrelin and GLP-1 in human fMRI. Physiol Behav, 2023. 263: p. 114111. [DOI] [PubMed] [Google Scholar]
- 104.Boswell RG and Grilo CM, General impulsivity in binge-eating disorder. CNS Spectr, 2021. 26(5): p. 538–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hege MA, et al. , Attentional impulsivity in binge eating disorder modulates response inhibition performance and frontal brain networks. Int J Obes (Lond), 2015. 39(2): p. 353–60. [DOI] [PubMed] [Google Scholar]
- 106.Carr MM, et al. , Impulsivity and compulsivity in binge eating disorder: A systematic review of behavioral studies. Prog Neuropsychopharmacol Biol Psychiatry, 2021. 110: p. 110318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Valdivia S, et al. , Escalation in high fat intake in a binge eating model differentially engages dopamine neurons of the ventral tegmental area and requires ghrelin signaling. Psychoneuroendocrinology, 2015. 60: p. 206–16. [DOI] [PubMed] [Google Scholar]
- 108.Bake T, Hellgren KT, and Dickson SL, Acute ghrelin changes food preference from a high-fat diet to chow during binge-like eating in rodents. J Neuroendocrinol, 2017. 29(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Suchankova P, Engel JA, and Jerlhag E, Sub-chronic Ghrelin Receptor Blockade Attenuates Alcohol- and Amphetamine-Induced Locomotor Stimulation in Mice. Alcohol Alcohol, 2016. 51(2): p. 121–7. [DOI] [PubMed] [Google Scholar]
- 110.Dunn DP, et al. , Role of mesolimbic ghrelin in the acquisition of cocaine reward. Neurosci Lett, 2019. 709: p. 134367. [DOI] [PubMed] [Google Scholar]
- 111.Schuette LM, Gray CC, and Currie PJ, Microinjection of Ghrelin into the Ventral Tegmental Area Potentiates Cocaine-Induced Conditioned Place Preference. J Behav Brain Sci, 2013. 3(8): p. 276–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.You ZB, et al. , Involvement of the ghrelin system in the maintenance and reinstatement of cocaine-motivated behaviors: a role of adrenergic action at peripheral beta1 receptors. Neuropsychopharmacology, 2022. 47(8): p. 1449–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.You ZB, et al. , Involvement of the ghrelin system in the maintenance of oxycodone self-administration: converging evidence from endocrine, pharmacologic and transgenic approaches. Mol Psychiatry, 2022. 27(4): p. 2171–2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.D’Cunha TM, et al. , A role for leptin and ghrelin in the augmentation of heroin seeking induced by chronic food restriction. Psychopharmacology (Berl), 2020. 237(3): p. 787–800. [DOI] [PubMed] [Google Scholar]
- 115.Charalambous C, et al. , Alterations in Rat Accumbens Dopamine, Endocannabinoids and GABA Content During WIN55,212–2 Treatment: The Role of Ghrelin. Int J Mol Sci, 2020. 22(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Koopmann A, et al. , The association of the appetitive peptide acetylated ghrelin with alcohol craving in early abstinent alcohol dependent individuals. Psychoneuroendocrinology, 2012. 37(7): p. 980–6. [DOI] [PubMed] [Google Scholar]
- 117.Landgren S, et al. , Association of pro-ghrelin and GHS-R1A gene polymorphisms and haplotypes with heavy alcohol use and body mass. Alcohol Clin Exp Res, 2008. 32(12): p. 2054–61. [DOI] [PubMed] [Google Scholar]
- 118.Farokhnia M, et al. , Exogenous ghrelin administration increases alcohol self-administration and modulates brain functional activity in heavy-drinking alcohol-dependent individuals. Mol Psychiatry, 2018. 23(10): p. 2029–2038. [DOI] [PubMed] [Google Scholar]
- 119.Landgren S, et al. , Expression of the gene encoding the ghrelin receptor in rats selected for differential alcohol preference. Behav Brain Res, 2011. 221(1): p. 182–8. [DOI] [PubMed] [Google Scholar]
- 120.Suchankova P, et al. , Ghrelin receptor (GHS-R1A) antagonism suppresses both alcohol consumption and the alcohol deprivation effect in rats following long-term voluntary alcohol consumption. PLoS One, 2013. 8(8): p. e71284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Cepko LC, et al. , Ghrelin alters the stimulatory effect of cocaine on ethanol intake following mesolimbic or systemic administration. Neuropharmacology, 2014. 85: p. 224–31. [DOI] [PubMed] [Google Scholar]
- 122.Kaur S and Ryabinin AE, Ghrelin receptor antagonism decreases alcohol consumption and activation of perioculomotor urocortin-containing neurons. Alcohol Clin Exp Res, 2010. 34(9): p. 1525–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Pawar GR, et al. , Ghrelin alleviates depression-like behaviour in rats subjected to high-fat diet and diurnal rhythm disturbance. Am J Transl Res, 2022. 14(10): p. 7098–7108. [PMC free article] [PubMed] [Google Scholar]
- 124.Cerit H, et al. , Divergent associations between ghrelin and neural responsivity to palatable food in hyperphagic and hypophagic depression. J Affect Disord, 2019. 242: p. 29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kazmierczak M and Nicola SM, The Arousal-motor Hypothesis of Dopamine Function: Evidence that Dopamine Facilitates Reward Seeking in Part by Maintaining Arousal. Neuroscience, 2022. 499: p. 64–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Clifford S, et al. , Impact of food restriction and cocaine on locomotion in ghrelin- and ghrelin-receptor knockout mice. Addict Biol, 2011. 16(3): p. 386–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hyland L, et al. , Central ghrelin receptor stimulation modulates sex motivation in male rats in a site dependent manner. Horm Behav, 2018. 97: p. 56–66. [DOI] [PubMed] [Google Scholar]
- 128.Hansson C, et al. , Ghrelin influences novelty seeking behavior in rodents and men. PLoS One, 2012. 7(12): p. e50409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Thiebaud N, et al. , The incretin hormone glucagon-like peptide 1 increases mitral cell excitability by decreasing conductance of a voltage-dependent potassium channel. J Physiol, 2016. 594(10): p. 2607–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Alhadeff AL, Rupprecht LE, and Hayes MR, GLP-1 neurons in the nucleus of the solitary tract project directly to the ventral tegmental area and nucleus accumbens to control for food intake. Endocrinology, 2012. 153(2): p. 647–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Merchenthaler I, Lane M, and Shughrue P, Distribution of pre-pro-glucagon and glucagon-like peptide-1 receptor messenger RNAs in the rat central nervous system. J Comp Neurol, 1999. 403(2): p. 261–80. [DOI] [PubMed] [Google Scholar]
- 132.Graham DL, et al. , A novel mouse model of glucagon-like peptide-1 receptor expression: A look at the brain. J Comp Neurol, 2020. 528(14): p. 2445–2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Cork SC, et al. , Distribution and characterisation of Glucagon-like peptide-1 receptor expressing cells in the mouse brain. Mol Metab, 2015. 4(10): p. 718–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hernandez NS, et al. , Glucagon-like peptide-1 receptor activation in the ventral tegmental area attenuates cocaine seeking in rats. Neuropsychopharmacology, 2018. 43(10): p. 2000–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kastin AJ, Akerstrom V, and Pan W, Interactions of glucagon-like peptide-1 (GLP-1) with the blood-brain barrier. J Mol Neurosci, 2002. 18(1–2): p. 7–14. [DOI] [PubMed] [Google Scholar]
- 136.Egecioglu E, Engel JA, and Jerlhag E, The glucagon-like peptide 1 analogue, exendin-4, attenuates the rewarding properties of psychostimulant drugs in mice. PLoS One, 2013. 8(7): p. e69010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Egecioglu E, Engel JA, and Jerlhag E, The glucagon-like peptide 1 analogue Exendin-4 attenuates the nicotine-induced locomotor stimulation, accumbal dopamine release, conditioned place preference as well as the expression of locomotor sensitization in mice. PLoS One, 2013. 8(10): p. e77284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Egecioglu E, et al. , The glucagon-like peptide 1 analogue Exendin-4 attenuates alcohol mediated behaviors in rodents. Psychoneuroendocrinology, 2013. 38(8): p. 1259–70. [DOI] [PubMed] [Google Scholar]
- 139.Fortin SM and Roitman MF, Central GLP-1 receptor activation modulates cocaine-evoked phasic dopamine signaling in the nucleus accumbens core. Physiol Behav, 2017. 176: p. 17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Vallof D, et al. , The glucagon-like peptide 1 receptor agonist liraglutide attenuates the reinforcing properties of alcohol in rodents. Addict Biol, 2016. 21(2): p. 422–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Fortin SM, Chartoff EH, and Roitman MF, The Aversive Agent Lithium Chloride Suppresses Phasic Dopamine Release Through Central GLP-1 Receptors. Neuropsychopharmacology, 2016. 41(3): p. 906–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Konanur VR, et al. , Phasic dopamine responses to a food-predictive cue are suppressed by the glucagon-like peptide-1 receptor agonist Exendin-4. Physiol Behav, 2020. 215: p. 112771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Mietlicki-Baase EG, et al. , The food intake-suppressive effects of glucagon-like peptide-1 receptor signaling in the ventral tegmental area are mediated by AMPA/kainate receptors. Am J Physiol Endocrinol Metab, 2013. 305(11): p. E1367–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Richard JE, et al. , Activation of the GLP-1 receptors in the nucleus of the solitary tract reduces food reward behavior and targets the mesolimbic system. PLoS One, 2015. 10(3): p. e0119034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Yang Y, et al. , Long term exendin-4 treatment reduces food intake and body weight and alters expression of brain homeostatic and reward markers. Endocrinology, 2014. 155(9): p. 3473–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wang XF, et al. , Endogenous Glucagon-like Peptide-1 Suppresses High-Fat Food Intake by Reducing Synaptic Drive onto Mesolimbic Dopamine Neurons. Cell Rep, 2015. 12(5): p. 726–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Poulin JF, et al. , Mapping projections of molecularly defined dopamine neuron subtypes using intersectional genetic approaches. Nat Neurosci, 2018. 21(9): p. 1260–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hernandez NS, et al. , GLP-1 receptor signaling in the laterodorsal tegmental nucleus attenuates cocaine seeking by activating GABAergic circuits that project to the VTA. Mol Psychiatry, 2021. 26(8): p. 4394–4408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Vallof D, Kalafateli AL, and Jerlhag E, Brain region specific glucagon-like peptide-1 receptors regulate alcohol-induced behaviors in rodents. Psychoneuroendocrinology, 2019. 103: p. 284–295. [DOI] [PubMed] [Google Scholar]
- 150.Vestlund J and Jerlhag E, The glucagon-like peptide-1 receptor agonist, exendin-4, reduces sexual interaction behaviors in a brain site-specific manner in sexually naive male mice. Horm Behav, 2020. 124: p. 104778. [DOI] [PubMed] [Google Scholar]
- 151.Brigande AM, Darwich JG, and Currie PJ, Mesolimbic exendin-4 attenuates reward salience evoked by neuropeptide Y and ghrelin. Behav Brain Res, 2022. 440: p. 114249. [DOI] [PubMed] [Google Scholar]
- 152.Colvin KJ, et al. , Brain Site-Specific Inhibitory Effects of the GLP-1 Analogue Exendin-4 on Alcohol Intake and Operant Responding for Palatable Food. Int J Mol Sci, 2020. 21(24). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Dickson SL, et al. , The glucagon-like peptide 1 (GLP-1) analogue, exendin-4, decreases the rewarding value of food: a new role for mesolimbic GLP-1 receptors. J Neurosci, 2012. 32(14): p. 4812–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Howell E, et al. , Glucagon-Like Peptide-1 (GLP-1) and 5-Hydroxytryptamine 2c (5-HT(2c)) Receptor Agonists in the Ventral Tegmental Area (VTA) Inhibit Ghrelin-Stimulated Appetitive Reward. Int J Mol Sci, 2019. 20(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Lopez-Ferreras L, et al. , GLP-1 modulates the supramammillary nucleus-lateral hypothalamic neurocircuit to control ingestive and motivated behavior in a sex divergent manner. Mol Metab, 2019. 20: p. 178–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.van Bloemendaal L, et al. , GLP-1 receptor activation modulates appetite- and reward-related brain areas in humans. Diabetes, 2014. 63(12): p. 4186–96. [DOI] [PubMed] [Google Scholar]
- 157.van Bloemendaal L, et al. , Brain reward-system activation in response to anticipation and consumption of palatable food is altered by glucagon-like peptide-1 receptor activation in humans. Diabetes Obes Metab, 2015. 17(9): p. 878–86. [DOI] [PubMed] [Google Scholar]
- 158.Schmidt HD, et al. , Glucagon-Like Peptide-1 Receptor Activation in the Ventral Tegmental Area Decreases the Reinforcing Efficacy of Cocaine. Neuropsychopharmacology, 2016. 41(7): p. 1917–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Dixon TN, McNally GP, and Ong ZY, Glucagon-Like Peptide-1 Receptor Signaling in the Ventral Tegmental Area Reduces Alcohol Self-Administration in Male Rats. Alcohol Clin Exp Res, 2020. 44(10): p. 2118–2129. [DOI] [PubMed] [Google Scholar]
- 160.Shirazi RH, Dickson SL, and Skibicka KP, Gut peptide GLP-1 and its analogue, Exendin-4, decrease alcohol intake and reward. PLoS One, 2013. 8(4): p. e61965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.McKay NJ, et al. , Glucagon-like peptide-1 receptor agonists suppress water intake independent of effects on food intake. Am J Physiol Regul Integr Comp Physiol, 2011. 301(6): p. R1755–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.McKay NJ, Galante DL, and Daniels D, Endogenous glucagon-like peptide-1 reduces drinking behavior and is differentially engaged by water and food intakes in rats. J Neurosci, 2014. 34(49): p. 16417–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Colvin KJ, et al. , Differential effects of intra-ventral tegmental area ghrelin and glucagon-like peptide-1 on the stimulatory action of D-amphetamine and cocaine-induced ethanol intake in male Sprague Dawley rats. Behav Brain Res, 2022. 421: p. 113726. [DOI] [PubMed] [Google Scholar]
- 164.Eban-Rothschild A, et al. , Arousal State-Dependent Alterations in VTA-GABAergic Neuronal Activity. eNeuro, 2020. 7(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Taylor NE, et al. , Optogenetic activation of dopamine neurons in the ventral tegmental area induces reanimation from general anesthesia. Proc Natl Acad Sci U S A, 2016. 113(45): p. 12826–12831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Brierley DI, et al. , Central and peripheral GLP-1 systems independently suppress eating. Nat Metab, 2021. 3(2): p. 258–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Hayes MR, Skibicka KP, and Grill HJ, Caudal brainstem processing is sufficient for behavioral, sympathetic, and parasympathetic responses driven by peripheral and hindbrain glucagon-like-peptide-1 receptor stimulation. Endocrinology, 2008. 149(8): p. 4059–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Hansotia T, et al. , Extrapancreatic incretin receptors modulate glucose homeostasis, body weight, and energy expenditure. J Clin Invest, 2007. 117(1): p. 143–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Kern KA and Mietlicki-Baase EG, Distributed amylin receptor signaling and its influence on motivated behavior. Physiol Behav, 2020. 222: p. 112958. [DOI] [PubMed] [Google Scholar]
- 170.Lutz TA, Control of energy homeostasis by amylin. Cell Mol Life Sci, 2012. 69(12): p. 1947–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Boyle CN, Lutz TA, and Le Foll C, Amylin - Its role in the homeostatic and hedonic control of eating and recent developments of amylin analogs to treat obesity. Mol Metab, 2018. 8: p. 203–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Christopoulos G, et al. , Multiple amylin receptors arise from receptor activity-modifying protein interaction with the calcitonin receptor gene product. Mol Pharmacol, 1999. 56(1): p. 235–42. [DOI] [PubMed] [Google Scholar]
- 173.Morfis M, et al. , Receptor activity-modifying proteins differentially modulate the G protein-coupling efficiency of amylin receptors. Endocrinology, 2008. 149(11): p. 5423–31. [DOI] [PubMed] [Google Scholar]
- 174.Mietlicki-Baase EG, et al. , Amylin receptor signaling in the ventral tegmental area is physiologically relevant for the control of food intake. Neuropsychopharmacology, 2013. 38(9): p. 1685–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Kalafateli AL, et al. , An amylin and calcitonin receptor agonist modulates alcohol behaviors by acting on reward-related areas in the brain. Prog Neurobiol, 2021. 200: p. 101969. [DOI] [PubMed] [Google Scholar]
- 176.Tilakaratne N, et al. , Amylin receptor phenotypes derived from human calcitonin receptor/RAMP coexpression exhibit pharmacological differences dependent on receptor isoform and host cell environment. J Pharmacol Exp Ther, 2000. 294(1): p. 61–72. [PubMed] [Google Scholar]
- 177.Paxinos G, et al. , In vitro autoradiographic localization of calcitonin and amylin binding sites in monkey brain. J Chem Neuroanat, 2004. 27(4): p. 217–36. [DOI] [PubMed] [Google Scholar]
- 178.Sexton PM, et al. , In vitro autoradiographic localization of amylin binding sites in rat brain. Neuroscience, 1994. 62(2): p. 553–67. [DOI] [PubMed] [Google Scholar]
- 179.van Rossum D, et al. , Autoradiographic distribution and receptor binding profile of [125I]Bolton Hunter-rat amylin binding sites in the rat brain. J Pharmacol Exp Ther, 1994. 270(2): p. 779–87. [PubMed] [Google Scholar]
- 180.Becskei C, et al. , Immunohistochemical mapping of calcitonin receptors in the adult rat brain. Brain Res, 2004. 1030(2): p. 221–33. [DOI] [PubMed] [Google Scholar]
- 181.Boccia L, et al. , Amylin brain circuitry. Peptides, 2020. 132: p. 170366. [DOI] [PubMed] [Google Scholar]
- 182.Mietlicki-Baase EG, et al. , Amylin modulates the mesolimbic dopamine system to control energy balance. Neuropsychopharmacology, 2015. 40(2): p. 372–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Kalafateli AL, Aranas C, and Jerlhag E, Activation of the amylin pathway modulates cocaine-induced activation of the mesolimbic dopamine system in male mice. Horm Behav, 2021. 127: p. 104885. [DOI] [PubMed] [Google Scholar]
- 184.Whiting L, et al. , The area postrema (AP) and the parabrachial nucleus (PBN) are important sites for salmon calcitonin (sCT) to decrease evoked phasic dopamine release in the nucleus accumbens (NAc). Physiol Behav, 2017. 176: p. 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Cao J, et al. , A structural basis for amylin receptor phenotype. Science, 2022. 375(6587): p. eabm9609. [DOI] [PubMed] [Google Scholar]
- 186.Kalafateli AL, Aranas C, and Jerlhag E, Effects of sub-chronic amylin receptor activation on alcohol-induced locomotor stimulation and monoamine levels in mice. Psychopharmacology (Berl), 2020. 237(11): p. 3249–3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Turek VF, et al. , Mechanisms of amylin/leptin synergy in rodent models. Endocrinology, 2010. 151(1): p. 143–52. [DOI] [PubMed] [Google Scholar]
- 188.Potes CS, et al. , Involvement of the extracellular signal-regulated kinase 1/2 signaling pathway in amylin’s eating inhibitory effect. Am J Physiol Regul Integr Comp Physiol, 2012. 302(3): p. R340–51. [DOI] [PubMed] [Google Scholar]
- 189.Mietlicki-Baase EG, et al. , Cooperative interaction between leptin and amylin signaling in the ventral tegmental area for the control of food intake. Am J Physiol Endocrinol Metab, 2015. 308(12): p. E1116–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Omrani A, et al. , Identification of Novel Neurocircuitry Through Which Leptin Targets Multiple Inputs to the Dopamine System to Reduce Food Reward Seeking. Biol Psychiatry, 2021. 90(12): p. 843–852. [DOI] [PubMed] [Google Scholar]
- 191.Reiner DJ, et al. , Amylin Acts in the Lateral Dorsal Tegmental Nucleus to Regulate Energy Balance Through Gamma-Aminobutyric Acid Signaling. Biol Psychiatry, 2017. 82(11): p. 828–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Mietlicki-Baase EG, et al. , Amylin receptor activation in the ventral tegmental area reduces motivated ingestive behavior. Neuropharmacology, 2017. 123: p. 67–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Smith SR, et al. , Pramlintide treatment reduces 24-h caloric intake and meal sizes and improves control of eating in obese subjects: a 6-wk translational research study. Am J Physiol Endocrinol Metab, 2007. 293(2): p. E620–7. [DOI] [PubMed] [Google Scholar]
- 194.Smith SR, et al. , Sustained weight loss following 12-month pramlintide treatment as an adjunct to lifestyle intervention in obesity. Diabetes Care, 2008. 31(9): p. 1816–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Friedman JM, Leptin and the endocrine control of energy balance. Nat Metab, 2019. 1(8): p. 754–764. [DOI] [PubMed] [Google Scholar]
- 196.Schwartz MW, et al. , Cerebrospinal fluid leptin levels: relationship to plasma levels and to adiposity in humans. Nat Med, 1996. 2(5): p. 589–93. [DOI] [PubMed] [Google Scholar]
- 197.Harrison L, et al. , Fluorescent blood-brain barrier tracing shows intact leptin transport in obese mice. Int J Obes (Lond), 2019. 43(6): p. 1305–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Duquenne M, et al. , Leptin brain entry via a tanycytic LepR-EGFR shuttle controls lipid metabolism and pancreas function. Nat Metab, 2021. 3(8): p. 1071–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Burguera B, et al. , The long form of the leptin receptor (OB-Rb) is widely expressed in the human brain. Neuroendocrinology, 2000. 71(3): p. 187–95. [DOI] [PubMed] [Google Scholar]
- 200.Hommel JD, et al. , Leptin receptor signaling in midbrain dopamine neurons regulates feeding. Neuron, 2006. 51(6): p. 801–10. [DOI] [PubMed] [Google Scholar]
- 201.Murakami T, Enjoji M, and Koyama S, Leptin attenuates D(2) receptor-mediated inhibition of putative ventral tegmental area dopaminergic neurons. Physiol Rep, 2018. 6(7): p. e13631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Mercuri NB, et al. , Monoamine oxidase inhibition causes a long-term prolongation of the dopamine-induced responses in rat midbrain dopaminergic cells. J Neurosci, 1997. 17(7): p. 2267–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Margolis EB, et al. , Midbrain dopamine neurons: projection target determines action potential duration and dopamine D(2) receptor inhibition. J Neurosci, 2008. 28(36): p. 8908–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Korotkova TM, et al. , Effects of arousal- and feeding-related neuropeptides on dopaminergic and GABAergic neurons in the ventral tegmental area of the rat. Eur J Neurosci, 2006. 23(10): p. 2677–85. [DOI] [PubMed] [Google Scholar]
- 205.Liu J, et al. , Selective deletion of the leptin receptor in dopamine neurons produces anxiogenic-like behavior and increases dopaminergic activity in amygdala. Mol Psychiatry, 2011. 16(10): p. 1024–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.You ZB, et al. , Reciprocal Inhibitory Interactions Between the Reward-Related Effects of Leptin and Cocaine. Neuropsychopharmacology, 2016. 41(4): p. 1024–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Shen M, et al. , Mesolimbic leptin signaling negatively regulates cocaine-conditioned reward. Transl Psychiatry, 2016. 6(12): p. e972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Leshan RL, et al. , Ventral tegmental area leptin receptor neurons specifically project to and regulate cocaine- and amphetamine-regulated transcript neurons of the extended central amygdala. J Neurosci, 2010. 30(16): p. 5713–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Leinninger GM, et al. , Leptin acts via leptin receptor-expressing lateral hypothalamic neurons to modulate the mesolimbic dopamine system and suppress feeding. Cell Metab, 2009. 10(2): p. 89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Schiffino FL, et al. , Activation of a lateral hypothalamic-ventral tegmental circuit gates motivation. PLoS One, 2019. 14(7): p. e0219522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Siemian JN, et al. , Lateral hypothalamic LEPR neurons drive appetitive but not consummatory behaviors. Cell Rep, 2021. 36(8): p. 109615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Scarpace PJ, et al. , Leptin overexpression in VTA trans-activates the hypothalamus whereas prolonged leptin action in either region cross-desensitizes. Neuropharmacology, 2013. 65: p. 90–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Matheny M, et al. , Targeted leptin receptor blockade: role of ventral tegmental area and nucleus of the solitary tract leptin receptors in body weight homeostasis. J Endocrinol, 2014. 222(1): p. 27–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.van den Heuvel JK, et al. , Differential modulation of arcuate nucleus and mesolimbic gene expression levels by central leptin in rats on short-term high-fat high-sugar diet. PLoS One, 2014. 9(1): p. e87729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Fulton S, et al. , Leptin regulation of the mesoaccumbens dopamine pathway. Neuron, 2006. 51(6): p. 811–22. [DOI] [PubMed] [Google Scholar]
- 216.Opland D, et al. , Loss of neurotensin receptor-1 disrupts the control of the mesolimbic dopamine system by leptin and promotes hedonic feeding and obesity. Mol Metab, 2013. 2(4): p. 423–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Tye KM, et al. , Amygdala circuitry mediating reversible and bidirectional control of anxiety. Nature, 2011. 471(7338): p. 358–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Liu J, Guo M, and Lu XY, Leptin/LepRb in the Ventral Tegmental Area Mediates Anxiety-Related Behaviors. Int J Neuropsychopharmacol, 2015. 19(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.de Vrind VAJ, et al. , Leptin Receptor Expressing Neurons in the Substantia Nigra Regulate Locomotion, and in The Ventral Tegmental Area Motivation and Feeding. Front Endocrinol (Lausanne), 2021. 12: p. 680494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Fernandes MF, et al. , Leptin Suppresses the Rewarding Effects of Running via STAT3 Signaling in Dopamine Neurons. Cell Metab, 2015. 22(4): p. 741–9. [DOI] [PubMed] [Google Scholar]
- 221.Villanueva EC and Myers MG Jr., Leptin receptor signaling and the regulation of mammalian physiology. Int J Obes (Lond), 2008. 32 Suppl 7(Suppl 7): p. S8–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Trinko R, et al. , Erk1/2 mediates leptin receptor signaling in the ventral tegmental area. PLoS One, 2011. 6(11): p. e27180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Morton GJ, et al. , The action of leptin in the ventral tegmental area to decrease food intake is dependent on Jak-2 signaling. Am J Physiol Endocrinol Metab, 2009. 297(1): p. E202–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Thompson JL and Borgland SL, Presynaptic leptin action suppresses excitatory synaptic transmission onto ventral tegmental area dopamine neurons. Biol Psychiatry, 2013. 73(9): p. 860–8. [DOI] [PubMed] [Google Scholar]
- 225.Bruijnzeel AW, Qi X, and Corrie LW, Anorexic effects of intra-VTA leptin are similar in low-fat and high-fat-fed rats but attenuated in a subgroup of high-fat-fed obese rats. Pharmacol Biochem Behav, 2013. 103(3): p. 573–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Jung CH and Kim MS, Molecular mechanisms of central leptin resistance in obesity. Arch Pharm Res, 2013. 36(2): p. 201–7. [DOI] [PubMed] [Google Scholar]
- 227.Matheny M, et al. , Region-specific diet-induced and leptin-induced cellular leptin resistance includes the ventral tegmental area in rats. Neuropharmacology, 2011. 60(2–3): p. 480–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Bruijnzeel AW, et al. , Effects of insulin and leptin in the ventral tegmental area and arcuate hypothalamic nucleus on food intake and brain reward function in female rats. Behav Brain Res, 2011. 219(2): p. 254–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Bach P, Koopmann A, and Kiefer F, The Impact of Appetite-Regulating Neuropeptide Leptin on Alcohol Use, Alcohol Craving and Addictive Behavior: A Systematic Review of Preclinical and Clinical Data. Alcohol Alcohol, 2021. 56(2): p. 149–165. [DOI] [PubMed] [Google Scholar]
- 230.Novak CM, Burghardt PR, and Levine JA, The use of a running wheel to measure activity in rodents: relationship to energy balance, general activity, and reward. Neurosci Biobehav Rev, 2012. 36(3): p. 1001–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Scarpace PJ, Matheny M, and Zhang Y, Wheel running eliminates high-fat preference and enhances leptin signaling in the ventral tegmental area. Physiol Behav, 2010. 100(2): p. 173–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Shapiro A, et al. , The act of voluntary wheel running reverses dietary hyperphagia and increases leptin signaling in ventral tegmental area of aged obese rats. Gerontology, 2011. 57(4): p. 335–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Hebebrand J, et al. , Clinical Trials Required to Assess Potential Benefits and Side Effects of Treatment of Patients With Anorexia Nervosa With Recombinant Human Leptin. Front Psychol, 2019. 10: p. 769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Verhagen LA, Luijendijk MC, and Adan RA, Leptin reduces hyperactivity in an animal model for anorexia nervosa via the ventral tegmental area. Eur Neuropsychopharmacol, 2011. 21(3): p. 274–81. [DOI] [PubMed] [Google Scholar]
- 235.Norton L, et al. , Insulin: The master regulator of glucose metabolism. Metabolism, 2022. 129: p. 155142. [DOI] [PubMed] [Google Scholar]
- 236.Hersom M, et al. , The insulin receptor is expressed and functional in cultured blood-brain barrier endothelial cells but does not mediate insulin entry from blood to brain. Am J Physiol Endocrinol Metab, 2018. 315(4): p. E531–E542. [DOI] [PubMed] [Google Scholar]
- 237.Rhea EM, Rask-Madsen C, and Banks WA, Insulin transport across the blood-brain barrier can occur independently of the insulin receptor. J Physiol, 2018. 596(19): p. 4753–4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Figlewicz DP, et al. , Expression of receptors for insulin and leptin in the ventral tegmental area/substantia nigra (VTA/SN) of the rat. Brain Res, 2003. 964(1): p. 107–15. [DOI] [PubMed] [Google Scholar]
- 239.Mizoguchi A, et al. , High-fat Feeding Causes Inflammation and Insulin Resistance in the Ventral Tegmental Area in Mice. Neuroscience, 2021. 461: p. 72–79. [DOI] [PubMed] [Google Scholar]
- 240.Naef L, et al. , Insulin in the ventral tegmental area reduces cocaine-evoked dopamine in the nucleus accumbens in vivo. Eur J Neurosci, 2019. 50(3): p. 2146–2155. [DOI] [PubMed] [Google Scholar]
- 241.Rice ME and Patel JC, Somatodendritic dopamine release: recent mechanistic insights. Philos Trans R Soc Lond B Biol Sci, 2015. 370(1672). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Adell A and Artigas F, The somatodendritic release of dopamine in the ventral tegmental area and its regulation by afferent transmitter systems. Neurosci Biobehav Rev, 2004. 28(4): p. 415–31. [DOI] [PubMed] [Google Scholar]
- 243.Mebel DM, et al. , Insulin in the ventral tegmental area reduces hedonic feeding and suppresses dopamine concentration via increased reuptake. Eur J Neurosci, 2012. 36(3): p. 2336–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Fordahl SC and Jones SR, High-Fat-Diet-Induced Deficits in Dopamine Terminal Function Are Reversed by Restoring Insulin Signaling. ACS Chem Neurosci, 2017. 8(2): p. 290–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Speed N, et al. , Impaired striatal Akt signaling disrupts dopamine homeostasis and increases feeding. PLoS One, 2011. 6(9): p. e25169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Figlewicz DP, et al. , Diabetes causes differential changes in CNS noradrenergic and dopaminergic neurons in the rat: a molecular study. Brain Res, 1996. 736(1–2): p. 54–60. [DOI] [PubMed] [Google Scholar]
- 247.Figlewicz DP, et al. , Dopamine transporter mRNA is increased in the CNS of Zucker fatty (fa/fa) rats. Brain Res Bull, 1998. 46(3): p. 199–202. [DOI] [PubMed] [Google Scholar]
- 248.Figlewicz DP, et al. , Intraventricular insulin increases dopamine transporter mRNA in rat VTA/substantia nigra. Brain Res, 1994. 644(2): p. 331–4. [DOI] [PubMed] [Google Scholar]
- 249.Patterson TA, et al. , Food deprivation decreases mRNA and activity of the rat dopamine transporter. Neuroendocrinology, 1998. 68(1): p. 11–20. [DOI] [PubMed] [Google Scholar]
- 250.Labouebe G, et al. , Insulin induces long-term depression of ventral tegmental area dopamine neurons via endocannabinoids. Nat Neurosci, 2013. 16(3): p. 300–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Castillo PE, et al. , Endocannabinoid signaling and synaptic function. Neuron, 2012. 76(1): p. 70–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Liu S, et al. , Effect of insulin on excitatory synaptic transmission onto dopamine neurons of the ventral tegmental area in a mouse model of hyperinsulinemia. Nutr Diabetes, 2013. 3(12): p. e97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Konner AC, et al. , Role for insulin signaling in catecholaminergic neurons in control of energy homeostasis. Cell Metab, 2011. 13(6): p. 720–8. [DOI] [PubMed] [Google Scholar]
- 254.Kamei N and Takeda-Morishita M, Brain delivery of insulin boosted by intranasal coadministration with cell-penetrating peptides. J Control Release, 2015. 197: p. 105–10. [DOI] [PubMed] [Google Scholar]
- 255.Fan LW, et al. , Rapid transport of insulin to the brain following intranasal administration in rats. Neural Regen Res, 2019. 14(6): p. 1046–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Kullmann S, et al. , Central Insulin Modulates Dopamine Signaling in the Human Striatum. J Clin Endocrinol Metab, 2021. 106(10): p. 2949–2961. [DOI] [PubMed] [Google Scholar]
- 257.Alsaadi HM and Van Vugt DA, Insulin sensitivity affects corticolimbic brain responses to visual food cues in polycystic ovary syndrome patients. Horm Mol Biol Clin Investig, 2015. 24(2): p. 101–15. [DOI] [PubMed] [Google Scholar]
- 258.Van Vugt DA, et al. , Effect of insulin sensitivity on corticolimbic responses to food picture in women with polycystic ovary syndrome. Obesity (Silver Spring), 2013. 21(6): p. 1215–22. [DOI] [PubMed] [Google Scholar]
- 259.Tiedemann LJ, et al. , Central insulin modulates food valuation via mesolimbic pathways. Nat Commun, 2017. 8: p. 16052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Tiedemann LJ, et al. , Insulin sensitivity in mesolimbic pathways predicts and improves with weight loss in older dieters. Elife, 2022. 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Figlewicz DP, et al. , Insulin acts at different CNS sites to decrease acute sucrose intake and sucrose self-administration in rats. Am J Physiol Regul Integr Comp Physiol, 2008. 295(2): p. R388–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Iniguez SD, et al. , Insulin receptor substrate-2 in the ventral tegmental area regulates behavioral responses to cocaine. Behav Neurosci, 2008. 122(5): p. 1172–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Russo SJ, et al. , IRS2-Akt pathway in midbrain dopamine neurons regulates behavioral and cellular responses to opiates. Nat Neurosci, 2007. 10(1): p. 93–9. [DOI] [PubMed] [Google Scholar]
- 264.Phillips RA 3rd, et al. , An atlas of transcriptionally defined cell populations in the rat ventral tegmental area. Cell Rep, 2022. 39(1): p. 110616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Grove JCR, et al. , Dopamine subsystems that track internal states. Nature, 2022. 608(7922): p. 374–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.de Jong JW, Fraser KM, and Lammel S, Mesoaccumbal Dopamine Heterogeneity: What Do Dopamine Firing and Release Have to Do with It? Annu Rev Neurosci, 2022. 45: p. 109–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Damian M, et al. , GHSR-D2R heteromerization modulates dopamine signaling through an effect on G protein conformation. Proc Natl Acad Sci U S A, 2018. 115(17): p. 4501–4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Kern A, et al. , Apo-ghrelin receptor forms heteromers with DRD2 in hypothalamic neurons and is essential for anorexigenic effects of DRD2 agonism. Neuron, 2012. 73(2): p. 317–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Lockie SH and Andrews ZB, The hormonal signature of energy deficit: Increasing the value of food reward. Mol Metab, 2013. 2(4): p. 329–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.de Macedo IC, de Freitas JS, and da Silva Torres IL, The Influence of Palatable Diets in Reward System Activation: A Mini Review. Adv Pharmacol Sci, 2016. 2016: p. 7238679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Murray S, et al. , Hormonal and neural mechanisms of food reward, eating behaviour and obesity. Nat Rev Endocrinol, 2014. 10(9): p. 540–52. [DOI] [PubMed] [Google Scholar]
- 272.Bittencourt A, et al. , High fat diet-induced obesity causes a reduction in brain tyrosine hydroxylase levels and non-motor features in rats through metabolic dysfunction, neuroinflammation and oxidative stress. Nutr Neurosci, 2022. 25(5): p. 1026–1040. [DOI] [PubMed] [Google Scholar]
- 273.Sun R, et al. , Inflammation in VTA Caused by HFD Induces Activation of Dopaminergic Neurons Accompanied by Binge-like Eating. Nutrients, 2022. 14(18). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.South T, Westbrook F, and Morris MJ, Neurological and stress related effects of shifting obese rats from a palatable diet to chow and lean rats from chow to a palatable diet. Physiol Behav, 2012. 105(4): p. 1052–7. [DOI] [PubMed] [Google Scholar]
- 275.Barry RL, et al. , Brief exposure to obesogenic diet disrupts brain dopamine networks. PLoS One, 2018. 13(4): p. e0191299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Bourdy R, et al. , The endocannabinoid system is modulated in reward and homeostatic brain regions following diet-induced obesity in rats: a cluster analysis approach. Eur J Nutr, 2021. 60(8): p. 4621–4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Davis JF, et al. , Exposure to elevated levels of dietary fat attenuates psychostimulant reward and mesolimbic dopamine turnover in the rat. Behav Neurosci, 2008. 122(6): p. 1257–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Li Y, et al. , High-fat diet decreases tyrosine hydroxylase mRNA expression irrespective of obesity susceptibility in mice. Brain Res, 2009. 1268: p. 181–189. [DOI] [PubMed] [Google Scholar]
- 279.Quansah Amissah R, et al. , Neuronal activities during palatable food consumption in the reward system of binge-like eating female rats. Physiol Behav, 2021. 242: p. 113604. [DOI] [PubMed] [Google Scholar]
- 280.Mazzone CM, et al. , High-fat food biases hypothalamic and mesolimbic expression of consummatory drives. Nat Neurosci, 2020. 23(10): p. 1253–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]