

Abstract
Mutations in the gene MYOC account for approximately 5% of cases of primary open angle glaucoma (POAG). MYOC encodes for the protein myocilin, a multimeric secreted glycoprotein composed of N-terminal coil-coil (CC) and leucine zipper (LZ) domains that are connected via a disordered linker to a 30 kDa olfactomedin (OLF) domain. More than 90% of glaucoma-causing mutations are localized to the OLF domain. While myocilin is expressed in numerous tissues, mutant myocilin is only associated with disease in the anterior segment of the eye, in the trabecular meshwork. The prevailing pathogenic mechanism involves a gain of toxic function whereby mutant myocilin aggregates intracellularly instead of being secreted, which causes cell stress and an early timeline for TM cell death, elevated intraocular pressure, and subsequent glaucomatous neurodegeneration. In this review, we focus on the work our lab has conducted over the past ~15 years to enhance our molecular understanding of myocilin-associated glaucoma, which includes details of the molecular structure and the nature of the aggregates formed by mutant myocilin. We conclude by discussing open questions, such as predicting phenotype from genotype alone, the elusive native function of myocilin, and translational directions enabled by our work.
Keywords: glaucoma, trabecular meshwork, molecular biophysics, protein misfolding, chaperone, amyloid, toxic gain of function, myocilin
1. Introduction
Glaucoma, the leading cause of irreversible blindness (Tham et al., 2014), is a group of chronic optic neuropathies often accompanied by increased intraocular pressure (IOP) (Kwon et al., 2009). This primary risk factor, increased IOP, is caused by an imbalance between production and outflow of aqueous humor (AH), a nutrient-rich fluid produced by the ciliary body that nourishes the anterior chamber of the eye (Goel et al., 2010). AH drains through the trabecular meshwork (TM), a key tissue for proper maintenance of IOP (Llobet et al., 2003). The TM is located in the iridocorneal angle, the angle formed between the iris and cornea (Goel et al., 2010). The most common glaucoma subtype, primary open angle glaucoma (POAG), is characterized by glaucomatous retinal degeneration with an open iridocorneal angle. Adult-onset POAG (>40 years old) and juvenile-onset OAG (3–40 years old) glaucoma affect ~60 million people worldwide (Allison et al., 2020; Tham et al., 2014).
In this review, we focus on the subset of POAG attributed to mutations in the gene encoding for myocilin (MYOC). Myocilin-associated glaucoma is estimated to account for 2–4% of adult-onset POAG and 10% JOAG cases and is characterized by elevated IOP (≥ 21 mmHg) and severe disease progression (Aldred et al., 2004; Fingert et al., 1999). Many new exciting insights have been gleaned since ~2002, when the last review on myocilin was published in this journal (Tamm, 2002). We will center our review on biochemical and cellular studies conducted in our lab since 2010 (Figure 1), which examine molecular details of myocilin structure and the amyloid nature of its pathogenic misfolding, using techniques common to molecular biophysics and structural biology (Table 1). Open questions, such as the still-elusive biological function of myocilin, new translational directions enabled by our work, differentiation among benign polymorphisms and disease-causing mutations, and potential links between wild type (WT) myocilin and glaucoma, will also be explored.
Figure 1. Timeline of myocilin-associated glaucoma research from the Lieberman Lab.

Of our 30+ publications related to myocilin, notable advances in our understanding of myocilin-associated glaucoma have included the discovery that disease-causing mutations are destabilized and primed to aggregate into the highly structured aggregate amyloid, and that mutant OLF interacts with chaperones in a way that promotes further aggregation instead of degradation. We have also structurally characterized both the N- and C-terminal domains, developed conformationally specific antibodies and established that Ca2+-depleted environments promote myocilin misfolding. Most recently, we have developed a quantitative metric to differentiate between pathogenic and benign mutations.
Table 1.
Biophysical techniques used for myocilin expression and characterization.
| Technique | Description | Purpose |
|---|---|---|
| X-ray Crystallography | Structural technique that uses the diffraction effect of X-rays incident upon a crystalline sample to obtain the atomic structure of the molecules generating the crystal lattice | Determination of myocilin domain structures at atomic level resolution |
| Small angle X-ray scattering | Low resolution structural technique that uses the scattering of X-rays incident on a solution sample and modeling to obtain a molecular envelope. | Low resolution molecular shape of a protein. |
| TEM/AFM | Imaging techniques for nanometer sized particles. | Visualization of full length myocilin, and amyloid/aggregate morphologies |
| ThT fluorescence | A fluorescent dye used to detect amyloid. | An increase in fluorescence over background indicates formation of amyloid-like aggregates |
| Recombinant protein expression | Gene on a plasmid is introduced into a heterologous host to produce a protein in large quantities | Allows high yields of purified proteins to be obtained for biophysical characterization |
| Differential Scanning Fluorimetry (DSF) | Assay that uses a solvatochromic dye and monitors increase in fluorescence as a sample is unfolded by slowly heating in an RT- qPCR instrument. | Provides a value for thermal stability. |
| Circular dichroism (CD) | Compares the difference between right and left circularly polarized light incident on a chiral sample. | Gives information about protein secondary structural elements and fingerprint for tertiary structure. Can also give thermal stability values if spectra are acquired at different temperatures. |
| ANS fluorescence | A fluorophore used as a read out for hydrophobicity. | Reveals if hydrophobic residues are exposed, e.g. if a protein is not compact. |
2. Genetics and clinical features of myocilin-associated glaucoma
2.1. Prevalence and clinical presentation of myocilin-associated glaucoma
The prevalence of glaucoma in the general population is <1%, but first degree relatives of patients with POAG have a 4–16% chance of developing the disease (Leighton, 1976), suggesting there is an underlying genetic predisposition. MYOC was the first glaucoma locus to be discovered and has remained the strongest genetic link to glaucoma. An autosomal-dominant inheritance pattern is observed for myocilin-associated glaucoma, along with a diagnosis earlier than 40 years of age, IOPs greater than 25 mmHg, and an average cup-disc ratio >0.3 (Gordon et al., 2002). Historically, myocilin-associated POAG has been seen across large pedigrees with ages of diagnosis as young as 4 years old (Rozsa et al., 1998) and with IOPs reaching as high as 77 mmHg (Alward et al., 1998).
2.2. Initial discovery
Three studies with overlapping timeframes led to myocilin initially being identified with three different gene names; GLC1A, TIGR, and MYOC. In initial studies connecting myocilin mutations to glaucoma, Stone and coworkers looked at numerous individuals, including those with a family history of glaucoma, glaucoma-patients with no family history of the disease, unrelated patients with retinal disease, and volunteers with no history of glaucoma whose IOPs were less than 20 mmHg. A genetic linkage analysis conducted by Sheffield et al. on a 37-member family with a high prevalence of JOAG identified a glaucoma-relevant region in 1q21–31 of chromosome 1; this was later replicated in several other families (Meyer et al., 1994; Richards et al., 1994; Sheffield et al., 1993). Thirteen of the glaucoma patients carried one of three coding mutations within GLC1A, leading to G364V, Q368X or Y437H protein variants. All affected family members carried Y437H or G364V mutations, whereas only one non-glaucoma control patient had a mutation (Q368X) (Sheffield et al., 1993; Stone et al., 1997). Stone et al. named the discovered gene GLC1A, in which “GLC” indicated glaucoma, “1” indicated primary OAG and “A” indicated that this was the first locus identified for this disease (Stone et al., 1997). This gene would later be named MYOC (myocilin).
The second study leading to the initial discovery of myocilin involved the study of elevated IOP as a potential side effect of topical corticosteroid application (Kersey and Broadway, 2006). IOP rises after weeks of continuous treatment with corticosteroids, such as dexamethasone (DEX) or prednisolone, and returns to baseline once treatment is terminated (Francois, 1954). TM cells treated with DEX, a model for steroid-induced glaucoma, secrete a 55 kDa protein (Polansky et al., 1997). Polanksy et al. named this protein trabecular meshwork inducible glucocorticoid response protein (TIGR) (Polansky et al., 1997). TIGR was later found to be identical to GLC1A, and later called myocilin.
The third project that led to the initial discovery of myocilin was seeking to better understand the genetic and molecular basis of light reception, as well as isolate potential retinal disease candidate genes by cloning a human retina cDNA library (Kubota et al., 1997). A 55 kDa protein was identified from this library expressed primarily in the photoreceptor cells of the retina and was flagged as a potential candidate gene for inherited retinal diseases like Usher syndrome. On the basis of sequence homology to myosin (see Section 4.1), Kubota et al. named this protein myocilin. In 1998, the Human Genome Organization Genome Database Nomenclature Committee permanently assigned the name MYOC (myocilin) (Johnson, 2000).
2.3. Myocilin genetics
Since the initial discovery of myocilin mutations, population and familial case-control genetic studies have identified numerous missense, synonymous, stop gained and frameshift mutations in myocilin (Fingert et al., 1999; Fingert et al., 2002; Hewitt et al., 2008; Liu and Allingham, 2017; Tamm, 2002). Myocilin variants (>300) with associated clinical metrics are collated online in the website myocilin.com (Hewitt et al., 2008). The vast majority of known glaucoma-causing mutations are missense coding mutations (Fingert et al., 1999; Fingert et al., 2002; Hewitt et al., 2008; Resch and Fautsch, 2009).
In general, disease-causing mutations are expected to be found at low allele frequencies within a population (Karczewski et al., 2020). Therefore, mutations in MYOC are not generally found by glaucoma-genome-wide association studies (Craig et al., 2020). In other words, the more common a myocilin mutation is found in the general population, the less likely it is to be pathogenic. Of the ~100 population-based mutations from genome sequencing data listed in gnomAD (Karczewski et al., 2020), (https://gnomad.broadinstitute.org/gene/ENSG00000034971?dataset=gnomad_r2_1), the most common OLF-localized non-synonymous coding variants (K398R and E352K) are not considered pathogenic. Q368X, which halts translation midway through the structural OLF domain, may be a genetic modifier (Craig et al., 2020). Interestingly, some glaucoma-related mutations cluster within specific ethnicities, for example G246R/Italian and Y479H/Spanish, while others have been identified in multiple families of different nationalities, for example P370L (Table 2). Conversely, some myocilin mutations not directly implicated in disease are also found in specific populations, such as E352K, which is found predominantly in individuals of African or African American descent (Scelsi et al., 2021). Notably, individuals of African descent are ~2.8x higher risk for glaucoma than other ethnic groups (Restrepo and Cooke Bailey, 2017), but no data to date implicates the myocilin variant E352K in disease (Scelsi et al., 2023).
∂Table 2.
Myocilin mutations with strong support for pathogenicity, adapted from Scelsi 2021.
| Mutation | Ethnicity | Definition |
|---|---|---|
| Cys245Tyr | Chinese | Pathogenic |
| Gly246Arg | French | Pathogenic |
| Gly252Arg | Australian, Chinese, Scottish, US/Caucasian | Pathogenic |
| Arg272Gly | US/Caucasian | Pathogenic |
| Glu323Lys | Panamanian | Pathogenic |
| Gly364Val | US/Cauacsian | Pathogenic |
| Gly367Arg | Japanese, German, Irish, Indian, Swiss, Australian, Scottish, Chinese, French, Canadian, Italian/French | Pathogenic |
| Gly367Gln368delinsVal | Italian | Pathogenic |
| Pro370Leu | Colombian, Chinese, Iranian, Japanese, French- Canadian, Canadian, Indian, Brazilian, Greek, French, US/Caucasian | Pathogenic |
| Thr377Met | Croatian, US/Caucasian, Indian, Australian, African American, Finnish, Greek, Dutch/Croatian, English, Greek/Macedonian, Japanese, Moroccan | Pathogenic |
| Asp380Ala | Spanish, English | Pathogenic |
| Lys423Glu | Brazilian, Italian, French-Canadian, Canadian | Pathogenic |
| Val426Phe | Spanish, US/Caucasian | Pathogenic |
| Cys433Arg | Brazilian | Pathogenic |
| Tyr437His | Chinese, US/Caucasian | Pathogenic |
| Ile477Asn | US/Caucasian | Pathogenic |
| Ile477Ser | French | Pathogenic |
| Tyr479His | Spanish | Pathogenic |
| Asn480Lys | Andean, Malay, French, Dutch, Indian | Pathogenic |
| Pro481Thr | US | Pathogenic |
| Pro481Leu | African American, Canadian | Pathogenic |
| Ile499Phe | French | Pathogenic |
| Ile499Ser | US/Caucasian | Pathogenic |
| Ser502Pro | English | Pathogenic |
| Val251Ala | German, Chinese, US/Caucasian | Likely pathogenic |
| Pro254Arg | Chinese | Likely pathogenic |
| Leu255Pro | Korean | Likely pathogenic |
| Pro274Arg | Indian | Likely pathogenic |
| Gln337Arg | Scottish, Indian | Likely pathogenic |
| Ser341Pro | Chinese, Korean | Likely pathogenic |
| Ala363Thr | Japanese | Likely pathogenic |
| Phe369Leu | Japanese | Likely pathogenic |
| Tyr371Asp | Uzbek | Likely pathogenic |
| Glu385Lys | Hispanic | Likely pathogenic |
| Thr438Ile | Australian, French, Danish | Likely pathogenic |
| Asn450Tyr | Chinese | Likely pathogenic |
| Leu486Phe | Chinese | Likely pathogenic |
3. Myocilin expression and detection
3.1. Myocilin expression in the trabecular meshwork
In accordance with its secretion signal sequence (see Section 4.1), myocilin is secreted from the cell to the extracellular space. Myocilin is expressed throughout the body with particularly high levels of expression in the TM (Adam et al., 1997). Above baseline constitutive expression, myocilin expression in the TM can be induced by corticosteroids such as DEX as shown by Polansky et al. (Polansky et al., 1997). As a result, increased expression of myocilin in response to DEX treatment is commonly used to distinguish TM cells from cells in the anterior segment (Keller et al., 2018; Nguyen et al., 1998; Polansky et al., 2000). Interestingly, although the progressive induction of myocilin protein expression occurs on a timescale concurrent with the clinical manifestation of corticosteroid-induced glaucoma (days-weeks) (Polansky et al., 1997) and the dose response of myocilin induction also mimics the increased IOP and outflow resistance seen for patients who receive glucocorticoid therapy (Armaly, 1963a, b), a direct relationship between myocilin and steroid-induced IOP elevation has been ruled out by the finding that DEX treatment can still induce IOP elevation in Myoc−/− mice (Patel et al., 2017). Myocilin overexpression as a result of steroid treatment is likely a secondary response, perhaps from the stress of high IOP or tissue damage (Borras et al., 2002; Polansky et al., 1997; Tamm et al., 1999).
3.2. Posttranslational modifications
Several posttranslational modifications have been reported for myocilin, although their connection to protein synthesis and function remain unclear. First, the secretion signal was confirmed experimentally (Nguyen et al., 1998; Shepard et al., 2003). Second, human myocilin appears as a 55/57-kDa doublet on denaturing and reducing gel electrophoresis (Jacobson et al., 2001; Nguyen et al., 1998; Shepard et al., 2001) due to incomplete N-glycosylation at N57 (Shepard et al., 2003). Third, some evidence has been put forth for intracellular endoproteolytic processing of myocilin (Aroca-Aguilar et al., 2005), specifically by calpain II in adherent HEK293T cells (Sanchez-Sanchez et al., 2007). We also observed an accumulation of proteolyzed species during the purification of myocilin expressed from suspension Expi293F cells. Mass spectrometry analysis revealed imprecise cleavage in the linker at numerous sites between residues 203–207 (Martin et al., 2021), ~20 residues earlier than reported for calpain II (Aroca-Aguilar et al., 2005). While a calpain-like enzyme may be the source of proteolytic cleavage, it is also possible that cleavage occurred by less selective enzymes such as those of the serine or metalloprotease families. To our knowledge, proteolytic cleavage has not been reported in myocilin isolated from TM cells, suggesting that the proteome and related regulation may be different, but any relevance to function or pathogenesis remains unknown.
3.3. Myocilin detection in research
Several anti-myocilin antibodies are commercially available, and our lab has evaluated epitopes for four. All four antibodies recognize enriched endogenous myocilin secreted from human TM (hTM) media. RRID:AB_776605 recognizes the far N-terminal region, RRID:AB_2721107 recognizes CC, and RRID:AB_2148649 recognizes LZ. The final antibody, RRID:AB_2148737, binds OLF (Patterson-Orazem et al., 2018). Due to the limited epitope variety, our lab has developed new antibodies (Patterson-Orazem et al., 2021), which have been licensed to Absolute Antibody and will be available commercially in the near future.
4. Myocilin structure
4.1. Domain organization
Initial insights into myocilin came from domain analysis of its protein sequence. Mature myocilin is a modular protein. The first 32 amino acids were predicted to comprise a secretion signal sequence (Kubota et al., 1997; Nielsen et al., 1997), which has been experimentally confirmed (see Section 3.2). Myocilin is composed of two protein domains that are connected via a 60-residue unstructured linker (Figure 2A). The N-terminal domain of myocilin (~25 kDa) has a predicted coiled-coil (CC) region similar to its namesake, the muscle protein myosin. Within the myocilin CC there is also a subdomain of coiled-coil called a leucine zipper (LZ) in which leucine residues appear in a particular pattern in the sequence (Kubota et al., 1997). The C-terminal domain was identified as a ~30 kDa olfactomedin (OLF) domain due to high sequence homology to olfactomedins from bullfrogs (Snyder et al., 1991; Yokoe and Anholt, 1993). This OLF domain is where the majority of glaucoma-related mutations are found (Fingert et al., 1999; Fingert et al., 2002; Hewitt et al., 2008; Resch and Fautsch, 2009).
Figure 2. Structural characterization of full length myocilin and individual components (Donegan et al., 2015; Hill et al., 2017).

(A) Myocilin domain map. (B) Myocilin C-term OLF domain is composed of a 5-bladed β-propeller with a di-nuclear metal center (Ca2+ in yellow; Na2+ in pink). (C) Crystal structure of mouse LZ, confirming the interdigitated leucine residues characteristic of a canonical leucine zipper. (D) Model of full length myocilin; Y-shaped dimer-of-dimers containing an N-term tetramer domain with two branching parallel CC dimers, connected to a C-term OLF domain with a di-nuclear metal center via a disordered linker region. (E) Visualization of three 2D class-averages of full-length myocilin consistent with tetramer model. Reprinted from (Martin et al., 2021) with permission. (F) Disease-causing variants mapped onto the OLF crystal structure. Reprinted from (Scelsi et al., 2023) with permission.
4.2. Model of full-length myocilin
Currently, there is no high-resolution structure of full-length myocilin. Based on our divide-and-rebuild approach, we infer that full-length myocilin is composed of an N-term tetrameric stalk with two parallel dimer-of dimer branches that are connected to pairs of C-term OLF domains via a disordered linker (Figure 2D). Previous studies of myocilin have detected both tetrameric (Gobeil et al., 2004; Nguyen et al., 1998), dimeric (Dismuke et al., 2012; Fautsch and Johnson, 2001; Gobeil et al., 2004; Nguyen et al., 1998; Russell et al., 2001; Stamer et al., 2006), and higher-order states (Dismuke et al., 2012; Fautsch et al., 2004; Russell et al., 2001). We have also seen evidence for a weak/transient OLF dimer that can be captured by chemical cross-linking (Huard et al., 2019). Our studies are further consistent with the interpretation that tetramerization occurs because of the residues present in the N-terminal CC stalk and does not involve disulfide bonds. Rather, our data indicate that disulfide bonds serve to stabilize the dimers that comprise LZ, culminating in the structurally resolved disulfide bond between two copies of Cys185. The tetrameric arrangement generated by the N-terminal domain yields four OLF domains per folded myocilin. OLF domains of two myocilin protomers are paired by their N-terminal disulfides, yet have some flexibility in their interactions due to the long linker. Two pairs of OLF dimers are separated by the angle between two LZ domains at the interface between CC and LZ. It is likely that this arrangement increases OLF avidity for binding partners, but one that requires both flexibility and distance.
Recently, we obtained the first low-resolution direct evidence for the proposed Y-shaped tetramer (Martin et al., 2021). We purified full length myocilin from Expi293F cells, which we visualized using negative stain transmission electron microscopy (TEM) (Table 1). Well-dispersed ~10 nm particles were observed on the TEM grids. Single particle reconstruction was used to generate low resolution 2D class averages. Each class had a dense sphere matching the OLF domain, and there was an extended structure consistent with the N-terminal domain (Figure 2E). In addition to the Y-shaped tetramer, there was also a potential octameric species that was linked at the N-term, as well as smaller species that are likely the result of partial cleavage.
A structure of full-length myocilin is an active area of research. Even if homogeneity could be improved, crystallization of full-length myocilin is unlikely to be successful due to the unstructured linker, which introduces conformational variability that impedes crystal growth. Cryogenic electron microscopy is an alternative, though the tetramer of myocilin is near the lower limit of particle size currently visualized using this method, and the linker may introduce additional complexities in data processing. Computational models, including Alphafold (Jumper et al., 2021), do not yield confident models outside of experimentally-characterized OLF, likely due to the fact that myocilin is a multimer and each protomer has an unstructured linker. Since protein structure and function are inextricably linked, an experimental structure of full-length myocilin will be an important step in generating new ideas about myocilin function.
4.3. Structural arrangement of CC/LZ
Our lab studied the organization of the CC/LZ region, in the absence of the OLF domain, by purifying different CC subdomains. We used small angle X-ray scattering (Table 1) to show that the CC domain forms a Y-shaped biopolymer. We solved the crystal structure of the LZ domain from mouse, revealing a dimer capped by a disulfide bond (Figure 2C). LZ is largely a prototypical leucine zipper with a few exceptions, such as one instance where a serine is present in place of a leucine (Figure 2C). Our data lead to the model of a parallel tetramer base with two branching LZ dimers.
4.4. OLF structure
4.4.1. Polypeptide arrangement
The crystal structure of the human myocilin OLF domain (Table 1) was solved by our lab, revealing the molecular arrangement of the polypeptide chain as well as additional details of the metal center (Donegan et al., 2015). OLF is a 40 Å wide 5-bladed β-propeller. Each blade (named A-E) of the β-propeller is composed of four antiparallel β-strands that are arranged radially around a water-filled cavity. The blades are notably asymmetric, and the central cavity contains the metal center (Figure 2B and see Section 4.1.2.2). Extensive interactions between discontinuous outer strands and a single disulfide bond act to stabilize the propeller in a closed circular conformation.
4.4.2. Metallocenter
Our lab first established that the OLF domain of myocilin has a metal binding site. Metal analysis initially measured a molar ratio of 1:1 Ca2+:OLF polypeptide and identified residue D380 as a metal-coordinating ligand (Donegan et al., 2012). Although the presence of stoichiometric calcium in OLF was known, the OLF crystal structure revealed atomic details of the metallocenter located in the internal cavity, in an unexpected dinuclear arrangement. The calcium ion is coordinated by side chains of D380, N428 and D478 as well as the carbonyl backbones of A429 and I477. The Ca2+ ion appears to act as an ionic tether for blades B, C, D and E (Figure 2B). The second metal ion, which is 3.4 Å deeper into the cavity, best modeled as Na+ in the X-ray structure, but the true identity of the metal is not yet known. A glycerol molecule, introduced during harvesting of the crystals, was also found in the hydrophilic cavity above the Ca2+ site. Even though loops on the top and bottom of the domain are well ordered in the crystal structure, the presence of an internal glycerol molecule in the protein suggests that molecules from the protein surface can access the central cavity (Donegan et al., 2015). The molecular role of calcium remains unclear, except that calcium is essential for proper folding of WT OLF. Namely, when WT OLF was expressed in E. coli under strictly calcium-free conditions, no soluble, properly folded monomer was detected (Saccuzzo et al., 2022).
4.4.3. Structures of OLF homologs
The myocilin OLF domain is just one branch of the olfactomedin protein family (Zeng et al., 2005). The OLF 5-bladed β-propeller is now confirmed for all other olfactomedin domain-containing proteins. The dinuclear metal center is also common to OLFs, aside from the gliomedin subfamily (Hill et al., 2015). Interestingly, the interior portion of OLF contains the most highly conserved residues, whereas the surface has the highest degree of sequence divergence. It is possible that the metal center in the central cavity plays a functional role. Enzymatic function at the metal center has also not been ruled out, although there are no hints from other systems.
5. Pathogenic OLF variants
More than 90% of known glaucoma-causing mutations are missense coding mutations located in the OLF domain (Fingert et al., 1999; Fingert et al., 2002; Hewitt et al., 2008; Resch and Fautsch, 2009). Structural studies have shown that disease-causing OLF variants are distributed throughout all 5 blades of the OLF domain (Figure 2F) (Donegan et al., 2015). Prior to the availability of the OLF structure, it was not clear whether disease variants would be clustered in the tertiary structure, as residues far in sequence can be close in space in a folded protein. We attempted to highlight parts of the structure in which pathogenic mutations appeared, such as the long loop connecting blade B to C, the metal center, and the hydrophobic belt (Donegan et al., 2015). However, this was not strict clustering. The radial nature of the OLF propeller limits interactions of any given amino acid to others within ~30 residues, i.e. the typical number of residues comprising a propeller blade, in either direction in the sequence.
5.2. OLF variants with WT-like structure
We have solved crystal structures for a number of OLF variants, albeit none that are disease-associated. We have tried extensively to obtain crystals of moderate variants such as D380A, but disease mutants are not amenable to crystallization. X-ray crystallography requires millions of identical copies of a protein to arrange into a well-defined 3-dimensional crystalline lattice that can robustly diffract X-rays. Less stable proteins, like disease-associated OLF variants (see Section 6.4), likely exhibit too much conformational variability for well-defined crystals to grow.
However, we have solved structures of OLF variants that are structurally indistinguishable from WT. For example, combined with supporting data, E396D (Donegan et al., 2015), T293K, R296H, L303I, V329M, S331L, E352K, T353I, K398R, A445V, V449I, and K500R (Scelsi et al., 2023), are likely benign polymorphisms. Only R296H exhibited an observable local perturbation in its structure, likely because the His sidechain is shorter than that of Arg. In addition to similarities of their polypeptide chains, all of these variants possess native metal centers (Scelsi et al., 2023).
5.3. OLF variants with non-native structure
OLF variants that involve mutating metal binding residues result in altered structures, first detected for the disease-associated variant D380A (Hill et al., 2014). As expected, substitution of calcium ligand D478 for Ser, Ala, or Asn eliminates calcium binding. However, unlike disease-associated D380A, variants D478S and D478N are stable and were amenable to crystallization. The structures not only reveal changes in the metal binding site but also show disordered loops, side helix unwinding, and a shift in blade A (Figure 2B). Notably, no additional structural changes were observed for the double mutant D380A/D478S, which has WT-like stability (Hill et al., 2019b). Interestingly, WT-OLF structure can be reverted by introducing triads of residues found naturally in sequences of invertebrate chordate olfactomedin homologs (see Section 4.1.2.5) at positions of D380, D478, and N428, even if some triad arrangements preclude metal binding (Hill et al., 2019a).
6. Pathogenic mechanism: a toxic gain of function due to myocilin misfolding
6.1. Initial evidence for a gain of function mechanism
Autosomal dominant disorders, such as glaucoma, can be caused by 3 general mechanisms: haploinsufficiency, gain of function, or a dominant negative mechanism (Wiggs and Vollrath, 2001). Haploinsufficiency was quickly ruled out for myocilin-associated glaucoma, as a patient with a known interstitial deletion of 1q23 to 1q25 (Franco et al., 1991), and therefore only 1 functional copy of MYOC, did not present with clinical evidence for glaucoma (Wiggs and Vollrath, 2001). Dominant-negative, which involves a mutant gene product impairing WT function, was also ruled out. In early studies with Chinese families, members both with and without a POAG diagnosis were found to be either homozygous (Lam et al., 2000) or heterozygous (Pang et al., 2002) for the null mutation Arg46Stop. This truncation mutation results in the deletion of ~90% of the protein. We now know Arg46X is one of the most common gene variants in gnomAD, predominant in Asian populations. In addition, deletion of murine myocilin does not affect IOP or TM architecture, and mice are viable and fertile (Kim et al., 2001). Taken together, the absence of myocilin does not cause glaucoma.
Thus, even early on, a toxic gain of function mechanism for mutant myocilin seemed most likely. Initial speculation on how mutant myocilin contributed to glaucoma was that excess secreted myocilin collected in the TM, obstructing aqueous outflow. However, this idea was ruled out by studies showing mutant myocilin experiences decreased cellular secretion (Figure 3A) (Caballero and Borras, 2001; Caballero et al., 2000; Jacobson et al., 2001; Joe et al., 2003) , and this intracellular accumulation is toxic (Yam et al., 2007). Primary TM cells expressing mutant myocilin are not widely available. To our knowledge, there is just one histochemical study reporting endoplasmic reticulum-localized accumulation of mutant (Y437H) myocilin from a donor eye (van der Heide et al., 2018). Since transfecting TM cells is low efficiency and goes against community standards (Keller et al., 2018), additional support for the toxic gain of function came from a cellular assay using model cell likes in which disease-associated myocilin variants accumulated as an insoluble material in the detergent Triton X-100, and benign polymorphisms remained soluble (Zhou and Vollrath, 1999). These aggregates appear to be localized to the endoplasmic reticulum in HEK293 cells transfected with mutant myocilin (Liu and Vollrath, 2004). Consistent with the idea that myocilin-associated glaucoma is a protein conformational disorder where myocilin is susceptible to misfolding, cellular secretion was rescued for some variants by decreasing the temperature of the culture media (Vollrath and Liu, 2006). Although this assay is not highly quantitative, cellular secretion and accumulation of a Triton X-100 insoluble species remains the predominant method to characterize mutant myocilins in cells (Figure 3B).
Figure 3. Proposed pathway for myocilin secretion from TM cells.

(A) In the case of WT myocilin (left), the protein is properly folded and gets secreted into the aqueous humor. Glaucomatous mutations (right) cause myocilin to misfold, promoting aberrant interactions with chaperones that leads to aggregation and sequestration within the ER. This accumulation of mutant myocilin causes ER stress, which results in eventual cell death. (B) Dot blot analysis (top) of secreted myocilin from spent media from transfected HEK293T cells compared to Western blot (bottom) analysis of detergent-soluble and -insoluble lysates for WT myocilin compared to severe disease-causing variants P370L and Y437H. Reprinted from (Hill et al., 2019b) with permission.
6.2. Mutant myocilin intracellular aggregates have hallmarks of amyloid
Cells have elaborate processes by which incorrectly folded proteins are triaged and degraded (Houck et al., 2012). Therefore, the observation that pathogenic mutant myocilin accumulates as insoluble material in the detergent Triton X-100 (Zhou and Vollrath, 1999) indicated to us that the aggregates may not be amorphous, but rather a templated fibrillar material called amyloid. Generally, amyloid formation by a folded protein involves partial unfolding to an aggregation-prone state that first groups together to form oligomers, which come together to form protofibrils, and further template mature fibril formation (Figure 4A). Amyloids are recalcitrant to resolubilization in harsh reagents like Triton X-100 and are common to protein misfolding diseases such as Alzheimer, Parkinson, and Amyotrophic Lateral Sclerosis, among ~50 others (Dobson, 2017).
Figure 4. Proposed aggregation pathway for mutant myocilin.

(A) Aggregation into amyloid begins with myocilin deviating from its native state into an aggregation prone partially-folded state, due to a destabilizing mutation or other destabilizing condition. This partially-folded state exposes amyloid-prone regions (APRs) that template fibrillization via misfolded oligomers, which assemble to form protofibrils, and then serve as templates for the mature end-point fibrils. (B) Representative nucleation-dependent fibrillization of OLF incubated at 37 °C, monitored by Thioflavin T (ThT) fluorescence. Overlay of differential interference contrast and ThT fluorescence confirms intracellular amyloid accumulation. (C) Intracelluar ThT fluorescence of myocilinP370L. Reprinted with permission from (Hill et al., 2014; Orwig et al., 2012).
To test whether aggregates formed by mutant myocilins were forming a templated amyloid architecture, we conducted experiments using purified proteins and experiments in cells. After multiple days of gentle rocking at 37°C, purified OLF converts from a folded protein to one with characteristic sigmoidal growth to a state with high fluorescence of Thioflavin T (ThT), a dye known to selectively fluoresce in the presence of amyloid (Table 1, Figure 4B). In collaboration with Douglas Vollrath, we expressed both WT and mutant myocilin in Chinese hamster ovary (CHO) cells. CHO cells expressing WT myocilin displayed minimal ThT fluorescence, signifying low levels of amyloid aggregation. In contrast, CHO cells expressing myocilinP370L, a severe glaucoma-causing variant, exhibited a strong ThT fluorescent signal (Figure 4C) (Orwig et al., 2012). More recent cellular studies using HEK293T cells from our lab have shown colocalization between a newer commercial amyloid detecting dye called Proteostat and the ER marker calnexin when cells expressed myocilinP370L. Thus, cellular data indicate that instead of being secreted, pathogenic mutant myocilins accumulate amyloid aggregates in the ER (Scelsi et al., 2023).
6.3. Mutant myocilin cannot be removed by chaperone-mediated degradation
Failure of the ubiquitin-proteasome pathway to clear mutant myocilin aggregation of misfolded proteins within the ER can trigger ER stress and lead to cell death (Liu and Vollrath, 2004). Our laboratory, in collaboration with Brian Blagg and the late Chad Dickey, established that mutant myocilin is a client of glucose-regulated protein 94 (Grp94) (Suntharalingam et al., 2012). Grp94 triages mutant myocilin through the ER-associated degradation (ERAD) process, and mutant myocilin is properly ubiquitinated, but ERAD ultimately fails. When Grp94 is depleted by siRNA, mutant myocilin is degraded. Additional studies identified that interactions between OLF and Grp94 are is localized to the N-terminal nucleotide-binding domain. The presence of the isolated N-terminal domain of Grp94 (Grp94N) increases the rate of OLF aggregation when assayed in vitro (Figure 5A) (Huard et al., 2019).
Figure 5. Inhibition of Grp94 prevents aberrant interaction with mutant myocilin.
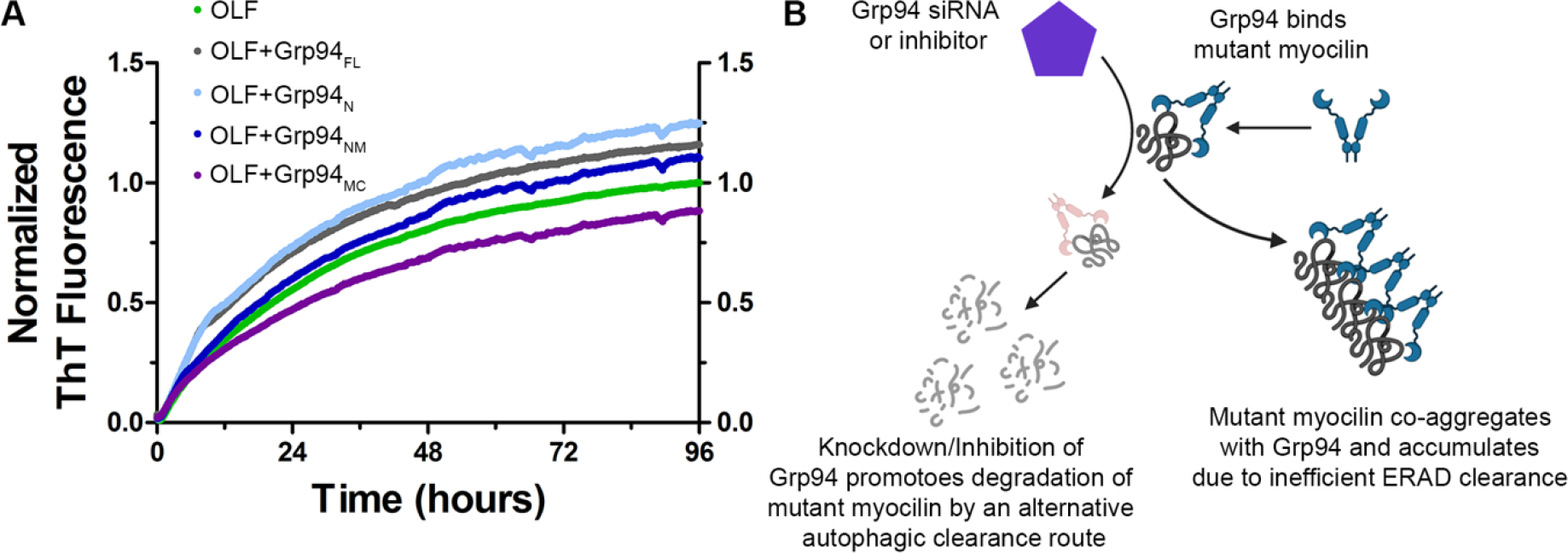
(A) Grp94 interacts with mutant myocilin to accelerate aggregation. Grp94 was divided into its component domains N, M, and C, expressed and purified for aggregation experiments. N=N-terminal ATP-binding domain, FL=full-length, M=middle domain, C=C-terminal dimerization domain. Grp94N enhances the rate of OLF aggregation to a similar extent as Grp94FL, whereas Grp94MC appears to stabilize OLF against aggregation, as indicated by ThT fluorescence. Printed with permission from (Huard et al., 2019). (B) Model showing prevention of Grp94 interactions with mutant myocilin via Grp94 inhibitors allows misfolded myocilin to be cleared via an alternate autophagic pathway, preventing aggregation.
The aberrant interaction between mutant myocilin and Grp94 can be inhibited via selective inhibition of Grp94 with small molecules (Figure 5B) (Huard et al., 2018; Stothert et al., 2014). This action shunts myocilin away from ERAD towards the autophagy system, a more robust clearance pathway, where it is efficiently degraded (Suntharalingam et al., 2012). One of the most selective Grp94 inhibitors, 4BrBnIm, was even effective at lowering IOP to baseline in a mutant myocilin mouse model (Stothert et al., 2017). Taken together, inhibiting Grp94 may be a therapeutic route for myocilin-associated glaucoma (see section 7.2).
6.4. Disease-associated OLFs are thermally destabilized and adopt non-native structures
In general, amyloid fibril formation by a folded protein, such as the myocilin OLF domain, is promoted by an initial partial unfolding event, introduced by a mutation or other environmental perturbations (Figure 4A). This scenario exposes segments of the polypeptide chain prone to amyloid aggregation, called amyloid-prone regions (APRs), for further templating of the aggregate. Using a fluorescence-based stability assay (Table 1), our lab established that glaucoma-causing mutations within OLF experience compromised thermal stability, the first indication that disease variants are adopting non-native, partially-folded protein states. In our initial studies, we noted that the least stable variants are correlated with earlier onset glaucoma (Burns et al., 2011), but we have since revisited this idea (see Section 7.1.1). In contrast to OLF, variants within CC/LZ do not affect stability and are not prone to aggregation (Hill et al., 2017).
Our lab conducted an in-depth molecular study to compare the solution characteristics of recombinantly expressed and purified WT OLF and four representative variants: a benign polymorphism (K398R), alongside mild (A427T), moderate (D380A) and severe (I499F) disease-causing variants. Circular dichroism spectroscopy (CD, Table 1) indicated that the four variants retain secondary structure, but the disease variants exhibit deviations in tertiary structure compared to WT OLF (Figure 6A). Anilinonapthalene-8-sulfonic acid (ANS) fluorescence (Table 1) showed the disease variants had higher levels of exposed hydrophobic regions compared to WT (Figure 6B) (Hill et al., 2014). All disease variants form ThT positive fibrils under conditions in which WT does not aggregate (Figure 6C). Amyloid fibrillization can only be induced in WT OLF through perturbations such as agitation (Figure 4B), acidic pH, or high temperatures (Orwig and Lieberman, 2011; Orwig et al., 2012), where presumably WT is also able to access a non-native state.
Figure 6. OLF disease variants adopt non-native, aggregation-prone states.
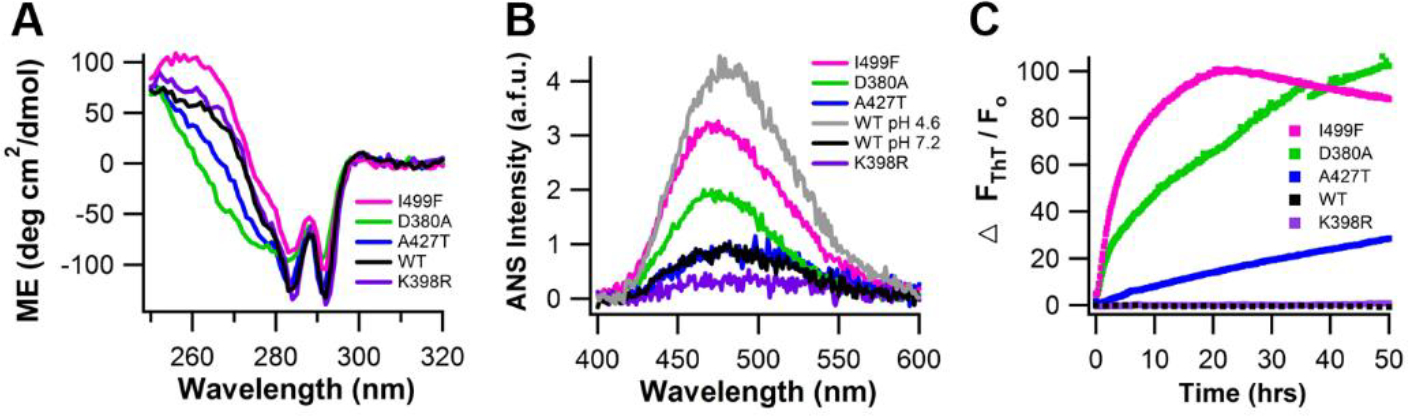
(A) Tertiary structure measured by near-UV CD. (B) ANS fluorescence of disease-mutants, with overlay of wild-type OLF at physiological pH (7.2) and denaturing pH (4.6). (C) ThT fluorescence monitored for 50 hr at 36°C. Reprinted with permission (Hill et al., 2014).
The effect of a non-synonymous mutation on OLF is complex and difficult to predict (see Section 7.1.1). While a single point mutation can lead to a destabilized, aggregation prone disease variant, not all mutations in OLF are destabilizing. First, polymorphism variants structurally characterized in Section 5.2 are indistinguishable from WT OLF in their laboratory characteristics. Most of the polymorphisms we characterized are located on the OLF surface where they experience fewer interactions compared those occurring between the blades where the hydrophobic residues are compactly arranged. The finding that V329M is WT-like was surprising because of its internal location where we intuited that a larger side chain might not fit given its internal location. Second, we predicted that all mutations involving ligands to Ca2+ would behave like D380A, a disease variant that destabilizes OLF and abolishes Ca2+ binding. This seemed like a reasonable hypothesis given that the metallocenter is centrally located in a highly conserved region of the protein, and tethers 4 blades in the propeller with ionic interactions, which would be lost upon mutation. However, D478S and related mutations replacing Asp with Asn or Ala, result in a more stable protein that does not aggregate, even though loss of metal binding causes structural perturbations (Section 5.3, (Hill et al., 2019b)). Finally, we pushed the effect of mutation on stability and misfolding to an extreme in a protein engineering study in collaboration with Sarel Fleishman’s lab. We challenged their computational prediction program PROSS (Protein Repair One Stop Shop) to predict mutations that would stabilize OLF without perturbing the metallocentre. The resulting OLF variant, harboring 21 mutations throughout the sequence, is the most stable of all OLFs measured to date (Goldenzweig et al., 2016) and retains native structure (Hill et al., 2019b).
6.5. Morphology of OLF aggregates and identification of amyloid prone peptide regions
To better understand the molecular details of myocilin aggregation beyond the initial observation (Orwig et al., 2012), we biophysically characterized OLF fibrils in vitro. WT OLF can be driven to fibril formation under mildly destabilizing conditions in neutral pH buffer with salt (Hill et al., 2014). Two main fibril morphologies were identified using atomic force microscopy (AFM). The first morphology, straight fibrils typical of amyloid aggregates, is generated by incubating WT OLF at 37°C with very gentle agitation (Figure 7A). The second morphology, which form as a result of incubating WT OLF at a slightly elevated temperature of 42°C, appears as a circular rope-like structure ~300 nm or larger in diameter (Figure 7B). Both morphologies share Fourier-transform infrared resonance (FTIR) signatures of amyloid. Namely, there is a shift between FTIR signatures of a native β-sheet in the monomer sample compared to a feature corresponding amyloid in the aggregated sample. Fibrils formed by A427T and I499F have the straight morphology similar to fibrils of WT OLF that was gently agitated at 37 °C, whereas D380A aggregates had a circular morphology that was similar to those formed by WT OLF incubated at 42°C (Hill et al., 2014). Thus, in addition to identifying two different morphologies of mature fibrils common to OLF and disease variants, we have developed a highly reproducible aggregation assay. This assay uses WT OLF aggregation as a proxy for disease variants that has found utility across numerous studies in the lab (Huard et al., 2018; Huard et al., 2019; Stothert et al., 2014).
Figure 7. Morphology of OLF fibrils visualized by AFM.

(A) Straight fibrils, common to many amyloids, formed as a result of incubating WT OLF at 37°C with gentle rocking. (B) Unique micron-length lasso oligomers, formed by incubating WT OLF at 42°C. (C) Straight fibril morphology observed for P1. (D) Closed loop morphology observed for P3. Scale bars for (C)-(D) are 300 nm. Reprinted with permission from (Hill et al., 2014).
With ample biophysical and cellular evidence for amyloid formation by myocilin, we identified APRs within OLF, namely peptide stretches with high amyloid propensity that might be responsible for templating fibril formation in the full-length OLF domain. The OLF protein sequence was evaluated by multiple amyloid prediction algorithms (Ahmed and Kajava, 2013; Oliveberg, 2010; Trovato et al., 2007; Tsolis et al., 2013). Three consensus peptides within OLF emerged as top contenders, and two (P1 and P3) were validated to aggregate experimentally (Hill et al., 2014). Aggregates formed by APR P1 had a similar straight morphology to those seen for WT OLF gently agitated at 37 °C (Figure 7A, C), and APR P3 aggregates had a circular morphology mimicking fibrils formed by WT OLF incubated at 42°C (Figure 7B, D), when visualized by AFM (Table 1) (Hill et al., 2014). Taken together, these data suggest that particular APR stretches in the OLF protein sequence are responsible for its fibrillization properties. The extent to which the detailed molecular arrangement of the isolated APRs P1 (Gao et al., 2021) and/or P3 relate to pathogenic myocilin aggregates is an active area of inquiry.
6.6. Mouse OLF aggregates faster than human OLF
Recent findings in the amyloid field have implicated intermediate aggregates as the likely toxic species rather than end-point fibrils (Hartley et al., 1999; Walsh et al., 1999). Mouse OLF aggregates faster than human OLF in vitro, so we posit that the increased aggregation rate for mouse OLF aggregation decreases the lifetime of the toxic misfolded intermediate, lessening the potential for damage (Patterson-Orazem et al., 2019). The crystal structures of human and mouse OLF, which is 87% identical in sequence to human OLF, are superimposable. The main difference is that mouse OLF has a more neutral and varied surface compared to the largely negative surface of human OLF. These differences may explain why expressing mutant mouse myocilin is insufficient to elicit the robust IOP elevation phenotype in humans with the same mutations (Senatorov et al., 2006). Indeed, to date, elevated IOP has only been observed when mutant myocilinY437H is introduced either as a transgene (Zode et al., 2011) or by adenovirus (McDowell et al., 2012).
7. Open questions and future directions
7.1. Predicting phenotype from genotype
The explosion of genome sequencing data has yielded new myocilin mutations, all of which are relatively rare (<<1% of the population). The vast majority of these mutations are novel, with no clinical data available, placing them in the category of uncertain pathogenicity. Pathogenicity of a given mutation within OLF is best supported by a combination of data from the clinic and laboratory. Conversely, the lack of either clinical data or laboratory characterization leads to less definitive assignments of pathogenicity. Computational algorithms developed to predict the effect of a mutation on resultant protein function are tailored to loss-of-function mutations. These algorithms do not perform well in the case of gain of function mutations (Flanagan et al., 2010), such as the case for myocilin-associated glaucoma (Scelsi et al., 2021).
The ability to confidently assign any given myocilin mutation as pathogenic would be transformative as it would motivate genotyping for early diagnosis and monitoring or intervention for glaucoma, a painless disease that often goes diagnosed until well-advanced and visual field is lost (Kendrick, 1999). Experimentally, we have observed paradoxical consequences of mutation, particularly when the metallocenter is perturbed: D380A is misfolded and pathogenic, whereas D478S/A/N variants are more stable and do not aggregate, despite adopting a non-native fold. Further, the ability to predict phenotype from genotype would motivate the development of new precision medicines for myocilin-associated glaucoma.
7.1.1. OLF domain
In a recent literature search, we reviewed evidence for pathogenicity for 97 OLF-resident myocilin variants. Criteria we considered for pathogenicity were multiple generations of sequencing data demonstrating an autosomal dominant inheritance pattern, a clinical diagnosis of early onset POAG, and supporting data indicating a misfolding phenotype (see Section 6). Of these, 23 missense mutations and one indel variant met all criteria for pathogenicity and 7 met criteria for benign. The remaining missense variants, 57 in all, were of uncertain pathogenicity, with 23 deemed likely pathogenic but lacked data in one or more of the criteria (Table 2) (Scelsi et al., 2021). Analysis conducted independently by the Clinical Genome Resource (ClinGen) variant curation expert panel (VCEP) for glaucoma considered 15 factors for pathogenicity, including allele frequencies, with rarer mutations deemed more likely pathogenic, computational evidence for a mutation having a detrimental effect on the gene product, evidence of a damaging effect in vitro or in vivo, and presence in control populations (benign) versus co-segregation in multiple generations within families (pathogenic) (Burdon et al., 2022). Classifications of non-synonymous OLF variants by ClinGen agree with our analysis. The only difference was Q368X; the ClinGen assessment labeled this variant as pathogenic whereas ours was of uncertain significance. Going forward, having a standardized set of rules defined by ClinGen provides a consistent framework for classifying new OLF variants across labs worldwide (Burdon et al., 2022).
Despite these efforts, predicting pathogenicity for new variants, particularly in the absence of clinical data, remains a challenge. In a recent study, we obtained laboratory data for 16 of the most frequently reported OLF mutations in gnomAD. Many of these variants had segregated in glaucoma patients in early studies but have since appeared in increasing allele frequencies in large-scale genome sequencing data suggesting they may be benign. Through this in-depth analysis, just one of the 16 variants exhibited multiple lines of evidence for pathogenicity (E300K). However, by considering all available laboratory data, we established the first quantitative, highly statistically significant metric that distinguishes disease variants from benign ones: OLF variants with a thermal stability <47°C can be classified as disease-causing (Figure 8, (Scelsi et al., 2023)). While a straightforward metric, the drawback is that each new mutation must be characterized for thermal stability in the lab. Additional efforts are needed in this area.
Figure 8. Clustering of disease and benign variants by thermal stability.

There is a cutoff between disease (blue) and benign (green) variants by thermal stability (Tm) at 47 °C. p<0.001, ***; p<0.0001, ****. Reprinted from (Scelsi et al., 2023).
7.1.2. CC, LZ, Linker
Few mutations have been documented outside of the OLF domain, including only one in the linker region (Hewitt et al., 2008), and pathogenicity assignments are not as well supported in the literature as they are for variants localized to the OLF domain. To our knowledge, only one study evaluated cellular trafficking and adhesion properties for three N-terminal variants: L95P, considered a neutral polymorphism in CC, and R82C and R126W, considered disease variants in CC and LZ, respectively. Unlike OLF, all three N-terminal variants were secreted, but R82C and L95P were partially sequestered intracellularly (Gobeil et al., 2006). Our lab characterized the above three variants plus R128W in LZ. Purified CC/LZ reversibly unfolds and refolds, and mutations in the LZ have no detectable misfolding phenotype. Only R82C and L95P in CC promote any kind of misfolding, observed as a change in retention time in size exclusion chromatography suggestive of quaternary structural changes (Hill et al., 2017). Additional variants of unknown pathogenicity are documented only in gnomAD. Whether such variants are causal for glaucoma and by what mechanism remains an open question.
7.2. Development of disease-modifying treatments for myocilin-associated glaucoma
Due to its clear association with glaucoma combined with an understanding of its molecular pathogenesis, MYOC is an attractive target for a glaucoma therapy. Based on our research, two conceptual pharmacological approaches have emerged. First, it should be possible to stabilize mutant myocilin to prevent aggregation, allowing mutant myocilin to escape detection by ER quality control and thus be secreted; a so-called pharmacological chaperone (Convertino et al., 2016). In this scenario, TM cells would then engulf mutant myocilin and degrade it upon phagocytosis (Matsumoto and Johnson, 1997). To this end, we developed a high throughput screen to identify small molecule ligands for OLF and showed that treatment of a stable HEK293T cell line expressing I477N mutant myocilin with identified molecules led to increased secretion (Orwig et al., 2014). Second, it should be possible to intervene during the process of mutant myocilin aggregation to promote degradation. Specifically, our work has shown that targeting Grp94 with a small molecule prevents its interaction with mutant myocilin and promotes mutant myocilin degradation (Stothert et al., 2017). We are continuing to pursue more potent and selective small molecules that inhibit Grp94, as well as other strategies such as comprehending the complement of chaperones that recognize mutant myocilin, which might reveal additional therapeutic avenues. Excitingly, the success of any of these directions would represent the first disease-modifying treatment for the disease.
7.3. Biological function of myocilin
A major outstanding question regarding myocilin is its native function. No severe phenotype is observed in Myoc−/− mice, though muscle heterotrophy has been documented in these mice (Judge et al., 2020). Thus, either the function of myocilin not biologically important or functionally redundant. Given that TM cells robustly express myocilin (Tamm et al., 1999), and cells do not expend energy producing relatively high levels of proteins that are not needed, our current hypothesis is that when myocilin is absent the function is compensated by another protein.
Discovering the native function of myocilin, or even enhancing our knowledge of the function of olfactomedin domain proteins in the TM, could help with our understanding of the general mechanism involved in regulating aqueous humor outflow, and what role myocilin might play in this task. The unique conformation of full-length myocilin confers its function. Full-length myocilin, particularly due to CC/LZ, is sticky (Hill et al., 2017; Russell et al., 2001). The N-terminal CC/LZ-containing region has been implicated in potential functional roles such as interacting with matrix components (Filla et al., 2002; Ueda et al., 2002; Wentz-Hunter et al., 2004), associating with membranes (Stamer et al., 2006), potential cell-adhesion interactions (Goldwich et al., 2009; Wentz-Hunter et al., 2004), and stickiness could contribute to mechanical stiffness of the TM extracellular tissue (Wang et al., 2017). Moreover, proteolytic cleavage of myocilin (Martin et al., 2021) may also be functionally relevant, based on precedent from other olfactomedin domain family members that dissociate from the N-terminal domains for function (Furutani et al., 2005; Maertens et al., 2007). However, to our knowledge, proteolytic cleavage has not been observed for myocilin secreted from TM cells. The long linker that extends from the N-terminal domain (Hill et al., 2017) to OLF may assist OLF in accessing binding partners, suggesting a role for protein-protein interactions. A change in multimerization, e.g. to octamer or dimer, may also enhance binding partner interactions. Multimerization could be triggered by changes to the redox conditions of the TM or aqueous humor. The functional relevance of the central dinuclear metal site within OLF is supported by high evolutionary conservation, and enzymatic function has not been ruled out (Donegan et al., 2015).
One effort in our lab to increase the likelihood of uncovering myocilin function has been to develop antibodies that bind to different domains and epitopes within myocilin. Using state-of-the-art protein engineering techniques in collaboration with Jennifer Maynard’s lab, we developed antibodies that bind LZ. Antibody 2H2 successfully immunoprecipitated full-length folded myocilin from media of hTM cells and detected myocilin in fixed TM cells lacking DEX treatment (Patterson-Orazem et al., 2021). Antibodies that target OLF are in near completion. Experiments that probe different epitopes on myocilin in the same sample should reveal currently inaccessible information. Finally, by disseminating these reagents to the vision research community, we hope new insights will be gleaned on the role of myocilin in the TM.
7.4. Possible role for WT myocilin in idiopathic cases of glaucoma
Based on parallels from other protein misfolding disease such as amyotrophic lateral sclerosis (Mathis et al., 2019), Alzheimer’s Disease, and Creutzfeldt-Jakob Disease (Goldman et al., 2004), we anticipate that better understanding of glaucoma caused by inherited mutations in MYOC will inform more general pathogenic mechanisms underlying sporadic glaucoma. Proteostasis declines with age (Hipp et al., 2019) and ER stress has been documented in the glaucomatous TM cells and tissue (Peters et al., 2015). ER stress triggers the unfolded protein response (UPR), which activates downstream responses to try and improve both protein folding and quality control pathways. Chronic ER stress eventually overrides UPR and results in apoptosis (Hetz and Saxena, 2017). Combined with the knowledge that WT OLF can misfold with mild environmental stressors, it appears plausible that WT myocilin could misfold under conditions known to cause glaucoma.
Our lab performed an initial investigation of the effect of ER stress on WT myocilin misfolding. When we treated two cell lines with the ATPase channel blocker thapsigargin to halt calcium import into the ER and thereby cause ER stress, WT myocilin ceased to secrete. Instead, WT myocilin was retained intracellularly and co-localized with the amyloid-detecting dye Proteostat (Table 1) and the ER marker calnexin like previously described for misfolding disease variants. This observation raises the possibility that ER-stress induced aggregation of WT myocilin may trigger a feedback loop that exacerbates ER stress (Saccuzzo et al., 2022). Ongoing efforts include studying the effect of ER stress on myocilin in TM cells. Even if intracellular misfolding of WT myocilin is not acutely responsible for sporadic glaucoma, the ability to track the extent of myocilin misfolding as a measure of proteostasis decline could be useful in the study of mechanisms related to glaucoma.
8. Conclusions
In summary, research conducted over the last 20 years have resulted in a better fundamental understanding of myocilin, particularly from a structural, biophysical, and cellular perspective. Work from our lab since 2010 has contributed high-resolution structures of the OLF domain, CC/LZ region, and a model for full-length myocilin as a unique Y-shaped tetramer. We have also established that glaucoma-causing OLF mutations destabilize myocilin in a way that primes it for formation of aggregates with hallmark of amyloid, and that blocking the aberrant interactions with Grp94 leads to mutant myocilin degradation. By leveraging our structural insights, we have generated new myocilin antibodies that hold promise for improved detection of myocilin in different research contexts and by combining clinical data with our biophysical studies, we have established a quantitative thermal stability cutoff that can be used for differentiating between pathogenic and benign variants. Our studies show the various ways that basic molecular science research, even with its reductionist approach, can contribute to solving complex biomedical problems. We are optimistic our insights will help guide future efforts by us and others to understand the underlying pathogenesis of glaucoma and uncover new directions for glaucoma therapeutics.
Acknowledgements.
Work on myocilin has been supported by NIH R01EY021205, R21EY031093. EGS was supported in part by T32 EY007092 and GAANN P200A210014. We thank Dr. Hannah Youngblood for critical reading of the manuscript.
Footnotes
Declarations of interest: RLL is a co-inventor of WO 2021/237213 A1 and US-2022-0026445-A1
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adam MF, Belmouden A, Binisti P, Brezin AP, Valtot F, Bechetoille A, Dascotte JC, Copin B, Gomez L, Chaventre A, Bach JF, Garchon HJ, 1997. Recurrent mutations in a single exon encoding the evolutionarily conserved olfactomedin-homology domain of TIGR in familial open-angle glaucoma. Hum Mol Genet 6, 2091–2097. [DOI] [PubMed] [Google Scholar]
- Ahmed AB, Kajava AV, 2013. Breaking the amyloidogenicity code: methods to predict amyloids from amino acid sequence. FEBS Lett 587, 1089–1095. [DOI] [PubMed] [Google Scholar]
- Aldred MA, Baumber L, Hill A, Schwalbe EC, Goh K, Karwatowski W, Trembath RC, 2004. Low prevalence of MYOC mutations in UK primary open-angle glaucoma patients limits the utility of genetic testing. Hum Genet 115, 428–431. [DOI] [PubMed] [Google Scholar]
- Allison K, Patel D, Alabi O, 2020. Epidemiology of Glaucoma: The Past, Present, and Predictions for the Future. Cureus 12, e11686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alward WL, Fingert JH, Coote MA, Johnson AT, Lerner SF, Junqua D, Durcan FJ, McCartney PJ, Mackey DA, Sheffield VC, Stone EM, 1998. Clinical features associated with mutations in the chromosome 1 open-angle glaucoma gene (GLC1A). N Engl J Med 338, 1022–1027. [DOI] [PubMed] [Google Scholar]
- Armaly MF, 1963a. Effect of Corticosteroids on Intraocular Pressure and Fluid Dynamics. I. The Effect of Dexamethasone in the Normal Eye. Arch Ophthalmol 70, 482–491. [DOI] [PubMed] [Google Scholar]
- Armaly MF, 1963b. Effect of Corticosteroids on Intraocular Pressure and Fluid Dynamics. Ii. The Effect of Dexamethasone in the Glaucomatous Eye. Arch Ophthalmol 70, 492–499. [DOI] [PubMed] [Google Scholar]
- Aroca-Aguilar JD, Sanchez-Sanchez F, Ghosh S, Coca-Prados M, Escribano J, 2005. Myocilin mutations causing glaucoma inhibit the intracellular endoproteolytic cleavage of myocilin between amino acids Arg226 and Ile227. J Biol Chem 280, 21043–21051. [DOI] [PubMed] [Google Scholar]
- Borras T, Rowlette LL, Tamm ER, Gottanka J, Epstein DL, 2002. Effects of elevated intraocular pressure on outflow facility and TIGR/MYOC expression in perfused human anterior segments. Invest Ophthalmol Vis Sci 43, 33–40. [PubMed] [Google Scholar]
- Burdon KP, Graham P, Hadler J, Hulleman JD, Pasutto F, Boese EA, Craig JE, Fingert JH, Hewitt AW, Siggs OM, Whisenhunt K, Young TL, Mackey DA, Dubowsky A, Souzeau E, 2022. Specifications of the ACMG/AMP variant curation guidelines for myocilin: Recommendations from the clingen glaucoma expert panel. Hum Mutat 43, 2170–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns JN, Turnage KC, Walker CA, Lieberman RL, 2011. The stability of myocilin olfactomedin domain variants provides new insight into glaucoma as a protein misfolding disorder. Biochemistry 50, 5824–5833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caballero M, Borras T, 2001. Inefficient processing of an olfactomedin-deficient myocilin mutant: potential physiological relevance to glaucoma. Biochem Biophys Res Commun 282, 662–670. [DOI] [PubMed] [Google Scholar]
- Caballero M, Rowlette LL, Borras T, 2000. Altered secretion of a TIGR/MYOC mutant lacking the olfactomedin domain. Biochim Biophys Acta 1502, 447–460. [DOI] [PubMed] [Google Scholar]
- Convertino M, Das J, Dokholyan NV, 2016. Pharmacological Chaperones: Design and Development of New Therapeutic Strategies for the Treatment of Conformational Diseases. ACS Chem Biol 11, 1471–1489. [DOI] [PubMed] [Google Scholar]
- Craig JE, Han X, Qassim A, Hassall M, Cooke Bailey JN, Kinzy TG, Khawaja AP, An J, Marshall H, Gharahkhani P, Igo RP Jr., Graham SL, Healey PR, Ong JS, Zhou T, Siggs O, Law MH, Souzeau E, Ridge B, Hysi PG, Burdon KP, Mills RA, Landers J, Ruddle JB, Agar A, Galanopoulos A, White AJR, Willoughby CE, Andrew NH, Best S, Vincent AL, Goldberg I, Radford-Smith G, Martin NG, Montgomery GW, Vitart V, Hoehn R, Wojciechowski R, Jonas JB, Aung T, Pasquale LR, Cree AJ, Sivaprasad S, Vallabh NA, consortium N, Eye UKB, Vision C, Viswanathan AC, Pasutto F, Haines JL, Klaver CCW, van Duijn CM, Casson RJ, Foster PJ, Khaw PT, Hammond CJ, Mackey DA, Mitchell P, Lotery AJ, Wiggs JL, Hewitt AW, MacGregor S, 2020. Multitrait analysis of glaucoma identifies new risk loci and enables polygenic prediction of disease susceptibility and progression. Nat Genet 52, 160–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dismuke WM, McKay BS, Stamer WD, 2012. Myocilin, a component of a membrane-associated protein complex driven by a homologous Q-SNARE domain. Biochemistry 51, 3606–3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobson CM, 2017. The Amyloid Phenomenon and Its Links with Human Disease. Cold Spring Harb Perspect Biol 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donegan RK, Hill SE, Freeman DM, Nguyen E, Orwig SD, Turnage KC, Lieberman RL, 2015. Structural basis for misfolding in myocilin-associated glaucoma. Hum Mol Genet 24, 2111–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donegan RK, Hill SE, Turnage KC, Orwig SD, Lieberman RL, 2012. The glaucoma-associated olfactomedin domain of myocilin is a novel calcium binding protein. J Biol Chem 287, 43370–43377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fautsch MP, Johnson DH, 2001. Characterization of myocilin-myocilin interactions. Invest Ophthalmol Vis Sci 42, 2324–2331. [PubMed] [Google Scholar]
- Fautsch MP, Vrabel AM, Peterson SL, Johnson DH, 2004. In vitro and in vivo characterization of disulfide bond use in myocilin complex formation. Mol Vis 10, 417–425. [PubMed] [Google Scholar]
- Filla MS, Liu X, Nguyen TD, Polansky JR, Brandt CR, Kaufman PL, Peters DM, 2002. In vitro localization of TIGR/MYOC in trabecular meshwork extracellular matrix and binding to fibronectin. Invest Ophthalmol Vis Sci 43, 151–161. [PubMed] [Google Scholar]
- Fingert JH, Heon E, Liebmann JM, Yamamoto T, Craig JE, Rait J, Kawase K, Hoh ST, Buys YM, Dickinson J, Hockey RR, Williams-Lyn D, Trope G, Kitazawa Y, Ritch R, Mackey DA, Alward WL, Sheffield VC, Stone EM, 1999. Analysis of myocilin mutations in 1703 glaucoma patients from five different populations. Hum Mol Genet 8, 899–905. [DOI] [PubMed] [Google Scholar]
- Fingert JH, Stone EM, Sheffield VC, Alward WL, 2002. Myocilin glaucoma. Surv Ophthalmol 47, 547–561. [DOI] [PubMed] [Google Scholar]
- Flanagan SE, Patch AM, Ellard S, 2010. Using SIFT and PolyPhen to predict loss-of-function and gain-of-function mutations. Genet Test Mol Biomarkers 14, 533–537. [DOI] [PubMed] [Google Scholar]
- Franco B, Lai LW, Patterson D, Ledbetter DH, Trask BJ, van den Engh G, Iannaccone S, Frances S, Patel PI, Lupski JR, 1991. Molecular characterization of a patient with del(1)(q23-q25). Hum Genet 87, 269–277. [DOI] [PubMed] [Google Scholar]
- Francois J, 1954. [Cortisone and eye strain]. Ann Ocul (Paris) 187, 805–816. [PubMed] [Google Scholar]
- Furutani Y, Manabe R, Tsutsui K, Yamada T, Sugimoto N, Fukuda S, Kawai J, Sugiura N, Kimata K, Hayashizaki Y, Sekiguchi K, 2005. Identification and characterization of photomedins: novel olfactomedin-domain-containing proteins with chondroitin sulphate-E-binding activity. Biochem J 389, 675–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Saccuzzo EG, Hill SE, Huard DJE, Robang AS, Lieberman RL, Paravastu AK, 2021. Structural Arrangement within a Peptide Fibril Derived from the Glaucoma-Associated Myocilin Olfactomedin Domain. J Phys Chem B 125, 2886–2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobeil S, Letartre L, Raymond V, 2006. Functional analysis of the glaucoma-causing TIGR/myocilin protein: integrity of amino-terminal coiled-coil regions and olfactomedin homology domain is essential for extracellular adhesion and secretion. Exp Eye Res 82, 1017–1029. [DOI] [PubMed] [Google Scholar]
- Gobeil S, Rodrigue MA, Moisan S, Nguyen TD, Polansky JR, Morissette J, Raymond V, 2004. Intracellular sequestration of hetero-oligomers formed by wild-type and glaucoma-causing myocilin mutants. Invest Ophthalmol Vis Sci 45, 3560–3567. [DOI] [PubMed] [Google Scholar]
- Goel M, Picciani RG, Lee RK, Bhattacharya SK, 2010. Aqueous humor dynamics: a review. Open Ophthalmol J 4, 52–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldenzweig A, Goldsmith M, Hill SE, Gertman O, Laurino P, Ashani Y, Dym O, Unger T, Albeck S, Prilusky J, Lieberman RL, Aharoni A, Silman I, Sussman JL, Tawfik DS, Fleishman SJ, 2016. Automated Structure- and Sequence-Based Design of Proteins for High Bacterial Expression and Stability. Mol Cell 63, 337–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman JS, Miller BL, Safar J, de Tourreil S, Martindale JL, Prusiner SB, Geschwind MD, 2004. When sporadic disease is not sporadic: the potential for genetic etiology. Arch Neurol 61, 213–216. [DOI] [PubMed] [Google Scholar]
- Goldwich A, Scholz M, Tamm ER, 2009. Myocilin promotes substrate adhesion, spreading and formation of focal contacts in podocytes and mesangial cells. Histochem Cell Biol 131, 167–180. [DOI] [PubMed] [Google Scholar]
- Gordon MO, Beiser JA, Brandt JD, Heuer DK, Higginbotham EJ, Johnson CA, Keltner JL, Miller JP, Parrish RK 2nd, Wilson MR, Kass MA, 2002. The Ocular Hypertension Treatment Study: baseline factors that predict the onset of primary open-angle glaucoma. Arch Ophthalmol 120, 714–720; discussion 829–730. [DOI] [PubMed] [Google Scholar]
- Hartley DM, Walsh DM, Ye CP, Diehl T, Vasquez S, Vassilev PM, Teplow DB, Selkoe DJ, 1999. Protofibrillar intermediates of amyloid beta-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J Neurosci 19, 8876–8884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, Saxena S, 2017. ER stress and the unfolded protein response in neurodegeneration. Nat Rev Neurol 13, 477–491. [DOI] [PubMed] [Google Scholar]
- Hewitt AW, Mackey DA, Craig JE, 2008. Myocilin allele-specific glaucoma phenotype database. Hum Mutat 29, 207–211. [DOI] [PubMed] [Google Scholar]
- Hill SE, Cho H, Raut P, Lieberman RL, 2019a. Calcium-ligand variants of the myocilin olfactomedin propeller selected from invertebrate phyla reveal cross-talk with N-terminal blade and surface helices. Acta Crystallogr D Struct Biol 75, 817–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill SE, Donegan RK, Lieberman RL, 2014. The glaucoma-associated olfactomedin domain of myocilin forms polymorphic fibrils that are constrained by partial unfolding and peptide sequence. J Mol Biol 426, 921–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill SE, Donegan RK, Nguyen E, Desai TM, Lieberman RL, 2015. Molecular Details of Olfactomedin Domains Provide Pathway to Structure-Function Studies. PLoS One 10, e0130888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill SE, Kwon MS, Martin MD, Suntharalingam A, Hazel A, Dickey CA, Gumbart JC, Lieberman RL, 2019b. Stable calcium-free myocilin olfactomedin domain variants reveal challenges in differentiating between benign and glaucoma-causing mutations. J Biol Chem 294, 12717–12728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill SE, Nguyen E, Donegan RK, Patterson-Orazem AC, Hazel A, Gumbart JC, Lieberman RL, 2017. Structure and Misfolding of the Flexible Tripartite Coiled-Coil Domain of Glaucoma-Associated Myocilin. Structure 25, 1697–1707 e1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hipp MS, Kasturi P, Hartl FU, 2019. The proteostasis network and its decline in ageing. Nat Rev Mol Cell Biol 20, 421–435. [DOI] [PubMed] [Google Scholar]
- Houck SA, Singh S, Cyr DM, 2012. Cellular responses to misfolded proteins and protein aggregates. Methods Mol Biol 832, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huard DJE, Crowley VM, Du Y, Cordova RA, Sun Z, Tomlin MO, Dickey CA, Koren J 3rd, Blair L, Fu H, Blagg BSJ, Lieberman RL, 2018. Trifunctional High-Throughput Screen Identifies Promising Scaffold To Inhibit Grp94 and Treat Myocilin-Associated Glaucoma. ACS Chem Biol 13, 933–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huard DJE, Jonke AP, Torres MP, Lieberman RL, 2019. Different Grp94 components interact transiently with the myocilin olfactomedin domain in vitro to enhance or retard its amyloid aggregation. Sci Rep 9, 12769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson N, Andrews M, Shepard AR, Nishimura D, Searby C, Fingert JH, Hageman G, Mullins R, Davidson BL, Kwon YH, Alward WL, Stone EM, Clark AF, Sheffield VC, 2001. Non-secretion of mutant proteins of the glaucoma gene myocilin in cultured trabecular meshwork cells and in aqueous humor. Hum Mol Genet 10, 117–125. [DOI] [PubMed] [Google Scholar]
- Joe MK, Sohn S, Hur W, Moon Y, Choi YR, Kee C, 2003. Accumulation of mutant myocilins in ER leads to ER stress and potential cytotoxicity in human trabecular meshwork cells. Biochem Biophys Res Commun 312, 592–600. [DOI] [PubMed] [Google Scholar]
- Johnson DH, 2000. Myocilin and glaucoma: A TIGR by the tail? Arch Ophthalmol 118, 974–978. [PubMed] [Google Scholar]
- Judge SM, Deyhle MR, Neyroud D, Nosacka RL, D’Lugos AC, Cameron ME, Vohra RS, Smuder AJ, Roberts BM, Callaway CS, Underwood PW, Chrzanowski SM, Batra A, Murphy ME, Heaven JD, Walter GA, Trevino JG, Judge AR, 2020. MEF2c-Dependent Downregulation of Myocilin Mediates Cancer-Induced Muscle Wasting and Associates with Cachexia in Patients with Cancer. Cancer Res 80, 1861–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Zidek A, Potapenko A, Bridgland A, Meyer C, Kohl SAA, Ballard AJ, Cowie A, Romera-Paredes B, Nikolov S, Jain R, Adler J, Back T, Petersen S, Reiman D, Clancy E, Zielinski M, Steinegger M, Pacholska M, Berghammer T, Bodenstein S, Silver D, Vinyals O, Senior AW, Kavukcuoglu K, Kohli P, Hassabis D, 2021. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, Gauthier LD, Brand H, Solomonson M, Watts NA, Rhodes D, Singer-Berk M, England EM, Seaby EG, Kosmicki JA, Walters RK, Tashman K, Farjoun Y, Banks E, Poterba T, Wang A, Seed C, Whiffin N, Chong JX, Samocha KE, Pierce-Hoffman E, Zappala Z, O’Donnell-Luria AH, Minikel EV, Weisburd B, Lek M, Ware JS, Vittal C, Armean IM, Bergelson L, Cibulskis K, Connolly KM, Covarrubias M, Donnelly S, Ferriera S, Gabriel S, Gentry J, Gupta N, Jeandet T, Kaplan D, Llanwarne C, Munshi R, Novod S, Petrillo N, Roazen D, Ruano-Rubio V, Saltzman A, Schleicher M, Soto J, Tibbetts K, Tolonen C, Wade G, Talkowski ME, Genome Aggregation Database C, Neale BM, Daly MJ, MacArthur DG, 2020. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller KE, Bhattacharya SK, Borras T, Brunner TM, Chansangpetch S, Clark AF, Dismuke WM, Du Y, Elliott MH, Ethier CR, Faralli JA, Freddo TF, Fuchshofer R, Giovingo M, Gong H, Gonzalez P, Huang A, Johnstone MA, Kaufman PL, Kelley MJ, Knepper PA, Kopczynski CC, Kuchtey JG, Kuchtey RW, Kuehn MH, Lieberman RL, Lin SC, Liton P, Liu Y, Lutjen-Drecoll E, Mao W, Masis-Solano M, McDonnell F, McDowell CM, Overby DR, Pattabiraman PP, Raghunathan VK, Rao PV, Rhee DJ, Chowdhury UR, Russell P, Samples JR, Schwartz D, Stubbs EB, Tamm ER, Tan JC, Toris CB, Torrejon KY, Vranka JA, Wirtz MK, Yorio T, Zhang J, Zode GS, Fautsch MP, Peters DM, Acott TS, Stamer WD, 2018. Consensus recommendations for trabecular meshwork cell isolation, characterization and culture. Exp Eye Res 171, 164–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendrick R, 1999. Gradual painless visual loss: glaucoma. Clin Geriatr Med 15, 95–101, vi-vii. [PubMed] [Google Scholar]
- Kersey JP, Broadway DC, 2006. Corticosteroid-induced glaucoma: a review of the literature. Eye (Lond) 20, 407–416. [DOI] [PubMed] [Google Scholar]
- Kim BS, Savinova OV, Reedy MV, Martin J, Lun Y, Gan L, Smith RS, Tomarev SI, John SW, Johnson RL, 2001. Targeted Disruption of the Myocilin Gene (Myoc) Suggests that Human Glaucoma-Causing Mutations Are Gain of Function. Mol Cell Biol 21, 7707–7713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota R, Noda S, Wang Y, Minoshima S, Asakawa S, Kudoh J, Mashima Y, Oguchi Y, Shimizu N, 1997. A novel myosin-like protein (myocilin) expressed in the connecting cilium of the photoreceptor: molecular cloning, tissue expression, and chromosomal mapping. Genomics 41, 360–369. [DOI] [PubMed] [Google Scholar]
- Kwon YH, Fingert JH, Kuehn MH, Alward WL, 2009. Primary open-angle glaucoma. N Engl J Med 360, 1113–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam DS, Leung YF, Chua JK, Baum L, Fan DS, Choy KW, Pang CP, 2000. Truncations in the TIGR gene in individuals with and without primary open-angle glaucoma. Invest Ophthalmol Vis Sci 41, 1386–1391. [PubMed] [Google Scholar]
- Leighton DA, 1976. Survey of the first-degree relatives of glaucoma patients. Trans Ophthalmol Soc U K (1962) 96, 28–32. [PubMed] [Google Scholar]
- Liu Y, Allingham RR, 2017. Major review: Molecular genetics of primary open-angle glaucoma. Exp Eye Res 160, 62–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Vollrath D, 2004. Reversal of mutant myocilin non-secretion and cell killing: implications for glaucoma. Hum Mol Genet 13, 1193–1204. [DOI] [PubMed] [Google Scholar]
- Llobet A, Gasull X, Gual A, 2003. Understanding trabecular meshwork physiology: a key to the control of intraocular pressure? News Physiol Sci 18, 205–209. [DOI] [PubMed] [Google Scholar]
- Maertens B, Hopkins D, Franzke CW, Keene DR, Bruckner-Tuderman L, Greenspan DS, Koch M, 2007. Cleavage and oligomerization of gliomedin, a transmembrane collagen required for node of ranvier formation. J Biol Chem 282, 10647–10659. [DOI] [PubMed] [Google Scholar]
- Martin MD, Huard DJE, Guerrero-Ferreira RC, Desai IM, Barlow BM, Lieberman RL, 2021. Molecular architecture and modifications of full-length myocilin. Exp Eye Res 211, 108729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathis S, Goizet C, Soulages A, Vallat JM, Masson GL, 2019. Genetics of amyotrophic lateral sclerosis: A review. J Neurol Sci 399, 217–226. [DOI] [PubMed] [Google Scholar]
- Matsumoto Y, Johnson DH, 1997. Trabecular meshwork phagocytosis in glaucomatous eyes. Ophthalmologica 211, 147–152. [DOI] [PubMed] [Google Scholar]
- McDowell CM, Luan T, Zhang Z, Putliwala T, Wordinger RJ, Millar JC, John SW, Pang IH, Clark AF, 2012. Mutant human myocilin induces strain specific differences in ocular hypertension and optic nerve damage in mice. Exp Eye Res 100, 65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer A, Valtot F, Bechetoille A, Rouland JF, Dascotte JC, Ferec C, Bach JF, Chaventre A, Garchon HJ, 1994. [Linkage between juvenile glaucoma and chromosome 1q in 2 French families]. C R Acad Sci III 317, 565–570. [PubMed] [Google Scholar]
- Nguyen TD, Chen P, Huang WD, Chen H, Johnson D, Polansky JR, 1998. Gene structure and properties of TIGR, an olfactomedin-related glycoprotein cloned from glucocorticoid-induced trabecular meshwork cells. J Biol Chem 273, 6341–6350. [DOI] [PubMed] [Google Scholar]
- Nielsen H, Engelbrecht J, Brunak S, von Heijne G, 1997. Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng 10, 1–6. [DOI] [PubMed] [Google Scholar]
- Oliveberg M, 2010. Waltz, an exciting new move in amyloid prediction. Nat Methods 7, 187–188. [DOI] [PubMed] [Google Scholar]
- Orwig SD, Chi PV, Du Y, Hill SE, Cavitt MA, Suntharalingam A, Turnage KC, Dickey CA, France S, Fu H, Lieberman RL, 2014. Ligands for glaucoma-associated myocilin discovered by a generic binding assay. ACS Chem Biol 9, 517–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orwig SD, Lieberman RL, 2011. Biophysical characterization of the olfactomedin domain of myocilin, an extracellular matrix protein implicated in inherited forms of glaucoma. PLoS One 6, e16347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orwig SD, Perry CW, Kim LY, Turnage KC, Zhang R, Vollrath D, Schmidt-Krey I, Lieberman RL, 2012. Amyloid fibril formation by the glaucoma-associated olfactomedin domain of myocilin. J Mol Biol 421, 242–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang CP, Leung YF, Fan B, Baum L, Tong WC, Lee WS, Chua JK, Fan DS, Liu Y, Lam DS, 2002. TIGR/MYOC gene sequence alterations in individuals with and without primary open-angle glaucoma. Invest Ophthalmol Vis Sci 43, 3231–3235. [PubMed] [Google Scholar]
- Patel GC, Phan TN, Maddineni P, Kasetti RB, Millar JC, Clark AF, Zode GS, 2017. Dexamethasone-Induced Ocular Hypertension in Mice: Effects of Myocilin and Route of Administration. Am J Pathol 187, 713–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson-Orazem AC, Hill SE, Fautsch MP, Lieberman RL, 2018. Epitope mapping of commercial antibodies that detect myocilin. Exp Eye Res 173, 109–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson-Orazem AC, Hill SE, Wang Y, Dominic IM, Hall CK, Lieberman RL, 2019. Differential Misfolding Properties of Glaucoma-Associated Olfactomedin Domains from Humans and Mice. Biochemistry 58, 1718–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson-Orazem AC, Qerqez AN, Azouz LR, Ma MT, Hill SE, Ku Y, Schildmeyer LA, Maynard JA, Lieberman RL, 2021. Recombinant antibodies recognize conformation-dependent epitopes of the leucine zipper of misfolding-prone myocilin. J Biol Chem 297, 101067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JC, Bhattacharya S, Clark AF, Zode GS, 2015. Increased Endoplasmic Reticulum Stress in Human Glaucomatous Trabecular Meshwork Cells and Tissues. Invest Ophthalmol Vis Sci 56, 3860–3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polansky JR, Fauss DJ, Chen P, Chen H, Lutjen-Drecoll E, Johnson D, Kurtz RM, Ma ZD, Bloom E, Nguyen TD, 1997. Cellular pharmacology and molecular biology of the trabecular meshwork inducible glucocorticoid response gene product. Ophthalmologica 211, 126–139. [DOI] [PubMed] [Google Scholar]
- Polansky JR, Fauss DJ, Zimmerman CC, 2000. Regulation of TIGR/MYOC gene expression in human trabecular meshwork cells. Eye (Lond) 14 ( Pt 3B), 503–514. [DOI] [PubMed] [Google Scholar]
- Resch ZT, Fautsch MP, 2009. Glaucoma-associated myocilin: a better understanding but much more to learn. Exp Eye Res 88, 704–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Restrepo NA, Cooke Bailey JN, 2017. Primary Open-Angle Glaucoma Genetics in African Americans. Curr Genet Med Rep 5, 167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards JE, Lichter PR, Boehnke M, Uro JL, Torrez D, Wong D, Johnson AT, 1994. Mapping of a gene for autosomal dominant juvenile-onset open-angle glaucoma to chromosome Iq. Am J Hum Genet 54, 62–70. [PMC free article] [PubMed] [Google Scholar]
- Rozsa FW, Shimizu S, Lichter PR, Johnson AT, Othman MI, Scott K, Downs CA, Nguyen TD, Polansky J, Richards JE, 1998. GLC1A mutations point to regions of potential functional importance on the TIGR/MYOC protein. Mol Vis 4, 20. [PubMed] [Google Scholar]
- Russell P, Tamm ER, Grehn FJ, Picht G, Johnson M, 2001. The presence and properties of myocilin in the aqueous humor. Invest Ophthalmol Vis Sci 42, 983–986. [PubMed] [Google Scholar]
- Saccuzzo EG, Martin MD, Hill KR, Ma MT, Ku Y, Lieberman RL, 2022. Calcium dysregulation potentiates wild-type myocilin misfolding: implications for glaucoma pathogenesis. J Biol Inorg Chem 27, 553–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Sanchez F, Martinez-Redondo F, Aroca-Aguilar JD, Coca-Prados M, Escribano J, 2007. Characterization of the intracellular proteolytic cleavage of myocilin and identification of calpain II as a myocilin-processing protease. J Biol Chem 282, 27810–27824. [DOI] [PubMed] [Google Scholar]
- Scelsi HF, Barlow BM, Saccuzzo EG, Lieberman RL, 2021. Common and rare myocilin variants: Predicting glaucoma pathogenicity based on genetics, clinical, and laboratory misfolding data. Hum Mutat 42, 903–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scelsi HF, Hill KR, Barlow BM, Martin MD, Lieberman RL, 2023. Quantitative differentiation of benign and misfolded glaucoma-causing myocilin variants on the basis of protein thermal stability. Dis Model Mech 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senatorov V, Malyukova I, Fariss R, Wawrousek EF, Swaminathan S, Sharan SK, Tomarev S, 2006. Expression of mutated mouse myocilin induces open-angle glaucoma in transgenic mice. J Neurosci 26, 11903–11914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffield VC, Stone EM, Alward WL, Drack AV, Johnson AT, Streb LM, Nichols BE, 1993. Genetic linkage of familial open angle glaucoma to chromosome 1q21-q31. Nat Genet 4, 47–50. [DOI] [PubMed] [Google Scholar]
- Shepard AR, Jacobson N, Fingert JH, Stone EM, Sheffield VC, Clark AF, 2001. Delayed secondary glucocorticoid responsiveness of MYOC in human trabecular meshwork cells. Invest Ophthalmol Vis Sci 42, 3173–3181. [PubMed] [Google Scholar]
- Shepard AR, Jacobson N, Sui R, Steely HT, Lotery AJ, Stone EM, Clark AF, 2003. Characterization of rabbit myocilin: Implications for human myocilin glycosylation and signal peptide usage. BMC Genet 4, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder DA, Rivers AM, Yokoe H, Menco BP, Anholt RR, 1991. Olfactomedin: purification, characterization, and localization of a novel olfactory glycoprotein. Biochemistry 30, 9143–9153. [DOI] [PubMed] [Google Scholar]
- Stamer WD, Perkumas KM, Hoffman EA, Roberts BC, Epstein DL, McKay BS, 2006. Coiled-coil targeting of myocilin to intracellular membranes. Exp Eye Res 83, 1386–1395. [DOI] [PubMed] [Google Scholar]
- Stone EM, Fingert JH, Alward WL, Nguyen TD, Polansky JR, Sunden SL, Nishimura D, Clark AF, Nystuen A, Nichols BE, Mackey DA, Ritch R, Kalenak JW, Craven ER, Sheffield VC, 1997. Identification of a gene that causes primary open angle glaucoma. Science 275, 668–670. [DOI] [PubMed] [Google Scholar]
- Stothert AR, Suntharalingam A, Huard DJ, Fontaine SN, Crowley VM, Mishra S, Blagg BS, Lieberman RL, Dickey CA, 2014. Exploiting the interaction between Grp94 and aggregated myocilin to treat glaucoma. Hum Mol Genet 23, 6470–6480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stothert AR, Suntharalingam A, Tang X, Crowley VM, Mishra SJ, Webster JM, Nordhues BA, Huard DJE, Passaglia CL, Lieberman RL, Blagg BSJ, Blair LJ, Koren J 3rd, Dickey CA, 2017. Isoform-selective Hsp90 inhibition rescues model of hereditary open-angle glaucoma. Sci Rep 7, 17951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suntharalingam A, Abisambra JF, O’Leary JC 3rd, Koren J 3rd, Zhang B, Joe MK, Blair LJ, Hill SE, Jinwal UK, Cockman M, Duerfeldt AS, Tomarev S, Blagg BS, Lieberman RL, Dickey CA, 2012. Glucose-regulated protein 94 triage of mutant myocilin through endoplasmic reticulum-associated degradation subverts a more efficient autophagic clearance mechanism. J Biol Chem 287, 40661–40669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamm ER, 2002. Myocilin and glaucoma: facts and ideas. Prog Retin Eye Res 21, 395–428. [DOI] [PubMed] [Google Scholar]
- Tamm ER, Russell P, Epstein DL, Johnson DH, Piatigorsky J, 1999. Modulation of myocilin/TIGR expression in human trabecular meshwork. Invest Ophthalmol Vis Sci 40, 2577–2582. [PubMed] [Google Scholar]
- Tham YC, Li X, Wong TY, Quigley HA, Aung T, Cheng CY, 2014. Global prevalence of glaucoma and projections of glaucoma burden through 2040: a systematic review and meta-analysis. Ophthalmology 121, 2081–2090. [DOI] [PubMed] [Google Scholar]
- Trovato A, Seno F, Tosatto SC, 2007. The PASTA server for protein aggregation prediction. Protein Eng Des Sel 20, 521–523. [DOI] [PubMed] [Google Scholar]
- Tsolis AC, Papandreou NC, Iconomidou VA, Hamodrakas SJ, 2013. A consensus method for the prediction of ‘aggregation-prone’ peptides in globular proteins. PLoS One 8, e54175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda J, Wentz-Hunter K, Yue BY, 2002. Distribution of myocilin and extracellular matrix components in the juxtacanalicular tissue of human eyes. Invest Ophthalmol Vis Sci 43, 1068–1076. [PubMed] [Google Scholar]
- van der Heide CJ, Alward WLM, Flamme-Wiese M, Riker M, Syed NA, Anderson MG, Carter K, Kuehn MH, Stone EM, Mullins RF, Fingert JH, 2018. Histochemical Analysis of Glaucoma Caused by a Myocilin Mutation in a Human Donor Eye. Ophthalmol Glaucoma 1, 132–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollrath D, Liu Y, 2006. Temperature sensitive secretion of mutant myocilins. Exp Eye Res 82, 1030–1036. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Hartley DM, Kusumoto Y, Fezoui Y, Condron MM, Lomakin A, Benedek GB, Selkoe DJ, Teplow DB, 1999. Amyloid beta-protein fibrillogenesis. Structure and biological activity of protofibrillar intermediates. J Biol Chem 274, 25945–25952. [DOI] [PubMed] [Google Scholar]
- Wang K, Read AT, Sulchek T, Ethier CR, 2017. Trabecular meshwork stiffness in glaucoma. Exp Eye Res 158, 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wentz-Hunter K, Kubota R, Shen X, Yue BY, 2004. Extracellular myocilin affects activity of human trabecular meshwork cells. J Cell Physiol 200, 45–52. [DOI] [PubMed] [Google Scholar]
- Wiggs JL, Vollrath D, 2001. Molecular and clinical evaluation of a patient hemizygous for TIGR/MYOC. Arch Ophthalmol 119, 1674–1678. [DOI] [PubMed] [Google Scholar]
- Yam GH, Gaplovska-Kysela K, Zuber C, Roth J, 2007. Aggregated myocilin induces russell bodies and causes apoptosis: implications for the pathogenesis of myocilin-caused primary open-angle glaucoma. Am J Pathol 170, 100–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoe H, Anholt RR, 1993. Molecular cloning of olfactomedin, an extracellular matrix protein specific to olfactory neuroepithelium. Proc Natl Acad Sci U S A 90, 4655–4659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng LC, Han ZG, Ma WJ, 2005. Elucidation of subfamily segregation and intramolecular coevolution of the olfactomedin-like proteins by comprehensive phylogenetic analysis and gene expression pattern assessment. FEBS Lett 579, 5443–5453. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Vollrath D, 1999. A cellular assay distinguishes normal and mutant TIGR/myocilin protein. Hum Mol Genet 8, 2221–2228. [DOI] [PubMed] [Google Scholar]
- Zode GS, Kuehn MH, Nishimura DY, Searby CC, Mohan K, Grozdanic SD, Bugge K, Anderson MG, Clark AF, Stone EM, Sheffield VC, 2011. Reduction of ER stress via a chemical chaperone prevents disease phenotypes in a mouse model of primary open angle glaucoma. J Clin Invest 121, 3542–3553. [DOI] [PMC free article] [PubMed] [Google Scholar]