

Abstract
Objectives:
To elucidate mechanisms contributing to skeletal muscle calcinosis in patients with juvenile dermatomyositis.
Methods:
A well-characterized cohorts of JDM (n = 68), disease controls (polymyositis, n = 7; juvenile SLE, n = 10, and RNP + overlap syndrome, n = 12), and age-matched health controls (n = 17) were analyzed for circulating levels of mitochondrial (mt) markers including mtDNA, mt-nd6, and anti-mitochondrial antibodies (AMAs) using standard qPCR, ELISA, and novel-in-house assays, respectively. Mitochondrial calcification of affected tissue biopsies was confirmed using electron microscopy and energy dispersive X-ray analysis. A human skeletal muscle cell line, RH30, was used to generate an in vitro calcification model. Intracellular calcification is measured by flow cytometry and microscopy. Mitochondria were assessed for mtROS production and membrane potential by flow cytometry and real-time oxygen consumption rate by Seahorse bioanalyzer. Inflammation (interferon-stimulated genes) was measured by qPCR.
Results:
In the current study, patients with JDM exhibited elevated levels of mitochondrial markers associated with muscle damage and calcinosis. Of particular interest are AMAs predictive of calcinosis. Human skeletal muscle cells undergo time- and dose-dependent accumulation of calcium phosphate salts with preferential localization to mitochondria. Calcification renders skeletal muscle cells mitochondria stressed, dysfunctional, destabilized, and interferogenic. Further, we report that inflammation induced by interferon-alpha amplifies mitochondrial calcification of human skeletal muscle cells via the generation of mitochondrial reactive oxygen species (mtROS).
Conclusions:
Overall, our study demonstrates the mitochondrial involvement in the skeletal muscle pathology and calcinosis of JDM and mtROS as a central player in the calcification of human skeletal muscle cells. Therapeutic targeting of mtROS and/or upstream inducers, such as inflammation, may alleviate mitochondrial dysfunction, leading to calcinosis. AMAs can potentially identify patients with JDM at risk for developing calcinosis.
Keywords: Juvenile dermatomyositis, Calcinosis, Mitochondria, Interferon, Mitochondrial reactive oxygen species
1. Introduction
Juvenile dermatomyositis (JDM), a systemic vasculopathy, is the most common inflammatory myopathy of childhood, with an incidence of 3.2 cases/per million children/year in the United States [1]. Proximal muscle weakness, raised serum concentrations of muscle enzymes, and pathognomonic skin rash characterize the disease [2].
One of the debilitating consequences of JDM is calcinosis, the accumulation of calcium salt crystals in soft tissues like skeletal muscle, skin, and blood vessels, reported in 30% of JDM patients [3]. Calcification contributes to the substantial morbidity of JDM, causing severe pain, cutaneous ulcerations, secondary infections, and joint contractures that may lead to severe disability [4]. Pathological calcified deposits in JDM, although similar to bone in composition, are distinct in having a high mineral-to-matrix ratio, suggesting different mechanisms of mineralization in JDM and bone [5]. The mineral primarily observed in JDM samples is bone-like hydroxyapatite (HA) [6]. The type of calcification seen in JDM is dystrophic, which by definition occurs in necrotic or degenerated tissues while the systemic calcium and phosphorous levels are normal [4]. Accordingly, calcific lesions in patients with JDM often occur at pressure points of daily trauma [7] and do not regress with calcium depletion therapy or modulators of calcium/phosphate metabolism [8]. Some mechanisms of insoluble calcium phosphate depositions in JDM include inflammation and an imbalance between promoters and inhibitors of mineralization [8]. Calcinosis is frequently seen in JDM children who have the pro-inflammatory TNF-α-308 promoter allele or children with persistent cutaneous inflammation, untreated disease of long duration, or chronic symptoms [4,9]. There is emerging evidence that early institution of immunosuppressive therapy may prevent the development and progression of calcinosis in children [8]. However, the mechanistic insights into the relationship between inflammation and pathological calcification remains unexplored. If discovered, they could lead to the development of novel therapies for calcinosis and the identification of biomarkers required for disease management.
Mitochondria, known for their remarkable calcium uptake and storage abilities, play a crucial role in maintaining cellular calcium homeostasis by scavenging excessive cytosolic calcium as calcium phosphate crystals [10]. Mitochondria mediate the biological mineralization process in bone [11]. Mitochondrial oxidative stress induced by LPS-mediated inflammation was shown to cause mitochondrial calcium overload even in baseline cytosolic calcium levels in vascular endothelial cells [12]. Hence, we reasoned that studying mitochondrial calcium dynamics in inflammatory conditions could provide insights into the mechanistic link between inflammation and pathologic calcification, particularly in skeletal muscle.
In the current study, by utilizing electron microscopy and energy dispersive X-ray analysis, we demonstrate calcified mitochondria intracellularly in the patient muscle cell, and in the extracellular space, presumed as being released from damaged/dying muscle tissue. Consistently, analysis of clinical samples demonstrated that patients with JDM have elevated levels circulating mitochondrial markers. Anti-mitochondrial antibodies were predictive of calcinosis. In an in vitro model of calcification, we report a novel mechanism of calcification in skeletal muscle cells mediated by mtROS downstream of the inflammatory influence of (IFN-α). Calcification in calcifying skeletal muscle cells was further shown to mediate cGAS-STING-dependent type I IFN responses, thus generating a positive feedback loop of calcification and inflammation in skeletal muscle and establishing muscle as a source of type I IFN. Our findings suggest that therapeutic agents targeting mtROS and/or upstream mediators of mtROS induction, such as type I interferons (IFNs) may prevent mitochondrial impairment and subsequent mitochondrial calcification/extrusion in skeletal muscle cells, reducing calcinosis.
2. Materials and methods
2.1. Human subjects and ethics approval
Children with JDM (n = 68), healthy controls (n = 16), and age-matched disease controls (n = 29) were included in the study after age-appropriate informed consent was obtained (Institutional Review Board #2008–13457, and #2001–11715; #2010–14117 as a consent IRB). For patient characteristics, see Table 1; information on disease controls is given in Supplementary Table 1. Myositis-specific autoantibodies were assessed at the Oklahoma Medical Research Foundation Clinical Immunology Laboratory. Calcifications were confirmed by radiography or computed tomography. Disease activity score (DAS) were obtained at the time of the physical examination [13]. Diagnostic criteria for patients include Bohan and Peter classification criteria for polymyositis and dermatomyositis diagnosis [14,15], American College of Rheumatology diagnostic criteria for SLE [16], Myositis Specific Antibodies (MSA) were assayed using Oklahoma Medical Research Foundation Myositis panel, OMRF panel [17,18].
Table 1.
JDM patient characteristics.
| Patient group | JDM (all) | Anti-MDA5 | Anti-Mi-2 | Anti-TIF1-γ | Anti-MJ/NXP2 | Negative |
|---|---|---|---|---|---|---|
|
| ||||||
| Number (#) | 68 | 4 | 10 | 21 | 19 | 14 |
| Age (year)a | 10.7 (2.4–24.5) | 13.3 (5.8–22.2) | 9.9 (2.4–24.5) | 13.2 (4.1–18.5) | 9.5 (3.1–23.2) | 7.8 (3.4–16.9) |
| Disease duration (months) | 42.7 (0.6–186.4) | 42.2 (16.2–145.9) | 14.9 (1.8–167.3) | 58.7 (2.6–186.4) | 70.5 (1.9–169.5) | 18.8 (0.6–95.0) |
| Gender (female, %) | 74 | 100 | 91 | 71 | 63 | 71 |
| Ethnicity (white, %) | 72 | 75 | 64 | 76 | 68 | 79 |
| Calcinosis, now, % | 18.8 | 0.0 | 9.1 | 23.8 | 31.6 | 7.1 |
| Calcinosis, history, % | 39.1 | 25.0 | 18.2 | 52.4 | 52.7 | 21.4 |
| DAS total | 6 (0–17) | 3 (3–3) | 7 (0–14) | 7 (0–17) | 6 (1–16) | 7.5 (0–16) |
| DAS skin | 5 (0–9) | 3 (1–3) | 5 (0–7) | 5 (0–9) | 5 (0–8) | 4.5 (0–9) |
| DAS muscle | 2 (0–9) | 0 (0–2) | 2 (0–9) | 2.5 (0–9) | 2 (0–9) | 3 (0–9) |
| Immunosuppressive treatment, % | 54 | 100 | 40 | 50 | 56 | 54 |
| Oral prednisone | 36 | 25 | 40 | 28 | 31 | 54 |
| IV Solu-Medrol | 18 | 25 | 20 | 11 | 31 | 8 |
| Methotrexate | 34 | 50 | 20 | 28 | 44 | 38 |
| IVIG | 10 | 0 | 0 | 22 | 6 | 8 |
| Hydroxychloroquine | 25 | 50 | 10 | 17 | 31 | 31 |
| Cyclosporine | 11 | 0 | 10 | 17 | 13 | 8 |
| CellCept | 30 | 50 | 10 | 33 | 31 | 31 |
DAS: Disease Activity Score; IV: Intravenous; IVIG: Intravenous Immunoglobulin.
All values, except when noted, are median and range.
2.2. Electron microscopy
Tissue samples were preserved in 2% paraformaldehyde and 0.5% glutaraldehyde in 0.05 M cacodylate buffer (pH 7.4) at 4 °C for 4–18 h. Following fixation, the tissues were treated with reduced osmium tetroxide followed by dehydration in ethyl alcohol. The Spurr’s resin (Electron Microscopy Sciences) was prepared and mixed 1:1 with propylene oxide and incubated overnight on a rotator. A change of Spurr’s resin:propylene oxide at 3:1 was made, and samples were infiltrated overnight. The final embedding was completed in pure Spurr’s resin (hard mixture) for 12–18 h and polymerized at 60 °C. Embedded samples were thin-sectioned (60–80 nm thickness) for electron microscopy using a diamond knife (Diatome) on a Reichert Ultracut E and analyzed using a Philips CM-12 transmission electron microscope at 80 kV accelerating voltage. Mineral crystal size and arrangement were studied by tilting the specimen stage through 0–20° of tilt and/or by darkfield imaging [19]. Individual crystals could be measured in the darkfield mode and by using the 002-diffraction line from selected-area diffraction.
2.3. Energy dispersive X-ray (EDAX) method for calcium content analysis
JDM muscle fibers previously identified on diagnostic MRI were preserved in 2% glutaraldehyde buffered to pH 7.4 in sodium cacodylate buffer. Samples were processed for transmission electron microscopy after polymerization in EPON resin, and thin sections were placed on copper grids. Each sample section was exposed to electron beam currents variable from 50 to 100 Kilovoltage (KV) to generate X-rays from the sample to be collected by the Energy Dispersive X-ray (EDAX) [20] detector within the microscope column. The same specimen was photographed to identify the crystalline particle size and shape within the mitochondria. Different magnifications were used to determine which beam intensity provided the most reliable signal intensity. The final analysis was done at 10,000× magnification. EDAX analysis was done on 25 to 50 mitochondria to obtain significant data for analysis. Relative osmium content represented by Os (Osmium) label measures lipid content, K line measures the total protein content under, which relates to tissue content and thickness, and calcium content is represented by K alpha.
2.4. Cell culturing
A rhabdomyosarcoma cell line, RH30 (SJCRH30) was obtained from the American Type Culture Collection and maintained at 37 °C and 5% CO2 incubator in complete growth medium composed of RPMI-1640 medium (Invitrogen) supplemented with 10% fetal bovine serum, 1X penicillin/streptomycin and 1 mM glutamine [21]. At 80–90% confluency, cellswere routinely subcultured using 0.25% trypsin, 0.03% EDTA to detach cells.
2.5. In vitro induction of calcification
RH30 cells are cultured in 24-well tissue culture plate at the concentration of 200,000 cells per well and allowed to adhere overnight (o/n) before o/n treatment with complete growth medium supplemented with following concentrations of CaCl2 and Na3PO4: Very Low CaP: 0.7575 mM CaCl2 and 5.63 mM Na3PO4, Low CaP: 1.095 mM CaCl2 and 6.13 mM Na3PO4; Moderate CaP: 1.77 mM CaCl2 and 6.63 mM Na3PO4 for different time points. Different reagents used in calcification experiments include Ru360 (557,440, Sigma-Aldrich), Antimycin A (A8674, Sigma-Aldrich), Carbonyl cyanide 4-(trifluoromethoxy) phenyl-hydrazone Ready Made Solution, FCCP (SML2959, Sigma-Aldrich), Universal Type I IFN Protein (Recombinant Human IFN-alpha A/D [BglII]), 11,200–1, R&D systems), H-151 (inh-h151, InvivoGen), and VBIT-4 (S3544, Selleckchem).
2.6. Flow cytometry analysis of calcification (hydroxyapatite complexes)
Formation of hydroxyapatite complexes is quantified by using OsteoImage™ Mineralization Assay (Lonza, PA-1503) following manufacturer’s instructions except that stained cells are analyzed by flow cytometry. Briefly, following CaP treatment, trypsinized cells from each treatment well are transferred into a U-bottomed 96-well plate for staining. Staining procedure involves fixation with 2% paraformaldehyde (PFA) for 20 min at room temperature (RT) followed by wash with osteowash buffer (1X in ddH2O) and staining with OsteoImage (1:100) in dark for 30 min at RT followed by three washes with 5 min incubation in between. Hydroxyapatite complexes are quantified by flow cytometry.
2.7. Microscopy analysis of calcification (hydroxyapatite complexes)
For microscopy analysis, RH30 cells are seeded on Ibidi μ-slide 8 well (Ibidi GmbH, Germany) plates at a density of 25,000 cells/well and incubated o/n to allow for cell adherence and followed by o/n treatment with CaP medium (1.77 mM CaCl2 and 6.63 mM Na3PO4). For staining, cells are fixed by incubating 20 min with 4% paraformaldehyde at RT, followed by 1 h treatment with permeabilization buffer (0.03% Triton™ X-100 diluted in 1X PBS with 7.5% normal goat serum). Following permeabilization cells are stained with Anti-TOM22 antibody (Tom22, Miltenyi Biotec 130–124-256) at a final concentration of 0.5 μg/ml, followed by 2x washes with osteowash buffer and staining with OsteoImage™ (1:100) as per the manufacturer’s instructions. In the last step, cells are stained with DAPI (1:1000) diluted in 1X osteowash buffer. Note that OsteoImage™ is incompatible with PBS and hence all cells are resuspended in osteowash buffer for imaging with confocal microscopy. Images were taken on a Leica SP8X confocal microscope in the W. M. Keck Microscopy Center, University of Washington. Three channels were used to look at DAPI, TOM22 Antibody, anti-human/mouse, APC (130–124-256, clone 1C9–2 Miltenyi Biotech Inc.), and OsteoImage, FITC (Lonza, PA-1503). Z stacks were taken of samples using the highest resolution lighting settings, a 63X oil immersion objective, zoom factor 2, and a 0.5 pin hole. Images were further processed with lightning deconvolution software and then each channel was converted to TIFF files in Fiji. Post-imaging processing was performed using ImageJ software (https://imagej.nih.gov/ij/index.html).
2.8. RNAi-mediated MICU1 knockdown
Lentivirus production and infection are performed following the reported detailed protocol [22]. Briefly, RH30 cells were plated in 75 cm flasks in 15 ml of complete growth media. 5 ml of the media was removed the next day and cells were incubated with 5 ml of lentivirus for expression of control shLacZ (TRCN0000072237) and two shMICU1 (TRCN0000053368 and TRCN0000053370), in the presence of 10 μg/ml polybrene for 6 h. Media was refreshed after 6 h. Cells were split two days later and selected with 2 μg/ml puromycin. MICU1 knockdown was confirmed by western blotting. HSP60 antibody: Cell Signaling Technology #12165; MICU1 antibody: MyBioSource #MBS9400198; MCU antibody: Cell Signaling Technology # 14,997.
2.9. Western blot
20 μg of protein from each sample was loaded on a 4–12% Bolt Bis-Tris gel (Thermo Fisher Scientific #NW04125BOX), samples were run using Bolt MES SDS running buffer (Thermo Fisher Scientific #B0002). Proteins were transferred to Trans-Blot Turbo Mini PVDF membranes (BioRad #1704156) using the mix transfer setting of a Trans-Blot Turbo Transfer System. Membranes were blocked with 5% milk prepared in TBST for an hour at RT. Antibodies were diluted 1:2000 in 5% milk in TBST and incubated overnight at 4 °C. HRP-conjugated secondary antibodies were used at 1:10,000 dilution in 5% milk in TBST and protein bands were imaged using Clarity Max ECL Substrate (BioRad #1705062) and an iBrighty Imaging system.
2.10. Mitochondrial isolation and purity check
Cells (RH30, HepG2), grown until 80% confluency, were resuspended in homogenization buffer (210 mM mannitol, 70 mM sucrose, 5 mM Tris-HCl pH 7.5, and 1 mM EDTA) prior to homogenization using Dounce homogenizer. The homogenized sample was centrifuged twice at 700 g for 10 min at 4 °C to remove cellular debris, followed by pelleting of mitochondria at 3,000 g for 10 min at 4 °C. Isolated mitochondria were treated with DNase (200 μg/ml, Millipore) for 60 min at 37 °C prior to analysis for purity by flow cytometry staining with MitoTracker Red CMXRos (ThermoFisher, diluted 1:4000), MitoView Green (Biotium, diluted 1:2000), anti-cardiolipin (diluted 1:100), and APC-conjugated anti-TOM22 antibodies (Miltenyi Biotech, diluted 1:10). Cardiolipin antibodies were detected using a FITC-conjugated polyclonal goat anti-human IgG antibody (diluted 1:100, Jackson Immunoresearch). Isolated mitochondria were used for downstream applications within 24 h.
2.11. Mitochondrial ROS and membrane potential measurement
mtROS analysis of RH30 cells was done using MitoSOX™ Red Mitochondrial Superoxide Indicator (mitoSOX) (Invitrogen™, M36008). Briefly, adhered RH30 cells were cultured in complete RPMI medium supplemented with or without CaCl2 and Na3PO4 for 4 h following which trypsinized cells from each treatment well are transferred into a U-bottomed 96-well plate for staining with mitoSOX (5 μM in warm 1X PBS) for 30 min at 37 °C and analysis by flow cytometry. Membrane potential is analyzed by MitoTracker Red CMXRos. Briefly, cultured, treated and trypsinized RH30 cells are stained with 250 nM of MitoTracker Red CMXRos (ThermoFisher, diluted 1:4000) for 20 min at 37 °C and 5% CO2 incubator followed by 2X washes in warm 1X PBS and analysis by flow cytometry.
2.12. Viability analysis (flow cytometry)
Viability analysis was done using APC-conjugated Annexin V (Biolegend Cat. No. 640920 and propidium iodide (Miltenyi Biotec Cat. No. 130–093-233). Briefly, adhered RH30 cells following trypsinization are transferred into a U-bottomed 96-well plate for staining. Cells are stained for 20 min at room temperature with APC-conjugated Annexin V (Biolegend cat # 640,920; 1:100 dilution) and propidium iodide (Miltenyi Biotec cat# 130–093-233; 1:100 dilution) diluted in Annexin V Binding Buffer (Cat. No. 422201) followed by 2X washes in warm Annexin V Binding Buffer and analysis by flow cytometry.
2.13. Oxygen consumption rate (OCR)
OCR in RH30 cells is measured with a Seahorse XF24 analyzer. RH30 cells (200,000 cells/well) cultured in complete RPMI medium supplemented with or without CaCl2 and Na3PO4 for 24 h are transferred to Seahorse XF24 plate (75,000 cells/well) and let to adhere for 6 h in the complete RPMI medium without CaP. Before the experiment, media was switched to pre-warmed Seahorse XF base medium (1 mM pyruvate, 2 mM glutamine, and 1 mg/ml glucose, pH to 7.4) for 1 h. OCRs were determined over 3-min increments that include a 3 min mixing step, a 30 s wait period, followed by a 3 min respiration measurement. Three measures of basal OCR were recorded before the sequential addition of the following compounds, each followed by three measurements of OCR: oligomycin (5 μM), carbonilcyanide p-triflouromethoxy phenylhydrazone (FCCP, 4.5 μM), and rotenone/antimycin A (1 μM each) phases of the mitochondrial stress test.
2.14. Metabolite profiling by LC-MS
Cell pellets collected from RH30 cells cultured in complete RPMI medium supplemented with or without CaCl2 and Na3PO4 for 24 h were subjected to metabolite profiling using targeted LC-MS metabolite analysis performed on a duplex-LC-MS system, at University of Washington’s Northwest Metabolomics Research Center (Seattle, Washington, US) [23]. Data analysis and over representative analysis (ORA) is performed in MetaboAnalyst version 5.0 (https://www.metaboanalyst.ca/) and volcano plot is drawn using VolcaNoseR web app [24].
2.15. mtDNA analysis
The StepOnePlus™ Real-Time PCR System was used to perform quantitative PCR. Primer sequences for mitochondrial gene, Cytochrome c oxidase subunit II, Cox II, Forward: 5′-CCCCACATTAGGCTTAAAAACAGAT-3′, Reverse: 5′-TATACCCCCGGTCGTGTAGC-3′) [25] targets were reported previously. PCR reaction was set up in a volume of 20 μL that had 1X final of Power SYBR Green PCR Master Mix, one pair of primers directed against nuclear or mitochondrial genes at a final concentration of 100 nM, 5 μL of supernatants and 3 μL of PCR-grade water. Each PCR reaction was set up at the following thermoprofile: incubation for 2 min at 50 °C, followed by a first denaturation step of 10 min at 95 °C and 40 cycles of 95 °C for 15 s, 55 °C (COXII) for 1 min; and 60 °C for 1 min. Absolute quantification of gene targets is derived from standard curve built by serial dilutions of synthesized nuclear and mitochondrial genes. In each run, non-template negative controls were also included. Post-amplification melting curve analysis was performed to confirm the specificity of the amplification.
2.16. Quantification of ISG gene expression
RH30 cells were lysed, and RNA was extracted using Zymo Quick-RNA Whole Blood (cat# R1201; Zymo Research, Irvine, CA) following a protocol meant to isolate RNA from cells, and cDNA was prepared using the High-Capacity cDNA Reverse Transcription Kit for qRT-PCR (cat# 4368814; Applied Biosystems™). qRT-PCR was performed on the StepOnePlus™ Real-Time PCR System with Power SYBR™ Green PCR Master Mix (cat# 4367659; Applied Biosystems™) with a template cDNA of 3 ng/μL. Gapdh was used as the housekeeping gene and the media treated condition Ct was used in the delta-delta calculations for the determination of the fold gene expression. The following primers were used: Human ISG15 (Forward: 5′-GAG AGG CAG CGA ACT CAT CT-3’; Reverse: 5′-CTT CAG CTC TGA CAC CGA CA-3′), Human IFI44L (Forward: 5′-GAA CTG GAC CCC ATG AAG G-3′, Reverse: 5′-ACT CTC ATT GCG GCA CAC C-3′) and Human GAPDH (Forward: 5′- CAA CGG ATT TGG TCG TAT T-3′, Reverse: 5′-GAT GGC AAC AAT ATC CAC TT-3′).
2.17. AMA analysis by flow cytometry
For analysis of immunoglobulin binding to isolated mitochondria (HepG2), mitochondria were stained with MitoTracker Red prior to incubation with serum (diluted 1:100) for 30 min at room temperature. Mitochondria were pelleted (3000 g 10 min at 4 °C) to remove non-bound antibodies and incubated with anti-human IgG-FITC (diluted 1:100, Jackson ImmunoResearch) for 30 min at room temperature. Mitochondria were washed in PBS and analyzed by flow cytometry. Only MitoTracker-positive particles, e.g., mitochondria, were included in the analysis.
2.18. AMA-M2 ELISA
Euroimmun anti-M2–3 E ELISA (IgG) (Order No. EA 1622–9601 G) was used to measure AMA against M2 antigen according to manufacturer’s instructions. Serum was diluted 1:100 in sample buffer. Ratio-based analysis with calibrator 2 was used to identify antibodies against M2 antigens and quality controls are included to ensure assay functionality. The ratio of ≤1 and ≥ 1 is considered negative and positive, respectively.
2.19. Statistical analysis
For non-parametric analysis, the Mann-Whitney U test and Wilcoxon matched-pairs signed rank test were used as applicable and data are represented as the median. P values were considered significant at *P < 0.05; **P < 0.01; ***P < 0.001; and ****P < 0.0001.
3. Results
3.1. Evidence of mitochondrial calcification in patients with JDM
Previously, we have provided evidence of infiltrating phagocytes, including neutrophils and macrophages, phagocytosing calcium crystals in the calcified muscle tissue of patients with JDM [26]. In the current study, we extended those findings to the organellar localization where transmission electron microscopy demonstrated early deposition of dense, elongated crystals in the mitochondria of affected JDM muscle tissue (Fig. 1A, Yellow arrows). Energy Dispersive X-ray Analysis (EDAX) coupled to an FEI transmission electron microscope (CM-12) was used to analyze the calcium content of these mineralized mitochondria. As shown in Fig. 1B, hard tissue, i.e., affected skeletal muscle tissue with crystalline particles in mitochondria, showed higher calcium content per area compared to tissue with non-crystalline particles (K alpha 28.23% vs. 3.42% at 10,000× magnification), confirming calcific nature of mineral deposits in the mitochondria. The analysis of the bone sample (no mitochondria) as a positive control for calcium showed 150.01% calcium content.
Fig. 1.
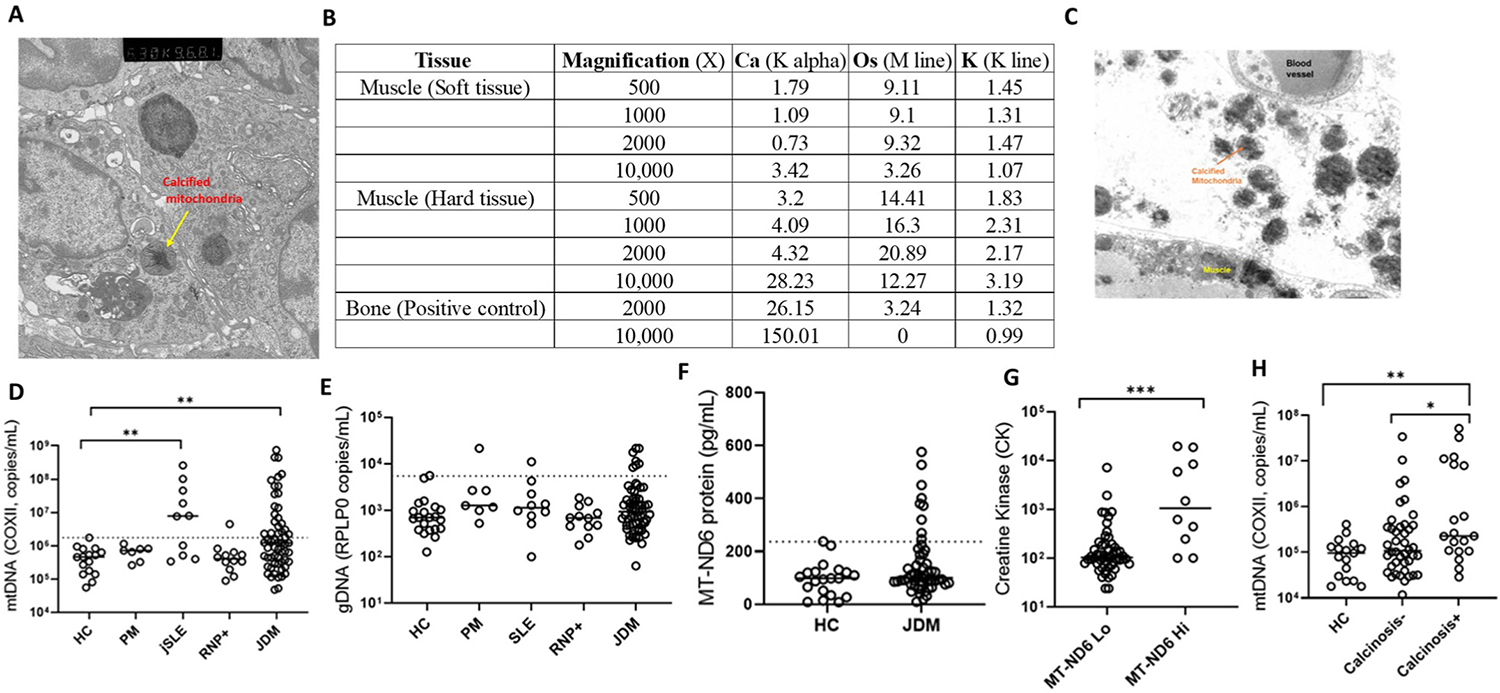
Mitochondria in JDM. (A) Electron microscopy image of degenerated muscle fiber with intracellular mitochondrial calcification. Yellow arrows: Calcified mitochondria with dense, elongated crystals. (B) EDAX mineral analysis to evaluate calcium content of mitochondria; soft tissue (patient muscle tissue without mineral deposits) and hard tissue (patient muscle tissue with mineralized mitochondria) and a positive control, bone (no mitochondria). K alpha: Calcium content; Os (osmium): Lipid content; K line: Total protein content. (C) Electron microscopy image demonstrating the presence of calcified mitochondria in the extracellular space in muscle tissue. qPCR analysis of plasma quantifying (D) mtDNA and (E) genomic DNA in different disease groups PM (n = 7), jSLE (n = 10), RNP + overlap syndrome (n = 12) and JDM (n = 60) compared to age-matched healthy controls (HC; n = 16). Dashed line indicates the 95th percentile of HC levels. (F) Plasma levels of MT-ND6 protein as measured by ELISA in JDM patients (n = 50) compared to age-matched healthy controls (n = 20). Dashed line indicates the 95th percentile of HCs. (G) Creatine Kinase level relative to MT-ND6 levels. MT-ND6 levels are grouped into Low (Lo, n = 54) and high (Hi, n = 10) based on 95th percentile of HC MT-MD6 levels as measured by ELISA. (H) Comparison of mtDNA levels in calcinosis − (n = 42), calcinosis + (n = 19) patients and healthy controls, HC (n = 17). All graphs: Median is shown. Statistical analysis: Non-parametric Mann-Whitney test. *p < 0.05, **p < 0.01, and ***p < 0.001. JDM, juvenile dermatomyositis, HC, age-matched healthy controls, PM, polymyositis, jSLE, juvenile systemic lupus erythematosus, RNP + overlap syndrome, ribonucleoprotein + overlap syndrome, MT-ND6, mitochondrial-NADH dehydrogenase 6, gDNA, genomic DNA, mtDNA, mitochondrial DNA.
Overall, this novel evidence of mitochondrial calcification in the degenerated muscle fibers of patients with JDM warrants a more in-depth characterization of mitochondrial involvement in the pathobiology of the disease.
3.2. Evidence of the association of extracellular mitochondria in JDM with muscle damage and calcinosis
In addition to intracellular calcified mitochondria in the degenerated muscle fibers of JDM patients (Fig. 1A), we also found evidence of extracellular mitochondria in JDM as shown in the electron microscopy image of muscle biopsy from an untreated JDM patient (Fig. 1C). On further analysis, elevated levels of mtDNA (Fig. 1D) but not genomic DNA (Fig. 1E) were found in the plasma of JDM patients and positive control jSLE compared to age-matched healthy controls. In addition, a subgroup of JDM patients also showed elevated levels of mitochondrial protein, MT-ND6 (Mitochondrial Encoded NADH: Ubiquinone Oxidoreductase Core Subunit 6) (Fig. 1F). Further analysis revealed that elevated levels of MT-ND6 and mtDNA levels associate with CK (marker of muscle damage) (Fig. 1G) and calcinosis (Fig. 1H), respectively, suggesting mitochondrial extrusion in JDM patients, potentially from damaged and calcified muscle tissues.
3.3. Anti-mitochondrial antibodies (AMA) in children with JDM
Given the presence of extracellular mitochondria and their remnants in JDM (Fig. 1A–H), we hypothesized that patients may develop AMAs. To test for the presence of AMA in JDM, purified mitochondria (Supplementary Figs. 1A and B) were incubated with plasma from JDM patients and analyzed for IgG binding by flow cytometry. Consistent with the role of mitochondria in SLE disease pathogenesis [27,28], positive disease control, jSLE patients had elevated levels of AMA compared to controls (p = 0.0003, Fig. 2A) and similarly increased levels of AMA are observed in JDM patients with select MSA (MDA5+, p = 0.04; p155/p140+, p = 0.02; Fig. 2A). Although, the JDM patient cohort showed only a trend for increased levels of AMA IgG, a statistically significant difference was observed for the percentage of AMA positivity (95th percentile of the healthy individuals) in the JDM cohort and in patients with select MSA compared to age-matched healthy controls (Fig. 2B). Levels of AMA were particularly elevated in JDM patients with diagnosis of calcinosis (p < 0.0001, Fig. 2C). Of note, in a small set of longitudinally followed patients, levels of AMAs were elevated prior to clinical detection of calcinosis, suggesting a potential prognostic utility of AMAs in predicting calcinosis development (p < 0.01, Fig. 2D). Only one of the JDM patients has reported AMAs, namely AMA against M2 antigen (Supplementary Fig. 2), the most sensitive and specific diagnostic marker of primary biliary cirrhosis [29]. Therefore, we now report the presence of novel AMAs in JDM and recognize the need for their characterization to elucidate their clinical and pathological relevance.
Fig. 2.
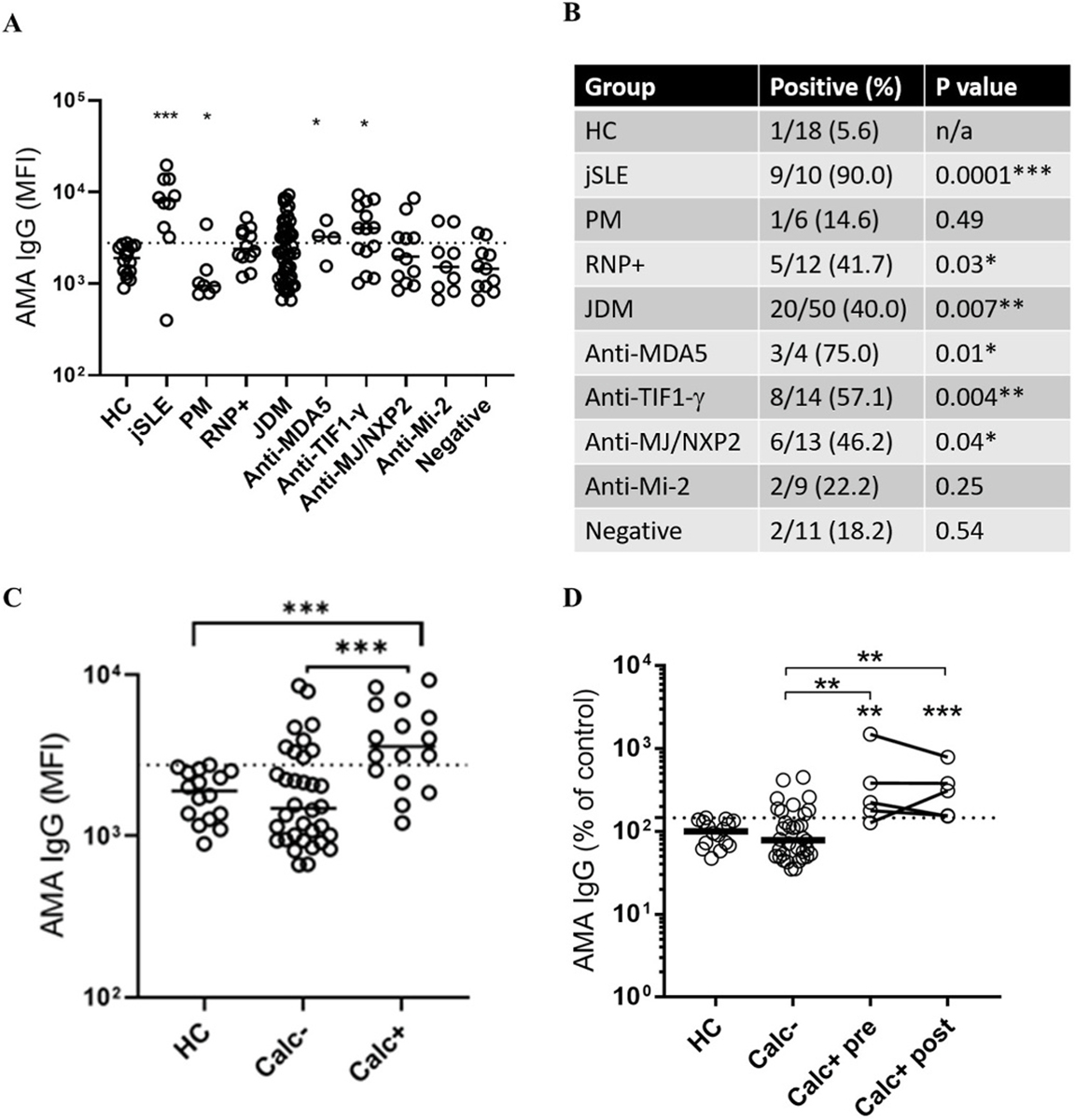
Anti-mitochondrial antibodies in JDM are associated with calcinosis. Levels of anti-mitochondrial antibodies (AMA) were assessed using an in-house flow cytometry assay. (A) Comparison of AMA levels in healthy controls (HC), juvenile SLE (jSLE), polymyositis (PM), RNP + overlap syndrome, and juvenile dermatomyositis (JDM). Further stratification was done based on myositis-specific autoantibodies (MSA). Data are presented as (A) median fluorescence intensity (MFI)and (B) percent positive patients, based on 95th percentile of healthy controls (dotted line). (C) Patients were stratified based on prior history of calcinosis (Calc+) or not (Calc−) and assessed for AMA levels. (D) AMA levels measured in JDM patients without history of calcinosis (Calc−), as well as patients with history of calcinosis with blood samples taken either prior (Calc + pre) or after (Calc + post) clinical manifest calcification. All graphs: Median is shown. Statistical analysis: A and C: Non-parametric Mann-Whitney test. B: Chi2 test. D: Wilcoxon matched-pairs signed rank test. *p < 0.05, **p < 0.01, and ***p < 0.001. The ‘floating’ significance asterisks signify comparisons to healthy controls, whereas the lined significance compares the groups under the line.
3.4. Human skeletal muscle cells undergo calcification
To investigate mechanisms of calcification that may primarily rely on mitochondria, we used the rhabdomyosarcoma cell line RH30, which has metabolically functional mitochondria [21,30]. Given that calcium deposits of JDM calcinosis are primarily of HA in nature [4,5], we quantified calcium phosphate complexes in RH30 cells using Osteoimage™ [31], a fluorescent staining reagent highly specific for HA as represented in the schematic (Fig. 3A). As shown in Fig. 3B, a dose-dependent increase in HA complex formation was observed in RH30 cells when cultured in RPMI 1640 supplemented with increasing Ca2+ and ion concentrations (Fig. 3B, Moderate vs. High calcium-phosphate concentrations, P ≤ 0.001). Osteoimage staining for the detection of HA complexes is also confirmed in purified mitochondria from RH30 cells (Supplementary Fig. 1C). Considering that we observed a significant effect on the viability of RH30 cells at high Ca2+ and ion concentrations (3.12 mM CaCl2 and 7.63 Na3PO4) compared to moderate levels (1.77 mM Cacl2 and 6.63 mM Na3PO4; referred to as CaP henceforth) (Fig. 3C, P ≤ 0.001), we used moderate Ca2+ and ion concentrations to culture RH30 cells in all subsequent experiments. In addition, a time-dependent increase in HA was observed in RH30 cells following their culture in moderate CaP media (24 h vs. 48 h, Fig. 3D, P ≤ 0.001). Thus, we have established an in vitro system to explore mechanisms of calcification in human skeletal muscle cells.
Fig. 3.
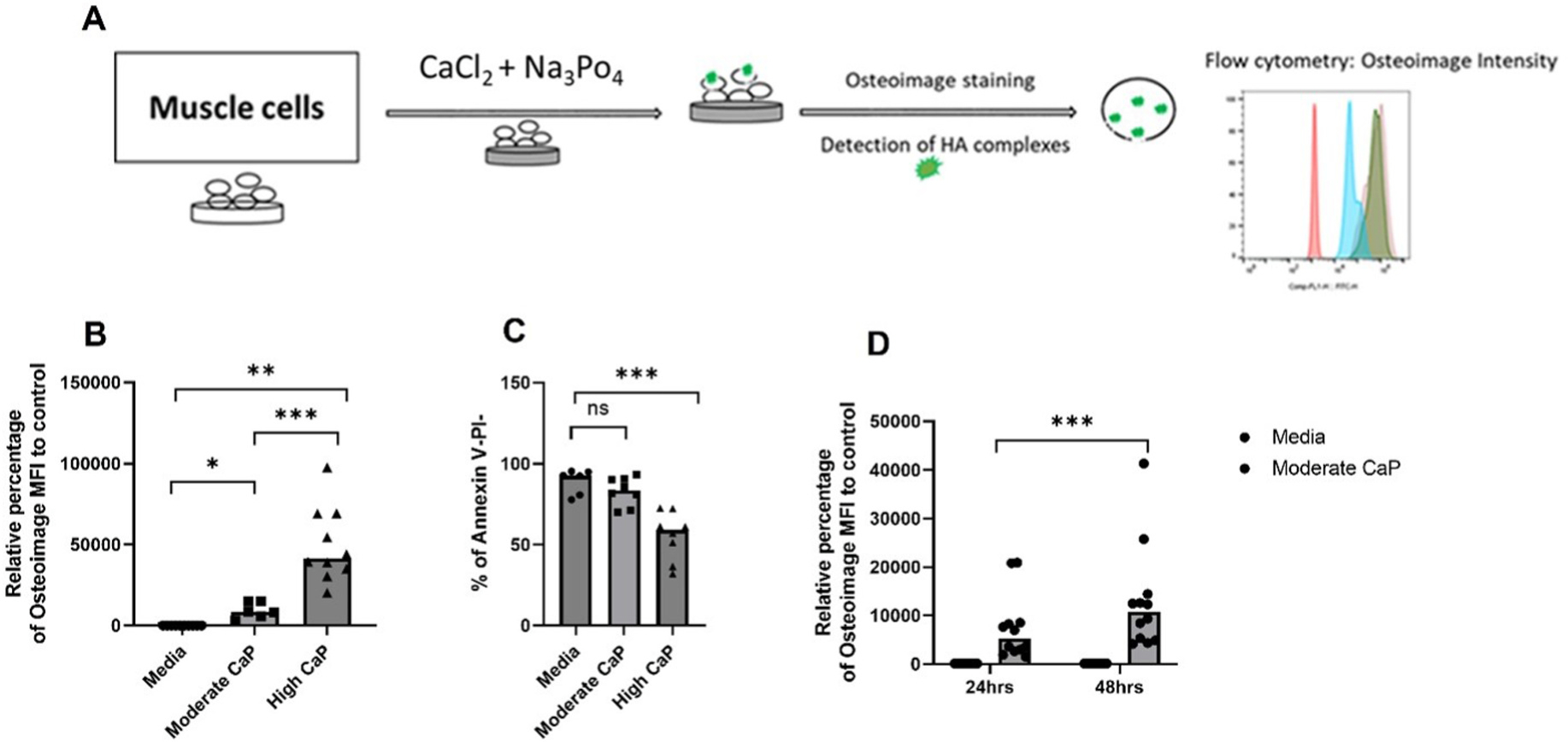
Dose- and time-dependent increase in hydroxyapatite (HA) calcium phosphate complexes in human skeletal muscle cells. (A) Schematic representation of calcification induction in RH30 cells and the quantification of HA complexes by flow cytometry. (B) Dose-dependent increase in osteoimage MFI of RH30 cells cultured in moderate (1.77 mM CaCl2 and 6.63 mM Na3PO4) or high (3.12 mM CaCl2 and 7.63 mM Na3PO4) calcium phosphate (CaP) medium for 24 h (n = 3 independent experiments). (C) Viability, as determined by Annexin V and Propidium (PI) iodide staining (live cells: Annexin V-PI-) of RH30 cells cultured in moderate and high CaP medium for 24 h (n = 3 independent experiments). (D) Time-dependent increase in osteoimage MFI in RH30 cells cultured in moderate CaP medium for 24 or 48 h (n = 6 independent experiments). All graphs: Median is shown. Statistical analysis: (B) Non-parametric Wilcoxon matched-pairs signed rank test was used for comparison between media and CaP conditions. CaP conditions are compared using non-parametric Mann-Whitney test. (C,D) Non-parametric Mann-Whitney test. (E) Non-parametric Wilcoxon matched-pairs signed rank test. *p < 0.05; **P ≤ 0.01; ***P ≤ 0.001.
3.5. Intracellular calcification of human skeletal muscle cells occurs in mitochondria
We primarily assessed the role of mitochondria as the intracellular sites for HA complexes in RH30 cells considering that the sequestration of Ca2+ in the form of calcium phosphate complexes including HA is the major calcium buffering mechanism in mitochondria [32]. The active accumulation of Ca2+ in mitochondria and their retention as calcium phosphate complexes depends on the presence of a steep mitochondrial membrane potential, ΔΨm, (~150–180 mV, and that the mitochondrial matrix is negative in charge) [33]. Therefore, we first assessed the formation of HA complexes in RH30 in the absence or presence of the mitochondrial uncoupler, p-(trifluoromethoxyl) [phenyl-hydrazone] (FCCP), known to dissipate ΔΨm. As shown in Fig. 4A, FCCP significantly suppressed the formation of HA complexes in RH30 cells compared to untreated RH30 cells (Fig. 4A, P ≤ 0.01), without affecting the viability of cells, but with a dissipation of ΔΨm (Supplementary Figs. 3A and B). Confocal microscopy analysis of RH30 cells further substantiated the presence of HA complexes in mitochondria as shown in Fig. 4B, where an overlap of HA complexes (osteoimage+) with a mitochondrial marker, anti-TOM22 was observed. Ru360 [34], a selective inhibitor of mitochondrial calcium uniporter (MCU) [35], the primary pathway of Ca2+ entry into mitochondria caused a significant suppression of HA complex formation in RH30 cells (Fig. 4C). Viability data for Ru360-treated RH30 cells are shown in Supplementary Fig. 3C. Further, knockdown of mitochondrial calcium uptake 1 (MICU1), a Ca2+ sensing gatekeeper of MCU that functions to prevent mitochondrial calcium overload [36], resulted in exaggerated degree of calcification in RH30 cells (Fig. 4D). Immunoblot analysis confirmed MICU1 knockdown efficiency in RH30 cells (Supplementary Fig. 4). Altogether, functional data, imaging, pharmacological inhibition, and genetic knockdown approaches suggest the mitochondrial localization of HA complexes in RH30 cells in this useful model of in vitro calcification.
Fig. 4.
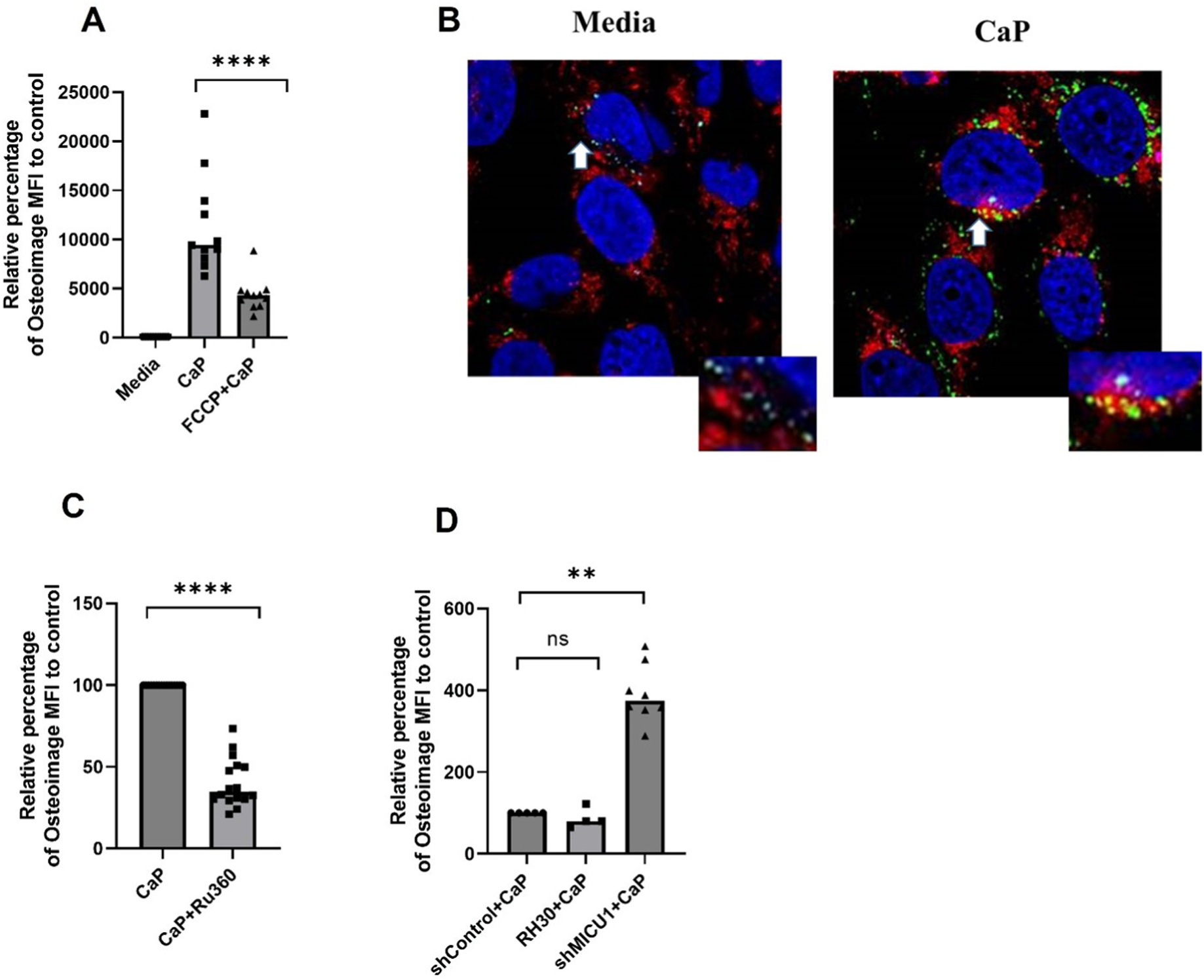
Mitochondrial calcification of human skeletal muscle cells cultured in moderate calcium phosphate medium. (A) Osteoimage flow cytometry analysis of RH30 cells cultured in complete media alone, or complete culture media supplemented with 1.77 mM CaCl2 and 6.63 mM Na3PO4 in the presence or absence of FCCP (5 μM) (n = 4 independent experiments) for 24 h. (B) Confocal images of RH30 cells stained for osteoimage, Green (HA), Anti-Tom22 (mitochondrial outer membrane protein, Red), and DAPI (nuclear stain, Blue) (representative of two independent experiments). White arrows highlight zoomed regions. (C) RH30 cells were pretreated with Ru360 (1 μM) 60 min before exposure to CaP medium for 24 h s followed by osteoimage analysis by flow cytometry (n = 3 independent experiments). (D) Relative percentage of calcification (osteoimage MFI) in MICU1 knockdown RH30 cells (shMICU1) compared to vector control (shControl) (n = 2 independent experiments). All graphs: Median is shown. Statistical analysis: (A) Non-parametric Mann-Whitney test. (C,D) Non-parametric Wilcoxon matched-pairs signed rank test. **P ≤ 0.01; ****≤0.0001.
3.6. Calcification causes mitochondrial dysfunction in human skeletal muscle cells
We found that calcifying RH30 cells exhibited mitochondrial hyperpolarization (Fig. 5A) and increased mtROS generation (Fig. 5B). This was accompanied by impaired mitochondrial respiration, reflected as a decreased rate of oxygen consumption (Fig. 5C–F). Additionally, defects in mitochondrial functions of calcifying RH30 cells were confirmed using a targeted LC-MS metabolite analysis approach. Some of the key downregulated metabolites identified in targeted metabolomics analysis, include glycerol 3-P, ADP, NAD, 5-methyl uridine, pyroglutamic acid and 3-hydroxybutyric acid (3HBA) (Supplementary Figs. 5B–G). Of note, adenine and cis-Acotinate include significantly upregulated metabolites in CaP-treated RH30 cells (Supplementary Figs. 5H and I). An over representation analysis (ORA) performed on significantly decreased metabolites in CaP-treated cells (Supplementary Fig. 5A) revealed several mitochondrial-related pathways including mitochondrial respiration in the top 25 enriched metabolites sets (Fig. 5G), suggesting that calcifying human skeletal muscle cells had mitochondrial dysfunction.
Fig. 5.
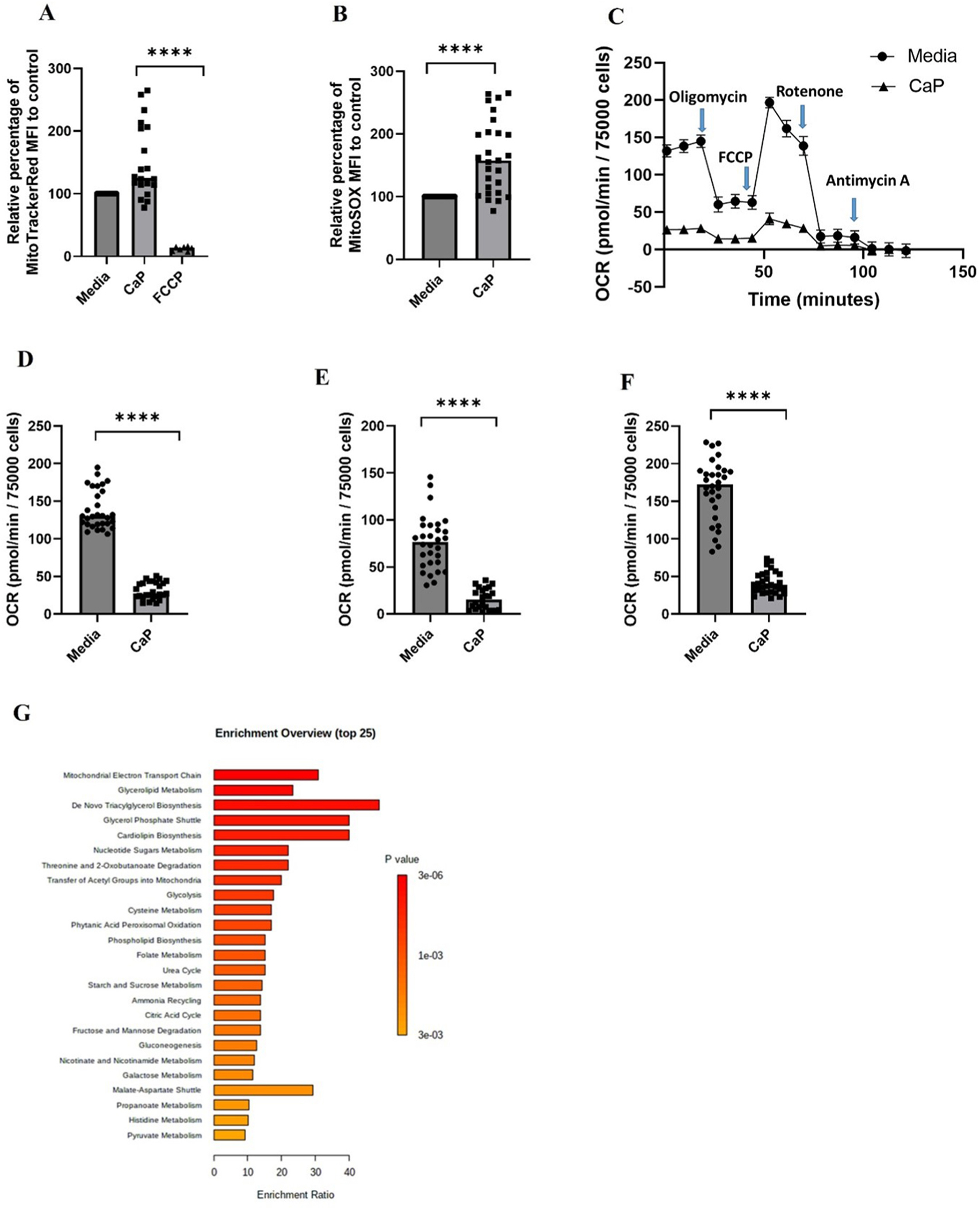
Calcification causes mitochondrial dysfunction in calcifying human skeletal muscle cells. (A) Mitochondrial membrane potential analysis using MitoTracker Red CMXRos, a membrane-potential dependent mitochondrial dye (n = 2 independent experiments). Briefly, RH30 cells were cultured in complete RPMI medium supplemented with or without 1.77 mM CaCl2 and 6.63 mM Na3PO4 for 4 h and analyzed for membrane potential by flow cytometry analysis. (B) mtROS analysis of RH30 cells using mitoSOX by flow cytometry (n = 6 independent experiments). Briefly, RH30 cells were cultured in the presence or absence of with 1.77 mM CaCl2 and 6.63 mM Na3PO4 for 4 h followed by staining with mitoSOX (5 μM in warm 1X PBS) for 30 min at 37 °C. (C) Representative mitochondrial oxygen consumption rates in RH30 cells cultured in complete RPMI medium supplemented with or without CaP medium for 24 h, under basal conditions, oligomycin (5 μM), FCCP (4.5 μM), rotenone (1 μM) or antimycin A (1 μM) are shown as measured using Seahorse XF24 analyzer (n = 2 independent experiments). (D) Average basal respiration (n = 2 independent experiments), (E) Average ATP-linked respiration (n = 2 independent experiments), and (F) Maximal respiration (n = 2 independent experiments) are shown. (G) Results from over representative analysis (ORA) using hypergeometric test to evaluate whether a particular metabolite set is represented more than expected by chance within significantly decreased metabolites in CaP-treated cells compared to media only (online Supplemental Fig. S5). ORA is performed in MetaboAnalyst version 5.0. Enrichment Ratio is computed by Hits/Expected, where hits = observed hits; expected = expected hits. One-tailed p values are provided after adjusting for multiple testing. All graphs: Median is shown. Statistical analysis (A,D,E,F) Non-parametric Mann-Whitney test. (B) Non-parametric Wilcoxon matched-pairs signed rank test. ****≤0.0001.
3.7. Human type I IFN-α (hIFN-α) amplifies mitochondrial calcification of human skeletal muscle cells in mtROS-dependent manner
LPS-induced oxidative stress was shown to augment the activity of MCU, leading to mitochondrial Ca2+ overload [12]. To determine if mtROS also amplifies HA complex formation in RH30 cells, we first performed the induction of calcification experiments in the absence or presence of antimycin A (AA), a complex III inhibitor of mitochondrial ETC and a known inducer of mtROS [37]. AA in a dose-dependent manner amplified calcification in RH30 cells (Supplementary Fig. 6A) with significant mtROS induction and membrane potential, respectively (Supplementary Figs. 6B and C). Overall, these experiments with AA suggested that mtROS can augment HA complex formation in skeletal muscle cells.
Given relevance of type I hIFN to the pathogenesis of JDM muscle and skin [38,39], we investigated the effect of hIFN-α on calcification of RH30 cells. Based on the evidence that hIFN-α induces mtROS [40], we hypothesized that hIFN-α could also amplify HA complex formation in RH30 cells in an mtROS-dependent manner. Treatment with hIFN-α caused a noticeable induction of mtROS in RH30 cells in a dose-dependent manner (CaP vs + 30 U/mL hIFN-α, Fig. 6A, P ≤ 0.05) overlapping with significant amplification of calcification by hIFN-α (CaP vs + 30 U/mL hIFN-α, Fig. 6B, P ≤ 0.05) consistent with our proposition of the mtROS role in calcification. The tested doses did not have a significant effect on the viability of RH30 cells (Supplementary Fig. 6D). Higher concentrations of hIFN-α did not further amplify mitochondrial calcification (data not shown) why subsequent experiments were performed with 30 U/mL. Similar to spontaneous calcification, the presence of mitochondrial uncoupler, FCCP (CaP+30 U/mL hIFN-α vs CaP + 30 U/mL hIFN-α + FCCP, Fig. 6C, P ≤ 0.01) attenuated hIFN-α amplified calcification. Next, the role of hIFN-α-induced mtROS in amplifying mitochondrial calcification of RH30 cells was substantiated by exposing RH30 cells to hIFN-α in the absence or presence of MitoTempo concentration (10 μM) that significantly scavenged hIFN-α-induced mtROS (Fig. 6D, P ≤ 0.05) that paralleled with a marked decrease in hIFN-α-induced calcification (Fig. 6E, P ≤ 0.01). Overall, we confirmed the role of hIFN-α, a pathogenic factor implicated in the pathogenesis of JDM, and as an amplifier of mitochondrial calcification of human skeletal muscle cells with mtROS as a central player in our in vitro induction of calcification studies.
Fig. 6.
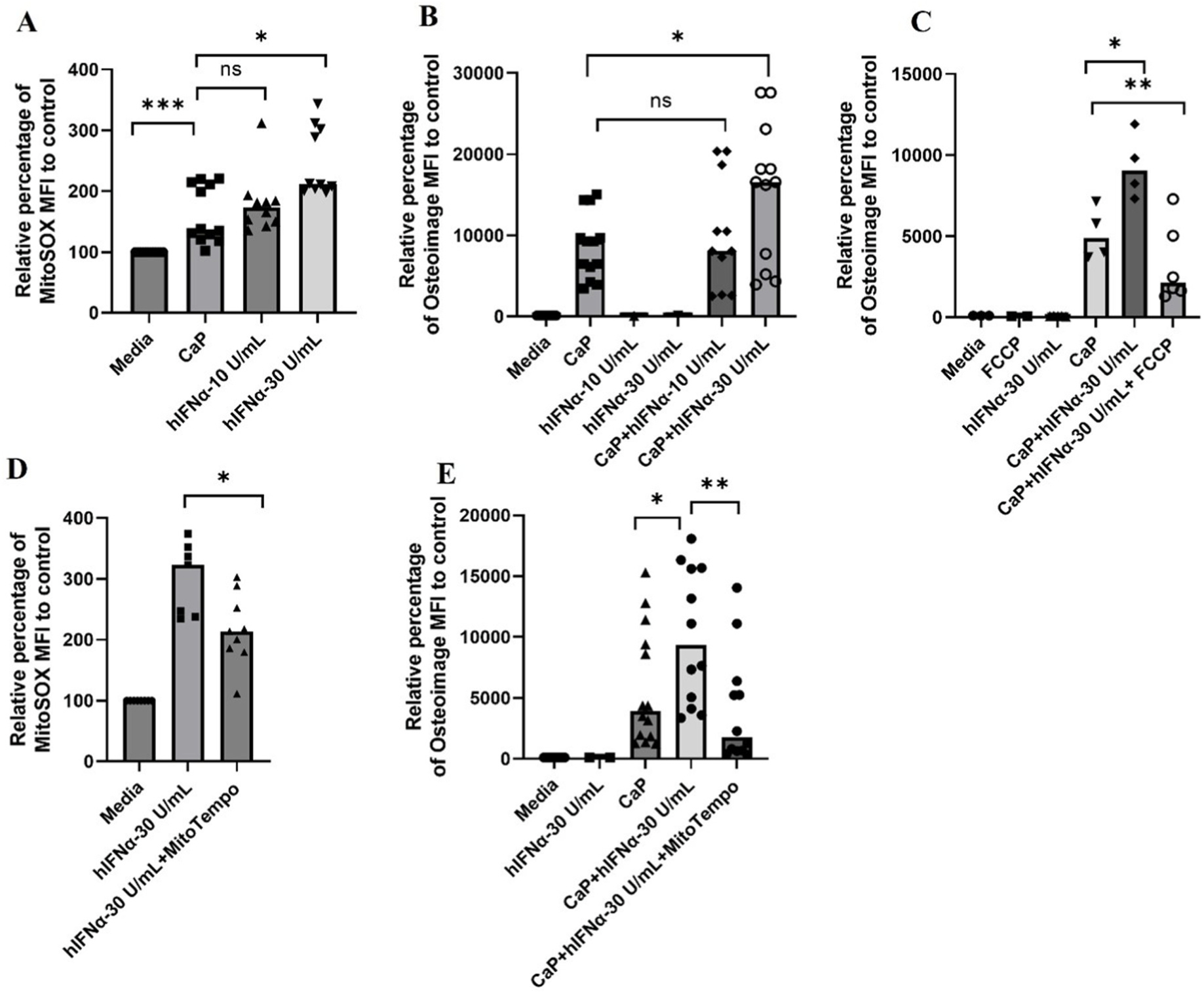
Intracellular mitochondrial calcification of human skeletal muscle cells amplified by hIFN-α occurs in an mtROS-dependent manner. (A) mtROS analysis of RH30 cells using mitoSOX by flow cytometry (n = 4 independent experiments). Briefly, RH30 cells were cultured in complete RPMI medium supplemented with 1.77 mM CaCl2 and 6.63 mM Na3PO4 in the presence or absence of different concentrations of hIFN-α for 4 h followed by staining with mitoSOX (5 μM in warm 1X PBS) for 30 min at 37 °C. (B) Dose-dependent increase in mitochondrial calcification of RH30 cells by hIFN-α (n = 5 independent experiments). RH30 cells are cultured for 24 h in with 1.77 mM CaCl2 and 6.63 mM Na3PO4 and −/+ human type I IFN-α and then analyzed for HA complex formation by osteoimage flow cytometry analysis. (C) Attenuation of calcification in RH30 cells by treatment of cells with FCCP (5 μM) (n = 2 independent experiments). RH30 cells are cultured for 24 h in complete RPMI medium supplemented with or without 1.77 mM CaCl2 and 6.63 mM Na3PO4 in the absence or presence of human type I IFN-α −/+ FCCP (5 μM) and then analyzed for HA complex formation by osteoimage flow cytometry analysis. (D) MitoSOX analysis of RH30 cells pre-treated with mitoTempo (10 μM) for 30 min prior to the hIFN-α treatment and/or CaP media. Cells were analyzed for mtROS using mitoSOX after 4 h of incubation (n = 3 independent experiments). (E) Osteoimage staining flow cytometry analysis of HA complexes in RH30 cells cultured for 24 h in complete RPMI medium supplemented with or without CaCl2 and Na3PO4 in the absence or presence of hIFN-α and mitoTempo (10 μM). RH30 cells are pre-treated MitoTempo 30 min prior to the addition of hIFN-α and 1.77 mM CaCl2 and 6.63 mM Na3PO4 (n = 5 independent experiments). All graphs: Median is shown. Statistical analysis (A) Non-parametric Wilcoxon matched-pairs signed rank test was used for comparison between media and CaP conditions. (A,B, D,E,F) CaP conditions are compared using non-parametric Mann-Whitney test. *P ≤ 0.05; **P ≤ 0.01.
3.8. Mitochondrial calcification-induced inflammation in human skeletal muscle cells propagates calcification independent of exogenous hIFNα
Mitochondrial dysfunction following calcification (Fig. 5) suggests potential mitochondrial damage that could result in cytosolic release of mtDNA contributing to calcification-induced inflammation. We performed a preliminary investigation with purified mitochondria from RH30 cells and exposed them to calcium phosphate medium. Elevated levels of mtDNA (Supplementary Fig. 7A) and increased cardiolipin exposure (Supplementary Fig. 7B) are observed in the supernatants and mitochondria of CaP-treated mitochondrial isolations, respectively. Consistent with the pro-inflammatory nature of calcified mitochondria, a significant induction of interferon-stimulated gene expression, ISGs, was observed in calcifying RH30 cells (IFI44L and ISG15), as shown in Fig. 7A and B. As a positive control for ISG expression, RH30 cells were cultured in media supplemented with hIFNα−30 U/mL and analyzed for ISG gene expression (Supplementary Figs. 8A and B).
Fig. 7.
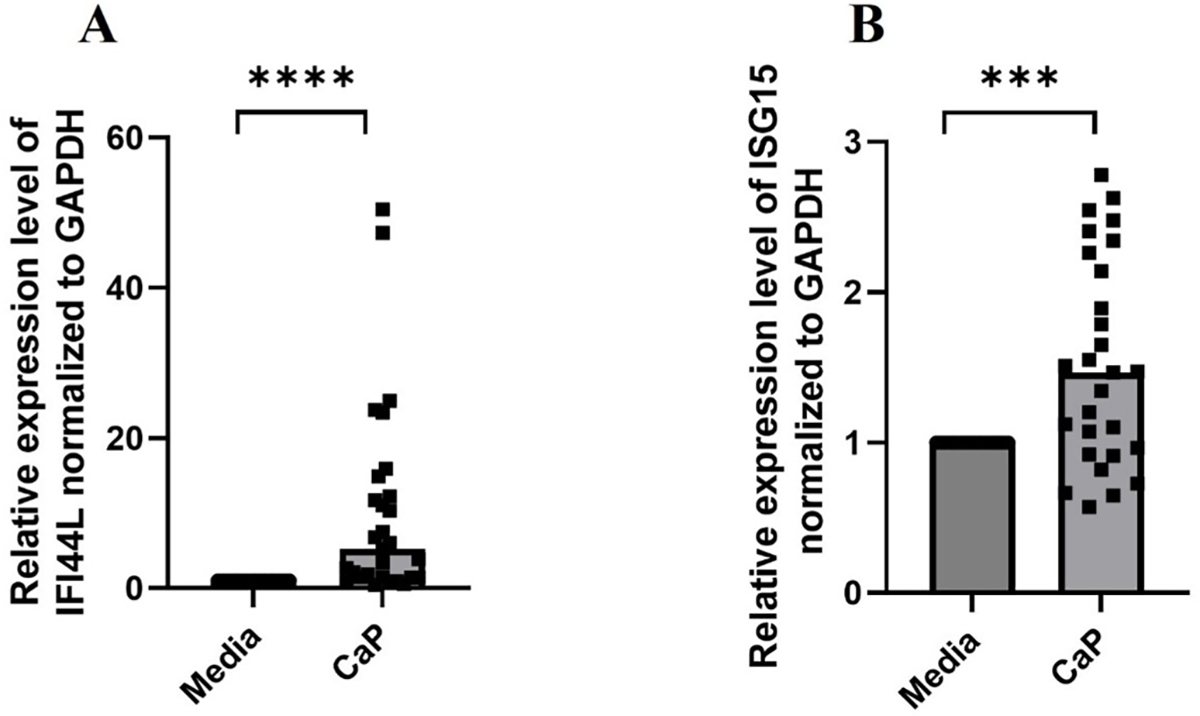
In vitro calcification induces inflammation in human skeletal muscle cells. (A) IFI44L and (B) ISG15 mRNA levels in RH30 cells cultured in complete RPMI medium supplemented with or without 1.77 mM CaCl2 and 6.63 mM Na3PO4 for 24 h prior to RNA isolation, cDNA preparation followed by qPCR (n = 4 independent experiments). All graphs: Median is shown. Statistical analysis: Non-parametric Wilcoxon matched-pairs signed rank. ****P ≤ 0.0001.
Given the release of mtDNA into the extra mitochondrial space upon calcification (Supplementary Fig. 7A), which in the context of cell would be cytosol, we next assessed the involvement of cGAS-STING pathway in calcification-induced inflammation. As shown in Fig. 8A, treatment of RH30 cells with H-151, a covalent antagonist of stimulator of interferon genes (STING) [41], led to a dose-dependent decrease in ISG expression. RH30 cells transfected with calf thymus DNA are used as a positive control (Supplementary Fig. 9). Based on the observation that exogenously added hIFNα amplified calcification in RH30 cells (Fig. 6B, C, and E), we hypothesized that calcification-induced endogenous inflammation potentiates calcification in the conditions of moderate CaP exposure. Consistently, attenuation of inflammation (ISG15 expression, Fig. 8A) paralleled with a significant decrease in calcification (Fig. 8B).
Fig. 8.
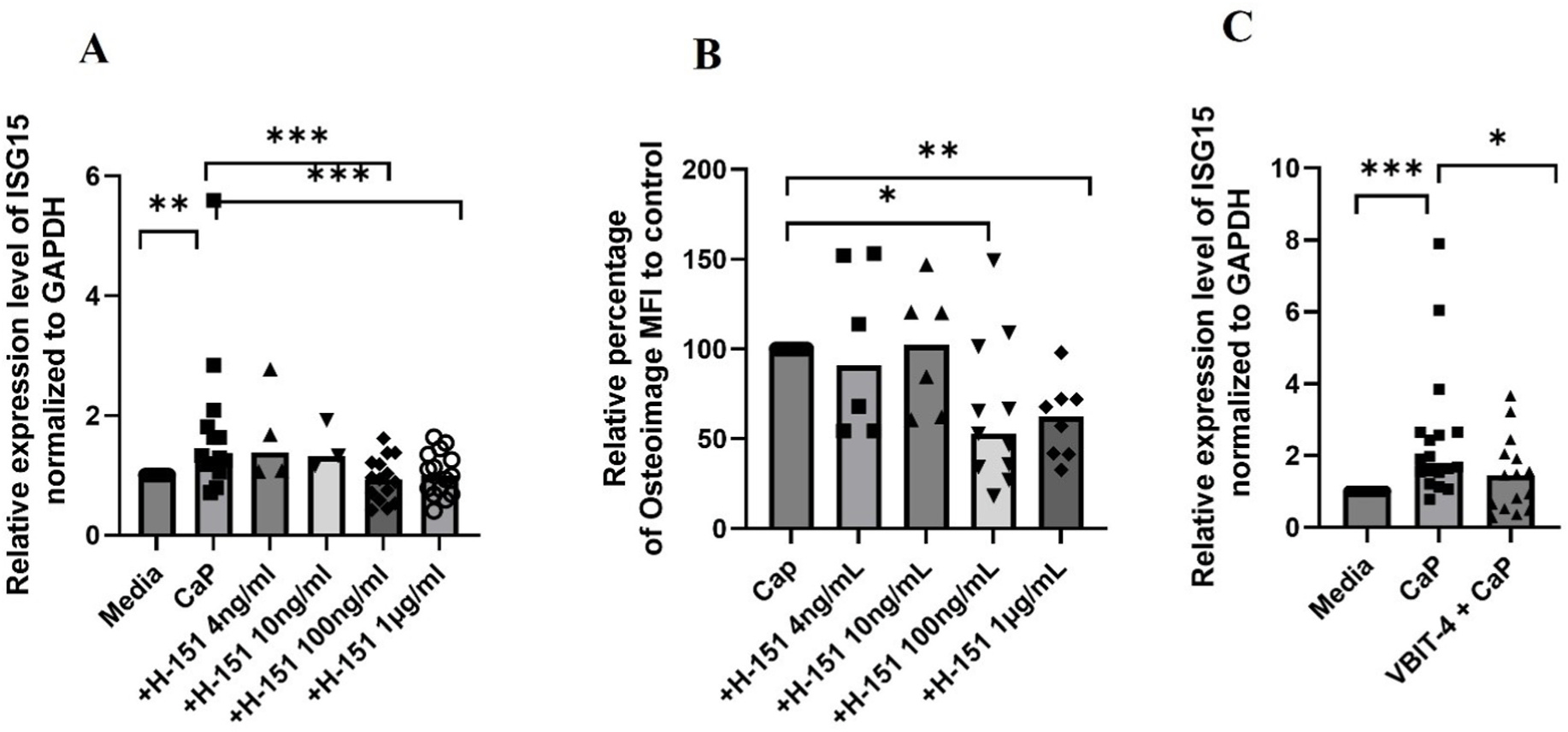
cGAS-STING-mediated ISG expression and voltage-dependent anion channel (VDAC)-mediated cytosolic release of mtDNA in calcifying human skeletal muscle cells. RH30 cells pretreated with or without different concentrations of H-151 for 60 min in RPMI medium followed by the treatment of 1.77 mM CaCl2 and 6.63 mM Na3PO4 media for 24 h and then analyzed for (A) ISG expression by qPCR (n = 4 independent experiments (B) for HA complex formation by osteoimage flow cytometry analysis (n = 3 independent experiments) or for gene expression. (C) RH30 cells pretreated with or without VBIT-4 (1 μM) for 30 min in RPMI medium followed by the treatment of 1.77 mM CaCl2 and 6.63 mM Na3PO4 media for 24 h and then analyzed by qPCR for ISG expression (n = 3 independent experiments). All graphs: Median is shown. Statistical analysis (A,B,C) Non-parametric Wilcoxon matched-pairs signed rank test. *P < 0.05, **P ≤ 0.01; ***P ≤ 0.001, ***≤0.0001.
3.9. Voltage-dependent anion channel (VDAC)-mediated cytosolic release of mtDNA in calcifying human skeletal muscle cells
We next investigated the mechanisms of mtDNA release leading to interferon responses using our in vitro calcification model. It was recently demonstrated that, in living cells, a mitochondrial outer membrane pore, formed by the oligomerization of VDAC1, facilitates cytosolic mtDNA leakage [42]. Therefore, using calcifying cells, we pursued mtDNA release using VDAC1 oligomers as a potential mechanism. We employed VBIT-4, an inhibitor of VDAC1 oligomerization, shown to block the cytosolic release of mtDNA from mitochondrial endonuclease g-deficient mouse fibroblasts [42]. VBIT-4 treatment significantly diminished ISG expression in RH30 cells (Fig. 8C). The concentration of the VBIT-4 did not affect the ISG expression induced by hIFN-α, suggesting that the observed significant decrease in ISG expression in calcifying RH30 cells is not due to general suppression of IFN responses (Supplementary Fig. 10).
4. Discussion
Although calcinosis remains the characteristic debilitating feature of patients with JDM, the pathophysiological mechanisms remain unknown. In an in vitro proof-of-concept study, we report mtROS as a central and therapeutically targetable regulator of inflammation-driven mitochondrial calcification of human skeletal muscle cells (study model; graphical abstract).
In this study, we demonstrate in vivo evidence of mitochondrial calcification in patients with JDM. The persistence of damaged mitochondria in JDM tissues suggested potential defects of mitophagy, a mitochondrial quality control mechanism, in these patients. This proposition corroborates with our previous transcriptomic study where we saw an RNA seq signature defective of autophagy in the peripheral blood and muscle of patients with JDM [39]. To muscle, defective autophagy of damaged mitochondria could not only mean myofiber apoptosis manifesting into myopathy but also accumulation of harmful calcified mitochondria extracellularly that can function as nucleation sites for tissue calcinosis and/or stimuli for inflammation [26]. There is growing body of literature on the role of defective mitophagic clearance of mtDNA in driving type I interferon responses reminiscent of autoimmune diseases in animal models [43] and so are the genetic association studies implicating defective autophagy genes in lupus and other autoimmune diseases [44,45]. However, what remains to be investigated are mechanisms leading to defective autophagy/mitophagy in various autoimmune diseases including JDM. In mesenchymal stem cells, chronic inflammation was found to cause mitochondrial Ca2+ overload mediated by excess Ca2+ transfer from ER and defective mitophagy resulting in the accumulation of damaged mitochondria [46]. Further, it would be interesting to explore if JDM patients with calcinosis would benefit from therapeutic interventions such as NAD+ supplementation, a known inducer of mitophagy restoring the mitochondrial dysfunction in effected tissues [47].
While physiological Ca2+ levels promote mitochondrial bioenergetics generating ATP, excessive accumulation of mitochondrial Ca2+ leads to mitochondrial dysfunction and cell death manifesting into skeletal muscle abnormalities and degeneration. The importance of mitochondrial Ca2+ for skeletal muscle function is substantiated in global MCU−/− mice that exhibited metabolic and functional alterations of skeletal muscle coinciding with the absence of mitochondrial Ca2+ uptake [48]. Human subjects with MICU1 mutations, (thus a loss of MCU gatekeeping resulting in elevated levels of resting mitochondrial Ca2+ levels) had symptoms comprised of early-onset proximal muscle weakness [49]. Similarly, in our in vitro calcification model, pharmacological inhibition of MCU and genetic knockdown of MICU1 led to perturbed calcification of skeletal muscle cells suggesting the primary involvement of mitochondria. Additionally, as reported in the literature, the role of other organelles, including the crosstalk of the endoplasmic reticulum (ER) with mitochondria leading to cellular calcification, cannot be ruled out [50].
As shown by us as well as others, several mechanisms, including buffering of excess calcium as calcium phosphate complexes of HA [32, 51], operate in mitochondria to prevent mitochondria from the toxicity of matrix-free Ca2+ overload. However, excess calcium-phosphate complexes can induce a cristae remodeling and thus form physical barriers for electron chain complexes resulting in mitochondrial dysfunction [52]. Accordingly, calcified skeletal muscle cells in our model exhibited hyperpolarization, elevated mtROS and compromised oxidative capacity, as confirmed in the functional and metabolomics studies.
The type of calcification seen in JDM is dystrophic by definition, given that calcification occurs predominantly in injured/degenerated tissues despite normal circulating calcium and phosphate levels, suggesting a complex interplay of systemic and localized mechanisms in the tissue calcinosis of JDM. Consistently, in an injury-driven dystrophic calcification murine model induced by cardiotoxin, we have observed that calcifications of injured skeletal muscle were transient (<28 days) [53]. Importantly, in cardiotoxin-induced calcification model, mitochondria were found to be nucleation sites for skeletal muscle calcification; a finding reproduced in our in vitro calcification model, where mitochondria were found to be primary sites of HA accumulation. The animal study demonstrated that in the absence of additional factors altering the formation and the removal of mineralization deposits, calcification is self-limiting. Among several factors implicated in JDM pathogenesis, including hypoxia, viral infections, and autoantibodies, the impact of inflammation on calcinosis has been widely investigated from the perspective of therapeutic interventions. In vitro, type I IFN was shown to mediate muscle atrophy with an upregulation of atrophy-associated genes and impaired muscle repair in primary human skeletal muscle cells, suggesting the direct pathogenic role of type I IFN in the muscle pathology of dermatomyositis [54]. Additionally, pathologically low levels of systemic mineralization inhibitors such as fetuin-A in the conditions of chronic inflammation were also proposed to contribute to ectopic calcification in JDM [55].
Given the induction of mitochondrial oxidative stress by type I IFN [40], we asked if type I IFN can also induce mtROS in a skeletal muscle cell line and whether such an induced mtROS can mediate/potentiate the calcification process. Mechanisms reported to promote mitochondrial calcium overload in the conditions of inflammation include oxidative stress-induced modifications of Ca2+ conducting ions channels, including mtROS-mediated S-glutathionylation of MCU leading to oligomerization of MCU and augmented channel activity [12] and disrupted mitochondria-ER coupling affecting inter organellar Ca2+ exchange [46]. Adding to the knowledge concerning the role of inflammation in mitochondrial Ca2+ homeostasis, we demonstrated that human recombinant IFN-α could amplify calcification of human skeletal muscle cells with mtROS-dependent manner. Mechanistically for JDM calcinosis, it could mean that type I IFN could predispose even uninjured muscle cells to calcify.
Crystal-mediated inflammation is well established in the literature, including neutrophil activation by calcium phosphate crystals as shown by us in JDM tissue and in vitro [26,56]. In the current study, we found that calcification makes mitochondria proinflammatory, such as increased exposure of cardiolipin, a characteristic of stressed mitochondria, and the release of mtDNA. Further exploration of the mechanisms with a pharmacological inhibitor of STING revealed the role of the cGAS-STING pathway in the induction of ISGs potentially by mtDNA released from destabilized calcified mitochondria. Importantly, impairment of ISG response also led to a significant reduction in the calcification, highlighting an autocrine amplification loop of inflammation and calcification in calcifying skeletal muscle cells. These findings add to the literature on muscle being the source of type I IFN in DM contributing to muscle damage and the role of mitochondria in perpetuating inflammation in muscle [57]. As a mechanism of mtDNA release from calcified mitochondria resulting in the engagement of cytosolic DNA sensor, cGAS, we demonstrated the role of VDAC-pore mediated mtDNA release, as reported in cells undergoing moderate stress without affecting their viability [42]. We ruled out BAX/BAK pore-mediated mtDNA release, considering that BAX/BAK pore activation in caspase efficient cells results in apoptosis [58].
Given the proof-of-concept nature of the investigation, there are inherent limitations to the study. All our in vitro studies were done on a rhabdomyosarcoma cell line, RH30, established from skeletal muscle precursor cells (myoblasts) that failed to differentiate [59] but retained the skeletal muscle characteristics [21]. Hence, study findings should be validated in primary human skeletal muscle cells and, more importantly, in patient muscle tissues, including in situ mitochondrial assessment, to address the biological relevance of the results to calcinosis in patients with JDM. We need animal models akin to our previously published dystrophic calcification model [53], with a study designed to investigate the role of inflammation and the associated mitochondrial dysfunction in calcinosis. Larger patient cohorts are needed to validate our biomarker studies, and detailed characterization is necessary to establish the identity of AMAs characteristic of JDM. Clinically inactive patients with JDM still exhibit robust immune activation signatures [39]. Not surprisingly, the current therapeutic approaches to JDM, including non-specific immunosuppression guided by clinical criteria, were met with limited success resulting in partial recovery, adverse effects, and frequent relapses. Hence, a deeper understanding of pathophysiology is needed to develop targeted therapies for JDM. Our study highlights the role of potential utility of mtROS scavengers and/or inhibition of upstream mediators of mitochondrial dysfunction as therapeutic strategies for skeletal muscle-associated calcinosis, as occurs in children and adults with dermatomyositis. Further, AMAs as predictive biomarkers can aid in the better management of calcinosis.
Supplementary Material
Acknowledgements
The authors are grateful for valuable advice from Drs. Keith Elkon and Tomas Mustelin, University of Washington.
Funding
This work was supported by Seattle Children’s Hospital Cure JM Center of Excellence grant, National Institutes of Health grant R21AR077565, National Institutes of Health grant R21AR079542, and National Institutes of Health grant R01HL158606 to CL, National Institutes of Health (NIH) UL1TR001422 and The Cure JM Foundation for Maintenance of the JM Repository; Jacque Den Uyl and other appreciated donors to LMP.
Footnotes
Declaration of competing interest
None declared.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jaut.2023.103061.
Data availability
Data will be made available on request.
References
- [1].Symmons DP, Sills JA, Davis SM, The incidence of juvenile dermatomyositis: results from a nation-wide study, Br. J. Rheumatol. 34 (1995) 732–736. [DOI] [PubMed] [Google Scholar]
- [2].Feldman BM, Rider LG, Reed AM, Pachman LM, Juvenile dermatomyositis and other idiopathic inflammatory myopathies of childhood, Lancet 371 (2008) 2201–2212. [DOI] [PubMed] [Google Scholar]
- [3].Chung MP, Richardson C, Kirakossian D, Orandi AB, Saketkoo LA, Rider LG, et al. , Calcinosis biomarkers in adult and juvenile dermatomyositis, Autoimmun. Rev. 19 (2020), 102533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Pachman LM, Boskey AL, Clinical manifestations and pathogenesis of hydroxyapatite crystal deposition in juvenile dermatomyositis, Curr. Rheumatol. Rep. 8 (2006) 236–243. [DOI] [PubMed] [Google Scholar]
- [5].Eidelman N, Boyde A, Bushby AJ, Howell PG, Sun J, Newbury DE, et al. , Microstructure and mineral composition of dystrophic calcification associated with the idiopathic inflammatory myopathies, Arthritis Res. Ther. 11 (2009) R159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Stock S, Ignatiev K, Lee P, Abbott K, Pachman L, Pathological calcification in juvenile dermatomyositis (JDM): microCT and synchrotron x-ray diffraction reveal hydroxyapatite with varied microstructures, Connect. Tissue Res. 45 (2004) 248–256. [DOI] [PubMed] [Google Scholar]
- [7].Blane CE, White SJ, Braunstein EM, Bowyer SL, Sullivan DB , Patterns of calcification in childhood dermatomyositis, AJR Am. J. Roentgenol. 142 (1984) 397–400. [DOI] [PubMed] [Google Scholar]
- [8].Davuluri S, Duvvuri B, Lood C, Faghihi-Kashani S, Chung L, Calcinosis in dermatomyositis: origins and possible therapeutic avenues, Best Pract. Res. Clin. Rheumatol. 36 (2022), 101768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Pachman LM, Liotta-Davis MR, Hong DK, Kinsella TR, Mendez EP, Kinder JM, et al. , TNFalpha-308A allele in juvenile dermatomyositis: association with increased production of tumor necrosis factor alpha, disease duration, and pathologic calcifications, Arthritis Rheum. 43 (2000) 2368–2377. [DOI] [PubMed] [Google Scholar]
- [10].Duvvuri B, Lood C, Mitochondrial calcification, Immunometabolism (2021) 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Boonrungsiman S, Gentleman E, Carzaniga R, Evans ND, McComb DW, Porter AE, et al. , The role of intracellular calcium phosphate in osteoblast-mediated bone apatite formation, Proc. Natl. Acad. Sci. U. S. A. 109 (2012) 14170–14175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Dong Z, Shanmughapriya S, Tomar D, Siddiqui N, Lynch S, Nemani N, et al. , Mitochondrial Ca(2+) uniporter is a mitochondrial luminal redox sensor that augments MCU channel activity, Mol. Cell 65 (2017) 1014–10128 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bode RK, Klein-Gitelman MS, Miller ML, Lechman TS, Pachman LM, Disease activity score for children with juvenile dermatomyositis: reliability and validity evidence, Arthritis Rheum. 49 (2003) 7–15. [DOI] [PubMed] [Google Scholar]
- [14].Bohan A, Peter JB, Polymyositis and dermatomyositis (second of two parts), N. Engl. J. Med. 292 (1975) 403–407. [DOI] [PubMed] [Google Scholar]
- [15].Bohan A, Peter JB, Polymyositis and dermatomyositis (first of two parts), N. Engl. J. Med. 292 (1975) 344–347. [DOI] [PubMed] [Google Scholar]
- [16].Hochberg MC, Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus, Arthritis Rheum-Us 40 (1997) 1725. [DOI] [PubMed] [Google Scholar]
- [17].Trieu EP, Targoff IN, SDS-PAGE for (35)S immunoprecipitation and immunoprecipitation western blotting, Methods Mol. Biol. 1855 (2019) 417–436. [DOI] [PubMed] [Google Scholar]
- [18].Mecoli CA, Albayda J, Tiniakou E, Paik JJ, Zahid U, Danoff SK, et al. , Myositis autoantibodies: a comparison of results from the Oklahoma medical research foundation myositis panel to the euroimmun research line blot, Arthritis Rheumatol. 72 (2020) 192–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bocciarelli DS, Morphology of crystallites in bone, Calcif. Tissue Res. 5 (1970) 261–269. [DOI] [PubMed] [Google Scholar]
- [20].Scimeca M, Bischetti S, Lamsira HK, Bonfiglio R, Bonanno E, Energy Dispersive X-ray (EDX) microanalysis: a powerful tool in biomedical research and diagnosis, Eur. J. Histochem. 62 (2018) 2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Gitterman DP, Wilson J, Randall AD, Functional properties and pharmacological inhibition of ASIC channels in the human SJ-RH30 skeletal muscle cell line, J. Physiol. 562 (2005) 759–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].MacEwen MJ, Markhard AL, Bozbeyoglu M, Bradford F, Goldberger O, Mootha VK, et al. , Evolutionary Divergence Reveals the Molecular Basis of EMRE Dependence of the Human MCU, vol. 3, Life Sci Alliance, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Guitart-Mampel M, Urquiza P, Carnevale Neto F, Anderson JR, Hambardikar V, Scoma ER, et al. , Mitochondrial inorganic polyphosphate (polyP) is a potent regulator of mammalian bioenergetics in SH-SY5Y cells: a proteomics and metabolomics study, Front. Cell Dev. Biol. 10 (2022), 833127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Goedhart J, Luijsterburg MS, VolcaNoseR is a web app for creating, exploring, labeling and sharing volcano plots, Sci. Rep. 10 (2020), 20560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Jiang WW, Masayesva B, Zahurak M, Carvalho AL, Rosenbaum E, Mambo E, et al. , Increased mitochondrial DNA content in saliva associated with head and neck cancer, Clin. Cancer Res. 11 (2005) 2486–2491. [DOI] [PubMed] [Google Scholar]
- [26].Duvvuri B, Pachman LM, Morgan G, Khojah AM, Klein-Gitelman M, Curran ML, et al. , Neutrophil extracellular traps in tissue and periphery in juvenile dermatomyositis, Arthritis Rheumatol. 72 (2020) 348–358. [DOI] [PubMed] [Google Scholar]
- [27].Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. , Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease, Nat. Med. 22 (2016) 146–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Becker Y, Loignon RC, Julien AS, Marcoux G, Allaeys I, Levesque T, et al. , Anti-mitochondrial autoantibodies in systemic lupus erythematosus and their association with disease manifestations, Sci. Rep. 9 (2019) 4530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Leung PS, Chuang DT, Wynn RM, Cha S, Danner DJ, Ansari A, et al. , Autoantibodies to BCOADC-E2 in patients with primary biliary cirrhosis recognize a conformational epitope, Hepatology 22 (1995) 505–513. [PubMed] [Google Scholar]
- [30].Chiu HY, Loh AHP, Taneja R, Mitochondrial calcium uptake regulates tumour progression in embryonal rhabdomyosarcoma, Cell Death Dis. 13 (2022) 419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Castillo Diaz LA, Elsawy M, Saiani A, Gough JE, Miller AF, Osteogenic differentiation of human mesenchymal stem cells promotes mineralization within a biodegradable peptide hydrogel, J. Tissue Eng. 7 (2016), 2041731416649789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Chalmers S, Nicholls DG, The relationship between free and total calcium concentrations in the matrix of liver and brain mitochondria, J. Biol. Chem. 278 (2003) 19062–19070. [DOI] [PubMed] [Google Scholar]
- [33].Chinopoulos C, Adam-Vizi V, Mitochondria as ATP consumers in cellular pathology, Biochim. Biophys. Acta 1802 (2010) 221–227. [DOI] [PubMed] [Google Scholar]
- [34].Matlib MA, Zhou Z, Knight S, Ahmed S, Choi KM, Krause-Bauer J, et al. , Oxygen-bridged dinuclear ruthenium amine complex specifically inhibits Ca2+ uptake into mitochondria in vitro and in situ in single cardiac myocytes, J. Biol. Chem. 273 (1998) 10223–10231. [DOI] [PubMed] [Google Scholar]
- [35].Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, et al. , Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter, Nature 476 (2011) 341–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Singh R, Bartok A, Paillard M, Tyburski A, Elliott M, Hajnoczky G, Uncontrolled mitochondrial calcium uptake underlies the pathogenesis of neurodegeneration in MICU1-deficient mice and patients, Sci. Adv. 8 (2022), eabj4716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Drose S, Brandt U, The mechanism of mitochondrial superoxide production by the cytochrome bc1 complex, J. Biol. Chem. 283 (2008) 21649–21654. [DOI] [PubMed] [Google Scholar]
- [38].Turnier JL, Pachman LM, Lowe L, Tsoi LC, Elhaj S, Menon R, et al. , Comparison of lesional juvenile myositis and lupus skin reveals overlapping yet unique disease pathophysiology, Arthritis Rheumatol. 73 (2021) 1062–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Roberson EDO, Mesa RA, Morgan GA, Cao L, Marin W, Pachman LM, Transcriptomes of peripheral blood mononuclear cells from juvenile dermatomyositis patients show elevated inflammation even when clinically inactive, Sci. Rep. 12 (2022) 275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Wang Y, Yu X, Song H, Feng D, Jiang Y, Wu S, et al. , The STAT-ROS cycle extends IFN-induced cancer cell apoptosis, Int. J. Oncol. 52 (2018) 305–313. [DOI] [PubMed] [Google Scholar]
- [41].Haag SM, Gulen MF, Reymond L, Gibelin A, Abrami L, Decout A, et al. , Targeting STING with covalent small-molecule inhibitors, Nature 559 (2018) 269–273. [DOI] [PubMed] [Google Scholar]
- [42].Kim J, Gupta R, Blanco LP, Yang S, Shteinfer-Kuzmine A, Wang K, et al. , VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease, Science 366 (2019) 1531–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Rai P, Janardhan KS, Meacham J, Madenspacher JH, Lin WC, Karmaus PWF, et al. , IRGM1 links mitochondrial quality control to autoimmunity, Nat. Immunol. 22 (2021) 312–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Zhou XJ, Lu XL, Lv JC, Yang HZ, Qin LX, Zhao MH, et al. , Genetic association of PRDM1-ATG5 intergenic region and autophagy with systemic lupus erythematosus in a Chinese population, Ann. Rheum. Dis. 70 (2011) 1330–1337. [DOI] [PubMed] [Google Scholar]
- [45].Xia Q, Wang M, Yang X, Li X, Zhang X, Xu S, et al. , Autophagy-related IRGM genes confer susceptibility to ankylosing spondylitis in a Chinese female population: a case-control study, Gene Immun. 18 (2017) 42–47. [DOI] [PubMed] [Google Scholar]
- [46].Zhai Q, Chen X, Fei D, Guo X, He X, Zhao W, et al. , Nanorepairers rescue inflammation-induced mitochondrial dysfunction in mesenchymal stem cells, Adv. Sci. 9 (2022), e2103839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Fang EF, Hou Y, Palikaras K, Adriaanse BA, Kerr JS, Yang B, et al. , Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease, Nat. Neurosci. 22 (2019) 401–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, et al. , The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter, Nat. Cell Biol. 15 (2013) 1464–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Logan CV, Szabadkai G, Sharpe JA, Parry DA, Torelli S, Childs AM, et al. , Loss-of-function mutations in MICU1 cause a brain and muscle disorder linked to primary alterations in mitochondrial calcium signaling, Nat. Genet. 46 (2014) 188–193. [DOI] [PubMed] [Google Scholar]
- [50].Marchi S, Patergnani S, Missiroli S, Morciano G, Rimessi A, Wieckowski MR, et al. , Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death, Cell Calcium 69 (2018) 62–72. [DOI] [PubMed] [Google Scholar]
- [51].Strubbe-Rivera JO, Chen J, West BA, Parent KN, Wei GW, Bazil JN, Modeling the effects of calcium overload on mitochondrial ultrastructural remodeling, Appl Sci (Basel) 11 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Malyala S, Zhang Y, Strubbe JO, Bazil JN, Calcium phosphate precipitation inhibits mitochondrial energy metabolism, PLoS Comput. Biol. 15 (2019), e1006719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Zhao Y, Urganus AL, Spevak L, Shrestha S, Doty SB, Boskey AL, et al. , Characterization of dystrophic calcification induced in mice by cardiotoxin, Calcif. Tissue Int. 85 (2009) 267–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Ladislau L, Suarez-Calvet X, Toquet S, Landon-Cardinal O, Amelin D, Depp M, et al. , JAK inhibitor improves type I interferon induced damage: proof of concept in dermatomyositis, Brain 141 (2018) 1609–1621. [DOI] [PubMed] [Google Scholar]
- [55].Marhaug G, Shah V, Shroff R, Varsani H, Wedderburn LR, Pilkington CA, et al. , Age-dependent inhibition of ectopic calcification: a possible role for fetuin-A and osteopontin in patients with juvenile dermatomyositis with calcinosis, Rheumatology (Oxford) 47 (2008) 1031–1037. [DOI] [PubMed] [Google Scholar]
- [56].Galozzi P, Bindoli S, Luisetto R, Sfriso P, Ramonda R, Scanu A, et al. , Regulation of crystal induced inflammation: current understandings and clinical implications, Expet Rev. Clin. Immunol. 17 (2021) 773–787. [DOI] [PubMed] [Google Scholar]
- [57].Meyer A, Laverny G, Allenbach Y, Grelet E, Ueberschlag V, Echaniz-Laguna A, et al. , IFN-beta-induced reactive oxygen species and mitochondrial damage contribute to muscle impairment and inflammation maintenance in dermatomyositis, Acta Neuropathol. 134 (2017) 655–666. [DOI] [PubMed] [Google Scholar]
- [58].McArthur K, Whitehead LW, Heddleston JM, Li L, Padman BS, Oorschot V, et al. , BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis, Science 359 (2018). [DOI] [PubMed] [Google Scholar]
- [59].Merlino G, Helman LJ, Rhabdomyosarcoma–working out the pathways, Oncogene 18 (1999) 5340–5348. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.