

Abstract
Interactions between AMPA receptors and synaptic scaffolding proteins are key regulators of synaptic receptor density and, thereby, synapse strength. Shank3 is one such scaffolding protein with high clinical relevance, as genetic variants and deletions of this protein have been linked to autism spectrum disorder. Shank3 acts as a master regulator of the postsynaptic density of glutamatergic synapses, interacting with ionotropic and metabotropic glutamate receptors and cytoskeletal elements to modulate synaptic structure. Notably, Shank3 has been shown to interact directly with the AMPAR subunit GluA1, and Shank3 knockout animals show deficits in AMPAR-mediated synaptic transmission. In this study, we sought to characterize the stability of GluA1-Shank3 interaction in response to chronic stimuli using a highly sensitive and specific proximity ligation assay. We found that GluA1-Shank3 interactions decrease in response to prolonged neuronal depolarization induced by elevated extracellular potassium, and that this reduced interaction is blocked by NMDA receptor antagonism. These results firmly establish the close interaction of GluA1 and Shank3 in cortical neurons in vitro, and that this select interaction is subject to modulation by depolarization.
Keywords: Shank3, GluA1, postsynaptic density, chronic depolarization, proximity ligation assay
1. Introduction
Dynamic changes in the quantity, composition, and conduction properties of ionotropic glutamate receptors in response to synaptic input form the molecular basis of synaptic plasticity. Modulation of surface receptor architecture occurs in part through protein interactions within the postsynaptic density (PSD), a specialized region at the postsynaptic membrane of glutamatergic synapses including structural proteins and AMPA, NMDA, and metabotropic glutamate receptors [1]. One hypothesized structural mediator between synaptic input and receptor localization and stabilization is the SH3 and multiple ankyrin repeat domains (Shank) family of synaptic scaffolding proteins [2].
Shank3 and other members of the Shank family are master regulators of PSD architecture, hypothesized to mediate postsynaptic structural changes in response to presynaptic input [3]. Although precise mechanisms have not been fully delineated, it is well-established that Shank3 is crucial for proper synaptic function. Shank3 deletion reduces AMPAR- and NMDAR-mediated EPSC amplitude and impairs long term potentiation (LTP) induction, culminating in altered cognitive and behavioral phenotypes in both animal models and human patients affected by SHANK3 haploinsufficiency [4–9]. Shank3 has been shown to interact directly with AMPA receptors, tetrameric ionotropic glutamate receptors that mediate fast excitatory neurotransmission, and previous work has shown that Shank3-AMPAR interaction is subunit specific. In one study, elevated extracellular zinc, mimicking glutamatergic synaptic zinc release, caused increased recruitment of AMPAR subunit GluA2 and dispersal of GluA1 from existing Shank3-positive postsynaptic puncta [10]. These authors postulated that zinc stimulation mediated the physiologic switch to GluA2-positive AMPARs via changes in colocalization with the underlying Shank3 scaffold.
Based on previous findings that Shank3-AMPAR interactions are putatively dependent on synaptic activity [10], we chose to study the effect of chronic neuronal depolarization on this receptor-scaffold protein interaction. Sustained periods of membrane depolarization may have important pathophysiologic correlates, such as those present during cortical spreading depolarization [11]. In this study, we used a highly sensitive and specific in situ proximity ligation assay to investigate the durability and stability of Shank3 interactions with AMPAR subunits GluA1 and GluA2 following prolonged neuronal depolarization. We found that chronic potassium exposure led to a specific decrease in endogenous GluA1-Shank3 interaction in mature dissociated rat cortical neurons, without altering synaptic expression of either protein. These findings suggest that sustained changes in neuronal activity can produce substantial alterations in specific components of the PSD.
2. Materials and Methods
2.1. Neuronal cultures
Cerebrocortical cultures were prepared from embryonic day 16 rats (Charles River Laboratory) and plated at a density of 680,000 cells per well in six well plates as described previously [12]. Neurons used for imaging experiments were plated and grown on glass coverslips. Except where noted otherwise, neurons were used at days 17–18 in vitro (DIV).
2.2. Quantitative RT-PCR
Cultures were rinsed twice with ice-cold phosphate-buffered saline (PBS) and total RNA was collected using TRIzol and isolated using the PureLink RNA Mini Kit (Invitrogen). RNA quality and quantity was determined using Nanodrop with 260/280 and 260/230 filter pairs before using the iScript cDNA synthesis kit (Bio-Rad). The following primer pairs were used to amplify the targets of interest: GluA1 sense (FW): 5’- GTGTCTTCTCCTTTCTTGACCCTTT-3’; GluA1 antisense (RV): 5’- CTCTTCGCTGTGCCATTCGTAG-3’; GluA2 sense (FW): 5’- AGAGAAAGAATACCCTGGAGCACA-3’; GluA2 antisense (RV): 5’- TCATCACTTGGACAGCATCATACG-3’; Shank3 sense (FW): 5’- TACAGCACTTGGAGCACCTG-3’; Shank3 antisense (RV): 5’- GTAATTGCGGACGTCCTTGT-3’; β-actin sense (FW): 5’- ACTCTTCCAGCCTTCCTTC-3’; β-actin antisense (RV): 5’- ATCTCCTTCTGCATCCTGTC-3’. Quantitative RT-PCR (qPCR) was performed using SsoAdvanced Universal SYBR Green Supermix with the CFX96 Touch Real-Time PCR detection system (Bio-Rad). Samples were collected from 4 separate rat dissociations (n=4) and were run in triplicate for each experiment. Shank3, GluA1, and GluA2 RNA levels are expressed as 2−ΔCt, where ΔCt represents the difference in mean threshold PCR amplification cycle between the RNA of interest and that of the β-actin control.
2.3. Protein Isolation
For both total lysate collection and synaptosome-enriched protein collection, cultures were first rinsed with ice-cold PBS. Cells were gently lifted from plates and lysates were centrifuged in the presence of cOmplete mini protease inhibitor cocktail (Roche) plus 1 mM phenylmethylsulfonyl fluoride (PMSF). Synaptosome-enriched protein was further isolated using the synPER synaptic protein extraction reagent (Thermo Fisher). The homogenate was first centrifuged at 1,200 x g for 10 minutes at 4°C. The resulting supernatant was isolated and centrifuged at 15,000 x g for 20 minutes at 4°C. The final supernatant containing cytosolic protein was removed, and the synaptosome-enriched pellet was resuspended in syn-PER plus protease inhibitor and PMSF. Protein concentration was measured using the Pierce BCA protein assay (Thermo Scientific).
2.4. Western blot
Sample preparation, running, and transfer conditions were optimized separately for experiments focused on Shank3, a high molecular weight (HMW) protein detected near 220 kD, and GluA1/2, which are detected at approximately 100 kD. Protein samples (20–30 μg per lane) were suspended in Laemmli reducing sample buffer (Bio-Rad) and heated at either 85°C (HMW) or 100°C (GluA1/2) for five minutes. Proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) using a Mini-PROTEAN TGX 4–20% gradient gel (Bio-Rad) (HMW) or 8% polyacrylamide gel (GluA1/2) with the Mini Protean 3 System (Bio-Rad). For HMW optimization, separated proteins were transferred to a 0.45 μm nitrocellulose membrane using transfer buffer containing 10% methanol and 0.02% SDS. Samples intended for GluA1/2 analysis were transferred to a 0.2 μm nitrocellulose membrane. All membranes were blocked with 1% bovine serum albumin (BSA) in PBS-Tween (PBS-T), then probed with rabbit anti-Shank3 (SYSY 162302, 1:1000), rabbit anti-GluA1 (Alomone AGC-004, 1:200), or mouse anti-GluA2 (Millipore MAB397, 1:1000) and mouse anti-β-actin (Sigma A5441, 1:5000), followed by Li-Cor IRDye-conjugated secondary antibodies 700CW (685 nm) and 800CW (780 nm). Protein bands were visualized using the Odyssey Infrared Imaging System (LI-COR), and densitometry analysis was performed using ImageJ (version 1.52q). Shank3, GluA1, and GluA2 protein levels were normalized to β-actin for all blots.
2.5. Experimental Design and Proximity Ligation Assay
On DIV17, cultures were treated overnight with growth media plus potassium chloride (KCl, 25 mM), MK801 (10 μM), zinc pyrithione (ZnPyr: 5 μM ZnCl2 plus 250 nM pyrithione), or tetrodotoxin (TTX, 500 nM). The concentration of KCl utilized in this study is not only non-toxic, but we and others have shown it to be protective against other forms of neuronal injury [13,14]. In a separate set of experiments, DIV18 cultures were exposed to 25 mM KCl for 15 minutes only and fixed immediately. Control coverslips were similarly treated in growth media alone. The nontoxic concentration of ZnPyr utilized was determined via a concentration-response toxicity assay (Supplementary Fig. 1). Briefly, DIV17-18 neurons were treated with ZnCl2 plus 250 nM pyrithione overnight in serum and phenol-free minimum essential medium. Toxicity was measured via lactate dehydrogenase (LDH) release (Sigma). Studies using zinc pyrithione were performed in serum-free minimum essential medium to minimize serum protein zinc. On DIV18, coverslips were fixed with 4% PFA/4% sucrose for 20 minutes at room temperature, rinsed with PBS, then blocked and permeabilized with 10% normal goat serum (NGS, Southern Biotech) plus 0.05% TritonX-100 for one hour at room temperature. Coverslips were stained with chicken anti-Map2 to visualize neuronal morphology (Invitrogen PA1-16751, 1:4000) for one hour at room temperature, followed by AlexaFluor 488 goat anti-chicken secondary antibody (Invitrogen A-11039, 1:1000). Coverslips were then incubated in primary antibodies targeting the protein pair of interest at 4°C overnight - either mouse anti-GluA1 (SYSY 182011, 1:300) or mouse anti-GluA2 (Millipore MAB397, 1:3000), plus rabbit anti-Shank3 (SYSY 162302, 1:2500) diluted in NaveniFlex primary antibody diluent (Navinci Diagnostics). The next day, the MR NaveniFlex proximity ligation assay (PLA, Navinci Diagnostics) was performed according to manufacturer instructions using the reaction C reagent containing the Atto647N fluorophore. The accuracy of the PLA was confirmed by omitting each primary antibody in successive control experiments (Supplementary Fig. 2). Coverslips from sister cultures were mounted on glass slides using Fluoromount G, then imaged using 60x oil magnification with a Nikon A1R laser scanning confocal microscope. All imaging and analyses were randomized by an independent aid and performed blinded to treatment condition. Moreover, fields of view were selected blind to PLA puncta (i.e., by visualizing MAP2 staining alone). For each coverslip, nine fields of view were systematically captured as Z-stack images using a 0.5 μm step size. Each field was analyzed as a maximum intensity projection image in ImageJ (version 1.52q). A minimum intensity brightness and contrast threshold was determined for each experiment to exclude background signal, then kept consistent throughout analysis for that independent experiment. Images were binarized and puncta ranging from 0.1 – 10 µm2, representing Shank3-GluA1/2 interaction, were counted automatically using the object count function. An average number of puncta per coverslip was calculated, then normalized to the control condition for each experiment. Each experiment (n) represents neurons collected from cultures from an independent rat dissociation.
2.6. Statistical Analyses
Statistics were performed using GraphPad Prism (version 9.1.0). Specific statistical tests and number of independent experiments are indicated in each figure legend. Significance was determined using a cutoff value of α=0.05. Error bars in bar graphs represent mean ± SEM.
3. Results
3.1. Developmental changes in GluA1, GluA2, and Shank3 expression
The aim of this study was to characterize the stability of postsynaptic Shank3-AMPAR interactions in response to prolonged neuronal depolarization. GluA1, GluA2, and members of the Shank family all show distinct developmental profiles in vivo [10,15]. Thus, to ensure adequate expression at the time of experimentation, we first characterized the developmental profiles of Shank3, GluA1, and GluA2 in our rat cerebrocortical culture model. As neurons matured, mRNA expression of the AMPAR subunits GluA1 and GluA2 increased (p=0.0065, p=0.0079, respectively). Expression of both GluA1 and GluA2 peaked after three weeks in vitro, having increased significantly from DIV5 to DIV19 (p=0.0050, p= 0.0066, respectively) (Fig. 1A–B). This developmental increase in GluA1 and GluA2 is consistent with prior in vivo findings, validating the physiologic relevance of the timepoints used in this investigation [16,17].
Figure 1:
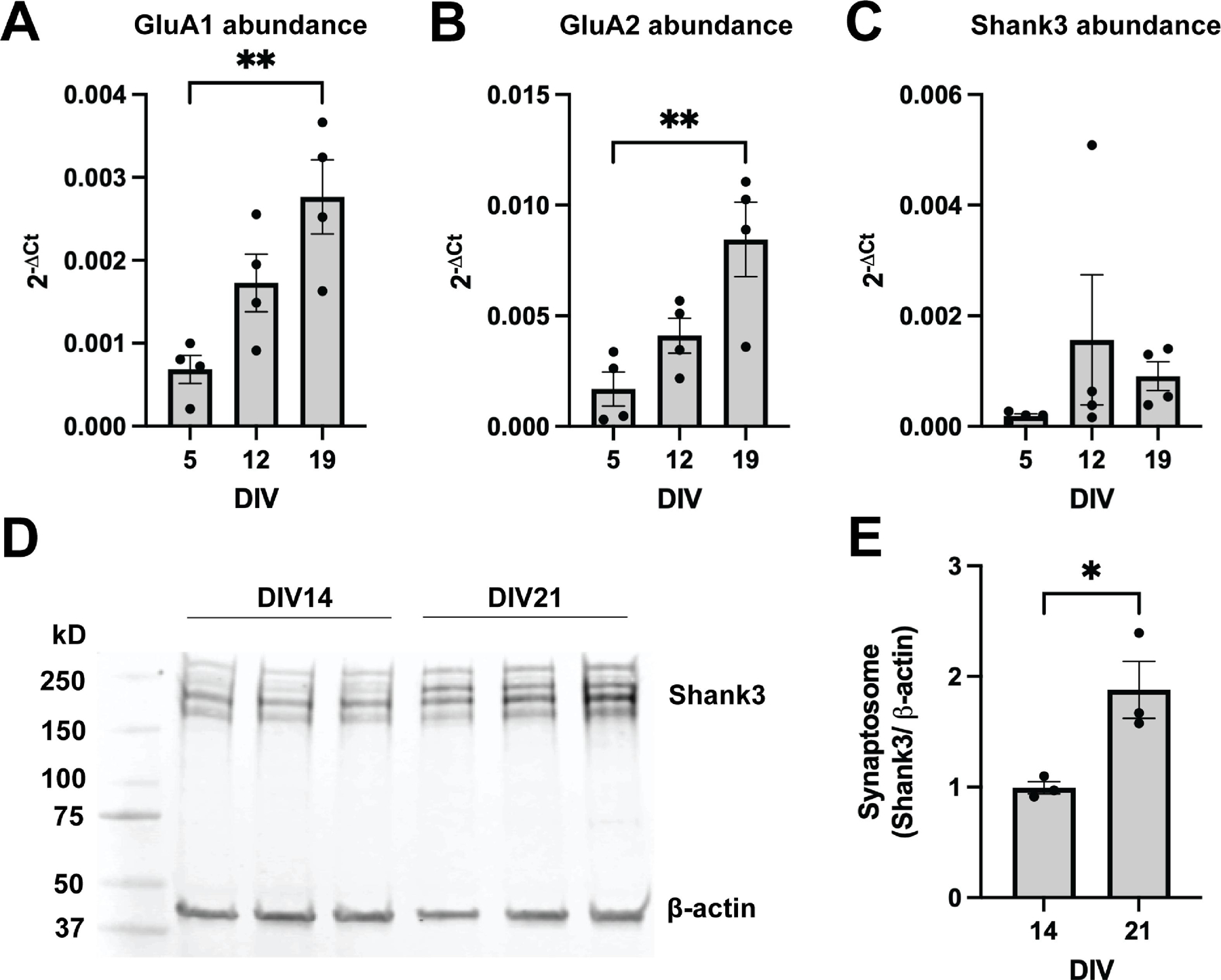
Developmental increases in AMPAR subunits GluA1 and GluA2, and Shank3 expression. (A) GluA1 mRNA abundance, represented as 2−ΔCt, increased across development (One-way ANOVA, p=0.0065, n=4), with a significant increase in expression at DIV19 compared to DIV5 (Tukey’s multiple comparisons, adjusted p=0.0050, n=4). (B) GluA2 mRNA abundance, represented as 2−ΔCt, increased across development (One-way ANOVA, p=0.0079, n=4), with a significant increase in expression at DIV19 compared to DIV5 (Tukey’s multiple comparisons, adjusted p=0.0066, n=4). (C) Shank3 mRNA abundance, represented as 2−ΔCt, was highly variable and did not change significantly across development (One-way ANOVA, p=0.4120, n=4). (D-E) Synaptosome-enriched Shank3 protein increased from the second week in culture (DIV14) to the third week in culture (DIV21) (unpaired t test, p=0.0285, n=3).
Next, we measured the mRNA expression and synaptic protein levels of the postsynaptic density (PSD) scaffolding protein Shank3. Shank3 mRNA expression was highly variable and did not significantly change with development (Fig. 1C) (p=0.4120). Consistent with other studies [18,19], our antibody detected several isoforms of Shank3 near the expected molecular weight of 220 kD, and, as such, summed quantification of the bands detected between 150–250 kD was performed. A loading artifact could be detected around 300 kD, and this band was not included in quantification (Fig. 1D). Because Shank3 is a master regulator of the PSD with known roles in spine induction [3,20], we were particularly interested in developmental changes in synaptic levels of Shank3. Quantification of synaptosome-enriched protein revealed an 87.9% increase in synaptic Shank3 from day 14 to day 21 in vitro (p=0.0285) (Fig. 1D–E). The observed increase in synaptic Shank3 is consistent with previously published findings [10]. Based on these results, we chose to study interactions of GluA1, GluA2 and Shank3 in mature dissociated neurons during the third week in vitro.
3.2. GluA1-Shank3 interaction decreases with prolonged depolarization in an NMDAR-dependent manner
Utilizing co-immunoprecipitation, GluA1 has been shown to interact directly with Shank3 via its C-terminal PDZ domain [21]. We confirmed this interaction using a proximity ligation assay (PLA), a highly sensitive and specific method allowing visualization and quantification of protein-protein interactions in situ, as long as the targets of interest are within 40 nm of each other (Fig. 2D–E) [22]. Given the inherent variability in spontaneous electrical activity of these primary cortical cultures [23], the number of GluA1-Shank3 interactions, as quantified by PLA fluorescent puncta, varied between biological replicates under control conditions (Fig. 2A). Thus, each experimental condition was analyzed as a percent of the mean number of puncta on the control coverslip, documenting the relative change in GluA1-Shank3 interaction following experimental intervention in sister cultures (Fig. 2B).
Figure 2:
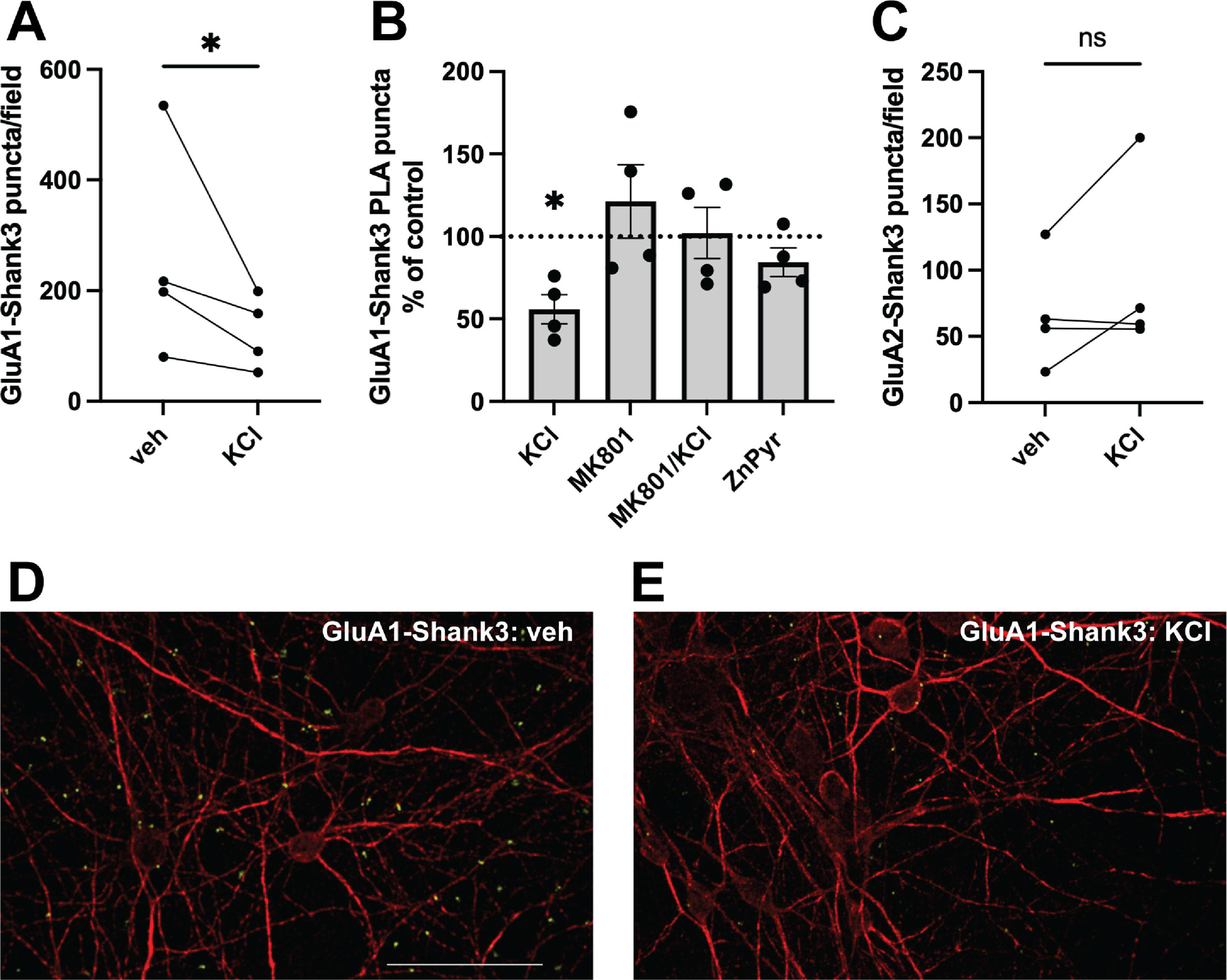
Prolonged depolarization causes a decrease in GluA1-Shank3 but not GluA2-Shank3 interaction. (A) Average number of GluA1-Shank3 PLA puncta per imaging field in sister cortical cultures incubated in the presence or absence of KCl treatment (ratio paired t test, p=0.0276, n=4). (B) PLA puncta quantified as a percent of control for each experimental condition. GluA1-Shank3 interaction decreased following overnight exposure to 25 mM KCl (one sample t test, p=0.0156, n=4). There was no significant change in GluA1-Shank3 interaction following overnight exposure to 10 μM MK801 alone (one sample t test, p=0.4126, n=4), co-exposure to 10 μM MK801 plus 25 mM KCl (one sample t test, p=0.9029, n=4), or 5 μM ZnCl2 plus 250 nM pyrithione (ZnPyr) (one sample t test, p=0.1714, n=4). (C) Average number of GluA2-Shank3 PLA puncta per imaging field in sister cortical cultures incubated in the presence or absence of KCl treatment (ratio paired t test, p=0.2645, n=4). (D-E) Representative images of PLA between GluA1 and Shank3 under control conditions (D) and following an overnight 25 mM KCl exposure (E). Neuronal morphology is shown with Map2 immunofluorescence (red), and PLA reactions are represented as discrete green puncta. Scale bar = 50 μm.
A previous investigation of activity-dependent AMPAR-Shank3 colocalization focused on the role of synaptic zinc signaling in the modulation of GluA1/2-Shank3 interactions [10]. Zinc is released along with glutamate at excitatory synapses, and enters postsynaptic cells via calcium-permeable AMPARs, NMDARs, and voltage-gated calcium channels [24]. Indeed, brief exposure to a very high concentration of KCl (90 mM, 120–180 seconds), mimicking depolarization, caused a near 50% increase in free zinc levels in postsynaptic spines of developing hippocampal neurons [10]. Additionally, Shank3 requires zinc binding its C-terminal sterile alpha motif (SAM) domain for localization and oligomerization at the PSD [25,26]. Given this background, existing knowledge on dynamic changes AMPAR-Shank3 colocalization has been focused on the consequences of elevated extracellular zinc [10]. In our investigation, we chose to study the effect of prolonged neuronal depolarization induced by a milder level of elevated extracellular KCl (25 mM). Thus, our stimulus may not only elicit zinc-dependent changes in AMPAR-Shank3 dynamics, but also models the broader consequences of sustained depolarization, including intracellular calcium influx, reduction of NMDAR blockade, and downstream changes in transcription programs [11].
We found that overnight exposure to 25 mM KCl caused a 43.7 ± 8.7% decrease in GluA1-Shank3 PLA puncta (p=0.0156) (Fig. 2B). Notably, a briefer KCl exposure (15 minutes) was also supportive of reduced GluA1-Shank3 interaction (Supplementary Fig. 3), but the effect was definitive after overnight depolarization. Prolonged depolarization may result in increased ambient or synaptically released glutamate and reduced magnesium block of NMDARs. As such, we tested whether blocking the NMDA receptor with MK801, a high affinity NMDAR channel blocker, would inhibit the KCl-induced decrease in GluA1-Shank3 interaction. We found that overnight treatment with MK801 (10 μm) had, in and of itself, no significant effect on GluA1-Shank3 interaction (Fig. 2B) (p=0.4126). However, when combined with KCl overnight, MK801 abolished the decrease in GluA1-Shank3 interaction (Fig. 2B) (p=0.9029). These results indicate that NMDAR activation is necessary for the depolarization-mediated reduction in GluA1-Shank3 interaction.
We next investigated the effect of zinc stimulation on GluA1-Shank3 interaction. Others have shown that zinc treatment causes dispersal of GluA1 from Shank3 in immature hippocampal neurons [10]. We thus evaluated whether exposure to zinc pyrithione altered the GluA1-Shank3 interaction in our cultures. Overnight exposure to sub-lethal concentrations of zinc (5 μm ZnCl2 plus 250 nM pyrithione; Supplementary Fig. 1) had no effect on the GluA1-Shank3 interaction (p=0.1714) (Fig. 2B). Finally, we explored whether silencing all neuronal activity with an overnight treatment with TTX (500 nM) would influence GluA1-Shank3 interactions. We found, however, that TTX had no effect on GluA1-Shank3 interaction (130.9 ± 21.74% of control, n=4, one sample t test, p=0.2508).
Along with dispersal of GluA1, brief zinc treatment has been reported to produce concomitant recruitment of GluA2-containing AMPARs to Shank3-positive post-synaptic puncta [10]. GluA1 and GluA2 have distinct conduction properties and C-terminal sequences, and are thus differentially trafficked to synapses [27,28]. We hypothesized that activity-dependent changes in scaffold interaction may differ for GluA2 and GluA1, and that sustained depolarization may result in increased GluA2-Shank3 interaction. We found that the PLA successfully detected GluA2-Shank3 interaction in our rat cerebrocortical neurons. However, in contrast to the KCl-induced depression of GluA1-Shank3 interaction, overnight depolarization did not cause a significant change in GluA2-Shank3 interaction (Fig. 2C) (p=0.2645), although an upward trend was suggested by the data. Thus, the KCl-induced changes in AMPAR-Shank3 interaction are specific to GluA1, and not a general disruption of AMPAR-Shank3 interaction.
3.3. Sustained depolarization does not alter synaptic expression of Shank3, GluA1 or GluA2
Finally, we investigated whether sustained depolarization changed the synaptic protein levels of Shank3, GluA1, or GluA2. We hypothesized that one potential reason for reduced GluA1-Shank3 interaction was a reduction in synaptic levels of either target, either via endocytosis and degradation, or re-localization away from the PSD, possibly as a result of homeostatic adaptation. We found no change in GluA1 synaptic expression (p=0.7685) following KCl treatment (Fig. 3A–B). It is noteworthy that although the expected molecular weight of GluA1 is ~100 kD, our antibody detected light bands around both 100 and 80 kD (Fig. 3A). There was, however, no difference in either band because of KCl exposure. We confirmed that there was no change in total GluA1 mRNA following KCl treatment (p=0.2982) (Fig. 3C). Similarly, KCl treatment did not alter Shank3 synaptic expression (p=0.4250) or mRNA abundance (p=0.3379) (Fig. 3D–F). Thus, the observed decrease in GluA1-Shank3 interaction likely cannot be attributed to reduced expression of either protein. We also measured GluA2 mRNA and synaptic protein abundance following KCl treatment. Interestingly, there was a small but significant 1.56 ± 0.15-fold increase in GluA2 mRNA abundance following KCl treatment when compared to control samples (p=0.0222) (Fig. 3I). However, this increase in mRNA abundance did not translate to a significant increase in synaptic GluA2 protein (p=0.4509) (Fig. 3G–H).
Figure 3:
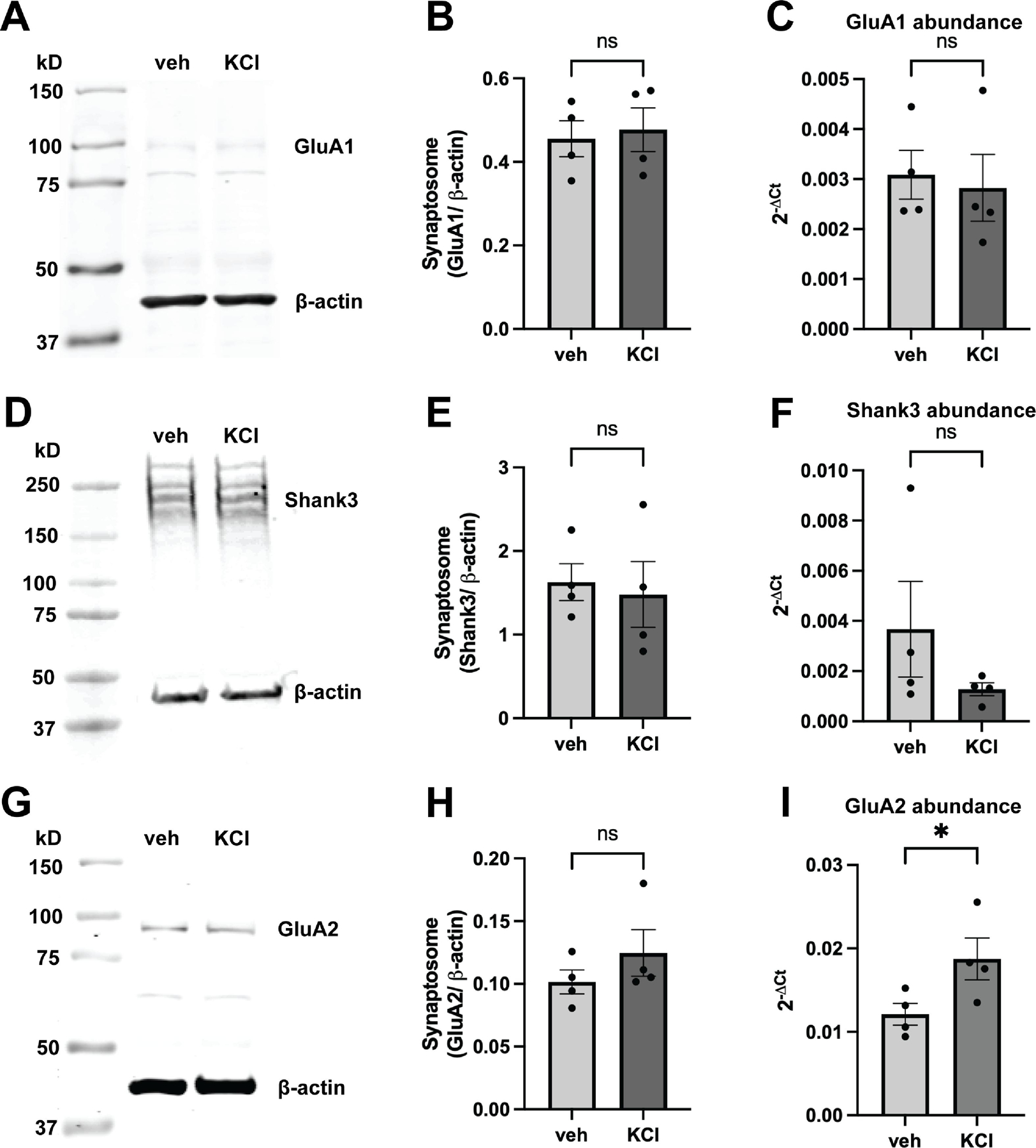
Effects of KCl treatment on synaptic GluA1, GluA2, or Shank3 protein levels and mRNA expression. (A-B) Overnight KCl treatment did not alter GluA1 synaptic protein levels (ratio paired t test, p= 0.7685, n=4) or (C) mRNA abundance, represented as 2−ΔCt (ratio paired t test, p=0.2982, n=4). The quantification represents summed intensity of the bands detected around 100 kD (expected molecular weight) and 80 kD. (D-E) Overnight KCl treatment also did not cause a change in Shank3 synaptic protein levels (ratio paired t test, p= 0.4250, n=4), or (F) mRNA expression (ratio paired t test, p=0.3379, n=4). (G-H) Overnight KCl treatment did not affect synaptic GluA2 protein levels (ratio paired t test, p=0.4509, n=4), but (I) did cause a 1.56-fold increase in GluA2 mRNA expression (ratio paired t test, p=0.0222, n=4).
4. Discussion
Establishing the complex mechanisms that regulate AMPAR trafficking, membrane insertion, and stabilization is key to understanding the molecular regulation of not only synaptic plasticity, but also synaptic pathophysiology [28,29]. One important contributor to receptor stabilization is interaction with scaffolding proteins. In this study, we demonstrate that cortical GluA1/2 expression and synaptic Shank3 expression increased with development in vitro (Fig. 1), and that endogenous levels of these proteins in mature cortical neurons have a close association, as detectable via proximity ligation assay (Fig. 2). GluA1-Shank3 interaction was stable following zinc and TTX treatments but decreased nearly 50% following sustained neuronal depolarization (Fig. 2). Notably, this reduced interaction was preventable with NMDAR antagonism, suggesting that the underlying mechanism mediating reduced interaction is at least partially attributable to NMDAR activity. Additionally, the altered AMPAR-Shank3 interaction was highly specific to the GluA1 subunit, as GluA2-Shank3 interaction did not change with sustained depolarization.
Shank proteins are one of many families of PSD components that interact with AMPARs and their auxiliary subunits to mediate AMPAR trafficking and stability. Two major mechanisms regulating synaptic AMPAR dynamics include endo- and exocytosis at extrasynaptic sites for delivery or removal from the plasma membrane, and lateral diffusion of constitutively mobile receptors through the membrane, leading to interaction with structural proteins for retention at the PSD [30,31]. The proposed model of AMPAR-scaffold interaction, called diffusion trapping, can be modulated by receptor and auxiliary subunit phosphorylation, or recruitment of synaptic scaffolds [32,33]. It is possible that the close associations that we are capturing with PLA are a snapshot of diffusion trapping interactions. We do not observe a change in synaptic levels of either protein and can thus hypothesize that the reduced interaction is not due to gross alterations in expression or degradation of either Shank3 or GluA1 (Fig. 3A–F). However, our findings do not eliminate the possibility of GluA1 endocytosis and retention within a pool that is still detectable in our synaptosomal preparation. Further investigations using methods like single particle tracking or phase separation would be a reasonable next step to better differentiate if the decreased GluA1-Shank3 interaction observed in our experiments is attributable to increased GluA1 endocytosis, or reduced diffusion trapping by Shank3. Notably, diffusion trapping is a process thought to occur on the order of seconds to minutes [32,34] and in this study we focus on a more long-term process.
Prolonged exposure to elevated extracellular potassium causes global depolarization of neurons in vitro, with subsequent reduction of NMDAR-Mg2+ blockade, elevation of intracellular calcium, activation of immediate early gene (IEG) programs, and alterations of intracellular signaling pathways involving CaMKs and MAPKs, among others [11]. It has been observed that cultured cortical neurons are spontaneously active [23], and that prolonged KCl treatment abrogates spontaneous firing [11]. The modeled state of elevated extracellular K+ and prolonged neuronal depolarization may be reflective of conditions present during spreading depolarizations (SD) [11]. SD describes a slowly propagating wave of neuronal depolarization including pronounced alterations in K+ balance [35], and has been observed clinically following traumatic brain injury [36,37], as well as during migraine with aura [38]. Notably, other SD models show that initiation and propagation of SD can be blocked with NMDAR antagonism similar to our study, reinforcing the finding that an NMDAR-activated pathway mediates the altered GluA1-Shank3 interaction we observe [39,40]. An alternative lens through which to interpret the stimulus of chronic KCl is synaptic downscaling [41]. If GluA1-Shank3 interaction promotes functional AMPAR-mediated ionic influx, then prolonged neuronal depolarization could reduce GluA1-Shank3 interaction in an attempt to maintain synaptic homeostasis [42]. Others have shown that modulation of another scaffolding protein – Homer1a – is involved in synaptic downscaling, and leads to reduced GluA2 tyrosine phosphorylation and reduced synaptic strength [43].
Finally, we observe a differential response of GluA1 and GluA2 interaction with Shank3 following prolonged depolarization. AMPAR subunits have unique electrophysiologic properties, developmental expression (Fig. 1A–B), and trafficking regulation [28]. For instance, GluA1 homomers require synaptic activity for surface insertion, while GluA2 homomers do not [27]. Interestingly, GluA1/2 heteromers, the dominant composition of AMPARs in CA1 pyramidal neurons [44], seem to be regulated by their GluA1 component and require synaptic activity for synapse delivery, whereas GluA2/3 heteromers, like GluA2 homomers, do not rely on synaptic activity for trafficking [27]. In our study, we do not know the total subunit composition of the AMPARs studied, but the differential response in Shank3 interaction suggests that the underlying process is highly specialized based on subunit-specific properties. One unexpected result was the observed increase in GluA2 mRNA expression only following KCl exposure (Fig. 3I). Almost all GluA2 subunits are Q/R edited so that AMPARs containing GluA2 are calcium impermeable [45]. It is thus possible that prolonged KCl-induced upregulation of GluA2 transcription is a homeostatic response to synaptic calcium influx overload. Alternatively, our GluA2 immunoblot and qPCR results may be explained by modulation of GluA2 mRNA post-transcription by GluA2-targeting miRNAs, which have been shown to regulate synaptic scaling [46,47].
5. Conclusions
Shank3 interacts closely with AMPAR subunits GluA1 and GluA2 in rat cerebrocortical neurons as measured by PLA. Sustained neuronal depolarization causes an NMDAR-dependent, selective decrease in GluA1-Shank3 interaction, without causing a change in the synaptic levels of these proteins. These findings are important for building understanding of receptor-scaffold interactions within the PSD in response to prolonged periods of neuronal depolarization, such as those occurring during spreading depression.
Supplementary Material
Highlights.
GluA1/2 and Shank3 expression and localization show developmental changes in vitro
Prolonged elevated extracellular KCl depresses GluA1 - Shank3 interaction
KCl-induced changes are prevented by an NMDA receptor channel blocker
KCl does not alter synaptic expression of GluA1 or Shank3
Acknowledgements
Thank you to Karen Harnett-Scott for technical assistance, and to Jenna Gale for support with data presentation. This work was supported in part by NIH grants NS043277 and NS117702 (EA), and by an American Academy of Neurology Medical Student Research Scholarship (2022, MMR).
Funding:
NIH NS117702 (EA); NIH NS043277 (EA); American Academy of Neurology Medical Student Research Scholarship (MMR)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CRediT authorship contribution statement
Madeline Ross: Conceptualization, Methodology, Investigation, Formal Analysis, Writing – original draft, reviewing and editing. Elias Aizenman: Conceptualization, Supervision, writing – reviewing and editing.
Declarations
The authors have no competing interests.
References
- [1].Vyas Y, Montgomery JM, The role of postsynaptic density proteins in neural degeneration and regeneration., Neural Regen. Res 11 (2016) 906–907. 10.4103/1673-5374.184481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Naisbitt S, Kim E, Tu JC, Xiao B, Sala C, Valtschanoff J, Weinberg RJ, Worley PF, Sheng M, Shank, a novel family of postsynaptic density proteins that binds to the NMDA receptor/PSD-95/GKAP complex and cortactin., Neuron 23 (1999) 569–582. 10.1016/s0896-6273(00)80809-0. [DOI] [PubMed] [Google Scholar]
- [3].Grabrucker AM, Schmeisser MJ, Schoen M, Boeckers TM, Postsynaptic ProSAP/Shank scaffolds in the cross-hair of synaptopathies., Trends Cell Biol 21 (2011) 594–603. 10.1016/j.tcb.2011.07.003. [DOI] [PubMed] [Google Scholar]
- [4].Betancur C, Buxbaum JD, SHANK3 haploinsufficiency: a “common” but underdiagnosed highly penetrant monogenic cause of autism spectrum disorders., Mol. Autism 4 (2013) 17. 10.1186/2040-2392-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bozdagi O, Sakurai T, Papapetrou D, Wang X, Dickstein DL, Takahashi N, Kajiwara Y, Yang M, Katz AM, Scattoni ML, Harris MJ, Saxena R, Silverman JL, Crawley JN, Zhou Q, Hof PR, Buxbaum JD, Haploinsufficiency of the autism-associated Shank3 gene leads to deficits in synaptic function, social interaction, and social communication., Mol. Autism 1 (2010) 15. 10.1186/2040-2392-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Fourie C, Vyas Y, Lee K, Jung Y, Garner CC, Montgomery JM, Dietary Zinc Supplementation Prevents Autism Related Behaviors and Striatal Synaptic Dysfunction in Shank3 Exon 13–16 Mutant Mice., Front. Cell. Neurosci 12 (2018) 374. 10.3389/fncel.2018.00374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Phelan K, McDermid HE, The 22q13.3 deletion syndrome (Phelan-McDermid syndrome)., Mol. Syndromol 2 (2012) 186–201. https://doi.org/000334260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Phelan K, Rogers RC, Phelan-McDermid Syndrome, in: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Stephens K, Amemiya A (Eds.), GeneReviews®, University of Washington, Seattle, Seattle (WA), 1993. 10.1016/B978-0-12-800109-7.00021-2. [DOI] [Google Scholar]
- [9].Wang X, McCoy PA, Rodriguiz RM, Pan Y, Je HS, Roberts AC, Kim CJ, Berrios J, Colvin JS, Bousquet-Moore D, Lorenzo I, Wu G, Weinberg RJ, Ehlers MD, Philpot BD, Beaudet AL, Wetsel WC, Jiang Y-H, Synaptic dysfunction and abnormal behaviors in mice lacking major isoforms of Shank3., Hum. Mol. Genet 20 (2011) 3093–3108. 10.1093/hmg/ddr212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ha HTT, Leal-Ortiz S, Lalwani K, Kiyonaka S, Hamachi I, Mysore SP, Montgomery JM, Garner CC, Huguenard JR, Kim SA, Shank and zinc mediate an AMPA receptor subunit switch in developing neurons., Front. Mol. Neurosci 11 (2018) 405. 10.3389/fnmol.2018.00405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Rienecker KDA, Poston RG, Saha RN, Merits and Limitations of Studying Neuronal Depolarization-Dependent Processes Using Elevated External Potassium., ASN Neuro 12 (2020) 1759091420974807. 10.1177/1759091420974807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hartnett KA, Stout AK, Rajdev S, Rosenberg PA, Reynolds IJ, Aizenman E, NMDA receptor-mediated neurotoxicity: a paradoxical requirement for extracellular Mg2+ in Na+/Ca2+-free solutions in rat cortical neurons in vitro., J. Neurochem 68 (1997) 1836–1845. 10.1046/j.1471-4159.1997.68051836.x. [DOI] [PubMed] [Google Scholar]
- [13].Yu SP, Yeh CH, Sensi SL, Gwag BJ, Canzoniero LM, Farhangrazi ZS, Ying HS, Tian M, Dugan LL, Choi DW, Mediation of neuronal apoptosis by enhancement of outward potassium current., Science 278 (1997) 114–117. 10.1126/science.278.5335.114. [DOI] [PubMed] [Google Scholar]
- [14].Aizenman E, Stout AK, Hartnett KA, Dineley KE, McLaughlin B, Reynolds IJ, Induction of neuronal apoptosis by thiol oxidation: putative role of intracellular zinc release., J. Neurochem 75 (2000) 1878–1888. 10.1046/j.1471-4159.2000.0751878.x. [DOI] [PubMed] [Google Scholar]
- [15].Kumar SS, Bacci A, Kharazia V, Huguenard JR, A developmental switch of AMPA receptor subunits in neocortical pyramidal neurons, J. Neurosci 22 (2002) 3005–3015. 10.1523/JNEUROSCI.22-08-03005.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Arai Y, Mizuguchi M, Takashima S, Developmental changes of glutamate receptors in the rat cerebral cortex and hippocampus., Anat. Embryol 195 (1997) 65–70. 10.1007/s004290050025. [DOI] [PubMed] [Google Scholar]
- [17].Tzakis N, Holahan MR, Investigation of GluA1 and GluA2 AMPA receptor subtype distribution in the hippocampus and anterior cingulate cortex of Long Evans rats during development., IBRO Rep 8 (2020) 91–100. 10.1016/j.ibror.2020.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Tao-Cheng J-H, Toy D, Winters CA, Reese TS, Dosemeci A, Zinc stabilizes shank3 at the postsynaptic density of hippocampal synapses., PLoS ONE 11 (2016) e0153979. 10.1371/journal.pone.0153979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hagmeyer S, Mangus K, Boeckers TM, Grabrucker AM, Effects of trace metal profiles characteristic for autism on synapses in cultured neurons., Neural Plast 2015 (2015) 985083. 10.1155/2015/985083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Roussignol G, Ango F, Romorini S, Tu JC, Sala C, Worley PF, Bockaert J, Fagni L, Shank expression is sufficient to induce functional dendritic spine synapses in aspiny neurons., J. Neurosci 25 (2005) 3560–3570. 10.1523/JNEUROSCI.4354-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Uchino S, Wada H, Honda S, Nakamura Y, Ondo Y, Uchiyama T, Tsutsumi M, Suzuki E, Hirasawa T, Kohsaka S, Direct interaction of post-synaptic density-95/Dlg/ZO-1 domain-containing synaptic molecule Shank3 with GluR1 alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor., J. Neurochem 97 (2006) 1203–1214. 10.1111/j.1471-4159.2006.03831.x. [DOI] [PubMed] [Google Scholar]
- [22].Zhu X, Zelmer A, Wellmann S, Visualization of Protein-protein Interaction in Nuclear and Cytoplasmic Fractions by Co-immunoprecipitation and In Situ Proximity Ligation Assay., J. Vis. Exp (2017). 10.3791/55218. [DOI] [PMC free article] [PubMed]
- [23].Dichter MA, Rat cortical neurons in cell culture: culture methods, cell morphology, electrophysiology, and synapse formation., Brain Res 149 (1978) 279–293. 10.1016/0006-8993(78)90476-6. [DOI] [PubMed] [Google Scholar]
- [24].Krall RF, Tzounopoulos T, Aizenman E, The function and regulation of zinc in the brain., Neuroscience 457 (2021) 235–258. 10.1016/j.neuroscience.2021.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Baron MK, Boeckers TM, Vaida B, Faham S, Gingery M, Sawaya MR, Salyer D, Gundelfinger ED, Bowie JU, An architectural framework that may lie at the core of the postsynaptic density., Science 311 (2006) 531–535. 10.1126/science.1118995. [DOI] [PubMed] [Google Scholar]
- [26].Boeckers TM, Liedtke T, Spilker C, Dresbach T, Bockmann J, Kreutz MR, Gundelfinger ED, C-terminal synaptic targeting elements for postsynaptic density proteins ProSAP1/Shank2 and ProSAP2/Shank3., J. Neurochem 92 (2005) 519–524. 10.1111/j.1471-4159.2004.02910.x. [DOI] [PubMed] [Google Scholar]
- [27].Shi S, Hayashi Y, Esteban JA, Malinow R, Subunit-specific rules governing AMPA receptor trafficking to synapses in hippocampal pyramidal neurons., Cell 105 (2001) 331–343. 10.1016/s0092-8674(01)00321-x. [DOI] [PubMed] [Google Scholar]
- [28].Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R, Glutamate receptor ion channels: structure, regulation, and function., Pharmacol. Rev 62 (2010) 405–496. 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Diering GH, Huganir RL, The AMPA receptor code of synaptic plasticity., Neuron 100 (2018) 314–329. 10.1016/j.neuron.2018.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Choquet D, Opazo P, The role of AMPAR lateral diffusion in memory., Semin. Cell Dev. Biol 125 (2022) 76–83. 10.1016/j.semcdb.2022.01.009. [DOI] [PubMed] [Google Scholar]
- [31].Maynard SA, Ranft J, Triller A, Quantifying postsynaptic receptor dynamics: insights into synaptic function., Nat. Rev. Neurosci 24 (2023) 4–22. 10.1038/s41583-022-00647-9. [DOI] [PubMed] [Google Scholar]
- [32].Opazo P, Labrecque S, Tigaret CM, Frouin A, Wiseman PW, De Koninck P, Choquet D, CaMKII triggers the diffusional trapping of surface AMPARs through phosphorylation of stargazin., Neuron 67 (2010) 239–252. 10.1016/j.neuron.2010.06.007. [DOI] [PubMed] [Google Scholar]
- [33].Tan HL, Queenan BN, Huganir RL, GRIP1 is required for homeostatic regulation of AMPAR trafficking., Proc Natl Acad Sci USA 112 (2015) 10026–10031. 10.1073/pnas.1512786112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Makino H, Malinow R, AMPA receptor incorporation into synapses during LTP: the role of lateral movement and exocytosis., Neuron 64 (2009) 381–390. 10.1016/j.neuron.2009.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Leao AAP, Spreading depression of activity in the cerebral cortex, Journal of Neurophysiology 7 (1944) 359–390. 10.1152/jn.1944.7.6.359. [DOI] [PubMed] [Google Scholar]
- [36].Carlson AP, Davis HT, Jones T, Brennan KC, Torbey M, Ahmadian R, Qeadan F, Shuttleworth CW, Is the Human Touch Always Therapeutic? Patient Stimulation and Spreading Depolarization after Acute Neurological Injuries., Transl. Stroke Res 14 (2023) 160–173. 10.1007/s12975-022-01014-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Fritch CD, Qeadan F, Shuttleworth CW, Carlson AP, Spreading depolarization occurs in repeating, recognizable, patient-specific patterns after human brain injury., Brain Inj 35 (2021) 299–303. 10.1080/02699052.2020.1861480. [DOI] [PubMed] [Google Scholar]
- [38].Major S, Huo S, Lemale CL, Siebert E, Milakara D, Woitzik J, Gertz K, Dreier JP, Direct electrophysiological evidence that spreading depolarization-induced spreading depression is the pathophysiological correlate of the migraine aura and a review of the spreading depolarization continuum of acute neuronal mass injury., Geroscience 42 (2020) 57–80. 10.1007/s11357-019-00142-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Gill R, Andiné P, Hillered L, Persson L, Hagberg H, The effect of MK-801 on cortical spreading depression in the penumbral zone following focal ischaemia in the rat., J. Cereb. Blood Flow Metab 12 (1992) 371–379. 10.1038/jcbfm.1992.54. [DOI] [PubMed] [Google Scholar]
- [40].Marrannes R, Willems R, De Prins E, Wauquier A, Evidence for a role of the N-methyl-D-aspartate (NMDA) receptor in cortical spreading depression in the rat., Brain Res 457 (1988) 226–240. 10.1016/0006-8993(88)90690-7. [DOI] [PubMed] [Google Scholar]
- [41].Siddoway B, Hou H, Xia H, Molecular mechanisms of homeostatic synaptic downscaling., Neuropharmacology 78 (2014) 38–44. 10.1016/j.neuropharm.2013.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB, Activity-dependent scaling of quantal amplitude in neocortical neurons., Nature 391 (1998) 892–896. 10.1038/36103. [DOI] [PubMed] [Google Scholar]
- [43].Hu J-H, Park JM, Park S, Xiao B, Dehoff MH, Kim S, Hayashi T, Schwarz MK, Huganir RL, Seeburg PH, Linden DJ, Worley PF, Homeostatic scaling requires group I mGluR activation mediated by Homer1a., Neuron 68 (2010) 1128–1142. 10.1016/j.neuron.2010.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Lu W, Shi Y, Jackson AC, Bjorgan K, During MJ, Sprengel R, Seeburg PH, Nicoll RA, Subunit composition of synaptic AMPA receptors revealed by a single-cell genetic approach., Neuron 62 (2009) 254–268. 10.1016/j.neuron.2009.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Burnashev N, Monyer H, Seeburg PH, Sakmann B, Divalent ion permeability of AMPA receptor channels is dominated by the edited form of a single subunit., Neuron 8 (1992) 189–198. 10.1016/0896-6273(92)90120-3. [DOI] [PubMed] [Google Scholar]
- [46].Silva MM, Rodrigues B, Fernandes J, Santos SD, Carreto L, Santos MAS, Pinheiro P, Carvalho AL, MicroRNA-186-5p controls GluA2 surface expression and synaptic scaling in hippocampal neurons., Proc Natl Acad Sci USA 116 (2019) 5727–5736. 10.1073/pnas.1900338116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Ho VM, Dallalzadeh LO, Karathanasis N, Keles MF, Vangala S, Grogan T, Poirazi P, Martin KC, GluA2 mRNA distribution and regulation by miR-124 in hippocampal neurons., Mol. Cell. Neurosci 61 (2014) 1–12. 10.1016/j.mcn.2014.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.