

Abstract
Sulfur-(hetero)arylation of sulfenamides with commercially-abundant (hetero)aryl iodides by Ullmann-type coupling with inexpensive copper(I) iodide as the catalyst is reported. A broad scope of reaction inputs was demonstrated, including both aryl and alkyl sulfenamides and highly sterically hindered aryl and 5- and 6-membered ring heteroaryl iodides. Relevant to many bioactive high oxidation state sulfur compounds, the (hetero)arylation of S-methyl sulfenamides is reported, including for complex aryl iodides. Smiles rearrangement of electron-deficient S-heteroaryl sulfilimines is also disclosed.
Graphical Abstract

There has been much recent attention towards the application of sulfoximines and sulfondiimines as new sulfur(VI) functional groups in agrochemicals and drugs (Scheme 1A).1 These sulfur species can display four different substituents on the configurationally stable sulfur atom and are uniquely poised for the exploration of new chemical space. The favorable physical and metabolic stability and aqueous solubility profiles for sulfoximines further the appeal of incorporating these functional groups into new bioactive compounds.1b
Scheme 1.

Bioactive Sulfoximines and Sulfondiimines and Their Methods for Preparation
Our group and others have recently developed sulfur-carbon bond formation reactions utilizing readily accessible sulfenamide starting inputs for the convergent and efficient synthesis of sulfilimines (the aza-analogues of sulfoxides) as intermediates to sulfoximines and sulfondiimines (Scheme 1B).2–8 Our initial report detailed the catalytic enantioselective S-alkylation of N-acyl sulfenamides with diazo coupling partners,2 and we and others have since reported additional methods for sulfenamide S-alkylation.4 Our group and others have also focused on the development of methods for the S-arylation of sulfenamides. To date, readily available boronic acids5 and diaryliodonium salts6 have been utilized as effective coupling partners (Scheme 1C). With the continued interest in incorporating high oxidation state sulfur functional groups in drugs and agrochemicals,9 we sought to expand the scope of coupling partners for S-arylation with a particular focus on commercially available inputs. We envisioned that aryl halides would be especially useful coupling partners given their broad commercial availability and extensive use in established cross-coupling methodologies.
Herein, we detail a new method for the copper-catalyzed sulfur-(hetero)arylation of sulfenamides with (hetero)aryl iodides under Ullmann-type10 reaction conditions (Scheme 1D). Notably, the number of commercially available (hetero)aryl iodides is >10-fold more than for (hetero)aryl boronic acids and >1000-fold more than for diaryliodonium salts.11 Both aryl and alkyl sulfenamides coupled efficiently. Highly sterically hindered aryl iodides and pyridyl and thienyl iodides with different substitution patterns were effective coupling partners. In contrast, for previously reported S-arylation reactions, pyridyl and 5-membered heterocyclic inputs were either shown to be unsuitable or were not reported (Scheme C).5,6 We also describe the preparation of S-methyl sulfenamides in a single step from commercially available materials and the first examples of their utilization for C–S bond formation, including for complex aryl iodide drugs and advanced drug intermediates. Significantly, many of the reported bioactive high oxidation state sulfur compounds incorporate an S-methyl substituent. Finally, we characterize an unprecedented Smiles rearrangement of electron-deficient S-heteroaryl sulfilimines.
After an extensive evaluation of reaction parameters, we determined that sulfenamide 1a coupled effectively with aryl iodide 2a when catalyzed by 20 mol % of inexpensive copper(I) iodide in the presence of Na2CO3 and molecular sieves in DMSO at 110 °C (Table 1, entry 1). Incomplete conversion to product was observed at a lower loading of copper (Table 1, entry 2), while a comparable yield was obtained at a higher loading (Table 1, entry 3). A 68% yield was observed upon excluding molecular sieves likely due to the hygroscopicity of the DMSO solvent (Table 1, entry 4). Other sieves types were also less effective (Table 1, entry 5). An excess of the aryl iodide provided the highest yield as decreasing the equivalents of 2a resulted in incomplete conversion (Table 1, entry 6). Relevant for circumstances in which a precious aryl iodide is employed, a 45% yield of 3a was still obtained when using a limiting amount of aryl iodide 2a and an excess of sulfenamide 1a (Table 1, entry 7).
Table 1.
Reaction Parameters for S-Arylation to Form 3aa

| ||
|---|---|---|
| Entry | Variation from standard conditions | Yield 3ab |
| 1 | None | 88% |
| 2 | 10 mol % CuI | 58% |
| 3 | 40 mol % CuI | 85% |
| 4 | No sieves | 68% |
| 5 | 4Å sieves powder | 78% |
| 6 | 1.5 equiv of 2a | 53% |
| 7 | 2a limiting, 2.0 equiv of 1a | 45% |
| 8 | K2CO3 as base | 34% |
| 9 | Li2CO3 as base | 71% |
| 10 | Under air | 27% |
| 11 | 100 °C | 72% |
| 12 | 120 °C | 86% |
| 13 | 0.2 M concentration | 69% |
| 14 | DMF as solvent | 26% |
| 15 | Dioxane as solvent | 0% |
| 16 | With 1,10-phenanthroline (30 mol %) | 35% |
| 17 | With p-bromotoluene | 41% |
| 18 | With p-tolyl triflate | 0% |
Reactions performed on 0.1 mmol scale of 1a.
Yields determined by 1H NMR using trimethyl(phenyl)silane as a standard.
The base employed has a significant effect on the reaction outcome as the use of K2CO3 led to mostly sulfenamide decomposition (Table 1, entry 8), while Li2CO3 gave a reduced 71% yield of 3a due to incomplete conversion (Table 1, entry 9). Performing the reaction under air resulted in a drastically reduced yield (Table 1, entry 10). A lower reaction temperature led to incomplete conversion to product (Table 1, entry 11), while a higher temperature provided a comparable yield (Table 1, entry 12). Doubling the reaction concentration to 0.2 M also resulted in a lower yield of 3a (Table 1, entry 13). Other high-boiling solvents such as DMF or dioxane give little or no product, respectively (Table 1, entries 14 and 15). While often beneficial in Ullmann-type couplings,10 ligands such as 1,10-phenanthroline resulted in a dramatically lower yield of the desired product 3a (Table 1, entry 16). Finally, using p-bromotoluene as a coupling partner instead of iodide 2a lowered the yield (Table 1, entry 17), while no product was generated when using p-tolyl triflate as the reactant (Table 1, entry 18).
We next evaluated the scope of S-arylation with respect to the aryl iodide inputs 2 (Scheme 2). Under the standard reaction conditions, coupling sulfenamide 1a with 4-iodotoluene (2a), provided sulfilimine 3a in 85% isolated yield. The corresponding aryl bromide was less reactive (see Table 1, entry 17), but 3a could still be isolated in 42% yield. Electron-rich 4-iodoanisole provided sulfilimine 3b in excellent yield, while electron-deficient 4-iodobenzotrifluoride and 4-iodobenzoate methyl ester provided sulfilimines 3c and 3d in 76% and 55% yields, respectively. Aryl chlorides are inert under these reaction conditions, and therefore, sulfilimine product 3e bearing a handle for further functionalization could be obtained in 84% yield. The methylenedioxy motif was also incorporated in 3f in good yield. Functional group compatibility was further demonstrated through the efficient coupling of aryl iodides featuring aldehyde (3g) and nitrile (3h) substituents.
Scheme 2. Scope of Aryl Iodide Inputs 2a.

aReactions performed at 0.2 mmol scale of 1a and isolated yields are reported.
To explore the effect of steric interactions on the aryl iodide input, we coupled 2-iodotoluene, which provided 3i without any reduction in yield relative to less sterically hindered aryl iodides. While 2,6-dimethylphenyl iodide also coupled to give sulfilimine 3j, it was obtained in a reduced 44% yield due to the bis-ortho substitution on this very hindered aryl iodide input.
The effective coupling of heteroaryl iodides is noteworthy. Sulfilimine 3k with a 3-pyridyl S-substituent was obtained in 82% yield. Additionally, 2-acetylamino-5-iodopyridine, despite incorporating strongly metal chelating functionality that often inhibits transition metal-catalyzed reactions, still gave sulfilimine 3l albeit in 42% yield. S-Thiophenyl sulfilimines 3m and 3n were obtained in good yields establishing that 5-membered heteroaryl iodides are also effective coupling partners. Moreover, the formation of styrenyl sulfilimine 3o in 78% yield demonstrated the viability of vinyl iodide reactants.
The scope of S-arylation was next investigated with respect to the sulfenamide inputs 1 (Scheme 3). An electron-rich aryl sulfenamide provided product 3p in quantitative yield. In contrast, electron-deficient aryl sulfenamide inputs are incompatible with this transformation. For example, the desired sulfilimine 3q was not obtained, though complete consumption of the sulfenamide input occurred. A sterically hindered o-naphthyl sulfenamide provided 3r in a synthetically useful yield of 49%. Alkyl sulfenamides were particularly effective inputs and reacted at a significantly lower reaction temperature of 70 °C, relative to aryl sulfenamides. For example, α-branched alkyl sulfenamides provided the S-cyclohexyl sulfilimine 3s in 75% yield and sulfilimine 3t incorporating an N-Boc piperidine in 51% yield. Benzyl and cyclopropyl sulfenamide inputs also resulted in good yields of 3u and 3v, respectively.
Scheme 3. Scope of Sulfenamide Inputs 1a.

aReactions performed at 0.2 mmol scale of 1 unless otherwise noted and isolated yields are reported. b70 °C. cAr-I limiting, 2.0 equiv of N-(methylthio)pivalamide, 40 mol % CuI, 80 °C.
The utilization of S-methyl sulfenamides for C–S bond formation has not been reported, despite the prevalence of bioactive high oxidation state sulfur compounds that incorporate an S-methyl substituent.1 In this context, we prepared N-(methylthio)pivalamide by reacting commercially available S-methyl methanethiosulfonate and pivalamide under basic conditions in 89% yield (see SI Section VII). Subsequent reaction of N-(methylthio)pivalamide with 4-iodotoluene gave 3w in an excellent 92% yield, and the corresponding N-benzoyl sulfilimine 3x and N-acetyl sulfilimine 3y were obtained in 90% and 41% yields, respectively. In contrast, when coupling the corresponding N-acyl S-phenyl sulfenamides, we found that acyl groups other than the pivaloyl group resulted in significant decomposition presumably because higher reaction temperatures were required for the less nucleophilic N-acyl S-phenyl sulfenamides (for a list of incompatible substrates see SI Section VIII).
Given the biological relevance of high oxidation state sulfur compounds bearing an S-methyl substituent, we explored the coupling of N-(methylthio)pivalamide with a panel of challenging aryl iodide inputs. S-Methyl sulfilimine products from methyl 4-iodobenzoate (3z), sterically hindered 2,6-dimethylphenyl iodide (3aa), and 2-acetylamino-5-iodopyridine (3ab) were all obtained in significantly higher yields relative to when the less nucleophilic S-phenyl sulfenamide 1a was employed (see Scheme 2). The scalability of this transformation was also demonstrated with the preparation of 3aa on a 1.0 mmol scale.
Aryl iodides are common intermediates for the synthesis of drugs. We therefore sought to demonstrate the coupling of complex drugs and advanced drug intermediates with N-(methylthio)pivalamide to generate sulfilimine products bearing an S-methyl substituent (Scheme 3). For example, the aryl iodide precursor to empaglifozen, bearing aryl chloride and ether functionalities, was successfully coupled to provide sulfilimine product 3ad in 92% yield. Importantly, under slightly modified reaction conditions, the aryl iodide could be used as the limiting reagent with an excess of the readily prepared N-(methylthio)pivalamide to afford 3ad in near-quantitative yield. A precursor to axitinib, with an N-tetrahydropyranyl protected indazole and 2-pyridylalkenyl functionalities, also coupled effectively under both conditions to provide 3ae in excellent yields. Finally, sulfilimine 3af was obtained in 85% yield employing limiting amounts of the densely functionalized drug trametinib, which incorporates acidic N-H functionality.
When assessing the scope of our newly developed transformation with respect to N-heteroaryl iodide inputs, we observed an unexpected reaction outcome upon coupling sulfenamide 1a with 2-iodopyridine (4) (Scheme 4). At elevated reaction temperatures, no desired sulfilimine was obtained. Rather, the unexpected pyridone product 5 was formed exclusively with its structure unambiguously assigned via X-ray crystallography (Scheme 4A). Notably, and of importance for synthetic utility, 2-iodopyridine (4) coupled effectively with methyl sulfenamide 1w in desired fashion to provide sulfilimine product 3ac in 92% yield at 70 °C (see Scheme 3). In contrast, when the highly electron-deficient 2-iodopyrimidine (6) was coupled with methyl sulfenamide 1w, only 7 was observed (Scheme 4B). We postulate that 7 is formed via a Smiles rearrangement from the initial S-pyrimidyl intermediate.12 Upon heating the isolated and purified N-thiomethyl imidate 7 to 130 °C in DMSO, isomerization occurred to give the pyrimidinone product 8 (Scheme 4C), which is structurally analogous to product 5 (Scheme 4A). The isomerization of 7 to 8 parallels a common side reaction for carboxylic acid coupling reactions mediated by carbodiimides wherein an initial O-acyl isourea isomerizes to the more stable N-acyl urea.13
Scheme 4. Discovery and Investigation of Rearrangement Productsa.
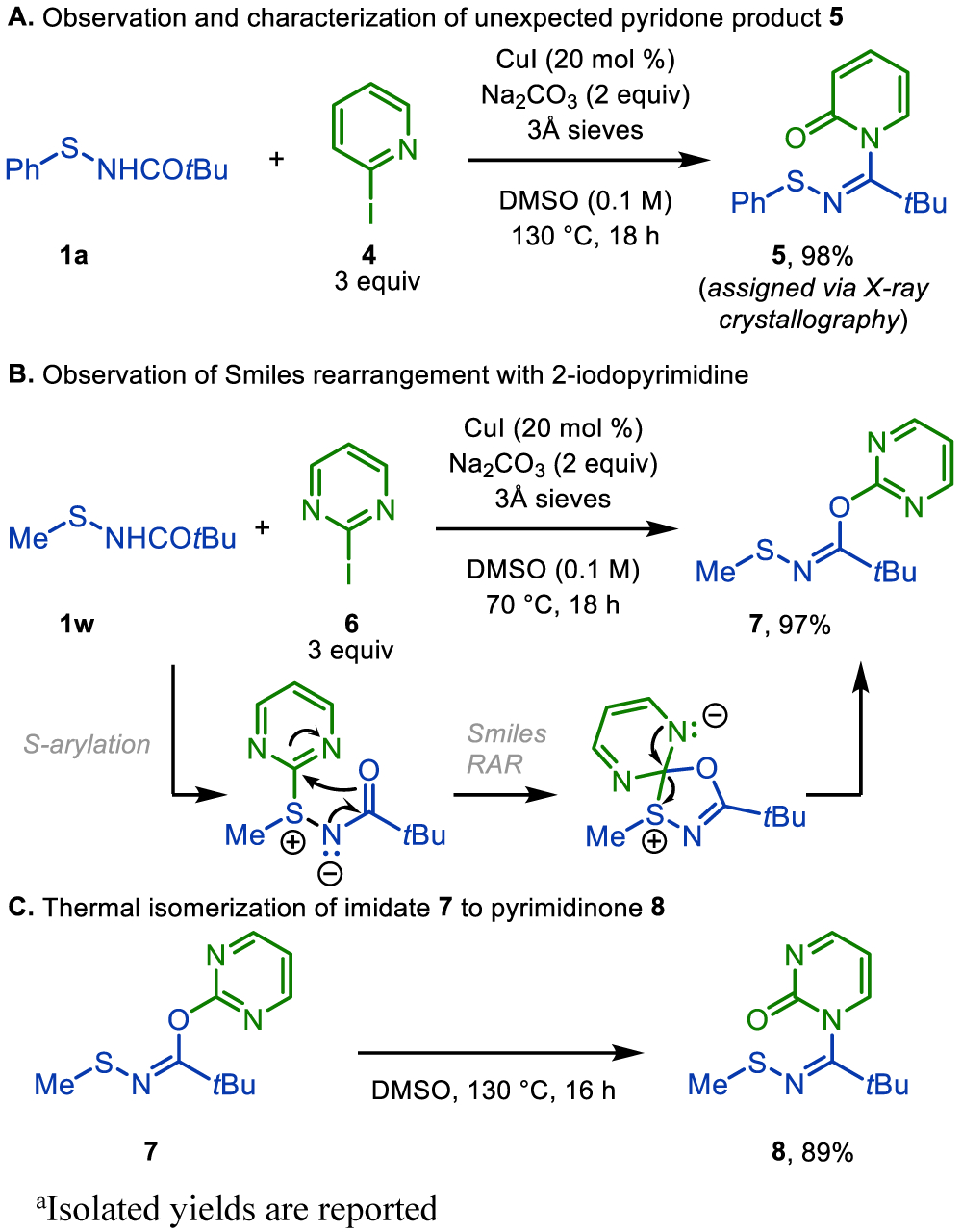
aIsolated yields are reported
The sulfilimine products 3 are versatile intermediates to high oxidation state sulfur(VI) functionalities. We and many others have previously reported a variety of transformations of N-acyl sulfilimines, including their conversion to sulfoximines and sulfondiimines.2,4–7 Here we chose to investigate the conversion of the highly sterically hindered, and consequently potentially challenging, sulfilimine 3aa to sulfoximines (Scheme 5). Sulfur-oxidation14 was successfully carried out to provide the N-protected sulfoximine 9 in high yield. The N-pivaloyl group was then cleaved under basic hydrolytic conditions to provide the free sulfoximine 10 in high yield.
Scheme 5. Conversion of a Sterically Hindered Sulfilimine to Sulfoximinesa.

aIsolated yields are reported.
In conclusion, a new method for the sulfur-arylation of sulfenamides is described. This transformation employs commercially abundant (hetero)aryl halides as coupling partners and is catalyzed by inexpensive copper(I) iodide under simple Ullmann-type reaction conditions. A broad range of coupling partners were shown to be effective, including 5- and 6-membered heterocyclic iodides, highly sterically hindered aryl iodides, and complex aryl iodides as exemplified by a drug and advanced drug intermediates. The first use of S-methyl sulfenamides in S–C bond formation was also demonstrated and should provide efficient access to high oxidation state sulfur compounds that incorporate S-methyl substituents. We expect that this method will be useful for the discovery and preparation of new bioactive sulfur(VI) compounds.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NIH Grant R35GM122473 (to J.A.E.). N.S.G. gratefully acknowledges the National Science Foundation Graduate Research Fellowship Program.
Footnotes
The authors declare no competing financial interests.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Experimental procedures, characterization data, and NMR spectra (PDF)
X-ray crystal data are available free of charge from the Cambridge Crystallographic Data Centre (https://www.ccdc.cam.ac.uk/) under CCDC reference no. 2265755 (compound 5).
Data Availability Statement
The data underlying this study are available in the published article and its online supplementary material.
REFERENCES
- (1).For relevant reviews see following:; (a) Lücking U Sulfoximines: A Neglected Opportunity in Medicinal Chemistry. Angew. Chem. Int. Ed 2013, 52, 9399–9408; [DOI] [PubMed] [Google Scholar]; (b) Frings M; Bolm C; Blum A; Gnamm C Sulfoximines from a Medicinal Chemist’s Perspective: Physicochemical and in vitro Parameters Relevant for Drug Discovery. Eur. J. Med. Chem 2017, 126, 225–245; [DOI] [PubMed] [Google Scholar]; (c) Lücking U Neglected sulfur(VI) pharmacophores in drug discovery: exploration of novel chemical space by the interplay of drug design and method development. Org. Chem. Front 2019, 6, 1319–1324. [Google Scholar]; (d) Mäder P; Kattner L Sulfoximines as Rising Stars in Modern Drug Discovery? Current Status and Perspective on an Emerging Functional Group in Medicinal Chemistry. J. Med. Chem 2020, 63, 14243–14275; [DOI] [PubMed] [Google Scholar]; (e) Han Y; Xing K; Zhang J; Tong T; Shi Y; Cao H; Yu H; Zhang Y; Liu D; Zhao L Application of sulfoximines in medicinal chemistry from 2013 to 2020. Eur. J. Med. Chem 2021, 209, 112885. [DOI] [PubMed] [Google Scholar]; (f) Passia MT; Schöbel J-H; Bolm C Sulfondiimines: synthesis, derivatisation and application. Chem. Soc. Rev 2022, 51, 4890–4901. [DOI] [PubMed] [Google Scholar]
- (2).Greenwood NS; Champlin AT; Ellman JA Catalytic Enantioselective Sulfur Alkylation of Sulfenamides for the Asymmetric Synthesis of Sulfoximines. J. Am. Chem. Soc 2022, 144, 17808–17814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Liu JT; Brandes DS; Greenwood NS; Ellman JA Synthesis of N-Acylsulfenamides from Amides and N-Thiosuccinimides. Synthesis 2022, eFirst, s-0041–1738430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).(a) Chen Y; Fang D-M; Huang H-S; Nie X-K; Zhang S-Q; Cui X; Tang Z; Li G-X Synthesis of Sulfilimines via Selective S–C Bond Formation in Water. Org. Lett 2023, 25, 2134–2138. [DOI] [PubMed] [Google Scholar]; (b) Huang G; Lu X; Yang K; Xu X Redox-Neutral Synthesis of Sulfilimines through the S-Alkylation of Sulfenamides. Org. Lett 2023, 25, 3173–3178. [DOI] [PubMed] [Google Scholar]; (c) Champlin AT; Ellman JA, Preparation of Sulfilimines by Sulfur-Alkylation of N-Acyl Sulfenamides with Alkyl Halides. J. Org. Chem 2023, 88, 7607–7614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).(a) Liang Q; Wells LA; Han K; Chen S; Kozlowski MC; Jia T Synthesis of Sulfilimines Enabled by Copper-Catalyzed S-Arylation of Sulfenamides. J. Am. Chem. Soc 2023, 145, 6310–6318. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Greenwood NS; Ellman JA Sulfur-Arylation of Sulfenamides via Chan–Lam Coupling with Boronic Acids: Access to High Oxidation State Sulfur Pharmacophores. Org. Lett 2023, 25, 2830–2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).(a) Huang G; Lu X; Liang F Redox-Neutral Strategy for Sulfilimines Synthesis via S-Arylation of Sulfenamides. Org. Lett 2023, 25, 3179–3183. [DOI] [PubMed] [Google Scholar]; (b) Zhou Q; Li J; Wang T; Yang X, Base-Promoted S-Arylation of Sulfenamides for the Synthesis of Sulfilimines. Org. Lett 2023, 23, 4335–4339. [DOI] [PubMed] [Google Scholar]
- (7).For leading references on iminations of sulfides, see:; (a) Basch T; Körber C Iron(II)-mediated Nitrene transfer from t-butyloxycarbonyl azide (BocN3) to sulfoxides, sulfides, and ketene acetals. Tetrahedron Lett. 1998, 39, 5015. [Google Scholar]; (b) Bach T; Körber C The Preparation of N-tert-Butyloxycarbonyl-(Boc-)protected Sulfoximines and Sulfimines by an Iron(II)-Mediated Nitrene Transfer from BocN3 to Sulfoxides and Sulfides. Eur. J. Org. Chem 1999, 1999, 1033. [Google Scholar]; (c) Lebel H; Huard K; Lectard S N-Tosyloxycarbamates as a Source of Metal Nitrenes: Rhodium-Catalyzed C−H Insertion and Aziridination Reactions. J. Am. Chem. Soc 2005, 127, 14198. [DOI] [PubMed] [Google Scholar]; (d) García Mancheño O; Bolm C Comparative Study of Metal-Catalyzed Iminations of Sulfoxides and Sulfides. Chem.-Eur. J 2007, 13, 6674. [DOI] [PubMed] [Google Scholar]; (e) Lebel H; Huard K De Novo Synthesis of Troc-Protected Amines: Intermolecular Rhodium-Catalyzed C−H Amination with N-Tosyloxycarbamates. Org. Lett 2007, 9, 639. [DOI] [PubMed] [Google Scholar]; (f) Manchenño OG; Dallimore J; Plant A; Bolm C Synthesis of Sulfoximines and Sulfilimines with Aryl and Pyrazolylmethyl Substituents. Adv. Synth. Catal 2010, 352, 309. [Google Scholar]; (g) Wang J; Frings M; Bolm C Enantioselective Nitrene Transfer to Sulfides Catalyzed by a Chiral Iron Complex. Angew. Chem., Int. Ed 2013, 52, 8661. [DOI] [PubMed] [Google Scholar]; (h) Wang J; Frings M; Bolm C Iron-Catalyzed Imidative Kinetic Resolution of Racemic Sulfoxides. Chem.-Eur. J 2014, 20, 966. [DOI] [PubMed] [Google Scholar]; (i) Lebel H; Piras H; Bartholoméüs J Rhodium-Catalyzed Stereoselective Amination of Thioethers with N-Mesyloxycarbamates: DMAP and Bis(DMAP)CH2Cl2 as Key Additives. Angew. Chem., Int. Ed 2014, 53, 7300. [DOI] [PubMed] [Google Scholar]; (j) Bizet V; Buglioni L; Bolm C Light-Induced Ruthenium-Catalyzed Nitrene Transfer Reactions: A Photochemical Approach Towards N-Acyl Sulfimides and Sulfoximines. Angew. Chem., Int. Ed 2014, 53, 5639. [DOI] [PubMed] [Google Scholar]; (k) Zenzola M; Doran R; Luisi R; Bull JA Synthesis of Sulfoximine Carbamates by Rhodium-Catalyzed Nitrene Transfer of Carbamates to Sulfoxides. J. Org. Chem 2015, 80, 6391. [DOI] [PubMed] [Google Scholar]; (l) Lebel H; Piras H; Borduy M Iron-Catalyzed Amination of Sulfides and Sulfoxides with Azides in Photochemical Continuous Flow Synthesis. ACS Catal. 2016, 6, 1109. [Google Scholar]; (m) Lai C; Mathieu G; Gabrielli Tabarez LP; Lebel H Batch and Continuous-Flow Iron(II)-Catalyzed Synthesis of Sulfilimines and Sulfoximines using N-Mesyloxycarbamates. Chem.- Eur. J 2019, 25, 9423. [DOI] [PubMed] [Google Scholar]; (n) Christian AH; Jia S; Cao W; Zhang P; Meza AT; Sigman MS; Chang CJ; Toste FD A Physical Organic Approach to Tuning Reagents for Selective and Stable Methionine Bioconjugation. J. Am. Chem. Soc 2019, 141, 12657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).For an elegant alternative approach to sulfilimines, see:; Tsuzuki S; Kano T Asymmetric Synthesis of Chiral Sulfimides through the O-Alkylation of Enantioenriched Sulfinamides and Addition of Carbon Nucleophiles. Angew. Chem., Int. Ed 2023, 62, e202300637. [DOI] [PubMed] [Google Scholar]
- (9).Tilby MJ; Willis MC How do we address neglected sulfur pharmacophores in drug discovery? Expert Opin. Drug Discovery 2021, 16, 1227–1231. [DOI] [PubMed] [Google Scholar]
- (10).(a) Ley SV; Thomas AW Modern Synthetic Methods for Copper-Mediated C(aryl)-O, C(aryl)-N, and C(aryl)-S Bond Formation. Angew. Chem. Int. Ed 2003, 42, 5400. [DOI] [PubMed] [Google Scholar]; (b) Monnier F; Taillefer M Catalytic C-C, C-N, and C-O Ullmann-Type Coupling Reactions. Angew. Chem. Int. Ed 2009, 48, 6954. [DOI] [PubMed] [Google Scholar]; (c) Sambiagio C; Marsden SP; Blacker AJ; McGowan PC Copper catalysed Ullmann type chemistry: from mechanistic aspects to modern development. Chem. Soc. Rev 2014, 43, 3525. [DOI] [PubMed] [Google Scholar]
- (11).A SciFinder substructure search followed by filtering for commercial availability was performed for each class of inputs.
- (12).Holden CM; Greaney MF, Modern Aspects of the Smiles Rearrangement. Chem. Eur. J 2017, 23, 8992–9008. [DOI] [PubMed] [Google Scholar]
- (13).(a) Valeur E; Bradley M, Amide bond formation: beyond the myth of coupling reagents. Chem. Soc. Rev 2009, 38, 606–631. [DOI] [PubMed] [Google Scholar]; (b) ElFaham A; Albericio F, Peptide Coupling Reagents, More than a Letter Soup. Chem. Rev 2011, 111, 6557–6602. [DOI] [PubMed] [Google Scholar]
- (14).Hendriks CMM; Lamers P; Engel J; Bolm C Sulfoxide to-Sulfilimine Conversions: Use of Modified Burgess-Type Reagents. Adv. Synth. Catal 2013, 355, 3363–3368. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this study are available in the published article and its online supplementary material.