

Abstract
Perfluorooctanoic acid (PFOA) is an artificial fluorinated organic compound that has generated increased public attention due to its potential health hazards. Unsafe levels of PFOA exposure can affect reproduction, growth and development. During tooth enamel development (amelogenesis), environmental factors including fluoride can cause enamel hypoplasia. However, the effects of PFOA on ameloblasts and tooth enamel formation remain largely unknown. In the present study we demonstrate several PFOA-mediated cell death pathways (necrosis/necroptosis, and apoptosis) and assess the roles of ROS-MAPK/ERK signaling in PFOA-mediated cell death in mouse ameloblast-lineage cells (ALC).
ALC cells were treated with PFOA. Cell proliferation and viability were analyzed by MTT assays and colony formation assays, respectively. PFOA suppressed cell proliferation and viability in a dose dependent manner. PFOA induced both necrosis (PI-positive cells) and apoptosis (cleaved-caspase-3, γH2AX and TUNEL-positive cells). PFOA significantly increased ROS production and up-regulated phosphor-(p)-ERK. Addition of ROS inhibitor N-acetyl cysteine (NAC) suppressed p-ERK and decreased necrosis, and increased cell viability compared to PFOA alone, whereas NAC did not change apoptosis. This suggests that PFOA-mediated necrosis was induced by ROS-MAPK/ERK signaling, but apoptosis was not associated with ROS. Addition of MAPK/ERK inhibitor PD98059 suppressed necrosis and increased cell viability compared to PFOA alone. Intriguingly, PD98059 augmented PFOA-mediated apoptosis. This suggests that p-ERK promoted necrosis but suppressed apoptosis. Addition of the necroptosis inhibitor Necrostatin-1 restored cell viability compared to PFOA alone, while pancaspase inhibitor Z-VAD did not mitigate PFOA-mediated cell death. These results suggest that 1) PFOA-mediated cell death was mainly caused by necrosis/necroptosis by ROS-MAPK/ERK signaling rather than apoptosis, 2) MAPK/ERK signaling plays the dual roles (promoting necrosis and suppressing apoptosis) under PFOA treatment. This is the initial report to indicate that PFOA could be considered as a possible causative factor for cryptogenic enamel malformation. Further studies are required to elucidate the mechanisms of PFOA-mediated adverse effects on amelogenesis.
Keywords: PFOA, ROS, Ameloblast, Apoptosis, Necrosis, MAPK, ERK
1. Introduction
Per- and poly-fluoroalkyl substances (PFAS) are a large group of man-made non-biodegradable compounds. Since the 1940s, PFAS have been produced and used in numerous commercial and industrial applications, including food containers and non-stick pans due to their unique hydrophobic and oleophobic properties. PFAS have generated increased public attention due to their potential environmental health hazards. PFAS compounds are seemingly ubiquitous in the oceans, streams, and groundwater. Elevated concentrations of PFAS in drinking water have been reported in numerous regions in the US, especially near industrial sites that produced or used PFAS. A recent study has shown that total PFAS concentrations in residential drinking water in Alaska ranged from not detected to 120 ng/L (ppt) (Babayev et al., 2022). Two water samples from nearby major source of PFAS contamination (Department of Transportation, and Gustavus (GST) airport in Gustavus, Alaska) had elevated PFAS concentrations of 14,600 and 228 ng/L (ppt), respectively. PFAS are a potential threat to living organisms due to their biopersistency, bioaccumulation and biomagnification in the food chain. The Centers for Disease Control and Prevention (CDC) has reported detectable serum PFAS levels in 97 % of the U.S. population. Epidemiological studies have demonstrated a positive association between human serum PFAS and an increase of cholesterol, liver damage and chronic kidney disease (Owumi et al., 2021; Shearer et al., 2021). Although it remains controversial, PFAS have been reported as thyroid and endocrine disruptors with the potential to cause adverse effects on thyroid gland function and the reproductive system (Coperchini et al., 2020; Mokra, 2021).
The most common PFAS; perfluorooctanoic acid (PFOA) and perfluorooctanesulfonic acid (PFOS) can affect the level of sex hormones and estrogen receptor activation (Kang et al., 2016). A recent study has reported that PFOA can promote breast cancer cell migration and invasion by activating PI3K/AKT and MAPK/ERK pathways via two estrogen receptors, ERα and G protein-coupled estrogen receptor (GPER) (Liu et al., 2023). While depending on cell type and stimuli, ERK activation is widely associated with cell proliferation, differentiation, and anti-apoptotic functions. ERK activity also will mediate different antiproliferative events and cell death, including apoptosis, autophagy, and senescence in vitro and in vivo (Cagnol and Chambard, 2010). PFOA activates MAPK/ERK signaling to induce hepatomegaly and hepatocarcinogenesis in rodents (Upham et al., 2009) and the p-p38/p38 MAPK ratio increased in testes and primary sertoli cells after PFOA exposure (Lu et al., 2016).
The US EPA (Environmental Protection Agency) guideline in 2016 set a limit of PFOA in drinking water as 70 parts per trillion (ppt, ng/L). In June 2022, the EPA updated the interim limit of PFOA to 0.004 ppt (EPA US, 2022), which is thousands of times stricter, suggesting that PFOA is more problematic than initially believed. PFOA has been detected in various human autopsy tissues, including bone, lung, liver and kidney (Perez et al., 2013). The median PFOA concentration in serum among the United States general population is 2.08 ng/mL (0.005 μM) as of 2011–2018 (CDC). Occupational studies have shown that serum PFOA levels in high-exposure workers ranged from 0.01 to 92.03 μg/mL (0.02 μM to 222 μM) (Olsen and Zobel, 2007) which is much higher than the general population. Epidemiological studies have demonstrated that serum PFOA concentrations are associated with kidney cancer (Shearer et al., 2021) and rheumatoid arthritis (Qu et al., 2022). Furthermore, PFOA can be transferred from mother to fetus via cord blood. A recent study has shown a correlation with PFOA concentrations in umbilical cord blood and cognitive functions of children (4–40 months) (Oh et al., 2022), suggesting that PFOA can cause adverse health effects on fetal development. Animal models demonstrate that PFOA is absorbed in oral, inhalation and dermal exposure. Rodent experiments show that the Lowest-observed-adverse-effect (LOAEL) doses correspond to serum PFOA levels of 20–51 μg/mL (48–123 μM) in rats and 10–14 μg/mL (24–33.8 μM) in mice (Loveless et al., 2006).
During tooth enamel development (amelogenesis), environmental factors (e.g., trauma, infections and chemicals) can cause enamel hypoplasia, including dental fluorosis and molar incisor hypomineralization (MIH). The optimal exposure of fluoride prevents tooth decay, but too much ingestion of fluoride during tooth development affects the function of enamel forming ameloblasts resulting in dental fluorosis (Suzuki et al., 2014). MIH is a developmental enamel defect caused by reduced mineralization, but the causative mechanism of MIH is still unclear (Almuallem and Busuttil-Naudi, 2018). Recent epidemiology studies have shown a relation between PFOA and dental caries prevalence (Puttige Ramesh et al., 2019; Wiener and Waters, 2019). Furthermore, animal models show possible adverse effects of PFOA in craniofacial development. For instance, PFOA is reported to cause craniofacial deformities in zebrafish (Jantzen et al., 2016) while perfluorooctane sulfonate (PFOS), another widely studied PFAS increases incidence of cleft palate in rodents (Era et al., 2009). Fluorotelomer alcohols (FTOHs), which are precursors of PFOA and PFOS, induced ameloblast degeneration leading to tooth malformation in rodents (Ladics et al., 2008; Mukerji et al., 2015; Serex et al., 2014). During PFOA metabolism from FTOHs, free fluorine is released from FTOHs to form PFOA as a final metabolite (Wang et al., 2005). Fluorine generated from FTOHs could play critical roles in ameloblast degeneration since excessive fluoride induces dental fluorosis. On the other hand, the final metabolite PFOA is fully fluorinated with the strongest carbon-fluorine (C-F) bonds, and is difficult to degrade to release free fluoride. The mechanism of how the final metabolite PFOA affects ameloblasts and enamel development remains largely unknown.
Here, we assessed the molecular mechanisms of PFOA-cytotoxicity in mouse ameloblast-lineage cells (ALC cells) in vitro. To the best of our knowledge, this is the first study to indicate that PFOA is potentially a causative factor for cryptogenic abnormalities in amelogenesis, including MIH of which the etiology is unknown.
2. Materials and methods
2.1. Cell culture
The mouse ameloblast-lineage cells (ALC cells), which are an immortalized cell line from the mouse mandibular molar (Nakata et al., 2003) were used in this study. ALC cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM) (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10 % fetal bovine serum. Indicated concentrations of PFOA (Sigma-Aldrich, St. Louis, MO, USA) were diluted with cell culture medium. All inhibitors used in this study were added 1 h prior to PFOA treatment. N-acetyl cysteine (NAC) (Thermo Fisher Scientific), which inhibits reactive oxygen species (ROS) production, was diluted with cell culture medium. The necroptosis inhibitors, RIP1 (Necrostatin-1) and PD98059 (ERK inhibitor) (Selleck Chemicals, Houston, TX, USA) were diluted with dimethyl sulfoxide (DMSO). A final concentration of 0.04 % DMSO (vehicle) served as a control and was equivalent to the amount of DMSO in each sample.
2.2. Cell proliferation assay
To quantify cell proliferation, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assays were performed as previously described (Deng et al., 2020). Briefly, cells were cultured in 96-well plates overnight. After PFOA treatment for 24 h, MTT reagent was added to each well and incubated for 3 h (Supplementary Fig. 1A). Afterwards the media was removed and replaced with DMSO and the cells were allowed to lyse while rotating on an orbital shaker for 15 min. OD was measured at 590 nm by a microplate reader (SYNERGY LX, BioTek, Winooski, VT, USA). Each experiment was performed in triplicate.
2.3. Colony formation assay
To examine cell viability, we performed a colony formation assay (Fujiwara et al., 2021). Briefly, ALC cells were treated with PFOA for 24h. Afterwards, cells were washed and re-seeded at 200 cells/well in 6 well plates and cultured for 7 days (Supplementary Fig. 1A). Formed colonies were washed with PBS followed by fixation with neutral buffered formalin and stained with 0.01 % crystal violet. The colony number was quantified by ImageJ software (National Institutes of Health, Bethesda, MD, USA). Each experiment was performed in triplicate.
2.4. Propidium iodide staining
To identify necrotic cells following PFOA treatment, the cells were stained using propidium iodide (PI) according to the manufacturer’s instructions (Invitrogen, Waltham, MA, USA). Briefly, ALC cells (2 × 106 cells) treated with PFOA were fixed with buffered formalin and then washed with PBS. The washed cells were post-fixed with methanol, washed again with PBS and then collected by centrifugation. The cell pellet was loosened with tapping and incubated with 3 μM PI solution for 15 min at room temperature. After a final washing of PI stained cells, fluorescence intensity was measured by RED filter (Ex/Em = 530/590) with a microplate reader (SYNERGY LX, BioTek).
2.5. ROS assay
ROS production was measured using cell permeable reagent 2’,7’-dichlorofluorescin diacetate (DCFDA) (Abcam, Waltham, MA, USA) as previously described (Suzuki et al., 2015). Briefly, ALC cells (2.5 × 103 cells/well) were seeded in a 96-well plate and incubated overnight. Afterwards, cells were washed with the buffer and incubated with DCFDA. Then, cells were treated with PFOA for 3 h. Fluorescence (Ex/Em = 485/528) was measured with a microplate reader (SYNERGY LX, BioTek).
2.6. TUNEL assay
We performed TUNEL assay for the identification of apoptotic cells following PFOA treatment using the quantitative colorimetric apoptosis kit according to the manufacturer’s instructions (Cat No. 4822–96-K-049, R&D Systems, Minneapolis, MN, USA). Briefly, ALC cells were treated with PFOA (500 μM) for 24 h and fixed with buffered formalin and methanol. Samples were treated with proteinase K solution, quenched by hydrogen peroxide, and labeled with terminal deoxynucleotidyl transferase (TdT). Cells that had incorporated labeled nucleotides were identified by horseradish peroxidase (HRP) and TACS-sapphire reagent. OD was measured at 450 nm using a microplate reader (SYNERGY LX, BioTek).
2.7. Western blot analysis
Western blot was performed as described previously (Fujiwara et al., 2021). Total protein from ALC cells treated with/without PFOA were extracted and equal amounts of protein were subjected to Immunoblot analysis. The following antibodies were used. Primary antibodies: rabbit anti-cleaved caspase 3, rabbit anti-γH2AX, rabbit anti-phospho-ERK, rabbit anti-total ERK antibodies (Cell Signaling Technology, Boston, MA, USA). Mouse anti-β-actin (Cell Signaling Technology) was used as a loading control protein. Secondary antibodies: HRP-conjugated anti--rabbit or mouse IgG secondary antibodies (Cell Signaling Technology). Signal was detected using the ChemiDoc™ Imaging System (Bio-Rad). Band densities were quantified by Image Lab™ software (Bio-Rad). Representative images are shown in the results section. Data of relative protein level are presented as means ± SD. Each experiment was performed in triplicate.
2.8. Statistical analysis
Data were analyzed by Tukey’s multiple comparisons test with one-way analysis of variance (ANOVA) and the Student’s t-test for comparing between two groups. Prism 9 (Graph Pad, San Diego, CA, USA) was used in the analysis. Significance was assessed at P < 0.05.
3. Results
3.1. PFOA induces cell death in ALC cells
We assessed the PFOA effects on cell proliferation in ALC cells by MTT assay. ALC cells were treated with PFOA at the indicated concentrations for 24 h. Fig. 1A shows that PFOA decreased cell proliferation in a dose-dependent manner. Cell growth was significantly decreased with PFOA at 500 μM (IC50) and 600 μM (IC65) compared to the control (**P < 0.01). To access whether PFOA induces cell death or merely suppresses cell growth, cell viability was determined using the colony formation assay and by determining the number of non-viable necrotic cells with propidium iodide (PI) staining. Lower doses of PFOA at 100–300 μM did not significantly decrease colony formation compared to the control (Supplementary Fig. 2). PFOA treatment (500 and 600 μM) significantly suppressed colony formation compared to the control (*P < 0.05) (Fig. 1B). PI-positive necrotic cells were increased two-fold by PFOA (500 μM) compared to the control (*P < 0.05) (Fig. 1C). These results suggest that PFOA at 500 μM (IC50) induced cell death in ALC cells. Henceforth, we assessed PFOA biological effects at IC50 in this study.
Fig. 1.
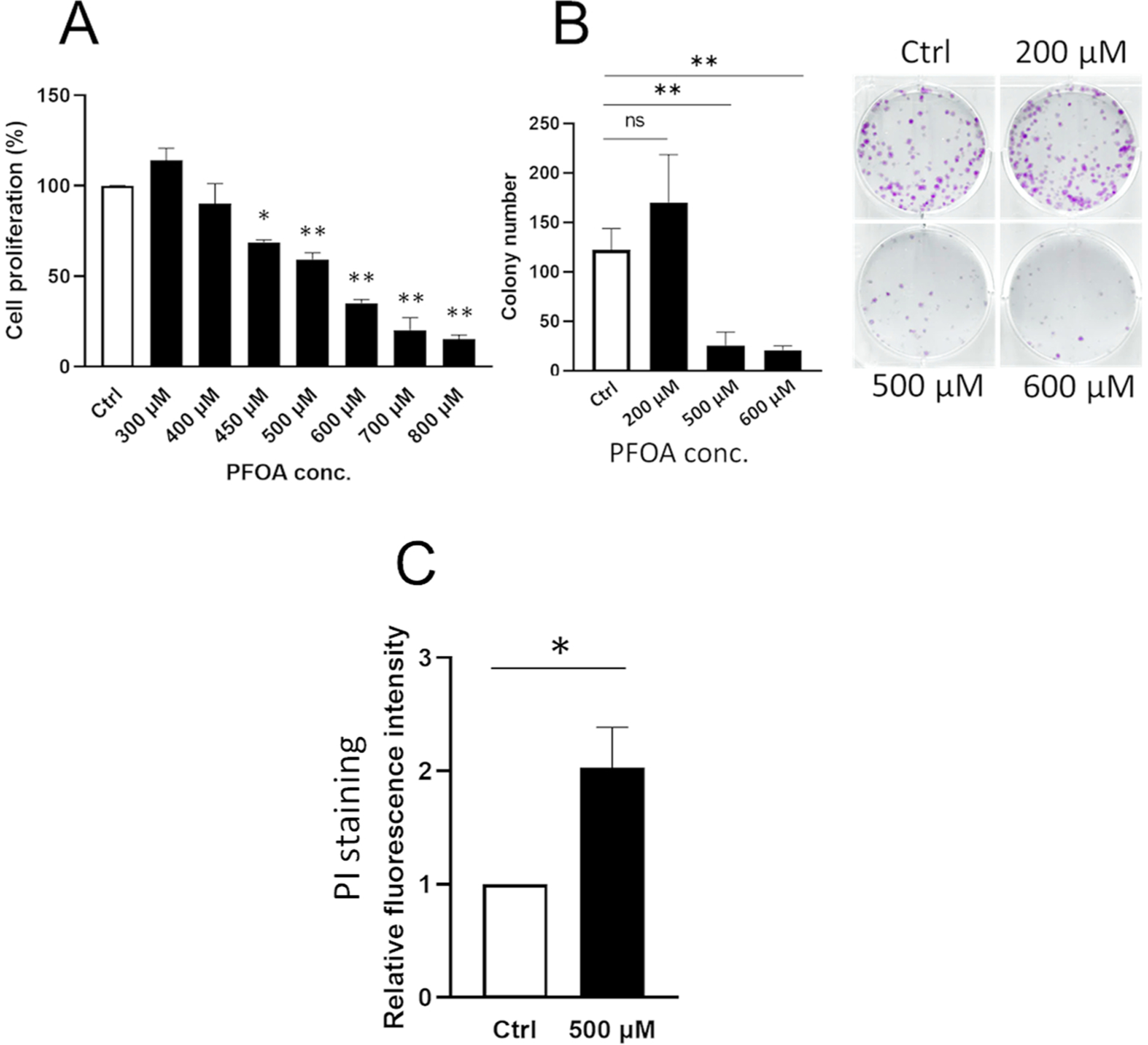
PFOA suppresses cell proliferation and induces cell death in ameloblast lineage cells (ALC). ALC cells were treated with PFOA (300–800 μM) for 24 h. (A) Cell proliferation was assessed by MTT assays. PFOA decreased cell proliferation in a dose-dependent manner compared with the control. (B) Cell viability was determined by colony formation assay. PFOA treatment (500 and 600 μM) significantly decreased colony numbers compared to the control. Representative images of colonies are shown at each dose. (C) PFOA mediated necrotic cell death was measured by propidium iodide (PI) staining. PFOA treatment (500 μM) significantly increased PI-positive necrotic cells compared to the control. Data are presented as means ± SD from three independent experiments. Statistically significant differences are indicated as follow: **; P < 0.01 vs Ctrl, *; P < 0.05 vs Ctrl, N.S.; no significance.
3.2. PFOA-mediated cell death is associated with ROS
Previous studies reported that PFOA treatment increased ROS production in mammalian cells. (Lopez-Arellano et al., 2019; Owumi et al., 2021). To assess if PFOA-mediated cell death is related to ROS, ROS production was measured in ALC cells. Fig. 2A shows that PFOA treatment (500 and 600 μM) for 3 h significantly increased ROS production compared to the control (**P < 0.01). ROS inhibitor N-acetyl cysteine (NAC) at 5 and 10 mM significantly suppressed PFOA-mediated ROS production compared to PFOA alone (**P < 0.01) (Fig. 2B). Fig. 2C shows that PFOA-mediated colony suppression was significantly reversed by NAC (5 and 10 mM) compared to PFOA alone (**P < 0.01), suggesting that PFOA-mediated cell death is caused by ROS production.
Fig. 2.
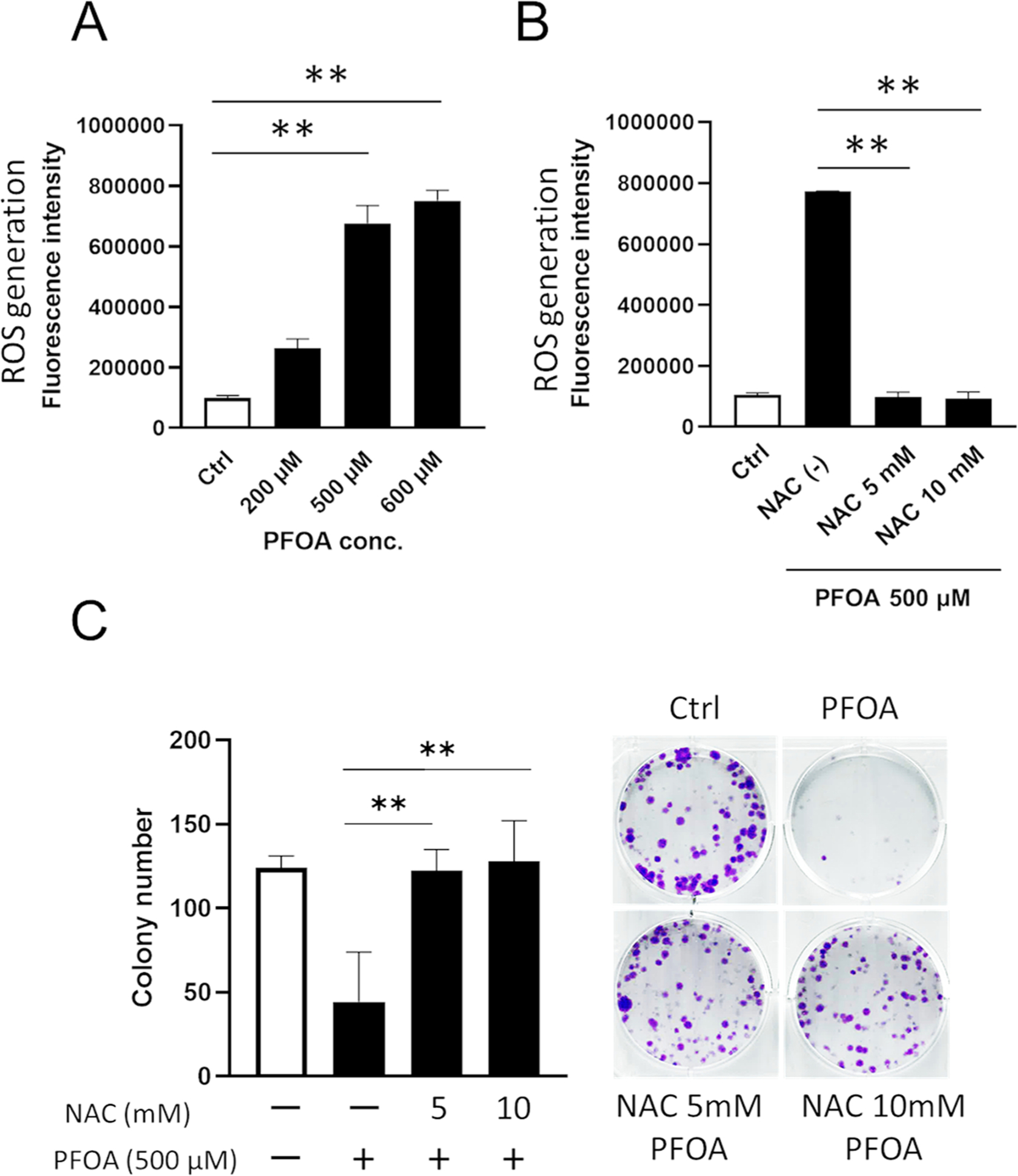
PFOA-mediated cell death is associated with ROS. (A) ALC cells were treated with PFOA for 3 h. Reactive oxidative species (ROS) production was measured by DCFDA. PFOA (500 and 600 μM) significantly increased ROS production compared to the control. (B) ALC cells were treated with ROS inhibitor, N-acetyl-L-cysteine (NAC) for 1 h prior to PFOA treatment (500 μM) for 3 h and ROS production was measured. NAC addition significantly decreased ROS production compared to PFOA alone. (C) Cell viability was determined by colony formation assay. PFOA with NAC treatment (5 and 10 mM) significantly reversed colony number compared to PFOA alone. The pictures shown are representative images of colonies. Open bars show controls. Data are presented as means ± SD from three independent experiments. Statistically significant differences are indicated as follow: **; P < 0.01, *; P < 0.05.
3.3. PFOA activates ERK signaling via ROS production leading to cell death
ROS can activate MAPK signaling (ERK and JNK), which regulates cell proliferation (Zhang and Liu, 2002), whereas ERK can activate apoptosis and mediate cell death (Sugiura et al., 2021). To assess the involvement of MAPK/ERK signaling in PFOA-induced cell death, phosphor-(p)-ERK and total-(t)-ERK protein level were determined by Western blotting. Fig. 3A shows that PFOA (500 μM) significantly increased the ratio of p-ERK/t-ERK compared to the vehicle control, DMSO (**P < 0.01). This PFOA-mediated ERK activation was significantly suppressed by NAC (5 and 10 mM) compared to PFOA alone (**P < 0.01). These results indicate that PFOA activates ERK signaling via ROS production. Next, we assessed the role of ERK signaling in PFOA-mediated cell death using the ERK inhibitor, PD98059. Fig. 3B shows that PFOA with PD98059 (5 and 40 μM) significantly reversed colony formation compared to PFOA alone (*P < 0.05, **P < 0.01, respectively). PI staining showed that PFOA with PD98059 (40 μM) significantly decreased the number of PI-positive necrotic cells compared to PFOA alone (*P < 0.05) (Fig. 3C). These results suggest that PFOA activates ERK by way of ROS which leads to cell death in ALC cells.
Fig. 3.

PFOA activates MAP/ERK signaling via ROS production. (A) ALC cells were treated with NAC (5 and 10 mM) for 1 h prior to PFOA (500 μM) treatment for 24 h. Phosphorylated-(p)-ERK (42 and 44 kDa) and total ERK (42 and 44 kDa) were detected by Western blot. The relative protein level was normalized by the loading control β-actin (44 kDa). PFOA significantly increased p-ERK/total ERK ratio compared to the control. This increase was suppressed by NAC addition. The blot shows the representative image from three biological replicates. (B) Cell viability was determined by colony formation assay. ALC cells were treated with ERK inhibitor PD98059 (5 and 40 μM) for 1 h prior to PFOA treatment for 24 h. PD98059 addition significantly increased PFOA-mediated colony formation compared to PFOA alone. The pictures shown are representative images of colonies. (C) PD98059 (40 μM) significantly decreased PI-positive cells (necrotic cell population) compared to PFOA alone. Open bars show controls. Data are presented as means ± SD from three independent experiments. Statistically significant differences are indicated as follow: **; P < 0.01, *; P < 0.05.
3.4. PFOA-induced apoptosis is not associated with ROS-mediated cell death
We assessed if apoptosis is involved in PFOA-induced cell death. PFOA at 500 μM and 600 μM (but not at 200 μM) significantly increased protein level of cleaved caspase-3 (Fig. 4A) and γH2AX (DNA-damage response marker) (Fig. 4B) compared to the control. PFOA (500 μM) significantly increased apoptosis by TUNEL assay compared to the control (Fig. 4C) (*P < 0.05), while there was no significant change at 200 μM (Supplementary Fig. 3). These results suggest that PFOA (IC50) induced apoptosis in ALC cells.
Fig. 4.
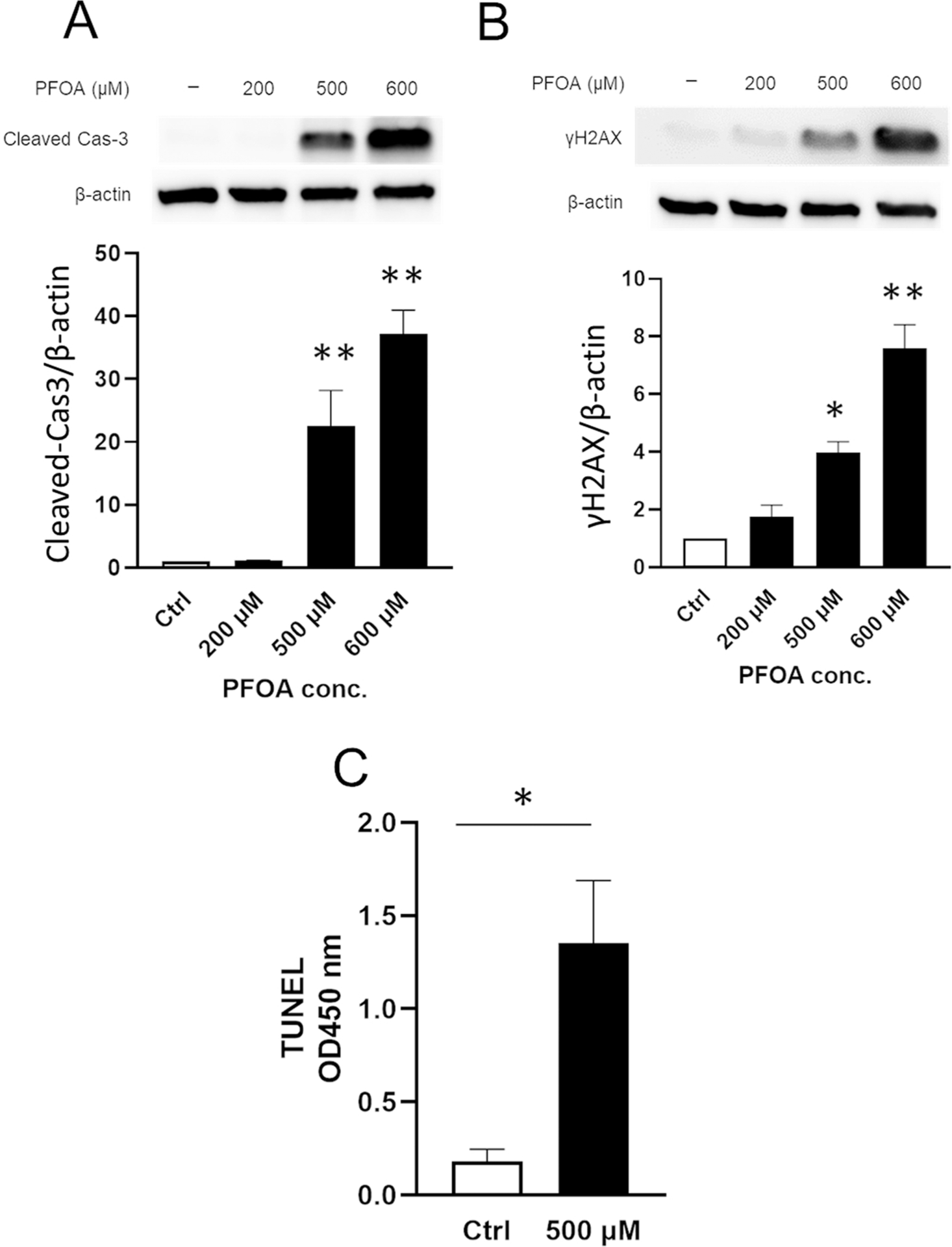
PFOA induces apoptosis. ALC cells were treated with PFOA at indicated concentrations for 24 h. Cleaved caspase-3 (17 kDa) and γH2AX (15 kDa) were detected by Western blot. Relative protein level was normalized by the loading control β-actin (44 kDa). PFOA treatment significantly increased (A) cleaved caspase-3 level and (B) γ-H2AX level compared to the control. Blots shown are representative images from three biological replicates. (C) ALC cells were treated with PFOA for 24 h. Apoptotic DNA fragmentation was detected by TUNEL assay. TUNEL-positive apoptotic cells were significantly increased by PFOA treatment. Data are presented as means ± SD from three independent experiments. Statistically significant differences are indicated as follow: **; P < 0.01 vs Ctrl, *; P < 0.05 vs Ctrl.
Next, we investigated whether ROS is associated with PFOA-induced apoptosis using NAC. Fig. 5 shows that NAC did not change the protein level of PFOA-induced cleaved caspase-3 (Fig. 5A) nor γH2AX (Fig. 5B). However, intriguingly, ROS inhibition by NAC significantly decreased PI-positive cell population compared to PFOA alone (Fig. 5C) (*P < 0.05), indicating that PFOA-mediated ROS induced necrosis, but ROS is not associated with apoptosis. Next, we performed the colony formation assay using pan-caspase inhibitor, Z-VAD-fmk (Z-VAD) to clarify if apoptosis plays a crucial role in PFOA-mediated cell death. PFOA with Z-VAD did not change the colony formation compared to PFOA alone (Supplementary Fig. 4). These results suggest that PFOA-induced apoptosis was independent of ROS production and other factors may contribute to PFOA-induced apoptosis. Multiple pathways are possibly activated by PFOA, and PFOA-mediated cell death might be mainly caused by ROS-mediated necrosis rather than apoptosis.
Fig. 5.
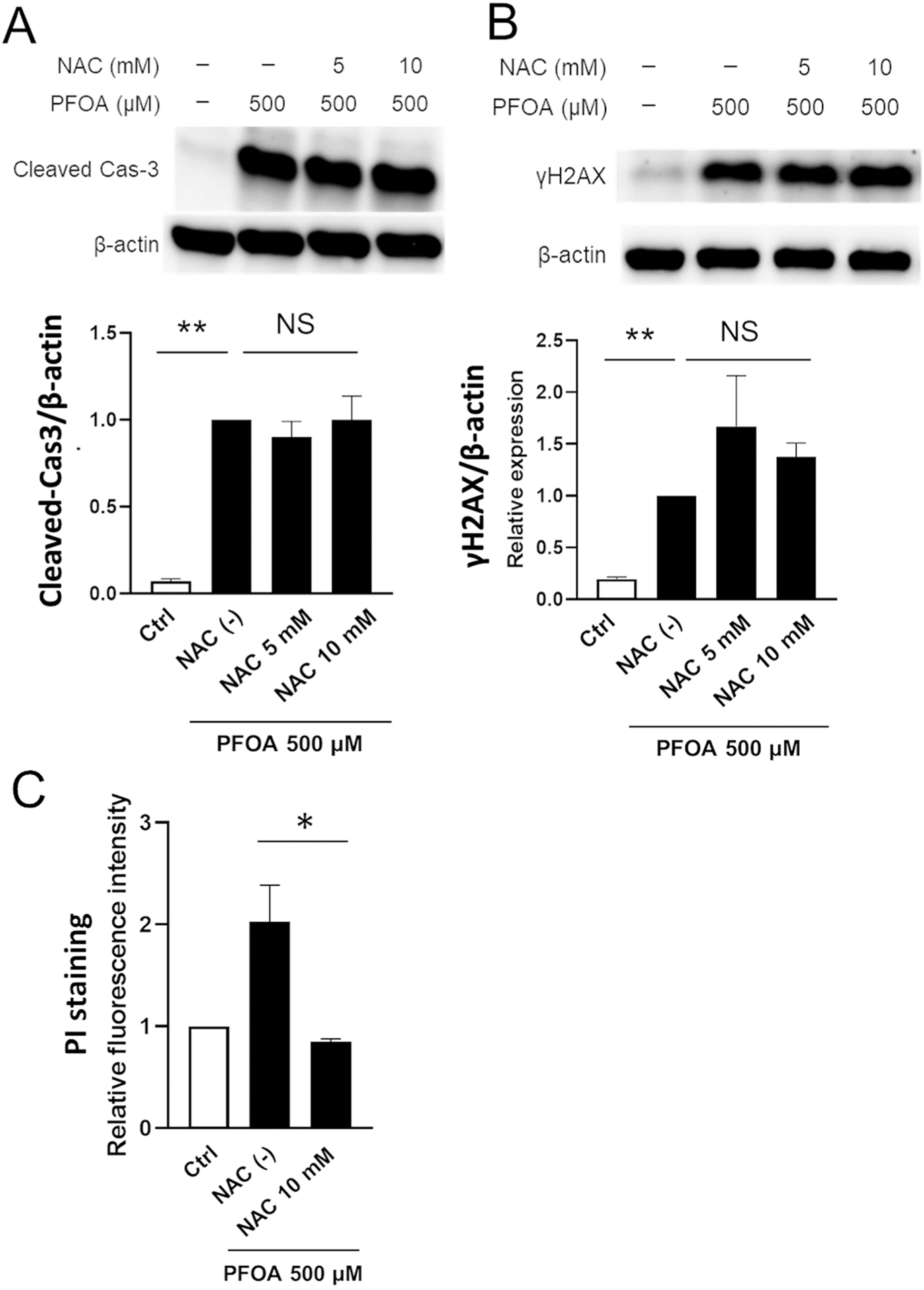
PFOA-induced apoptosis is not associated with ROS-mediated cell death. ALC cells were treated with NAC (5 and 10 mM) for 1 h prior to PFOA (500 μM) for 24 h. Cleaved caspase-3 (17 kDa) and γH2AX (15 kDa) were detected by Western blot. Relative protein level was normalized by the loading control β-actin (44 kDa). NAC with PFOA treatment did not change (A) cleaved caspase-3 levels nor (B) γ-H2AX levels compared to PFOA alone. Blots shown are representative images from three biological replicates. (C) Dead cells were measured by PI staining. NAC addition did not change apoptosis, but significantly decreased PI-positive necrotic cells compared to PFOA alone. Data are presented as means ± SD from three independent experiments. Statistically significant differences are indicated as follow: **; P < 0.01, *; P < 0.05, N.S.; no significance.
3.5. Dual roles of ERK in PFOA-mediated necrosis and apoptosis
Fig. 3 shows that inhibition of ERK in ALC cells by PD98059 decreased PI-positive cells and rescued PFOA-mediated cell death, indicating that PFOA-mediated necrosis is induced via ERK pathway.
Next, we further assessed the role of ERK pathway in PFOA-induced apoptosis. We hypothesized that ERK pathway promotes both necrosis and apoptosis and that the ERK inhibitor PD98059 would reduce both PFOA-mediated necrosis and apoptosis, thereby rescuing the cells from cell death. However, contrary to our expectation, PFOA with PD98059 (40 μM) significantly increased the protein levels of cleaved caspase-3 (Fig. 6A) (**P < 0.01) and γH2AX (Fig. 6B) (**P < 0.01). These results suggest that ERK activation plays a suppressive role in PFOA-mediated apoptosis. Although inhibition of ERK by PD98059 significantly increased the apoptotic pathway (Fig. 6), the inhibitor significantly decreased PI-positive cells and rescued cell viability (Fig. 3). These results suggest that PFOA-activated ERK has dual roles (Fig. 8) and ERK-mediated necrosis plays a more critical role in PFOA-mediated cell death than apoptosis.
Fig. 6.

ERK inhibitor PD98059 augments PFOA-mediated apoptosis. ALC cells were treated with ERK inhibitor, PD98059 (5 and 40 μM) for 1 h prior to PFOA (500 μM) treatment for 24 h. Cleaved caspase-3 (17 kDa) and γH2AX (15 kDa) were detected by Western blot. Relative protein level was normalized by the loading control β-actin (44 kDa). PD98059 with PFOA significantly increased (A) cleaved caspase-3 levels and (B) γ-H2AX levels compared to PFOA alone. Blots shown are representative images from three biological replicates. Open bars show controls. Data are presented as means ± SD from three independent experiments. **; P < 0.01.
Fig. 8.
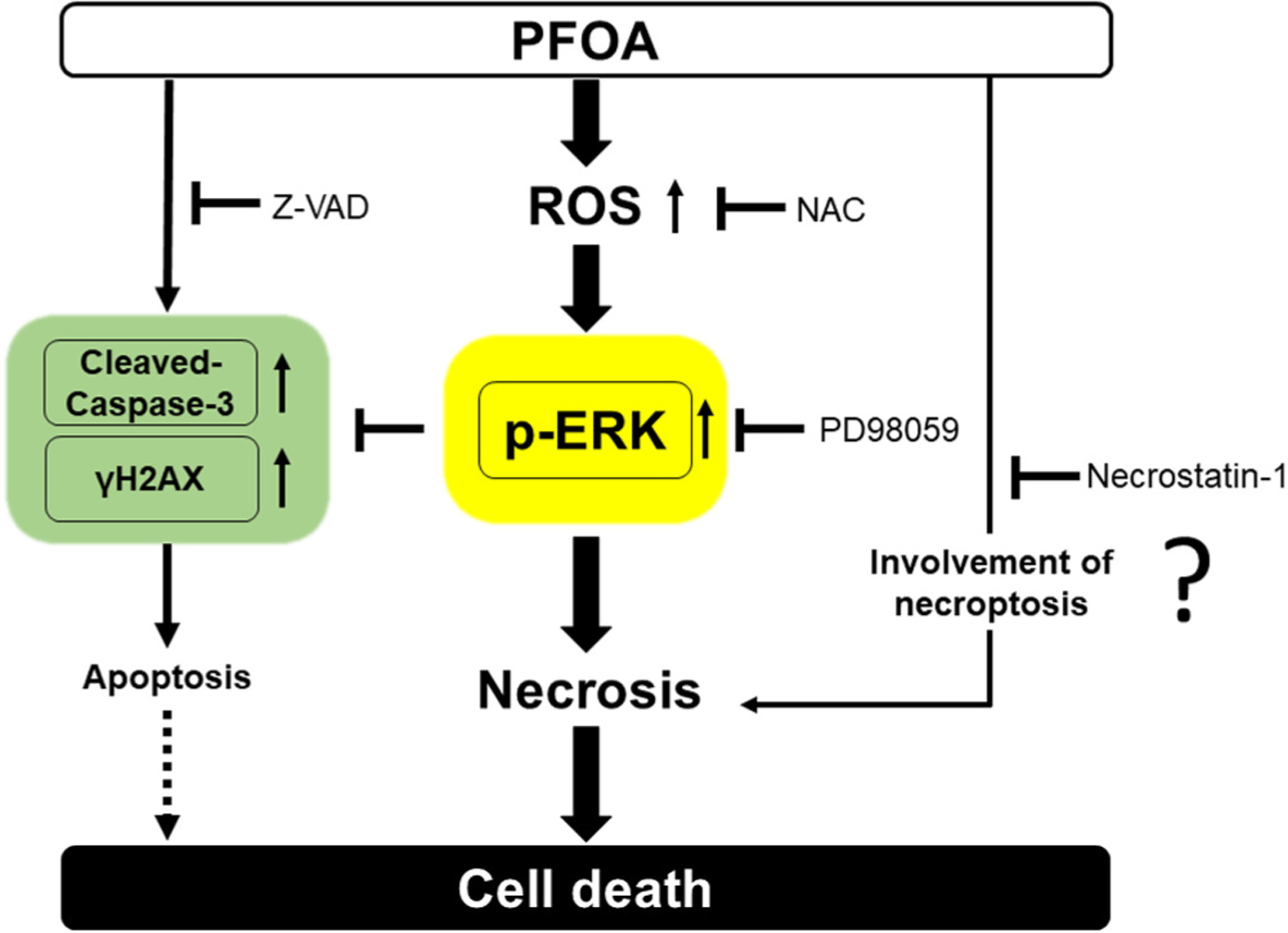
Schema of PFOA-mediated cell death pathways via ROS-MAPK/ERK Signaling. PFOA induces three death pathways apoptosis/necrosis/necroptosis in ALC cells. PFOA increases ROS generation to upregulate phosphor-(p)-ERK leading to necrosis. However, use of the ROS inhibitor NAC suppresses ERK activation and decreases necrosis to mitigate cell death by PFOA. Furthermore, PFOA induces apoptosis by increasing cleaved-caspase-3 and the DNA damage marker γH2AX, neither of which are dependent on ROS production. Use of the ERK inhibitor PD98059 suppresses ERK activation and decreases necrosis, while PD98059 augments PFOA-mediated apoptosis. The necroptosis inhibitor Necrostation-1 attenuates cell death, whereas the Pan-caspase inhibitor Z-VAD did not. Taken together, we hypothesize that PFOA-mediated cell death is mainly caused by necrosis/necroptosis rather than apoptosis.
3.6. PFOA-mediated cell death is associated with necroptosis
Necroptosis is a programmed form of necrosis and ROS can serve as an initiator of necroptosis (Zhang et al., 2017). To assess if the necroptosis pathway is involved in PFOA-mediated cell death, we performed the colony formation assay using the necroptosis inhibitor, Necrostation-1. Treatment of cells with PFOA along with Necrostation-1 (20 and 40 μM) significantly improved colony formation compared to PFOA alone (Fig. 7A) (**P < 0.01). Moreover, Fig. 7B shows that Necrostation-1 (40 μM) along with PFOA significantly decreased PI-positive cells compared to PFOA alone (**P < 0.01). These results suggest that necroptosis pathway is associated with PFOA-mediated cell death.
Fig. 7.

Necroptosis inhibitor Necrostation-1 mitigates PFOA-mediated cell death. ALC cells were treated with RIP1 inhibitor (necroptosis inhibitor; Necrostation-1) for 1 h prior to PFOA treatment (500 μM) for 24 h. (A) Cell viability was determined by colony formation assay. PFOA with Necrostation-1 (20 and 40 μM) significantly increased colony formation compared to PFOA alone. The picture shown is a representative image of colonies. (B) PFOA with Necrostation-1 (40 μM) significantly decreased PI-positive necrotic cells compared to PFOA alone. Open bars show controls. Data are presented as means ± SD from three independent experiments. Statistically significant differences are indicated as follow: **; P < 0.01.
4. Discussion
PFOA is one of per- and polyfluoroalkyl substances (PFAS) that have generated increased public attention due to their widespread presence in the environment. Although the results contradict, recent epidemiology studies have shown the relation between PFOA and dental caries prevalence. One concluded that PFOA was not associated with the prevalence of dental caries, while the other demonstrated that PFOA had an association with dental caries experience (Wiener and Waters, 2019). Animal models have shown that PFOA caused craniofacial deformities in zebrafish (Jantzen et al., 2016) in rodents (Balmanno and Cook, 2009). PFOA precursor, FTOHs induced ameloblast degeneration leading to tooth malformation in rodents (Ladics et al., 2008; Mukerji et al., 2015; Serex et al., 2014). During FTOHs metabolism, fluorine is generated to form the final metabolite PFOA (Wang et al., 2005). The released fluorine could play a critical role in ameloblast degeneration, and lead to a dental fluorosis-like phenotype. However, mechanistically how the final metabolite PFOA can affect enamel development (amelogenesis) is not understood. In the present study, we assessed if PFOA itself affects ameloblast-lineage cell line (ALC) cells in vitro.
Serum PFOA levels in high-exposure workers ranged from 0.02 μM to 222 μM (Olsen and Zobel, 2007). Lowest-observed-adverse-effect (LOAEL) doses correspond to serum PFOA levels of 48–123 μM in rats (Loveless et al., 2006).
A significant part of the PFOA in serum is bound to albumin and other proteins. This interaction limits the level of PFOA that moves by passive transport into tissue cells. It has been reported in vitro, that PFOA enters the cell membrane of Fisher rat thyroid line-5 (FRTL-5) cells by passive transport. Analysis of FRTL-5 cells exposed to 100 μM PFOA in culture media resulted in an intracellular calculated mean amount of 2.3 nM PFOA (Coperchini et al., 2015). Coperchini et al. found that the intracellular PFOA concentration in the cell pellet was 0.0023 % of the medium PFOA concentration (100 μM). This finding demonstrates that levels of PFOA entering cells in vitro are lower than the concentration found in culture media. Zhang et al. (Zhang et al., 2020) found that addition of required fetal bovine serum to culture media used for in vitro cellular assays contains albumin which binds PFOA and reduces PFOA cellular toxicity. We also expect a PFOA concentration gradient between ALC cells and culture medium in our experimental setting. Therefore, the available PFOA able to move by passive transport into ALC cells is likely much lower than that in the surrounding media. Taken together, we consider the concentration of 500 μM PFOA in culture medium is within a valid biological range for in vitro settings.
PFOA acts as an endocrine disruptor (Coperchini et al., 2020; Mokra, 2021) and promotes cancer proliferation, migration, invasion and causes drug resistance (Pierozan et al., 2022), suggesting that PFOA biological effects vary and depend on the types of tissue and cell. In the present study, PFOA at 500 μM (IC50 in ALC cells) induced cytotoxic effects, inducing apoptosis and necrosis in ALC cells, while PFOA at 500 μM did not affect cell viability in mouse spermatogonical cells (GC-1 cells) (Lin et al. 2020). This suggests that 500 μM PFOA is not resulting in non-specific physical damage due to the concentration, but rather inducing a cell type-dependent effect. Moreover, intervention by ROS inhibitor (NAC) or by MAPK inhibitor (PD98059) 1 h before PFOA
treatment, rescued the PFOA-induced cytotoxicity in this study. This suggests that PFOA-induced cytotoxicity is a reversible reaction via cell signaling cascades rather than irreversible cell damage caused by non-specific physical damage (e.g., direct cell membrane disruption).
Previous studies have shown that PFOA can cause oxidative stress with ROS generation. PFOA induces oocyte apoptosis and necrosis in mice (Lopez-Arellano et al., 2019) and ROS inhibitor NAC abates PFOA-mediated hepatorenal toxicities in rats (Owumi et al., 2021). Concordant with these studies, our results show that PFOA significantly increased ROS production and induced three death pathways, necrosis, programmed cell death (apoptosis) and necroptosis in ALC cells (Fig. 8). Moreover, PFOA activated MAPK/ERK signaling, and the ROS inhibitor NAC suppressed ERK and inhibited necrosis to mitigate PFOA cytotoxicity. However, NAC did not change PFOA-mediated apoptosis. Taken together, this suggests that 1) PFOA activates MAPK/ERK via ROS, but the apoptosis pathway is induced by other factors outside of ROS, 2) ROS-MAPK/ERK-mediated necrosis is critical for PFOA cytotoxicity rather than apoptosis. Indeed, a recent study showed that PFOA induced mitochondrial-mediated apoptosis via endoplasmic reticulum (ER) stress in human liver L02 cells (Wang et al., 2022) and PFOA (500 and 750 μM) increased ER stress marker Chop in immature mouse Leydig cell line TM3 (Han and Park, 2023). ROS can activate cell stress responses such as oxidative stress and ER stress in ameloblasts (Suzuki et al., 2015). ROS-mediated cell stress response could participate in PFOA-induced cytotoxicity in addition to the ROS-MAPK/ERK.
ERK inhibitor PD98059 significantly decreased necrosis and ameliorated PFOA cytotoxicity. However, intriguingly, PD98059 significantly augmented PFOA-mediated apoptosis. These results suggest that ERK activation increased necrosis but simultaneously, ERK plays an anti-apoptotic role under PFOA treatment. ERK activation is widely associated with cell proliferation, differentiation, and anti-apoptotic functions while depending on the cell type and stimulus, ERK activity also will mediate different antiproliferative events and cell death, including apoptosis, autophagy, and senescence in vitro and in vivo (Cagnol and Chambard, 2010). Anti-tumor agent edelfosine-induced cell death in human glioblastoma U118 cells occurs mainly through necrosis/necroptosis, whereas apoptosis was scarce. Inhibition of ERK phosphorylation led to a dramatic increase in edelfosine-induced apoptosis (Melo-Lima et al., 2015), which is in accordance with our results showing that ERK inhibition shifts the cell response to PFOA from necrosis to apoptosis. Although ERK inhibition significantly increased cleaved-caspase-3, ERK inhibition reduced necrotic cells and mitigated PFOA-mediated cell death. Pan-caspase inhibitor z-VAD-fmk did not rescue PFOA-mediated cell death, while necroptosis inhibitor Necrostation-1 ameliorated PFOA-mediated cell death. These results suggest that PFOA-mediated cell death was mainly through necrosis/necroptosis rather than the apoptosis pathway.
The environmental toxicant cadmium causes cell death by apoptosis in a human lymphoblastoid cell line and chemical inhibitors of caspases 3 and 8 blocked cleavage of PARP but not cell death, suggesting the existence of a caspase-independent death caused by cadmium (Coutant et al., 2006). PFOA-mediated cell death may be caused by a caspase-independent mechanism, whereas our data raise questions about the role of caspase activation/apoptosis pathway in PFOA-mediated cytotoxicity. Caspases were originally identified as important mediators of inflammatory response and apoptosis. However, caspase activation has other roles. For example, preconditioning tissue in an in vivo model of ischemic tolerance showed widespread caspase-3 cleavage, without cell death, in which heat-shock protein 70 was upregulated and was essential for neuroprotection (McLaughlin et al., 2003). Moreover, bone morphogenetic protein-4 (BMP-4) induced the growth arrest and differentiation in osteoblastic MC3T3-E1 cells via caspase-8, caspase-2, and caspase-3 activation, despite no apoptosis or necrosis (Mogi and Togari, 2003). These studies indicate that caspases have various roles such as cell protection and cell differentiation. Recent studies have unveiled caspase roles in regulating necrotic cell death, including necroptosis and pyroptosis (Yuan et al., 2016). Caspase-8 suppresses the activation of necroptosis and caspase-3 induces pyroptosis. Caspase roles in PFOA-mediated necroptosis remain obscure, but caspases may involve crosstalk between PFOA-mediated apoptosis and necroptosis. Further studies will be required to understand the role of caspase in PFOA-treated ALC cells.
Although this study provides new insight demonstrating possible cytotoxic molecular mechanisms of PFOA in amelogenesis, there are certain limitations to our findings. Our results in this in vitro study suggest that in addition to fluoride, PFOA itself could affect enamel formation. However, more research is required 1) to identify the level and mechanism by which PFOA contributes to enamel malformation in vivo and 2) to determine pathophysiology of PFOA on ameloblasts in addition to induction of cell death pathway in vivo.
5. Conclusion
This is the initial report suggesting the possible PFOA cytotoxicity to cells responsible for enamel development. We demonstrated that three cell death pathways were activated by PFOA and the role of ROS-MAPK/ERK signaling in PFOA cytotoxicity. PFOA increased necrosis via ROS-MAPK/ERK signaling but apoptosis was not associated with ROS. PFOA-mediated cell death was mainly caused by necrosis/necroptosis rather than apoptosis. PFOA could be considered as a possible causative factor for cryptogenic abnormalities in anelogenesis, including Molar Incisor Hypomineralisation (MIH) of which the etiologies are largely unknown. Further studies are required to elucidate the mechanisms of PFOA-mediated adverse effects on amelogenesis.]
Supplementary Material
Acknowledgments
We thank Dr. Whitford M Gary for suggesting this work.
Funding
Research reported in this publication was supported by the National Institute of Dental and Craniofacial Research of the National Institutes of Health; R01DE027648 (MS) and K02DE029531 (MS).
Footnotes
CRediT authorship contribution statement
Natsumi Fujiwara: Data curation, Investigation, Methodology, Validation, Writing – review & editing. Shohei Yamashita: Data curation, Investigation, Methodology, Validation, Writing – review & editing. Motoki Okamoto: Data curation, Investigation, Methodology, Validation, Writing – review & editing. Marion A. Cooley: Supervision, Validation, Writing – review & editing. Kazumi Ozaki: Supervision, Validation, Writing – review & editing. Eric T. Everett: Supervision, Validation, Writing – review & editing. Maiko Suzuki: Formal analysis, Validation, Funding acquisition, Investigation, Project administration, Writing – review & editing.
Appendix A. Supporting information
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.ecoenv.2023.115089.
Declaration of competing for financial interests
The authors declare they have no known competing financial interests or personal relationships that could appear to influence the work reported in this paper.
Data Availability
Data will be made available on request.
References
- Almuallem Z, Busuttil-Naudi A, 2018. Molar incisor hypomineralisation (mih) - an overview. Br.Dent. J 225, 601–609. [DOI] [PubMed] [Google Scholar]
- Babayev M, Capozzi SL, Miller P, McLaughlin KR, Medina SS, Byrne S, Zheng G, Salamova A, 2022. Pfas in drinking water and serum of the people of a southeast alaska community: a pilot study. Environ. Pollut 305, 119246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balmanno K, Cook SJ, 2009. Tumour cell survival signalling by the erk1/2 pathway. Cell Death Differ. 16 (3), 368–377. [DOI] [PubMed] [Google Scholar]
- Cagnol S, Chambard JC, 2010. Erk and cell death: Mechanisms of erk-induced cell death–apoptosis, autophagy and senescence. FEBS J. 277 (1), 2–21. [DOI] [PubMed] [Google Scholar]
- Coperchini F, Pignatti P, Lacerenza S, Negri S, Sideri R, Testoni C, de Martinis L, Cottica D, Magri F, Imbriani M, et al. , 2015. Exposure to perfluorinated compounds: in vitro study on thyroid cells. Environ. Sci. Pollut. Res. Int 22 (3), 2287–2294. [DOI] [PubMed] [Google Scholar]
- Coperchini F, Croce L, Ricci G, Magri F, Rotondi M, Imbriani M, Chiovato L, 2020. Thyroid disrupting effects of old and new generation pfas. Front. Endocrinol 11, 612320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutant A, Lebeau J, Bidon-Wagner N, Levalois C, Lectard B, Chevillard S, 2006. Cadmium-induced apoptosis in lymphoblastoid cell line: Involvement of caspase-dependent and -independent pathways. Biochimie 88 (11), 1815–1822. [DOI] [PubMed] [Google Scholar]
- Deng H, Fujiwara N, Cui H, Whitford GM, Bartlett JD, Suzuki M, 2020. Histone acetyltransferase promotes fluoride toxicity in ls8 cells. Chemosphere 247, 125825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EPA US, 2022. Interim drinking water health advisory: Perfluorooctanoic acid (pfoa) casrn 335–67–1.
- Era S, Harada KH, Toyoshima M, Inoue K, Minata M, Saito N, Takigawa T, Shiota K, Koizumi A, 2009. Cleft palate caused by perfluorooctane sulfonate is caused mainly by extrinsic factors. Toxicology 256 (1–2), 42–47. [DOI] [PubMed] [Google Scholar]
- Fujiwara N, Whitford GM, Bartlett JD, Suzuki M, 2021. Curcumin suppresses cell growth and attenuates fluoride-mediated caspase-3 activation in ameloblast-like ls8 cells. Environ. Pollut 273, 116495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JW, Park HJ, 2023. Perfluorooctanoic acid induces cell death in tm3 cells via the er stress-mitochondrial apoptosis pathway. Reprod. Toxicol 118, 108383. [DOI] [PubMed] [Google Scholar]
- Jantzen CE, Annunziato KA, Bugel SM, Cooper KR, 2016. Pfos, pfna, and pfoa sub-lethal exposure to embryonic zebrafish have different toxicity profiles in terms of morphometrics, behavior and gene expression. Aquat. Toxicol 175, 160–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JS, Choi JS, Park JW, 2016. Transcriptional changes in steroidogenesis by perfluoroalkyl acids (pfoa and pfos) regulate the synthesis of sex hormones in h295r cells. Chemosphere 155, 436–443. [DOI] [PubMed] [Google Scholar]
- Ladics GS, Kennedy GL, O’Connor J, Everds N, Malley LA, Frame SR, Gannon S, Jung R, Roth T, Iwai H, et al. , 2008. 90-day oral gavage toxicity study of 8–2 fluorotelomer alcohol in rats. Drug Chem. Toxicol 31 (2), 189–216. [DOI] [PubMed] [Google Scholar]
- Lin T, Zhang Y, Ding X, Huang T, Zhang W, Zou W, Kuang H, Yang B, Wu L, Zhang D, 2020. Perfluorooctanoic acid induces cytotoxicity in spermatogonial gc-1 cells. Chemosphere 260, 127545. [DOI] [PubMed] [Google Scholar]
- Liu Q, Liu Y, Li X, Wang D, Zhang A, Pang J, He J, Chen X, Tang NJ, 2023. Perfluoroalkyl substances promote breast cancer progression via eralpha and gper mediated pi3k/akt and mapk/erk signaling pathways. Ecotoxicol. Environ. Saf 258, 114980. [DOI] [PubMed] [Google Scholar]
- Lopez-Arellano P, Lopez-Arellano K, Luna J, Flores D, Jimenez-Salazar J, Gavia G, Teteltitla M, Rodriguez JJ, Dominguez A, Casas E, et al. , 2019. Perfluorooctanoic acid disrupts gap junction intercellular communication and induces reactive oxygen species formation and apoptosis in mouse ovaries. Environ. Toxicol 34 (1), 92–98. [DOI] [PubMed] [Google Scholar]
- Loveless SE, Finlay C, Everds NE, Frame SR, Gillies PJ, O’Connor JC, Powley CR, Kennedy GL, 2006. Comparative responses of rats and mice exposed to linear/branched, linear, or branched ammonium perfluorooctanoate (apfo). Toxicology 220 (2–3), 203–217. [DOI] [PubMed] [Google Scholar]
- Lu Y, Luo B, Li J, Dai J, 2016. Perfluorooctanoic acid disrupts the blood-testis barrier and activates the tnfalpha/p38 mapk signaling pathway in vivo and in vitro. Arch. Toxicol 90 (4), 971–983. [DOI] [PubMed] [Google Scholar]
- McLaughlin B, Hartnett KA, Erhardt JA, Legos JJ, White RF, Barone FC, Aizenman E, 2003. Caspase 3 activation is essential for neuroprotection in preconditioning. Proc. Natl. Acad. Sci. USA 100 (2), 715–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo-Lima S, Lopes MC, Mollinedo F, 2015. Erk1/2 acts as a switch between necrotic and apoptotic cell death in ether phospholipid edelfosine-treated glioblastoma cells. Pharmacol. Res 95-96, 2–11. [DOI] [PubMed] [Google Scholar]
- Mogi M, Togari A, 2003. Activation of caspases is required for osteoblastic differentiation. J. Biol. Chem 278 (48), 47477–47482. [DOI] [PubMed] [Google Scholar]
- Mokra K, 2021. Endocrine disruptor potential of short- and long-chain perfluoroalkyl substances (pfass)-a synthesis of current knowledge with proposal of molecular mechanism. Int. J. Mol. Sci 22 (4), 2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukerji P, Rae JC, Buck RC, O’Connor JC, 2015. Oral repeated-dose systemic and reproductive toxicity of 6:2 fluorotelomer alcohol in mice. Toxicol. Rep 2, 130–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakata A, Kameda T, Nagai H, Ikegami K, Duan YQ, Terada K, Sugiyama T, 2003. Establishment and characterization of a spontaneously immortalized mouse ameloblast-lineage cell line. Biochem. Biophys. Res. Co 308 (4), 834–839. [DOI] [PubMed] [Google Scholar]
- Oh J, Shin HM, Nishimura T, Rahman MS, Takahashi N, Tsuchiya KJ, 2022. Perfluorooctanoate and perfluorooctane sulfonate in umbilical cord blood and child cognitive development: Hamamatsu birth cohort for mothers and children (hbc study). Environ. Int 163, 107215. [DOI] [PubMed] [Google Scholar]
- Olsen GW, Zobel LR, 2007. Assessment of lipid, hepatic, and thyroid parameters with serum perfluorooctanoate (pfoa) concentrations in fluorochemical production workers. Int. Arch. Occup. Environ. Health 81 (2), 231–246. [DOI] [PubMed] [Google Scholar]
- Owumi S, Bello T, Oyelere KA, 2021. N-acetyl cysteine abates hepatorenal toxicities induced by perfluorooctanoic acid exposure in male rats. Environ. Toxicol. Pharmacol 86, 103667. [DOI] [PubMed] [Google Scholar]
- Pierozan P, Cattani D, Karlsson O, 2022. Tumorigenic activity of alternative per- and polyfluoroalkyl substances (pfas): mechanistic in vitro studies. Sci. Total Environ 808, 151945. [DOI] [PubMed] [Google Scholar]
- Puttige Ramesh N, Arora M, Braun JM, 2019. Cross-sectional study of the association between serum perfluorinated alkyl acid concentrations and dental caries among us adolescents (nhanes 1999–2012). BMJ Open 9 (2), e024189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu J, Zhao Y, Zhang L, Hu S, Liao K, Zhao M, Wu P, Jin H, 2022. Evaluated serum perfluoroalkyl acids and their relationships with the incidence of rheumatoid arthritis in the general population in hangzhou, china. Environ. Pollut 307, 119505. [DOI] [PubMed] [Google Scholar]
- Serex T, Anand S, Munley S, Donner EM, Frame SR, Buck RC, Loveless SE, 2014. Toxicological evaluation of 6:2 fluorotelomer alcohol. Toxicology 319, 1–9. [DOI] [PubMed] [Google Scholar]
- Shearer JJ, Callahan CL, Calafat AM, Huang WY, Jones RR, Sabbisetti VS, Freedman ND, Sampson JN, Silverman DT, Purdue MP, et al. , 2021. Serum concentrations of per- and polyfluoroalkyl substances and risk of renal cell carcinoma. J. Natl. Cancer Inst 113 (5), 580–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura R, Satoh R, Takasaki T, 2021. Erk: A double-edged sword in cancer. Erk-dependent apoptosis as a potential therapeutic strategy for cancer. Cells 10 (10), 2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, Shin M, Simmer JP, Bartlett JD, 2014. Fluoride affects enamel protein content via tgf-beta1-mediated klk4 inhibition. J. Dent. Res 93 (10), 1022–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, Bandoski C, Bartlett JD, 2015. Fluoride induces oxidative damage and sirt1/autophagy through ros-mediated jnk signaling. Free Radic. Biol. Med 89, 369–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upham BL, Park JS, Babica P, Sovadinova I, Rummel AM, Trosko JE, Hirose A, Hasegawa R, Kanno J, Sai K, 2009. Structure-activity-dependent regulation of cell communication by perfluorinated fatty acids using in vivo and in vitro model systems. Environ. Health Perspect 117 (4), 545–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N, Szostek B, Buck RC, Folsom PW, Sulecki LM, Capka V, Berti WR, Gannon JT, 2005. Fluorotelomer alcohol biodegradation-direct evidence that perfluorinated carbon chains breakdown. Environ. Sci. Technol 39 (19), 7516–7528. [DOI] [PubMed] [Google Scholar]
- Wang Q, Chen W, Zhang B, Gao Z, Zhang Q, Deng H, Han L, Shen XL, 2022. Perfluorooctanoic acid induces hepatocellular endoplasmic reticulum stress and mitochondrial-mediated apoptosis in vitro via endoplasmic reticulum-mitochondria communication. Chem. Biol. Interact 354, 109844. [DOI] [PubMed] [Google Scholar]
- Wiener RC, Waters C, 2019. Perfluoroalkyls/polyfluoroalkyl substances and dental caries experience in children, ages 3–11 years, national health and nutrition examination survey, 2013–2014. J. Public Health Dent 79 (4), 307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Najafov A, Py BF, 2016. Roles of caspases in necrotic cell death. Cell 167 (7), 1693–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Zhang H, Chen B, Luan T, 2020. Fetal bovine serum attenuating perfluorooctanoic acid-inducing toxicity to multiple human cell lines via albumin binding. J. Hazard. Mater 389, 122109. [DOI] [PubMed] [Google Scholar]
- Zhang W, Liu HT, 2002. Mapk signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 12 (1), 9–18. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Su SS, Zhao S, Yang Z, Zhong CQ, Chen X, Cai Q, Yang ZH, Huang D, Wu R, et al. , 2017. Rip1 autophosphorylation is promoted by mitochondrial ros and is essential for rip3 recruitment into necrosome. Nat. Commun 8, 14329. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.