

Abstract
Immuno-oncology therapies have been of great interest with the goal of inducing sustained tumor regression, but clinical results have demonstrated the need for improved and widely applicable methods. An antigen-free method of cancer immunotherapy can stimulate the immune system to recruit lymphocytes and produce immunostimulatory factors without prior knowledge of neoantigens, while local delivery reduces the risk of systemic toxicity. To improve the interactions between tumor cells and cytotoxic lymphocytes, a gene delivery nanoparticle platform was engineered to reprogram the tumor microenvironment (TME) in situ to be more immunostimulatory by inducing tumor-associated antigen-presenting cells (tAPCs) to activate cytotoxic lymphocytes against the tumor. Biodegradable, lipophilic poly(beta-amino ester) (PBAE) nanoparticles were synthesized and used to co-deliver mRNA constructs encoding a signal 2 co-stimulatory molecule (4-1BBL) and a signal 3 immuno-stimulatory cytokine (IL-12), along with a nucleic acid-based immunomodulatory adjuvant. Nanoparticles are combined with a thermoresponsive block copolymer for gelation at the injection site for local NP retention at the tumor. The reprogramming nanoparticle gel synergizes with immune checkpoint blockade (ICB) to induce tumor regression and clearance in addition to resistance to tumor rechallenge at a distant site. In vitro and in vivo studies reveal increases in immunostimulatory cytokine production and recruitment of immune cells as a result of the nanoparticles. Intratumoral injection of nanoparticles encapsulating mRNA encoding immunostimulatory agents and adjuvants via an injectable thermoresponsive gel has great translational potential as an immuno-oncology therapy that can be accessible to a wide range of patients.
Keywords: Nanoparticles, immunotherapy, nonviral gene delivery, immuno-oncology, mRNA, immunoengineering
Introduction
The immunotherapy space for oncology applications has been growing substantially since the development of immune checkpoint blockades, chimeric antigen receptor (CAR) T-cell therapies, and improved gene delivery technologies (1, 2). Despite this, leveraging the immune system for cancer treatment remains challenging, given tumor escape mechanisms and barriers to ex vivo modification of cells. Cancer immunotherapies aim to kick-start the immune system to effectively target and kill cancer cells within the tumor. The tumor microenvironment is composed of a heterogenous population of cells in addition to tumor cells, including immune cells (3). Among these are cytotoxic CD8+ T cells, which have the ability to infiltrate the tumor and kill cancer cells. Thus, it is crucial for immunotherapies to stimulate these effector T cells to ultimately reduce tumor burden. Productive CD8+ T-cell activation requires the coordinated engagement of three signals: signal 1 interaction between the T-cell receptor (TCR) and a tumor antigen-loaded major histocompatibility complex class I molecule (MHC I); signal 2 between a co-stimulatory molecule receptor on the T cell and its corresponding ligand; and signal 3 soluble cytokines recognized by receptors on the T cell (4) (Schematic 1). These interactions can occur between antigen-presenting cells (APCs) and CD8+ T cells, leading to specific tumor cell killing. Biomimetic scaffold and microparticle methods have been used to present the signals necessary to recruit and stimulate T cells of interest, but these methods require harvesting of patient cells, ex vivo modification, and reintroduction of manipulated cells, which is costly and inefficient and is associated with high regulatory burden (5, 6). Microparticle- or scaffold-based antigen presentation methods (7, 8) that can be injected for T cell stimulation in situ still pose challenges, given the need for a patient-matched MHC complex, efficient loading of signals 1-3, and meticulously engineered degradation and release profiles of encapsulated molecules. Lastly, designing these technologies for each tumor type and patient can limit feasibility given the need to screen for unique antigens (9, 10).
Schematic 1. Overview of the mRNA-containing nanoparticle (NP) fabrication with cationic poly(beta-amino ester)s (PBAEs),

mRNA encoding for therapeutic signal 2 and 3, and immunostimulatory adjuvant. Resulting mRNA NPs are combined with PLGA-PEG-PLGA thermoresponsive polymers and injected intratumorally. The NPs transfect tumor-associated cells to express signal 2 and secrete signal 3 to behave immunologically like antigen presenting cells. These programmed tumor-associated antigen-presenting cells (tAPCs) can engage and activate T effector cells to attack cancer cells.
Alternatively, nanoparticle gene delivery technologies can induce the expression of various molecules in situ via non-invasive administration strategies (11–14). Compared to viral vectors, non-viral vectors like polymeric nanoparticles exhibit lower immunogenicity, thus allowing redosing, high cargo capacity, and ease of manufacturing for clinical translation (15, 16). In particular, poly(beta-amino ester)s (PBAEs), a class of degradable cationic polymers, can easily self-assemble with negatively charged nucleic acids, while the high buffering capacity of their amine groups allows efficient endosomal escape, which is crucial for delivery of intact cargo. PBAEs degrade via hydrolysis, allowing safe clearance from the body, and can be modified with other compounds such as lipid-conjugated polyethylene glycol (PEG) for precise engineering of the surface properties to optimize bioavailability in vivo (17). Here, we report the in vitro and in vivo use of these PBAEs to deliver mRNA constructs encoding signals 2 and 3 to the tumor microenvironment (TME) in order to cause immunostimulation. Because this activation occurs in the context of the tumor, where signal 1—the tumor antigen in MHC I—is present, this results in tumor-specific activation of a T cell-mediated immune response. At the same time, this antigen-agonistic therapy can be used as an off-the-shelf product applicable to many types of solid tumors. Expression of the delivered signals 2 and 3 in the TME is transient, with the goal of catalyzing the immune response cascade with long-lasting effects without the need to permanently edit transfected cells.
Building on previous work on in situ generation of tumor-associated APCs (tAPCs) using DNA nanoparticles (18), the mRNA nanoparticles used prevent any risk of genomic integration and increase the level of gene expression in the tumor. Further, to facilitate mRNA delivery, a new lipophilic polymer was used for the first time for enhanced local transfection of tumors (19). Here, PBAE NPs were used to deliver two therapeutic mRNA sequences encoding costimulatory signal 2 molecule 4-1BB ligand (4-1BBL) and soluble cytokine interleukin 12 (IL-12), which have been shown to cause anti-tumor immunity when induced in conjunction at the site of a tumor (18). Costimulatory signal 4-1BBL is normally expressed on APCs and causes activation and proliferation of Th1 helper CD4+ and cytotoxic CD8+ cells (20, 21). Secreted, soluble IL-12 has been previously shown to increase CD8+ T cell survival, proliferation, and cytolysis, as well as induce immunostimulatory interferon gamma (IFNγ) production (22). IFNγ has been previously shown to induce upregulation of MHC antigen presentation (23–25) which is advantageous in tumor microenvironments where MHC I is downregulated as an immune escape mechanism. Delivery of the cytokine via locally injected mRNA nanoparticles limits its expression beyond the tumor site, maintaining high levels of local expression and minimizing adverse effects often associated with cytokine delivery (26). This is crucial to the use of IL-12, as early clinical trials showed that this cytokine has anti-tumor potential (27–29) but causes severe toxicity when administered systemically, including causing deaths during a clinical trial (30, 31), and showed only moderate efficacy in larger studies, even when given at doses that still caused some adverse events. Adjuvants were also incorporated in the nanoparticle formulation in order to further activate immune cells and produce inflammatory signals (32–34). Here, the use of TLR9-agonist CpG oligodeoxynucleotide (CpG ODN) (35–37), STING (stimulator of interferon genes)-activating cyclic dinucleotide (CDN) (38–41), and TLR3-agonist polyinosinic-polycytidylic acid (poly(I:C)) (42–45) were explored and their effect on transfection, viability, stimulation of IFNγ production, and recruitment of cells to the TME.
Because this nanoparticle formulation is antigen-agnostic, transfection restricted to the tumor site is crucial to eliciting tumor-specific T cell activation. The triblock thermoresponsive copolymer poly(lactic-co-glycolic) acid-poly(ethylene glycol)-poly(lactic-co-glycolic) acid (PLGA-PEG-PLGA) facilitates this, as it is liquid and injectable at 4°C but forms a gel structure at 37°C (46), thereby sequestering co-formulated nanoparticles at the tumor injection site. Intratumoral (i.t.) administration of these nanoparticle formulations can therefore be applied to various solid, accessible tumors while avoiding the risks associated with systemic administration of immunotherapies.
In this study, we designed an mRNA PBAE nanoparticle (NP) for reprogramming tumor cells to tAPCs by delivering genes encoding 4-1BBL and IL-12 along with an immunostimulatory adjuvant. The transfection efficacy of these nanoparticles and the TME immune landscape after nanoparticle injections were analyzed to explore the contributions of each therapeutic mRNA molecule and adjuvant. When combined with anti-PD1 immune checkpoint blockade (ICB), the PBAE NPs with 4-1BBL, IL12, and CpG adjuvant reduce E0771 breast tumor burden in a murine model while extending animal survival and protecting the animal from tumor re-challenge at a distant site. These results are also shown in a different tumor model, MC38 colorectal carcinoma, demonstrating its potential applicability to two tumor types as an off-the-shelf immunotherapy.
Materials and Methods
Poly(Beta-Amino Ester) (PBAE) Synthesis and Chemical Characterization.
The PBAE was synthesized using previously described methods (19) to form the polymeric structure shown in Fig. S1A. The PBAE is composed of a diacrylate backbone monomer [bisphenol A glycerolate (1 glycerol/phenol) diacrylate], aminoalcohol sidechain monomers 1-dodecylamine and 4-(2-aminoethyl)morpholine (80% and 20%, respectively), and an amine-terminated endcap diethylentriamine. To characterize the synthesized polymer, gel permeation chromatography (GPC) (Agilent Technologies, Santa Clara, CA) was used to determine polymer molecular weight. PBAE was dissolved in dimethylformamide (DMF) at 10mg/mL for GPC analysis, and molecular weight was quantified against linear polystyrene standard. The structure of the diethyl ether precipitated PBAE dissolved in CDCl3 was confirmed by 1H NMR spectra and analyzed using TopSpin 3.5 software.
In Vitro Transfection of Tumor Cells.
Murine tumor cells 4T1, E0771, and MC38 were seeded in 96-well flat-bottom plates (5×103 cells/well) in 100 μL/well of complete growth medium composed of RPMI 1640 (Gibco, Waltham, MA), 10% fetal bovine serum (FBS), and 1% penicillin/streptomycin and allowed to adhere overnight in an incubator at 37°C and 5% CO2. For transfection on the following day, nanoparticles (NPs) were complexed via self-assembly by diluting PBAE in 25 mM sodium acetate buffer (pH 5, NaAc). mRNA [green fluorescent protein (GFP), firefly luciferase (fLuc), 4-1BB ligand (4-1BBL), and/or interleukin-12 (IL-12) (TriLink Biotechnologies, San Diego, CA)] and/or adjuvant [CpG oligodeoxynucleotide (CpG ODN), cyclic dinucleotide (CDN), or polyinosine-polycytidylic acid (poly(I:C)) (InvivoGen, San Diego, CA)] at the ratios specified below were also diluted together in NaAc, then mixed at 30 w/w (mass ratio of polymer to total nucleic acid) for 5 mins on for self-assembly into NPs. Particles were then added to cells at the specified dose and incubated with the cells for 24 h. For qualitative assessment of transfection and toxicity, cells were imaged with a Zeiss AxioObserver A1 inverted fluorescence/phase contrast microscope. For quantitative analysis by flow cytometry, cells were trypsinized and then resuspended in PBS buffer with 2% FBS, 0.25 μg/mL 7-aminoactinomycin D (7AAD), and 0.2% sodium azide. Cells were then analyzed on an Attune NxT Flow Cytometer (ThermoFisher Scientific). Transfection was calculated as a percentage of GFP-positive cells among total cells and also as the geometric mean GFP fluorescence intensity compared to untreated control. For 4-1BBL expression, cells were trypsinized and stained with PE anti-4-1BBL (CD137L) (BioLegend, San Diego, CA) (Table S1) diluted in PBS buffer with 2% FBS, 0.2% sodium azide. For IL-12 secretion, supernatant was collected after the 24-h transfection and measured using an ELISA MAX Deluxe Set Mouse IL-12 (p70) (BioLegend, San Diego, CA) and read using a Biotek Synergy 2 Plate Reader (Biotek, Winooski, VT). Head-to-head transfection of 4T1, E0771, and MC38 cells by PBAE vs Lipofectamine MessengerMAX was carried out by formulating Lipofectamine and GFP mRNA according to the manufacturer’s protocols and formulating PBAE/GFP mRNA NPs at 30 w/w and dosed at 100 ng/well (0.83 ng/μL) mRNA for both materials. Cells were incubated for 24 h for flow analysis of GFP geometric mean intensity via flow cytometry.
Characterization of Nanoparticles.
For transmission electron microscopy (TEM), NPs were formulated as above and diluted 1:10 serially in Millipore water. Five microliters of each sample was added to a carbon film 400 mesh copper grid (Electron Microscopy Sciences; Hatfield, PA), dried overnight, then imaged via a Hitachi 7600 transmission electron microscope (Hitachi High-Tech; Tokyo, Japan). DLS (Zetasizer NSP, Malvern, Worcestershire, UK) was used to measure the size of NPs in suspension after complexing. For zeta potential surface charge, NPs were diluted 6x with PBS (pH 7.4) and measured using the Zetasizer NSP. PEGylated NPs were co-formulated with 1,2-dimyristoyl-rac-glycero-3-methoxypolyethylene glycol-2000 (DMG PEG2k) by mixing the lipid-PEG with polymer in 100% ethanol, with DMG-PEG comprising 0-5% of the polymer mass. After mixing the PBAE+PEG with mRNA in NaAc to form NPs, the total volume was dialyzed against 1x PBS at 4°C for 60-75 min in a SpectraPor Float-A-Lyzer G2 dialysis device with 50 kDa MW cutoff (Repligen, Waltham, MA) to replace ethanol and other buffers with isotonic PBS buffer. The size and charge of freshly made NPs, with and without PEG, were measured and then assessed again after a freeze-thaw cycle at −80°C to assess aggregation. The NP encapsulation efficiency of mRNA and CpG when incubated at 37°C in PBS for 24 h was assessed using the gel electrophoresis assay. The NPs were formed with Cy5-labeled mRNA (TriLink Biotechnologies, San Diego, CA) and FITC-labeled CpG ODN (ODN 1826; InvivoGen, San Diego, CA). As control, NPs were incubated in 10 mg/mL heparin solution for complete dissociation to calculate % encapsulation of the nucleic acid cargos for the NP formulations incubated in PBS. Samples were loaded in an 1% agarose (UltraPure Agarose, Invitrogen, Carlsbad, CA) gel, and the gel was run for 20 min at 100 V and imaged with iBright FL1500 Imaging System (Thermo Fisher Scientific, Carlsbad, CA) using fluorescence mode to compare encapsulation efficiency for NPs in PBS compared to free mRNA and CpG (NPs in presence of heparin). For storage stability studies, NPs were formed and either kept stored in suspension at 4°C, −20°C, or −80°C or lyophilized and then stored at room temperature (RT), 4°C, −20°C, and −80°C. At each timepoint, the stored aliquot thawed then resuspended in complete media, then added to seeded 4T1 cells at 100 ng mRNA/well.
In Vitro Immune Activation by Transfected Tumor Cells.
For co-culture transfection studies, E0771 and MC38 cells were seeded in flat-bottom 96-well plates as described above. On the day of transfection, C57BL/6 mice were euthanized humanely and their spleens collected. Spleens were dissociated physically and filtered through a 40-μm cell strainer. The suspension was centrifuged at 500 × g for 5 mins, and the pellet was resuspended in 1 mL of ammonium-chloride-potassium (ACK) lysis buffer (Quality Biological, Gaithersburg, MD) before diluting in 20 mL of 1× PBS. The pellet was finally resuspended in 1x PBS, counted, and diluted in complete growth media to seed 50k splenocytes/well in the tumor-seeded plate, using splenocytes from a syngeneic strain for each cell type, in a final volume of 90 μL fresh media per well. NPs were made at 30 w/w as described above, then added to the tumor/splenocyte co-culture at a dose of 300 ng mRNA/well in a final volume of 60 μL/well before incubating at 37°C and 5% CO2. The supernatant media was collected at day 3, and IFNγ was measured using an IFNγ mouse ELISA kit (Thermo Fisher Scientific, Carlsbad, CA) and BioLegend LEGENDplex Mouse Inflammation Panel (13-plex) kit (BioLegend, San Diego, CA).
In Vitro and In Vivo Characterization of Thermoresponsive Triblock Copolymer.
PLGA-PEG-PLGA triblock copolymers AK091 (Mw ~1,500:1,500:1,500 Da, LA:GA 6:1) and AK012 (Mw ~1,000:1,000:1,000 Da, LA:GA 50:50) were purchased from PolySciTech (West Lafayette, IN). AK091 was dissolved in ultrapure sterile water at 33.4% w/v and AK012 at 26.1% w/v by agitation at 4°C for up to 3 days. The dissolved copolymer was stored in small aliquots at −20°C and thawed overnight at 4°C before use. For in vivo use, thermogel block copolymers AK091 and AK012 were mixed at a 1:1 (v/v) ratio and mixed with 10x PBS for a final concentration of 1x PBS, referred to hereafter as copolymer. NPs for in vivo use were PEGylated and dialyzed as described above, and the volume was retrieved and adjusted with additional 1×PBS to reach the final desired mRNA concentration of 200 ng/μL. The NPs were mixed 1:1 (v/v) with the copolymer/PBS solution on ice, and the mixture was then injected i.t. into anesthetized mice (25 μL injection volume) for all in vivo screens and studies.
All animal studies were approved by and conducted according to the regulations of the Johns Hopkins University Animal Care and Use Committee. For characterization of off-target effects in vivo, female 6- to 8-week-old BALB/c mice were injected with 5 χ 105 4T1 cells in the mammary fat pad or C57BL/6 mice were injected with 1 x 106 MC38 cells and allowed to grow for 7-10 days until tumors were palpable. Mice were then injected i.t. with NPs containing fLuc mRNA with or without adjuvant (2.5 μg mRNA per tumor). Mice were then imaged by IVIS Spectrum (Perkin Elmer, Waltham, MA) for whole-body bioluminescence at 12 h and 24 h. D-luciferin potassium salt solution (Cayman Chemical Company, Ann Arbor, MI) was injected subcutaneously at 150 mg/kg, and mice were imaged 7 min afterward. IVIS images were analyzed using Living Image software (Perkin Elmer, Waltham, MA). Blood serum samples were collected from the mice and analyzed for liver enzymes using the Alanine Aminotransferase Activity Assay and Aspartate Aminotransferase (AST) Activity Assay Kit (Sigma-Aldrich, St. Louis, MO). Tissues were then collected from the mice, dissociated, and analyzed using plate-reader based Promega Luciferase Assay System (Promega, Madison, WI).
For studies analyzing NP release from the copolymer, GFP mRNA was conjugated with Cy5 using Label IT Nucleic Acid Labeling Kit Cy5 (Mirus Bio, Marietta, GA) according to the manufacturer’s instructions. NPs were formulated with Cy5-mRNA at 30 w/w with 1% DMG-PEG2k, dialyzed, then adjusted for a final concentration of 5 μg mRNA/100 μL. NPs were then mixed 1:1 with copolymer as described, and 100 μL of the mixture was pipetted into a tube of 500 μL PBS at 37°C to form the gel (n=5 tubes), then stored in an incubator at 37°C. At each timepoint, the supernatant was removed and replaced with fresh PBS at 37°C. The fluorescence released into the supernatant was measured using a Biotek Synergy 2 plate reader (647 nm/670 nm excitation/emission).
In Vivo Antitumor Efficacy of tAPC Reprogramming Nanoparticles in E0771 and MC38 Tumor Models.
Female 6-to 8-week-old C57BL/6 mice were inoculated with 1 × 106 E0771 cells or 5 × 105 MC38 cells per mouse, respectively, as described above, and allowed to grow for 7-10 days until the tumor was palpable. Groups with similar tumor size were assigned before the start of treatment based on initial tumor measurements using calipers. The experimenter measuring tumor size over time was blinded to group assignments. For E0771, mice (n=8) were injected i.t. three times at two-day intervals with PBAE NPs, with 2.5 μg mRNA and 25 μL volume injected per tumor per dose. In some groups, mice were also injected i.p. with 200 μg anti-PD1 antibody (Table S1) in 100 μL volume on the first two treatment days. Groups included: fLuc NPs (control), fLuc+CpG NPs, 4-1BBL+IL-12 NPs, and 4-1BBL+IL-12+CpG NPs. Each NP type was tested with or without i.p. anti-PD1. For MC38 (n=7), the same procedure was followed with the study arms including fLuc NPs, 4-1BBL+IL12 NPS, and 4-1BBL+IL12+CpG NPs only. Tumor area was measured every 2-3 days, and mice were euthanized when tumor area (length × width) reached 200 mm2 or when tumors ulcerated and caused signs of distress to the mice. Survival was tracked based on the human euthanasia criteria.
Mice in the E0771 and MC38 study that cleared their primary tumor and survived the study were identified as long-term survivors. These survivors were re-challenged with a subcutaneous injection of 1 × 106 E0771 tumor cells on the opposite flank at day 92 or 5 × 105 MC38 tumor cells on the opposite flank at day 60. Naïve, age-matched female C57BL/6 control mice were inoculated with the same tumor injection on day 92 for E0771 tumors and day 60 for MC38 tumors. No additional NP nor anti-PD1 was administered to any mice in the re-challenge study. Throughout the studies, tumor size was measured and survival was tracked as described above.
For studies on immune cell recruitment and activation due to the adjuvant, mice were inoculated with E0771 tumors following the procedure above, and NPs and anti-PD1 were administered when tumors became palpable, with NPs encapsulating fLuc mRNA and CpG at two mRNA:adjvant ratios. Mice were euthanized 36 h following NP injection, and tumors were excised for ex vivo analysis. Tumor tissue was dissociated physically, enzymatically digested in 2 mg/mL Collagenase D from Clostridium histolyticum (Millipore Sigma, Burlington, MA) and incubated at 37°C for 1 h with shaking, then stained with the antibodies in Table S1. Tumor cells were then analyzed on an Attune NxT Flow Cytometer (ThermoFisher Scientific, Carlsbad, CA).
For ex vivo immune cell characterization following treatment with reprogramming mRNA/adjuvant NPs, E0771 tumors were treated with NPs encapsulating fLuc mRNA, fLuc+CpG, 4-1BBL+IL12, 4-1BBL+IL12+CpG and anti-PD1 as described above. An additional 4-1BBL+IL12 DNA (4-1BBL (pUNO1 backbone) and pIL-12elasti-N1) group formulated with 5-3-49 PBAE at 30 w/w is added to compare results to previous work; details of NP formulation are provided in Tzeng et al. (18). Lastly, an additional control NP group of 4-1BBL+IL12+CpG was added without the thermoresponsive gel. Tumors were excised three days after after the last treatment and dissociated by mincing and incubating with 2 mg/mL collagenase D and in 1×PBS for 1 h. Red blood cell lysis was carried out using ACK buffer and samples were filtered and staining for analysis via flow cytometry using the antibodies in Table S1. Additionally, tumor samples were lysed and analyzed using the BioLegend LEGENDplex Mouse Inflammation Panel (13-plex) kit (BioLegend, San Diego, CA). Blood serum samples were collected from the mice and analyzed for liver enzymes using the Alanine Aminotransferase Activity Assay and Aspartate Aminotransferase (AST) Activity Assay Kit (Sigma-Aldrich, St. Louis, MO).
Statistical Analysis.
Statistical analysis was performed using GraphPad Prism 9 (GraphPad, La Jolla, CA). Data are expressed as mean ± SEM with a minimum n (biological replicates) = 3-5 wells or mice per time point and per treatment group. Specific numbers for each experiment are listed in the figure captions. Statistical comparisons among experimental groups were performed using one- or two-way ANOVA with a Tukey multiple comparisons test or an unpaired two-tailed t-test, unless indicated otherwise in the figure caption and comparisons were considered significant with p < 0.05 indicated as *: p < 0.05, **: p < 0.001, ***: p < 0.0001, and ns: not significant. In all animal studies, groups were assigned to minimize initial differences among groups, and subjective measurements were conducted by a blinded experimenter.
Results
PBAE/mRNA nanoparticles transfect multiple cancer cell lines in vitro
A PBAE structure called 7-90,c12-63 80% (19), recently described to be safe and effective at transfecting cells in vivo after intravenous (i.v.) administration, was synthesized using Michael addition (Fig. S1A). The polymer structure was confirmed by NMR (Fig. S1A). The synthesized polymer was characterized using GPC to reveal a molecular weight of 5,937 Da (number-weighted average (Mn)) and polydispersity of 2.051 (Fig. S1B). Nanoparticles (NPs) formed using this structure were also imaged with transmission electron microcopy (TEM) to determine their dry size, which was approximately 100-150 nm in diameter (Fig. S1C). To demonstrate one advantage of the 7-90,c12-63 80% PBAE, a head-to-head transfection against gold-standard Lipofectamine MessengerMAX was carried out. Results show statistically significantly higher geometric mean intensity of GFP in 4T1 breast cancer and E0771 breast cancer, with similar intensity in MC38 colon adenocarcinoma cells (Fig. S1D). 4T1, E0771, and MC38 cells were transfected with NPs encapsulating mRNA encoding reporter gene GFP, and images of transfected cells are shown demonstrating GFP expression (Fig. 1A). IL-12 mRNA was titrated in the NP formulation and exhibited a dose response with the amount of exogenously expressed and secreted IL-12 in the cell media at 24 h post-transfection (Fig. 1B). Flow cytometry results showed dose dependence of exogenous 4-1BBL expression for the 4T1 cell line, but similar 4-1BBL expression on E0771 and MC38 cells across 4-1BBL mRNA doses (Fig. 1C).
Figure 1. PBAE mRNA nanoparticles transfect three different tumor cell lines in vitro.
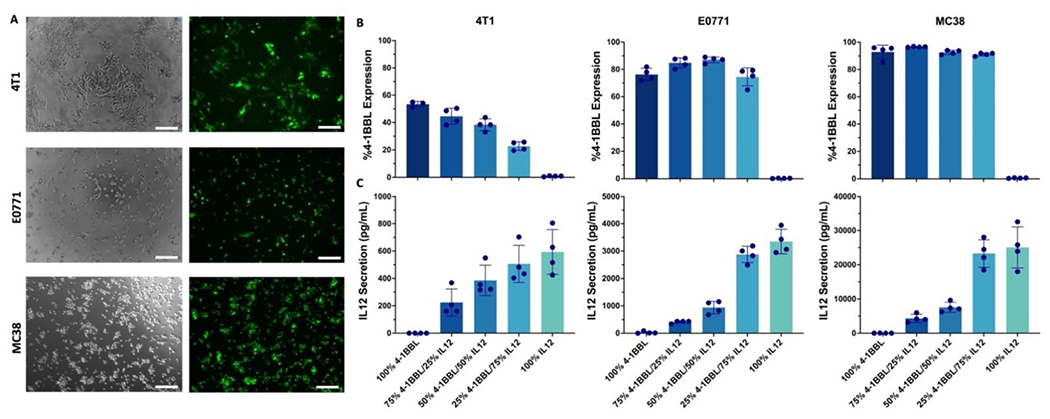
(A) Representative images of 4T1, E0771, and MC38 cells 24 h after transfection with 7-90,c12-63 80% NPs at 30 w/w. Brightfield and GFP filters are shown, scale bar: 200 nm). (B) Flow cytometry was used to measure surface presentation of 4-1BBL after transfecting 4T1, E0771, and MC38 with mRNA NPs with varying mRNA ratios (n=4 wells per group). (C) Sandwich ELISA was used to measure secretion of IL-12 after transfecting 4T1, E0771, and MC38 with mRNA NPs with varying mRNA ratios (n=4 wells per group). Mean ± SE is shown for all graphs.
Nanoparticles co-injected with in situ-gelling copolymer transfect tumors specifically at the injection site
The thermoresponsive triblock copolymer used in the in vivo experiments were made by blending AK091 and AK012 PLGA-PEG-PLGA structures as described in Mohammadi et al. (46) to form a mixture that remained liquid and injectable at room temperature and formed a gel at physiological temperature (37°C) (Fig. 2A). Images of the final copolymer mixture are shown in a vial at 4°C in liquid phase versus gel phase at 37°C (Fig. 2B). NPs used for in vivo studies were formulated with an additional 1 mass% 1,2-dimyristoyl-rac-glycero-3-methoxypolyethylene glycol-2000 (DMG-PEG2k) (percentage of polymer mass) and dialyzed against isotonic PBS to improve particle stability. For long-term storage, particles were frozen at −80°C until use, and dynamic light scattering (DLS) performed before and after freeze-thaw revealed that NP diameter remained stable for the PEG-containing formulations (332 +/− 6 nm [mean±SEM] before freezing and 320 +/− 10 nm after thawing) with p-value 0.3765 (n.s., not significant) via an unpaired two-tailed t-test, but those without PEG aggregated while freezing, with diameters measuring 2100 +/− 200 nm after thawing (Fig. S1C).
Figure 2. Block co-polymer thermoresponsive gel demonstrates sequestered expression and release in vivo and in vitro.

(A) Block co-polymer structure shown (PLGA-PEG-PLGA LG 50:50 (w:w)). (B) The commercially available block co-polymers are engineered to be liquid and injectable at 4C but form a gel-like hydrogel network at body temperature (37°C). (C) Lead formulation PBAE fLuc mRNA NPs (30 w/w 1m% DMG-PEG2k 2.5 μg/tumor) combined with or without the block co-polymer is injected (single) 4T1 intratumorally and imaged by IVIS after 12 and 24 h to show biodistribution of expression (n=5 mice per group). (D) In vitro degradation and release of 1:1 block co-polymer and PBAE Cy5 labeled GFP mRNA nanoparticle formulated with 1 mass% DMG-PEG2k (n=5). Mean ± SE is shown.
For in vivo use, firefly luciferase (fLuc) mRNA NPs were complexed with PBAE at 30 or 60 w/w with 0, 1, or 5 mass percent of DMG-PEG2k included. NPs were injected i.t., and mice were imaged after 12 h by In Vivo Imaging System (IVIS) to identify a formulation with the highest expression at the tumor site and minimal expression at off-target sites like the spleen and liver (Fig. S2A). A separate experiment with more replicates was repeated comparing fLuc mRNA NP injections with and without scaffold using a highly sensitive ex vivo analysis kit (Fig. S2B). 4T1-bearing Balb/c mice were used for all in vivo imaging studies examining on-target gene delivery due to their lack of pigment, allowing more accurate detection of bioluminescence. The luciferase expression of NPs fabricated at 30w/w and 1% DMG-PEG2k was high and consistent in the tumor with minimal off-target expression.
Using the optimized formulation, NPs formulated with fLuc mRNA at 30 w/w with 1% DMG-PEG2k and a final dose of 2.5 μg mRNA per injection were injected i.t. into orthotopic 4T1 tumors with or without the copolymer gel solution (Fig. 2C). At 12 h and 24 h, IVIS images were taken to show that fLuc expression was confined the tumor site when NPs were co-injected with the copolymer gel, whereas those injected without copolymer gel revealed off-target expression in the liver and spleen. To demonstrate release of NPs from the copolymer gel, PEGylated NPs with Cy5-labeled mRNA were co-formulated with the copolymer and pipetted into tubes with warm PBS and incubated at 37°C. The PBS surrounding the gel was measured at each timepoint and showed sustained release of NPs out of the gel over 24 days before the copolymer gel fully degraded (Fig. 2D).
Adjuvant-containing nanoparticles remain effective at transfection while heightening the immune response at the injection site
Adjuvants were added to the NPs to boost the immune response by stimulating TLR9, TLR3, or STING pathways to produce pro-inflammatory cytokines. Because the adjuvants used here are nucleic acid-based, they were added to the NPs by simple mixing with mRNA prior to complexation with the PBAE. To ensure that the addition of the antigen would not negatively affect the formulation, the size, charge, transfection efficacy, and toxicity of the new NPs was assessed in vitro and assessed expression in vivo. NPs were formulated at 30 w/w and varying mass ratios of GFP mRNA to adjuvant (1:1, 3:1, 10:1) using three different adjuvants: CpG (TLR9 agonist), CDN (STING agonist), and poly(I:C) (TLR3 agonist). These NPs were used to transfect E0771 and MC38 cells. CpG 3:1, CpG 101:1 and CDN 10:1 had the least effect on GFP expression in E0771 cells measured by the percent of transfected cells (Fig. 3A), and when using normalized geometric mean intensity of the GFP expression, the CpG 3:1 and CDN 10:1 groups were not significantly different compared to the control group with no adjuvant. Lastly, CpG 10:1 and CDN 10:1 did not cause increased toxicity compared to the no-adjuvant control. In MC38 cells, CpG 3:1, CpG 10:1, CDN 3:1, CDN 10:1 and poly(I:C) 10:1 performed statistically similarly to the no-adjuvant control, measured by percent of GFP expression (Fig. 3B). Measured by GFP intensity, the CpG 3:1, CpG 10:1 CDN 3:1, CDN 10:1 and poly(I:C) 10:1 groups showed the greatest transfection. CpG 3:1, CpG 10:1: CDN 3:1, and poly(1:C) 10:1 all also did not cause additional toxicity to MC38 cells compared to minimal toxicity shown in no-adjuvant control NPs. Top adjuvant-containing formulations were then assessed in vivo by IVIS using fLuc mRNA NPs co-formulated with copolymer (Fig. 3C). When injected intratumorally into 4T1 tumors, CpG 3:1 induced luciferase expression in 4 out of 5 mice with minimal to no off-target expression at the liver and the spleen. When compared to fLuc mRNA NPs without adjuvant, other groups, such as CDN 10:1, showed consistently high expression at the tumor but resulted in expression at the liver/spleen, while others, such as CpG 10:1, CDN 3:1, and poly(I:C) 30:1, did not express consistently at the tumor via IVIS. To analyze the fLuc expression in the tumor with higher sensitivity, we repeated the experiment with more replicates and excised the tumors for luminescence measurement, comparing fLuc mRNA on its own or fLuc mRNA co-delivered with CpG at 3:1 or 10:1 ratios of mRNA:CpG (Fig. 3D). Statistical analysis reveals no significant differences among the groups, though there is a clear trend toward lower mRNA expression in the groups co-delivering CpG. The slight decrease in expression after addition of adjuvant molecules may be due to innate activation of the transfected cells (47–49). From these studies, CpG 3:1 was selected as a formulation that did not negatively affect in vitro cell viability or transfection and had minimal effect on in vivo mRNA expression.
Figure 3. Formulation optimization with various GFP mRNA:adjuvant ratios in vitro and fLuc mRNA:adjuvant ratios in vivo.
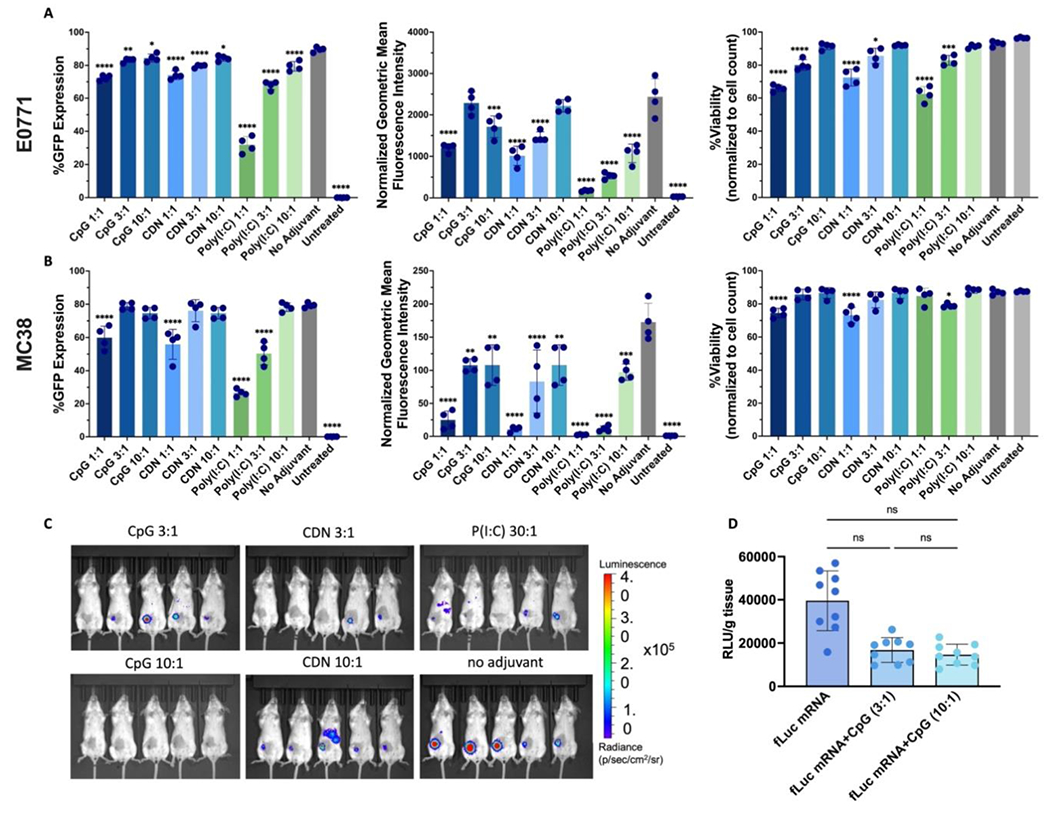
Percent GFP expression, normalized geometric mean of expression, and viability of NP transfection (30 w/w, 150ng/well) in (A) E0771 and (B) MC38 cells. (C) IVIS imaging at 12 h of PBAE fLuc mRNA NPs (30 w/w 1m% PEG 2.5 μg/tumor) + block co-polymer with varying adjuvants and mRNA:adjuvant ratios 4T1 tumors (n=5 mice per group). (D) Relative luminescence units (RLU)/g tissue readings of tumors ex vivo analysis comparing gel-embedded NPs carrying mRNA only or mRNA along with CpG at two ratios (n=9 mice per group). For A-B, statistically significant differences were measured by one-way ANOVA with Dunnett post-tests comparing to the control no adjuvant nanoparticle group. For D, statistically significant differences were measured by one-way ANOVA with Tukey’s post-tests comparing all groups to each other. All bar graphs show mean ± SE. Four (n=4) replicates were used for each formulation group. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Adjuvant- and mRNA-containing NPs formulated for in vivo usage with 1% DMG-PEG2k and purified by dialysis were then fully characterized in vivo. NPs that were either PEGylated and dialyzed or dialyzed only were fabricated and used to transfect E0771 and MC38 cells. Though the dialysis process resulted in lower in vitro transfection levels, PEGylated particles performed statistically better for transfection compared to non-PEGylated particles using a t-test and Dunnett’s multiple comparisons test, and viability remained high across all formulation groups (Fig. S3A-F). The particles were also analyzed via DLS, and those with adjuvant were found to maintain similar size and charge to those without adjuvant, with diameters within 9.5% and positive zeta potential within 15% of the no-adjuvant control particles (Supp Fig. 4). To ensure the PBAE NPs efficiently encapsulate mRNA and CpG, NPs were formulated with labeled nucleic acid cargo and incubated in PBS at 37°C (Fig. 3G). Following this, NPs were mixed with heparin (5 mg/mL final concentration) to disrupt the NP and release the components. Gel electrophoresis showed that both mRNA and CpG remained stably bound in the NPs for at least 24 h at physiological temperature in the absence of cells, and both components were shown to be intact after dissociation.
To assess the effect of adjuvants in vivo, E0771 orthotopic tumors were injected with PEGylated fLuc mRNA NPs with either no adjuvant or 10:1 or 3:1 CpG, co-formulated with the thermoresponsive copolymer. After 36 h, tumors were excised and processed for flow cytometry analysis using antibody markers to identify immune cell populations (Fig. S5). Notably, 3:1 mRNA:CpG NPs statistically significantly increased the infiltration of CD45+ immune cells in the tumor compared to fLuc mRNA NPs with no adjuvant and 10:1 CpG NPs. Tumors treated with 3:1 CpG exhibited increased neutrophil infiltration compared to those treated with 10:1 CpG, but this difference was not significantly different from those seen in mice treated with no-adjuvant control particles. Trends also suggested a small, statistically insignificant increase in dendritic cells found in the tumors treated with 3:1 CpG compared to the other two groups. Furthermore, T cells and macrophages also increased significantly in the 3:1 CpG group compared to the no-adjuvant control and 10:1 CpG.
To investigate mechanisms and immune stimulation by NPs carrying both adjuvant and immunostimulatory mRNA sequences, primary splenocytes isolated from mice with E0771 or MC38 tumor cells were co-cultured, and then the co-culture was transfected with NPs. The NP groups included fLuc mRNA NPs as a control, 50% 4-1BBL+50% fLuc mRNA NPs, 50% IL-12+50% fLuc mRNA NPs, 50% 4-1BBL+50% IL12 mRNA NPs, as well as each of those groups with 3:1 CpG. Cytokine secretion into the media was measured via IFNγ ELISA (Fig. 6) and multiplex bead-based flow cytometry (Fig. 4, Table S2) after 3 days as an indicator of immune activation and inflammation. The E0771 results demonstrate increased IFNγ secretion in groups treated with 4-1BBL+IL12 and 4-1BBL+IL12+CpG NPs, which are not significantly different from each other in vitro (Fig. 4, Fig. S6). After measuring other cytokines involved in inflammation and immune activation, it was found that the group treated with 4-1BBL+IL12+CpG NPs compared to 4-1BBL+IL12 NPs showed the highest level of TNF-α, whereas IL-27 and IFN-β secretion remained similar between the two groups. In MC38 cells, high IFNγ secretion is seen when co-cultured cells are treated with 4-1BBL+IL12 mRNA NPs and fLuc+IL12 +/−CpG mRNA NPs, showing the highest level of IFNγ secretion, with the former being significantly higher with CpG compared to without. TNF-α and IL-27 secretion, but not IFN-β, was higher in MC38 cells when treated with 4-1BBL+IL12+CpG NPs than when treated with 4-1BBL+IL12 NPs without adjuvant. Taken together, these results show that not only do 4-1BBL and IL-12 act synergistically, but also the two mRNAs act synergistically with CpG in this in vitro setting.
Figure 6. Ex vivo analysis of therapeutic study for immune analysis in E0771 tumors treated with mRNA NPs and anti-PD-1.

(A) Serum analysis collected 72 h after NP injections analyzed using bead-based LEGENDplex immunoassay for IFN-γ, IL-1α, TNF-α, and IL-6. (B) Flow cytometry analysis of CD45+ immune cells, CD3+ T cells, FoxP3+ Tregs, CD8+ T cells, CD62L, and CD44 for CD8+ T cell phenotype as % of tumor. All formulations are injected in tumors with copolymer gel unless noted as “no gel.” Significant differences in the growth rate were measured by one-way ANOVA with Tukey’s post-test. All graphs show mean ± SE. Five (n=5 or 4) replicates were used per group. *=p<0.05, **=p<0.01, ***=p<0.001, ****=p<0.0001.
Figure 4. Cytokine secretion on d3 after co-culture of tumor cells and splenocytes/immune cells in vitro after NP administration.

Supernatant from E0771 and MC38 cells co-cultured with splenocytes analyzed using bead-based LEGENDplex immunoassay for IFN-γ, TNF-α, IL-27, and IFN-β. Statistically significant differences were measured by one-way ANOVA with Tukey’s post-tests comparing each group to each other. Only comparisons between 4-1BBL+IL-12 NPs and 4-1BBL+IL-12+CpG are shown here, full report can be found in Table S2. All bar graphs show mean ± SE. Four (n =4) well replicates were used per group. *=p<0.05, **=p<0.01, ***=p<0.001, ****=p<0.0001
Nanoparticles co-delivering mRNA and adjuvant have anti-tumor efficacy in multiple mouse models of cancer
To assess the capacity of the NP treatment to reduce tumor burden, E0771 tumor cells were inoculated subcutaneously in the right mammary fat pad of female C57BL/6 mice and treated with NPs and/or anti-PD1 (Fig. 5A). To demonstrate safety and low toxicity of the NPs, serum was collected 72 h after the last injection and analyzed for alanine transaminase (ALT) and aspartate transaminase (AST), which showed minimal and non-significant changes compared to untreated mice (Fig. S7). This suggests that these treatments are safe and do not cause excessive systemic toxicity, measured by activity of liver enzymes in circulation. Tumor area measurements show steady growth, with differences between groups beginning on day 14. The groups that showed sustained tumor growth statistically significant compared to our complete group, 4-1BBL+IL12+CpG NPs with anti-PD1, are mice left untreated or treated with fLuc NPs or 4-1BBL+IL12+CpG NPs. Groups with tumor size reduction are fLuc NPs with anti-PD-1, fLuc+CpG NPs with anti-PD-1, 4-1BBL+IL12 NPs with anti-PD1, and 4-1BBL+IL12+CpG NPs with anti-PD1 (Fig. 5B).
Figure 5. Intratumoral injection of tAPC reprogramming NPs elicits a strong and long-term anti-tumor effect.

(A) Schematic of NP and anti-PD-1 (aPD1) dosing regimen. Tumor area measurements for (B) E0771 and (D) E0771 survivors rechallenge. Kaplan-Meier survival plots for (D) E0771 and (E) E0771 survivors rechallenge. For A, statistically significant differences in the growth rate were measured by two-way ANOVA with Dunnett’s post-test. For B and E, differences in survival were calculated by the Mantel-Cox log-rank test compared to controls Group 1 (fLuc NPs) and naïve, respectively, with Bonferroni p-value correction for multiple comparisons. All graphs show mean ± SE. Seven or eight (n=7-8) mice were used per group. *=p<0.05, **=p<0.01, ***=p<0.001, ****=p<0.0001
Interestingly, all groups with anti-PD1 showed some tumor reduction compared to the control group, but the group that improved survival the most (Fig. 5C) was the group that received 4-1BBL+IL12+CpG NPs and anti-PD1, with 5 out of 8 mice completely clearing their tumor and surviving long-term. Other groups with long-term survivors include the group treated with 4-1BBL+IL12 NPs and anti-PD1 (3 out of 7), fLuc+CpG NPs and anti-PD1 (3 out of 8), and 4-1BBL+IL12+CpG NPs (1 out of 8). These 12 survivors were then re-challenged with E0771 tumor cells injected on the left flank and compared to 8 naïve age-matched mice inoculated with the same E0771 tumor cells. None of the survivor mice formed a tumor in response to the re-challenge. Interestingly, this includes mice that were treated with our NPs and/or anti-PD1 as well as those treated with a control condition, indicating that simple exposure to the tumor may be sufficient to provide this long-lasting and distant immune response as long as the initial tumor was able to be cleared by the immune system; however, our NP treatment in combination with anti-PD1 did significantly increase the likelihood of surviving long enough to be re-challenged.
To analyze the immune response to the treatment, E0771 tumors were again treated with fLuc NPs, fLuc+CpG NPs, 4-1BBL+IL12 NPs, and 4-1BBL+IL12+CpG NPs, all also treated i.p. with anti-PD1. Ex vivo analysis of tumors was conducted 7 days after starting treatment via flow cytometry for cytokines and cell infiltration (Fig. 6). Tumor lysates and serum were analyzed for cytokines. As expected, high levels of cytokines indicating inflammation and immune activation were measured in the tumors in groups treated with particles, with significantly elevated levels of IFNγ, TNF-α, and IL-6 measured for the 4-1BBL+IL12+CpG NP group compared to controls, and significantly elevated IFNγ in the group treated with DNA NPs (Fig. 6A). Although the group treated with NPs without the thermostabilizing copolymer did show slightly elevated—though not significantly higher—cytokine levels in the tumor, this group was associated with significantly higher levels of IFNγ, IL-1α, TNF-α, and IL-6 in the serum (Fig. 6A), likely due to the increased off-target systemic transfection expected in this group. Flow cytometry analysis of digested tumor tissues revealed a trend of increased CD45+ immune cell infiltration and CD3+ T cells in the treatment groups compared to untreated, incomplete mRNA NPs with fLuc mRNA, 4-1BBL+IL12 DNA NPs with gel, and 4-1BBL+IL12+CpG NPs injected without gel (Fig. 6B). Further analysis showed significant increases in cytotoxic CD8+ T cell infiltration in tumors treated with 4-1BBL+IL12+CpG NPs injected with gel compared to without (Fig. 6B, Fig. S8). Phenotyping of the T cell populations within the tumor demonstrate some FoxP3+ Tregs and a high number of effector CD8+ T cells in the treated groups.
To demonstrate the broad applicability of the NPs, these results were replicated in MC38 colon adenocarcinoma tumors using a subset of the experimental groups to show proof of concept. Injection of the MC38 flank tumors with fLuc mRNA NPs co-formulated with copolymer showed expression of fLuc sequestered at the tumor site (Fig. 7A). To test anti-tumor efficacy, MC38 tumor-bearing mice were treated with fLuc mRNA NPs, 4-1BBL+IL12 mRNA NPs, and 4-1BBL+IL12+CpG mRNA NPs by i.t. injection. Mice treated with 4-1BBL+IL12 mRNA NPs showed significantly slower tumor growth compared to those treated with fLuc NPs and 4-1BBL+IL12 NPs (Fig. 7B). Survival was tracked, and 4/7 mice were found to survive long-term in the group treated with 4-1BBL+IL12+CpG mRNA NPs, while only one mouse survived in the 4-1BBL+IL12 mRNA NP groups. On the other hand, all the mice treated with fLuc mRNA NPs did not survive (Fig. 7C). The remaining five mice were rechallenged with MC38 tumor cells inoculated on the opposite flank at day 60. The four mice treated with 4-1BBL+IL12+CpG NPs and the one mouse treated with 4-1BBL+IL12 NPs also survived a re-challenge on the opposite flank and did not grow a second tumor.
Figure 7. Demonstrated proof-of-concept in an additional, non-breast cancer tumor model MC38.

(A) IVIS imaging of fLuc mRNA NPs + block co-polymer injected intratumorally in flank MC38 tumors at 12 h (n=7 mice). (B) Area of MC38 tumors following dose regimen of NPs + block co-polymer and (C) survival with rechallenge indicated with an orange arrow. Statistically significant differences in the growth rate were measured by two-way ANOVA with Dunnet’s post-test. Differences in survival were calculated by the Mantel-Cox log-rank test compared to control fLuc NPs group with Bonferroni correction for multiple comparisons. All graphs show mean ± SE. Seven (n=7) mice were used per group. *=p<0.05, **=p<0.01, ***=p<0.001, ****=p<0.0001
Discussion
Nanoparticle (NP) delivery of mRNA encoding immunologically relevant and stimulating molecules have the potential to address the low efficacy and detrimental side effects of immunotherapies currently in the clinic. Here, we describe the development of a non-viral mRNA NP formulation to deliver and co-express two immunostimulatory genes, signals 2 and 3 (4-1BBL and IL12), along with an adjuvant to reprogram tumor-associated cells to APC-like cells in situ. This treatment causes regression of murine E0771 breast cancer and MC38 colon adenocarcinoma solid tumor models in vivo and elicits a durable immune response, which prevents the formation of a second tumor re-challenge (Schematic 1). Given that our therapy is antigen-agnostic, it is crucial that the NPs do not travel to distant organs, such as the spleen, and cause off-target immune activation in healthy organs that could lead to detrimental effects. To localize the NP administration to the TME and limit off-target effects, our NPs were co-formulated with a thermoresponsive triblock copolymer that underwent gelation at 37°C and prevented particles from flowing away from the injection site. The strategy could be beneficial in the clinic for patients with readily accessible solid tumors, and the fact that long-term surviving mice resisted tumor formation distant from the initial tumor site suggests that local injection of NPs into a primary tumor could have widespread effects on other distant tumors.
This study furthered the work reported previously (18), demonstrating the ease with which nucleic acid cargos can be formulated into NPs in a tunable way by simple ratio mixing at defined ratios. The incorporation of a copolymer sequestered the transfection at the tumor microenvironment while allowing release of NPs over time (Fig. 2C-D), an important safety consideration for translational applications, and the addition of an adjuvant increased immune stimulation and demonstrated improved effect both in vitro and in vivo while limiting signs of systemic toxicity (Fig. S7) as a new local tumor immunotherapy modality. Incorporation of the thermoresponsive block copolymer in our formulations demonstrate sequestered expression in the tumor (Fig. 3), as well as undetectable levels of systemic inflammation (Fig. 6) compared to formulations without the copolymer, further supporting the safety of our antigen-agnostic NPs.
The co-culture of immune cells with transfected tumor cells demonstrated the importance of having both 4-1BBL and IL12 mRNA in the NP to induce immune activation by tAPCs across two cell lines, including the secretion of IFNγ (23–25), TNF-α (50), IL-27 (51), and IFN-β (52), which have been implicated in anti-tumor mechanisms and some of which have been used in the clinic (Fig. 4). Additionally, after E0771 tumors were treated i.t. with NPs, most of the mice treated with 4-1BBL+IL12+CpG NPs in addition to anti-PD1 survived long-term, demonstrating the immunostimulatory nature of this therapy (Fig. 5 A-C). Groups treated with 4-1BBL+IL12 NPs with anti-PD1 and fLuc+CpG NPs with anti-PD1 also performed significantly better than the controls, suggesting the importance of the use of dual therapeutic mRNA molecules and/or immunostimulatory adjuvants in combination with the type of systemic ICB currently used in the clinic. These survivors resisted tumor re-challenge at a distant site, suggesting that the immune response to our treatment was not only long-lasting but also systemic (Fig. 5D-E). To ensure this modality is effective in an additional tumor model, mice were inoculated with MC38 tumors and treated with the same NPs (Fig. 7B-C). Most (57%) of the mice treated with 4-1BBL+IL12+CpG NPs survived long-term, a greater percentage than that seen in mice treated with NPs containing only 4-1BBL and IL12 mRNA (14%), showing the importance of each therapeutic molecule in the formulation. Immune analysis of excised tumors revealed that the anti-tumor effects of our NPs were likely driven by cytotoxic CD8+ effector T cells, which were found in elevated numbers in the tumor, and showed increased local levels of interferons, TNF-α, and IL-6 (Fig. 6). When the thermoresponsive copolymer was used during injection, it also prevented high systemic levels of these inflammatory molecules.
The PBAE NP technology offers a “plug-and-play” platform with the option to exchange mRNA molecules and adjuvants. The high cargo capacity of polymeric NP formulations allows the encapsulation of multiple nucleic acid molecules, such as two mRNAs and an adjuvant, and efficient multiplexed delivery into the same cell (Fig. 1C). To activate T cells as a downstream result of transfecting tumor-associated cells, it is crucial for signals 1-3 to all be present for coordinated interaction with the T cell. The ability to adjust the identity and ratios of these molecules allows us to tune the expression levels of multiple genes after transfection (Fig. 1B-C, Fig. 3) and alter the effects of the NPs on the immune response in the TME in a significant and quantifiable way (Fig. S5). This flexibility in formulation greatly broadens the potential uses of this technology, particularly because of the ease with which these different NPs can be altered and manufactured. Related to this, it is also showed that NPs formulated with 1% DMG-PEG2k remained stable in size and charge after a freeze-thaw cycle at −80°C, allowing long-term storage and transfection efficiency (Fig. S2C, 9), also a critical parameter for a technology to be practically manufactured and implemented.
Polymeric PBAE NPs offer many advantages as a gene delivery modality over competing technologies like viral vectors and lipid nanoparticles (LNPs) (13, 15, 16). PBAEs have been a long-standing polymer in gene delivery, but recent work has continued to improve the biomaterial. Specifically, charge-altering releasable transporters (CARTs) (53–55) have been designed with oligo(α-amino ester) that exhibit a cationic charge at low pH maintaining the nanoparticle complex, such as in the endosome, but then undergo a charge-neutralizing rearrangement at physiological pH 7.4, such as in the cytosol. This results in a highly controlled self-immolative reaction, releasing the mRNA cargo and allowing safe neutral small molecule degradation. These CART-mRNA complexes have shown >90% transfection in vitro across various cell lines, including Hela, J774, HEK-293, and HepG2, with further improved transfection when formulated with lipophilic blocks. On the other hand, LNPs have gained considerable traction in recent years given the FDA-approved Moderna (NCT04470427) (56) and BioNTech/Pfizer (NCT04368728) (57) LNP-mRNA SARS-CoV-2 vaccines (13). These vaccine formulations have demonstrated safety and greater public in addition to regulatory acceptance of mRNA gene delivery, and this current work, which adapts a prior study by our group on DNA delivery to tumor cells, may bring this technology closer to translation by its use of mRNA in place of DNA.
Others have shown the use of these LNP complexes for immunostimulatory factor-encoding mRNA intratumoral gene delivery exhibiting anti-cancer effects (58). Viral vectors have also been utilized for intratumoral delivery (59) with some clinical trials specifically transferring the IL-12 gene (NCT00849459) (60). The genes 4-1BBL and IL-12 have been previously explored together in other work (61–63), and the authors found, as we did, that this combination of genes is effective at generating an anti-tumor immune response. However, previous studies have relied on viruses as gene delivery vectors (63), which are in general far more immunogenic than synthetic polymers like PBAEs, leading to the risk of toxicity and the inability to administer repeated doses if needed. Others have delivered modified gene products systemically, which was effective in a murine model (61, 62), but systemic delivery of soluble cytokines for cancer has been found repeatedly to have toxic side effects (30, 64, 65). Local, intratumoral delivery of immunotherapies such as cytokines and gene delivery vectors can offer a robust anti-tumor effect, minimizing systemic toxicity while influencing the immune system globally (66). Other groups have explored the local, intratumoral delivery of nucleic acids, specifically plasmid DNA, encoding the IL-12 cytokine (NCT00323206, NCT01502293, NCT02345330, NCT01579318, NCT02493361, NCT03132675, NCT01440816) (67–76). These studies have shown low to moderate anti-tumor effect with some anti-metastatic effect, but delivery of these formulations requires electroporation, which has been associated with disadvantages such as pain and cell and tissue damage (77, 78). PBAEs allows for high cargo capacities and safe redosing due to their minimal immunogenicity, critical for therapies that deliver various factors and require multiple injections. Fabrication and manufacture of the PBAE NPs can be scaled up, and their larger cargo capacity and flexibility in molecule encapsulation allows them to be used for a broad range of applications. With translational promise, the NPs described in this report leverage and stimulate the patient’s own immune system in situ at the local tumor microenvironment to attack the malignant cells and reduce solid tumor burden, leading to a patient- and tumor-specific treatment without the need for individual tumor antigen profiling.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (NIH) [P41EB028239 (NIBIB), R21AI160738 (NIAID), and R37CA246699 (NCI)]. S.Y.N (DGE-1746891) and S.R.S. (DGE-1746891) thank the National Science Foundation (NSF) for fellowship support. S.Y.N. thanks the ARCS Metro-Washington Chapter for fellowship support. The authors thank the Bloomberg~Kimmel Institute for Cancer Immunotherapy for their support. Figure graphics were created with BioRender.com.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing Interest Statement: J.J.G., S.Y.N., and S.Y.T. are inventors on patent applications related to this work filed by Johns Hopkins University. S.Y.T and J.J.G are co-founders and Managers of OncoSwitch Therapeutics. J.J.G. is a co-founder of WyveRNA Therapeutics. Any potential conflicts of interest are managed by the Johns Hopkins University Committee on Outside Interests.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data Availability
The data presented are available upon request.
References
- 1.Kraehenbuehl L, Weng CH, Eghbali S, Wolchok JD, Merghoub T, Enhancing immunotherapy in cancer by targeting emerging immunomodulatory pathways. Nat. Rev. Clin. Oncol. 2021 191 19, 37–50 (2021). [DOI] [PubMed] [Google Scholar]
- 2.Taefehshokr S, et al. , Cancer immunotherapy: Challenges and limitations. Pathol. - Res. Pract 229, 153723 (2022). [DOI] [PubMed] [Google Scholar]
- 3.Tang H, Qiao J, Fu YX, Immunotherapy and tumor microenvironment. Cancer Lett. 370, 85–90 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Raskov H, Orhan A, Christensen JP, Gögenur I, Cytotoxic CD8+ T cells in cancer and cancer immunotherapy. Br. J. Cancer 2020 1242 124, 359–367 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martin JD, Cabral H, Stylianopoulos T, Jain RK, Improving cancer immunotherapy using nanomedicines: progress, opportunities and challenges. Nat. Rev. Clin. Oncol. 2020 174 17, 251–266 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fesnak AD, June CH, Levine BL, Engineered T cells: the promise and challenges of cancer immunotherapy. Nat. Rev. Cancer 2016 169 16, 566–581 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rhodes KR, Green JJ, Nanoscale artificial antigen presenting cells for cancer immunotherapy. Mol. Immunol 98, 13–18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheung AS, Zhang DKY, Koshy ST, Mooney DJ, Scaffolds that mimic antigen-presenting cells enable ex vivo expansion of primary T cells. Nat. Biotechnol. 2018 362 36, 160–169 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blass E, Ott PA, Advances in the development of personalized neoantigen-based therapeutic cancer vaccines. Nat. Rev. Clin. Oncol. 2021 184 18, 215–229 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lang F, Schrörs B, Löwer M, Türeci Ö, Sahin U, Identification of neoantigens for individualized therapeutic cancer vaccines. Nat. Rev. Drug Discov. 2022 214 21, 261–282 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mitchell MJ, et al. , Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 2020 202 20, 101–124 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Neshat SY, Tzeng SY, Green JJ, Gene delivery for immunoengineering. Curr. Opin. Biotechnol 66, 1–10 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meyer RA, Neshat SY, Green JJ, Santos JL, Tuesca AD, Targeting strategies for mRNA delivery. Mater. Today Adv 14, 100240 (2022). [Google Scholar]
- 14.Yin H, Kauffman KJ, Anderson DG, Delivery technologies for genome editing. Nat. Rev. Drug Discov 16, 387–399 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Nie W, Chen J, Wang B, | Gao Xiang, Nonviral vector system for cancer immunogene therapy. MedComm – Biomater. Appl 1 (2022). [Google Scholar]
- 16.Kavanagh EW, Green JJ, Toward Gene Transfer Nanoparticles as Therapeutics. Adv. Healthc. Mater 11, 2102145 (2022). [DOI] [PubMed] [Google Scholar]
- 17.Grun MK, et al. , PEGylation of poly(amine-co-ester) polyplexes for tunable gene delivery. Biomaterials 272, 120780 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tzeng SY, et al. , In situ genetic engineering of tumors for long-lasting and systemic immunotherapy. Proc. Natl. Acad. Sci. U. S. A 117, 4043–4052 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rui Y, et al. , High-throughput and high-content bioassay enables tuning of polyester nanoparticles for cellular uptake, endosomal escape, and systemic in vivo delivery of mRNA. Sci. Adv 8, 2855 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bukczynskit J, Wen T, Ellefsen K, Gauldie J, Watts TH, Costimulatory ligand 4-1BBL (CD137L) as an efficient adjuvant for human antiviral cytotoxic T cell responses. Proc. Natl. Acad. Sci. U. S. A 101, 1291 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reithofer M, et al. , 4-1BB costimulation promotes bystander activation of human CD8 T cells. Eur. J. Immunol 51, 721–733 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Henry CJ, Ornelles DA, Mitchell LM, Brzoza-Lewis KL, Hiltbold EM, IL-12 Produced by Dendritic Cells Augments CD8+ T cell Activation through the Production of the Chemokines CCL1 and CCL17. J. Immunol 181, 8576 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang Y, Xiang Z, Ertl HCJ, Wilson JM, Upregulation of class I major histocompatibility complex antigens by interferon gamma is necessary for T-cell-mediated elimination of recombinant adenovirus-infected hepatocytes in vivo. Proc. Natl. Acad. Sci 92, 7257–7261 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Früh K, Yang Y, Antigen presentation by MHC class I and its regulation by interferon γ. Curr. Opin. Immunol 11, 76–81 (1999). [DOI] [PubMed] [Google Scholar]
- 25.Jorgovanovic D, Song M, Wang L, Zhang Y, Roles of IFN-γ in tumor progression and regression: a review. Biomark. Res. 2020 81 8, 1–16 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nguyen KG, et al. , Localized Interleukin-12 for Cancer Immunotherapy. Front. Immunol 11, 2510 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Younes A, et al. , Phase II clinical trial of interleukin-12 in patients with relapsed and refractory non-Hodgkin’s lymphoma and Hodgkin’s disease. Clin. Cancer Res 10, 5432–5438 (2004). [DOI] [PubMed] [Google Scholar]
- 28.Little RF, et al. , Activity of subcutaneous interleukin-12 in AIDS-related Kaposi sarcoma. Blood 107, 4650–4657 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bajetta E, et al. , Pilot study of subcutaneous recombinant human interleukin 12 in metastatic melanoma. Clin. cancer Res. an Off. J. Am. Assoc. Cancer Res 4, 75–85 (1998). [PubMed] [Google Scholar]
- 30.Leonard JP, et al. , Effects of single-dose interleukin-12 exposure on interleukin-12-associated toxicity and interferon-gamma production. Blood 90, 2541–2548 (1997). [PubMed] [Google Scholar]
- 31.Lasek W, Zagożdżon R, Jakobisiak M, Interleukin 12: still a promising candidate for tumor immunotherapy? Cancer Immunol. Immunother 63, 419–435 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Braunstein MJ, Kucharczyk J, Adams S, Targeting Toll-Like Receptors for Cancer Therapy. Target. Oncol. 2018 135 13, 583–598 (2018). [DOI] [PubMed] [Google Scholar]
- 33.Luchner M, Reinke S, Milicic A, TLR Agonists as Vaccine Adjuvants Targeting Cancer and Infectious Diseases. Pharm. 2021, Vol. 13, Page 142 13, 142 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim H, et al. , Polymeric nanoparticles encapsulating novel TLR7/8 agonists as immunostimulatory adjuvants for enhanced cancer immunotherapy. Biomaterials 164, 38–53 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Jahrsdörfer B, Weiner GJ, CpG oligodeoxynucleotides as immunotherapy in cancer. Update Cancer Ther. 3, 27–32 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fu J, et al. , Cationic polymers for enhancing CpG oligodeoxynucleotides-mediated cancer immunotherapy. Eur. Polym. J 113, 115–132 (2019). [Google Scholar]
- 37.Jin Y, Zhuang Y, Dong X, Liu M, Development of CpG oligodeoxynucleotide TLR9 agonists in anti-cancer therapy. 10.1080/14737140.2021.1915136 21, 841–851 (2021). [DOI] [PubMed] [Google Scholar]
- 38.Zheng H, et al. , Polymersome-mediated cytosolic delivery of cyclic dinucleotide STING agonist enhances tumor immunotherapy. Bioact. Mater 16, 1–11 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leach DG, et al. , STINGel: Controlled release of a cyclic dinucleotide for enhanced cancer immunotherapy. Biomaterials 163, 67–75 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gogoi H, Mansouri S, Jin L, The Age of Cyclic Dinucleotide Vaccine Adjuvants. Vaccines 2020, Vol. 8, Page 453 8, 453 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wilson DR, et al. , Biodegradable STING agonist nanoparticles for enhanced cancer immunotherapy. Nanomedicine Nanotechnology, Biol. Med 14, 237–246 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sultan H, et al. , Poly-IC enhances the effectiveness of cancer immunotherapy by promoting T cell tumor infiltration. J. Immunother. Cancer 8, e001224 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Di S, et al. , Combined adjuvant of poly I:C improves antitumor effects of CAR-T cells. Front. Oncol 9, 241 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fang L, et al. , Light-controllable charge-reversal nanoparticles with polyinosinic-polycytidylic acid for enhancing immunotherapy of triple negative breast cancer. Acta Pharm. Sin. B 12, 353–363 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cheng YS, Xu F, Anticancer function of polyinosinic-polycytidylic acid. Cancer Biol. Ther 10, 1219–1223 (2010). [DOI] [PubMed] [Google Scholar]
- 46.Mohammadi M, et al. , Injectable drug depot engineered to release multiple ophthalmic therapeutic agents with precise time profiles for postoperative treatment following ocular surgery. Acta Biomater. 73, 90–102 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mauger DM, et al. , mRNA structure regulates protein expression through changes in functional half-life. Proc. Natl. Acad. Sci 116, 24075–24083 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li B, Luo X, Dong Y, Effects of Chemically Modified Messenger RNA on Protein Expression. Bioconjug. Chem 27, 849–853 (2016). [DOI] [PubMed] [Google Scholar]
- 49.Moradian H, Roch T, Anthofer L, Lendlein A, Gossen M, Chemical modification of uridine modulates mRNA-mediated proinflammatory and antiviral response in primary human macrophages. Mol. Ther. - Nucleic Acids 27, 854–869 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Freeman AJ, Kearney CJ, Silke J, Oliaro J, Unleashing TNF cytotoxicity to enhance cancer immunotherapy. Trends Immunol. 42, 1128–1142 (2021). [DOI] [PubMed] [Google Scholar]
- 51.Liu Z, Yu J, Carson WE 3rd, Bai X-F, The role of IL-27 in the induction of anti-tumor cytotoxic T lymphocyte response. Am. J. Transl. Res 5, 470–480 (2013). [PMC free article] [PubMed] [Google Scholar]
- 52.V Medrano RF, Hunger A, Mendonça SA, Barbuto JAM, Strauss BE, Immunomodulatory and antitumor effects of type I interferons and their application in cancer therapy. Oncotarget 8, 71249–71284 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McKinlay CJ, et al. , Charge-altering releasable transporters (CARTs) for the delivery and release of mRNA in living animals. Proc. Natl. Acad. Sci. U. S. A 114, E448–E456 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McKinlay CJ, Benner NL, Haabeth OA, Waymouth RM, Wender PA, Enhanced mRNA delivery into lymphocytes enabled by lipid-varied libraries of charge-altering releasable transporters. Proc. Natl. Acad. Sci. U. S. A 115, E5859–E5866 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Haabeth OAW, et al. , mRNA vaccination with charge-altering releasable transporters elicits human T cell responses and cures established tumors in mice. Proc. Natl. Acad. Sci. U. S. A 115, E9153–E9161 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.A Study to Evaluate Efficacy, Safety, and Immunogenicity of mRNA-1273 Vaccine in Adults Aged 18 Years and Older to Prevent COVID-19 - Full Text View - ClinicalTrials.gov (October 26, 2022).
- 57.Study to Describe the Safety, Tolerability, Immunogenicity, and Efficacy of RNA Vaccine Candidates Against COVID-19 in Healthy Individuals - Full Text View - ClinicalTrials.gov (October 26, 2022).
- 58.Hewitt SL, et al. , Durable anticancer immunity from intratumoral administration of IL-23, IL-36γ, and OX40L mRNAs. Sci. Transl. Med 11 (2019). [DOI] [PubMed] [Google Scholar]
- 59.Walther W, et al. , Novel jet-injection technology for nonviral intratumoral gene transfer in patients with melanoma and breast cancer. Clin. Cancer Res 14, 7545–7553 (2008). [DOI] [PubMed] [Google Scholar]
- 60.Gene Therapy in Treating Women With Metastatic Breast Cancer - Full Text View - ClinicalTrials.gov (October 26, 2022).
- 61.Xu D-P, et al. , The systemic administration of Ig-4-1BB ligand in combination with IL-12 gene transfer eradicates hepatic colon carcinoma. Gene Ther. 12, 1526–1533 (2005). [DOI] [PubMed] [Google Scholar]
- 62.Chen SH, et al. , Rejection of disseminated metastases of colon carcinoma by synergism of IL-12 gene therapy and 4-1BB costimulation. Mol. Ther 2, 39–46 (2000). [DOI] [PubMed] [Google Scholar]
- 63.Dowell AC, Oldham KA, Bhatt RI, Lee SP, Searle PF, Long-term proliferation of functional human NK cells, with conversion of CD56(dim) NK cells to a CD56 (bright) phenotype, induced by carcinoma cells co-expressing 4-1BBL and IL-12. Cancer Immunol. Immunother 61, 615–628 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Car BD, Eng VM, Lipman JM, Anderson TD, The toxicology of interleukin-12: a review. Toxicol. Pathol 27, 58–63 (1999). [DOI] [PubMed] [Google Scholar]
- 65.Atkins MB, et al. , Phase I evaluation of intravenous recombinant human interleukin 12 in patients with advanced malignancies. Clin. cancer Res. an Off. J. Am. Assoc. Cancer Res 3, 409–417 (1997). [PubMed] [Google Scholar]
- 66.Aznar MA, et al. , Intratumoral Delivery of Immunotherapy—Act Locally, Think Globally. J. Immunol 198, 31–39 (2017). [DOI] [PubMed] [Google Scholar]
- 67.Mahvi DM, et al. , Intratumoral injection of IL-12 plasmid DNA--results of a phase I/IB clinical trial. Cancer Gene Ther. 14, 717–723 (2007). [DOI] [PubMed] [Google Scholar]
- 68.Phase I Trial of Intratumoral pIL-12 Electroporation in Malignant Melanoma - Full Text View - ClinicalTrials.gov (October 26, 2022).
- 69.Algazi A, et al. , Intratumoral delivery of tavokinogene telseplasmid yields systemic immune responses in metastatic melanoma patients. Ann. Oncol 31,532–540 (2020). [DOI] [PubMed] [Google Scholar]
- 70.Trial of pIL-12 Electroporation in Squamous Cell Carcinoma of the Head and Neck (IL12HNSCC) - Full Text View - ClinicalTrials.gov (October 26, 2022).
- 71.Trial of pIL-12 Electroporation Malignant Melanoma - Full Text View - ClinicalTrials.gov (October 26, 2022).
- 72.Phase II Intratumoral IL12 Plasmid Electroporation in Cutaneous Lymphoma - Full Text View - ClinicalTrials.gov (October 26, 2022).
- 73.Jacobs L, et al. , Intratumoral DNA-based delivery of checkpoint-inhibiting antibodies and interleukin 12 triggers T cell infiltration and anti-tumor response. Cancer Gene Ther. 29, 984–992 (2022). [DOI] [PubMed] [Google Scholar]
- 74.Trial of pIL-12/MK-3475 in Metastatic Melanoma - Full Text View - ClinicalTrials.gov (October 26, 2022).
- 75.Tavo and Pembrolizumab in Patients With Stage III/IV Melanoma Progressing on Pembrolizumab or Nivolumab Treatment - Full Text View - ClinicalTrials.gov (October 26, 2022).
- 76.IL-12 Gene and in Vivo Electroporation-Mediated Plasmid DNA Vaccine Therapy in Patients With Merkel Cell Cancer - Full Text View - ClinicalTrials.gov (October 26, 2022).
- 77.Lambricht L, et al. , Clinical potential of electroporation for gene therapy and DNA vaccine delivery. Expert Opin. Drug Deliv 13, 295–310 (2016). [DOI] [PubMed] [Google Scholar]
- 78.Bodles-Brakhop AM, Heller R, Draghia-Akli R, Electroporation for the Delivery of DNA-based Vaccines and Immunotherapeutics: Current Clinical Developments. Mol. Ther 17, 585–592 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data presented are available upon request.