

Summary
Cancer alters the function of multiple organs beyond those targeted by metastasis1,2. Here, we show that inflammation, fatty liver, and dysregulated metabolism are hallmarks of systemically-affected livers in murine models and patients with extrahepatic metastasis. We identified tumor-derived extracellular vesicles and particles (EVPs) as crucial mediators of cancer-induced hepatic reprogramming, which could be reversed by reducing tumor EVP secretion through Rab27 loss. Importantly, all EVP subpopulations, exosomes and principally exomeres, could dysregulate hepatic function. Tumor EVP fatty acid cargo, particularly palmitic acid, induced tumor necrosis factor alpha (TNFα) secretion by Kupffer cells, generating a pro-inflammatory microenvironment, suppressing fatty acid metabolism and oxidative phosphorylation, and promoting fatty liver formation. Strikingly, Kupffer cell ablation or TNFα blockade markedly decreased tumor-induced fatty liver generation. Tumor implantation or pre-treatment with tumor EVPs diminished cytochrome P450 gene expression and attenuated drug metabolism in a TNFα-dependent manner. Importantly, fatty liver and decreased cytochrome P450 expression were also observed, at diagnosis, in tumor-free livers of pancreatic cancer patients who later developed extrahepatic metastasis, highlighting the clinical relevance of our findings. Notably, tumor EVP education enhanced chemotherapy side effects, including bone marrow suppression and cardiotoxicity, suggesting liver metabolic reprograming by tumor-derived EVPs may limit chemotherapy tolerance in cancer patients. Our results reveal how tumor-derived EVPs dysregulate hepatic function and their targetable potential, along with TNFα inhibition, in preventing fatty liver formation and enhancing chemotherapy efficacy.
Cancer is a systemic disease mediated by soluble factor release, including EVPs1,3. Tumor-derived EVPs not only establish pre-metastatic niches (PMNs), but also reprogram cell function in multiple metastasis-free organs3,4. Liver functional reprograming in the absence of metastasis during cancer progression has been overlooked. Importantly, we and others have identified the liver as one of the primary organs targeted by tumor-derived EVPs5–9, prompting us to investigate their roles in altering liver function. Here, by applying multi-omic analyses to multiple murine tumor models and liver tissues from cancer patients, we demonstrate that tumor EVP-packaged saturated fatty acids induce metabolic dysregulation, which, in turn, promotes fatty liver formation and diminishes the drug-metabolizing capacity of the liver.
Distant tumors alter liver metabolism
To investigate how cancer affects the liver, we orthotopically implanted murine B16F10 melanoma or K7M2 osteosarcoma, which preferentially metastasize to the lung10,11. At 3-4 weeks (wk), we observed no liver metastasis in either model, but detected lung metastases in the K7M2 model by histology (Extended Data Fig. 1a–c). Furthermore, melanoma-specific Trpm1 and mCherry were detected by qRT-PCR in the lungs but not livers of B16F10 and K7M2-mCherry tumor-bearing mice, respectively (Extended Data Fig. 1d), ruling out liver micrometastases.
Notably, RNA sequencing of metastasis-free livers of control and tumor-bearing mice revealed concordant transcriptional changes with 469 upregulated and 317 downregulated genes (q<0.05) in both models (Fig. 1a, Extended Data Fig. 1e). Gene set enrichment analysis (GSEA) revealed dysregulated immune homeostasis and metabolism, including links to inflammation and TNFα signaling via NF-κB and decreased oxidative phosphorylation (OXPHOS) and fatty acid metabolism (Fig. 1b).
Figure 1. Distant primary tumors induce metabolic dysfunction in the liver.
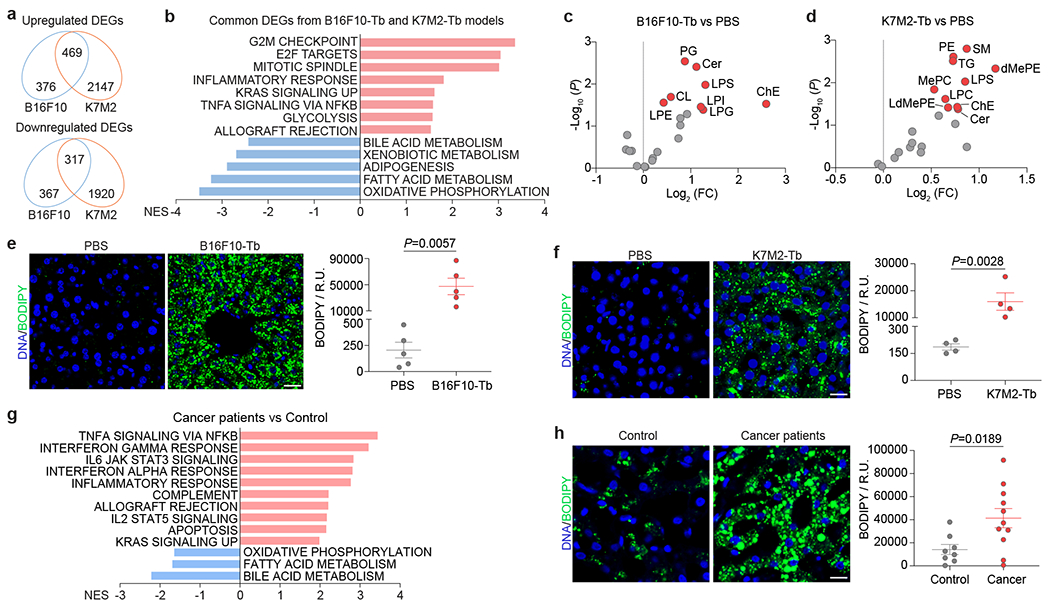
a, Venn diagram analysis of DEGs (q<0.05) in livers from B16F10-Tb (n=3) and K7M2-Tb mice (n=5), compared to their respective PBS-injected controls. b, GSEA of the common DEGs in (a) using hallmark gene sets, and the significantly changed signaling pathways with FDR<0.1 are shown. Gene lists for signaling pathways are shown in Supplementary Table 6. c,d, Volcano plots showing lipid classes (labeled in red, P<0.05) significantly enriched in the livers of B16F10-Tb (c) and K7M2-Tb (d) mice, compared to their respective PBS-injected controls. (n=5 each). e,f, Representative images (left) and associated statistical analysis (right) of BODIPY staining of the livers from B16F10-Tb (n=5) (e) and K7M2-Tb (n=4) (f) mice, and their respective PBS-injected controls. g. GSEA of the normalized gene expression values of tumor-free livers from PDAC patients (n=5) compared to control subjects (n=8) with benign lesions using hallmark gene sets. Downregulated signaling pathways with FDR<0.05 and top 10 upregulated signaling pathways with FDR<0.05 are shown. Gene lists for signaling pathways are shown in Supplementary Table 7. h, Representative images (left) and associated statistical analysis (right) of BODIPY staining of the livers from PDAC patients (n=11) and control subjects (n=8) with benign lesions. Scale bars, 20 μm. P values were determined by the two-tailed, unpaired Student’s t-test (c-f,h). Data are mean ± s.e.m. Tb, tumor-bearing. NES, normalized enrichment score. FC, fold change. R.U., relative unit.
Additional orthotopic low-metastatic melanoma B16F1 and non-metastatic (67NR) and highly-metastatic (lung-tropic 4T1)12 breast cancer, strikingly showed similar alterations, i.e., immune response and metabolism-associated pathways, in metastasis-free livers (Extended Data Fig. 1a,b,e–h), indicating multiple cancer types systemically and uniformly reprogram liver function.
To evaluate liver metabolic dysfunction, we performed metabolomic mass spectrometry (MS) (Supplementary Tables 1 and 2) and partial least squares-discriminant analysis (PLS-DA) which showed segregated metabolite profiles with amino acids, carbohydrates, cofactors and vitamins, citric acid cycle, lipids, and nucleotides significantly elevated in metastasis-free livers of B16F10 and K7M2 tumor-bearing mice compared to their respective controls (Extended Data Fig. 2a–d). Furthermore, metabolite set enrichment analysis identified lipid- and amino acid-associated metabolite sets, including phosphatidylethanolamine biosynthesis, phosphatidylcholine biosynthesis, glycine and serine metabolism, urea cycle, and arginine and proline metabolism pathways, suggesting metabolic rewiring of metastasis-free livers in both models (Extended Data Fig. 2e, f).
As lipid-associated metabolic pathways were impacted, we conducted lipidomics MS (Supplementary Tables 3, 4). PLS-DA revealed distinct liver lipid profiles in B16F10 and K7M2 tumor-bearing mice relative to their respective controls (Extended Data Fig. 3a,b). Multiple lipid classes, including phospholipids (cardiolipin [CL], lysophosphatidylethanolamine [LPE], lysophosphatidylglycerol [LPG], lysophosphatidylinositol [LPI], lysophosphatidylserine [LPS], and phosphatidylglycerol [PG] for B16F10; lysophosphatidylcholine [LPC], LPS, and phosphatidylethanolamine [PE] for K7M2), sphingolipids (ceramide [Cer] for B16F10; Cer and sphingomyelin [SM] for K7M2), and neutral lipids (cholesterol ester [ChE] for B16F10; ChE and triglyceride [TG] for K7M2), as well as multiple ChE and TG species (for B16F10 and K7M2) were significantly elevated in the livers of tumor-bearing mice (Fig. 1c,d, Extended Data Fig. 3c,d).
Liver accumulation of cholesterol, triglycerides, and ceramides is linked to non-alcoholic fatty liver disease (NAFLD) 13. We performed BODIPY staining to quantify lipid droplet formation in the livers of B16F10, K7M2, B16F1, 67NR, and 4T1 allograft tumor models and a genetic model of spontaneous melanoma14 (Extended Data Fig. 1a), none of which develop liver metastasis. We found elevated lipid droplet accumulation in the livers of tumor-bearing mice compared to controls (Fig. 1e,f, Extended Data Fig. 3e), showing that fatty liver formation is common in multiple cancer models.
We also employed a genetic murine model of pancreatic ductal adenocarcinoma (PDAC) 15 to determine whether fatty liver disease co-exists with liver PMN formation. Livers of 14-wk tumor-bearing mice, prior to tumor cell arrival at 20-wk, represent the PMN (Extended Data Fig. 3f). Lipid droplets did not accumulate in PMN livers (Extended Data Fig. 3g), suggesting that PMN formation precludes fatty liver generation. Thus, tumors with extrahepatic metastatic tropism, but not those with liver metastatic propensity, induce fatty liver generation.
To determine the clinical relevance of our findings, we analyzed metastasis-free livers of mice implanted with human SK-MEL-192 melanoma or seven patient-derived osteosarcoma xenografts (PDXs) (Supplementary Table 5a; Extended Data Fig. 3h,i,j). Strikingly, all models displayed fatty livers (Extended Data Fig. 3k). Concordant with murine studies, RNA-seq analysis of liver biopsies from newly diagnosed patients with localized PDAC, who later developed extrahepatic (peritoneal or lung, but not liver) metastasis versus controls with non-cancerous lesions (Supplementary Table 5b), revealed that immune homeostasis (i.e., TNFα signaling via NF-κB) and metabolic function (i.e., fatty acid metabolism and OXPHOS) were significantly altered in these patients compared to controls (Fig. 1g). Fatty liver disease was noted in both biopsied and autopsied patients with PDAC who presented with extrahepatitic metastases (Supplementary Table 5b, Fig. 1h), despite similar body mass index in cancer patients and controls (Extended Data Fig. 3l). Thus, in the absence of liver metastasis, remote tumors can disrupt liver metabolism and induce fatty liver formation in cancer patients.
Tumor EVPs reprogram liver metabolism
Liver is a primary organ targeted by tumor EVPs5–9. We investigated whether tumor-secreted factors, specifically EVPs, targeted the liver to initiate metabolic dysfunction and fatty liver formation. We isolated a heterogeneous population of vesicles (small exosomes, Exo-S, 60-80 nm; large exosomes, Exo-L, 90-120 nm) and particles (exomeres, <50nm)5, from B16F10 and K7M2 tissue explants (TEs) by ultracentrifugation16,17 and characterized them by transmission electron microscopy and nanoparticle tracking analysis (Extended Data Fig. 4a). Near-infrared dye labeled B16F10- and K7M2-TE-EVPs were intravenously injected into naïve mice and 24h later, whole organ imaging confirmed EVP liver uptake (Fig. 2a,b).
Figure 2. Tumor-derived EVPs induce liver metabolic dysfunction.
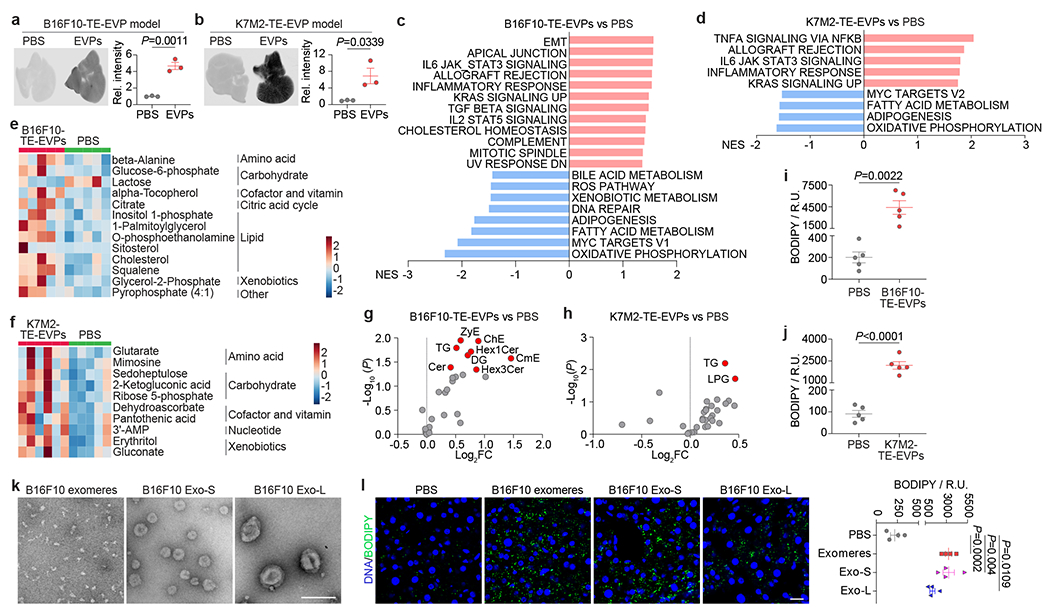
a,b, Representative LI-COR Odyssey images (left) and associated quantification of relative signal intensity (right) of the livers from mice 24 h post intravenously injection of 10 μg of CellVue NIR815-labeled B16F10-TE-EVPs (a) and K7M2-TE-EVPs (b), and PBS controls. n=3 each. c,d, GSEA of gene expression profiles, which were ranked based on the sign of log2FC*(−log10P value), in livers from mice educated for 4 weeks with B16F10-TE-EVPs (n=5) (c), or K7M2-TE-EVPs (n=3) (d), compared with PBS-educated controls, using hallmark gene sets, and the significantly changed signaling pathways with FDR<0.05 are shown. Gene lists for signaling pathways are shown in Supplementary Tables 12 and 13. e,f, Heatmaps showing the metabolites significantly changed in the livers from B16F10-TE-EVP- (e) and K7M2-TE-EVP- (f) educated mice, compared to PBS-educated controls. n=5 B16F10-TE-EVP-educated mice and controls; n=6 K7M2-TE-EVP-educated mice, and n=5 controls. g,h, Volcano plots showing the significantly enriched lipid classes (labeled in red, P<0.05) in the livers from B16F10-TE-EVP- (g) and K7M2-TE-EVP- (h) educated mice, compared to PBS-educated controls. n=7 B16F10-TE-EVP-educated mice, and n=5 controls; n=5 K7M2-TE-EVP-educated mice and controls. i,j, Quantification of BODIPY staining of the livers from B16F10-TE-EVP- (i) and K7M2-TE-EVP- (j) educated mice, and PBS-educated controls. n=5 each. k, Representative TEM images of B16F10 exomeres, Exo-S and Exo-L. This experiment was repeated three times independently with similar results. l, Representative images (left) and associated quantification (right) of BODIPY staining of the livers from mice educated with PBS, B16F10 exomeres, B16F10 Exo-S, or B16F10 Exo-L for 4 weeks. n=4 each. Scale bar, 200 nm for (k) and 20 μm for (l). P values were determined by the two-tailed, unpaired Student’s t-test (a,b,g-j,l). Data are mean ± s.e.m. EMT, epithelial mesenchymal transition. ROS, reactive oxygene species.
To investigate TE-EVP role in liver metabolic “education”, we intravenously injected 10 μg of B16F10- or K7M2-TE-EVPs, the equivalent of physiological concentrations, into mice, every other day for 4 weeks (Extended Data Fig. 4b,c) 10. The livers of TE-EVP- or PBS control-educated mice were subjected to RNA-seq, metabolomic, and lipidomic MS (Fig. 2c–h, and Supplementary Tables 8–11). Transcriptomic analysis revealed increased inflammatory responses and decreased metabolism (i.e., OXPHOS and fatty acid metabolism) in TE-EVP-educated livers (Fig. 2c,d, Extended Data Fig. 4d). Metabolomic MS identified similar dysregulated metabolites in TE-EVP-educated and tumor-bearing mouse livers (Fig. 2e,f and Extended Data Fig. 4e–h). Lipids (e.g o-phosphoethanolamine, cholesterol, and sitosterol) and lipid metabolism-associated pathways (e.g., phosphatidylethanolamine, phosphatidylcholine, and steroid biosynthesis) were enriched in B16F10-TE-EVP-educated livers (Fig. 2e, Extended Data Fig. 4g). Groups of amino acids (β-alanine for B16F10 model, glutarate and mimosine for K7M2 model) and carbohydrates (glucose-6-phosphate and lactose for B16F10 model; sedoheptulose, 2-ketogluconic acid, and ribose 5-phosphate for K7M2 model) were also enriched in TE-EVP-educated livers (Fig. 2e,f). Lipidomics MS further revealed that ceramide and cholesterol ester were upregulated in B16F10-TE-EVP-educated livers and triglyceride was upregulated in both B16F10- and K7M2-TE-EVP-educated livers (Fig. 2g,h and Extended Data Fig. 4i–l). Consistently, lipid droplets accumulated in TE-EVP-educated livers compared to controls, although to a lesser extent than in tumor-bearing mouse livers (Fig. 2i,j, Extended Data Fig. 4m,n). Taken together, these data show that TE-EVPs recapitulate tumor function in systemically disrupting liver metabolism and inducing fatty liver formation.
Distinct EVP subsets induce fatty livers
Upon examining whether tumor cell-specific EVPs were responsible for liver dysfunction, B16F10 and K7M2 cell line-derived EVPs (CL-EVPs) were found to target liver primarily (Extended Data Fig. 5a–d) and like TE-EVPs, they activated immune response and suppressed metabolic pathways, with amino acid, carbohydrate, and lipid enrichment and accumulation of lipid droplets (Extended Fig. 5e–p). These data suggest tumor CL-EVPs are sufficient to dysregulate liver metabolism and induce fatty liver formation and thus were used for further studies.
To determine if non-EVP soluble factors could contribute to fatty liver generation, we intraperitoneally injected mice with control media or EVP-depleted conditioned media (CM) from B16F10 or K7M2 cells (concentrated from the same volume required to isolate 10 μg of EVPs) every other day for 4 weeks, but observed no fatty liver formation (Extended Data Fig. 5q). Furthermore, simultaneous education with EVP-depleted CM and B16F10-CL-EVPs did not enhance fatty liver formation (Extended Data Fig. 5r), suggesting that tumor-derived EVPs, but not soluble factors, are sufficient to promote fatty liver generation.
To examine if tumor CL-EVPs are essential for cancer-induced fatty liver formation, we deleted Rab27a, a key regulator of EVP secretion10,18, in B16F10 and K7M2 cells via knock-out/KO or knock-down/KD, respectively. Consistent with previous results10, Rab27a ablation suppressed tumor cell EVP secretion without altering morphology (Extended Data Fig. 6a–h). Rab27a loss reduced primary tumor growth in both models despite unaltered proliferation in vitro (Extended Data Fig. 6i–l). BODIPY staining showed significantly decreased fatty liver induction in Rab27a-KO/KD tumor-bearing mice compared to their respective controls with comparable tumor burden (Extended Data Fig. 6m–p), indicating that tumor-derived EVPs are crucial for liver metabolic reprogramming.
As EVPs are heterogeneous, we evaluated which subpopulation(s) (i.e., exomeres, Exo-S, and Exo-L, isolated by asymmetric-flow field-flow fractionation [AF4], Fig. 2k) 5 from B16F10 cells, could induce hepatic lipid droplet accumulation after 4-week education. All three subpopulations induced inflammation, altered liver metabolism (Extended Data Fig. 6q–t), and fatty liver formation (promoted by exomeres and Exo-S to a greater extent than Exo-L) (Fig. 2l). Notably, Rab27a loss reduced the production of exomeres, Exo-S (to a lesser extent) and Exo-L (to the least extent) from B16F10 cells and decreased B16F10 tumor-induced fatty liver generation (Extended Data Fig. 6o,p,u), suggesting a major contribution of exomeres to fatty liver induction, consistent with our previous findings of exomere uptake by the liver5.
Kupffer cells drive lipid accumulation
To identify liver cells targeted by tumor EVPs, we intravenously injected fluorescently-labelled B16F10- or K7M2-CL-EVPs into mice. Flow cytometry analysis of livers 24h post-injection revealed that EVPs from both models were predominantly uptaken by Cd45+ immune cells and Cd31+ vascular endothelial cells, but not desmin+ stellate cells, albumin+ hepatocytes, or Lyve1+ lymphatic and sinusoidal endothelial cells (Extended Data Fig. 7a–d). Specifically, more than 90% of Cd45+ immune cells taking up EVPs were Cd11b+F4/80+ Kupffer cells (KCs), a finding confirmed by immunofluorescence microscopy (Fig. 3a,b, Extended Data Fig. 7e).
Figure 3. Uptake of tumor-derived EVPs by KCs induces fatty liver formation.
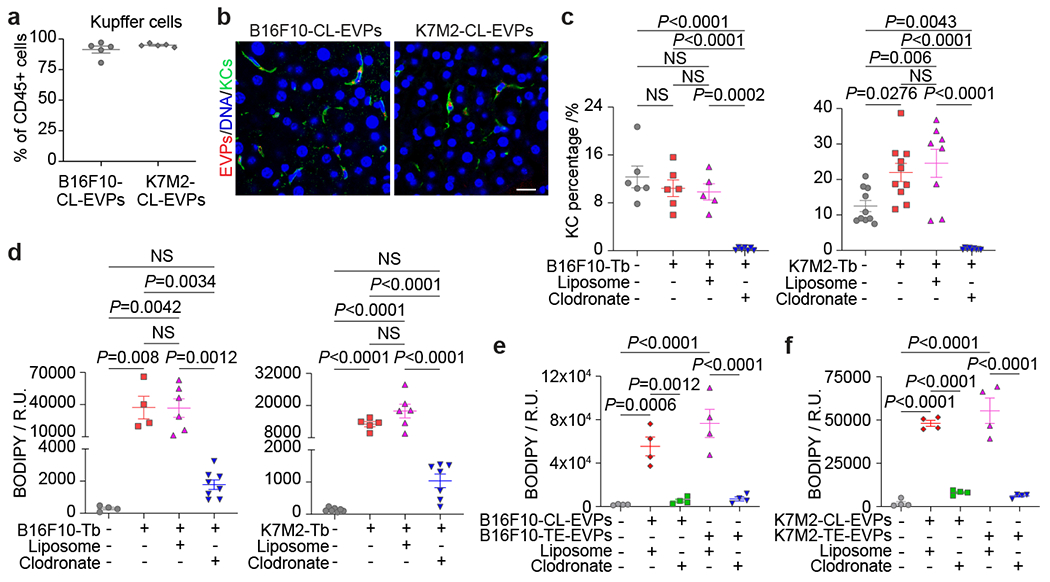
a, Flow cytometry analysis of the percentage of KCs in the Cd45+ immune cells. n=5 each. b, Representative immunofluorescence images of the co-localization of KCs (green) and B16F10-CL-EVPs (red, left), or K7M2-CL-EVPs (red, right). DNA in blue. Scale bar, 20 μm. This experiment was repeated three times independently with similar results. c,d, Flow cytometry analysis of the percentage of KCs (c) and quantification of BODIPY staining (d) in the livers from PBS-injected control mice, B16F10- or K7M2-Tb mice, and the tumor-bearing mice treated with liposome or clodronate as illustrated in Extended Data Fig. 8a. In (c), B16F10-Tb model (n=6 controls and Tb mice, n=5 Tb mice treated with liposome, n=7 Tb mice treated with clodronate); K7M2-Tb model (n=10 controls and Tb mice, n=8 Tb mice treated with liposome, n=9 Tb mice treated with clodronate). In (d), B16F10-Tb model (n=4 controls and Tb mice, n=6 Tb mice treated with liposome, n=8 Tb mice treated with clodronate); K7M2-Tb model (n=7 controls, n=5 Tb mice, n=6 Tb mice treated with liposome, n=7 Tb mice treated with clodronate). e,f, Quantification of the BODIPY staining of precision-cut liver slices from liposome- or clodronate- treated mice after treatment with B16F10-CL-EVPs or B16F10-TE-EVPs (e), or K7M2-CL-EVPs or K7M2-TE-EVPs (f), following the procedure illustrated in Extended Data Fig. 8h. n=4 each. P values were determined by the one-way ANOVA with Tukey’s test (c-f). Data are mean ± s.e.m. NS, not significant.
We hypothesized that KCs, which promote NAFLD development19, are key for tumor EVP-induced fatty liver formation, so we depleted KCs in B16F10- and K7M2-tumor-bearing mice via intravenous injection of clodronate beginning on day 12 or 15 post tumor implantation. KC depletion efficiency was confirmed by flow cytometry and immunofluorescence analyses (Fig. 3c, Extended Data Fig. 8a–d). Without affecting primary tumor growth, hepatic lipid droplet deposition was dramatically reduced by KC depletion in both models compared to liposome-injected controls (Fig. 3d, Extended Data Fig. 8e,f). Next, we depleted KCs in naïve mice via clodronate treatment and generated viable precision-cut liver slices for EVP treatment ex vivo. Indeed, neither CL- nor TE-derived EVPs from B16F10 or K7M2 models could induce lipid droplet formation in KC-ablated liver slices (Fig. 3e,f, Extended Data Fig. 8g–j). Collectively, these findings indicate that KCs are necessary for tumor EVP-induced fatty liver formation.
EVP-educated KCs upregulate TNFα
To investigate whether tumor EVP-targeted KCs release specific factors to mediate fatty liver formation, after validating the functional integrity of isolated KCs, we showed that multiple cytokines and chemokines, including IL-1α, TNFα, CCL2, CCL3, CXCL1, CXCL2 and CXCL13, were enriched in KCs isolated from B16F10 and K7M2 tumor-bearing mice (Extended Data Fig. 9a–d), indicating an inflammatory liver microenvironment. We then isolated KCs from naïve mice and treated them ex vivo with 10 μg/mL of B16F10- or K7M2-CL-EVPs, or PBS every 24h for 3 days. Cytokine array analysis of KC conditioned media showed increased secretion of TNFα, CCL2, CCL3, CXCL1, and CXCL2 from CL-EVP-treated KCs compared to control PBS-treated KCs (Extended Data Fig. 9e,f).
Remarkably, B16F10-CL-EVP-treated KCs released 100-fold more TNFα than control KCs, which was confirmed by ELISA (Extended Data Fig. 9e,g). Furthermore, for both B16F10 and K7M2 models, plasma TNFα levels were significantly higher in tumor-bearing mice and CL- or TE-EVP-educated mice than in control mice (Fig. 4a,b). Tnf gene expression was also upregulated in KCs isolated from tumor-bearing mice and naïve mice after CL- or TE-EVP treatment in vitro (Extended Data Fig. 9h,i). Since TNFα was not detected in EVP-depleted CM of B16F10 and K7M2 cells, we ruled out the tumor as the TNFα source (Extended Data Fig. 9j).
Figure 4. Tumor EVP-packaged palmitic acid induces TNFα secretion from KCs and promotes fatty liver generation.
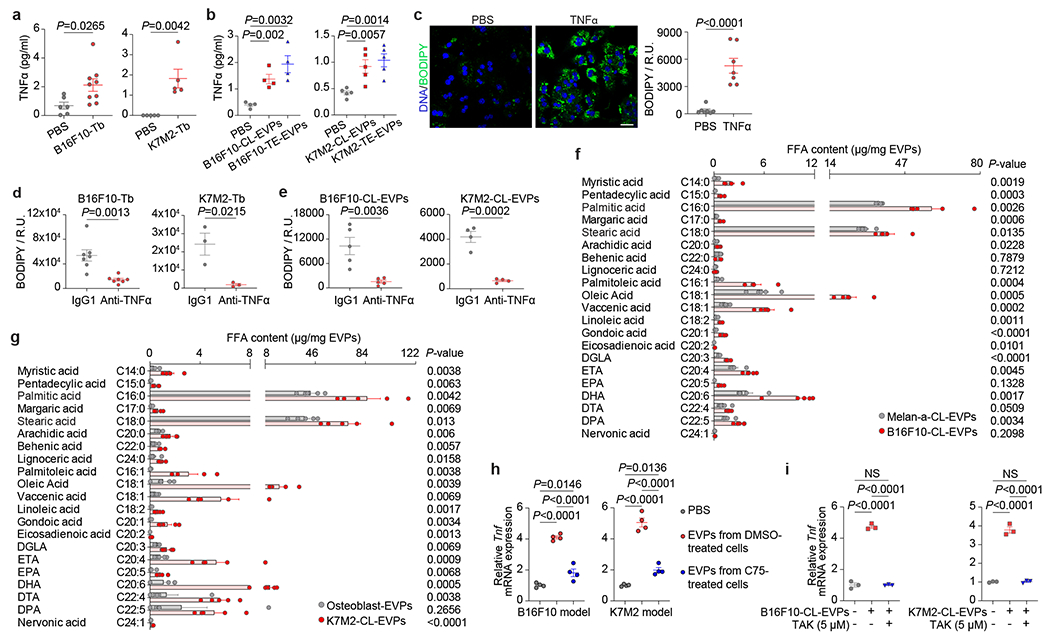
a, TNFα concentration determined by ELISA in the plasma from B16F10-Tb (left) and K7M2-Tb mice (right), and PBS-injected controls. B16F10-Tb model (n=6 controls, n=9 Tb mice), K7M2-Tb model (n=5). b, TNFα concentration in the plasma of B16F10-CL-EVP- and B16F10-TE-EVP-educated mice (n=4 each) (left), K7M2-CL-EVP- and K7M2-TE-EVP-educated mice (n=5 each) (right), and PBS-educated controls. c, Representative images (left) and associated statistical analysis (right) of BODIPY staining of the primary hepatocytes 48 h post treatment with recombinant murine TNFα protein (25 ng/ml), or PBS control. n=7 independent experiments. d,e, Statistical analysis of BODIPY staining of the livers from B16F10- and K7M2-Tb mice (d), and B16F10-CL-EVP- and K7M2-CL-EVP-educated mice (e), treated with anti-TNFα antibody or IgG1 isotype control. B16F10-Tb model (n=7), K7M2-Tb model (n=3), B16F10-CL-EVP model (n=5), K7M2-CL-EVP model (n=4). f, g, Quantitative analysis of the long-chain free fatty acids in the EVPs derived from B16F10 and Melan-a cells (f), and K7M2 cells and primary murine osteoblasts (g). n=5 per group. h, qRT-PCR analysis of the Tnf expression in KCs 4 h post treatment with PBS, 10 μg of EVPs derived from DMSO or C75 (40 μM) treated B16F10 cells (left) or K7M2 cells (right). n=4 independent experiments. i, qRT-PCR analysis of Tnf expression in KCs which were pre-treated with DMSO or TAK (5 μM) for 1 h, and subsequently treated with PBS, 10 μg of B16F10-CL-EVPs (left) or K7M2-CL-EVPs (right) with or without TAK (5 μM) for 4 h. n=3 independent experiments. P values were determined by the two-tailed, unpaired Student’s t-test (a-g), or one-way ANOVA with Tukey’s test (h,i). Data are mean ± s.e.m. Scale bar, 20 μm. NS, not significant.
As TNFα promotes NAFLD development20, we speculated that TNFα drives liver dysregulation. We treated primary hepatocytes with recombinant TNFα which induced lipid droplet formation (Fig. 4c). Transcriptomic profiling revealed significant downregulation of lipid and fatty acid catabolic signatures in TNFα-treated primary hepatocytes (Extended Data Fig. 9k). We administered a pan-TNFα-blocking antibody intraperitoneally in B16F10 and K7M2 tumor-bearing mice and observed reduced hepatic lipid droplet accumulation without affecting tumor growth (Fig. 4d, Extended Data Fig. 9l–n). Similar results were obtained in anti-TNFα treated B16F10- or K7M2-CL-EVP-educated mice (Fig. 4e, Extended Data Fig. 9l,o), indicating that KC-secreted TNFα is required for tumor EVP-induced fatty liver formation.
EVP-packaged palmitic acid activates KCs
To identify the EVP cargo responsible for KC activation, we performed metabolomics MS on B16F10-TE-EVPs and control skin TE- EVPs. Long-chain saturated fatty acids, i.e., palmitic acid (PA) and stearic acid (SA), were significantly enriched in the B16F10-TE-EVPs (Extended Data Fig. 10a and Supplementary Table 14). Quantitative MS of free fatty acids confirmed enrichment of multiple saturated (with PA displaying the highest concentration) and unsaturated fatty acids in B16F10-TE-EVPs as well as B16F10- and K7M2-CL-EVPs compared with their respective controls (i.e., EVPs isolated from the mouse skin TE, melanocyte Melan-a, and primary osteoblasts) (Fig. 4f,g, Extended Data Fig. 10b, Supplementary Tables 15–17).
As PA induces KC secretion of pro-inflammatory cytokines, including TNFα, during NAFLD progression21,22, we asked if tumor EVP-packaged PA stimulated TNFα secretion in KCs. We first showed that PA treatment induced Tnf expression in KCs compared to controls (Extended Data Fig. 10c). Tumor cells treated with the fatty acid synthase inhibitor C75 (Ref23) significantly reduced free fatty acids, including PA and longer chain fatty acids, in EVPs (Extended Data Fig. 10d), which impaired EVP-induced KC Tnf expression (Fig. 4h). Moreover, primary osteoblast-EVPs, which package less PA, failed to induce Tnf expression in KCs, lipid droplet accumulation ex vivo or in vivo, despite efficient osteoblast-EVP uptake in the liver (Extended Data Fig. 10e–g). Neither skin (for B16F10) nor bone (for K7M2) normal TE-EVP controls could induce Tnf expression in KCs or promote lipid droplet accumulation in liver slices (Extended Data Fig. 10e,h). However, Melan-a EVPs induced Tnf expression in KCs despite low PA levels, due to 12-O-tetradecanoylphorbol-13-acetate present from the culture media (Extended Data Fig. 10e,i,j). Taken together, these data support a critical role for tumor EVP-packaged PA in inducing KC TNFα secretion.
Previous studies showed that saturated fatty acids promote Toll-like receptor 4 (Tlr4)-dependent signaling24,25, thus we assessed if Tlr4 was required for EVP-PA-induced Tnf expression in KCs. KCs were pretreated with TAK, a Tlr4 inhibitor, for 1h, followed by PA treatment in the presence of TAK or DMSO control for another 4h. Inhibition of Tlr4 diminished PA-induced Tnf expression in KCs (Extended Data Fig. 10c). Importantly, Tlr4 inhibition also suppressed Tnf induction mediated by B16F10- and K7M2- CL- and TE-EVPs (Fig. 4i, Extended Data 10k), suggesting that EVP-packaged PA activation of inflammatory pathways in KCs requires Tlr4 functional integrity.
EVPs decrease liver drug metabolism
In patients with advanced cancers, low tolerance for standard chemotherapy is often observed, likely due to impaired liver detoxification26,27. We hypothesized that tumor EVP-induced fatty liver and inflammation could suppress liver drug metabolism. Indeed, recombinant TNFα downregulated core Cyp genes corresponding to human cytochrome P450 homologs, Cyp1a2, Cyp2b10, Cyp2c38, Cyp2d9/2d10, and Cyp3a11, in murine hepatocytes (Fig. 5a and Supplementary table 18). These genes were also downregulated in hepatocytes isolated from B16F10 tumor-bearing mice and B16F10-CL-EVP-educated mice, compared to controls (Fig. 5b,c). Notably, CYP enzymes metabolize ~70-80% of prescribed drugs28, and GSEA revealed downregulation of the drug-metabolizing cytochrome P450 signaling pathway in primary hepatocytes after TNFα treatment and in the livers of both B16F10 tumor-bearing mice and pancreatic cancer patients without liver metastasis (but who later developed extrahepatic metastasis) (Fig. 5d).
Figure 5. Tumor EVPs suppress liver drug metabolism and enhance chemotoxicity.
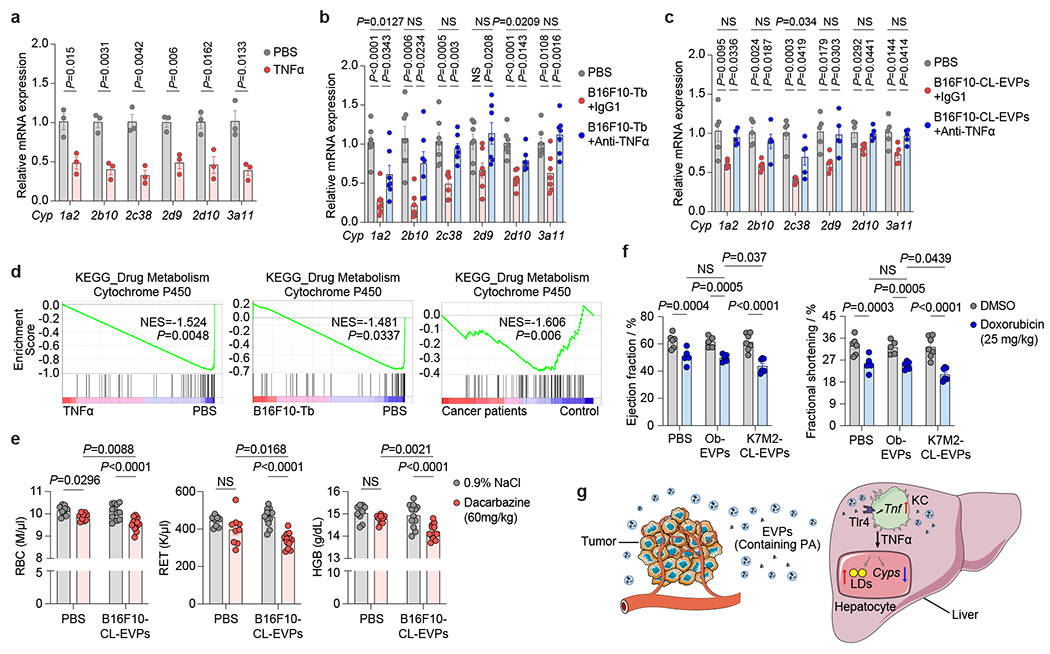
a, qRT-PCR analysis of the expression of Cyp genes in the primary hepatocytes treated with PBS or recombinant murine TNFα protein (25 ng/ml) for 24 h. n=3 independent experiments. b,c, qRT-PCR analysis of the expression of Cyp genes in hepatocytes isolated from B16F10-Tb mice (n=7) (b) and B16F10-CL-EVP-educated mice (n=5) (c) treated with anti-TNFα antibody or IgG1 isotype control, compared to their respective PBS-injected controls. d, GSEA of gene expression profiles in the TNFα-treated murine primary hepatocytes (top), in the livers from B16F10-Tb mice (middle) and cancer patients (bottom), compared to their respective controls. n=3 per group for TNFα treatment and B16F10-Tb mice, and PBS controls; n=5 cancer patients and n=8 control subjects. e, Analysis of red blood cell (RBC), reticulocyte (RET) and hemoglobin (HGB) counts in PBS- (n=9) or B16F10-CL-EVP-educated mice (n=12) after treatment with dacarbazine (60 mg/kg) or 0.9% NaCl. f, Analysis of left ventricular ejection fraction and fractional shortening (M-mode) in PBS-, osteoblast (Ob)-EVP-, or K7M2-CL-EVP-educated mice after treatment of doxorubicin (cumulative dose of 25 mg/kg) or DMSO. PBS groups (n=6), K7M2-CL-EVP groups (n=7), Ob-EVP group treated with DMSO (n=5) or doxorubicin (n=6). g, A schematic illustration of the working model. Tumor-derived EVPs specifically target KCs in the liver. EVP-packaged saturated fatty acids, such as palmitic acid (PA), stimulate the secretion of TNFα from KCs depending on Tlr4 functional integrity. This produces a pro-inflammatory microenvironment in the liver, thereby systemically inducing fatty liver formation and suppressing the drug-metabolizing activity of the liver. P values were determined by the two-tailed, unpaired t-test (a), one-way ANOVA with Tukey’s test (b,c), or two-way ANOVA with Fisher’s LSD test (e,f). Data are mean ± s.e.m. NS, not significant. LDs, lipid droplets.
Next, we showed that the capacity of each CYP enzyme to metabolize specific substrates was indeed decreased in liver slices from tumor-bearing mice compared to controls by measuring the levels of metabolized products (Extended Data Fig. 11a,b). Using methoxsalen (a Cyp1a2 inhibitor) and phenobarbital (a Cyp2b10 inducer) as negative and positive controls, respectively, we further showed ex vivo B16F10-CL-EVP treatment was sufficient to suppress drug metabolism in liver slices from naïve mice (Extended Data Fig. 11c,d). Notably, TNFα blockade rescued hepatocyte expression of Cyp genes in B16F10 tumor-bearing and B16F10-CL-EVPs educated mice (Figure 5b,c) and reversed drug metabolism impairment in B16F10 tumor-bearing mouse-derived liver slices and naïve mouse-derived liver slices treated with B16F10-CL-EVPs (Extended Data Fig. 11b,d). These results suggest that tumor EVP-induced secretion of TNFα by KC impairs liver drug metabolism via CYP enzyme suppression.
EVPs enhance chemotherapy toxicities
As impaired drug metabolism induced by tumor EVPs may impact chemotherapy toxicity, we evaluated toxicity of dacarbazine and doxorubicin, two drugs metabolized by CYP enzymes used for melanoma and osteosarcoma treatment, respectively29–31. We intraperitoneally injected dacarbazine (60 mg/kg) or 0.9% NaCl into mice pre-educated with PBS or B16F10-CL-EVPs every other day for a week while continuing B16F10-CL-EVP or PBS education (Extended Data Fig. 11e). One day after the last injection, we observed a 2.8% decrease in red blood cells and no significant decrease in reticulocyte numbers or hemoglobin levels in PBS-educated mice, however, dacarbazine toxicity was significantly enhanced, with a 5.6% reduction in red blood cells, a 26.9% reduction in reticulocytes and a 4.9% reduction in hemoglobin in EVP-educated mice (Fig. 5e).
Next, we intraperitoneally injected doxorubicin (1 mg/kg) or DMSO into mice pre-educated with PBS, primary osteoblast-EVPs or K7M2-CL-EVPs every 24 h for 25 days while continuing PBS or EVP education. One day after the last injection, echocardiography revealed doxorubicin decreased the left ventricular ejection fraction from 62.1% and 61.5% to 50.8% and 49.7% (an 18.2% and 19.2% reduction) in PBS and osteoblast-EVP educated mice, respectively, with a further decrease, from 61.3% to 43.7% (a 28.7% reduction) in K7M2-CL-EVP educated mice (Fig. 5f, Extended Data Fig. 11e–g, and Supplementary Videos 1–6), into a range approaching heart failure (ejection fraction of 40% or less versus above 55% for normal cardiac function32). Doxorubicin also decreased left ventricular fractional shortening from 32.9% and 32.2% to 25.4% and 24.8% (a reduction of 22.8% and 23%, respectively) in PBS- and osteoblast-EVP-educated mice, with a further decrease from 32.3% to 21% (a 35% reduction) in K7M2-CL-EVP-educated mice (Fig. 5f, Extended Data Fig. 11f,g). Thus, tumor EVPs enhance chemotherapy toxicity by impairing liver drug metabolism, consequently reducing chemotherapy tolerance in cancer patients.
Discussion
Cancer cells secrete EVPs, systemically reprograming local and distant organ function1,3,6,7,10. EVPs promote PMN formation as well as pathological events in other organs not destined for metastasis3,4,6,33. Here, we show remote tumor-derived EVPs induce inflammation and downregulate lipid catabolism in tumor-free livers, reprogramming metabolism in mice and cancer patients (Fig. 5g). Mechanistically, tumor EVPs packaging PA elicit TNFα secretion in KCs thus driving fatty liver formation. Blocking EVP secretion by Rab27a ablation in cancer cells, KC depletion, or TNFα blockade rescues the phenotypes. Importantly, we showed that exomeres, the predominant EVP targeting the liver5, primarily contribute to fatty liver formation.
Interestingly, we noticed that fatty liver generation and liver PMN formation are inversely correlated, as liver-tropic tumor EVPs stimulate KC TGFβ secretion to activate stellate cells and produce a fibrotic PMN6,7, while tumors with extrahepatic metastatic tropism induce fatty liver via EVP-mediated TNFα secretion by KCs. Furthermore, we observed that exosome subpopulations target PMN organs7, whereas exomeres predominantly target the liver5, driving fatty liver formation. Further dissection of EVP cargo, and functional heterogeneity within each subpopulation is critical to decipher the mechanisms promoting PMN formation and metastasis versus the paraneoplastic effects of cancer. Our study also highlights a novel mechanism for EVP-packaged PA in altering non-metastatic liver function, which is distinct from PA effects in PMNs34.
EVP-induced fatty liver may provide energy to support tumor growth systemically at other sites, consistent with reports that NAFLD increases the risk of extrahepatic cancers35,36. Beyond tumor support, liver metabolic dysregulation could have multi-organ systemic effects, promoting cardiovascular disease through increased cholesterol and triglyceride production as seen here, impairing immune function, such as T cell function, or promoting cachexia.
Our work highlights the need for cancer treatments to take into account the systemic effects of cancers. Employing strategies that restore liver function is critical to minimize side effects and enhance treatment response to chemotherapeutic drugs metabolized by the liver. Since we showed that tumor EVP-induced TNFα secretion downregulates Cyp genes and compromises liver drug metabolism, integrating chemotherapy with FDA-approved TNFα inhibitors may restore liver function in cancer patients. Strategies reducing tumor EVP production should also be considered since they will not only prevent liver metabolic dysfunction but also inhibit PMN formation. Lastly, restoration of normal liver function will also prevent further systemic pathologies (e.g., cardiovascular disease) associated with cancer.
Materials and methods
Cell lines and cell culture
B16-F10, K7M2, B16-F1, 4T1 and 293T cells were purchased from American Type Culture Collection (ATCC), SK-MEL-192 cell line was obtained from Memorial Sloan Kettering Cancer Center (MSKCC). These cells were cultured in DMEM (Corning, #10-013-CV) supplemented with 10% FBS (Gibco, #10438-026) and 1× penicillin/streptomycin (100 U/ml of penicillin and 100 μg/ml of streptomycin, Thermo Fisher Scientific, #15070-063).
The 67NR cell line was obtained from Dr. Fred Miller (Karmanos Cancer Institute) 12, and cultured in RPMI (Corning, #10-040-CV) supplemented with 10% FBS and 1× penicillin/streptomycin. The murine melanocyte Melan-a was obtained from The Wellcome Trust Functional Genomics Cell Bank and cultured in RPMI supplemented with 10% FBS, 1× penicillin/streptomycin and 0.2 μM of TPA (Sigma, P1585). Primary osteoblasts were isolated from mouse bones, and primary hepatocytes and Kupffer cells (KCs) were isolated from mouse livers as described below. When collecting conditioned media for EVP isolation, FBS was first depleted of EVPs by ultracentrifugation at 100,000g for 4 h. Cells were cultured in EVP-depleted media for 3 days and supernatant was collected before cells reached confluency for EVP isolation. Cells were maintained in a humidified 37 °C incubator with 5% CO2, and cell lines routinely tested and confirmed to be negative for mycoplasma.
Generation of Rab27a-KO B16F10, Rab27a-KD K7M2 and mCherry-expressing K7M2 cell lines
Rab27a-KO in B16F10 cells was achieved by infecting cells using lentivirus (lentiCRISPR vector [Addgene, #52961] carrying guide RNA targeting mouse Rab27a at 5’-CCTGAAATCAATGCCCACTG-3’). The single guide RNA targeting sequence was chosen using GPP sgRNA Designer (https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design). Lentivirus-infected cells were single-cell-sorted into a 96-well plate for clonal growth. Rab27a depletion was confirmed by western blotting analysis and Rab27a gene KO editing was verified by Sanger sequencing. As a control, B16F10 cells were infected with lentiCRISPR empty-vector virus. Rab27a-KD in K7M2 cells was achieved by infecting cells using lentivirus (pLKO.1 shRab27a, TRCN0000381753, Sigma). Knockdown efficiency of Rab27a in K7M2 cells was confirmed by western blotting analysis. As a control, K7M2 cells were infected with pLKO.1 shRNA empty-vector virus. mCherry-expressing K7M2 (K7M2-mCherry) cell line was generated by infecting cells using lentivirus (LeGO-C, Addgene, #27348).
Lentivirus was produced by co-transfection of the lentiviral expression vector and viral packaging/envelope plasmids, including pRRE (Addgene, #12251), pMD2g (Addgene, #12259) and pRSV-REV (Addgene, #12253) into 293T cells using Lipofectamine LTX/PLUS (Invitrogen, #15338030).
Cell proliferation assay
Cell proliferation was assessed according to the manufacturer’s instructions (cell counting kit-8, CCK-8). In brief, cells were plated in a 96-well plate at a concentration of 5,000 cells/well. After cells attached to the plate (time point set as 0 h), 10 μl of CCK-8 (Dojindo, CK04) was added into the cell culture medium (100 μl) and incubated at 37 °C for 30 min. The absorbance at 450 nm was measured using a microplate reader (accuSkan GO, Thermo Fisher Scientific). Cell proliferation assay was also performed at time point of 24 h, 48 h, 72 h and 96 h, respectively.
Western blotting
Western blotting was done as previously described2. In brief, cells were lysed in RIPA buffer (Millipore, Sigma, R0278) supplemented with Halt Protease Inhibitor Cocktail (Thermo Fisher Scientific, #87786). Proteins were separated by SDS–PAGE (Invitrogen, XP00100BOX) and transferred onto a polyvinylidene difluoride (PVDF) membrane (Bio-Rad, #1620177). After membranes were blocked with 5% BSA (EMD, Millipore, #12659) for 30 min, they were probed with primary antibodies, including anti-Rab27a (Cell Signaling, #69295) and anti-β-Actin (Cell Signaling, #4970) overnight at 4 °C, followed by incubation with HRP-conjugated secondary antibodies for 1 h at room temperature. The membranes were then developed using ECL substrate (Bio-Rad) and visualized using the Bio-Rad ChemiDoc Touch Imaging System.
Isolation of primary hepatocytes and KCs from murine livers
Plates were coated with collagen I (30 μg/ml in sterile water, Thermo Fisher Scientific, A10483-01) overnight prior to seeding cells. Mice were anesthetized using 2% isoflurane/1.5% oxygen inhalation, and the livers were first perfused with 30 ml of HBSS (Ca2+ and Mg2+ free, Thermo Fisher Scientific, #14175-095) containing 0.5 mM of EGTA (Sigma, E8145) and 25 mM of HEPES (Gibco, #15630-080), and then digested with 50 ml of collagenase solution (DMEM containing 100 U/ml of collagenase type 2 [Worthington Biochemical Corporation, LS004176], 15 mM of HEPES, 1× penicillin/streptomycin). After collagenase digestion, livers were removed and whole liver cells were released into DMEM containing 1× non-essential amino acids solution (Thermo Fisher Scientific, #11140-050), 1× penicillin/streptomycin and 1× L-Glutamine (Gibco, #25030-081) by rupturing the Glisson’s Capsule, and filtered through a 70 μm cell strainer (Thermo Fisher Scientific, #08-771-2) to remove undigested tissues. The flow-through cells were subjected to centrifugation at 50g for 3 min at 4 °C, and the pellet was collected for primary hepatocyte isolation and the supernatant was collected for KC isolation, respectively. For primary hepatocyte isolation, the pellet was washed twice in 30 ml of Hepatocyte Wash Medium (Thermo Fisher Scientific, #17704-024) by centrifugation at 50g for 2 min at 4 °C, and the hepatocytes were pelleted down at 50g for 2 min at 4 °C and resuspended in Hepatocyte Culture Medium (Lonza, CC-3198) and cultured in collagen I-coated plates in a humidified 37 °C incubator with 5% CO2. For KC isolation, the supernatant was subjected to centrifugation at 300g for 10 min at 4 °C to pellet non-parenchymal cells, which were resuspended in 450 μl of MACS buffer (PBS containing 2% FBS). 50 μl of anti-F4/80 microbeads (Miltenyi Biotec, #130-110-443) was added to the cell suspension, which was gently rocked at 4 °C for 15 min in the dark, and then the cells were washed twice with MACS buffer and pelleted by centrifugation at 300g for 10min at 4 °C. Cells were then resuspended in 0.5 ml of MACS buffer and applied onto an LS column (Miltenyi Biotec, #130-042-401). After washing with MACS buffer, the KCs retained in the LS column were collected according to the manufacturer’s protocol. KCs were resuspended in DMEM containing 1× non-essential amino acids solution, 1× penicillin/streptomycin, 1× L-Glutamine and 10% EVP-depleted FBS, and cultured in the collagen I-coated 6-well plates in a humidified 37 °C incubator with 5% CO2. Cells were washed with PBS 1-2 h after plating to remove dead cells and either collected for RNA extraction or cultured in fresh medium overnight for downstream experiments. qRT-PCR analysis of the expression of Cyp genes (in hepatocytes) and Tnf (in KCs) was performed as described below.
TNFα treatment of primary hepatocytes
Primary hepatocytes cultured in the collagen I-coated 6-well plates or 8-well chamber slides (Millipore, Sigma) were treated with PBS or 25 ng/ml of recombinant murine TNFα (aa 80-235) protein (R&D system, 410-MT-025/CF) for 24 h or 48 h, followed by qRT-PCR analysis of the Cyp gene expression or BODIPY staining, respectively.
Treatment of KCs
For EVP treatment, KCs were treated with PBS or 10 μg/ml of EVPs derived from cell lines or tissue explants for 4 h; or pretreated with DMSO or 5 μM of TAK (MedChemExpress, HY-11109) for 1 h, followed by treatment with EVPs (10 μg/ml) in the presence or absence of TAK (5 μM) for 4 h. KCs were then collected for RNA extraction and qRT-PCR analysis.
For TPA treatment, KCs were treated with DMSO or 0.2 μM of TPA for 4h, followed by RNA extraction and qRT-PCR analysis.
For palmitic acid (PA) treatment, stock PA (Millipore Sigma, P5585) was made up in 100% ethanol at 200 mM and diluted into 0.2 μm-filter sterilized PA-carrier medium (DMEM containing 5% EVP-depleted FBS, 5% fatty acid-free BSA [Sigma Aldrich, A3803], 1× non-essential amino acids solution, 1× penicillin/streptomycin and 1× L-Glutamine) at a final concentration of 200 μM. As a control, 100% ethanol was diluted into PA-carrier medium at 1:1000 dilution. To facilitate the conjugation of PA to BSA, the mixture was incubated at 37 °C for 30 min with gentle shaking. KCs were pretreated with DMSO or 5 μM of TAK for 1 h, washed once with PBS and then incubated in medium carrying 200 μM of PA or control ethanol with or without TAK (5 μM) for 4 h. KCs were then collected for RNA extraction and qRT-PCR analysis.
RNA extraction and qRT-PCR analysis
Total RNA was extracted using TRIzol reagent (Thermo Fisher Scientific, #15596018) as previously described2, and RNA was further purified using RNeasy Mini kit including a DNase digest following the manufacturer’s instructions (Qiagen, #74014). Total RNA was then subjected to qRT-PCR analysis and RNA-seq as described below.
For qRT-PCR analysis, 100-500 ng of total RNA was used for cDNA synthesis using the high-capacity cDNA reverse transcription kit with RNase inhibitor (Thermo Fisher Scientific, #4374966) following the manufacturer’s instructions. Ten nanograms of cDNA were used for qRT-PCR reactions using SYBR Green Supermix (Bio-Rad, #1725271) and gene-specific primers. 18S rRNA was used as an internal control. qRT-PCR was performed on a CFX384 Touch Real-Time PCR System (Bio-Rad), and data were analyzed by CFX Manager (Version 3.1, Bio-Rad). The relative gene expression was quantified by 2−ΔΔCt method37. Primers used in qRT–PCR analysis are listed in Supplementary Table 27.
Primary osteoblast isolation and culture
Primary murine osteoblasts were isolated from BALB/c mouse bones as previously described38. Briefly, 7-week-old female mice were euthanized and bone tissues including tibia, femur, humerus were collected. Bone marrow was removed by flushing with basal medium (10 g/L of αMEM [BioConcept, #1-23P10-M] containing 2.2 g/L of NaHCO3, 1× penicillin/streptomycin, 2 mM of L-Glutamine, 0.375× MEM amino acids [BioConcept, #5-12K01-H] and 10% EVP-depleted FBS). Bone tissues were then cut into small pieces and seeded into a 10 mm dish with 10 ml of digestion medium (basal medium containing 1 mg/ml of collagenase II [Sigma, C6885]). After incubation at 37 °C for 90 min, the digestion medium was replaced by basal medium to allow the cells to migrate from bone pieces and attach to the dish. Three days later, cells were detached with collagenase I (Thermo Fisher Scientific, #17100-017) solution (1 mg/ml in 0.9% NaCl) followed by TrypLE Express Enzyme (Thermo Fisher Scientific, #12604013). When cells reached passage 3 to 5, the isolation of osteoblasts was confirmed by measuring of mineralization using Alizarin Red-S staining, and >97% of cultured cells were osteoblasts (data not shown). The supernatant from passage 3 to 5 osteoblasts was collected for EVP isolation.
EVP purification, characterization and labeling
To isolate EVPs from cultured cell lines, the cell culture and preparation of conditioned media were described above. For tissue explant culture, primary tumors (B16F10 or K7M2) and mouse skin were cut into millimeter-sized pieces and cultured overnight in RPMI supplemented with 1× penicillin/streptomycin, and the supernatant was collected and filtered using a 70 μm cell strainer to remove tissue debris. To purify EVPs from DMSO or C75 (MedChemExpress, HY-12364) treated cells, B16F10 or K7M2 cells were cultured in EVP-depleted media for 48 h, and then washed three times with PBS and cultured in EVP-depleted media containing DMSO or C75 (40 μM) for 48 h. Conditioned media was then collected for EVP purification. For EVP isolation from the circulation, 200 μl of mouse plasma was diluted into 800 μl of PBS to a total volume of 1 ml before centrifugation as described below.
EVP purification was performed as previously described16,17. In brief, the conditioned media of the cultured cells or tumor explants, or PBS-diluted mouse plasma, was sequentially centrifuged at 500g for 10 min, at 3,000g for 20 min and then at 12,000g for 20 min. EVPs were pelleted by ultracentrifugation of this supernatant at 100,000g for 70 min and washed with PBS once. EVPs were then collected by ultracentrifugation at 100,000g for 70 min and resuspended in PBS. The protein concentration of EVPs in PBS was measured by BCA Protein Assay Kit (Thermo Fisher Scientific, #23225). EVP size and particle number were analyzed using the LM10 nanoparticle characterization system (NanoSight, Malvern Instruments). EVPs were imaged by negative-stain transmission electron microscopy.
For EVP labeling, 10 μg of EVPs, or an equivalent volume of mock PBS, were first gently mixed with 0.4 μl of CellVue dye (NIR815 or Burgundy, LI-COR) diluted in 50 μl of Diluent C and incubated in the dark for 5 min; 25 μl of 35% BSA (Sigma, A7979) was added to the mixture to quench the labeling reaction and incubated for 1 min. The mixture was washed with PBS and EVPs were pelleted by ultracentrifugation at 100,000g for 70 min, and then resuspended in PBS for downstream experiments.
Transmission electron microscopy (TEM)
TEM was performed as previously described5,16. For negative staining TEM analysis, 5 μl of EVPs in PBS (0.1 μg/μl) were placed on a formvar/carbon-coated grid and allowed to settle for 1 min. The sample was blotted and negatively stained with four successive drops of aqueous uranyl acetate (1.5%), blotting between each drop. Following the last drop of stain, the grid was blotted and air-dried. Grids were imaged with a JEOL JSM 1400 (JEOL, USA, Ltd, Peabody, MA) transmission electron microscope operating at 100 kV. Images were captured on a Veleta 2k × 2k charge-coupled device camera (Olympus-SIS, Munich, Germany).
Asymmetric-flow field-flow fractionation (AF4)
EVP subpopulations (exomeres, < 50 nm with an average of 35 nm in diameter; Exo-S, 60-80 nm in diameter; Exo-L, 90-120 nm in diameter) were separated using AF4 as previously described5,39. Briefly, EVP samples were separated in a short channel (144 mm length, Wyatt Technology, Santa Barbara) with a 10 kDa molecular weight cutoff (MWCO) Regenerated Cellulose membrane (Millipore) on the accumulation bottom wall and a 490 μm spacer (channel thickness). The fractionation was operated by the Eclipse AF4 system (Wyatt Technology). EVP samples were spun at 12,000g for 5 min to remove aggregates before loading onto the AF4 system, and 100 μg of EVPs (1 μg/μl, and 100 μl in total) was injected using the autosampler (Agilent Technologies). Chemstation software (Agilent Technologies) with an integrated Eclipse module (Wyatt Technology) was used to operate the AF4 flow and Astra 6 (Wyatt Technology) was used for data acquisition and analysis.
Flow cytometry
Flow cytometry analyses were performed as previously described40. To identify the liver cells uptaking tumor-derived EVPs, mice were intravenously injected with 10 μg of CellVue Burgundy-labeled EVPs in 100 μl of PBS, or an equivalent volume of mock reaction mixture. At 24 h post injection, mice were sacrificed and a small lobe of murine liver was dissected and digested with Dispase/collagenase/DNase (Roche; 1.5 mg/ml of Dispase II [cat# 4942078001] and collagenase A [cat# 10103586001], 0.1 mg/ml of DNase I [cat# 10104159001]) for 30 min at 37 °C with gentle shaking (40 r.p.m.). Single-cell suspensions were obtained by pipetting and filtering through a 70 μm cell strainer. Cells were washed with flow cytometry buffer (PBS [Ca2+/Mg2+-free] containing 2% BSA and 2 mM of EDTA) and collected by centrifugation at 300g for 5 min. Cell pellets were incubated with ACK Lysing buffer (Gibco, A10492-01) at room temperature for 5 min to remove red blood cells. Cells were resuspended in flow cytometry buffer and one million of cells were incubated with TruStain FcX™ PLUS (anti-mouse CD16/32) antibody (Biolegend, clone S17011E) for 10 min on ice. Subsequently, the cell suspensions were incubated with antibodies (see Supplementary table 28) for 25 min on ice. For intracellular staining, cell suspensions were treated with 0.01% PFA for 10 min and 0.5% Tween20 for 15 min before incubating with antibodies. After antibody labeling, cells were washed with flow cytometry buffer and stained with 0.1 ng/ml of DAPI solution (Thermo Scientific, #62248). Flow cytometry was performed on a Cytek Aurora. Single cells were gated according to FSC and SSC to exclude doublets. Different cell populations were analyzed by gating live cells (DAPI-negative) and then measuring the expression of Cd45, Cd11b, F4/80, Cd31, albumin, Lyve1, and desmin. FlowJo Software was used for data analysis. To analyze the abundance of KCs in the livers of mice treated with liposome (Liposoma BV) or clodronate (Liposoma BV), livers were subjected to flow cytometry analyses of Cd45, Cd11b and F4/80-expressing cells following the method described above.
Tissue processing and immunofluorescence (IF) staining
Liver tissues were collected and fixed in 4% paraformaldehyde (PFA) overnight at 4 °C, immersed in 30% sucrose in PBS overnight at 4 °C, followed by 30% sucrose: OCT (1:1, v/v) incubation for overnight at 4 °C, and then embedded in OCT compound (Sakura, #4583) and frozen on dry ice. OCT-embedded liver tissues were sectioned at 10-μm thickness using LEICA CM3050 S, and slides were stored in −80 °C freezer or used immediately.
For IF staining, slides were dried and cryosections were blocked with universal Fc receptor blocker (Innovex, NB309-15) for 0.5 h, incubated in normal blocking and penetrating buffer (PBS containing 2% BSA and 0.1% Triton X-100), and then incubated with primary antibodies (see Supplementary table 28) overnight at 4 °C. Slides were then washed with PBS for 5 min, incubated with secondary antibodies (see Supplementary Table 28) for 1.5 h, stained with DAPI (1 μg/ml), and mounted with ProLong Diamond Antifade Mountant (Thermo Fisher Scientific, P36970). Images were captured using Confocal Microscope (Zeiss LSM 880).
For precision-cut liver slices, IF staining was performed on free-floating slices using the same protocol as for liver tissue cryosections.
For histological analysis of livers with H&E staining, liver tissues were fixed in 4% PFA overnight at 4 °C and subjected to paraffin embedding. Paraffin-embedded liver tissues were sectioned at 7-μm thickness, and sections were processed for H&E staining and mounted with VectaMount medium (Vector Laboratories, H-5000).
Precision-cut liver slices (PCLS)
Preparation and incubation of the murine PCLS were modified from the previously described protocols40–42. In brief, 6-8 weeks aged C57BL/6 or BALB/c mice (Jackson Laboratories) were anesthetized using 2% isoflurane/1.5% oxygen inhalation, and livers were dissected in Krebs-Henseleit Buffer (KHB, pH 7.4, Sigma, #K3753). Fresh liver was cored using an 8-mm biopsy punch (Acuderm INC). Cores were embedded in microwave-preheated 4% low-melting agarose (Lonza, #50101) in KHB once the agarose cooled to 37 °C. Once solidified, the agarose-embedded liver tissues were superglued to the specimen plate, submersed in the buffer tray containing ice-cold KHB, and cut into slices (250 μm of thickness) using a VT 1200s vibratome (Leica) at the speed of 0.08 mm/sec and amplitude of 2 mm. Groups of two PCLS were transferred onto the 0.4 μm pore polycarbonate membrane cell culture insert (Sigma, Z353086) in 6-well plate with 1.5 ml of medium (William’s E Medium (Thermo Fisher Scientific, #32551020) containing 2% EVP-free FBS, 1× penicillin/streptomycin, 1× L-glutamine and 2g/L glucose (Gibco, A24940-01)) per well, and cultured in a humidified 37 °C incubator with 5% CO2 with agitation (50 r.p.m.).
To visualize the EVP-induced lipid droplet accumulation, the PCLS from naïve mice were cultured overnight, and then treated with PBS or 10 μg/ml of EVPs for 48 h, followed by BODIPY staining as described below.
To measure drug metabolism after EVP administration, the PCLS from naïve mice were cultured overnight, followed by treatment with PBS or 10 μg/ml of B16F10-EVPs in the presence of anti-TNFα antibody (20 μg/ml, BP0058, Clone: XT3.11 from BioXCell) or IgG1 isotype control (20 μg/ml, BP0290 from BioXCell) for 24 h. Then, the PCLS were cultured in media containing substrates for the core Cyp enzymes in the presence of PBS or 10 μg/ml of B16F10-EVPs and co-treated with anti-TNFα antibody (20 μg/ml) or IgG1 isotype control (20 μg/ml) for another 24 h. One milliliter of media was collected to measure drug-metabolizing activity using liquid chromatography-tandem mass spectrometry (LC-MS/MS) as described below, and PCLS were weighed for normalization.
To measure drug metabolism in the livers of PBS-injected control and B16F10 tumor-bearing mice treated with anti-TNFα antibody or IgG1 isotype control (described below), the PCLS from PBS-injected control and anti-TNFα antibody or IgG1 isotype control treated B16F10-Tb mice were cultured overnight, and then cultured in the media containing substrates for the core Cyp enzymes for 24 h. Then, 1 ml of media was collected for drug-metabolizing activity assay using LC-MS/MS and PCLS were weighed for normalization. The substrates for Cyp enzymes and their concentrations are shown in Supplementary table 18.
Lipid droplet staining
For liver tissues, the cryosections were dried and rinsed first with Milli-Q water, followed by PBS. The cryosections were then incubated with autofluorescence quenching reagents (Vector Laboratories, SP-8400-15) for 10 min, and washed thoroughly with PBS for 5 min. Subsequently, the cryosections were stained with 2 μM of BODIPY 493/503 (Thermo Fisher Scientific, D3922) at 37 °C for 15 min and then washed with PBS for 5 min. For primary hepatocytes cultured on chamber slides, the culture media were removed, and hepatocytes were rinsed in PBS three times and stained with 2 μM of BODIPY 493/503 at 37 °C for 15 min, and then washed with PBS for 5 min. For PCLS, the culture media were removed, and PCLS were rinsed in PBS three times and incubated with autofluorescence quenching reagents for 10 min, and washed thoroughly with PBS for 5 min. PCLS were then stained with 2 μM of BODIPY 493/503 at 37 °C for 15 min and washed with PBS for 5 min, followed by 4% PFA fixation for 30 min and PBS washing for 5 min. All samples, including cryosections, hepatocytes and PCLS, were then counterstained with DAPI (1 μg/mL) for 5 min, washed with PBS for 5 min and mounted with ProLong Diamond Antifade Mountant. Lipid droplets were imaged by Confocal Microscope (Zeiss LSM 880) and BODIPY fluorescence intensities were quantified using ImageJ.
Cytokine array
For tumor-bearing mouse models, KCs isolated from control and tumor-bearing mice were first cultured for 2 h. Dead (unattached) cells were removed and viable cells were lysed in RIPA buffer (Sigma) for cytokine array analysis. For EVP education model, KCs isolated from naïve mice were cultured overnight, followed by treatment with PBS or 10 μg/ml of EVPs for 3 d. The conditioned media were then collected for cytokine array analysis. Cytokine array analysis was carried out using the Proteome Profiler Mouse Cytokine Array Kit, Panel A (R&D, ARY006) according to manufacturer’s instructions. Multiple exposure times (1-30 min) of the membranes to X-ray film were applied, and the pixel densities on the developed X-ray film was quantified using ImageJ.
Enzyme-linked immunosorbent assay (ELISA)
To measure KC-secreted IL-6 after treatment with lipopolysaccharide (LPS) in vitro, KCs isolated from naïve mice were cultured overnight, followed by treatment with LPS (1 μg/ml) for 0 h, 1 h, 2 h, 4 h, and 16 h, respectively. The culture media were then collected for IL-6 measurement using the mouse IL-6 ELISA kit (Thermo Fisher, KMC0061) according to the manufacturer’s instructions.
To measure KC-secreted TNFα after treatment with EVPs in vitro, KCs isolated from naïve mice were cultured overnight, followed by treatment with 10 μg/ml of EVPs or an equivalent volume of PBS for 3 d. The conditioned media was then collected for TNFα measurement using the mouse TNFα ELISA kit (R&D, MHSTA50) according to the manufacturer’s instructions.
To determine whether TNFα is secreted by cancer cells, 1 ml of EVP-depleted conditioned media obtained after EVP isolation (i.e., after first spin of 100,000g for 70 min as described above) from cultured B16F10 and K7M2 cells was subjected to TNFα measurement using the mouse TNFα ELISA kit according to the manufacturer’s instructions.
To measure TNFα in the circulation, the retroorbital blood samples were collected from tumor-bearing mice and EVP-educated mice, and their respective controls, using K2EDTA plasma collection tubes (BD, #365974). Plasma TNFα was then measured using the mouse TNFα ELISA kit according to the manufacturer’s instructions.
Murine studies
Treatment of mice was performed in accordance with institutional, IACUC and AAALAS guidelines. All the murine studies were performed according to Weill Cornell Medicine animal protocols #0709-666A and #2016-0057, except for studies using the genetic melanoma murine model, which were performed at The Ohio State University according to animal protocol #2012A00000134. In addition, patient-derived xenograft (PDX) models of primary osteosarcoma were generated in compliance with MSKCC animal protocol #14-02-002. All animal experiments were conducted in compliance with relevant ethical regulations regarding animal research. Animals were monitored for stress, illness or abnormal tissue growth, and euthanized if health deteriorated. Mice that died before the experimental endpoint were excluded from the analysis. Female C57BL/6, BALB/c, Nude (outbred) and NOD/SCID/IL2Rγnull (NSG) mice aged 6-8 weeks were obtained from The Jackson Laboratory. BALB/c mice aged 6-8 weeks were used for the implantation of murine breast cancer cell lines (67NR and 4T1) and murine osteosarcoma cell line (K7M2), as well as treatment with osteoblast-EVPs, K7M2-CL-EVPs, K7M2-TE-EVPs, bone-TE-EVPs; C57BL/6 mice aged 6-8 weeks were used for the implantation of murine melanoma cell lines (B16F1 and B16F10), and treatment with B16F10-CL-EVPs, B16F10-TE-EVPs and skin-TE-EVPs. Nude (outbred) mice aged 6-8 weeks were used for implantation of human melanoma cell line SK-MEL-192. NSG mice aged 6-8 weeks were used for PDX models. Mice were housed in the animal facility under conventional conditions with a light (12 hours dark/light circle)-, humidity (30%-70%)- and temperature (70 °F −74 °F)-controlled environment. No statistical method was used to pre-determine the sample size and no method of randomization was used to allocate animals to experimental groups.
For tumor cell implantation, 1×105 of 67NR or 4T1 cells in 50 μl of PBS were injected into the mammary fat pad of BALB/c mice; 5×105 of B16F1 or B16F10 cells in 100 μl of PBS were subcutaneously injected into C57BL/6 mice; 1×106 of K7M2 cells in 10 μl of PBS were injected into the tibias of BALB/c mice; 5×105 of SK-MEL-192 cells in 100 μl of PBS were subcutaneously injected into nude (outbred) mice. Tumor growth was monitored by measuring the tumor size with a digital caliper using the formula: tumor volume (mm3) = (width in mm)2 × (length in mm) /2. Mice were euthanized 3 weeks (for B16F1-, B16F10-, 67NR-, 4T1- and SK-MEL-192-Tb models), or 4 weeks (for K7M2-Tb model) post tumor cell implantation for tissue collection. The maximal tumor size in mice that was permitted by Weill Cornell Medicine IACUC is 2 cm in any one dimension, and this limit was not exceeded in any of the experiments. Mice injected with an equivalent volume of PBS following the same procedure were used as the control group.
For the genetic melanoma murine model, C57BL/6 TN mice43, carrying a Cre-inducible NrasQ61R oncogene, were treated topically with 20 mM of 4-hydroxytamoxifen (4-OHT) on postnatal days 1 and 2 to activate melanocyte-specific CRE recombination from the Tyr-CRE-ER(T2) transgene44. On postnatal day 3, pups received 1.5 SED (standard erythemal dose) of solar radiation as described45. Animals were monitored for spontaneous tumor formation and euthanized upon reaching exclusion criteria (tumor length ≥ 1.6cm or ulceration ≥ 2mm) and this limit was not exceeded in any of the experiments. Non-tumor bearing control mice carried the NrasQ61R, p16fl/f and Tyr-CRE-ER (T2) alleles but were not treated to induce CRE activity or tumor formation. To increase melanoma susceptibility, eight of the tumor-bearing mice were functionally null Mc1r, a gene important for eumelanin production.
To generate PDX models of human primary osteosarcoma, freshly surgically excised tumor specimens obtained from MSKCC patients were transplanted into female NOD/SCID/IL2Rγnull (NSG) mice. All patients provided informed consent for tissue donation according to the IRB approved protocol (MSKCC IRB #06-107 and #17-067), and de-identified patient information was described in Supplementary Table 5a. In brief, freshly obtained tumor tissue specimens were rinsed with PBS (EDTA-free), minced into 2-4 mm size pieces, and then transplanted subcutaneously into mice. Typically, PDX tumor growth became evident during the first 1-2 weeks after engraftment and mice were euthanized for tissue collection within a total of 4-8 weeks after engraftment. Primary tumor growth and metastases were followed carefully by small animal Magnetic Resonance Imaging (MRI). Mouse MRI was carried out on a 400 MHz Bruker 9.4T Biospec scanner equipped with a 530 mT/m ID 114 mm gradient (Bruker Biospin MRI GmbH, Ettlingen, Germany). Radio frequency (RF) excitation and acquisition was achieved by a Bruker 40 mm quadrature birdcage resonator. The mice were immobilized with 2% isoflurane (Baxter Healthcare Corp., Deerfield, IL) gas in oxygen. Animal respiration was monitored with a small animal physiological monitoring system (SA Instruments, Inc., Stony Brook, NY). Scout images along three orthogonal orientations were first acquired for animal positioning. Mouse thoracic imaging was performed using a respiratory gated gradient-echo sequence with the time of repetition (TR) 170 ms, time of echo (TE) 1.6 ms, 10 average, a slice thickness of 0.5 mm and an in-plane resolution of 198 μm × 146 μm. For mouse liver imaging, axial T2-weighted images using fast spin-echo rapid acquisition with relaxation enhancement (RARE) sequence was acquired with TR 3.1 s, TE 11 ms, RARE factor of 8, slice thickness of 0.7 mm, 3 averages and in-plane resolution of 113 μm × 168 μm. The maximal tumor size in mice that was permitted by MSKCC IACUC is 2 cm in any one dimension, and this limit was not exceeded in any of the experiments.
For the murine model of spontaneous pancreatic ductal adenocarcinoma (PDAC), tamoxifen-inducible KPC mice (Krastm4Tyj Trp53tm1Brn Tg[Pdx1-cre/Esr1*]#Dam/J, The Jackson Laboratory, JAX 032429 ) were used, as previously described15. Briefly, Trp53 excision was induced in pups via lactation upon administration of tamoxifen (6 mg/mouse) via oral gavage to the dam. Mice carrying the KrasG12D mutation in one allele (KrasWT/G12D) developed pancreatic intraepithelial neoplasia (PanIN) lesions at 8-10 weeks of age, which progressed into local PDAC at 10-14 weeks and invasive PDAC at 15-18 weeks, associated with distant metastasis to several sites including lymph nodes, liver, lungs, diaphragm, and spleen. KrasWT/WT mice from the same litter were used as healthy controls. For PDAC tumor-bearing mice, the tumor volume cannot be appreciated as tumors grow internally, but the mice will be euthanized if discomfort, including loss of appetite, inactivity, difficulty breathing, hunched posture and withdrawal from other mice. The exclusion criteria were not exceeded in any of the experiments.
For in vivo EVP biodistribution assessment, CellVue NIR815-labeled EVPs (10 μg) resuspended in 100 μl of PBS, or an equivalent volume of mock reaction mixture, were intravenously injected into naïve mice. At 24 h post injection, livers were collected and imaged using Odyssey CLx (LI-COR). Data quantification was performed using Image Studio (version 5.2).
For in vivo EVP education, 10 μg of EVPs, exomeres, Exo-S, or Exo-L, resuspended in 100 μl of PBS, were intravenously injected into mice every other day for 4 weeks, mimicking continuous and systemic EVP release from primary tumors7,10. Mice injected with an equivalent volume of PBS following the same procedure served as the control group. One day post the last injection, the EVP-educated mice and control mice were euthanized, and tissues were collected for downstream analysis.
For in vivo conditioned media (CM) education, EVP-depleted CM obtained after EVP isolation (i.e., the media supernatant after first spin of 100,000g for 70 min as described above) from cultured cancer cells was concentrated using Amicon Ultra-15 centrifugal filters with 10 KDa cutoff (Millipore, UFC901024) at 4,000g at 4 °C. In general, 5 ml of cell culture CM yields approximately 10 μg of EVPs. The same volume of EVP-depleted CM was further concentrated to a volume of 200 μl and used for each injection. Blank media (DMEM supplemented with 10% EVP-depleted FBS and 1× penicillin/streptomycin) was also concentrated to obtain control CM. Two hundred microliters of concentrated EVP-depleted CM, equivalent to 10 μg of EVPs, were intra-peritoneally injected into mice every other day for 4 weeks. Mice injected with 200 μl of control CM according to the same procedure served as the control group. One day after the last injection, CM-educated and control mice were euthanized, and tissues were collected for downstream analysis.
For in vivo education with both EVPs and CM, 10 μg of EVPs in 100 μl of PBS and 200 μl of concentrated CM were simultaneously injected into mice via intravenous injection and intraperitoneal injection, respectively, every other day for 4 weeks. One day post the last injection, the educated mice were euthanized, and tissues were collected for downstream analysis.
For KC depletion in naïve mice, liposome or clodronate was intravenously injected into mice at a dosage of 100 μl of suspension per 10 g of mouse weight. The concentration of clodronate in the suspension is 5 mg/ml. At 24 h post injection, mice were euthanized and livers were sectioned into PCLS using a VT 1200s vibratome. PCLS were then subjected to immunofluorescence staining to confirm the efficiency of KC depletion, or cultured for EVP treatment and subsequent BODIPY staining as described above.
For KC depletion in tumor-bearing mice, liposome or clodronate (100 μl of suspension per 10 g of mouse weight) was intravenously injected into B16F10-Tb mice on day 12, day 15, and day 19; or into K7M2-Tb mice on day 15, day 18, day 22, and day 25 post tumor cell implantation (see Extended Data Fig. 8a). Mice were euthanized at the endpoint, and liver tissues were collected for flow cytometry, immunofluorescence and BODIPY staining.
For in vivo neutralization assays, InVivoPlus anti-TNFα (BP0058, Clone: XT3.11) or InVivoPlus IgG1 Isotype control (BP0290) from BioXCell were intraperitoneally injected into the mice at a dosage of 200 μg/mouse every other day starting on day 12 post injection of B16F10 tumor cells; or on day 15 post injection of K7M2 tumor cells; or on day 15 post education of B16F10-CL-EVPs or K7M2-CL-EVPs (see Extended Data Fig. 9l). One day post the last injection, mice were euthanized and tissues were collected for downstream analysis.
Chemotoxicity analysis were performed on B16F10-CL-EVP and K7M2-CL-EVP education models. For the B16F10-CL-EVP education model, mice were intravenously injected with PBS or 10 μg of B16F10-CL-EVPs every other day for 4 weeks, and then intraperitoneally injected with dacarbazine (60 mg/kg) or 0.9% NaCl together with intravenous injection of PBS or 10 μg of B16F10-CL-EVPs every other day, for a total of 4 injections. One day after the last injection, retroorbital blood was collected for complete blood count (CBC) by the Laboratory of Comparative Pathology at MSKCC. For the K7M2-CL-EVP education model, mice were intravenously injected with PBS, 10 μg of osteoblast-EVPs or K7M2-CL-EVPs every other day for 4 weeks, and then intraperitoneally injected with doxorubicin (1 mg/kg) or DMSO every 24 h together with intravenous injection of PBS, 10 μg of osteoblast- or K7M2-CL-EVPs every other day for 25 days. The cumulative dose of doxorubicin was 25 mg/kg. One day after the last injection, mice were subjected to echocardiography. Dacarbazine (MedChemExpress, HY-B0078) was dissolved in 0.2 μm-filter sterilized 0.9% NaCl solution with sonication to prepare the dacarbazine suspension. Doxorubicin hydrochloride (MedChemExpress, HY-15142) was dissolved in DMSO to prepare the doxorubicin hydrochloride solution.
Echocardiography
Transthoracic echocardiography was performed using a VisualSonics Vevo 3100 system equipped with MX400 transducer (Visual Sonics). Measurements were performed on anesthetized mice under body-temperature controlled conditions. The echocardiographer was blinded to treatment conditions. Anesthesia was induced by 3% isoflurane and confirmed by lack of response to firm pressure on one of the hind paws.
During echocardiogram acquisition, isoflurane was reduced to 1.0-1.5% and adjusted to maintain a heart rate in the range of 400-500 beats per minute. Left ventricular ejection fraction, fractional shortening, and left ventricular end-diastolic diameter were obtained from parasternal long-axis M-mode scans at the basal-to-mid ventricular level. Following echocardiography, all mice recovered from anesthesia without difficulty. All parameters were measured on three separate heartbeats, and means were presented.
Percent fractional shortening was calculated by the formula (LVIDd – LVIDs)/LVIDd × 100%. Left ventricular ejection fraction (LVEF) was calculated automatically with VisualSonics Vevo LAB image analysis software from calculated LV volumes (LVOL) using the formula LVEF = [LVOLd – LVOLs]/LVOLd, where LVOLd or LVOLs were calculated by the cubed sphere formula LVOL = (7/(2.4+LVID) × LVID3) at end-diastole or end-systole, respectively.
Human studies
Fresh human liver tissues were obtained intraoperatively at MSKCC (IRB #15-015) from patients undergoing pancreatectomy for either localized resectable pancreatic cancer or non-cancerous pancreatic/peri-pancreatic lesions (details of included pathologies are shown in Supplementary Table 5b). None of the patients had any evidence of distant metastasis at the time of surgery. Patients with pancreatic cancer were followed for at least 21 months after surgery and site(s) of initial recurrence was identified. In addition, fresh, grossly normal liver tissues were obtained from patients undergoing hepatectomy for hepatic adenomas at MSKCC (IRB #06-107). Intraoperative liver biopsies were collected from patients with germline CDH1 mutations undergoing risk-reducing gastrectomy at the NIH (IRB #13-C-0076). Although these patients had very early-stage incidental gastric cancer in the stomach specimens, none had evidence of metastasis on imaging or by pathologic examination of sampled tissues (lymph nodes, skin, spleen, liver and jejunum). Lastly, unaffected liver specimens from decedents who have previously been diagnosed with pancreatic ductal adenocarcinoma were obtained from the University of Nebraska Medical Center (UNMC)’s Tissue Bank through the Rapid Autopsy Program (RAP) in compliance with IRB 091-01, and de-identified patient information was described in Supplementary Table 5b. To ensure specimen quality, organs were harvested within three hours post-mortem. All individuals provided informed consent for tissue donation according to protocols approved by the Institutional Review Boards of MSKCC, NIH and UNMC. All patient liver tissue-associated experiments were performed according to the protocol approved by the Institutional Review Boards of Weill Cornell Medicine (IRB #0604008488). The study is compliant with all relevant ethical regulations regarding research involving human participants.
Gas-chromatography mass-spectrometry (GC/MS) metabolomics and data analysis
Approximately 150 mg of liver tissues, or 150-500 μg of EVPs, were homogenized in 1 ml or 200 μl of methanol: chloroform (1:2, v/v) using Bead Rupture 24 (OMNI), respectively. The homogenized liver tissues or EVPs were then subjected to metabolite profiling MS at the Metabolomics Laboratory of the Roy J. Carver Biotechnology Center, University of Illinois at Urbana-Champaign. In brief, samples were dried in a vacuum and derivatized with 50 μl of methoxyamine hydrochloride (Sigma, MO, USA) (40 mg/ml in pyridine) at 50 °C for 1.5 h, then with 50 μl of MSTFA + 1%TMCS (Thermo, MA, USA) at 70 °C for 2 h, followed by incubation at room temperature for 2 h. 30 μl of 1 mg/ml hentriacontanoic acid was added to each sample prior to derivatization to serve as an internal standard. Metabolite profiles were acquired using a GC-MS system (Agilent Inc, CA, USA) consisting of an Agilent 7890 gas chromatograph, an Agilent 5975 MSD and 7683B autosampler as previously described46. Gas chromatography was carried out on a ZB-5MS (60 m × 0.32 mm I.D. and 0.25 μm film thickness) capillary column (Phenomenex, CA, USA). The inlet and MS interface temperatures were 250 °C, and the ion source temperature was adjusted to 230 °C. An aliquot of 2 μl was injected with the split ratio of 10:1. The helium carrier gas was kept at a constant flow rate of 2.4 ml/min. The temperature program was: isothermal heating at 70 °C for 5 min, followed by an oven temperature increase of 5 °C/min to 310 °C after which a final 10 min incubation at 310 °C. The mass spectrometer was operated in positive electron impact mode (EI) at 69.9 eV ionization energy at m/z 30-800 scan range. The spectra of all chromatogram peaks were evaluated using the AMDIS 2.71 (NIST, MD, USA) using a custom-built MS database (460 unique metabolites). Throughout the sample preparation, data-acquisition and data-preprocessing, samples were compared to the QCs to evaluate potential variation that may have arisen in the dataset throughout the analytical study. All known artificial peaks were identified and removed prior to data mining. To allow comparison between samples, all data were normalized to the internal standard in each chromatogram and sample volume. The instrument variability was within the standard acceptance limit (5%).
Data derived from the metabolomics MS were analyzed using MetaboAnalyst 4.0 (Ref. 47). Briefly, following imputation of missing values, i.e., removing features with >25% missing values and estimating the remaining missing values using KMN (feature-wise), data were normalized by Auto scaling (mean-centered and divided by standard deviation of each variable). Partial Least-Squares Discriminant Analysis (PLS-DA) was performed to visualize the discrimination of groups. Significantly changed metabolites (P<0.05 for tumor-bearing models and P<0.1 for EVP-education models) identified by t-test were utilized for generating heatmap and subsequent metabolite set enrichment analysis (MSEA) based on the Small Molecule Pathway Database (SMPDB).
Lipidomics MS and data analysis
Approximately 150 mg of liver tissues were homogenized in 1 ml of methanol: chloroform (1:2, v/v) using Bead Rupture 24, and then subjected to lipidomics MS as previously described5. In brief, samples were first sonicated with a Model Q700 QSonica sonicator equipped with an Oasis 180 Chiller (4 °C; amplitude, 95; process, 5 min; pulse on 30 s; plus off 55 s), centrifuged at 14,800 r.p.m. for 10 min at 4 °C, then 50 μl of the extract supernatant was spiked with 2 μl, 50 μg/ml, of internal standard mixture (Cer 18:1/12:0; PC 12:0/12:0; PE 14:0/14:0; PG 14:0/14:0; PS 14:0/14:0). Subsequently, the samples were analyzed by using the Thermo Q-Exactive MS system at the Metabolomics Laboratory of the Roy J. Carver Biotechnology Center, University of Illinois at Urbana-Champaign. The software Xcalibur 3.0.63 was used for data acquisition and analysis. A Dionex Ultimate 3000 series HPLC system (Thermo) was used, and LC separation was performed on a Thermo Accucore C30 column (2.1 × 150 mm, 2.6 μm) with mobile phase A (60% acetonitrile: 40% H2O with 10 mM ammonium formate and 0.1% formic acid) and mobile phase B (90% isopropanol: 10% acetonitrile with 10 mM ammonium formate and 0.1% formic acid) and a flow rate of 0.3 ml/min. The linear gradient was as follows: 0-2 min, 80% A; 4 min, 55% A; 12 min, 35% A; 18 min, 15% A; 20-28 min, 0% A; 28.1-36 min, 80% A. The autosampler was set to 15 °C and the column was kept at 45 °C. The injection volume was 10 μl. Mass spectra were acquired under both positive (sheath gas flow rate, 50; auxiliary gas flow rate, 13; sweep gas flow rate, 3; spray voltage, 3.5 kV; capillary temperature, 263 °C; auxiliary gas heater temperature, 425 °C) and negative (sheath gas flow rate, 50; auxiliary gas flow rate, 13; sweep gas flow rate, 3; spray voltage, −2.5 kV; capillary temperature, 263 °C; auxiliary gas heater temperature, 425 °C) electrospray ionization. The full scan mass spectrum resolution was set to 70,000 with a scan range of m/z 230-1,600 and the automatic gain control (AGC) target was 1E6 with a maximum injection time of 200 ms. For MS/MS scans, the mass spectrum resolution was set to 17,500 and the AGC target was 5E4 with a maximum injection time of 50 ms. The loop count was 10. The isolation window was 1.0 m/z with normalized collision energy (NCE) of 25 and 30 eV. For data analysis, LipidSearch (v.4.1.30, Thermo) was used for lipid identification. The lipid signal responses were normalized to the corresponding internal standard signal response. For those lipid classes without corresponding internal standard, positive lipid ion signals were normalized with the signal of internal standard Cer 18:1/12:0 and negative ion signals were normalized with the signal of internal standard PG 14:0/14:0.
The amount of lipid classes within a sample was calculated by adding that of each of the individual molecular species quantified within a specific lipid class as previously described5. For lipid species, following imputation of missing values, i.e., removing features with >25% missing values and estimating the remaining missing values using KMN (feature-wise), data were normalized by Auto scaling (mean-centered and divided by standard deviation of each variable). PLS-DA was performed to visualize the discrimination of groups using MetaboAnalyst 4.0. Statistical significance was determined by t-test and P value < 0.05 was considered statistically significant. Volcano plots were generated using Prism 9 software (GraphPad).
Free fatty acids (FFA) analysis
Approximately 300 μg of EVPs were dissolved in 600 μl of methanol: chloroform (1:2, v/v) for FFA analysis at the Metabolomics Laboratory of the Roy J. Carver Biotechnology Center, University of Illinois at Urbana-Champaign. Raw samples were converted to fatty acid methyl esters (FAMEs) using (trimethylsilyl)diazomethane hexanes with a 1 h incubation at room temperature. Derivatized samples were subjected to GC/MS analysis using the Agilent 7890B GC with 5977A/Extractor XL MS system. The gas chromatography separation was performed on a ZB-5MS (60 m × 0.32 mm I.D. and 0.25 μm film thickness) capillary column (Phenomenex, CA, USA). The inlet and MSD interface temperatures were both at 250 °C, with the ion source temperature adjusted to 230 °C. The helium carrier gas was kept at a constant flow rate of 2 ml/min. The temperature program was as follows: initial 5-min isothermal heating at 150 °C, followed by an oven temperature increase of 5 °C/min to 310 °C (3 min isothermal). Mass spectra were recorded in the m/z 50-800 scanning range. Data analysis was performed using Prism 9 software (GraphPad), and statistical significance (P value < 0.05) was determined by multiple unpaired t-test.
FFA content in the EVPs isolated from DMSO or C75 treated cells was determined using Free Fatty Acid Quantitation Kit (Sigma, MAK044). In brief, 50 μg of EVPs were homogenized in 250 μl of a 1% (w/v) Triton X-100 in chloroform solution. Samples were spun at 13,000g for 10 minutes to remove insoluble material, and the organic phase (lower phase) was collected and air dried at 50 °C to remove chloroform. Samples were vacuum-dried for 30 minutes to remove trace chloroform and dissolved in 200 μl of fatty acid assay buffer by vortexing extensively for 5 minutes. Fifty microliters of the solution were subjected to FFA quantification following the manufacturer’s instructions.
Drug-metabolizing activity analysis
Drug-metabolizing activity analysis was performed at the Metabolomics Laboratory of the Roy J. Carver Biotechnology Center, University of Illinois at Urbana-Champaign. The collected media were analyzed using the Triple 6500+ LC-MS/MS system (Sciex, Framingham, MA). Software Analyst 1.7.1 was used for data acquisition and analysis. The 1260 Infinity II HPLC system (Agilent Technologies, Santa Clara, CA) includes a degasser, an autosampler, and a binary pump. The LC separation was performed on an Agilent SB-Aq (4.6 × 50 mm, 5 μm) with mobile phase A (0.1% formic acid in water) and mobile phase B (0.1% formic acid in acetonitrile). The flow rate was 0.3 ml/min. The linear gradient was as follows: 0-1 min, 95% A; 10-14 min, 2% A; 14.1-20 min, 95% A. The autosampler was set at 10 °C and the injection volume was 10 μl. Mass spectra were acquired positive electrospray ionization (ESI) with the ion spray voltage of 5500 V. The source temperature was 400 °C. The curtain gas, ion source gas 1, and ion source gas 2 were 35, 50, and 65 psi, respectively. Multiple reaction monitoring (MRM) was used for quantitation: acetaminophen m/z 152.0 --> m/z 43.0; 4-hydroxytolbutamide m/z 287.0 --> m/z 188.0; dextrorphan m/z 258.1 --> m/z 115.0; 1-hydroxymidazolam m/z 342.2 --> m/z 168.1; hydroxybupropion m/z 256.1 --> m/z 167.0.
LC-MS/MS analysis of 12-O-tetradecanoylphorbol-13-acetate (TPA) in EVP samples
LC-MS/MS analysis of TPA in EVP samples was performed at the Duke Proteomics and Metabolomics Core Facility. Approximately 100 μg of EVPs resuspended in PBS were vortexed for 2 min, and subjected to ultrasonication for 20 min. The ultrasonicated EVPs were then centrifuged at 10,000 rpm for 10 min at 4 °C, and the supernatants were collected for LC-MS/MS injection. Samples were analyzed using the 6500+ QTRAP LC-MS/MS system (Sciex, Framingham, MA). Software Analyst 1.7.1 was used for data acquisition and analysis. The Sciex ExionLC UPLC system includes a degasser, an AD autosampler, an AD column oven, a controller, and an AD pump. The LC separation was performed on an Agilent Zorbax Eclipse plus C18 (2.1×50mm, 1.8 μm) with mobile phase A (0.2% formic acid in water) and mobile phase B (0.2% formic acid in acetonitrile). The flow rate was 0.4 ml/min. The linear gradient was as follows: 0-1 min, 60% A; 5-7 min, 0% A; 7.1-8.5 min, 60% A. The autosampler was set at 10 °C and the column was kept at 40 °C. The injection volume was 15 μl. Mass spectra were acquired under negative ESI with the ion spray voltage of −4500 V. The source temperature was 500 °C. The curtain gas, ion source gas 1, and ion source gas 2 were 32, 60, and 60 psi, respectively. MRM was used for quantitation: m/z 615.2 --> m/z 227.1.
RNA-seq and computational analysis
For RNA-seq of murine livers, total RNA was quantified using Nanodrop 2000c (Thermo Scientific), and RNA quality was assessed by Agilent 2100 Bioanalyzer (Agilent Technologies, Inc.). TruSeq stranded mRNA library preparation and RNA-seq using Illumina HiSeq 4000 or Illumina NovaSeq 6000 were performed in the Genomics Resources Core Facility at Weill Cornell Medicine. The raw sequencing reads in BCL format were processed through bcl2fastq 2.19 (Illumina) for FASTQ conversion and demultiplexing. RNA reads were aligned and mapped to the mm9 mouse reference genome by TopHat2 (Version2.0.11) (http://ccb.jhu.edu/software/tophat/index.shtml), and transcriptome reconstruction was performed by Cufflinks (Version 2.1.1) (http://cole-trapnell-lab.github.io/cufflinks/). The abundance of transcripts was measured with Cufflinks in Fragments Per Kilobase of exon model per Million mapped reads (FPKM). Gene expression profiles were constructed for differential expression, clustering, and principal component analyses using the DESeq2 package (https://bioconductor.org/packages/release/bioc/html/DESeq2.html). For differential expression analysis, pairwise comparisons between two or more groups were performed using parametric tests where read-counts follow a negative binomial distribution with a gene-specific dispersion parameter. Corrected p-values were calculated based on the Benjamini-Hochberg method to adjust for multiple testing.
RNA-seq of human liver biopsies was performed at Integrated Genomics Operation (IGO), MSKCC. Briefly, total RNA from 5 mg of frozen liver tissue was extracted using the RNeasy Mini Kit (QIAGEN, catalog no. 74104) according to the manufacturer’s instructions. After RiboGreen quantification and QC analysis of 1 μg of RNA using Total RNA Nano chip on Agilent Bioanalyzer 2100, samples were subjected to ribosomal RNA depletion and library preparation using the TruSeq Stranded Total RNA LT Kit (Illumina, catalog no. RS-122-1202) according to the manufacturer’s instructions with 6-8 cycles of PCR. Samples were barcoded and run on a HiSeq 2500 in Rapid or High Output Mode or a HiSeq 4000 in a 50bp/50bp paired end run, using the HiSeq Rapid SBS Kit v2, TruSeq SBS Kit v4, or HiSeq 3000/4000 SBS Kit, respectively (Illumina). Alignment metrics were calculated by GATK’s CollectRnaSeqMetrics and AlignmentSummaryMetrics to obtain raw STAR alignments and htseq count files. On average, 88 million paired reads were generated per sample and 32% of the data mapped to the human GENECODE 18 transcriptome. Standard pipeline analyses were performed by IGO and subsequent comparisons were performed using DESeq2, R software.
For B16F10 and K7M2 tumor-bearing models, a pre-ranked gene list was created with shared differently expressed genes (DEGs) (q < 0.05) in livers from both B16F10 and K7M2 models, where Log2FC values were used for ranking. Gene set enrichment analysis was then conducted for those pre-ranked gene lists. For all the other models, except RNA-seq data from human liver biopsies, genes were ranked based on the sign of log2FC*(−log10P value) and subjected to gene set enrichment analysis. For RNA-seq data from human liver biopsies, the normalized gene expression values were utilized for gene set enrichment analysis using weighted enrichment statistic and Signal2Noise method (metric for ranking genes). Gene set enrichment analysis was performed using standalone GSEA software (v4.1.0 & v4.3.2) (downloaded from http://www.gsea-msigdb.org/gsea/downloads.jsp) against the HALLMARK or KEGG gene sets48,49.
Statistical analysis
Statistical significance was determined by the two-tailed, unpaired Student’s t-test, or one-way ANOVA with post hoc Tukey’s test, or two-way ANOVA followed with Bonferroni’s multiple comparisons test, or two-way ANOVA followed with Fisher’s LSD test, or multiple unpaired t-test using Prism 9 software (GraphPad). The P-value and FDR q-value were determined by Kolmogorov-Smirnov test for gene set enrichment analysis48. P value < 0.05 was considered statistically significant. All values are mean ± standard error of the mean (s.e.m.). The number of independent biological replicates for each experiment and the sample size of each experimental group/condition are provided in the figure legends. ImageJ (version 1.53i) was used for image processing and analysis.
Extended Data
Extended Data Figure 1. Primary tumors dysregulate the metabolism of metastasis-free livers.
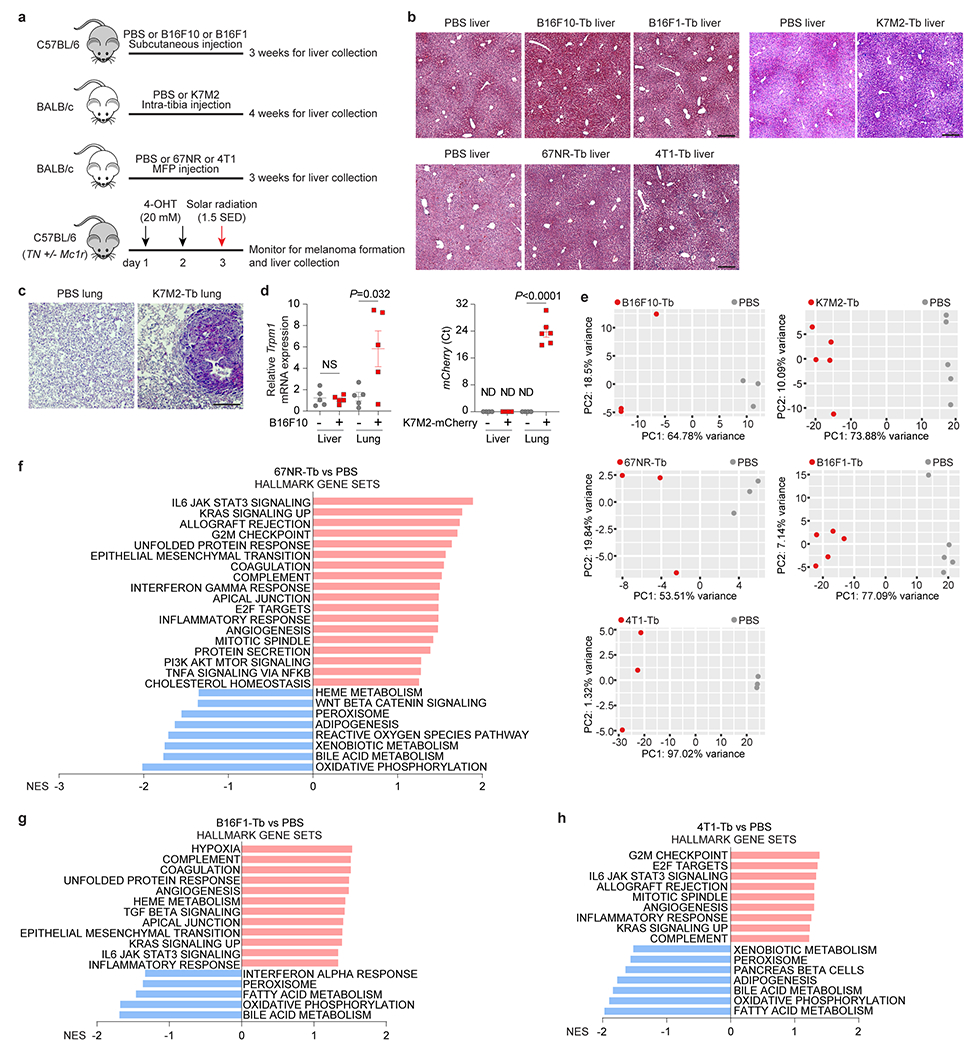
a, Schematic representation of murine tumor models utilized in this study. B16F10, K7M2, 67NR, B16F1 and 4T1 cells, were orthotopically injected into syngeneic mice. Age and sex-matching mice injected with PBS were used as controls of these experimental murine models. C57BL/6 TN mice carrying a Cre-inducible NrasQ61R oncogene were employed for generating spontaneous melanoma. Non-tumor bearing control mice carried the NrasQ61R, p16fl/fl and Tyr-CRE-ER(T2) alleles but were not treated to induce CRE activity or tumor formation. Functionally null Mc1r increased melanoma susceptibility. b, Representative H&E staining images of the livers from B16F10-Tb and B16F1-Tb mice (top, left), K7M2-Tb mice (top, right), 67NR-Tb and 4T1-Tb mice (bottom), and their respective controls. This experiment was repeated three times independently with similar results. c, Representative H&E staining images of lung metastases in the PBS control mice and K7M2-Tb mice. This experiment was repeated three times independently with similar results. d, qRT-PCR analysis of Trpm1 and mCherry expression in livers and lungs of mice implanted with B16F10 and mCherry-expressing K7M2 cells, respectively, compared to their controls. n=5 per group for B16F10-Tb model, n=4 control and n=6 K7M2-mCherry-Tb mice. NS, not significant; ND, not detected. e, Principal component analysis (PCA) of gene expression in the livers from mice implanted with B16F10, or K7M2, or 67NR, or B16F1, or 4T1 tumor cells, compared to their respective PBS-injected controls. Results showed that the gene expression profiles of livers from tumor-bearing mice independently segregated from their respective controls. n=3 mice per group for B16F10-Tb, 67NR-Tb and 4T1-Tb models; n=5 mice per group for K7M2-Tb and B16F1-Tb models. f-h, GSEA of the gene expression profiles, which were ranked based on the sign of log2FC*(−log10P value), in the livers from 67NR-Tb mice (f), B16F1-Tb mice (g), or 4T1-Tb mice (h), compared to their respective PBS-injected controls, using hallmark gene sets, and the significantly changed signaling pathways with FDR<0.2 are shown. n=3 mice per group for 67NR-Tb and 4T1-Tb models; n=5 mice per group for B16F1-Tb model. Gene lists for signaling pathways are shown in Supplementary Tables 19–21. Scale bars for (b,c), 200 μm. P values were determined by two-tailed, unpaired Student’s t-test (d).
Extended Data Figure 2. Tumors induce metabolic dysfunction of the metastasis-free livers.
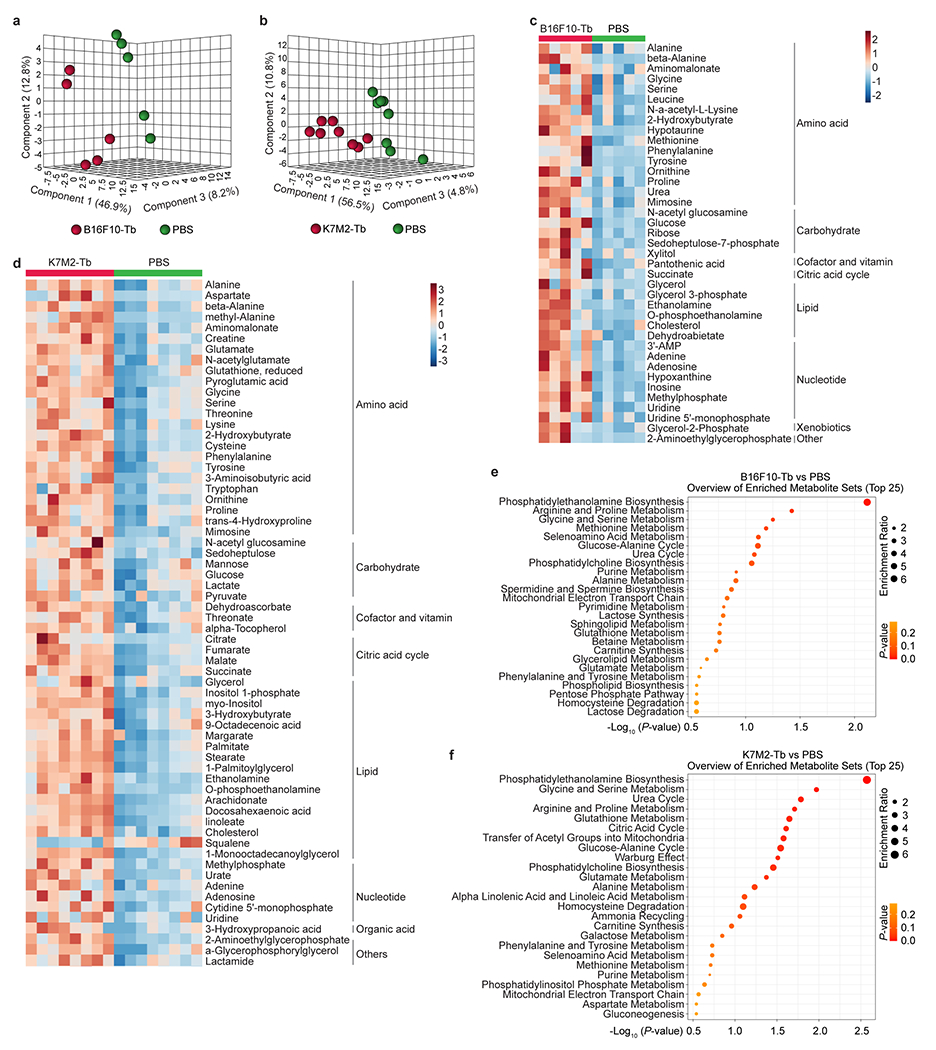
a,b, PLS-DA plots of metabolites detected in the livers from B16F10-Tb mice (a) and K7M2-Tb (b) mice, compared to their PBS-injected controls. Metabolomics MS data are shown Supplementary Tables 1 and 2. n=5 B16F10-Tb mice and controls; n=8 K7M2-Tb mice and controls. c,d, Heatmaps showing the significantly changed metabolites, classified into different groups, in the livers from B16F10-Tb mice (c) and K7M2-Tb mice (d), compared to their PBS-injected controls. n=5 B16F10-Tb mice and controls; n=8 K7M2-Tb mice and controls. e,f, Metabolite set enrichment analysis of metabolites (shown in c,d) significantly changed in the livers of B16F10-Tb (e) and K7M2-Tb (f) mice, compared to their respective PBS-injected controls. n=5 B16F10-Tb mice and controls; n=8 K7M2-Tb mice and controls. P values were determined by the hypergeometric test (e,f).
Extended Data Figure 3. Tumors induce lipid accumulation in metastasis-free livers.
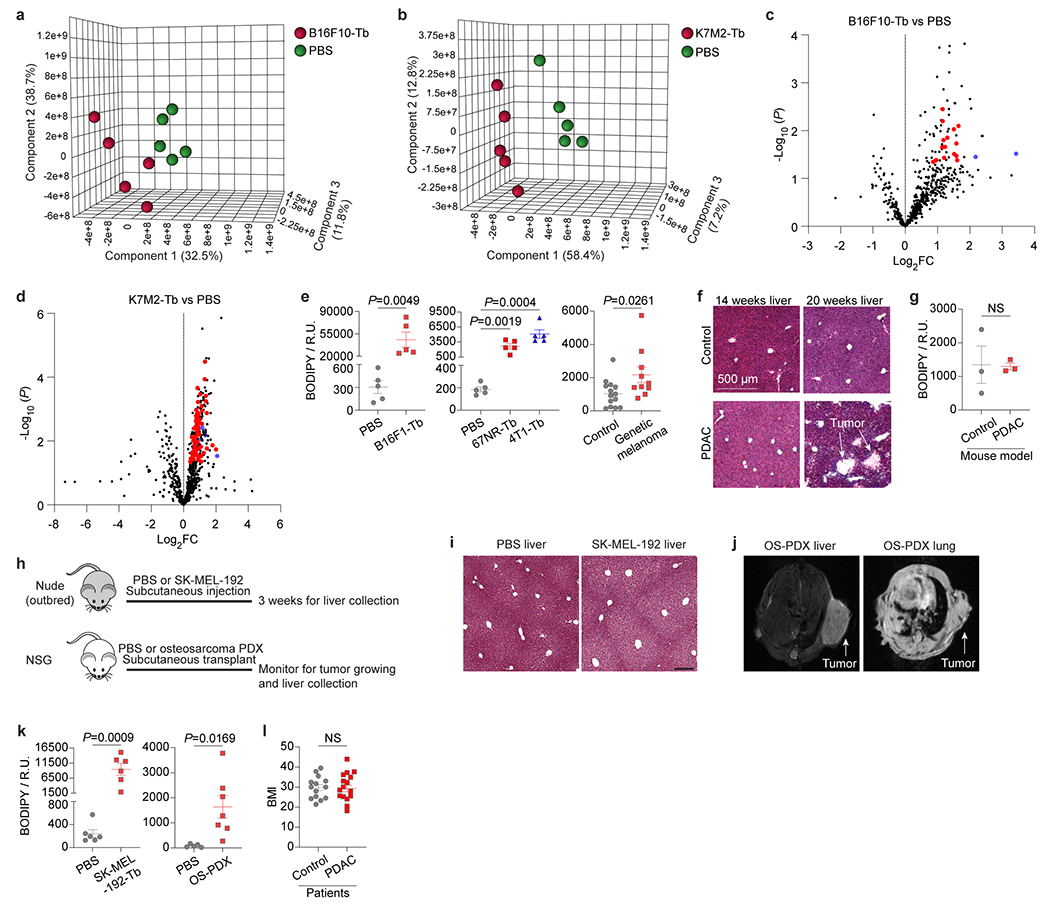
a,b, PLS-DA plots of the lipid species in the livers from B16F10-Tb mice (a) and K7M2-Tb mice (b), compared to their PBS-injected controls. Lipidomics MS data are shown in Supplementary Tables 3 and 4. n=5 mice per group. c,d, Volcano plots showing the significantly (P<0.05) enriched triglyceride species (red) and cholesteryl ester species (blue) in the livers from B16F10-Tb mice (c) and K7M2-Tb mice (d), compared to their PBS-injected controls. n=5 mice per group. e, Statistical analysis of BODIPY staining of livers from murine melanoma B16F1-Tb mice, murine breast cancer 67NR-Tb and 4T1-Tb mice, genetic melanoma-bearing mice, and their respective controls. n=5 mice per group for B16F1, 67NR and 4T1 models; n=14 control and n=10 tumor-bearing mice for the genetic melanoma model. f, Representative H&E staining images of livers from KPC mice. Liver metastasis was observed in 20-week, but not 14-week tumor bearing mice. g, Statistical analysis of BODIPY staining of the livers from 14-week PDAC tumor-bearing mice, compared to non-PDAC mouse controls. n=3 mice per group. h, Schematic representation of murine tumor models utilized in this study. SK-MEL-192 cells were subcutaneously injected into nude (outbred) mice. Patient-derived xenograft (PDX) models of osteosarcoma were generated by transplanting surgically excised primary tumor specimens from clinical patients into NOD/SCID/IL2Rγnull (NSG) mice. Age and sex-matching mice injected with PBS were used as controls of these experimental murine models. i, Representative H&E staining images of the livers from SK-MEL-192-Tb and control. This experiment was repeated three times independently with similar results. j, Representative MRI images showing lung metastasis, but not liver metastasis detected in osteosarcoma-PDX (OS-PDX) mouse models. This experiment was repeated seven times independently with similar results. k, Statistical analysis of BODIPY staining of livers from human melanoma SK-MEL-192-Tb mice, osteosarcoma-PDX tumor-bearing mice, and their respective controls. n=6 mice per group for SK-MEL-192 model; n=5 control and n=7 tumor-bearing mice for osteosarcoma-PDX model. l, Measurement of body mass index (BMI) of PDAC patients and control subjects with benign lesions. n=14 control subjects and n=16 PDAC patients. Scale bars, 500 μm for (f) and 200 μm for (i). P values were determined by the two-tailed, unpaired Student’s t-test (c-e,g,k,l). Data are mean ± s.e.m. NS, not significant.
Extended Data Figure 4. Tumor explant (TE)-derived EVPs dysregulate liver metabolism.
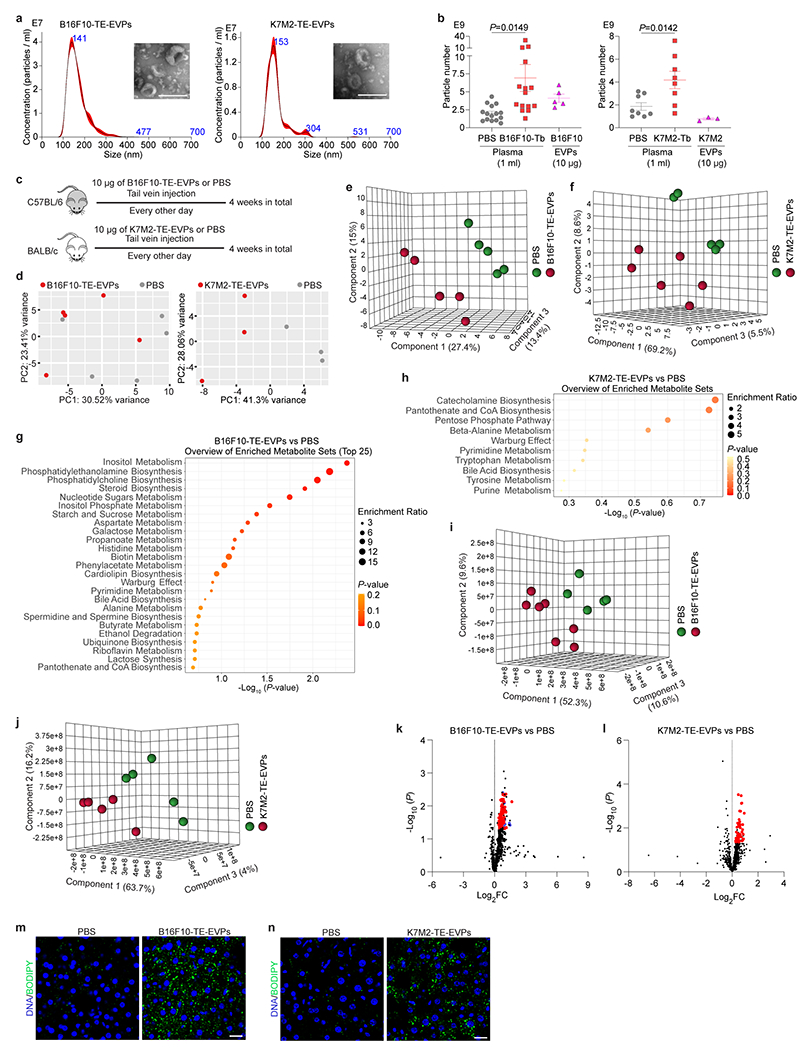
a, Representative nanoparticle tracking analysis (NTA) and TEM images (insertions) of B16F10-TE-EVPs (left) and K7M2-TE-EVPs (right). Shown are graphs and images representative of three independent experiments. b, Quantification of the particle numbers in 1 ml of plasma from B16F10-Tb (left) and K7M2-Tb (right) mice, compared to their PBS-injected controls, as well as in 10 μg of EVPs derived from B16F10 (left) and K7M2 (right) cells. n=15 control and B16F10-Tb mice; n=8 control and K7M2-Tb mice; n=5 and n=3 independent experiments for EVPs derived from B16F10 and K7M2 cells, respectively. c, Schematic illustration of the procedure of syngeneic mouse education with B16F10-TE-EVPs or K7M2-TE-EVPs, and PBS control. d, PCA of gene expression in the livers of mice educated with B16F10-TE-EVPs (left) and K7M2-TE-EVPs (right), compared to PBS-educated controls. n=5 mice per group for B16F10-TE-EVP education model; n=3 mice per group for K7M2-TE-EVP education model. e,f, PLS-DA plots of metabolites detected in the livers from B16F10-TE-EVP- (e) and K7M2-TE-EVP- (f) educated mice, compared to PBS-educated controls. Metabolomics data are shown in Supplementary Tables 8 and 9. n=5 B16F10-TE-EVP-educated mice and controls; n=6 K7M2-TE-EVP-educated mice and n=5 controls. g,h, Metabolite set enrichment analysis of the metabolites significantly changed in the livers from B16F10-TE-EVP- (g) and K7M2-TE-EVP- (h) educated mice, compared to PBS-educated controls (see lists of metabolites in Fig. 2e,f). n=5 B16F10-TE-EVP-educated mice and controls; n=6 K7M2-TE-EVP-educated mice and n=5 controls. i,j, PLS-DA plots of the lipid species in the livers from B16F10-TE-EVP- (i) and K7M2-TE-EVP- (j) educated mice, compared to PBS-educated controls. Lipidomics data are shown in Supplementary Tables 10 and 11. n=7 B16F10-TE-EVP-educated mice and n=5 controls; n=5 K7M2-TE-EVP-educated mice and controls. k,l, Volcano plots showing the significantly (P<0.05) enriched triglyceride species (red) and cholesteryl ester species (blue) in the livers from B16F10-TE-EVP- (k) and K7M2-TE-EVP- (l) educated mice, compared to PBS-educated controls. n=7 B16F10-TE-EVP-educated mice and n=5 controls; n=5 K7M2-TE-EVP-educated mice and controls. m,n, Representative images of BODIPY staining of the livers from B16F10-TE-EVP- (m) and K7M2-TE-EVP- (n) educated mice, and PBS-educated controls. Associated quantification of BODIPY staining was shown in Fig. 2i,j. Scale bars, 200 nm for (a) and 20 μm for (m,n). P values were determined by the two-tailed, unpaired Student’s t-test (b,k,l), or hypergeometric test (g,h). Data are mean ± s.e.m.
Extended Data Figure 5. Tumor cell line (CL)-derived EVPs dysregulate liver metabolism.
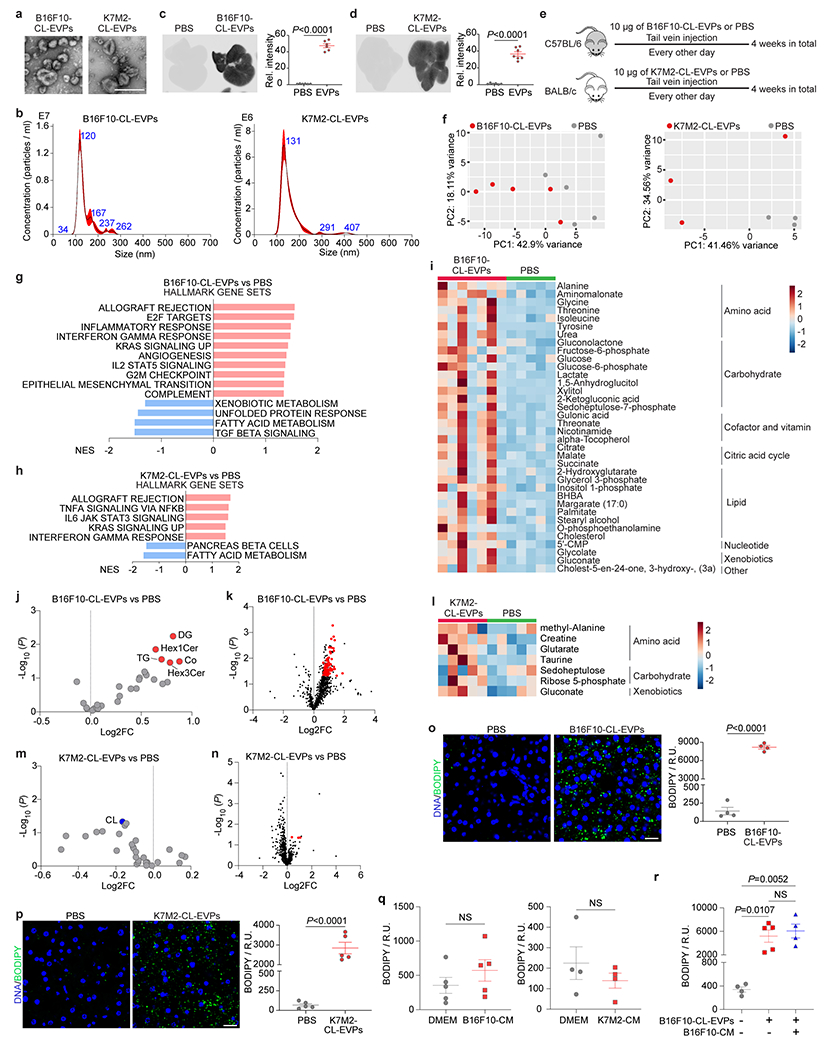
a, Representative TEM images of B16F10-CL-EVPs (left) and K7M2-CL-EVPs (right). Scale bar, 200 nm. This experiment was repeated three times independently with similar results. b, NTA of B16F10-CL-EVPs and K7M2-CL-EVPs. Shown are graphs representative of three independent experiments. c,d, Representative images (left) and associated statistical analysis of relative signal intensity (right) of the livers from mice 24 h post intravenous injection of 10 μg of CellVue NIR815-labeled B16F10-CL-EVPs (c) and K7M2-CL-EVPs (d), and their mock PBS-injected controls. n=6 mice injected with B16F10-CL-EVPs or PBS mock reaction; n=7 mice injected with K7M2-CL-EVPs and n=6 mice injected with PBS mock control. e, Schematic illustration of syngeneic mouse education with B16F10-CL-EVPs or K7M2-CL-EVPs, and PBS control. f, PCA of gene expression in the livers from mice educated with B16F10-CL-EVPs (left) and K7M2-CL-EVPs (right), compared to PBS-educated controls. n=5 mice per group for B16F10-CL-EVP education model; n=3 mice per group for K7M2-CL-EVP education model. g,h, GSEA of gene expression profiles, ranked based on the sign of log2FC*(−log10P value), in the livers from B16F10-CL-EVP- (g) and K7M2-CL-EVP- (h) educated mice, compared to PBS-educated controls, using hallmark gene sets. Significantly changed signaling pathways with nominal P<0.05 are shown. n=5 B16F10-CL-EVP-educated mice and controls; n=3 K7M2-CL-EVP-educated mice and controls. Gene lists for signaling pathways are shown in Supplementary Tables 22 and 23. i, Heatmap showing the metabolites significantly changed in the livers of B16F10-CL-EVP-educated mice, compared to PBS-educated controls. n=5 control and n=7 B16F10-CL-EVP-educated mice. Metabolomics MS data are shown in Supplementary Table 29. j,k, Volcano plots showing the significantly (P<0.05) enriched lipid classes (red) (j) and the significantly (P<0.05) enriched triglyceride species (red) (k) in the livers of B16F10-CL-EVP-educated mice, compared to PBS-educated controls. n=5 control and n=6 B16F10-CL-EVPs educated mice. Lipidomics MS data are shown in Supplementary Table 30. l, Heatmap showing the metabolites significantly changed in the livers of K7M2-CL-EVP-educated mice, compared to PBS-educated controls. n=5 K7M2-CL-EVP-educated mice and controls. Metabolomics MS data are shown in Supplementary Table 31. m,n, Volcano plots showing the significantly (P<0.05) changed lipid class (blue) (m), and the significantly (P<0.05) enriched triglyceride species (red) (n) in the livers from K7M2-CL-EVP-educated mice, compared to PBS-educated controls. n=5 control and n=4 K7M2-CL-EVP-educated mice. Lipidomics MS data are shown in Supplementary Table 32. o,p, Representative immunofluorescence (IF) images (left) and associated statistical analysis (right) of BODIPY staining of the livers from B16F10-CL-EVP- (o) and K7M2-CL-EVP- (p) educated mice, and PBS-educated controls. n=4 mice per group for B16F10-CL-EVP-education model; n=5 mice per group for K7M2-CL-EVP-education model. Scale bars, 20 μm. q, Quantification of BODIPY staining of the livers from B16F10- and K7M2-conditioned media (CM) -educated mice, compared to blank DMEM media-educated mice. n=5 mice per group for B16F10-CM model and n=4 mice per group for K7M2-CM model. r, Quantification of BODIPY staining of the livers from PBS-educated and B16F10-CL-EVP-educated mice, in the absence or presence of B16F10-CM co-education. n=4 mice educated with PBS, n=5 mice educated with B16F10-CL-EVPs alone, and n=4 mice educated with B16F10-CL-EVPs together with B16F10-CM. P values were determined by the two-tailed, unpaired Student’s t-test for (c,d,j,k,m-p) and one-way ANOVA with post hoc Tukey’s test for (r). Data are mean ± s.e.m. NS, not significant.
Extended Data Figure 6. Ablation of Rab27a expression suppresses EVP secretion from tumor cells.
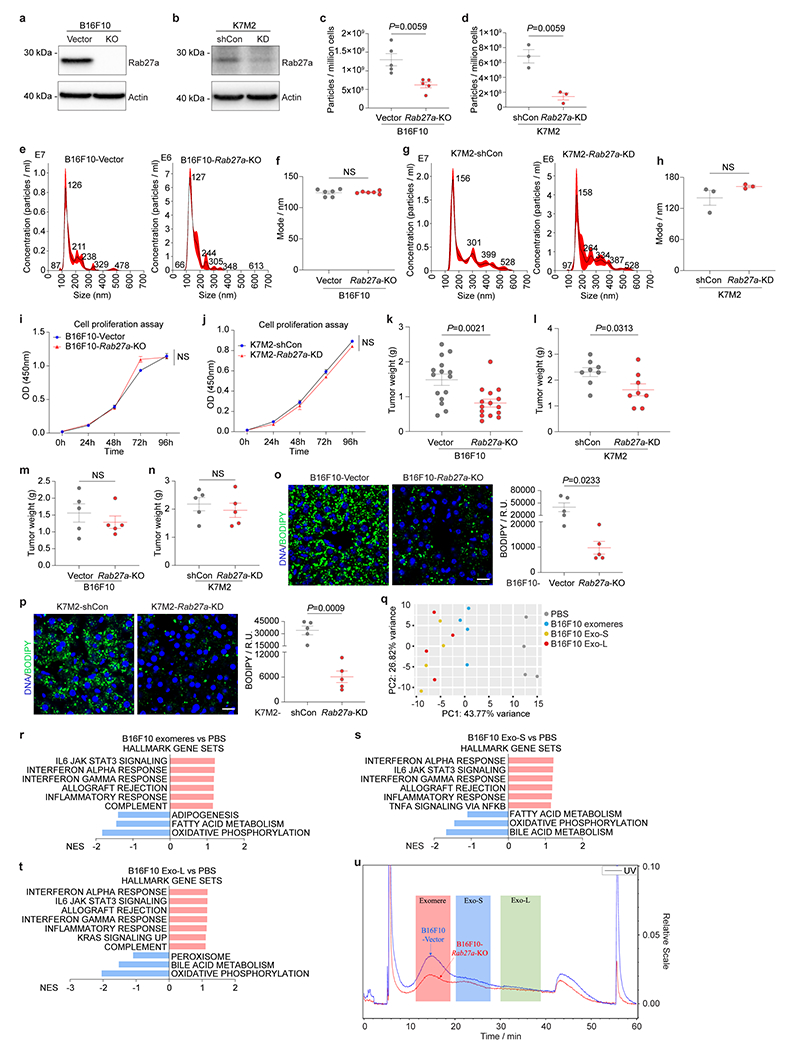
a, Western blot analysis of Rab27a expression in B16F10 cells infected with vector control or Rab27a-CRISPR KO virus. Actin was used as a loading control. Shown is representative data from three independent experiments. b, Western blot analysis of Rab27a expression in K7M2 cells infected with vector control (shCon) or Rab27a-shRNA KD virus. Actin was used as a loading control. Shown is representative data from three independent experiments. c, NTA of the numbers of EVPs secreted from B16F10-control (Vector) or B16F10-Rab27a-KO cells. n=5 independent experiments per group. d, NTA of the numbers of EVPs secreted from K7M2-control (shCon) or K7M2-Rab27a-KD cells. n=3 independent experiments per group. e,f, Representative NTA profiles (e) and associated analysis of the diameter mode (f) of EVPs isolated from B16F10-control (Vector) or B16F10-Rab27a-KO cells. n=6 independent experiments per group. g,h, Representative NTA profiles (g) and associated analysis of the diameter mode (h) of EVPs isolated from K7M2-control (shCon) or K7M2-Rab27a-KD cells. n=3 independent experiments per group. i, Proliferation of B16F10-control (Vector) and B16F10-Rab27a-KO cells. n=3 independent experiments. j, Proliferation of K7M2-control (shCon) and K7M2-Rab27a-KD cells. n=4 independent experiments. k, Statistical analysis of the weights of tumors from mice inoculated with B16F10-control (Vector) or B16F10-Rab27a-KO cells. n=15 mice per group. l, Statistical analysis of the weights of tumors from mice inoculated with K7M2-control (shCon) or K7M2-Rab27a-KD cells. n=8 mice per group. m,n, Statistical analysis showing the similar tumor burden from a subset of mice in k and l inoculated with B16F10-Rab27a-KO cells (m), or K7M2-Rab27a-KD cells (n) subjected to BODIPY staining of their livers as shown in (o,p). n=5 mice per group. o,p, Representative images (left) and associated statistical analysis (right) of BODIPY staining of the livers from B16F10-Rab27a-KO (o) and K7M2-Rab27a-KD (p) tumor bearing mice, and their respective controls. n=5 mice per group. Scale bars, 20 μm. q, PCA of the gene expression profiling in the livers from mice educated with control PBS, B16F10 exomeres, or B16F10 Exo-S, or B16F10 Exo-L for 4 weeks. n=4 mice per group. r-t, GSEA of gene expression profiles, which were ranked based on the sign of log2FC*(−log10P value), in the livers from B16F10 exomere- (r), Exo-S- (s), and Exo-L- (t) educated mice, compared to PBS-educated controls. (n=4 each). Signaling pathways significantly changed with nominal P<0.05 are shown. Gene lists for signaling pathways are shown in Supplementary Tables 24–26. u, EVPs isolated from equal numbers of B16F10-Vector control and B16F10-Rab27a-KO cells were resolved via AF4. Shown are real time measurement of UV on a relative scale (right axis), indicating the abundance of fractionated particles. Shaded areas mark the elution time periods for exomeres (red), Exo-S (blue) and Exo-L (green), respectively. As reflected by the UV signal, the production of exomeres, Exo-S (to a less extent) and Exo-L (to the least extent) were reduced in the B16F10-Rab27a-KO cells compared to the B16F10-Vector control cells. P values were determined by the two-tailed, unpaired Student’s t-test (c,d,f,h,k-p), or two-way ANOVA followed with Bonferroni’s multiple comparisons test (i,j). Data are mean ± s.e.m. NS, not significant. KO, knockout. KD, knockdown. For western blotting source data of (a,b), see Supplementary Figure 1.
Extended Data Figure 7. Tumor-derived EVPs are uptaken by KCs in the liver.
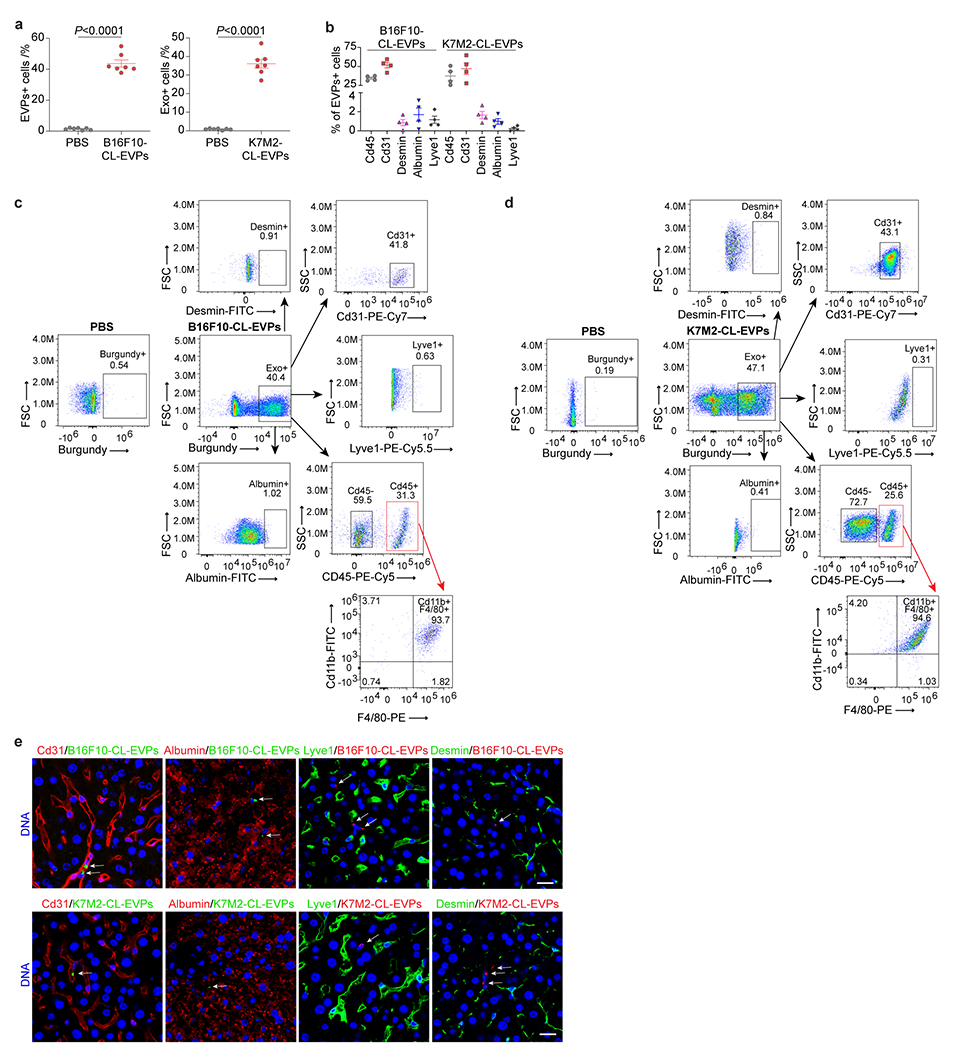
a, Flow cytometry analysis of the percentage of EVP positive cells in the livers from mice 24 h post intravenous injection of 10 μg of CellVue Burgundy-labeled B16F10-CL-EVPs (left) and K7M2-CL-EVPs (right), and PBS controls. n=7 mice per group. b, Flow cytometry analysis of the percentage of different cell types, including Cd45+ immune cells, Cd31+ vascular endothelial cells, desmin+ stellate cells, albumin+ hepatocytes, Lyve1+ lymphatic and sinusoidal endothelial cells, among the B16F10-CL-EVP and K7M2-CL-EVP positive cells shown in a. n=4 each. c,d, Representative flow cytometry gating strategy evaluating the percentage of different cell types, including Cd31+ vascular endothelial cells, albumin+ hepatocytes, Lyve1+ lymphatic and sinusoidal endothelial cells, desmin+ stellate cells, Cd45+ immune cells and Cd11b+F4/80+ KCs in Cd45+ immune cells, among the B16F10-CL-EVP- (c) and K7M2-CL-EVP- (d) positive cells shown in (a,b and Fig. 3a). e, Representative images of the immunofluorescence (IF) co-staining of EVPs (indicated by white arrows) with Cd31+ vascular endothelial cells, albumin+ hepatocytes, Lyve1+ lymphatic and sinusoidal endothelial cells, and desmin+ stellate cells 24 h post intravenous injection of B16F10-CL-EVPs (top) or K7M2-CL-EVPs (bottom). DNA in blue. Scale bars, 20 μm. This experiment was repeated three times independently with similar results. P values were determined by the two-tailed, unpaired Student’s t-test (a).
Extended Data Figure 8. Depletion of Kupffer cells alleviates tumor EVP-induced fatty liver formation without impairing tumor growth.
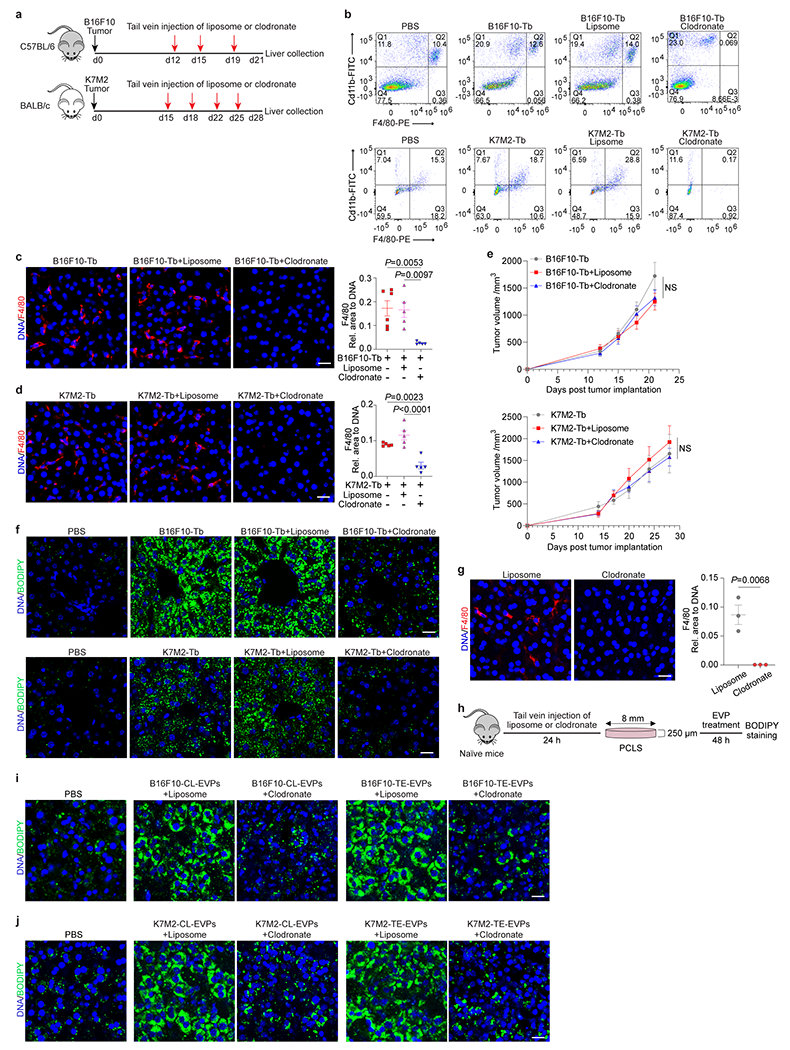
a, Schematic illustration of liposome or clodronate (100 μl of suspension per 10 g of mouse weight) treatment, delivered via intravenous injection to B16F10-Tb and K7M2-Tb mice. The concentration of clodronate in the suspension is 5 mg/ml. b, Representative flow cytometry gating strategy examining the abundance of KCs (Cd11b+F4/80+) in the livers from PBS-injected control mice, B16F10-Tb (up) or K7M2-Tb (bottom) mice, and tumor-bearing mice treated with liposome or clodronate. Associated quantification of KC abundance was shown in Fig. 3c. c,d, Representative IF images (left) and associated statistical analysis (right) of KC staining of the livers from B16F10-Tb (c) and K7M2-Tb (d) mice, and tumor-bearing mice treated with liposome or clodronate. F4/80 staining for KCs in red, and DNA in blue. For B16F10-Tb model, n=6 B16F10-Tb mice, n=5 B16F10-Tb mice treated with liposome or clodronate. For K7M2-Tb model, n=5 mice per group. e, Measurement of the tumor volumes from B16F10-Tb (left) or K7M2-Tb (right) mice, and tumor-bearing mice treated with liposome or clodronate as shown in (a). n=5 B16F10-Tb mice and n=7 B16F10-Tb mice treated with liposome or clodronate; n=3 K7M2-Tb mice and n=5 K7M2-Tb mice treated with liposome or clodronate. f, Representative IF images of BODIPY staining of the livers from PBS-injected control mice, B16F10- or K7M2-Tb mice, and the tumor-bearing mice treated with liposome or clodronate as shown in Fig. 3d. g, Representative IF images (left) and associated statistical analysis (right) of the precision-cut liver slices (PCLS) stained with F4/80 (red) and DAPI (blue) to show the KC depletion in mouse livers 24 h post treatment with liposome or clodronate (100 μl of suspension per 10 g of mouse weight). n=3 mice per group. h, Schematic illustration of precision-cut liver slices (PCLS) from naïve mice 24 h post treatment with liposome or clodronate (100 μl of suspension per 10 g of mouse weight) followed by EVP (10 μg/ml) treatment ex vivo for 48 h. Liver slices were then subjected to BODIPY staining. i,j, Representative IF images of BODIPY staining of PCLS sectioned from liposome- or clodronate- (100 μl of suspension per 10 g of mouse weight) treated mice (as shown in g) that were further treated with 10 μg/ml of B16F10-CL-EVPs or B16F10-TE-EVPs (i), K7M2-CL-EVPs or K7M2-TE-EVPs (j) ex vivo for 48 h (see also Fig. 3e,f). PCLS from naïve mice treated with PBS were used as controls. BODIPY in green and DNA in blue. Scale bars, 20 μm. P values were determined by the one-way ANOVA with post hoc Tukey’s test (c,d) , or two-way ANOVA followed with Bonferroni’s multiple comparisons test (e), or two-tailed, unpaired Student’s t-test (g). Data are mean ± s.e.m. NS, not significant.
Extended Data Figure 9. Tumor EVPs promote TNFα secretion from KCs.
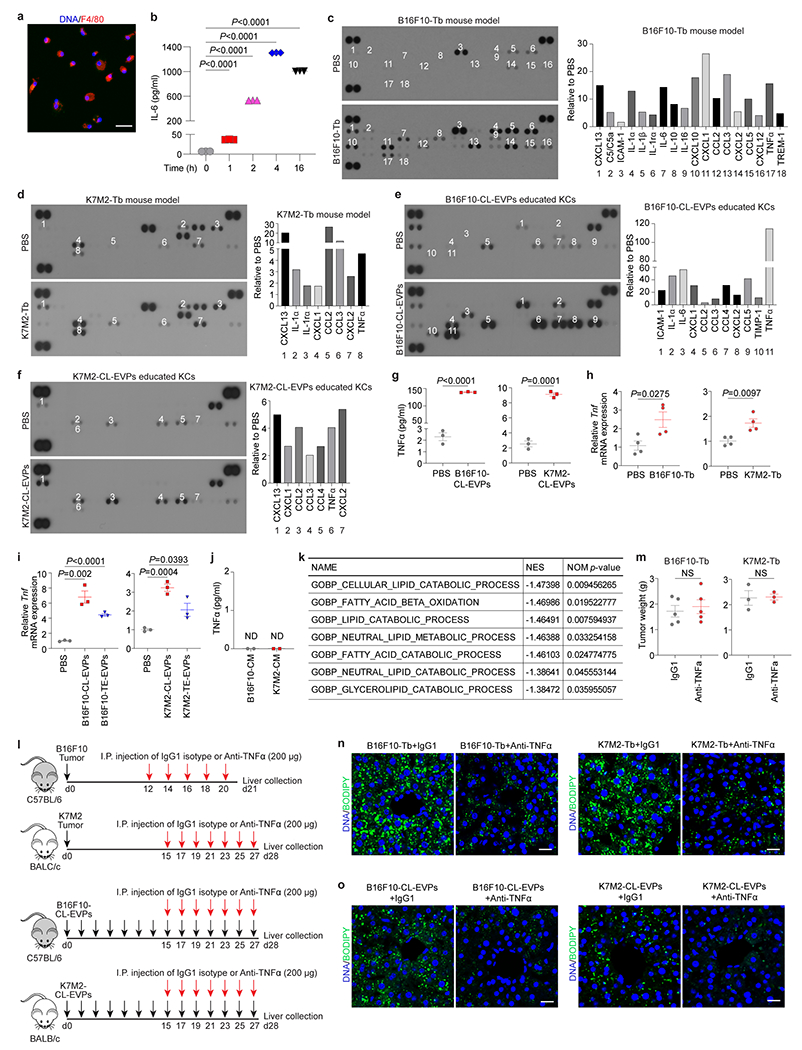
a, Representative IF image of the primary KCs isolated from naïve mice. F4/80 in red and DAPI in blue. Shown is representative image from three independent experiments. Scale bar, 20 μm. b, ELISA analysis of IL-6 secretion from naïve mice-derived KCs after treatment with LPS (1 μg/ml). n=3 independent experiments per group. c,d, Representative cytokine array blots and associated quantification charts for the cytokines and chemokines in the whole cell lysates of KCs isolated from B16F10-Tb mice (c) and K7M2-Tb mice (d), and their respective PBS-injected controls. e,f, Representative cytokine array blots and associated quantification charts for the cytokines and chemokines in the conditioned media of KCs isolated from naïve mice and educated with 10 μg/ml of B16F10-CL-EVPs (e) or K7M2-CL-EVPs (f) in vitro for 3 days, and their respective PBS-educated controls. g, TNFα ELISA on the conditioned medium (CM) of KCs educated with 10 μg/ml of B16F10-CL-EVP- (left), or K7M2-CL-EVP- (right), and their respective PBS controls for 3 days. n=3 independent experiments per group. h, qRT-PCR analysis of Tnf expression in KCs isolated from B16F10-Tb (left), or K7M2-Tb (right) mice, and their respective PBS-injected controls. n=4 mice per group. i, qRT-PCR analysis of Tnf expression in KCs treated with control PBS, EVPs derived from B16F10 cells or B16F10 tumor explants (left), or EVPs derived from K7M2 cells or K7M2 tumor explants (right) for 4 h. n=3 independent experiments per group. j, TNFα ELISA on EVP-depleted CM from B16F10 and K7M2 cells. n=2 independent experiments. ND, not detected. k, GSEA of gene expression profiles, ranked based on the sign of log2FC*(−log10P value), in hepatocytes 24 h post treatment with recombinant murine TNFα protein (25 ng/ml), compared to PBS control, using Gene Ontology gene sets. Shown are downregulated lipid catabolism-associated gene sets. NES, normalized enrichment score. NOM p-value, nominal p-value. l, Schematic Illustration of anti-TNFα antibody (200 μg/mouse) or IgG1 isotype control (200 μg/mouse) treatment of tumor-bearing mice or tumor EVP-educated mice. I.P., intraperitoneal. m, Weights of tumors from mice inoculated with B16F10 (left) or K7M2 (right) cells and treated with anti-TNFα antibody or IgG1 isotype control as shown in (l). n=5 mice per group for B16F10-Tb model and n=3 per group for K7M2-Tb model. n,o, Representative images of BODIPY staining of the livers from B16F10- and K7M2-Tb mice (n), and B16F10-CL-EVP- and K7M2-CL-EVP-educated mice (o), treated with anti-TNFα antibody or IgG1 isotype control (see also Fig. 4d,e). Scale bars, 20 μm. P values were determined by the one-way ANOVA with post hoc Tukey’s test (b), or two-tailed, unpaired Student’s t-test (g-i,m). Data are mean ± s.e.m. NS, not significant. For cytokine array blot source data of (c-f), see Supplementary Figure 2.
Extended Data Figure 10. Tumor EVP-packaged palmitic acid (PA) induces TNFα secretion from KCs.
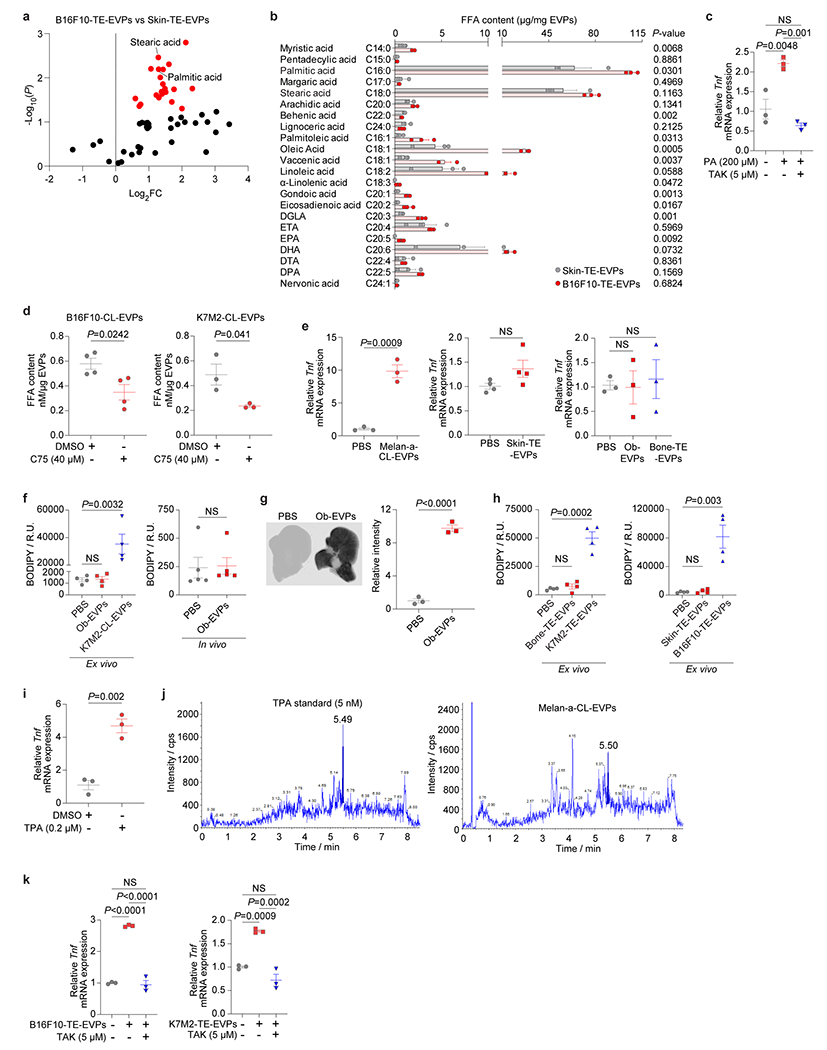
a, Volcano plot of metabolites significantly changed (highlighted in red, P<0.05) in B16F10-TE-EVPs versus skin-TE-EVPs. n=5 B16F10-TE-EVPs and n=4 skin-TE-EVPs. b, Quantitative MS analysis of the long-chain free fatty acids, including saturated and unsaturated fatty acids, in the B16F10-TE-EVPs and skin-TE-EVPs. n=3 per EVP group. c, qRT-PCR analysis of Tnf expression in KCs from naïve C57BL/6 mice treated with DMSO or the Tlr4 inhibitor TAK (5 μM) for 1 h followed by vehicle (100% ethanol diluted into PA-carrier medium at 1:1000 dilution) or 200 μM of PA with or without TAK (5 μM) for 4 h. n=3 mice per group. d, FFA content in the EVPs isolated from B16F10 (left) or K7M2 (right) cells treated with DMSO or C75 (40 μM) for 48 h. n=4 independent experiments for DMSO- or C75-treated B16F10 cells; n=3 independent experiments for DMSO- or C75-treated K7M2 cells. e, qRT-PCR analysis of Tnf expression in KCs isolated from naïve C57BL/6 (left and middle) or BALB/c (right) mice then treated with control PBS, Melan-a-CL-EVPs (left), skin-TE-EVPs (middle), osteoblast-EVPs (Ob-EVPs), or bone-TE-EVPs (right) in vitro for 4 h. n=3 independent experiments per group for treatment with Melan-a-CL-EVPs, Ob-EVPs, bone-TE-EVPs, and their PBS controls. n=4 independent experiments for treatment with skin-TE-EVPs and PBS. f, Quantification of BODIPY staining of precision-cut liver slices treated with Ob-EVPs or K7M2-CL-EVPs ex vivo for 48 h, or livers educated with Ob-EVPs in vivo for 4 weeks, compared to PBS-treated or PBS-educated controls, respectively. n=4 mice per group for ex vivo EVP treatment and n=5 mice per group for in vivo EVP education. g, Representative LI-COR Odyssey images (left) and associated statistical analysis of relative signal intensity (right) of the livers from mice 24 h post intravenously injection of 10 μg of CellVue NIR815-labeled Ob-EVPs and mock PBS control. n=3 mice per group. h, Quantification of BODIPY staining of the precision-cut liver slices treated with control PBS, bone-TE-EVPs or K7M2-TE-EVPs (left), skin-TE-EVPs or B16F10-TE-EVPs (right) ex vivo for 48 h. n=4 mice per group. i, qRT-PCR analysis of Tnf expression in the KCs isolated from C57BL/6 mice and treated with DMSO or 12-O-tetradecanoylphorbol-13-acetate (TPA) (0.2 μM) in vitro for 4 h. TPA induced KC Tnf expression, compared to DMSO control. n=3 independent experiments. j, Representative LC-MS/MS chromatograms of TPA standard (5 nM, left) and TPA detected in the Mela-a-CL-EVPs (right). The concentration of TPA detected in Mela-a-CL-EVPs was 6.5 nM per 100 μg of EVPs. k, qRT-PCR analysis of Tnf expression in KCs which were pre-treated with DMSO or TAK (5 μM) for 1 h, and subsequently treated with PBS, 10 μg of B16F10-TE-EVPs or K7M2-TE-EVPs with or without TAK (5 μM) for 4 h. n=3 independent experiments. P values were determined by the two-tailed, unpaired Student’s t-test (a,b,d-i,k), or one-way ANOVA with post hoc Tukey’s test (c), or Data are mean ± s.e.m. NS, not significant.
Extended Data Figure 11. Tumor EVPs suppress liver drug metabolism and enhance chemotoxicity.
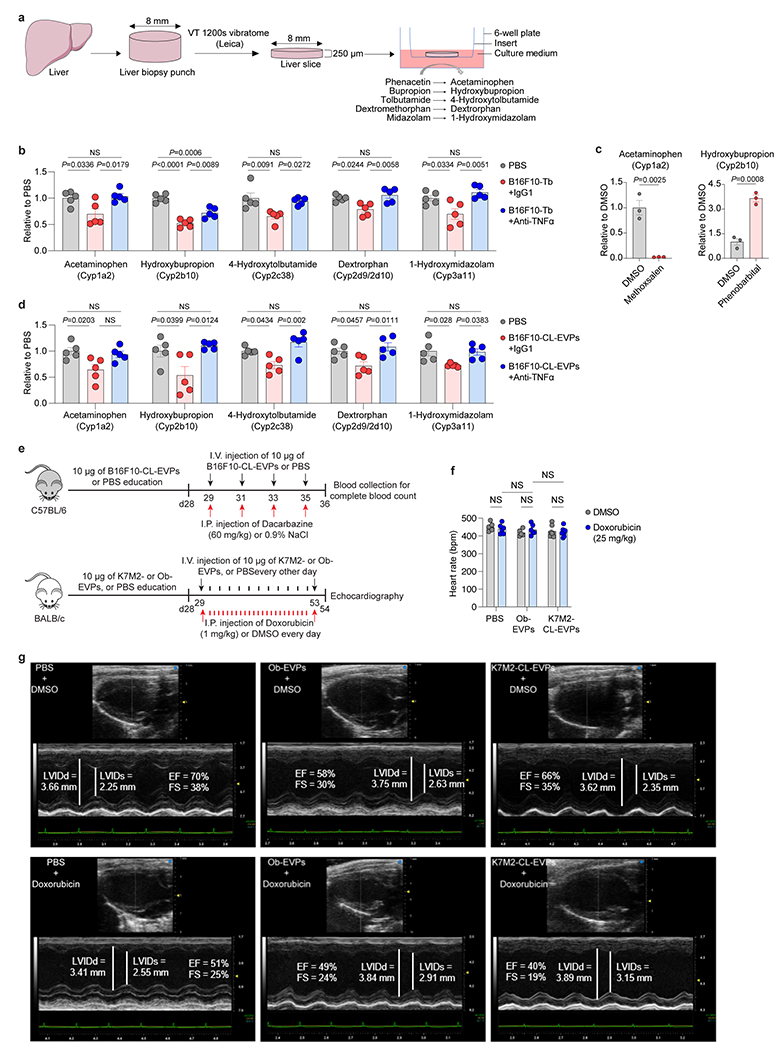
a, Schematic illustration of the procedure of drug-metabolizing activity analysis of the precision-cut liver slices. Substrates of Cytochrome P450 enzymes (including phenacetin, bupropion, tolbutamide, dextromethorphan and midazolam) were added to the media, and their corresponding metabolites (including acetaminophen, hydroxybupropion, 4-hydroxytolbutamide, dextrorphan and 1-hydroxymidazolam) were analyzed by LC/MS/MS. b, Drug-metabolizing activity of the core CYP enzymes in the precision-cut liver slices sectioned from PBS-injected mice, and B16F10-Tb mice treated with anti-TNFα antibody or IgG1 isotype control. n=5 mice per group. c, Drug-metabolizing activity of Cyp1a2 (left) or Cyp2b10 (right) in the precision-cut liver slices pre-treated with DMSO control or mathoxsalen (5 μM), or phenobarbital (0.1 mM) for 24 h. n=3 mice per group. d, Drug-metabolizing activity of the core CYP enzymes in the precision-cut liver slices sectioned from naïve mice pre-treated with PBS or 10 μg/ml of B16F10-CL-EVPs with co-treatment of anti-TNFα antibody (20 μg/ml) or IgG1 isotype (20 μg/ml) control for 24 h. n=5 mice per group. e, Schematic illustration of the procedure of chemotoxicity analysis using EVP-educated mice. For melanoma model (top), C57BL/6 mice were intravenously injected with PBS or 10 μg of B16F10-CL-EVPs every other day for 4 weeks, and then intraperitoneally injected with dacarbazine (60 mg/kg) or 0.9% NaCl together with intravenous injection of PBS or 10 μg of B16F10-CL-EVPs every other day for 4 times. Retroorbital blood of the mice was then collected for complete blood count. For osteosarcoma model (bottom), BALB/c mice were intravenously injected with PBS, 10 μg of osteoblast-EVPs (Ob-EVPs) or K7M2-CL-EVPs every other day for 4 weeks, and then intraperitoneally injected with doxorubicin (1 mg/kg) or DMSO every 24 h together with intravenous injection of PBS, 10 μg of Ob-EVPs or K7M2-CL-EVPs every other day for 25 days, and mice were then subjected to echocardiography. f, Statistical analysis of the heart rates of PBS-, Ob-EVP- or K7M2-CL-EVP-educated mice after treatment of DMSO or doxorubicin (cumulative dose of 25 mg/kg). No difference of heart rates was observed in different groups. n=6 mice for PBS groups and n=7 mice for K7M2-CL-EVP groups, n=5 and n=6 for DMSO and doxorubicin treated Ob-EVP-educated mice, respectively. g, Representative M-mode images of echocardiography for PBS-, Ob-EVP- or K7M2-CL-EVP-educated mice after treatment of DMSO or doxorubicin as described in (e). LVIDd, left ventricular internal dimension at end diastole. LVIDs, left ventricular internal dimension at end systole. EF, ejection fraction. FS, fractional shortening. P values were determined by the one-way ANOVA with post hoc Tukey’s test (b,d), or two-tailed, unpaired Student’s t-test (c), or two-way ANOVA followed with Fisher’s LSD test (f). Data are mean ± s.e.m. NS, not significant. Ob, osteoblast. I.V., intravenours. I.P., intraperitoneal.
Supplementary Material
Acknowledgments
The authors acknowledge the Genomics Resource Core Facility (Weill Cornell Medicine), Electron Microscopy and Histology Core Facility (Weill Cornell Medicine), Molecular Cytology Core Facility (Memorial Sloan Kettering Cancer Center, MSKCC), and Laboratory of Comparative Pathology (MSKCC) for their high-quality service. The authors also acknowledge technical support from Wyatt Technology. The authors thank members of the Lyden laboratory for insightful discussions. The authors gratefully acknowledge support from the National Cancer Institute (CA232093, CA163117 and CA207983 to D.L., and CA218513 to D.L. and H.Z.), the Thompson Family Foundation (to D.L. and D.K.), the Tortolani Foundation (to D.L. and J.B.), the Pediatric Oncology Experimental Therapeutics Investigator’s Consortium, the Malcolm Hewitt Weiner Foundation, the Manning Foundation, the Sohn Foundation, the AHEPA Vth District Cancer Research Foundation, the Children’s Cancer and Blood Foundation, the Hartwell Foundation (to D.L.), the National Institutes of Health (R01CA234614, 2R01AI107301, R01CA234614 and R01DK121072 to R.E.S.), the United States Department of Defense (W81XWH-21-1-0978 to R.E.S.), the Paul G. Allen Family Foundation UWSC13448 (to R.E.S.), the National Natural Science Foundation of China (No. 81902730 to J.L.), Guangdong Foundation of Medical Science and Technology (No. A2019213 to J.L.), China Scholarship Council (CSC No. 202008440567 to J.L.), the Swedish Cancer Society Pancreatic Cancer Fellowship (to L.B.), the Lions International Postdoctoral fellowship (to L.B.), the Sweden-America stipend (to L.B.), the fellowship from Alan and Sandra Gerry Metastasis and Tumor Ecosystems Center of Memorial Sloan Kettering Cancer Center (to C.P.Z.), the part of the research involved in developing osteosarcoma PDXs and tumor imaging was funded in part through the NIH/NCI Cancer Center Support Grant P30 CA008748 to MSKCC, the National Institutes of Health (R01CA237213 to C.E.B and V.P., and R01CA254036 to S.K.B.). R.E.S. is an Irma Hirschl Trust Research Award Scholar. Schematic models were generated in part using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license, with further modifications.
Footnotes
Competing interests
The authors declare no competing financial interests.
D.L. is on the scientific advisory board of Aufbau Holdings Ltd.
R.E.S. is on the scientific advisory board of Miromatrix and is a speaker and consultant for Alnylam Inc.
M.E. is a member of the research advisory board for brensocatib for Insmed; a member of the scientific advisory board for Vividion Therapeutics; a consultant for Protalix BioTherapeutics; and holds shares in Agios.
Data availability
RNA-seq raw data and associated processed data files that support the findings of this study have been deposited in the Gene Expression Omnibus under accession codes GSE199863 and GSE220446. Metabolomics data derived from livers of PBS-injected control and B16F10-Tb mice (Supplementary Table 1) have been deposited at Figshare (DOI: 10.6084/m9.figshare.22233187). Metabolomics data derived from livers of PBS-injected control and K7M2-Tb mice (Supplementary Table 2) have been deposited at Figshare (DOI: 10.6084/m9.figshare.22233253). Lipidomics data derived from livers of PBS-injected control and B16F10-Tb mice (Supplementary Table 3) have been deposited at Figshare (DOI: 10.6084/m9.figshare.22233265). Lipidomics data derived from livers of PBS-injected control and K7M2-Tb mice (Supplementary Table 4) have been deposited at Figshare (DOI: 10.6084/m9.figshare.22233274). Metabolomics data derived from livers of PBS- and B16F10-TE-EVP-educated mice (Supplementary Table 8) have been deposited at Figshare (DOI: 10.6084/m9.figshare.22233289). Metabolomics data derived from livers of PBS- and K7M2-TE-EVP-educated mice (Supplementary Table 9) have been deposited at Figshare (DOI: 10.6084/m9.figshare.22233298). Lipidomics data derived from livers of PBS- and B16F10-TE-EVP-educated mice (Supplementary Table 10) have been deposited at Figshare (DOI: 10.6084/m9.figshare.22233307). Lipidomics data derived from livers of PBS- and K7M2-TE-EVP-educated mice (Supplementary Table 11) have been deposited at Figshare (DOI: 10.6084/m9.figshare.22233316). Metabolomics data derived from skin-TE-EVPs and B16F10-TE-EVPs (Supplementary Table 14) have been deposited at Figshare (DOI: 10.6084/m9.figshare.22233352). Metabolomics data derived from livers of PBS- and B16F10-CL-EVP-educated mice (Supplementary Table 29) have been deposited at Figshare (DOI: 10.6084/m9.figshare.22233379). Lipidomics data derived from livers of PBS- and B16F10-CL-EVP-educated mice (Supplementary Table 30) have been deposited at Figshare (DOI: 10.6084/m9.figshare.22233403). Metabolomics data derived from livers of PBS- and K7M2-CL-EVP-educated mice (Supplementary Table 31) have been deposited at Figshare (DOI: 10.6084/m9.figshare.22233424). Lipidomics data derived from livers of PBS- and K7M2-CL-EVP-educated mice (Supplementary Table 32) have been deposited at Figshare (DOI: 10.6084/m9.figshare.22233445). Metabolomics data were analyzed using MetaboAnalyst 4.0 (https://www.metaboanalyst.ca/home.xhtml), and metabolite set enrichment analysis was performed based on the Small Molecule Pathway Database (SMPDB) (http://www.smpdb.ca/).
References
- 1.McAllister SS & Weinberg RA The tumour-induced systemic environment as a critical regulator of cancer progression and metastasis. Nat Cell Biol 16, 717–727 (2014). 10.1038/ncb3015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang G et al. Metastatic cancers promote cachexia through ZIP14 upregulation in skeletal muscle. Nat Med 24, 770–781 (2018). 10.1038/s41591-018-0054-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lucotti S, Kenific CM, Zhang H & Lyden D Extracellular vesicles and particles impact the systemic landscape of cancer. EMBO J 41, e109288 (2022). 10.15252/embj.2021109288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang G et al. Tumor induces muscle wasting in mice through releasing extracellular Hsp70 and Hsp90. Nat Commun 8, 589 (2017). 10.1038/s41467-017-00726-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang H et al. Identification of distinct nanoparticles and subsets of extracellular vesicles by asymmetric flow field-flow fractionation. Nat Cell Biol 20, 332–343 (2018). 10.1038/s41556-018-0040-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Costa-Silva B et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol 17, 816–826 (2015). 10.1038/ncb3169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoshino A et al. Tumour exosome integrins determine organotropic metastasis. Nature 527, 329–335 (2015). 10.1038/nature15756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang H et al. Exosome-delivered EGFR regulates liver microenvironment to promote gastric cancer liver metastasis. Nat Commun 8, 15016 (2017). 10.1038/ncomms15016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xie Z et al. Exosome-delivered CD44v6/C1QBP complex drives pancreatic cancer liver metastasis by promoting fibrotic liver microenvironment. Gut 71, 568–579 (2022). 10.1136/gutjnl-2020-323014 [DOI] [PubMed] [Google Scholar]
- 10.Peinado H et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med 18, 883–891 (2012). 10.1038/nm.2753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khanna C et al. Metastasis-associated differences in gene expression in a murine model of osteosarcoma. Cancer Res 61, 3750–3759 (2001). [PubMed] [Google Scholar]
- 12.Aslakson CJ & Miller FR Selective events in the metastatic process defined by analysis of the sequential dissemination of subpopulations of a mouse mammary tumor. Cancer Res 52, 1399–1405 (1992). [PubMed] [Google Scholar]
- 13.Tilg H, Adolph TE, Dudek M & Knolle P Non-alcoholic fatty liver disease: the interplay between metabolism, microbes and immunity. Nat Metab 3, 1596–1607 (2021). 10.1038/s42255-021-00501-9 [DOI] [PubMed] [Google Scholar]
- 14.Burd CE et al. Mutation-specific RAS oncogenicity explains NRAS codon 61 selection in melanoma. Cancer Discov 4, 1418–1429 (2014). 10.1158/2159-8290.CD-14-0729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maddipati R & Stanger BZ Pancreatic Cancer Metastases Harbor Evidence of Polyclonality. Cancer Discov 5, 1086–1097 (2015). 10.1158/2159-8290.CD-15-0120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoshino A et al. Extracellular Vesicle and Particle Biomarkers Define Multiple Human Cancers. Cell 182, 1044–1061 e1018 (2020). 10.1016/j.cell.2020.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bojmar L et al. Extracellular vesicle and particle isolation from human and murine cell lines, tissues, and bodily fluids. STAR Protoc 2, 100225 (2021). 10.1016/j.xpro.2020.100225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ostrowski M et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat Cell Biol 12, 19–30; sup pp 11-13 (2010). 10.1038/ncb2000 [DOI] [PubMed] [Google Scholar]
- 19.Stienstra R et al. Kupffer cells promote hepatic steatosis via interleukin-1beta-dependent suppression of peroxisome proliferator-activated receptor alpha activity. Hepatology 51, 511–522 (2010). 10.1002/hep.23337 [DOI] [PubMed] [Google Scholar]
- 20.De Taeye BM et al. Macrophage TNF-alpha contributes to insulin resistance and hepatic steatosis in diet-induced obesity. Am J Physiol Endocrinol Metab 293, E713–725 (2007). 10.1152/ajpendo.00194.2007 [DOI] [PubMed] [Google Scholar]
- 21.Feldstein AE et al. Free fatty acids promote hepatic lipotoxicity by stimulating TNF-alpha expression via a lysosomal pathway. Hepatology 40, 185–194 (2004). 10.1002/hep.20283 [DOI] [PubMed] [Google Scholar]
- 22.Korbecki J & Bajdak-Rusinek K The effect of palmitic acid on inflammatory response in macrophages: an overview of molecular mechanisms. Inflamm Res 68, 915–932 (2019). 10.1007/s00011-019-01273-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuhajda FP et al. Synthesis and antitumor activity of an inhibitor of fatty acid synthase. Proc Natl Acad Sci U S A 97, 3450–3454 (2000). 10.1073/pnas.050582897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rocha DM, Caldas AP, Oliveira LL, Bressan J & Hermsdorff HH Saturated fatty acids trigger TLR4-mediated inflammatory response. Atherosclerosis 244, 211–215 (2016). 10.1016/j.atherosclerosis.2015.11.015 [DOI] [PubMed] [Google Scholar]
- 25.Milanski M et al. Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: implications for the pathogenesis of obesity. J Neurosci 29, 359–370 (2009). 10.1523/JNEUROSCI.2760-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fearon KC, Glass DJ & Guttridge DC Cancer cachexia: mediators, signaling, and metabolic pathways. Cell Metab 16, 153–166 (2012). 10.1016/j.cmet.2012.06.011 [DOI] [PubMed] [Google Scholar]
- 27.Vasilogianni AM et al. Proteomics of colorectal cancer liver metastasis: A quantitative focus on drug elimination and pharmacodynamics effects. Br J Clin Pharmacol (2021). 10.1111/bcp.15098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jamwal R & Barlock BJ Nonalcoholic Fatty Liver Disease (NAFLD) and Hepatic Cytochrome P450 (CYP) Enzymes. Pharmaceuticals (Basel) 13 (2020). 10.3390/ph13090222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reid JM, Kuffel MJ, Miller JK, Rios R & Ames MM Metabolic activation of dacarbazine by human cytochromes P450: the role of CYP1A1, CYP1A2, and CYP2E1. Clin Cancer Res 5, 2192–2197 (1999). [PubMed] [Google Scholar]
- 30.Lewis IJ et al. Improvement in histologic response but not survival in osteosarcoma patients treated with intensified chemotherapy: a randomized phase III trial of the European Osteosarcoma Intergroup. J Natl Cancer Inst 99, 112–128 (2007). 10.1093/jnci/djk015 [DOI] [PubMed] [Google Scholar]
- 31.Bagdasaryan AA et al. Pharmacogenetics of Drug Metabolism: The Role of Gene Polymorphism in the Regulation of Doxorubicin Safety and Efficacy. Cancers (Basel) 14 (2022). 10.3390/cancers14215436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Savarese G, Stolfo D, Sinagra G & Lund LH Heart failure with mid-range or mildly reduced ejection fraction. Nat Rev Cardiol 19, 100–116 (2022). 10.1038/s41569-021-00605-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peinado H et al. Pre-metastatic niches: organ-specific homes for metastases. Nat Rev Cancer 17, 302–317 (2017). 10.1038/nrc.2017.6 [DOI] [PubMed] [Google Scholar]
- 34.Altea-Manzano P et al. A palmitate-rich metastatic niche enables metastasis growth via p65 acetylation resulting in pro-metastatic NF-kappaB signaling. Nat Cancer (2023). 10.1038/s43018-023-00513-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Allen AM, Hicks SB, Mara KC, Larson JJ & Therneau TM The risk of incident extrahepatic cancers is higher in non-alcoholic fatty liver disease than obesity - A longitudinal cohort study. J Hepatol 71, 1229–1236 (2019). 10.1016/j.jhep.2019.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mantovani A et al. Non-alcoholic fatty liver disease and increased risk of incident extrahepatic cancers: a meta-analysis of observational cohort studies. Gut 71, 778–788 (2022). 10.1136/gutjnl-2021-324191 [DOI] [PubMed] [Google Scholar]
- 37.Livak KJ & Schmittgen TD Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408 (2001). 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- 38.Chevalier C et al. Primary mouse osteoblast and osteoclast culturing and analysis. STAR Protoc 2, 100452 (2021). 10.1016/j.xpro.2021.100452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang H & Lyden D Asymmetric-flow field-flow fractionation technology for exomere and small extracellular vesicle separation and characterization. Nat Protoc 14, 1027–1053 (2019). 10.1038/s41596-019-0126-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rodrigues G et al. Tumour exosomal CEMIP protein promotes cancer cell colonization in brain metastasis. Nat Cell Biol 21, 1403–1412 (2019). 10.1038/s41556-019-0404-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.de Graaf IA et al. Preparation and incubation of precision-cut liver and intestinal slices for application in drug metabolism and toxicity studies. Nat Protoc 5, 1540–1551 (2010). 10.1038/nprot.2010.111 [DOI] [PubMed] [Google Scholar]
- 42.Paish HL et al. A Bioreactor Technology for Modeling Fibrosis in Human and Rodent Precision-Cut Liver Slices. Hepatology 70, 1377–1391 (2019). 10.1002/hep.30651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hennessey RC et al. Ultraviolet radiation accelerates NRas-mutant melanomagenesis: A cooperative effect blocked by sunscreen. Pigment Cell Melanoma Res 30, 477–487 (2017). 10.1111/pcmr.12601 [DOI] [PubMed] [Google Scholar]
- 44.Bosenberg M et al. Characterization of melanocyte-specific inducible Cre recombinase transgenic mice. Genesis 44, 262–267 (2006). 10.1002/dvg.20205 [DOI] [PubMed] [Google Scholar]
- 45.Weiss TJ et al. Cell-intrinsic melanin fails to protect melanocytes from ultraviolet-mutagenesis in the absence of epidermal melanin. Pigment Cell Melanoma Res (2022). 10.1111/pcmr.13070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Borgogna JC et al. The association of Chlamydia trachomatis and Mycoplasma genitalium infection with the vaginal metabolome. Sci Rep 10, 3420 (2020). 10.1038/s41598-020-60179-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chong J, Wishart DS & Xia J Using MetaboAnalyst 4.0 for Comprehensive and Integrative Metabolomics Data Analysis. Curr Protoc Bioinformatics 68, e86 (2019). 10.1002/cpbi.86 [DOI] [PubMed] [Google Scholar]
- 48.Subramanian A et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550 (2005). 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liberzon A et al. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1, 417–425 (2015). 10.1016/j.cels.2015.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq raw data and associated processed data files that support the findings of this study have been deposited in the Gene Expression Omnibus under accession codes GSE199863 and GSE220446. Metabolomics data derived from livers of PBS-injected control and B16F10-Tb mice (Supplementary Table 1) have been deposited at Figshare (DOI: 10.6084/m9.figshare.22233187). Metabolomics data derived from livers of PBS-injected control and K7M2-Tb mice (Supplementary Table 2) have been deposited at Figshare (DOI: 10.6084/m9.figshare.22233253). Lipidomics data derived from livers of PBS-injected control and B16F10-Tb mice (Supplementary Table 3) have been deposited at Figshare (DOI: 10.6084/m9.figshare.22233265). Lipidomics data derived from livers of PBS-injected control and K7M2-Tb mice (Supplementary Table 4) have been deposited at Figshare (DOI: 10.6084/m9.figshare.22233274). Metabolomics data derived from livers of PBS- and B16F10-TE-EVP-educated mice (Supplementary Table 8) have been deposited at Figshare (DOI: 10.6084/m9.figshare.22233289). Metabolomics data derived from livers of PBS- and K7M2-TE-EVP-educated mice (Supplementary Table 9) have been deposited at Figshare (DOI: 10.6084/m9.figshare.22233298). Lipidomics data derived from livers of PBS- and B16F10-TE-EVP-educated mice (Supplementary Table 10) have been deposited at Figshare (DOI: 10.6084/m9.figshare.22233307). Lipidomics data derived from livers of PBS- and K7M2-TE-EVP-educated mice (Supplementary Table 11) have been deposited at Figshare (DOI: 10.6084/m9.figshare.22233316). Metabolomics data derived from skin-TE-EVPs and B16F10-TE-EVPs (Supplementary Table 14) have been deposited at Figshare (DOI: 10.6084/m9.figshare.22233352). Metabolomics data derived from livers of PBS- and B16F10-CL-EVP-educated mice (Supplementary Table 29) have been deposited at Figshare (DOI: 10.6084/m9.figshare.22233379). Lipidomics data derived from livers of PBS- and B16F10-CL-EVP-educated mice (Supplementary Table 30) have been deposited at Figshare (DOI: 10.6084/m9.figshare.22233403). Metabolomics data derived from livers of PBS- and K7M2-CL-EVP-educated mice (Supplementary Table 31) have been deposited at Figshare (DOI: 10.6084/m9.figshare.22233424). Lipidomics data derived from livers of PBS- and K7M2-CL-EVP-educated mice (Supplementary Table 32) have been deposited at Figshare (DOI: 10.6084/m9.figshare.22233445). Metabolomics data were analyzed using MetaboAnalyst 4.0 (https://www.metaboanalyst.ca/home.xhtml), and metabolite set enrichment analysis was performed based on the Small Molecule Pathway Database (SMPDB) (http://www.smpdb.ca/).