

Abstract
We report the facile photochemical generation of a library of Ni(I)–bpy halide complexes (Ni(I)(Rbpy)X (R = t-Bu, H, MeOOC; X = Cl, Br, I) and benchmark their relative reactivity toward competitive oxidative addition and off-cycle dimerization pathways. Structure-function relationships between ligand set and reactivity are developed, with a particular emphasis on rationalizing previously uncharacterized ligand-controlled reactivity toward high energy and challenging C(sp2)–Cl bonds. Through a dual Hammett and computational analysis, the mechanism of the formal oxidative addition is found to proceed through an SNAr-type pathway, consisting of a nucleophilic two-electron transfer between the Ni(I) 3d(z2) orbital and the Caryl–Cl σ* orbital, which contrasts the mechanism previously observed for activation of weaker C(sp2)–Br/I bonds. The bpy substituent provides a strong influence on reactivity, ultimately determining whether oxidative addition or dimerization even occur. Here we elucidate the origin of this substituent influence as arising from perturbations to the effective nuclear charge (Zeff) of the Ni(I) center. Electron donation to the metal decreases Zeff, which leads to a significant destabilization of the entire 3d orbital manifold. Decreasing the 3d(z2) electron binding energies leads to a powerful two-electron donor to activate strong C(sp2)–Cl bonds. These changes also prove to have an analogous effect on dimerization, with decreases in Zeff leading to more rapid dimerization. Ligand-induced modulation of Zeff and the 3d(z2) orbital energy is thus a tunable target by which the reactivity of Ni(I) complexes can be altered, providing a direct route to stimulate reactivity with even stronger C–X bonds and potentially unveiling new ways to accomplish Ni-mediated photocatalytic cycles.
Graphical Abstract
1. Introduction
Photoredox catalysis has captivated the fields of organic and inorganic chemistry, with nickel(II)–bipyridine (bpy) aryl halide complexes retaining a prominent place as the metal-ligand scaffold of choice for numerous cross-coupling reactivity pathways.1–11 Among these, C–C and C–heteroatom couplings have been facilitated by Ni(II)–bpy complexes through either the use of an external photosensitizer (e.g., Ir(ppy)3) or via direct excitation of the Ni(II)–bpy aryl halide complex.12–18 Due to the diverse reactivity and intriguing photophysics of these complexes, much interest has been placed on understanding the underlying photophysical and thermal processes involved in photoredox mediated cross-coupling reactivity.19–28
While originally thought to proceed through a Ni(0)/Ni(II) cycle,6 recent work has instead supported a Ni(I)/Ni(III) cycle in the direct excitation pathway (Figure 1A).23,29,30 Through analysis of a library of Ni(II)–bpy aryl halide complexes, we have revealed that excited-state Ni(II)–Caryl bond homolysis from the S = 0 square planar Ni(II) ground state features a key ligand-to-metal charge transfer (LMCT) process. This LMCT results in electron excitation between the Ni–aryl σ and σ* orbitals, which lowers the bond order from one to zero, resulting in repulsive homolytic bond cleavage and the generation of an aryl radical and a three-coordinate Ni(I)–bpy halide species.27
Figure 1.
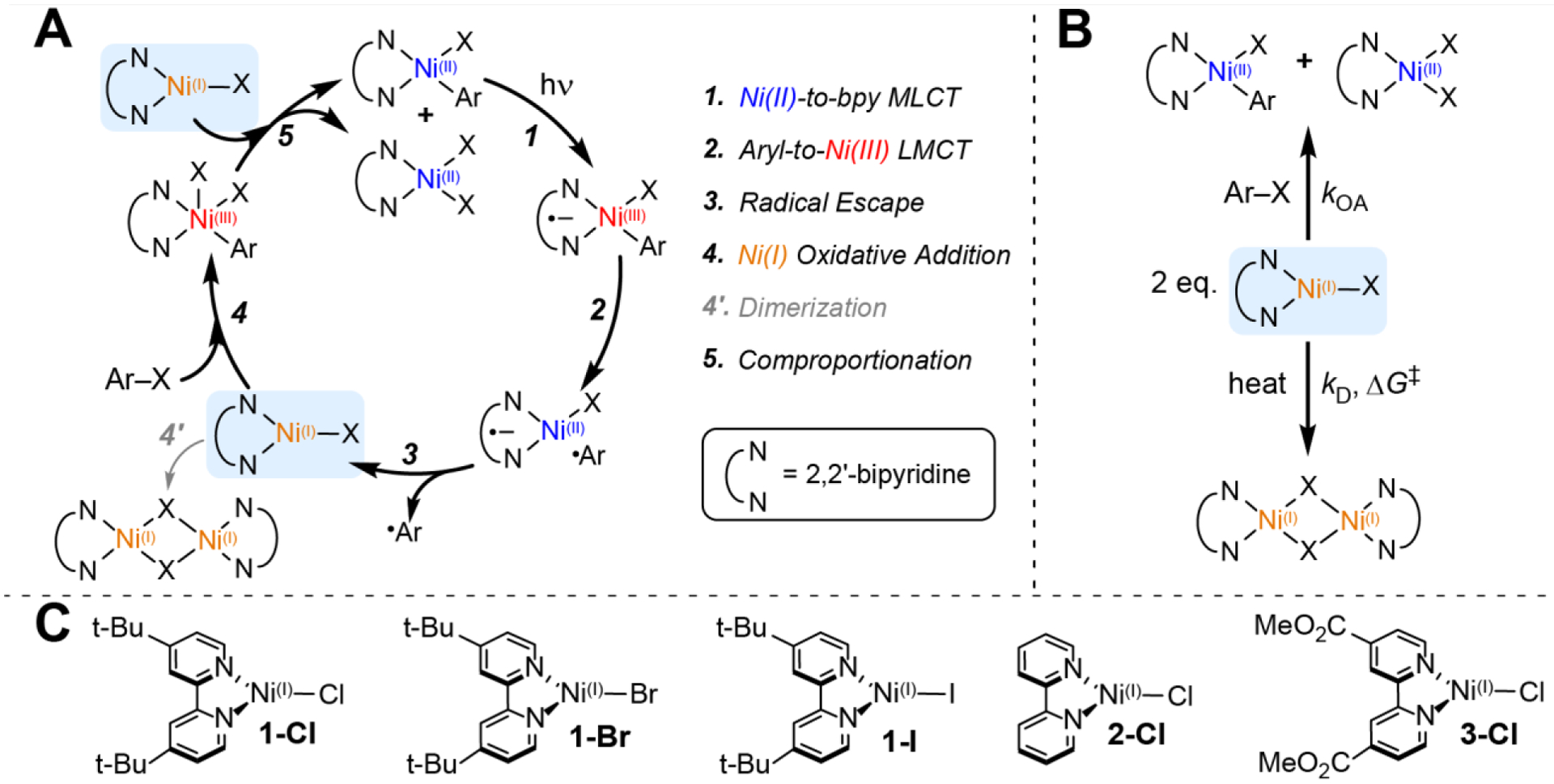
(A) Catalytic cycle displaying the photochemical activation of Ni(II)–bpy aryl halide complexes proposed in ref. 27 for the generation of Ni(I) and Ni(III) intermediates. (B) Two competing pathways for the Ni(I) complexes investigated here: oxidative addition and dimerization. (C) Photogenerated Ni(I)(Rbpy)X structures examined in this work (R = t-Bu, H, MeOOC; X = Cl, Br, I).
Related Ni(I)–bpy halide complexes have been prepared by alternate methods, including pulse radiolysis, electrolysis, and independent synthesis.30–33 Importantly, these compounds have demonstrated potency for the activation of aryl halide substrates. Following the work by Vicic et al. on Ni(I)–terpyridine complexes,34–36 Bird and MacMillan et al. reported a nickel(I)(4,4′-di-tert-butyl bipyridine)bromide complex (Ni(I)(t-Bubpy)Br) that exhibited rapid reactivity toward aryl iodides with second-order rate constants of ~104 M−1 s−1.31 The activation of C(sp2)–Br substrates was demonstrated by Doyle et al. using a nickel(I)(diethyl-2,2′-bipyridine-4,4′-dicarboxylate)chloride complex, Ni(I)(EtOOCbpy)Cl. Activation of the stronger aryl bromide bond proceeded with slower, but still catalytically relevant, rate constants of ~10−2 – 101 M−1 s−1.30 Notably, C(sp2)–Cl bonds could not be activated by this Ni(I) complex.
Careful kinetic analysis has been employed to understand the mechanism of this reactivity;30,37 the Ni(I) species has been suggested to undergo two-electron oxidative addition to form a short-lived five-coordinate Ni(III)–bpy aryl dihalide complex. This Ni(III) intermediate rapidly decays by comproportionation with another equivalent of Ni(I), forming Ni(II)–bpy aryl halide and Ni(II)–bpy dihalide complexes (Figure 1A).30 Using three-coordinate pyridinophane Ni(I) model complexes, Mirica et al. have also provided evidence for this Ni(I)/Ni(III) oxidative addition pathway.29 Therefore, these and related studies have argued for the importance of three-coordinate Ni(I)–bpy halide complexes to facilitate the oxidative addition step, without which the cross-coupling catalytic cycle would not close.21,23,38,39
While the reactivity of Ni(I)–bpy complexes has proven desirable for organic synthesis, their solution-phase stabilities vary widely, limiting their scope. Numerous Ni(I) dimerization products have been characterized, and the resultant polypyridyl species are no longer reactive toward oxidative addition with aryl halides.30,32,40,41 Dimerization thereby acts as an off-cycle sink with diminished catalytic activity (Figure 1A). Hazari et al. independently synthesized and characterized the dimeric [Ni(I)(t-Bubpy)Cl]2 complex. Once formed, the compound was stable in solution and exhibited no oxidative addition reactivity even with weak C(sp2)–I bonds.32 The related [Ni(I)(t-Bubpy)Br]2 complex was studied by Bird and MacMillan et al. and similarly showed no oxidative addition reactivity with aryl iodides.31 Additionally, tetrameric [Ni(I)(EtOOCbpy)Cl]4 and dimeric [Ni(I)(EtOOCbpy)Cl]2 complexes were studied by Doyle et al.30 In solution, both species can dissociate to form the monomeric Ni(I)(EtOOCbpy)Cl complex mentioned above. This three-coordinate species was reported to exist in solution-phase equilibria with Ni(II)Cl2 and Ni(0)(EtOOCbpy)2. However, the oxidative addition reactivity was favored from the Ni(I) complex, not the Ni(0) species, and no reactivity was found for the dimeric or tetrameric species.
Understanding the interplay between the oxidative addition and dimerization/oligomerization pathways available to three-coordinate Ni(I)–bpy halide complexes is critical for optimized ground-state and metallaphotoredox catalysis and reaction development (Figure 1B). There is also a fundamental knowledge gap related to specific structure-function relationships underlying the elementary oxidative addition step in Ni(I)–bpy complexes, a key component of the scope of bond activations that can be achieved. To the best of our knowledge, no systematic study has either compared the oxidative addition reactivity and solution-phase stability of Ni(I)–bpy complexes or rationalized the relative rates of oxidative addition upon structural perturbation.
Here we accomplish both of these aims through the facile photogeneration of a series of Ni(I)(Rbpy)X species (R = t-Bu, H, MeOOC; X = Cl, Br, I; Figure 1C). We provide direct evidence for C(sp2)–Cl bond activation with Ni(I)–bpy complexes, which thus far has only been implied as part of catalytic cycles.33,42 Furthermore, we broadly detail the kinetics and mechanisms of their oxidative addition reactivity toward C(sp2)–X (X = Cl, Br, I) bonds, and their thermal barriers for dimerization. Results reported herein implicate key ligand effects on the electronic structure of the Ni(I)–bpy halide complexes that result in tunable reactivity and variable solution-phase lifetimes. Ultimately, the ability of Ni(I)–bpy complexes to activate challenging C(sp2)–Cl bonds stems from bpy-induced modulation of the effective nuclear charge (Zeff) of the Ni(I) center. Related analyses reported here have afforded a molecular orbital-based picture of oxidative addition reactivity, which provides specific electronic structure benchmarks that need to be achieved to activate strong C(sp2)–X bonds and opens the door for the targeted synthesis of next-generation cross-coupling catalysts.
2. Results and Analysis
2.1. Photochemical Synthesis and Spectroscopic Characterization of Ni(I)–bpy halide complexes
Parent four-coordinate Ni(II)(Rbpy)(o-tolyl)X (R = tert-butyl, H, MeOOC; X = Cl, Br, I) complexes were synthesized according to a previous report.27 Three-coordinate Ni(I)(Rbpy)X species, which are important reaction intermediates in photoredox cross-coupling catalysis, can be accessed directly from these precursors by air- and moisture-free irradiation at 370 nm using purple LEDs. Light excitation drives a key LMCT process that results in Ni(II)–Caryl excited-state bond homolysis; this shorter wavelength enhances the quantum yield of Ni(I)(Rbpy)X generation relative to more commonly used, longer wavelength blue light sources (Table S2). Typical irradiation times were ~30 – 60 minutes; irradiation time variations among complexes depended on the rate of photo-driven decay of the starting Ni(II) complex and the rate of Ni(I) decomposition. Global kinetic analysis provides strong support for a first-order process in which the Ni(II)–bpy aryl halide parent complex is cleanly photolyzed to the Ni(I)–bpy halide (for more details, see Supporting Information Section S1.5, including Figures S6–S10).
To systematically probe the reactivity and stability of the photochemically generated Ni(I) complexes, the bpy ligand was varied with electron donating tert-butyl groups (Ni(I)(t-Bubpy)Cl, 1-Cl), electronically neutral H-atoms (Ni(I)(Hbpy)Cl, 2-Cl), and electron withdrawing methyl ester groups (Ni(I)(MeOOCbpy)Cl, 3-Cl). The halide was also varied by first synthesizing Ni(II)(t-Bubpy)(o-tolyl)Br and Ni(II)(t-Bubpy)(o-tolyl)I parent complexes, followed by irradiation to form Ni(I)(t-Bubpy)Br (1-Br) and Ni(I)(t-Bubpy)I (1-I), respectively. Note that 1-Br is the same as that examined by Bird and MacMillan et al. by pulse radiolysis,31 while 3-Cl is similar to that studied by Doyle et al.,30 the only difference being the appended methyl ester vs ethyl ester substituents, following our previous study.27
Having direct access to the Ni(I)–bpy halide complexes via photolysis allows for their characterization using UV-vis electronic absorption and EPR spectroscopies (Figure 2A and 2C, respectively). Irradiation of Ni(II) parent compounds and spectral analysis were done in tetrahydrofuran (THF). The primary UV-vis absorption band in the ~550 nm – 900 nm region of the three-coordinate Ni(I)(Rbpy)X species shifts strongly with changes to the bpy ligand from 660 nm (1-Cl) to 805 nm (3-Cl). Moderate shifts are also observed when changing the halide (640 nm for 1-I to 660 nm for 1-Cl, Figure 2A). To further corroborate the spectral assignment of UV-vis peak to the Ni(I)–bpy halides, we conducted reductive spectroelectrochemistry on Ni(t-Bubpy)Cl2 to form Ni(t-Bubpy)Cl; the resultant spectrum is identical to that of photogenerated 1-Cl (Figure 2B).
Figure 2.
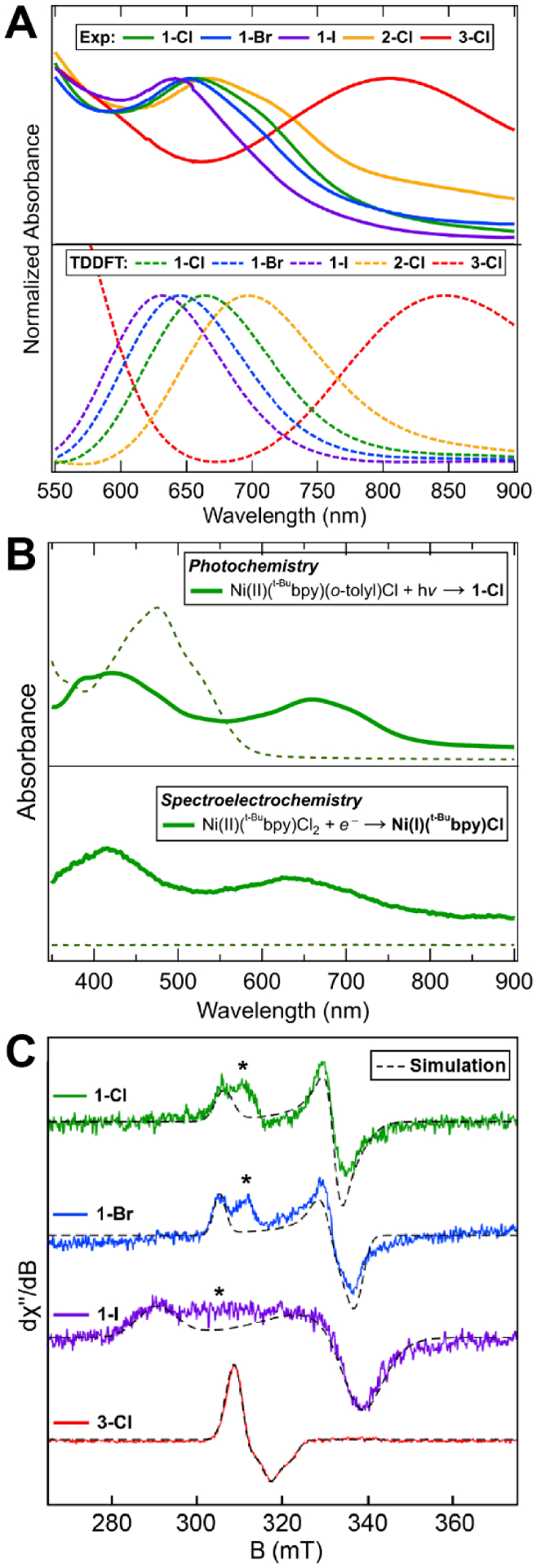
Spectroscopic characterization of the Ni(I)–bpy halides examined in this study. (A) UV-vis absorption spectra (THF) of the three-coordinate Ni(I)–bpy halide complexes, highlighting the Ni(I)-to-bpy MLCT transitions and the excellent spectral agreement between experiment and TDDFT calculations (dashed lines). (B) Top: UV-vis spectrum for the photochemical preparation of 1-Cl (solid line) from its Ni(II)–bpy aryl halide parent (dashed lines) using 370 nm LEDs. Bottom: Spectroelectrochemistry of Ni(II)(t-Bubpy)Cl2 (dashed lines) forming Ni(I)(t-Bubpy)Cl (solid line) in THF with TBAPF6 electrolyte. For more details, see Figure S13. (C) Frozen-glass X-band CW-EPR spectra for the S = 1/2 Ni(I) region (T = 5 K; solvent = 2-MeTHF; frequency for 1-Cl, 1-Br, 3-Cl = 9.637 GHz, for 1-I = 9.646 GHz; power = 2.2 mW; modulation amplitude = 8 G). Corresponding g values are given in Table 1. Starred peaks may correspond to THF coordination.
From TDDFT calculated spectra (bottom of Figure 2A; see Computational Details in Supporting Information Section S2.1), the intensity of the primary absorption band is attributed to Ni(I)-to-bpy metal-to-ligand charge transfer (MLCT) transitions (3d(xz/yz) → π*(1); note these orbitals are labeled according to their parallel (||) or perpendicular (⊥) orientation to Ni‒halide bond – see details in Supporting Information Section S2.4). Consistent with the experimental spectra, smaller shifts are observed for variations in the halide; the MLCT energies are more sensitive to bpy variation, with an energy trend of 1-Cl > 2-Cl > 3-Cl. Through analyses of the molecular orbital energy diagrams for these complexes (Figures S40–S44 and Table S7), the MLCT energy shift arises mainly from the stabilization of the bpy π* acceptor orbitals as a function of electron-with-drawing substituents.
While room temperature EPR analysis provided no resolvable signals, likely due to rapid spin relaxation times (Figure S31), spectra taken at 5 K in 2-methyl tetrahydrofuran (2-MeTHF) after irradiation provided signals characteristic of S = 1/2 Ni(I) species (Figure 2C), with g values in the range of gz = ~2.22 – 2.37, gx = ~2.04 – 2.17, gy = ~2.07 – 2.20, and giso = ~2.12 – 2.19 (Figure 2C; Table 1). The axial g tensor values are overall consistent with a single unpaired electron in the Ni(I) 3d(x2-y2) orbital. The EPR spectra and g values of compounds 1-Cl/Br and 3-Cl are congruent with previous reports for Ni(I) halide species with aromatic ligand backbones (e.g., phenanthroline, bipyridine, and bis(pyrazolyl)pyridines).30,39,42–45 The 14N hyperfine values employed in the 3-Cl simulation are of comparable magnitude to those previously reported for Ni(I)-neocuproine complexes (0, 50, and 170 MHz).32 Sufficiently high concentration samples of 2-Cl were precluded due to a precipitation (dimerization) pathway (vide infra, Section 2.4); as such, no EPR signal could be detected. The intermediate peak at ~310 mT in 1-Cl/Br/I is likely attributable to THF solvent coordination (see Supporting Information Section 2.7), as it does not appear in previous spectra recorded in toluene but does appear in toluene:THF solvent mixtures and in neat 2-MeTHF.42,43 These published data share excellent agreement with the spectra collected and presented in Figure 2C. Importantly, the relative intensities of the gz and intermediate peaks remain approximately constant with variations in photolysis time, indicating there is not a second Ni(I) species with a distinct kinetic profile (Figures S33–S35). The DFT calculated g values are in general agreement with the experimental data, with the exception of the gz value for 1-I and significantly more predicted rhombicity throughout (computed gx and gy in Table 1).
Table 1.
Experimental g values for the Ni(I)–bpy halide complexes in 2-MeTHF at 5 K. DFT computed g values are given in parentheses.
| Compound | gz,exp. (gz,calc.) |
gx,exp. (gx,calc.) |
gy,exp. (gy,calc.) |
giso,exp. (giso,calc.) |
|---|---|---|---|---|
| 1-Cl | 2.248 (2.238) |
2.050 (2.070) |
2.070 (2.174) |
2.123 (2.161) |
| 1-Br | 2.255 (2.246) |
2.042 (2.080) |
2.079 (2.166) |
2.125 (2.164) |
| 1-I | 2.370 (2.262) |
2.044 (2.094) |
2.075 (2.161) |
2.163 (2.172) |
| 2-Cl a | --- (2.238) |
--- (2.066) |
--- (2.190) |
--- (2.165) |
| 3-Cl | 2.217 (2.252) |
2.171 (2.057) |
2.195 (2.251) |
2.194 (2.187) |
Sufficiently high concentration samples of 2-Cl were precluded due to precipitation. As such, no EPR signal could be resolved experimentally. 14N hyperfine values of Ax = 146 MHz, Ay = 91 MHz, and Az = 64 MHz were fit for 3-Cl and required to model the line shape. Computational details are given in Supporting Information Section S2.1.
We also note the presence of additional complex signals in the half-field region of the concentrated samples of 1-Cl in the low-temperature (5 K) EPR (Figure S33–S35). Variable temperature (VT) UV-vis experiments indicate that these are attributable to a reversible concentration- and temperature-dependent speciation (see Supporting Information Section S1.9). That is, these additional species form rapidly upon freezing the EPR samples. Future studies should consider speciation changes that can occur upon freezing samples for low temperature spectroscopic characterization. That said, under standard catalysis conditions (~0.2 mM, room temperature) studied herein, the Ni(I)–bpy halide complexes corresponding to the S = 1/2 signals are the dominant species after irradiation is terminated (~95% Ni(I) by VT UV-vis analysis), with the additional species (~5%) being the starting Ni(II)–bpy aryl halide complex.
2.2. Oxidative Addition Kinetics with 2-Chloro-toluene
Having demonstrated near quantitative conversion of parent Ni(II) complexes to the three-coordinate Ni(I) complexes, we sought to gauge their relative reactivity toward oxidative addition. As expected, room temperature addition of excess 2-bromo-toluene or 2-iodo-toluene (0.2 mL) to all photogenerated Ni(I) complexes studied herein resulted in immediate color changes, with loss of the Ni(I)-bpy MLCT features observed in the UV-vis spectra (Figures S14–S15). The diagnostic S = 1/2 Ni(I) EPR signal is also quenched upon addition of aryl halide (Figure S36). Again, reactivity with C(sp2)–I and C(sp2)–Br bonds is consistent with previous reports.30,31 However, interestingly, many of the Ni(I) intermediates studied herein also react with 2-chloro-toluene – the exception being 3-Cl. As a control, addition of 0.2 mL of toluene did not result in UV-vis spectral changes, further implicating the reactivity of the Ni(I) species with the C(sp2)–Cl bond. The lack of reactivity of 3-Cl is also significant and is directly related to the bpy ligand, as discussed further in Section 2.3.
Taking1-Cl as a representative compound, we sought to confirm the C(sp2)–Cl reactivity by stoichiometric 1H NMR studies in d8-THF. However, to facilitate reaction turnover at a rate greater than the decay of the Ni(I) intermediate, a large excess of 2-chloro-toluene is necessary, which over-whelms the 1H NMR analysis. Using fewer equivalents of aryl halide, the reaction between 2-chloro-toluene and the photogenerated Ni(I) species is too slow and precludes definitive product speciation assignments (Supporting information Section S1.7). This can be circumvented using a fluorinated aryl halide and 19F NMR analysis. As depicted in Scheme 1, addition of 20 μL of 2-chloro-α,α,α-trifluorotoluene (~200-fold excess, 19F NMR peak at −63 ppm) to a solution of 1-Cl afforded a new peak in the 19F NMR spectrum at −58 ppm. Independent synthesis27 confirms this peak originates from the four-coordinate complex, Ni(II)(t-Bubpy)(CF3Ph)Cl (Figure S18). Thus, this experiment provides direct evidence of a Ni(I)–bpy halide complex activating an aryl chloride substrate. We now describe the relative reactivity of the Ni(I)–bpy complexes toward C(sp2)–Cl oxidative addition.
Scheme 1.
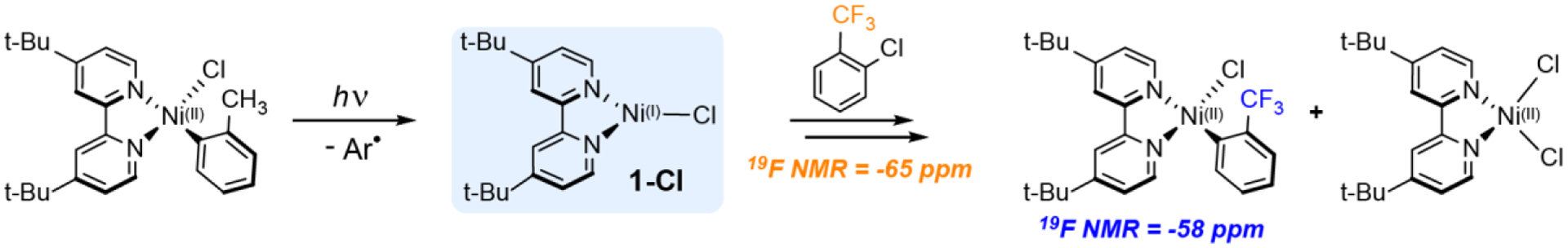
Oxidative addition reaction conducted with the photogenerated Ni(I)–bpy halide, 1-Cl. Incident wavelength = 370 nm; solvent = d8-THF. Formation of Ni(II)(t-Bubpy)(CF3Ph)Cl was verified by 19F NMR; the high spin Ni(II)–bpy dihalide is paramagnetic and not observed, but is invoked to account for the mass balance on the basis of previous work.30
Pseudo-first order kinetics investigations with quantitative addition of excess 2-chloro-toluene to the photogenerated Ni(I) complexes were carried out by monitoring the fast decay of the primary MLCT absorption feature via UV-vis spectroscopy (Figure 3A and S20A). The natural logarithm of the normalized peak absorbance varies linearly with time for ~3–5 half-lives, depending on the magnitude of starting Ni(I) absorption (Figure 3B and S20B). The slope of this correlation provides kobs values that vary linearly with 2-chloro-toluene concentration (Figure 3C and S20C), yielding second-order rate constants for oxidative addition, kOA (M−1 s−1) (Table 2). The bimolecular rate constants are on the order of 10−2 M−1 s−1, six orders of magnitude slower than the reaction of 1-Br with aryl iodides and two orders of magnitude slower than the reaction of Ni(I)(EtOOCbpy)Cl with aryl bromides.30,31 This reduction in rate constant for the activation of 2-chloro-toluene is attributable to the increased bond dissociation free energy (BDFE) of the carbon–halogen bond (BDFE of C(sp2)–Cl > C(sp2)–Br > C(sp2)–I, Table S15) and a steric effect of the ortho-methyl group (Figure 4).
Figure 3.
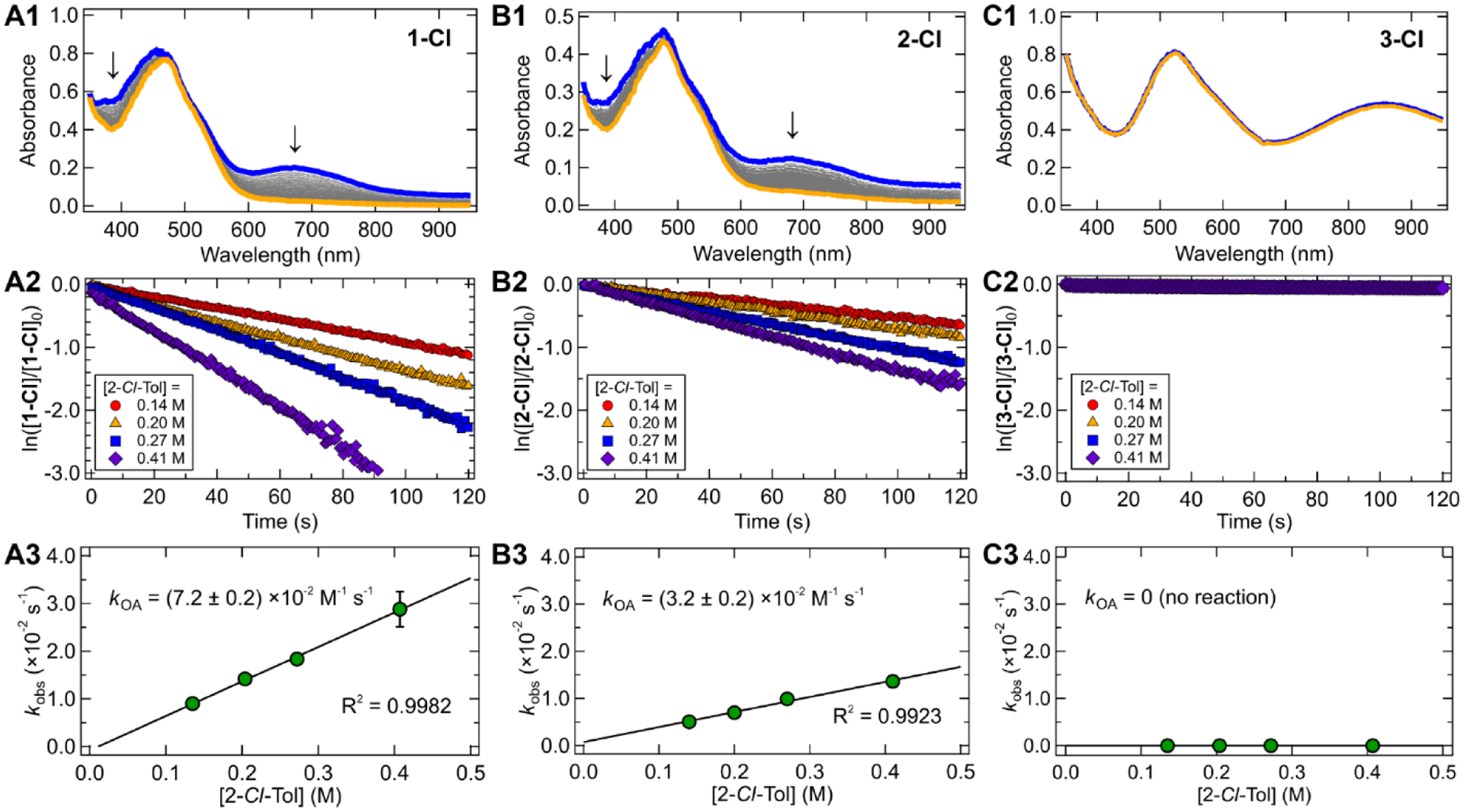
Experimental kinetic analysis of the oxidative addition reaction for 1-Cl, 2-Cl, and 3-Cl. (A1, B1, C1) UV-vis absorption plots in THF showing the addition of 2-chloro-toluene to photochemically generated Ni(I)–bpy halides (initial spectrum of Ni(I)–bpy halide, blue line (~10−4 M); after 2-chloro-toluene addition, orange line). Note the orange spectra depict unreacted and newly generated parent four-coordinate Ni(II)–bpy aryl halide (Figure S1–S2). (A2, B2, C2) Linear plots of ln([Ni(I)]/[Ni(I)]0) vs time showing the pseudo-first order nature of the oxidative addition reaction over several half-lives; [2-Cl-Tol] = 0.14 M (red circles), 0.20 M (orange triangles), 0.27 M (blue squares), and 0.41 M (purple diamonds). (A3, B3, C3) Plots of kobs vs [2-Cl-Tol]. Error bars are one standard deviation of three trials. Slope of the fitted line gives second-order rate constants, kOA (M−1 s−1). Complex 3-Cl showed no reaction with 2-chloro-toluene. Analogous data for 1-Cl, 1-Br, and 1-I are given in Figure S20.
Table 2.
Rate constants for room temperature oxidative addition for the reaction between 2-chloro-toluene and various Ni(I)(Rbpy)X species (R = t-Bu, H, MeOOC; X = Cl, Br, I).a
| Compound | σp (R) | σp (X) | kOA (x10−2 M−1 s−1) |
|---|---|---|---|
| 1-Cl | −0.20 | 0.23 | 7.2 ± 0.2 |
| 1-Br | −0.20 | 0.23 | 7.0 ± 0.4 |
| 1-I | −0.20 | 0.18 | 6.5 ± 0.6 |
| 2-Cl | 0.00 | 0.23 | 3.2 ± 0.2 |
| 3-Cl | 0.45 | 0.23 | no reaction |
Reactions were done under inert atmosphere in anhydrous THF and followed by UV-vis spectroscopy. Reported errors are one standard deviation of three trials. Hammett σp values taken from ref. 46.
Figure 4.
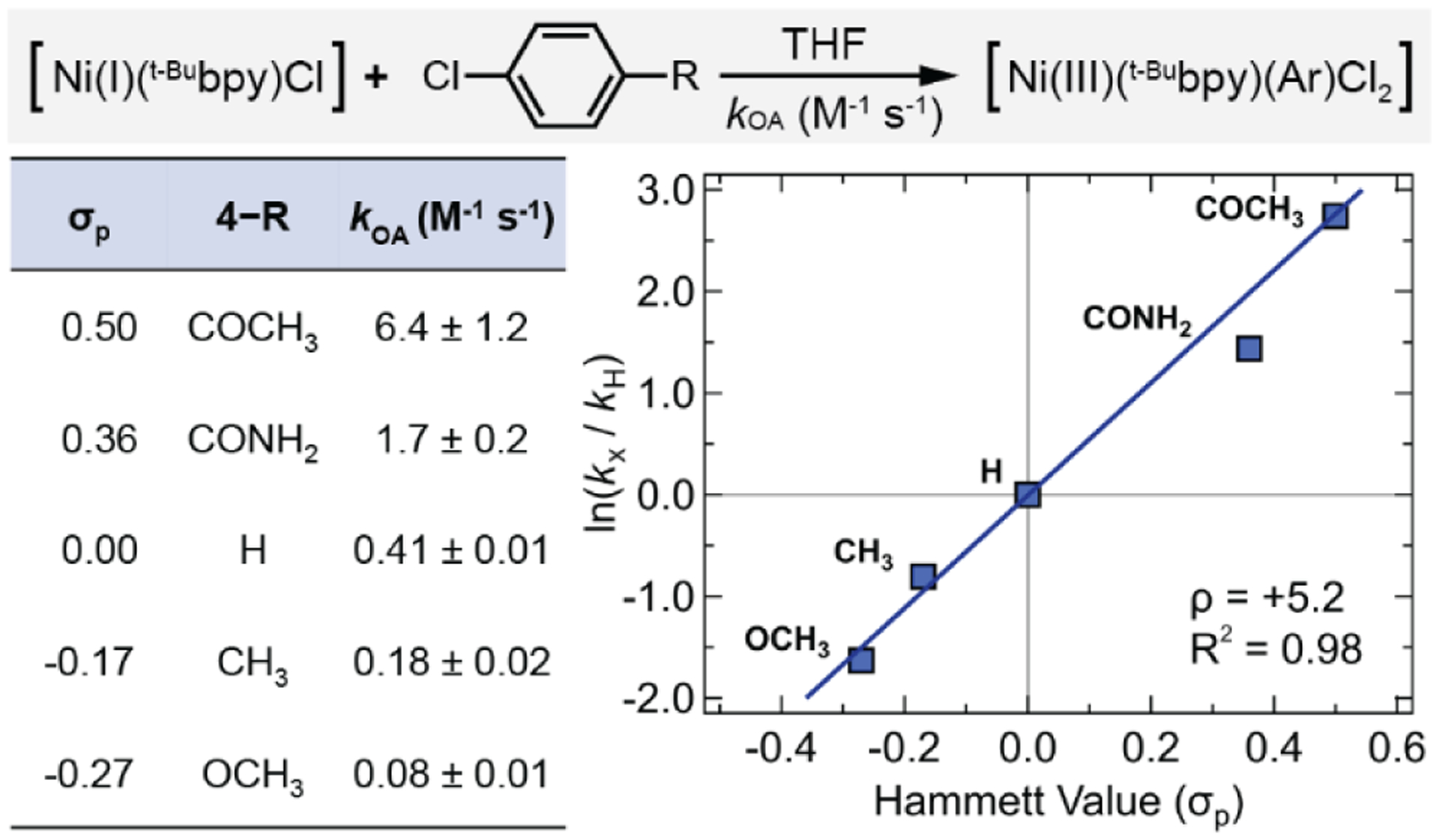
Hammett analysis for the reaction of 1-Cl with 4-substituted aryl chlorides. Recall that the reaction of 1-Cl with sterically hindered 2-chloro-toluene has kOA = 0.072 M−1 s−1. Hammett σp values taken from ref. 46. Reported standard errors are propagated from the linear least-squares analysis (Figures S21–S25).
Analysis of ligand effects reveals that changes to the halide manifest in minor changes in kOA (Figure S20). These results agree well with the electronic effects of the halide as predicted by the Hammett parameters (σp = 0.23 for Cl and Br, and 0.18 for I).46 More pronounced effects are observed upon variation of the bpy substituent. Substitution of the electron donating tert-butyl groups (σp = –0.20) for hydrogens (σp = 0.0) in going from 1-Cl to 2-Cl results in a two-fold decrease in kOA from 7.2 ± 0.2 × 10−2 M−1 s−1 to 3.2 ± 0.2 × 10−2 M−1 s−1. Furthermore, introduction of the electron with-drawing methyl ester groups (σp = 0.45) eliminates C(sp2)–Cl oxidative addition reactivity altogether. As described further below in Section 2.3, these differences in oxidative addition reactivity can be traced directly to ligand-induced differences in Zeff on the metal, as changes in Zeff tune the energy of the redox active molecular orbital (RAMO) involved in the oxidative addition reaction. Follow-up work is currently underway to push the limits of Ni(I)-facilitated oxidative addition reactivity by substituent-based modulation of Zeff.
2.3. Oxidative Addition Mechanistic Investigations
Having examined the effect of the substituents on the Ni(I)–bpy halides toward the C(sp2)–Cl oxidative addition reactivity, we next turned to the kinetic dependence of the substituents on the aryl chloride substrates. We performed a Hammett analysis on the reactivity of series of para-substituted aryl chlorides with 1-Cl. The rate constants (kOA) of the formal Ni(I)/Ni(III) oxidative addition process increased by two orders of magnitude when switching from electron donating to electron withdrawing substituents (σp = − 0.27 to 0.50, Figure 4). We further found the slope (ρ) of the plot of ln(kx/kH) vs σp was ~5, indicative of an SNAr type activation47 of the C(sp2)– Cl bond, wherein the Ni(I) metal center acts as a nucleophile, attacking the polarized C(sp2)– Cl bond at the electron-deficient carbon. While an activation pathway that proceeds by single electron transfer (SET) also has a fairly large ρ value (~4),48 the reduction potentials of the aryl chlorides are too negative (experimentally found to be less than –3.2 V vs Fc+/Fc, calculated as –3.5 V vs Fc+/Fc; for more details see Supporting Information Section S1.6 and S2.6) to be accessed by the Ni(I)–bpy halides (calculated Ni(II)/Ni(I) potential of 0.2 V vs Fc+/Fc).49
Interestingly, we note that ρ ~5 represents a change in the specific mechanism of C(sp2)– X bond activation by Ni(I) halides. Aryl bromides and aryl iodides exhibit Hammett slope values of ρ ~2–3, typical of a concerted oxidative addition pathway,30,31,50,51 but the activation of the stronger, more polarized C(sp2)–Cl bond is better described as nucleophilic aromatic substitution.
DFT calculations were used to further detail the mechanism of oxidative addition (i.e., step-wise vs concerted two-electron nucleophilic attack by Ni) and to identify the key frontier molecular orbitals involved in this elementary reaction step. Oxidative addition of 2-chloro-toluene to a three-coordinate Ni(I)(Rbpy)X species yields a five-coordinate Ni(III)(Rbpy)(o-tolyl)(Cl)(X) intermediate. Following the initial oxidative addition step, comproportionation of the Ni(III) complex with an additional Ni(I)(Rbpy)X species can take place with a calculated Gibbs free energy of ~–23 kcal mol−1, which produces a considerable driving force for the overall reaction. Comproportionation can occur via transfer of a halide from the five-coordinate intermediate, which is readily accessible through a barrierless transition state (Figure 5A). This step therefore results in the regeneration of a parent Ni(II)‒bpy aryl halide complex and the formation of a Ni(II) dihalide (i.e., the speciation determined experimentally by Diao et al. and Doyle et al. and 19F NMR and UV-vis studies presented herein (vide supra, Section 2.2)).30,39
Figure 5.
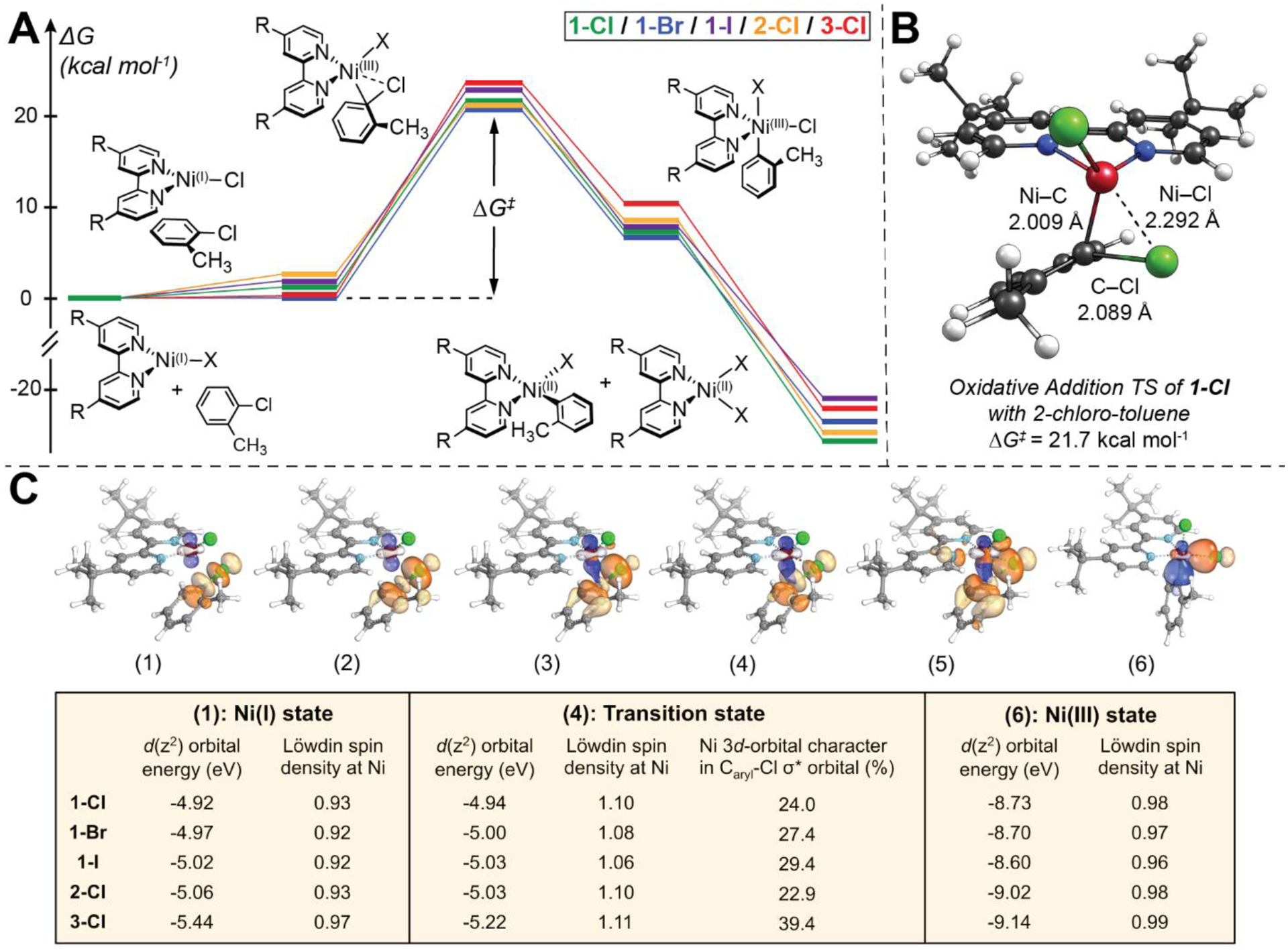
(A) DFT energetics of the oxidative addition reactions of Ni(I)(Rbpy)X with 2-chloro-toluene and (B) the transition state structure of 1-Cl with 2-chloro-toluene. Relative Gibbs free energy values were computed at the B3LYP-D3/def2-TZVP(CPCM)//BP86-D3/def2-TZVP(CPCM) level.52–54 Computed free energies are provided in Table S13. A related plot showing the DFT energetics of the oxidative addition with 2-bromo-toluene and 2-iodo-toluene is given in Figure S65. (C) Computational analysis of the oxidative addition process. The doubly occupied Ni 3d(z2) orbital transforms into a new Ni–Caryl σ-bond (blue), transferring electrons into the Caryl‒Cl σ* orbital (orange). This breaks the Caryl‒Cl σ bond, affording a new Ni‒Cl σ-bond. Tabulated are the energies of the β Ni 3d(z2) orbital for several steps along the IRC, the Ni 3d character in the Caryl‒Cl σ* orbital at the transition state, and the Löwdin spin density at the Ni center. Note energies of Ni 3d(z2) orbitals trend with the Ni(I) reactivity (see Table 2), while the other tabulated properties uncover the two-electron nature of the nucleophilic attack by the Ni(I) at the Caryl site. See Supporting Information Section S2.3 for more detailed IBO analysis.
The rate-determining step of the overall reaction is found to be oxidative addition, with calculated Gibbs free-energy barriers of ~21 – 23 kcal mol−1, consistent with the slow reaction rates at room temperature and the requirement of a large excess of 2-chloro-toluene (Figure 5A). The computed free energy barriers vary by only ~1.3 kcal mol−1 for 1-Cl, 1-Br, 1-I, and 2-Cl (i.e., within the general accuracy obtainable with DFT calculations).55,56 However, this observation is still in accord with the differences in kOA for the compounds observed experimentally (Table 2). Furthermore, the highest computed activation free energy is observed for 3-Cl (~23 kcal mol−1, red in Figure 5A), consistent with its lack of reactivity with 2-chloro-toluene.
We have also computationally reproduced the Hammett analysis for the reaction of 1-Cl with 4-substituted aryl chlorides and have found excellent agreement with experiment (calculated ρ ~5.8; see Supporting Information Section S2.6). Furthermore, the calculated bond lengths in the transition state show that the new Ni–Cl bond is ~0.3 Å longer than the new Ni–C bond (potentially implying the Ni–C bond is formed prior to the Ni–Cl bond and corroborating an SNAr-type mechanism, Figure 5B). Additionally, we observe that the experimental rate constants trend linearly and positively with the Löwdin atomic charges, the Mulliken atomic charges, and the natural population analysis (NPA) charges at the carbon in the Ar–Cl bond, but they do not trend well with the energy of the LUMO of the aryl chlorides (Figure S66). This observation is also indicative of an SNAr mechanism as the electrophilicity of this carbon is related to the rate and not the reduction potential of the aryl chlorides (as would be expected for SET).57
To further elucidate the origin of the different reactivity profiles for the different Ni(I) species, we analyzed the initial oxidative addition step in the context of intrinsic bond orbital (IBO) progression.58,59 IBO analysis allowed for the identification of orbital changes (δ-orb) along the intrinsic reaction coordinate (IRC), wherein the separated Ni(I) and aryl halide reactants come together to form the five-coordinate Ni(III) adduct. Although the unpaired electron is located in the Ni 3d(x2-y2) orbital (cf. EPR analysis in Section 2.1), the largest δ-orb effects are observed in the doubly-occupied Ni 3d(z2) orbital and Caryl–Cl σ orbital. These are transformed into new Ni‒Caryl and Ni‒Cl σ bonds, respectively (Figure S39). This change in bonding is the outcome of two-electron transfer from the occupied Ni 3d(z2) orbital into the virtual Caryl– Cl σ* orbital (Figure 5C). Direct observation of Ni-to-(Caryl–Cl) σ* backbonding at the transition state is evidenced by significant mixing of filled 3d orbital character into the unoccupied Caryl–Cl σ* orbital along the IRC (Figure 5C, middle). For example, from 1-Cl to 2-Cl to 3-Cl, the Löwdin Ni 3d character varies from 24.0 to 29.4 to 39.4. Previous analysis on Pd catalyzed aryl chloride activation found that the increase in Pd backbonding to the Caryl– Cl σ* orbital was related to an increase in the barrier for the reaction.60 This same trend is seen in our Ni-based C(sp2)–Cl bond activation. Thus, backbonding in the transition state may play an important role in activating stronger C(sp2)–Cl bonds by Ni(I) species. As discussed further below, this backbonding will increase with less negative electron binding energies on the metal.
At the transition state, the α- and β-type orbitals are transformed in conjunction, suggesting a two-electron nucleophilic attack by the Ni(I) 3d(z2) orbital on the Caryl atom, yielding a Ni(III) intermediate. This concerted two-electron transfer is supported by a small change of Löwdin spin density on the Ni center throughout the reaction (~1 unpaired electron throughout; see inset table in Figure 5C); one-electron transfer or stepwise two-electron transfer would reveal more substantial metal-based spin density changes along the IRC. The small increase in the spin density at the transition state arises from the slightly misaligned attack initiated by β Ni 3d(z2) orbital, which is ~0.8 eV higher in energy than the α Ni 3d(z2) orbital due to spin polarization arising from the presence of five vs four 3d electrons in α and β manifolds, respectively (Table S7). That Caryl–Cl bond-breaking is rate-determining is consistent with the more facile oxidative addition activation of 2-bromo-toluene and 2-iodo-toluene substrates (e.g., their reactivity is observed even with 3-Cl due to their weaker Caryl–halogen σ bonds and lower energy Caryl–Cl σ* orbitals, which facilitates backbonding from the metal).
From the preceding analysis, the reactivity of individual Ni(I) complexes should correlate with the energies of the Ni 3d(z2) orbitals. Indeed, the calculated Ni(I)–bpy 3d(z2) orbital energies trend with the kOA rate constants (Figure 5C and Table 2). For example, the most and least reactive complexes, 1-Cl and 3-Cl, have β 3d(z2) orbital energies of −4.92 eV and −5.44 eV, respectively (Δ = −0.52 eV). There is also a linear trend between the energy of the 3d(z2) orbital and the bpy σp Hammett parameters for 1-Cl, 2-Cl, and 3-Cl (Figure S45). The less-negative electron binding energies of 1-Cl will increase nucleophilicity and propensity for a two-electron reduction. As discussed below, this effect on the 3d orbital energies is not necessarily specific to 3d(z2) alone, specifically because the bpy-based σ interaction only involves the torus of the 3d(z2) orbital. Rather, the bpy ligand ultimately tunes the entire 3d orbital manifold via changes in Zeff of the metal, making the ligand substitution effects a key predictor of reactivity. In further support of these changes, the calculated Ni(I) 1s and 2p(x,y,z) orbital energies of 1-Cl, 2-Cl, and 3-Cl all trend linearly with bpy-based Hammett parameters and oxidative addition rate constants, with slopes similar to the 3d orbitals (Figure S45–S46). Moving forward, the calculated 3d orbital energies of three-coordinate Ni(I) intermediates will be a useful predictor for oxidative addition reactivity and will bracket relative reactivity for specific C(sp3)–X and C(sp2)–X bonds.
2.4. Thermodynamics of the Dimerization of Ni(I)–bpy halides
Having established the reactivity of the Ni(I) complexes toward oxidative addition, we sought to better understand their general stability in solution. Their stability is of particular interest, as unreactive halide-bridged dimers have been proposed to form in bpy and related systems.30–32,40 Photogeneration of the Ni(I) compounds was again achieved using 370 nm LEDs in THF. Even in the absence of aryl halide, the characteristic MLCT band of the Ni(I)–bpy halide complexes decayed over time; this decay was accelerated by increased temperature. In all cases, this decomposition results in the formation of a precipitate. However, we found that the precipitate of complexes 1-Cl/Br exhibited slight solubility in THF, likely owing to the bulky, non-polar tert-butyl substituents on the bipyridine. Taking 1-Cl as a representative complex, we thus collected the precipitation product and analyzed it by UV-vis, 1H NMR, and EPR (in THF, d8-THF, and 2-MeTHF, respectively; Figures S28–S30). We found that its spectra matched that of independently synthesized [Ni(I)(t-Bubpy)Cl]2, suggesting that the primary thermal decomposition product of the Ni(I)–bpy halides is their halide-bridged dimers (see Supporting Information Section S1.8). Furthermore, this thermal decay pathway of the Ni(I)–bpy halides can be monitored by the decrease of their characteristic UV-vis spectra over time (Figure 6, top). Linear fits were obtained when plotting the reciprocal of the absorbance change vs time (Figure 6, middle), consistent with a decay process that is second-order in nickel concentration. Together with the comparison to independent synthesis, this pathway is therefore assigned to Ni(I)–bpy halide dimerization.
Figure 6.
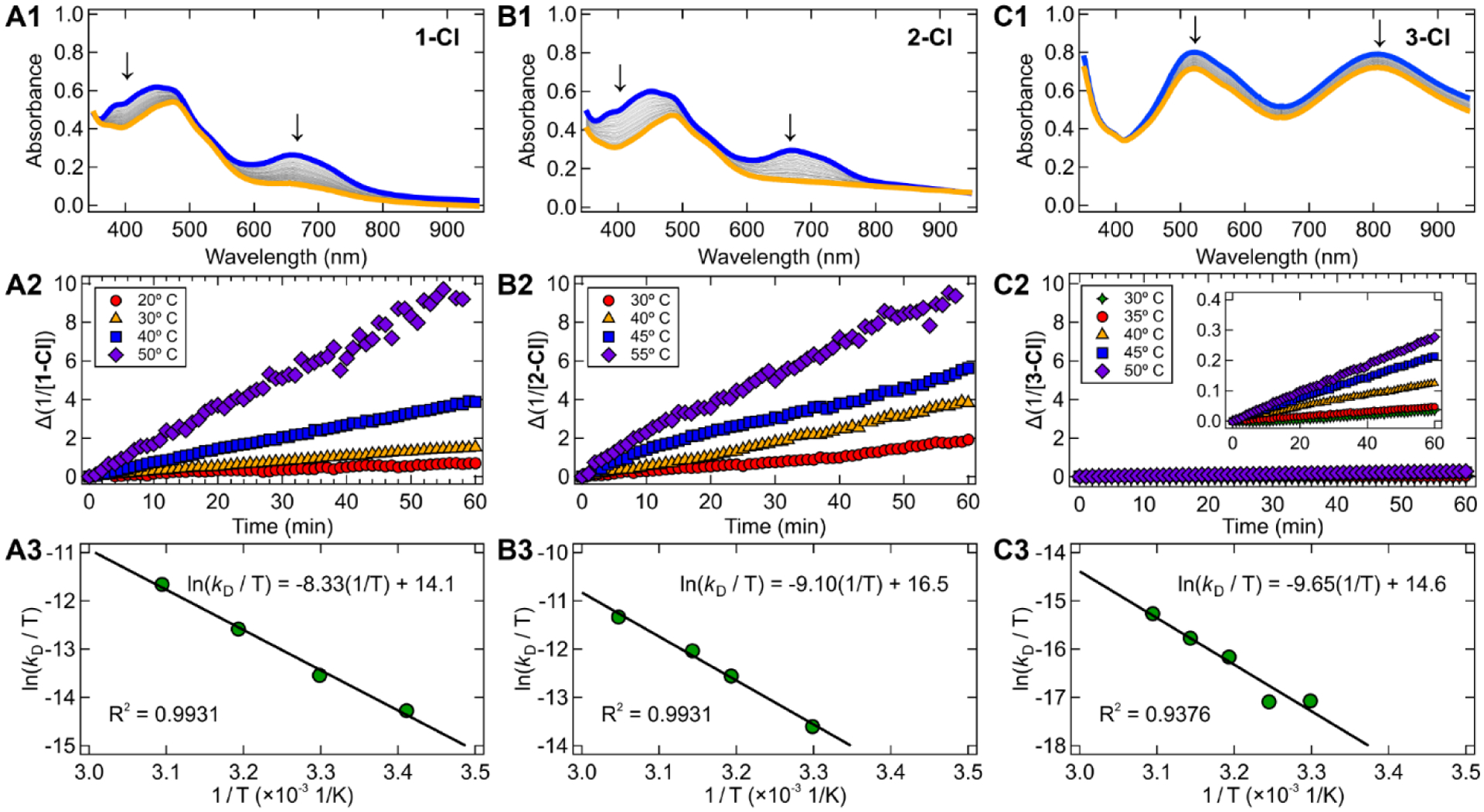
Experimental thermodynamic analysis of the dimerization reaction for 1-Cl, 2-Cl, and 3-Cl. (A1, B1, C1) UV-vis absorption plots in THF showing the starting spectra of the Ni(I)–bpy halide generated by irradiation with 370 nm light (blue line) and the final spectra after thermally induced dimerization (orange line). Note that in the orange spectra that some unreacted parent four-coordinate Ni(II)–bpy aryl halide remains (Figure S1). (A2, B2, C2) Linear plots of 1/([Ni(I)]-[Ni(I)]0) vs time showing the second-order nature of the dimerization; temperatures between 20 °C and 55 °C were chosen. (A3, B3, C3) Eyring plots of ln(kD/T) vs 1/T giving thermodynamics data. Prohibitively slow dimerization made low temperature data collection challenging for 3-Cl, so an intermediate temperature point was added. Analogous data for 1-Cl, 1-Br, and 1-I are given in Figure S10.
The slope of the linear fit yielded second-order dimerization rate constants, kD, which were observed to be temperature-dependent (Figure 6, bottom and Table 3). Eyring analysis of the temperature-dependent rate constants afforded enthalpic and entropic thermodynamic parameters, ΔH‡ and ΔS‡ (Table 3).61–63 Rather large enthalpic barriers are observed for all compounds, ranging from ~11 kcal mol−1 to 19 kcal mol−1. The most significant trend follows ΔH‡ of 3-Cl > 2-Cl > 1-Cl. ΔS‡ values ranged from ~ − 15 cal mol−1 K−1 to −38 cal mol−1 K−1.
Table 3.
Thermodynamic parameters for dimerization of Ni(I)–bpy halide complexes.a
| Compound | kD (x10−5 M−1 s−1)b | ΔH‡ (kcal mol−1) | ΔS‡ (cal mol−1 K−1) | ΔG‡(298 K) (kcal mol−1) |
|---|---|---|---|---|
| 1-Cl | 4.0 | 16.6 ± 1.0 | −19.3 ± 1.1 | 22.3 ± 0.5 |
| 1-Br | 3.6 | 11.1 ± 1.2 | −38.1 ± 4.1 | 22.4 ± 2.4 |
| 1-I | 1.8 | 15.8 ± 1.6 | −25.1 ± 2.5 | 23.3 ± 2.3 |
| 2-Cl | 3.8 | 18.1 ± 1.1 | −14.5 ± 0.9 | 22.4 ± 1.3 |
| 3-Cl | 0.1 | 19.2 ± 2.9 | −18.3 ± 5.8 | 24.6 ± 3.7 |
Dimerization is monitored under nitrogen atmosphere in anhydrous THF and followed by UV-vis spectroscopy. Reported standard errors are propagated from the linear least-squares analysis.
Second-order rate constants for the dimerization reaction (kD) are given at 30 °C (303 K), as no rate constant for 3-Cl was measurable at room temperature.
The fairly large, negative values of ΔS‡ are consistent with an associative (i.e., bimolecular) transition state. ΔG‡(298 K) values yielded small overall differences, however, falling in the range of ~22 kcal mol−1 to 25 kcal mol−1. The largest ΔG‡(298 K) is observed for 3-Cl; at ~25 kcal mol−1, this barrier is consistent with very slow decomposition at room temperature and is in agreement with the overall stability of the related Ni(I)(EtOOCbpy)Cl complex.30 We note, however, that dimer precipitation can offset the chemical equilibrium, acting as a thermodynamic sink and driving the Ni(I) monomer to dimer conversion.
We again turn to substituent-based electronic effects to rationalize the changes in dimerization rate constants and thermodynamics. The ΔH‡ for dimerization is lowest for 1-Cl/Br/I and higher for 2-Cl/3-Cl, a trend similar to that observed for oxidative addition (vide supra, Section 2.2). Electron withdrawing effects weaken the nucleophilicity of the Ni(I) center, lowering the propensity for dimerization. Conversely, electron donation from the ligands enhances the reactivity of the Ni(I) center for both oxidative addition and dimerization. The halide appears to have a lesser effect on the dimerization thermodynamics than the bpy, with 1-Br exhibiting the lowest enthalpic barrier of the three halide variants (Figure S27).
2.5. Dimerization Mechanistic Investigations
DFT calculations also support the decay mechanism of Ni(I)‒bpy halides through dimerization. The experimental second-order decay in Ni(I) allows for a direct 2 Ni(I)(Rbpy)X → [Ni(I)(Rbpy)X]2 pathway (Figure 7A).
Figure 7.
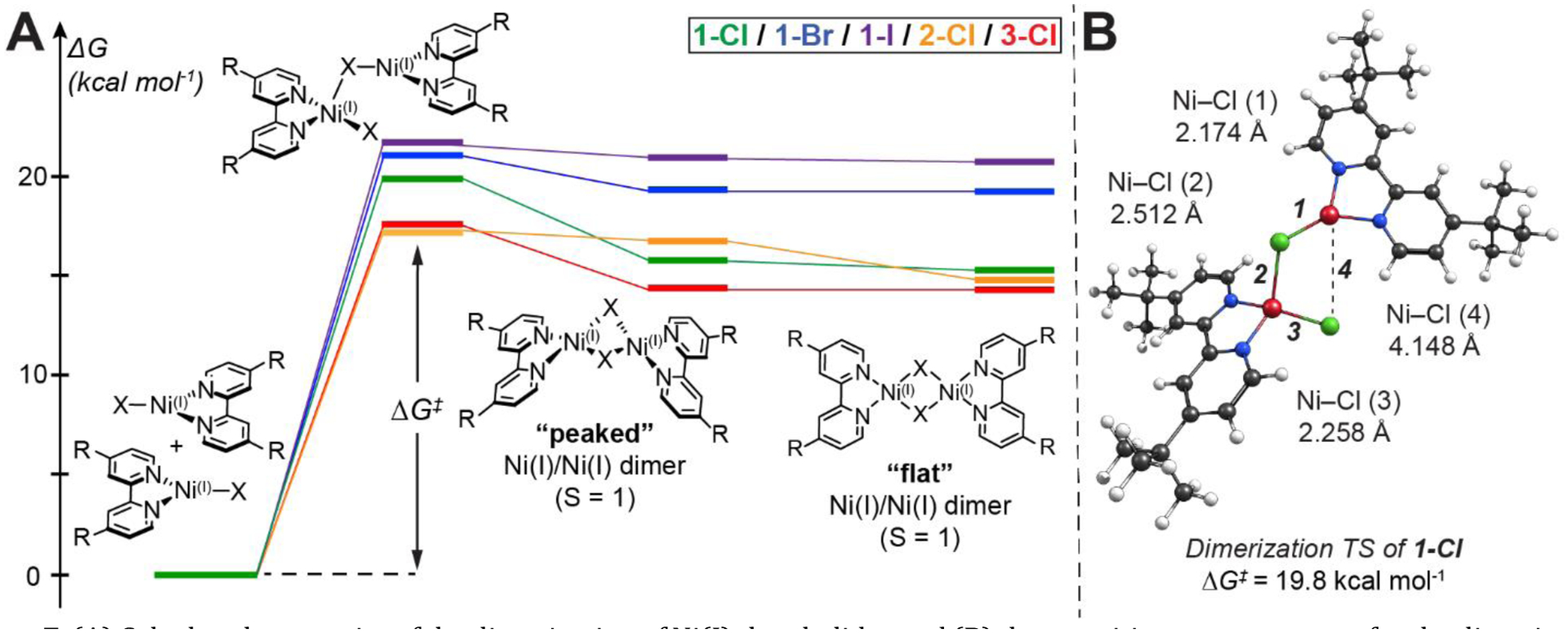
(A) Calculated energetics of the dimerization of Ni(I)‒bpy halides and (B) the transition state structure for the dimerization of 1-Cl. Relative Gibbs free energy values were computed at the B3LYP/def2-TZVP(CPCM)//BP86/def2-TZVP(CPCM) level. Computed free energies are tabulated in Table S16.
We note that the experimental kinetic analysis does not rule out the inclusion of an intermediate step, 2 Ni(I)(Rbpy)X → Ni(0)(Rbpy) + Ni(II)(Rbpy)X2 → [Ni(I)(Rbpy)X]2. In either case, the starting reactants are two Ni(I)(Rbpy)X species, and the ultimate decomposition product is the [Ni(I)(Rbpy)X]2 dimer, making the overall reaction free energy (ΔG) the same for both pathways. We have elected to computationally examine the simpler case which does not invoke a Ni(0) species.
Like the oxidative addition mechanism discussed in Section 2.3, DFT finds that dimer formation is initiated by nucleophilic attack by the doubly-occupied Ni 3d(z2) orbital. Rather than interacting with the C(sp2)–X σ* orbital of an aryl halide, the acceptor orbital for dimerization is an unoccupied halide p-orbital of another three-coordinate Ni(I) complex. This attack results in a transition state with a pseudo-tetrahedral geometry (i.e., a formal Ni(I)–bpy dihalide complex bridged via a halide to a second Ni(I)–bpy (Figure 7B)). The predicted barrier for this transformation is ~17 – 22 kcal mol−1 across the complexes. Two distinct dimerization product geometries were obtained by following the downhill reaction coordinate from the transition state, both featuring two bridging halides but with different orientations (i.e., “peaked” vs “flat” geometry in Figure 7A). At the DFT level, both dimer geometries feature two nickel sites in the Ni(I) oxidation state (Figure S68). Although both dimers are rather high in energy – alone not providing enough driving force for the dimerization – they show distinct similarities to the dimer structures obtained previously in the solid phase.30,32,64 Given the low solubility of the [Ni(I)(Rbpy)X]2 dimers in THF, we argue that precipitation drives the dimerization process in the forward direction and makes the dissociation of the dimer (i.e., the back-reaction) less favorable.
The free energies of activation are comparable to the experiment for 1-Cl/Br/I; however, the DFT transition states are predicted too low in energy for 2-Cl and 3-Cl (orange and red data in Figure 7A). The discrepancy between experimental vs DFT values may again be attributed to the better solubility of 1-Cl/Br in THF, influencing the reversibility of the speciation. The higher multiconfigurational character for Ni(I)‒bpy halides26,27 and likely for the dimeric structures (not captured by DFT) could also contribute to the differences between computed and experimental data. These should be present to a greater degree in complexes with electron-withdrawing substituents on the bpy (i.e., the electron withdrawing trend of 3-Cl > 2-Cl > 1-Cl). Therefore, it may be the case that the starting three-coordinate Ni(I)‒bpy halides and their corresponding dimers are predicted to be too high in energy for 2-Cl and 3-Cl due to insufficient account of ligand non-innocent radical character on the bpy ligand, which may result in smaller effective barriers for dimerization. Nevertheless, computational analysis provides a plausible mechanism for Ni(I)–bpy halide dimerization.
3. Discussion
Photoredox catalysis mediated by electronically excited nickel complexes, coupled to thermal reactivity pathways, can accomplish bond transformations with broad substrate versatility. Ni(I) intermediates are thought to be critical for specific bond activation steps facilitating reaction turnover (Figure 1A). Here we have demonstrated the facile photochemical generation of a library of Ni(I)(Rbpy)X complexes (R = t-Bu, H, MeOOC; X = Cl, Br, I). Through experimental and computational analyses, we have evaluated their competitive reactivity with challenging C(sp2)–Cl bonds and tendency toward dimerization and have developed structure-function relationships between ligand set and reactivity.
UV-vis electronic absorption and EPR spectroscopies have provided an initial characterization of the electronic structures of the photochemically generated Ni(I)–bpy complexes. By UV-vis spectroscopy, all of the Ni(I) species exhibit relatively low energy MLCT transitions (Figure 2A), the energies of which trend with the electronic effects of the bpy substituents. More electron-withdrawing functionalities lead to the shift of the most intense low-energy MLCT bands from ~660 nm to ~805 nm going from 1-Cl to 3-Cl. This shift is attributed to the stabilization of the bpy π* acceptor orbitals. In contrast, the variation of the halides shows a rather small influence on the MLCT energies.
EPR spectra reflect S = 1/2 Ni(I) ground state electronic configurations with giso values of ~2.12 – ~2.19 (Table 1). The giso observed here deviate somewhat from the free electron g value (2.0023) due to the presence of ground state orbital angular momentum. They are larger than those observed for methyl Ni(II)–terpyridine•– (giso = 2.02) and mesityl Ni(II)–phenanthroline•– (giso = 2.01) but smaller than observed for Ni(I)–bisoxazoline bromide (giso = 2.24).34,39,45 These giso values are consistent with greater ligand character for the terpyridine and phenanthroline alkyl complexes and greater Ni(I) character for the bisoxazoline bromide complex. While the giso of Ni(I)-bipyridine halide compounds would be consistent with Ni(I) character intermediate to these complexes, a full analysis of the aggregate giso values will require the additional assignment of the ligand field transitions that spin-orbit couple with the ground state, giving rise to orbital angular momentum.
Current efforts are underway to assign the UV-vis transitions across various isolable Ni(I) complexes to elucidate the amount of ligand vs metal character, as well as ligand field vs MLCT contributions to the observed g values. These may be useful to evaluate the degree of electron delocalization onto the bpy ligand. Indeed, multireference/multiconfigurational calculations predict significant multiconfigurational character (i.e., Ni(I)(bpy)X vs Ni(II)(bpy•–)X).26,27 A full analysis of the Ni(I) g values will require experimental characterization of the Ni-based ligand field transitions. The g values report on the spin-orbit coupling in the ground state of Ni 3d(x2-y2) SOMO parent-age. However, importantly, the electronic effects of the bpy ligand and its substituents extend beyond the Ni 3d(x2-y2) SOMO and allow for energetic tuning of the lower-energy, occupied Ni 3d orbitals that are actually involved in the elementary steps of oxidative addition and dimerization (as further discussed below).
When examining the Ni(I)–based reactivity toward C(sp2)–Cl oxidative addition, we find that complexes 1-Cl/Br/I, which feature the most electron donating tert-butyl substituent on bpy, exhibit the largest rate constants for the oxidative addition of 2-chloro-toluene (kOA ~7.0 ×10−2 M−1 s−1, Figure 3 and Table 2). Changing this bpy substituent to hydrogen atoms (2-Cl) reduces the rate constant by two-fold, while changing to the electron withdrawing methyl ester (3-Cl) completely eliminates oxidative addition reactivity. Furthermore, 1-Cl is competent towards the activation of a variety of aryl chlorides, with rate constants spanning two orders of magnitude (from kOA ~ 8.0 ×10−2 M−1 s−1 to kOA ~ 6.4 M−1 s−1). Hammett relationships indicate that the mechanism of the C(sp2)–Cl bond activation proceeds by a SNAr-type pathway, marking a discrete change in mechanism for the oxidative addition of aryl chlorides vs. aryl bromides or aryl iodides. These reactivity trends have been rationalized herein through analysis of the associated Ni-based 3d(z2) orbital involved in the nucleophilic two-electron transfer. In particular, we find linear trends between the 3d(z2) RAMO and the Hammett parameter of the bpy ligand (Figure S45), with electron donation and electron withdrawing substituents destabilizing or stabilizing the RAMO, respectively. Interestingly, the energetic (de)stabilization is observed to occur for the entire 3d orbital manifold (Figure 8B). This observation is consistent with bpy-induced modifications of the Zeff of the metal center; the calculated Ni(I) 1s and 2p(x,y,z) orbital energies also trend linearly with the observed rate constants (Figure S46). Increased Zeff on the metal stabilizes the 3d orbital manifold and perturbs the overall reactivity of the complex (Figure 8A) through the kinetic barrier (ΔG‡).
Figure 8.
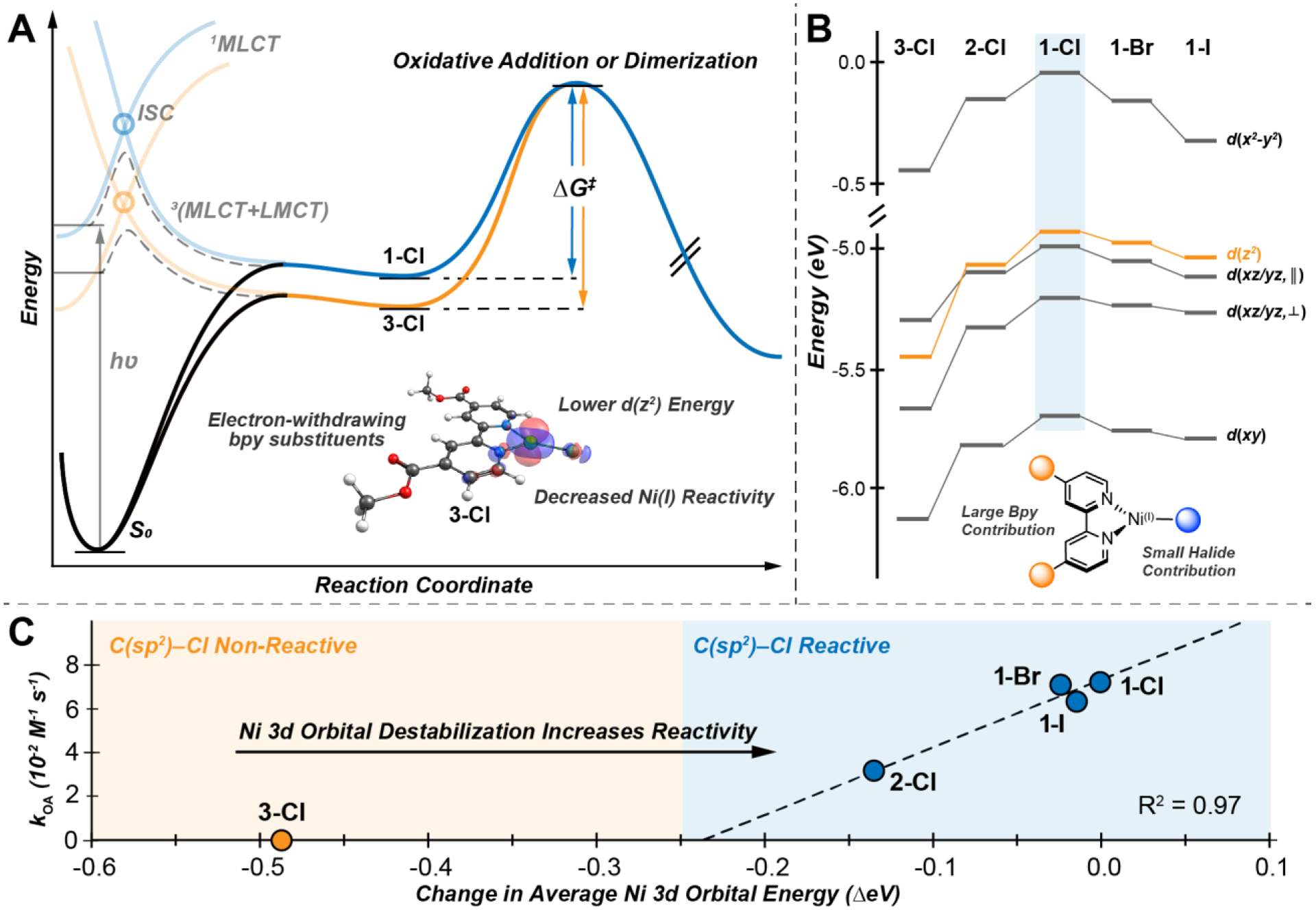
Reaction coordinate and state energy diagrams for the Ni(I)–bpy halide complexes. (A) Blue (1-Cl) and orange (3-Cl) lines depict the photoexcitation mechanism (the MLCT+LMCT ‘one-photon, two-electron’ excitation previously discussed in ref. 27) for the formation of Ni(I) complexes. Subsequent reaction coordinate diagrams for the Ni(I)-based oxidative addition (if aryl halide is present) or dimerization pathways are given for 1-Cl (blue) and 3-Cl (orange), highlighting the difference in kinetic barriers, ΔG‡, for the two complexes. (B) Molecular orbital diagram for the Ni(I) species, demonstrating the (de)stabilization of the β 3d orbital manifolds due to changes in Zeff; the chemically active, nucleophilic 3d(z2) orbital is highlighted in orange. (C) Plot of the oxidative addition rate constants, kOA, vs the average change in Ni 3d orbital energy relative to 1-Cl.
The bpy and halide substituents therefore present themselves as synthetic handles by which the reactivity of Ni(I)–bpy halide complexes can be adjusted and evaluated. For example, compound 3-Cl is selective toward the oxidative addition of C(sp2)–Br/I bonds, while 1-Cl demonstrates robust reactivity toward even the challenging C(sp2)–Cl functionality. The relative reactivity of 1-Cl/Br/I, 2-Cl, and 3-Cl allows for an estimate of the minimum energy of the Ni 3d(z2) orbital required to activate the challenging C(sp2)–Cl bond (Figure 8B). Furthermore, as the Ni 3d orbitals are stabilized in energy, the rate constant decreases linearly (R2 = 0.97, Figure 8C). Extrapolating this correlation to the x-axis provides an approximation of the minimum orbital energy required for oxidative addition reactivity. Notably, 3-Cl, which is not reactive toward C(sp2)–Cl bonds, falls well off the correlation for the other complexes. Thus, from this picture, in order to activate reactivity, one would need to move the 3-Cl point along the x-axis until it intersects with the slope for the other compounds (i.e., increase its 3d orbital energies). Correlations of this type over other Ni(I) species reacting with a variety of C–X bonds will be useful for generating robust reactivity predictions. It further indicates that tuning the energy of the Ni 3d orbitals is a key aspect for reactivity. X-ray absorption spectroscopies will prove useful to quantify ligand contributions to Zeff and the 3d(z2) RAMO.65 Outside of ligand contributions, which thus far appear to be non-specific and affect all 3d orbitals, it may be possible to utilize other contributions such as solvent or additive effects as another handle by which to specifically target the axially accessible 3d(z2) orbital and modulate the energy and reactivity (and thereby the selectivity) of these complexes toward oxidative addition. Current work is underway to further define the reactivity capabilities of these Ni(I)–bpy halide complexes.
Not only is the oxidative addition reactivity of these complexes tunable via the Ni 3d(z2) orbital, but their general solution-phase stability is as well. Analysis of the temperature-dependent kinetics indicates that 3-Cl has a markedly decreased tendency toward dimerization, with a measured room temperature free energy of activation, ΔG‡, of ~25 kcal mol−1. Conversely, 1-Cl readily dimerizes at room temperature suggesting that this decomposition pathway may be of concern during catalysis. These dimerization pathways also feature a nucleophilic attack by the Ni 3d(z2) orbital, but in this case, the acceptor is the unoccupied p orbital of the halide on a second complex. Thus, the competitive oxidative addition and dimerization reactivity pathways are both correlated with the Ni 3d(z2) orbital energy, suggesting it as a sensitive target for researchers to tune to discourage Ni(I) reactant decomposition and increase the catalytic turnover number. One can also envision similar concepts from a multiconfigurational bonding perspective. That is, increasing a ligand non-innocent Ni(II)(bpy•–)X configuration over the metal-centered Ni(I)(bpy)X configuration will deactivate the complex toward oxidative addition and dimerization. Future studies examining additional factors (e.g., steric effects, solvent coordination, etc.) that could suppress Ni(I) dimerization without affecting its performance toward oxidative addition would be of interest.
4. Conclusions
In summary, we report the straightforward photochemical generation of a series of Ni(I)–bpy halide complexes, which has allowed for detailed studies of their relative reactivity toward competitive pathways of oxidative addition and dimerization/oligomerization. Interestingly, many of the Ni(I)–bpy halide complexes studied herein are active toward oxidative addition of high energy C(sp2)–Cl bonds. Through time-resolved UV-vis kinetic analysis, we have determined rate constants for these oxidative addition reactions. A Hammett analysis on substituted aryl chlorides elucidated the specific mechanism for the C(sp2)–Cl bond activation step as proceeding through an SNAr-type pathway, wherein the Ni(I) complex acts as a nucleophile. Relatedly, computational studies have also supported this mechanism, well reproducing the experimental ρ value, and reveal a nucleophilic two-electron transfer from the 3d(z2) RAMO into the Caryl–X σ* orbital.
While the Ni(I)-mediated reactivity is not strongly dependent on the identity of the halide ligand, we find that it is strongly influenced by the identity of the bpy substituents. Electron withdrawing substituents increase the energetic barrier for oxidative addition and can even completely abolish reactivity with C(sp2)–Cl bonds. Electron donating substituents, however, promote reactivity. The role of the bpy ligand in reactivity has been traced directly to substituent-induced changes in the Ni(I)-based Zeff. For example, the most reactive complex, 1-Cl, has the Zeff decreased by electron donation from the tert-butyl substituents on the bpy. The enhanced electron density on the metal more effectively screens the nuclear charge through electron-electron Coulomb repulsion; this decrease in Zeff destabilizes the entire 3d-orbital manifold. Thus, the energy of the nucleophilic 3d(z2) orbital is modulated through changes in Zeff, and greater energetic destabilization results in greater reactivity due to decreases in electron binding affinities. While not evaluated here, the low coordination number and planarity of the bpy ligand also likely reduce steric contributions allowing for an ideal orbital overlap between the 3d(z2) and Caryl–X σ* orbitals to be achieved.
We have additionally utilized temperature-dependent reaction kinetics to determine the thermodynamics associated with Ni(I) complex dimerization. We again find that stabilization of the complexes is made possible via ligand effects, reporting room temperature dimerization barriers that range from moderate to untenable. Dimerization reactivity considerations parallel those of oxidative addition. Altogether, geometric changes to the ligand scaffold about the Ni(I) center in these and other catalytically relevant complexes have important implications for their relative electronic structures and reactivities. Modulation of the 3d(z2) orbital (by ligand substitution or through yet-to-be explored solvent coordination) represents a tunable target by which the reactivity of Ni(I)–bpy halide complexes can be altered, potentially unveiling new ways to drive Ni-mediated photocatalytic cycles and a direct route toward stimulating reactivity with even stronger C–X bonds.
Supplementary Material
SYNOPSIS:
We report the facile photochemical generation of a library of Ni(I)–bpy halide complexes and benchmark their relative kinetics toward competitive oxidative addition and off-cycle dimerization pathways. With a particular emphasis on rationalizing previously uncharacterized ligand-controlled reactivity toward high energy and challenging C(sp2)–Cl bonds, we developed structure-function relationships between ligand set and reactivity. The energies of the Ni-based orbitals provide a strong influence on reactivity, ultimately determining whether oxidative addition or dimerization occur.
ACKNOWLEDGMENT
D.A.C. is a National Science Foundation Graduate Research Fellow (DGE-1745301) and is supported by a National Academies of Science, Engineering, and Medicine Ford Foundation Pre-doctoral Fellowship. This project has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement No. 883987 (D.B.). N.P.K. acknowledges support from the Hertz Fellowship and from the National Science Foundation Graduate Research Fellowship under Grant No. DGE1745301. The Caltech EPR facility acknowledges support from the Beckman Institute and the Dow Next Generation Educator Fund. Support has been provided by the National Institutes of Health (National Institute of General Medical Sciences, R35-GM142595). The computations presented here were conducted in the Resnick High Performance Computing Center, a facility supported by Resnick Sustainability Institute at the California Institute of Technology.
The authors declare no competing financial interest.
Footnotes
Supporting Information.
The Supporting Information is available free of charge on the ACS Publications website at DOI:
Experimental and computational methods, UV–vis/photochemical data, NMR and EPR spectra, calculated spectra/properties, and additional comments.
Contributor Information
David A. Cagan, Division of Chemistry and Chemical Engineering, Arthur Amos Noyes Laboratory of Chemical Physics, California Institute of Technology, Pasadena, California 91125, United States.
Daniel Bím, Division of Chemistry and Chemical Engineering, Arthur Amos Noyes Laboratory of Chemical Physics, California Institute of Technology, Pasadena, California 91125, United States.
Brendon J. McNicholas, Division of Chemistry and Chemical Engineering, Arthur Amos Noyes Laboratory of Chemical Physics, California Institute of Technology, Pasadena, California 91125, United States
Nathanael P. Kazmierczak, Division of Chemistry and Chemical Engineering, Arthur Amos Noyes Laboratory of Chemical Physics, California Institute of Technology, Pasadena, California 91125, United States
Paul H. Oyala, Division of Chemistry and Chemical Engineering, Arthur Amos Noyes Laboratory of Chemical Physics, California Institute of Technology, Pasadena, California 91125, United States
Ryan G. Hadt, Division of Chemistry and Chemical Engineering, Arthur Amos Noyes Laboratory of Chemical Physics, California Institute of Technology, Pasadena, California 91125, United States
REFERENCES
- (1).Nicewicz DA; MacMillan DWC Merging Photoredox Catalysis with Organocatalysis: The Direct Asymmetric Alkylation of Aldehydes. Science 2008, 322 (5898), 77–80. 10.1126/science.1161976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Shaw MH; Twilton J; MacMillan DWC Photoredox Catalysis in Organic Chemistry. J. Org. Chem 2016, 81 (16), 6898–6926. 10.1021/acs.joc.6b01449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Twilton J; Le C; Zhang P; Shaw MH; Evans RW; MacMillan DWC. The Merger of Transition Metal and Photocatalysis. Nat. Rev. Chem 2017, 1 (7), 0052. 10.1038/s41570-017-0052. [DOI] [Google Scholar]
- (4).Cavalcanti LN; Molander GA Photoredox Catalysis in Nickel-Catalyzed Cross-Coupling. Top. Curr. Chem 2016, 374 (4), 39. 10.1007/s41061-016-0037-z. [DOI] [PubMed] [Google Scholar]
- (5).Larsen CB; Wenger OS Photoredox Catalysis with Metal Complexes Made from Earth-Abundant Elements. Chem. – Eur. J 2018, 24 (9), 2039–2058. 10.1002/chem.201703602. [DOI] [PubMed] [Google Scholar]
- (6).Welin ER; Le C; Arias-Rotondo DM; McCusker JK; MacMillan DWC Photosensitized, Energy Transfer-Mediated Organometallic Catalysis through Electronically Excited Nickel(II). Science 2017, 355 (6323), 380–385. 10.1126/science.aal2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Chan AY; Perry IB; Bissonnette NB; Buksh BF; Edwards GA; Frye LI; Garry OL; Lavagnino MN; Li BX; Liang Y; Mao E; Millet A; Oakley JV; Reed NL; Sakai HA; Seath CP; MacMillan DWC Metallaphotoredox: The Merger of Photoredox and Transition Metal Catalysis. Chem. Rev 2021. 10.1021/acs.chemrev.1c00383. [DOI] [PubMed] [Google Scholar]
- (8).Dongbang S; Doyle AG Ni/Photoredox-Catalyzed C(Sp3)–C(Sp3) Coupling between Aziridines and Acetals as Alcohol-Derived Alkyl Radical Precursors. J. Am. Chem. Soc 2022, 144 (43), 20067–20077. 10.1021/jacs.2c09294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Millet A; Cesana PT; Sedillo K; Bird MJ; Schlau-Cohen GS; Doyle AG; MacMillan DWC; Scholes GD Bioinspired Supercharging of Photoredox Catalysis for Applications in Energy and Chemical Manufacturing. Acc. Chem. Res 2022, 55 (10), 1423–1434. 10.1021/acs.accounts.2c00083. [DOI] [PubMed] [Google Scholar]
- (10).Kariofillis SK; Doyle AG Synthetic and Mechanistic Implications of Chlorine Photoelimination in Nickel/Photoredox C(sp3)–H Cross-Coupling. Acc. Chem. Res 2021, 54 (4), 988–1000. 10.1021/acs.accounts.0c00694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Zhu C; Yue H; Chu L; Rueping M Recent Advances in Photoredox and Nickel Dual-Catalyzed Cascade Reactions: Pushing the Boundaries of Complexity. Chem. Sci 2020, 11 (16), 4051–4064. 10.1039/D0SC00712A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Feng Y; Luo H; Zheng W; Matsunaga S; Lin L Light-Promoted Nickel-Catalyzed Aromatic Halogen Exchange. ACS Catal. 2022, 12 (18), 11089–11096. 10.1021/acscatal.2c03354. [DOI] [Google Scholar]
- (13).Speckmeier E; Maier TC ART─An Amino Radical Transfer Strategy for C(sp2)–C(sp3) Coupling Reactions, Enabled by Dual Photo/Nickel Catalysis. J. Am. Chem. Soc 2022, 144 (22), 9997–1005. 10.1021/jacs.2c03220. [DOI] [PubMed] [Google Scholar]
- (14).Shin J; Lee J; Suh J-M; Park K Ligand-Field Transition-Induced C–S Bond Formation from Nickelacycles. Chem. Sci 2021, 12 (48), 15908–15915. 10.1039/D1SC05113J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Zhang R; Li G; Wismer M; Vachal P; Colletti SL; Shi Z-C Profiling and Application of Photoredox C(sp3)–C(sp2) Cross-Coupling in Medicinal Chemistry. ACS Med. Chem. Lett 2018, 9 (7), 773–777. 10.1021/acsmedchemlett.8b00183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Lacker CR; DeLano TJ; Chen EP; Kong J; Belyk KM; Piou T; Reisman SE Enantioselective Synthesis of N-Benzylic Heterocycles by Ni/Photoredox Dual Catalysis. J. Am. Chem. Soc 2022, 144 (44), 20190–20195. 10.1021/jacs.2c07917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Cheung KPS; Sarkar S; Gevorgyan V Visible Light-Induced Transition Metal Catalysis. Chem. Rev 2022, 122 (2), 1543–1625. 10.1021/acs.chemrev.1c00403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Li R; Yang C-X; Niu B-H; Li L-J; Ma J-M; Li Z-L; Jiang H; Cheng W-M Visible Light-Induced Ni-Catalyzed C–Heteroatom Cross-Coupling of Aryl Halides via LMCT with DBU to Access a Ni(I)/Ni(III) Cycle. Org. Chem. Front 2022, 9 (14), 3847–3853. 10.1039/D2QO00607C. [DOI] [Google Scholar]
- (19).Gutierrez O; Tellis JC; Primer DN; Molander GA; Kozlowski MC Nickel-Catalyzed Cross-Coupling of Photoredox-Generated Radicals: Uncovering a General Manifold for Stereoconvergence in Nickel-Catalyzed Cross-Couplings. J. Am. Chem. Soc 2015, 137 (15), 4896–4899. 10.1021/ja513079r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Maity B; Scott TR; Stroscio GD; Gagliardi L; Cavallo L The Role of Excited States of LNiII/III(Aryl)(Halide) Complexes in Ni–Halide Bond Homolysis in the Arylation of Csp3–H Bonds. ACS Catal. 2022, 12 (21), 13215–13224. 10.1021/acscatal.2c04284. [DOI] [Google Scholar]
- (21).Tang T; Jones E; Wild T; Hazra A; Minteer SD; Sigman MS Investigating Oxidative Addition Mechanisms of Allylic Electrophiles with Low-Valent Ni/Co Catalysts Using Electroanalytical and Data Science Techniques. J. Am. Chem. Soc 2022, 144 (43), 20056–20066. 10.1021/jacs.2c09120. [DOI] [PubMed] [Google Scholar]
- (22).Almansa A; Jardel D; Massip S; Tassaing T; Schatz C; Domergue J; Molton F; Duboc C; Vincent J-M Dual Photoredox Ni/Benzophenone Catalysis: A Study of the NiII Precatalyst Photo-reduction Step. J. Org. Chem 2022, 87 (16), 11172–11184. 10.1021/acs.joc.2c01467. [DOI] [PubMed] [Google Scholar]
- (23).Ben-Tal Y; Lloyd-Jones GC Kinetics of a Ni/Ir-Photo-catalyzed Coupling of ArBr with RBr: Intermediacy of ArNiII(L)Br and Rate/Selectivity Factors. J. Am. Chem. Soc 2022, 144 (33), 15372–15382. 10.1021/jacs.2c06831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Shields BJ; Kudisch B; Scholes GD; Doyle AG Long-Lived Charge-Transfer States of Nickel(II) Aryl Halide Complexes Facilitate Bimolecular Photoinduced Electron Transfer. J. Am. Chem. Soc 2018, 140 (8), 3035–3039. 10.1021/jacs.7b13281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Ting SI; Garakyaraghi S; Taliaferro CM; Shields BJ; Scholes GD; Castellano FN; Doyle AG 3d-d Excited States of Ni(II) Complexes Relevant to Photoredox Catalysis: Spectroscopic Identification and Mechanistic Implications. J. Am. Chem. Soc 2020, 142 (12), 5800–5810. 10.1021/jacs.0c00781. [DOI] [PubMed] [Google Scholar]
- (26).Cagan DA; Stroscio GD; Cusumano AQ; Hadt RG Multireference Description of Nickel–Aryl Homolytic Bond Dissociation Processes in Photoredox Catalysis. J. Phys. Chem. A 2020, 124 (48), 9915–9922. 10.1021/acs.jpca.0c08646. [DOI] [PubMed] [Google Scholar]
- (27).Cagan DA; Bím D; Silva B; Kazmierczak NP; McNicholas BJ; Hadt RG Elucidating the Mechanism of Excited-State Bond Homolysis in Nickel–Bipyridine Photoredox Catalysts. J. Am. Chem. Soc 2022, 144 (14), 6516–6531. 10.1021/jacs.2c01356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Yuan M; Song Z; Badir SO; Molander GA; Gutierrez O On the Nature of C(sp3)–C(sp2) Bond Formation in Nickel-Catalyzed Tertiary Radical Cross-Couplings: A Case Study of Ni/Photo-redox Catalytic Cross-Coupling of Alkyl Radicals and Aryl Halides. J. Am. Chem. Soc 2020, 142 (15), 7225–7234. 10.1021/jacs.0c02355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Na H; Mirica LM Deciphering the Mechanism of the Ni-Photocatalyzed C‒O Cross-Coupling Reaction Using a Tridentate Pyridinophane Ligand. Nat. Commun 2022, 13 (1), 1313. 10.1038/s41467-022-28948-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Ting SI; Williams WL; Doyle AG Oxidative Addition of Aryl Halides to a Ni(I)-Bipyridine Complex. J. Am. Chem. Soc 2022, 144 (12), 5575–5582. 10.1021/jacs.2c00462. [DOI] [PubMed] [Google Scholar]
- (31).Till NA; Oh S; MacMillan DWC; Bird MJ The Application of Pulse Radiolysis to the Study of Ni(I) Intermediates in Ni-Catalyzed Cross-Coupling Reactions. J. Am. Chem. Soc 2021, 143 (25), 9332–9337. 10.1021/jacs.1c04652. [DOI] [PubMed] [Google Scholar]
- (32).Mohadjer Beromi M; Brudvig GW; Hazari N; Lant HMC; Mercado BQ Synthesis and Reactivity of Paramagnetic Nickel Polypyridyl Complexes Relevant to C(sp2)–C(sp3)Coupling Reactions. Angew. Chem. Int. Ed 2019, 58 (18), 6094–6098. 10.1002/anie.201901866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Kawamata Y; Vantourout JC; Hickey DP; Bai P; Chen L; Hou Q; Qiao W; Barman K; Edwards MA; Garrido-Castro AF; deGruyter JN; Nakamura H; Knouse K; Qin C; Clay KJ; Bao D; Li C; Starr JT; Garcia-Irizarry C; Sach N; White HS; Neurock M; Minteer SD; Baran PS Electrochemically Driven, Ni-Catalyzed Aryl Amination: Scope, Mechanism, and Applications. J. Am. Chem. Soc 2019, 141 (15), 6392–6402. 10.1021/jacs.9b01886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Jones GD; Martin JL; McFarland C; Allen OR; Hall RE; Haley AD; Brandon RJ; Konovalova T; Desrochers PJ; Pulay P; Vicic DA Ligand Redox Effects in the Synthesis, Electronic Structure, and Reactivity of an Alkyl−Alkyl Cross-Coupling Catalyst. J. Am. Chem. Soc 2006, 128 (40), 13175–13183. 10.1021/ja063334i. [DOI] [PubMed] [Google Scholar]
- (35).Ciszewski JT; Mikhaylov DY; Holin KV; Kadirov MK; Budnikova YH; Sinyashin O; Vicic DA Redox Trends in Terpyridine Nickel Complexes. Inorg. Chem 2011, 50 (17), 8630–8635. 10.1021/ic201184x. [DOI] [PubMed] [Google Scholar]
- (36).Vogt N; Sandleben A; Kletsch L; Schäfer S; Chin MT; Vicic DA; Hörner G; Klein A Role of the X Coligands in Cyclometalated [Ni(Phbpy)X] Complexes (HPhbpy = 6-Phenyl-2,2′-Bipyridine). Organometallics 2021, 40 (11), 1776–1785. 10.1021/acs.organomet.1c00237. [DOI] [Google Scholar]
- (37).Kalvet I; Guo Q; Tizzard GJ; Schoenebeck F When Weaker Can Be Tougher: The Role of Oxidation State (I) in P- vs N-Ligand-Derived Ni-Catalyzed Trifluoromethylthiolation of Aryl Halides. ACS Catal. 2017, 7 (3), 2126–2132. 10.1021/acscatal.6b03344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Diccianni JB; Diao T Mechanisms of Nickel-Catalyzed Cross-Coupling Reactions. Trends Chem. 2019, 1 (9), 830–844. 10.1016/j.trechm.2019.08.004. [DOI] [Google Scholar]
- (39).Lin Q; Diao T Mechanism of Ni-Catalyzed Reductive 1,2-Dicarbofunctionalization of Alkenes. J. Am. Chem. Soc 2019, 141 (44), 17937–17948. 10.1021/jacs.9b10026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Sun R; Qin Y; Ruccolo S; Schnedermann C; Costentin C; Nocera DG Elucidation of a Redox-Mediated Reaction Cycle for Nickel-Catalyzed Cross Coupling. J. Am. Chem. Soc 2019, 141 (1), 89–93. 10.1021/jacs.8b11262. [DOI] [PubMed] [Google Scholar]
- (41).Lin C-Y; Power PP Complexes of Ni(I): A “Rare” Oxidation State of Growing Importance. Chem. Soc. Rev 2017, 46 (17), 5347–5399. 10.1039/C7CS00216E. [DOI] [PubMed] [Google Scholar]
- (42).Yang L; Lu H-H; Lai C-H; Li G; Zhang W; Cao R; Liu F; Wang C; Xiao J; Xue D Light-Promoted Nickel Catalysis: Etherification of Aryl Electrophiles with Alcohols Catalyzed by a NiII-Aryl Complex. Angew. Chem. Int. Ed 2020, 59 (31), 12714–12719. 10.1002/anie.202003359. [DOI] [PubMed] [Google Scholar]
- (43).Song G; Li Q; Nong D-Z; Song J; Li G; Wang C; Xiao J; Xue D Ni-Catalyzed Photochemical C-N Coupling of Amides with (Hetero)Aryl Chlorides. Chem. – Eur. J 2023, Accepted Article, e202300458. 10.1002/chem.202300458. [DOI] [PubMed] [Google Scholar]
- (44).Arendt KM; Doyle AG Dialkyl Ether Formation by Nickel-Catalyzed Cross-Coupling of Acetals and Aryl Iodides. Angew. Chem. Int. Ed 2015, 54 (34), 9876–9880. 10.1002/anie.201503936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Ju L; Lin Q; LiBretto NJ; Wagner CL; Hu CT; Miller JT; Diao T Reactivity of (Bi-Oxazoline)Organonickel Complexes and Revision of a Catalytic Mechanism. J. Am. Chem. Soc 2021, 143 (36), 14458–14463. 10.1021/jacs.1c07139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Hansch Corwin.; Leo A; Taft RW. A Survey of Hammett Substituent Constants and Resonance and Field Parameters. Chem. Rev 1991, 91 (2), 165–195. 10.1021/cr00002a004. [DOI] [Google Scholar]
- (47).Portnoy M; Milstein D Mechanism of Aryl Chloride Oxidative Addition to Chelated Palladium(0) Complexes. Organometallics 1993, 12 (5), 1665–1673. 10.1021/om00029a026. [DOI] [Google Scholar]
- (48).Tsou TT; Kochi JK Mechanism of Oxidative Addition. Reaction of Nickel(0) Complexes with Aromatic Halides. J. Am. Chem. Soc 1979, 101 (21), 6319–6332. 10.1021/ja00515a028. [DOI] [Google Scholar]
- (49).Lin Q; Fu Y; Liu P; Diao T Monovalent Nickel-Mediated Radical Formation: A Concerted Halogen-Atom Dissociation Pathway Determined by Electroanalytical Studies. J. Am. Chem. Soc 2021, 143 (35), 14196–14206. 10.1021/jacs.1c05255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Amatore C; Pfluger F Mechanism of Oxidative Addition of Palladium(0) with Aromatic Iodides in Toluene, Monitored at Ultramicroelectrodes. Organometallics 1990, 9 (8), 2276–2282. 10.1021/om00158a026. [DOI] [Google Scholar]
- (51).Biscoe MR; Fors BP; Buchwald SL A New Class of Easily Activated Palladium Precatalysts for Facile C−N Cross-Coupling Reactions and the Low Temperature Oxidative Addition of Aryl Chlorides. J. Am. Chem. Soc 2008, 130 (21), 6686–6687. 10.1021/ja801137k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Weigend F; Ahlrichs R Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys 2005, 7 (18), 3297–3305. 10.1039/B508541A. [DOI] [PubMed] [Google Scholar]
- (53).Becke AD Density-functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys 1993, 98 (7), 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]
- (54).Lee C; Yang W; Parr RG Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37 (2), 785–789. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- (55).Cramer JC G. Truhlar D, Density Functional Theory for Transition Metals and Transition Metal Chemistry. Phys. Chem. Chem. Phys 2009, 11 (46), 10757–10816. 10.1039/B907148B. [DOI] [PubMed] [Google Scholar]
- (56).Mardirossian N; Head-Gordon M Thirty Years of Density Functional Theory in Computational Chemistry: An Overview and Extensive Assessment of 200 Density Functionals. Mol. Phys 2017, 115 (19), 2315–2372. 10.1080/00268976.2017.1333644. [DOI] [Google Scholar]
- (57).Tang T; Hazra A; Min DS; Williams WL; Jones E; Doyle AG; Sigman MS Interrogating the Mechanistic Features of Ni(I)-Mediated Aryl Iodide Oxidative Addition Using Electroanalytical and Statistical Modeling Techniques. J. Am. Chem. Soc 2023, 145 (15), 8689–8699. 10.1021/jacs.3c01726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Knizia G; Klein JEMN Electron Flow in Reaction Mechanisms—Revealed from First Principles. Angew. Chem. Int. Ed 2015, 54 (18), 5518–5522. 10.1002/anie.201410637. [DOI] [PubMed] [Google Scholar]
- (59).Knizia G Intrinsic Atomic Orbitals: An Unbiased Bridge between Quantum Theory and Chemical Concepts. J. Chem. Theory Comput 2013, 9 (11), 4834–4843. 10.1021/ct400687b. [DOI] [PubMed] [Google Scholar]
- (60).Vermeeren P; Sun X; Bickelhaupt FM Arylic C–X Bond Activation by Palladium Catalysts: Activation Strain Analyses of Reactivity Trends. Sci. Rep 2018, 8 (1), 10729. 10.1038/s41598-018-28998-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Eyring H The Activated Complex in Chemical Reactions. J. Chem. Phys 1935, 3 (2), 107–115. 10.1063/1.1749604. [DOI] [Google Scholar]
- (62).Lente G; Fábián I; J. Poë A. A Common Misconception about the Eyring Equation. New J. Chem 2005, 29 (6), 759–760. 10.1039/B501687H. [DOI] [Google Scholar]
- (63).Carvalho-Silva VH; Coutinho ND; Aquilanti V Temperature Dependence of Rate Processes Beyond Arrhenius and Eyring: Activation and Transitivity. Front. Chem 2019, 7 (380), 1–11. 10.3389/fchem.2019.00380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Day CS; Rentería-Gómez Á; Ton SJ; Gogoi AR; Gutierrez O; Martin R Elucidating Electron-Transfer Events in Polypyridine Nickel Complexes for Reductive Coupling Reactions. Nat. Catal 2023, 6 (3), 244–253. 10.1038/s41929-023-00925-4. [DOI] [Google Scholar]
- (65).Solomon EI; Szilagyi RK; DeBeer George S; Basumallick L Electronic Structures of Metal Sites in Proteins and Models: Contributions to Function in Blue Copper Proteins. Chem. Rev 2004, 104 (2), 419–458. 10.1021/cr0206317. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.