

Abstract
Although drugs targeting the orthosteric binding site of cannabinoid receptors (CBRs) have several therapeutic effects on human physiological and pathological conditions, they can also cause serious adverse effects. Only a few orthosteric ligands have successfully passed clinical trials. Recently, allosteric modulation has become a novel option for drug discovery, with fewer adverse effects and the potential to avoid drug overdose. In this review, we highlight novel findings related to the drug discovery of allosteric modulators (AMs) targeting CBRs. We summarize newly synthesized AMs and the reported/predicted allosteric binding sites. We also discuss the structural determinants of the AMs binding as well as the molecular mechanism of CBR allostery.
Keywords: cannabinoid receptors, allosteric modulation, allosteric binding site, drug discovery, human endocannabinoid systems
Teaser:
Unlocking the secrets of allostery: we review binding sites and molecular mechanisms for the design of novel allosteric modulators of cannabinoid receptors.
Introduction
The endocannabinoid system (ECS) is a lipid signaling system comprising CBRs, enzymes, and endocannabinoids expressed in the major domain of the mesocorticolimbic system.1,2 It not only has a vital role in multiple physiological processes, but also acts as a valuable therapeutic target for treating neurodegenerative diseases, autoimmune diseases, inflammatory diseases, psychosis, mood disorders, and pain, and for modulating smooth muscle motility as well as gut integrity in the gastrointestinal tract.3–7 Besides these positive implications, the ECS is also involved in reward processing and drug addiction.8 Therefore, ECS drugs are required to balance the therapeutic and adverse events of the ESC.
In the ECS, the seven-transmembrane (TM) cannabinoid receptor type 1 (CB1) and type 2 (CB2) are the most abundant receptors, classified as canonical CBRs. They also belong to the class A Rhodopsin-like superfamily G-protein-coupled receptors (GPCRs) and show ~44% amino acid similarity.9 The TM regions of CBRs exhibit 68% homology.10,11 CB1 is widely distributed throughout the central nervous system (CNS), whereas CB2 is highly expressed in peripheral tissues and immune cells, with limited expression in the CNS.11–14 Activation of CBRs is triggered by endocannabinoids or synthetic molecules binding to the orthosteric binding site (orthosteric ligands), leading to conformational selection comprising inactive and active states (Figure 1a). This is the common activation mechanism among GPCRs.15 Agonists preferentially stabilize the active state and inverse agonists stabilize the inactive state, whereas ‘neutral’ antagonists block the binding of agonists/inverse agonists that bind to an overlapping orthosteric site because of the similar affinity for both states (Figure 1a).15
Figure 1.
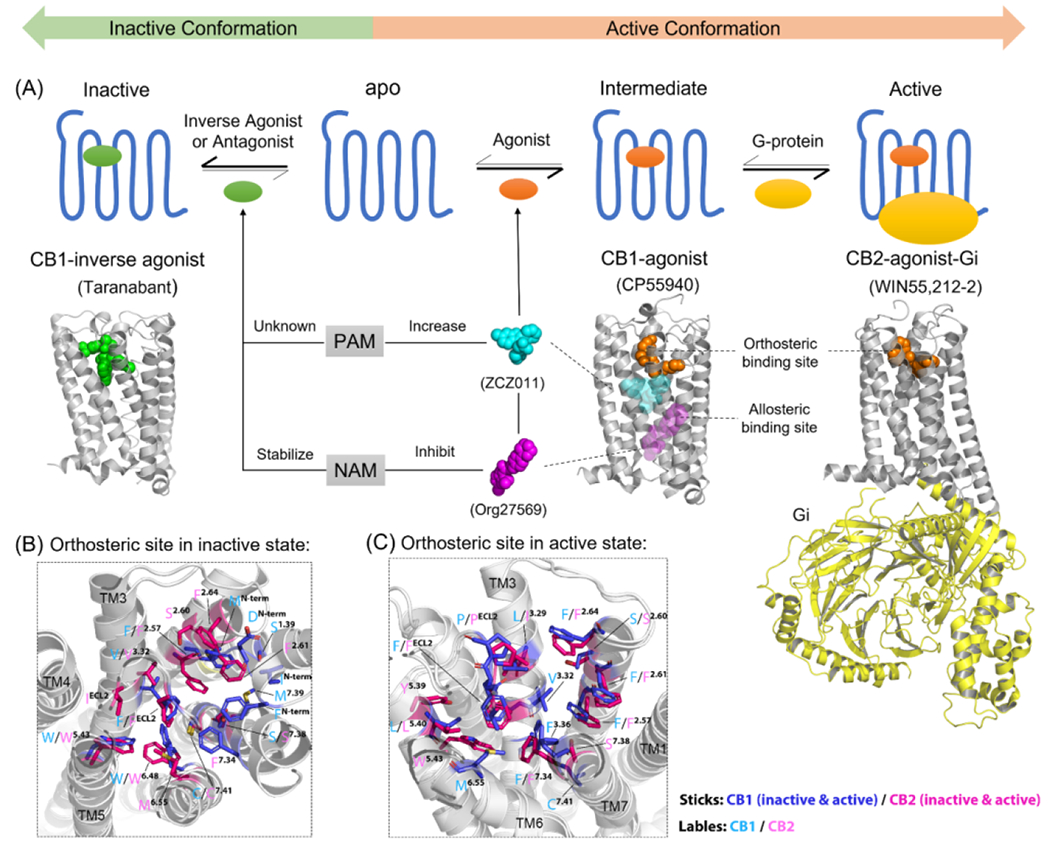
Active and inactive states of cannabinoid receptors (CBRs). (a) Brief overview of how orthosteric ligands and allosteric modulators (AMs) can alter the active/inactive states of CBRs in general. All the 3D structure of CBRs are shown in gray. The X-ray crystal structure of a CB1-inverse agonist (Taranabant, green sphere) complex [Protein Data Bank (PDB): 5U09] is used to represent the inactive conformation of CBR. The X-ray crystal structure of a CB1-agonist (CP55940, orange sphere) complex (PDB: 6kqi) is used to represent the intermediate state. The cryo-electron microscopy (cryo-EM) structure of a CB2-agonist (WIN55,212–2, orange sphere)-Gi protein (yellow cartoon) complex (PDB: 6pt0) is used to represent the active state. PAM (ZCZ011, cyan sphere) and NAM (Org27569, magenta sphere) were used as examples to show the allosteric binding sites on CBRs. However, there are various positions of the binding sites of positive allosteric modulators (PAMs) and negative allosteric modulators (NAMs); in addition, there is not enough evidence to identify whether PAM and NAM could bind to the same sites. The orthosteric binding sites of CBRs in (b) inactive and (c) active states. The 3D structures of CBRs are shown as white cartoons. The important ligand-interacting residues within 4 Å of the orthosteric ligands in the orthosteric binding site are shown in blue (inactive/active CB1R, PDB: 5U09/7wv9), and magenta (inactive/active CB2R, PDB: 5zty/6pt0) with cyan (CB1) and pink (CB2) labels. Figure based on information from 15.
Orthosteric ligands of CBRs have been widely studied since the main constituents of cannabis, 9-tetrahydrocannabinol (Δ9-THC) and cannabinoid (CBD), were successfully isolated during the 1960s.16 THC, 2-arachidonoylglycerol (2-AG), and anandamide (AEA) are endogenous cannabinoids produced by the body and show selectivity on CBRs. WIN55,212-2 and CP55940 are synthetic/exogenous cannabinoids that have high binding affinity on both CB1 and CB2. Although orthosteric ligands have different selectivity and binding affinity on CBRs, THC, 2-AG, AEA, WIN55,212-2, and CP55940 share the same orthosteric binding sites on CBRs, which are located within the TM regions close to the N terminus (Figure 1a). Detailed information on orthosteric binding sites in active and inactive states is shown in Figure 1B,C.
However, serious adverse events can result, especially with orthosteric ligands targeting the CB1 orthosteric binding site. CB1 agonists exert psychotropic side effects, and CB1 inverse agonists/antagonists cause psychiatric side effects, such as anxiety, depression, and suicidal tendencies.17 In clinical studies, CB2 agonists also show some side effects.18 Given the homology of CBRs, molecules targeting CB2 with low affinity and selectivity can cause off-target effects, such as CB1 psychotropic adverse effects.19 CB2 agonists can also cause on-target immune suppression upon chronic use.11 Most cannabis clinical candidates targeting the orthosteric binding site of CBRs were abandoned because of serious adverse effects.
Recently, allosteric modulation has emerged as a promising avenue for drug discovery because of its potential to avoid drug overdose and minimize adverse effects. AMs bind to the allosteric binding site on the receptor and mediate structural changes between inactive and active states, which is called ‘allosteric transition’ (Figure 1a).15 Positive allosteric modulators (PAMs) enhance the activity, negative allosteric modulators (NAMs) inhibit the activity, whereas neutral allosteric ligands (NALs) have no effects on the activity of orthosteric ligands, but competitively block the binding of PAMs or NAMs that bind to the same allosteric site (Figure 1a).15
One of the most popular research areas in the drug development of CBR AMs is finding or designing molecules that bind to a spatially distinct allosteric site to modulate CBR activity. Crystallographic or cryo-electron microscopy (cryo-EM) structures of CB1 and CB2 coupled with Gi protein released in 2019 and 2020, respectively showed the structural differences of CBRs between inactive and active states.20–22 In 2022, the reported crystal and cryo-EM structures of CB1 bound with ZCZ011 (PAM),23 together with the reported crystal structure of the CB1-ORG27569 (NAM) complex, released in 2019,24 provided deep insight into the molecular mechanism of CBR allosteric modulation.
This review highlights recent progress in the drug discovery of CBR AMs. We summarize newly synthesized AMs that target CBRs and the reported/predicted allosteric binding sites, and analyze the binding pattern of AMs. We also discuss the structural determinants of allosteric binding, important key binding residues in allosteric sites, as well as CBR allostery.
Allosteric modulators of the endocannabinoid system
Since the first AM of CBRs was identified in 2005,25 several AMs targeting CB1 or CB2 have been reported, increasing our understanding of the mechanisms behind allostery and enabling the development of potential therapeutic agents. Here, we summarize recently reported AMs targeting CBRs, including one peptide and six categories of AMs (Table 1).
Table 1.
CBR AMs reported between 2018 and 2022a
| Name | Biological target | Structure | In vitro biological assays | Type of allosteric modulator | Refs | |
|---|---|---|---|---|---|---|
| Orthosteric ligand used | Assay | |||||
| Osteogenic growth peptide | CB2R |
|
CP55,940 | Competition binding, cAMP simulation, [35S]GTPγS binding | Ago-PAM | 26 |
| Δ9-Tetrahydrocannabinolic acid | CB1R |
|
CP55,940 | Competition binding, cAMP simulation, [35S]GTPγS binding, β-arrestin recruitment, ERK1/2 phosphorylation | Ago-PAM | 29 |
| EC-21a | CB2R |
|
CP55,940, 2- AG, AEA | Competition binding, cAMP simulation, [35S]GTPγS binding, β-arrestin recruitment | PAM | 30 |
| CBD derivatives | CB2R |
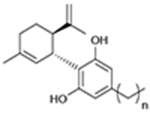
|
JWH-133 | cAMP simulation | NAM or PAM depend on carbon chain
length; orthosteric effect untested |
37 |
| n-Butyl-diphenylcarboxamides | CB1R |
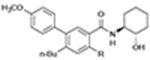
|
CP55,940 | Competition binding, [35S]GTPγS binding | PAM on CP55,940 binding, SAM on functional assays | 32 |
| Org27569 and PSNCBAM-1 hybrids | CB1R |
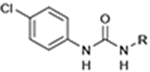
|
CP55,940 | Calcium mobilization, cAMP simulation, [35S]GTPγS binding | NAM | 40 |
| (−)-Cannabidiol-dimethylheptyl | CB1R, CB2R |
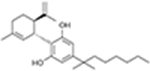
|
CP55,940 | Competition binding, [35S]GTPγS binding, β-arrestin recruitment | Ago-PAM for CB1R; NAM or PAM depend on different signaling pathways on CB2R | 34 |
The bitopic ligands, FD-22a and FD-24a are not included.
Endogenous allosteric modulators
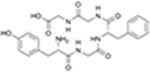
Osteogenic growth peptide
In 2022, the endogenous peptide osteogenic growth peptide (OGP) was reported by Raphael-Mizrahi et al. as an ago-PAM of CB2R (Table 1).26 Ago-PAMs bind to an allosteric site to enhance potency and/or efficacy of the orthosteric ligand, inducing receptor activation independent of an agonist. OGP has a positive role in maintaining bone mass, which might be associated with CB2 activation. The authors found that OGP alone decreased forskolin (FSK)-stimulated cAMP levels (logEC50 = −12.10) by activating Gi-coupled CB2 in CHO cells expressing human CB2, indicating its orthosteric agonist activity. However, the results of molecular docking and molecular dynamics (MD) simulations showed that OGP preferred binding to an allosteric site that mainly comprised an N terminus, extracellular loop 1 (ECL1), and extracellular loop 2 (ECL2), rather than the orthosteric site, and formed a stable conformation during the MD simulation in the presence of CP55940, which is a nonselective orthosteric ligand targeting CBRs. The results of competitive binding and [35S]GTPγS binding assays showed that OGP did not compete with CP55940 at the orthosteric binding site, but increased the 0.1 nM or 10 nM CP55940-induced [35S]GTPγS stimulation in a dose-dependent manner, indicating a PAM profile of OGP for CB2.26
Together, OGP acts as an ago-PAM of CB2, which itself decreased the FSK-stimulated cAMP level and showed PAM effects on CP55940-induced [35S]GTPγS binding (Table 1).26 However, further characterization is needed to illustrate the bell-shaped dose–response curve of OGP as well as its effect on orthosteric ligand binding and β-arrestin recruitment.
Recently, Hua et al. reported that cholesterol, the precursor of pregnenolone, could have allosteric effects on CB1 based on the crystal structure.21 However, more experiments are needed to further verify this hypothesis.
Exogenous allosteric modulators
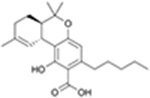
Δ9–tetrahydrocannabinolic acid
The precursor of Δ9-THC is Δ9-tetrahydrocannabinolic acid (Δ9-THCA-A), a nonpsychotropic phytocannabinoid in cannabis. Recent studies showed that it is a potent agonist of peroxisome proliferator-activated receptor-γ (PPARγ), and acts as an anti-inflammatory and neuroprotective agent.27,28
In 2020, Palomares et al. analyzed the role of Δ9-THCA-a in activating CBRs.29 Δ9-THCA-A competitively binds to both CB1 and CB2 with Ki values of 252 ± 140 nM and 506 ± 198 nM, respectively, showing its affinity for both two receptors. In a [35S] GTPγS binding assay, Δ9-THCA-A proved to be a partial agonist of CB1 (EC50 = 3.8 ± 0.5 μM), whereas it acted as an antagonist or an inverse agonist (IC50 = 1.3 ± 0.4 μM) of CB2.29
Further research investigated the possible allosteric activity of Δ9-THCA-A against CB1 (Table 1). CP55940-induced [35S] GTPγS binding occurred in the absence or presence of 0.1 nM Δ9-THCA-A. The results showed that the concentration of Δ9-THCA-A had no effect on [35S] GTPγS binding. Δ9-THCA-A significantly increased the efficacy of CP55940 [Emax (% over basal) = 125.1 versus Emax (%) = 151.5 and Emin (%) = −13.1 versus Emin (%) = 43.9]. Further validation showed that Δ9-THCA-A can enhance the agonist effect of CP55940 at the cAMP level and β-arrestin recruitment in the presence of CP55940, whereas it showed no effect on the cAMP level and a slight inducing effect on β-arrestin recruitment in the absence of CP55940. Possible effects of Δ9-THCA-A on phosphorylation of ERK1/2 were also evaluated. The results showed that both Δ9-THCA-A and CP55940 could trigger the phosphorylation of ERK1/2 when either was added alone.29
Thus, Δ9-THCA-A acts as an orthosteric partial agonist and a PAM of the CB1R (Table 1), but further experiments are needed to exclude the possible Δ9-THC contamination when preparing Δ9-THCA-A.29
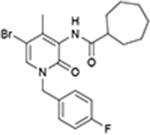
EC–21a and bitopic ligand series of FD–22a
In 2019, Ga et al. synthesized a PAM of CB2, creating a keystone of developing CB2 AMs (Table 1).30 In a radioligand binding assay, EC-21a significantly increased the binding of [3H]CP55940 at a low concentration and prevented its dissociation for ~2 h. In a functional assay, EC-21a potentiated CP55940 and 2-AG-induced [35S] GTPγS binding and had no effect on AEA. However, EC-21a showed no effect on [35S] GTPγS accumulation in the absence of orthosteric ligand, indicating its PAM profile of CB2 and probe dependence on [35S] GTPγS binding.30 EC-21a also augmented CP55940-induced cAMP inhibition by increasing both the potency and efficacy. In addition, EC-21a increased β-arrestin recruitment triggered by 10 nM CP55940.31 Structure–activity relationship (SAR) studies of this 2-oxopyridine analog showed that the cycloheptane-carboxamide group is necessary for PAM effects, and that replacement of the 4-fluoro benzyl group by aliphatic substituent or unsubstituted benzyl derivative fully abolished the PAM effects of compounds, indicating specific stereo-electronic requirements for this interaction.30
Several CB2 bitopic ligands have been designed by linking the pharmacophoric portion of EC-21a and FM-6b together that result in binding to both the orthosteric pocket and allosteric binding site. FM-6b is an orthosteric agonist of CB2R.32,33 Two synthesized bitopic molecules, FD-24a and FD-22a, inhibited FSK-stimulated cAMP accumulation induced by hCB2R activation (EC50 = 73 nM and 8.0 nM, respectively). However, they showed no enhancement of β-arrestin2 recruitment. The combined effect of FD-24a or FD-22a with increasing concentration of EC-21a was evaluated on the inhibition of FSK-stimulated cAMP accumulation. At a low concentration of EC-21a, the effect of FD-24a or FD-22a was not significantly increased. At high concentrations, EC-21a facilitated FD-24a agonism. These results suggest that FD-22a and FD-24a share the same allosteric binding pocket as EC-21a.33 The allosteric binding site of this bitopic ligand was also predicted using molecular docking, as discussed below. The results of the effects of FD-22a and FD-24a against 100 nM CB2 antagonist/inverse agonists showed that both molecules interact with both orthosteric and allosteric sites.33 However, FD-22a had higher affinity for the allosteric site and lower affinity for the orthosteric site compared with FD-24a. Finally, a [3H]CP55,940 competition binding assay was carried out to further investigate the characteristics of the most promising bitopic ligand, FD-22a; the results showed that it can only partially displace [3H]CP55,940 binding [Emin (% CP55,940) = 40 ± 6.4 and Ki = 1.4 nM], indicating the complex pharmacology of this bitopic ligand.33
Cannabidiol derivatives
Cannabidiol (CBD) is the second most abundant phytocannabinoid in cannabis.34 It is a NAM of CB1-dependent ERK1/2 and PLCβ3 phosphorylation, β-arrestin recruitment, and cAMP inhibition that reduces CP55940 binding (concentration <1 μM).35,36 Navarro et al. developed a series of CBD derivatives as potential AMs of CB2 (Table 1).37 Based on the NAM activity of CBD that was discovered previously,38 they applied MD simulations as well as molecular docking to identify an allosteric binding site of CBD in CB2, which is close to the orthosteric site. They also found that the NAM effect of CBD was completely abolished when they mutated key residues within this pocket. The authors then altered the length of the carbon chain of CBD to generate a series of CBD derivatives. The results of the FSK-stimulated cAMP accumulation assays indicated that two of the CBD derivatives increased the effect of orthosteric ligand JWH-133, whereas the other three decreased its orthosteric effect.37 In further work, the authors used MD simulation and site-direct mutagenesis to show that their predicted allosteric binding site lies on the access of orthosteric binding. These results suggest that it is possible to design allosteric modulators by using structure-based methods.39
n-Butyl-diphenylcarboxamides
In 2022, Gado et al. reported the design and synthesis of a novel n-butyl-diphenylcarboxamides analog originally expected as an antagonist analog of CBRs.32 However, two designed molecules were identified as PAMs for CB1 (Table 1). Compounds C7 and C10 increased CP55940 binding in the [3H]CP55940 competitive binding assay. In particular, C7 strongly increased CP55940 binding at a very low concentration. However, the results of [35S]GTPγS binding showed that C7 failed to significantly affect the action of CP55940 to stabilize the active state of CB1.32 Although more characterization regarding their effect on the β-arrestin signaling pathway is required, C7 and C10 are still noteworthy for their potential silent allosteric effect. Further docking studies utilizing those compounds indicated a preferred interaction with the binding site for NAMs and a possible location for the binding of PAMs,32 as discussed below.
Org27569 and PSNCBAM–1 hybrids
Org27569 and PSNCBAM-1 are well-known NAMs for CB1. They increase agonist binding and decrease antagonist binding and decrease agonist signaling in many functional assays.40,41 In 2021, Nguyen et al. designed and synthesized a series of CB1 NAM compounds (Table 1).40 They merged the key structural features of Org27569 and PSNCBAM-1, which contributed to their binding with CB1 in MD simulation and molecular docking studies. Org27569 and PSNCBAM-1 also maintained the binding pattern of those features. Several compounds displayed comparable effects with Org27569 and PSNCBAM-1 in functional assays. One of the new designs, compound 8, showed a similar potency to Org27569 and PSNCBAM-1 in reducing CP55940-induced CB1-mediated calcium mobilization (pIC50 = 6.7 compared with 6.1 for Org27569 and 7.5 for PSNCBAM-1).40 However, in [35S]GTPγS binding and cAMP accumulation assays, the NAM potency of compound 8 decreased (pIC50 = 5.2 and 5.5, respectively), whereas compound 9 had a similar potency to Org27569 only in a GTPγS binding assay (pIC50 = 5.9 compared with 6.0 for Org27569).40 This study successfully provided a novel scaffold of CB1 NAM for which SAR studies of CB1 NAMs are now required.
(−)–Cannabidiol–dimethylheptyl
In 2019, Tham et al. identified (−)-cannabidiol-dimethylheptyl (CBD-DMH) as an ago-PAM of CB1, whereas, for CB2R, it showed a PAM profile in cAMP modulation and a NAM profile of β-arrestin1 recruitment (Table 1).34 In a FSK-induced cAMP accumulation assay and β-arrestin1 recruitment on CB1s, CBD-DMH enhanced the potency and efficacy of CP55940 (with a KB of 121 nM and 237 nM, respectively), whereas it acted as a partial agonist when administered alone. The authors further verified that CBD-DMH could increase the orthosteric ligand binding of CP55940. Similar to Org27569, CBD-DMH increased the CP55940 binding at a low concentration (<1 μM) but reduced its binding above 1 μM. On CB2, CBD-DMH affected the CB2R-dependent cAMP inhibition induced by CP55940. As a result, CBD-DMH enhanced both the potency and efficacy of CP55940 with a KB of 38 nM. However, in a β-arrestin1 recruitment assay, CBD-DMH inhibited CP55940-induced β-arrestin1 recruitment, indicating a NAM profile for β-arrestin1 recruitment, but a PAM profile for G-protein-dependent pathway of CB2Rs. A radioligand binding assay verified that CBD-DMH can also increase the CP55940 binding in CB2 (Table 1).34
Identified allosteric sites and the key binding residues in CB1
CB1 reported/putative allosteric binding sites
To date, there are two X-ray crystal or cryo-EM structures of CB1-orthosteric ligand-AM complexes available [Org27569; Protein Data Bank (PDB): 6kqi and ZCZ011 (PDB: 7wv9, 7fee)], identified by Shao et al. in 2019 and Yang et al. in 2022. These two AMs bind to the extrahelical site of CB1 in the TM 2-3-4 surface.23,24 The distance between the two co-crystalized binding sites is ~7 Å (Figure 2). Org27569 is a NAM of CB1. Its indole ring packs against the indole side chain of W2414.50 a highly conserved residue among all class A GPCRs. The residues are named using the Ballesteros–Weinstein (B-W) numbering scheme, which is based on the highly conserved residues present in every TM helix of Class A GPCRs.42 The B-W numbering scheme comprises two numbers: the first represents the TM helix number (1–7), and the second denotes the residue position relative to the most conserved residue in that helix, which is defined as number 50.43 For example, 4.51 represents a residue located in TM4, one residue after the most conserved residue, W2414.50. The amide-linked piperidinylphenyl chain of Org27569 extends across the exterior side of the receptor and interacts with T2424.51 and I2454.54. Site-directed mutagenesis is a widely used technique to validate functions of residues. Table 2 summarizes all reported mutagenesis data for CBRs.23,24,44
Figure 2.
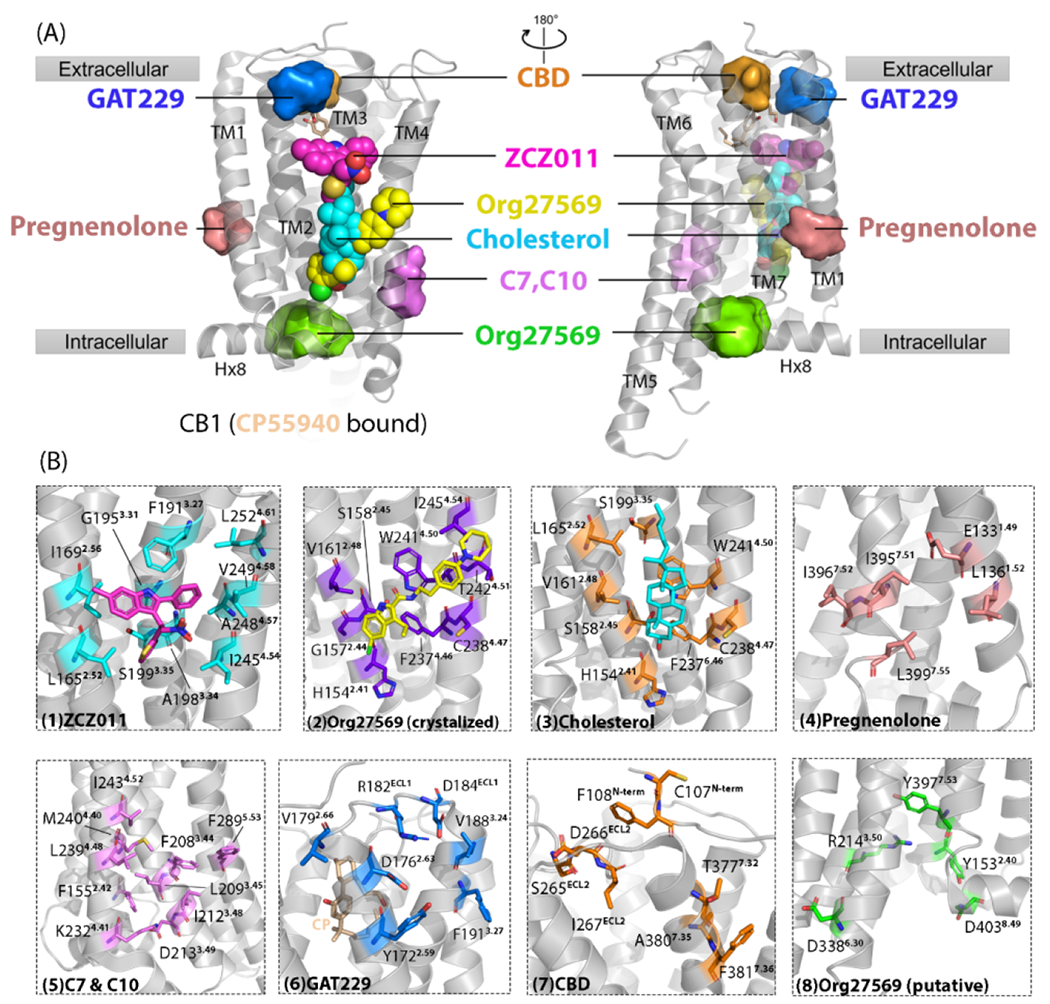
Cannabinoid receptor 1 (CB1) reported/putative allosteric binding sites. (a) Cryo-electron microscopy (cryo-EM) structure of CB1 (CP55940 bound) is shown in gray [Protein Data Bank (PDB): 7wv9]. CP55940 is represented as yellow sticks. Seven allosteric sites are shown. Three allosteric sites reported by high-resolution X-ray crystal or cryo-EM structures are shown as spheres: yellow sphere, Org27569 binding site; cyan sphere, cholesterol binding site; magenta sphere, ZCZ011 binding site. Other allosteric sites shown as surfaces are putative ones revealed via computational approaches, such as molecular dynamics simulations and molecular docking, and validated by site-directed mutagenesis. (b) Magnified details for each site shown in (a). From left to right, the detailed ligand-interacting residues in each allosteric site are: (1) ZCZ011 (cyan sticks) in cryo-EM structures (PDB: 7wv9), (2) co-crystalized Org27569 (purple sticks) (PDB: 6kqi), (3) co-crystallized cholesterol (orange sticks) (PDB: 5xra), (4) pregnenolone (salmon sticks), (5) c7 and C10 (violet sticks), (6) GAT229 (blue sticks), (7) CBD (orange sticks), and (8) Org27569 putative site (green sticks).
Table 2.
Summary of CBR site-directed mutagenesisa
| Position | Residue | Mutant(s) | Results |
|---|---|---|---|
| CB1 | |||
| N terminus | C98 | C98A | Abolished CBD NAM potency |
| C98S | Retained wild-type response | ||
| C107 | C107A | Abolished CBD NAM potency | |
| C107S | Retained wild-type response | ||
| CB2 | |||
| ICL2 | L222 | L222P, L222A, L222F | Lost Gs coupling, still coupled to Gi |
| Increased basal signaling through Gs | |||
| ICL3 | L341 | L341A | Attenuated Gi coupling, increased agonist potency, coupled to Gαs |
| A342 | A342L | ||
| TM2 | F155 | F155W | Reduced agonist-induced receptor activation; reduced CP55940-induced receptor activation; impaired PAM potency of ZCZ011 |
| F155V | Enhance Gi signaling activation ability | ||
| TM2 | I156 | I156A | Enhanced β-arrestin 1 recruitment |
| I156T | Reduced β-arrestin recruitment | ||
| TM2 | F170 | F170A | Dramatically decreased functionally affinity for rimonabant and AM6538; did not alter potency of CP55940 |
| F170W | No appreciable effect on antagonist binding | ||
| TM2 | F174 | F174A | Dramatically decreased functionally affinity for rimonabant and AM6538; did not alter potency of CP55940 |
| F174W | No appreciable effect on antagonist binding | ||
| TM2 | D163 | D163N | No effect on CP55940, AEA, Δ9-THC,
SR131416A binding; significantly attenuated WIN55,212-2 binding,
inhibited receptor internalization, no effect on CP55940 no effect on
CP55940 or WIN55,212-2 binding |
| D163Q | No effect on CP55940, AEA, Δ9-THC SR131416A binding; significantly attenuated WIN55,212-2 binding | ||
| TM2 | L165 | L165A | Strongly abolished PAM potency of ZCZ011 |
| I169 | I169A | Strongly abolished PAM potency of ZCZ011 | |
| TM3 | F191 | F191A | Partially reduced PAM potency of ZCZ011 |
| TM3 | S199 | S199A | Strongly abolished PAM potency of ZCZ011 |
| TM3 | R214 | R214A | Decreased ability to recruit Go proteins, did not affect basal recruitment or potency of 2-AG or WIN55,212-2 |
| TM3 | F200 | F200A | High basal activity in GTPγS binding assay |
| TM3 | T210 | T210A | Disabled Gi coupling |
| T210I | Decreased apparent thermostability | ||
| TM4 | K192 | K192A,K192Q,K192E | Decreased binding affinity of agonists: CP55940, HU-210, AEA |
| TM4 | F237 | F237L | Increased agonist CP55940 affinity, decreased inverse agonist SR141716A affinity |
| TM4 | I245 | I245A | Strongly abolished PAM potency of ZCZ011 |
| TM6 | G357 | G357F | Reduced flexibility of TM6 |
| TM7 | F379 | F379W | Loss of CP55940 potency |
| F379A | Greater loss of CP55940 potency | ||
| TM7 | N393 | N393A | Impaired β-arrestin signaling; no effect on G-protein signaling |
| TM7 | Y397 | Y397F | Impaired β-arrestin signaling; no effect on G-protein signaling |
| ECL2 | F268 | F268W | Dramatically decreased binding of all agonists; little impact on antagonist binding |
| F269 | F269A | ||
| H270 | H270A | ||
| I271 | I271A | ||
| ICL2 | P139 | P139F,P139M,P139L | Allowed CB2 to couple to Gs protein |
| TM1 | V36 | V36F | Impaired effect of JMH-133 of decreasing forskolin-induced cAMP level |
| V36M | Did not affect JMH-133 effect of decreasing forskolin-induced cAMP level | ||
| TM1 | F87 | Greatly decreased potency of CP55940 | |
| F91 | |||
| F94 | |||
| H95 | |||
| TM2 | D80 | D80Q, D80N | Did not affect CP55940, AEA, Δ9-THC, WIN55,212-2 binding |
| TM3 | V113 | V113E | Complete loss of ligand binding |
| V113L | Retain ligand binding | ||
| TM3 | T127 | T127A | Disrupted Gi protein coupling; slightly decreased agonist binding |
| TM2 | G78 | G78L | Improved protein homogeneity and thermostability; no impact on binding affinity of AM10257, SR144528, CP55940 |
| TM3 | T127 | T127A | |
| TM4 | T153 | T153L | |
| TM6 | R242 | R242E | |
| TM8 | G304 | G304E | |
| TM5 | S193 | S193G | No effect on competition binding of CP55940, SR144528, and WIN55,212-2 |
| TM5 | L201 | L201P | Disrupted CB2 signaling |
| TM6 | F259 | F259G | TM6 had higher flexibility |
| TM7 | F281 | F281K | Complete loss of CP55940 and SR144528 binding |
| TM7 | A282 | A282F | Significantly impaired effect of JMH-133 of decreasing forskolin-induced cAMP level |
| A282M | Did not affect JMH-133 effect of decreasing forskolin-induced cAMP level | ||
| ECL2 | F183 | F183Q, F183V | Significant loss of CP55940 and SR144528 binding |
Mutagenesis data related to the potency of allosteric modulators are highlighted in red.
F2374.46 is positioned to directly interact with Org27569 within 4.7 Å.24 F2374.46L attenuated the negative modulation of Org27569, and increased the orthosteric CP55940 binding compared with wild-type CB1 (Table 2).24 This allosteric site has also been validated through molecular docking and MD simulations. Moreover, the docked poses are slightly different compared with the co-crystalized pose.45 This co-crystalized allosteric site is also found partially overlapped with the binding site of cholesterol in the crystal structure of agonist-only CB1.46 Another predicted allosteric site for Org27569 was shown to be more energetically favored compared with the co-crystalized site in the analysis of 200 ns MD simulations of several CB1–CP55940 ternary complexes, which located at the intracellular side near TM2-6-7 and helix 8 (Hx8). Its position matches the binding sites of some intracellular antagonists of other class A GPCRs.45
Unlike the allosteric site of co-crystalized Org27569, ZCZ011, a CB1R PAM, binds to the upper leaflet of TM2-3-4 (Figure 2), which is 7.9 Å away from the orthosteric site. This PAM-binding site is mainly mediated by van der Waals and hydrophobic interactions. F1913.27 and G1953.31 anchored ZCZ011 via its thiophene ring, with F1913.27 forming a hydrogen bond and π-π stacking with ZCZ011. F1913.27A partially reduced the PAM potency. The L1652.52, I1692.56, S1993.35, and I2454.54 residues were also important for the binding and PAM activity of ZCZ011, because their alanine mutations strongly abolished the PAM potency (Table 2). The authors also proved the selectivity of ZCZ011 for CB1 compared with CB2R.23 Allosteric sites with similar position and volume were also reported in protease-activated receptor 2 (PAR2) and GPR40.47,48
Recently, a putative allosteric binding site for CBD in CB1 was discovered using computational approaches, which was only observed in agonist-binding CB1 but not in its inactive state. This allosteric site is in the extracellular side of TM3–7 above the orthosteric pocket (Figure 2), where CBD adopts a vertical disposition with its alkyl chain extending toward the TM region. Molecular docking results showed that the major residues (<5 Å) there were nonpolar, except for D104N-term and D266ECL2, which formed ion-dipole interactions with 1′,3′-hydroxyl groups of CBD. These groups also dipole interacted with the C98N-term-C107N-term disulfide bond.49 C98N-term-C107N-term alanine mutations abolished CBD NAM potency, while their serine mutations remained the wild-type response, proving their essential roles for NAM function (Table 2).50 For more information about the C98N-term-C107N-term disulfide bond, please see the CB1 structure modeled by Chung et al.49 Jakowiecki et al. also investigated the binding of CBD to CB1 and reconstructed the entire N terminus of CB1.51 CBD aromatic ring had a π-π stacking with F108N-term.49 IN addition, 25-ns MD simulations were performed on a CB1-THC-CBD complex, showing a hydrogen bond between CBD and I267ECL2.49 One of the CBD derivatives, CBD-DMH, was reported to have allosteric activity on CB1 as well as CB2. Through in silico molecular docking, radioligand binding assays, and functional assays, CBD-DMH was detected to bind to a region that bridged the orthosteric pocket and the allosteric site of CBD (as described above), sharing the ligand-interacting residues with the agonist.34 The position of this allosteric site was deeper than that of CBD.
Pregnenolone acts as an endogenous CB1-biased NAM.52 Its potential allosteric site on CB1 was detected in the intracellular (IC) TM1–7 and Hx8 exosite (Figure 2) using the Forced-Biased Metropolis Monte Carlo (FBMMC) simulated annealing program.52 Another allosteric site of CB1-only PAM, GAT229, was detected by FBMMC in the presence of orthosteric ligand CP55940. GAT229 was in the extracellular TM2-3 and ECL1 forming hydrogen bonds with D1762.63 (distance = 8.0 Å) and Y1722.59 (distance = 2.9 Å). Y1722.59 also had offset parallel π-π stacking with the GAT229 indole ring phenyl substituent (ring-ring central distance = 4.1 Å). Its indole ring also had cation-π stacking with R182 in the ECL1.53 The PAM site has also been acknowledged as the potential binding site for an ago-PAM, which is a GAT211 derivative, shown by docking studies.54 This site was at the same position as the cholesterol binding site in the human A2A adenosine receptor (A2AAR) (PDB: 4EIY), which is also a class A GPCR.55
C7 and C10 were found to increase the binding of CP55940, indicating a potential PAM function. Blind docking using the GOLD program was performed on all available CB1 structures, including both states. Two potential allosteric sites were discovered for C7 and C10. One was the co-crystalized allosteric site for Org27569, the same position as the cholesterol binding site in CB1 (PDB: 5XRA). The docking results showed that C7 is positioned deeper in this allosteric site compared with C10, interacting with W2414.50 as well as H1542.41.32 These were experimental key residues for NAM activity.24 However, because PAM/NAM induces different conformational changes in helix arrangement, it is hard to determine whether PAM/NAM shares the same site. Another allosteric site was found in TM2-3-4 with various volumes in different CB1 activation states (Figure 2). C7 and C10 were not able to engage polar interactions, such as with F2083.44, which stacked with PAMs shown in the molecular docking results.32 This putative allosteric site corresponds to an ago-PAM, AP8, co-crystalized binding site in GPR40.56
CB2 allosteric binding sites
To date, no cryo-EM or co-crystalized NAMs or PAMs for CB2 have been reported, leaving its allosteric site a mystery. However, several molecules have been identified with allosteric potency through in vitro and in vivo assays, which becomes a good entry point for studying the CB2 allosteric site. Computational approaches are widely applied to predict and analyze potential allosteric sites of CB2. Two putative allosteric sites were recently discovered. One is located on the extracellular side of TM1-2-7 (Figure 3), predicted by via different approaches.33,37,39,57,58 TBC is a highly selective ago-NAM of CB2, whereas DHGA shows NAM effects on CB2 in a probe-dependent manner. DHGA was reported as a mixture of two diastereomers with the difference only at the 4’-OH, separating DHGA into 4R and 4S stereoisomers.59 The molecular docking results indicated that both TBC and DHGA formed strong hydrophobic interactions within this site. Hydrophobic interactions strengthened after 200-ns MD simulations in the presence of CP55940. The results also revealed that ligands shifted slightly deeper into the allosteric site, where both TBC and DHGA interacted with Val361.35, Val922.62, Phe2817.35, and Ala2827.30 through strong hydrophobic interactions. DHGA also formed a hydrogen bond with Lys2797.33.57 Superimposition of the computer model of CB2-JWH-133 (agonist) to the crystal structure of CB1R-AM6538 (antagonist) complex also revealed this ‘extra site’. It lies adjacent to JWH-133 and near TM1–7, located at the same position as the one that TBC/DHGA binds to (Figure 3). Navarro, et al. also proposed that CBD binds to this allosteric site with its propenyl-methylcyclohexane moiety pointing toward the entrance channel of TM1–7 and the pentyl chain pointing toward the intracellular side.37 Methionine mutants of Val361.35 and Ala2827.30 completely impaired CBD NAM effects on JWH-133 because of a spatial block (Table 2). Ser2857.39 formed an essential hydrogen bond with the CBD metabenzenediol moiety. Its leucine mutant partially impaired the NAM activity of CBD (Table 2).37 This binding site is also consistent with a previously predicted allosteric ‘site H’.58
Figure 3.
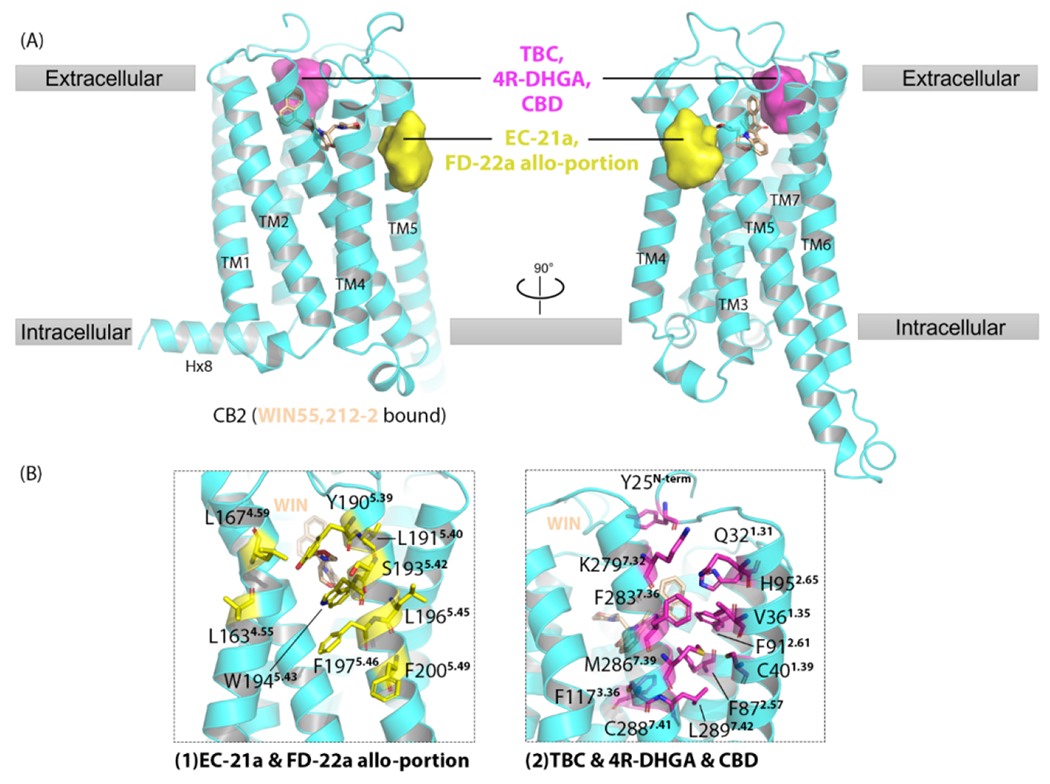
Cannabinoid receptor 1 (CB2) putative allosteric binding sites. (a) Cryo-electron microscopy (cryo-EM) structure of CB2 (WIN55,212–2–bound) is shown in cyan [Protein Data Bank (PDB): 6pt0]. WIN55,212–2 is represented as yellow sticks. Two allosteric sites are shown as surfaces, predicted by computational approaches, such as molecular dynamics simulations and as molecular docking, and then validated by site-directed mutagenesis. (b) Magnified details for each site shown in (a). (1) Allosteric site with residues shown in yellow sticks predicted for EC-21a and the allosteric portion of bitopic ligand FD-22a. (2) Allosteric site with residues shown in magenta sticks predicted for TBC, 4R-DHGA, and CBD.
Another putative allosteric binding site is on the receptor surface between TM4 and TM5. (Figure 3) The first identified PAM of CB2, EC-21a, docked into this site, where it mainly formed hydrophobic interactions, except for one hydrogen bond with S1935.42.33 Recently, the bitopic ligand, FD-22a, was found to bind to a cavity that bridges this allosteric site and the orthosteric site; it links the pharmacophoric portion of EC-21a with the CB2R orthosteric agonist FM-6b. The allosteric portion of FD-22a also formed a hydrogen bond with S1935.42.33 The glycine mutant of S1935.42 did not affect the binding of orthosteric agonists, CP55940, SR144528, and WIN55,212-2, indicating that S1935.42 does not interact with orthosteric ligands (Table 2).33 Based on these data, S1935.42 is a potential key residue in this allosteric site. The potential structural determinants for FD-22a to bind to this site are two key moieties: the triazole ring and the amide, the latter of which engages a hydrogen bond with S1935.42.33
Molecular mechanisms of CBR allostery
Although pharmacological and biochemical approaches have driven the drug discovery of AMs that target CBRs, CBR allostery or how allosteric drugs modulate the effects triggered by orthosteric ligands remain a mystery. Over the past 5 years, structural studies have contributed more to AM discovery of CBRs, providing some structural understanding of CBR allostery. The reported/putative CBR allosteric sites encompass three regions (Figure 4a): the extracellular vestibule, outside the 7TM regions, as well as the intracellular surface, which are all consistent with the common allosteric regions for GPCRs.15 Here, we analyze the allostery of each of these allosteric regions.
Figure 4.
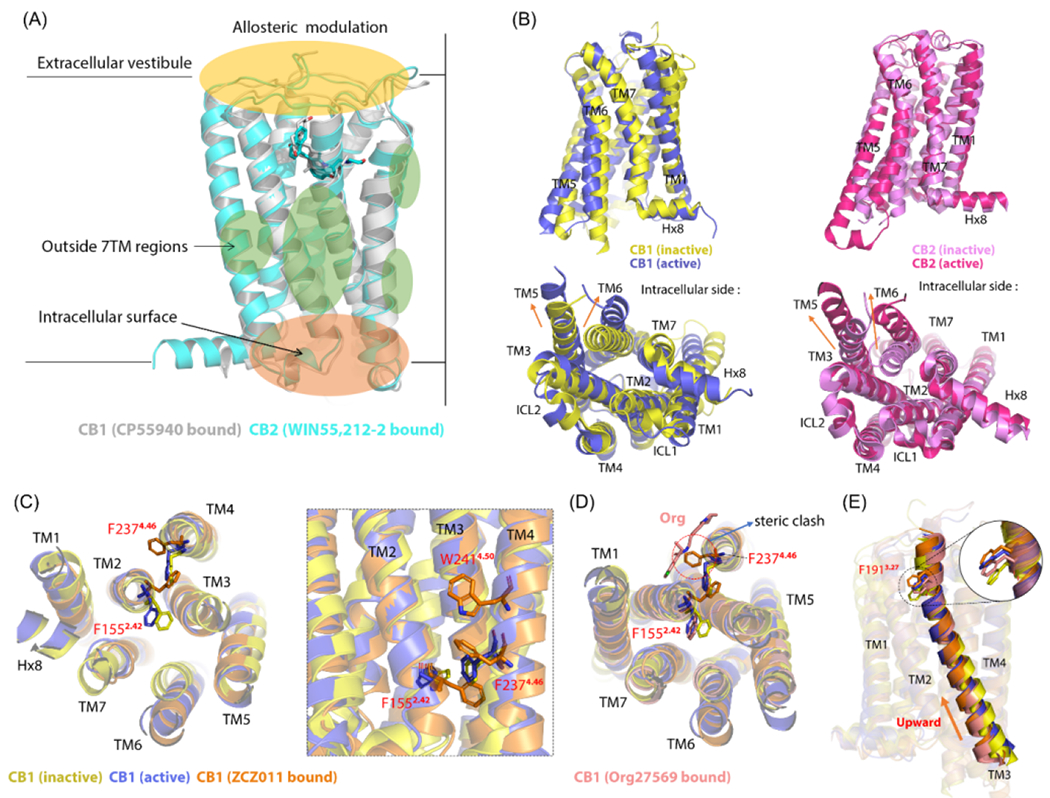
Molecular mechanism for cannabinoid receptor (CBR) allostery: 1. (a) CBR allosteric modulators (AMs) mainly bind to three regions: extracellular vestibule, outside transmembrane (TM) 7 regions, and intracellular surface. Cryo-electron microscopy (cryo-EM) overlapped structures of CP55940-bound CB1R [in grey; Protein Data Bank (PDB): 7wv9] and WIN55,212–2–bound CB2R (in cyan; PDB: 6pt0). (b) TM6 outward movement and TM5 arrangement in overlapped CB1R and CB2R in the active and inactive state. The crystal structures of CB1R in inactive state are shown in yellow (PDB: 5tyz), whereas the active state is shown in blue (PDB: 5xr8). The crystal structures of CB2R in the inactive state are shown in violet (PDB: 5zty), whereas the active state is shown in pink (PDB: 6pt0). Both CB1R and CB2R have significant outward movement of TM6. For the TM5 arrangement, an elongation occurs in CB1R, whereas an outward movement occurs in CB2R. (c) TM2 arrangement in ZCZ011-bound CB1R (in orange; PDB: 7wv9) compared with inactive and active CB1R with the reorientation of F1552.42 and F2374.46. The left panel shows the intracellular view, and the right panel shows the same residue positions from the view of the TM domain. W2414.50 forms an aromatic stack with F2374.46. (d) Org27569-bound CB1R (in salmon; PDB: 6kqi) causes a steric clash with F2374.46, thus influencing F1552.42 reorientation and impeding the TM2 arrangement. (e) Upward movement of TM3 in four overlapped CB1Rs (F1913.27 as reference). The sequence from top to bottom is: ZCZ011-bound CB1R, active CB1R, Org–bound CB1R, and inactive CB1R. Figure based on 15,21,23.
The only region that was found to bind to co-crystalized AMs is the region outside the 7TM domain extending to the bilayer. For many class A GPCRs (except CBRs), the allosteric site of this region is TM5-6-7.15 No reported/putative allosteric sites of CBRs entirely overlapped with this one; however, based on the biostructural data of X-ray crystallography or cryo-EM as well as computational approaches, such as MD simulations and molecular docking, the AMs bound to the outside 7TM domains were found to share common mechanisms:
TM2 arrangement
During CBR activation, agonist binding triggers a TM6 outward movement of ~6–8 Å (Figure 4b), and creates an intracellular transducer binding site, which further provides space for G protein coupling.60 The TM6 outward movement is a hallmark of GPCR activation.57 Before TM6 fully moved outward, a co-crystalized PAM (ZCZ011), which binds to the surface of CB1R TM2-3-4, was observed promoting the TM2 rearrangement: F1552.42 (CB1) reoriented from pointing inside to outside of the TM region, following by the ECL moving inward and the intracellular loop (ICL) rotating downward.20,23 This pushed F2374.46 (CB1) to point from inside to outside to avoid a collision and formed an aromatic stack with W2414.50 (Figure 4c).20,23 Meanwhile, TM6 was observed moving partially outward.23 This is a unique intermediate state found in CB1, in which the orthosteric binding site is contracted to fit around the agonist but the toggle switch residues do not reorient and TM6 movement is hindered.24 The TM6 then moved fully outward, and CB1 was stabilized in the active state.23 When co-crystalized NAM (Org27569) bound to CB1 in the presence of CP55940, it disturbed F2374.46 orient because of the steric clash, resulting in the failure of F1552.42 reorientation, thus impeding the TM2 rearrangement (Figure 4d). This further indirectly disfavored TM6 movement and stabilized CB1 in an inactive conformation.23,24 Molecular docking was performed for C7 and C10 (potential CB1R PAMs) in the co-crystalized Org27569 allosteric site. An intramolecular hydrogen bond between the methoxy moiety of the central phenyl and the amide nitrogen pushed C7 to a deeper region, interacting with both F2374.46 and H1542.41. C10 lacks the methoxy moiety and binds 1 Å higher compared with C7. C10 was also found to be less potent compared with C7 for increasing CP binding.32 The docking results for C7/C10 in their putative allosteric site in TM2-3-4 also showed similar F1552.42 reorientation by comparing these results to the crystal structure of inactive CB1, but C7/C10 lost potential PAM interactions. In experiments, C7/C10 did not influence CP-induced effects but increased CP binding.32
More studies will be helpful to discover the connection between the structural features and the experimental results. A similar TM2 arrangement is rare in other class A GPCR activations.23 The binding of ZCZ011 in CB1 also induced TM3 upward movement (F1913.27 as reference) (Figure 4e). The hydrogen bond and π-π stacking interaction with F1913.27 (CB1) contribute to stabilizing the fully active conformation of CB1R.23 However, no TM3 displacement has been reported in the binding of co-crystalized Org27569.
DRY motif and ionic lock in TM3
D3.49 R3.50 Y3.51 residues form a conversed motif among class A GPCRs, linking the orthosteric site and transducer binding site to help allosteric transition.15 These crucial residues are almost identical between the active conformations of CB1R and CB2R.21 The salt-bridge interaction (called ‘ionic lock’) between R3.50 and D6.30 is essential to maintain the inactive state of the CBR, which prevents G-protein coupling (Figure 5a,b).53,61 Interruptions of this salt bridge unlock the outward movement of TM6, triggering CBR activation. After the ‘ionic lock’ is broken, R3.50 of the DRY motif in CB2R extends toward TM7 and strengthens the interactions between them (Figure 5b), further forming a hydrogen bond with G352 from the α5 helix of Gi protein.57 Breakdown of the ‘ionic lock’ also leads to a reduction in both the TM3–7 and TM3–5 distance in CB1 and CB2. In CB1, Y2945.58 was also positioned toward Y3977.53 (Figure 5a) and formed a hydrogen bond with Gi protein in the agonist bound CB1 structures.32 Y3977.53 is an essential residue for β-arrestin recruitment by CB1.44
Figure 5.
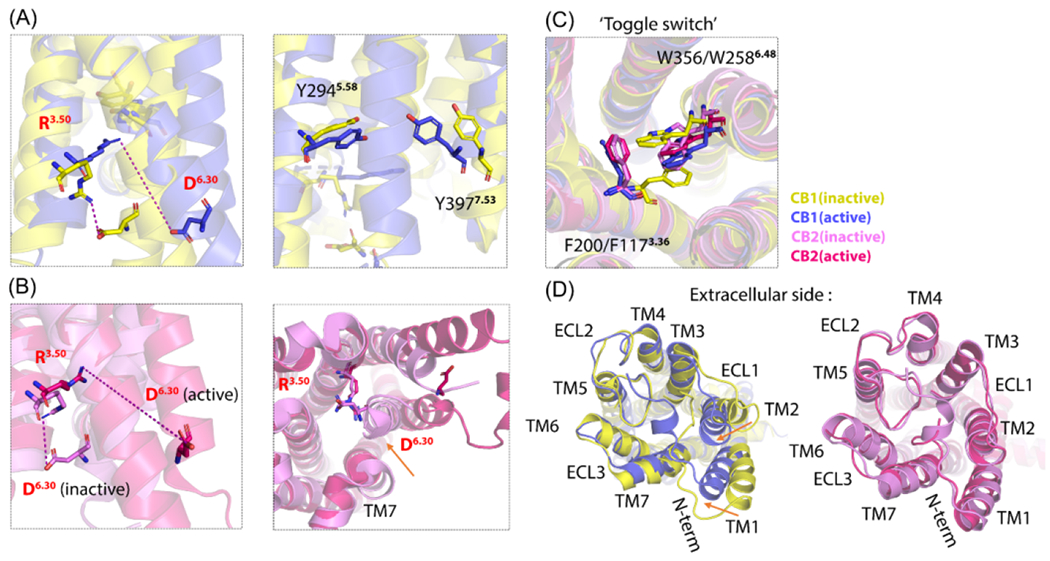
Molecular Mechanism for cannabinoid receptor (CBR) allostery: 2. (a) The ‘Ionic lock’ residues of overlapped CB1R inactive (in yellow; Protein Data Bank (PDB): 5tyz) and active conformations (in blue; PDB: 6kpg). When transferring from an inactive to active state, D6.30 moves far away from R3.50, indicating that breaking the ‘ionic lock’ unlocks transmembrane (TM)-6 outward movement. In the right panel, after the ‘ionic lock’ is broken, Y2945.58 is positioned toward Y3977.53. (b) The ‘Ionic lock’ residues of the overlapped CB2R inactive (in violet; PDB: 5zty) and active conformations (in pink; PDB: 6pt0). When transferring from an inactive to active state, D6.30 moves further away from R3.50 compared with CB1R, indicating that breaking the ‘ionic lock’ unlocks TM6 outward movement. In the right panel, after the ‘ionic lock’ is broken, R3.50 extends toward TM7. (c) The conformations of ‘toggle switch’ residues in four overlapped structures of CBRs’ inactive and active states. (d) The left panel shows the extracellular view of TM1–2 inward movement during CB1R activation, showing an obvious extracellular loop (ECL) closure. This might be because of the binding of covalent small molecules. In the right panel, CB2R has a minor ECL closure. Figure based on21,23.
TM5 rearrangement
During activation, the evident reduction in TM3–5 distance and the incremental distance of TM3–4 enlarged the putative allosteric site of C7 and C10, making D2133.49 accessible from this site compared with antagonist-bound CB2.32 The cytoplasmic portion of TM5 in CB1 elongated ~6 Å to extensively interact with the α5 helix of Gi protein, whereas TM5 in CB2 showed a larger movement (Figure 4b). G2105.59 (CB2) provides extra flexibility of TM5 compared with M2955.59 (CB1).21 The ‘toggle switch’ residues, F2003.36–W3566.48 in CB1 and F1173.36–W2586.48 in CB2, stabilize the inactive conformation of CBRs via π-π stacking (Figure 5c). TM3 and TM5 rearrangement can disrupt this interaction, changing CBRs to an active state.46,49,57,62 This is consistent with MD simulation results in a CB1-CP55940-CBD environment and two CB2-CP55940-TBC/DHGA environments, revealing that NAMs bind to the active conformation of CBRs, altering the orientation of the ‘toggle switch’ residues to form a stacking interaction, which further prompts the transition of the CBRs to inactive states.49,57
However, the CB2 ‘toggle switch’ residues remain controversial. Hua et al. proposed that a single ‘toggle switch’ residue, W2586.48, was enough to trigger CB2 activation as well as downstream signaling.21 Figure 5c shows little conformational difference of F1173.36 between the inactive and active state of CB2. The position of the putative CBD allosteric site in CB2 is closer to TM1-2 compared with CBD in CB1. The pentyl chain of CBD expands toward the intracellular hydrophobic cavity and interacts with F1173.36 to modulate CB2 activation.37 However, CBD was reported to act as a NAM of CBR only at nanomolar concentrations, whereas, at micromolar concentrations, CBD acted as an agonist.38,63 There is no evidence to link the structural features of CBD-bound CB2 to its dual properties. The active conformation of CB2 was found to have another TM5 arrangement in the presence of PAM EC-21a or bitopic ligand FD-22a: S1985.45 switched 180° to accommodate Gi protein coupling.33
Another region is the extracellular vestibule. CBD and GAT229 were predicted to bind there in CB1 (Figure 2a). TBC, DHGA, and CBD were predicted to bind to the same site in CB2 (Figure 3a) but not the same position as in CB1. Activation causes the closure of the extracellular vestibule in Gi-coupled class A GPCRs. The common mechanism for PAMs in this region is allosterically stabilizing the closed extracellular vestibule, whereas NAMs have an opposite effect in inactive conformations of class A GPCRs.15 The crystal structure of Org27569-bound CB1 showed a reorientation of F1552.42 and F2374.46, inducing the TM2 arrangement as mentioned above (Figure 4c). The rotation of the extracellular half of TM2 is also responsible for CB1 orthosteric site contraction and the closure of ECL.24,32 Metropolis Monte Carlo simulation results showed that GAT229 binds to the TM2-3-ECL1 putative allosteric site, and its indole ring anchors the ECL1 residue, R182, through a cation-π interaction. GAT229 also indirectly stabilized CB1 ECL2 as well as ECL3 in the active conformation, indicating the PAM function.53 The closure of the extracellular region is also reported in crystalized CB1 compared with antagonist-bound CB1, in which the extracellular half of TM1 and 2 moved inward by 6.6 and 6.8 Å, respectively (Figure 5d).49 The same closure also happens in activation of CB2. MD simulations showed that the binding of both TBC and DHGA promoted CP55940 to interact more with ECL2 (L182ECL2 and Y1905.39 as examples).57 One intraloop disulfide bond between C98N-term and C107N-term constrained the missing membrane proximal region (MPR) of the N terminus in a V-shape, which modulated access to the orthosteric site: the N-terminus tail opens in the active state but closes in the inactive state. Docking studies indicated that CBD interacted with C98N-term and C107N-term in agonist-bound CB1, but bound deeper in antagonist-bound CB1 and even overlapped with the orthosteric site.49
The third common allosteric region is in the intracellular surface. For class A GPCRs, NAM binding to this site stabilizes an inactive conformation of TM6 and sterically impedes interactions between receptor and Gi protein/β-attestin.15 However there is a lack of evidence showing that this is the location of the allosteric site in CBRs.
Combining this evidence, we summarize the primary mechanism of CBR allosteric modulation. In inverse agonist or antagonist-bound CBRs, ‘ionic lock’ (a salt-bridge between R3.50 and D6.30) and π-π stacking of ‘toggle switch’ residues (F3.36 and W6.48) are key structural features in CBR inactive conformations. A well-known 6-8 Å outward movement of TM6 triggered the fully active conformation of CBRs, promoting Gi coupling. The role of AMs in CBR activation is reflected in two main conformational changes, including TM2 and TM3–5 arrangements: (i) PAM promotes the TM2 arrangement with the subsequent ECL inward movement as well as ICL downward rotation. It further promotes the TM6 outward movement, changing the CBR conformation from partially active to fully active. The closed extracellular vestibules as well as loops also result from the TM2 arrangement. NAM impedes the TM2 arrangement mainly by steric clash, and further hinders the TM6 outward movement to keep the CBR in an intermediate state, especially for CB1.23,24,32 Similar structural changes have not been reported in CB2; and (ii) PAM binding induces a TM3 arrangement, breaking the ‘ionic lock’ to unlock the TM6 outward movement53 and decreasing the distances of TM3–5 and TM3–7. TM5 arrangement is also triggered. CB1 has TM5 elongation, whereas CB2 has a larger TM5 movement to provide space for G-protein binding.21 Meanwhile, the TM3 arrangement promotes the TM6 moving outward in CB1 and/or CB2. NAM has different effects. It promotes the ‘toggle switch’ residues to form π-π stacking, stabilizing CBR in an inactive conformation.49,53,57,61 However, there is still not enough evidence to validate this structural allosteric mechanism. In addition, few studies have analyzed all the conformational changes simultaneously, hindering a deeper understanding of CBR allostery. Although MD simulations contribute to revealing the structural features of CBR allostery, calculating the binding free energies obtained from a single trajectory could lead to biased results. It would be more reliable to run multiple trajectories with different initial velocity seeds to obtain binding free energies.57
Concluding remarks and outlook
CBRs are potential therapeutic targets involved in metabolic regulation, diabetes, cardiac physiology, ischemia, psychiatric disorders, neurological diseases, inflammatory diseases, osteoporosis, and more.64–68 However, drugs involved in orthosteric modulation on CB1 can cause mood alteration, suicidal ideation, and motor performance,69 and those targeting the CB2 orthosteric site can induce proinflammatory actions and immune suppression upon chronic use.70 The development of AMs targeting CBRs has become a popular area for drug design because of various crucial advantages of AMs over orthosteric ligands, including higher receptor subtype selectivity, saturable effects, probe dependence, and the possibility to avoid drug overdose.47 In this review, we summarized recently synthesized AMs that target CBRs.
Numerous exogenous AMs have been reported. For CB1, both Org27569 and PSNCBAM-1 are NAMs40,41 and Δ9-THCA-A acts as a PAM and a partial agonist of CB1.29 For CB2, EC-21a was the first synthesized PAM, and two bitopic ligands, FD-22a and FD-24a, were subsequently designed30 by linking one orthosteric portion, FM-6b, and one allosteric portion, EC-21a. However, they showed complex pharmacology in experiments.33 C7 and C10 are two n-butyl-diphenylcarboxamides with PAM activities targeting CB2.32 Given that CBD shows a NAM profile on both CB1 and CB2, several CBD derivatives were synthesized.38 However, more characterizations are required to identify their allosteric properties. CBD-DMH is an ago-PAM of CB1 but showed a controversial PAM and NAM profile of CB2.34 OGP is a recently discovered endogenous AM that is an ago-PAM of CB2.26 The description of newly emerged or potential AMs and their allosteric features will give researchers more direction for ligand-based AM design.
These AMs were also useful for promoting the discovery of allosteric binding sites of CBRs. To date, only two reported allosteric sites for CB1 have been determined by X-ray crystallography or cryo-EM. More computational approaches, such as molecular docking and MD simulations, have also been widely used to discover potential allosteric sites for AMs. We described detailed structural features for AM binding, including binding poses, residue interactions, and some mutagenesis data, all of which can be used to promote structure-based AM design. ‘Pocketome’ is a newly discussed concept for GPCRs introduced by Hedderich et al., who investigated 557 GPCRs and calculated their structures to find potential allosteric binding pockets.71 Comparing existing allosteric binding sites with putative sites to reveal more structural information would be useful for identifying AMs of CBRs.
With the development of CBR AMs, crystallography, cryo-EM, as well as computational approaches have allowed researchers to shed light on the mysterious molecular mechanisms of allosteric modulation. Several findings from computational approaches are consistent with those from crystallography and cryo-EM. Two major conformational changes during activation of CBRs are the TM6 outward movement and the broken of ‘ionic lock’ and ‘toggle switch’ interaction. After binding to CBR, AM changes the conformation of its binding region first and then indirectly triggers other conformational changes, such as helix rearrangements, residue reorientation, and movement of ECL/ICL. These structural features reflect the functions of AMs and can provide insight for designing AMs. Researchers can develop AMs based on either the shape or ligand-interacting residues of potential allosteric sites. In addition to experimental validation, researchers can also prevalidate their designed AMs by analyzing whether similar conformational changes occur after AM binds to the CBR. However, some structural features, such as the TM2 arrangement, are only reported in CB1.
We hope that researchers will pay attention to all these structural features during structural biological studies and computational studies for CBRs, which will help fill the gaps in understanding CBR allostery and further contribute to progress in AM design. The structural differences caused by the allosteric modulation of CB1 and CB2 could also shed light on the selectivity of CBRs. We hope that this review will help support the design of AMs that target CBRs from both structure-based and ligand-based perspectives.
Highlights.
This review paper presents an overview of the recent findings on the allosteric modulation of cannabinoid receptors.
Six categories of allosteric modulators have new designs that show positive/negative allosteric profiles or dual properties.
Seven allosteric binding sites for cannabinoid receptor 1 and two for cannabinoid receptor 2 have been reported via structural biology or predicted via computational approaches.
The allosteric modulators that bind to different regions of cannabinoid receptors are revealed to trigger similar conformational changes to stabilize the receptors in active/inactive states.
Acknowledgments
The authors would like to acknowledge Chih-Jung Chen and Chen Jiang for helpful suggestions for generating the figures. The authors would also like to express their gratitude to Yasmin Othman, who provided great support in refining the wording and grammar of the manuscript. The authors would like to acknowledge funding support to the Xie laboratory and CCGS Center from the National Institutes of Health, National Institute on Drug Abuse (P30 DA035758A1, R01 DA052329, and R56 AG074951).
Biographies
Author biographies

Jiayi Yuan received her BSc from Shenyang Pharmaceutical University in 2020. She obtained her MSc from the School of Pharmacy, University of Pittsburgh in 2022, where she is now pursuing her PhD degree. Her research interests include drug design and development of cannabinoid receptor 2 allosteric modulators and the development of in silico methods to facilitate vaccine design, including epitope prediction.

Bo Yang received his BSc from the East China University of Science and Technology. He is now studying for an MSc at the School of Pharmacy, University of Pittsburgh. His research focuses on the design and synthesis of allosteric modulators for human cannabinoid receptor 2 (CBR2) and using computational chemistry methods to aid the design of allosteric modulators for human CBR2.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors declare that they have no conflicts of interest.
References
- 1.Di Marzo V. New approaches and challenges to targeting the endocannabinoid system. Nat Rev Drug Discov. 2018; 17(9): 623–639. [DOI] [PubMed] [Google Scholar]
- 2.Manzanares J, Cabañero D, Puente N, García-Gutiérrez MS, Grandes P, Maldonado R. Role of the endocannabinoid system in drug addiction. Biochem Pharmacol. 2018; 157: 108–121. [DOI] [PubMed] [Google Scholar]
- 3.Basavarajappa BS, Shivakumar M, Joshi V, Subbanna S. Endocannabinoid system in neurodegenerative disorders. J Neurochem. 2017; 142(5): 624–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garani R, Watts JJ, Mizrahi R. Endocannabinoid system in psychotic and mood disorders, a review of human studies. Prog Neuropsychopharmacol Biol Psychiatry. 2021; 106: 110096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fowler CJ. The endocannabinoid system - current implications for drug development. J Intern Med. 2021; 290(1): 2–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jung KM, Lin L, Piomelli D. The endocannabinoid system in the adipose organ. Rev Endocr Metab Disord. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jansma J, Brinkman F, van Hemert S, El Aidy S. Targeting the endocannabinoid system with microbial interventions to improve gut integrity. Prog Neuropsychopharmacol Biol Psychiatry. 2021; 106: 110169. [DOI] [PubMed] [Google Scholar]
- 8.Spanagel R. Cannabinoids and the endocannabinoid system in reward processing and addiction: from mechanisms to interventions. Dialogues Clin Neurosci. 2020; 22(3): 241–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leo LM, Abood ME. CB1 Cannabinoid receptor signaling and biased signaling. Molecules. 2021; 26(17): 5413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993; 365(6441): 61–65. [DOI] [PubMed] [Google Scholar]
- 11.Morales P, Jagerovic N. Novel approaches and current challenges with targeting the endocannabinoid system. Expert Opinion on Drug Discovery. 2020; 15(8): 917–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wold EA, Chen J, Cunningham KA, Zhou J. Allosteric modulation of class A GPCRs: targets, agents, and emerging concepts. J Med Chem. 2019; 62(1): 88–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Navarro G, Morales P, Rodríguez-Cueto C, Fernández-Ruiz J, Jagerovic N, Franco R. Targeting cannabinoid CB2 receptors in the central nervous system. medicinal chemistry approaches with focus on neurodegenerative disorders. Front Neurosci. 2016; 10: 406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saleh N, Hucke O, Kramer G, Schmidt E, Montel F, Lipinski R, et al. Multiple binding sites contribute to the mechanism of mixed agonistic and positive allosteric modulators of the cannabinoid CB1 receptor. Angew Chem Int Ed Engl. 2018; 57(10): 2580–2585. [DOI] [PubMed] [Google Scholar]
- 15.Thal DM, Glukhova A, Sexton PM, Christopoulos A. Structural insights into G-protein-coupled receptor allostery. Nature. 2018; 559(7712): 45–53. [DOI] [PubMed] [Google Scholar]
- 16.Gaoni Y, Mechoulam R. Isolation, structure, and partial synthesis of an active constituent of hashish. Journal of the American Chemical Society. 1964; 86(8): 1646–1647. [Google Scholar]
- 17.Lu D, Immadi SS, Wu Z, Kendall DA. Translational potential of allosteric modulators targeting the cannabinoid CB(1) receptor. Acta Pharmacol Sin. 2019; 40(3): 324–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shang Y, Tang Y. The central cannabinoid receptor type-2 (CB2) and chronic pain. Int J Neurosci. 2017; 127(9): 812–823. [DOI] [PubMed] [Google Scholar]
- 19.Whiting ZM, Yin J, de la Harpe SM, Vernall AJ, Grimsey NL. Developing the cannabinoid receptor 2 (CB2) pharmacopoeia: past, present, and future. Trends Pharmacological Sciences. 2022; 43(9): 754–771. [DOI] [PubMed] [Google Scholar]
- 20.Krishna Kumar K, Shalev-Benami M, Robertson MJ, Hu H, Banister SD, Hollingsworth SA, et al. Structure of a signaling cannabinoid receptor 1-G protein complex. Cell. 2019; 176(3): 448–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hua T, Li X, Wu L, Iliopoulos-Tsoutsouvas C, Wang Y, Wu M, et al. Activation and signaling mechanism revealed by cannabinoid receptor-Gi complex structures. Cell. 2020; 180(4): 655–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xing C, Zhuang Y, Xu TH, Feng Z, Zhou XE, Chen M, et al. Cryo-EM structure of the human cannabinoid receptor CB2-G(i) signaling complex. Cell. 2020; 180(4): 645–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang X, Wang X, Xu Z, Wu C, Zhou Y, Wang Y, et al. Molecular mechanism of allosteric modulation for the cannabinoid receptor CB1. Nat Chem Biol. 2022; 18(8): 831–840. [DOI] [PubMed] [Google Scholar]
- 24.Shao Z, Yan W, Chapman K, Ramesh K, Ferrell AJ, Yin J, et al. Structure of an allosteric modulator bound to the CB1 cannabinoid receptor. Nat Chem Biol. 2019; 15(12): 1199–1205. [DOI] [PubMed] [Google Scholar]
- 25.Price MR, Baillie GL, Thomas A, Stevenson LA, Easson M, Goodwin R, et al. Allosteric modulation of the cannabinoid CB1 receptor. Molecular Pharmacology. 2005; 68(5): 1484. [DOI] [PubMed] [Google Scholar]
- 26.Raphael-Mizrahi B, Attar-Namdar M, Chourasia M, Cascio MG, Shurki A, Tam J, et al. Osteogenic growth peptide is a potent anti-inflammatory and bone preserving hormone via cannabinoid receptor type 2. Elife. 2022; 11: e65834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nadal X, Del Río C, Casano S, Palomares B, Ferreiro-Vera C, Navarrete C, et al. Tetrahydrocannabinolic acid is a potent PPARγ agonist with neuroprotective activity. British Journal of Pharmacology. 2017; 174(23): 4263–4276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Palomares B, Ruiz-Pino F, Garrido-Rodriguez M, Eugenia Prados M, Sánchez-Garrido MA, Velasco I, et al. Tetrahydrocannabinolic acid A (THCA-A) reduces adiposity and prevents metabolic disease caused by diet-induced obesity. Biochemical Pharmacology. 2020; 171: 113693. [DOI] [PubMed] [Google Scholar]
- 29.Palomares B, Garrido-Rodriguez M, Gonzalo-Consuegra C, Gómez-Cañas M, Saen-Oon S, Soliva R, et al. Delta(9) -Tetrahydrocannabinolic acid alleviates collagen-induced arthritis: role of PPARgamma and CB1 receptors. Br J Pharmacol. 2020; 177(17): 4034–4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gado F, Di Cesare Mannelli L, Lucarini E, Bertini S, Cappelli E, Digiacomo M, et al. Identification of the first synthetic allosteric modulator of the CB2 receptors and evidence of its efficacy for neuropathic pain relief. J Med Chem. 2019; 62(1): 276–287. [DOI] [PubMed] [Google Scholar]
- 31.Gado F, Mohamed KA, Meini S, Ferrisi R, Bertini S, Digiacomo M, et al. Variously substituted 2-oxopyridine derivatives: extending the structure–activity relationships for allosteric modulation of the cannabinoid CB2 receptor. European Journal of Medicinal Chemistry. 2021; 211: 113116. [DOI] [PubMed] [Google Scholar]
- 32.Gado F, Ceni C, Ferrisi R, Sbrana G, Stevenson LA, Macchia M, et al. CB1 receptor binding sites for NAM and PAM: a first approach for studying, new n-butyl-diphenylcarboxamides as allosteric modulators. European Journal of Pharmaceutical Sciences. 2022; 169: 106088. [DOI] [PubMed] [Google Scholar]
- 33.Gado F, Ferrisi R, Polini B, Mohamed KA, Ricardi C, Lucarini E, et al. Design, synthesis, and biological activity of new CB2 receptor ligands: From orthosteric and allosteric modulators to dualsteric/bitopic ligands. Journal of Medicinal Chemistry. 2022; 65(14): 9918–9938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tham M, Yilmaz O, Alaverdashvili M, Kelly ME, Denovan-Wright EM, Laprairie RB. Allosteric and orthosteric pharmacology of cannabidiol and cannabidiol-dimethylheptyl at the type 1 and type 2 cannabinoid receptors. British Journal of Pharmacology. 2019; 176(10): 1455–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Laprairie RB, Bagher AM, Rourke JL, Zrein A, Cairns EA, Kelly MEM, et al. Positive allosteric modulation of the type 1 cannabinoid receptor reduces the signs and symptoms of Huntington’s disease in the R6/2 mouse model. Neuropharmacology. 2019; 151: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Laprairie RB, Mohamed KA, Zagzoog A, Kelly MEM, Stevenson LA, Pertwee R, et al. Indomethacin enhances type 1 cannabinoid receptor signaling. Frontiers in Molecular Neuroscience. 2019; 12: 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Navarro G, Gonzalez A, Sánchez-Morales A, Casajuana-Martin N, Gómez-Ventura M, Cordomí A, et al. Design of negative and positive allosteric modulators of the cannabinoid CB2 receptor derived from the natural product cannabidiol. Journal of Medicinal Chemistry. 2021;64(13): 9354–9364. [DOI] [PubMed] [Google Scholar]
- 38.Navarro G, Reyes-Resina I, Rivas-Santisteban R, Sánchez de Medina V, Morales P, Casano S, et al. Cannabidiol skews biased agonism at cannabinoid CB1 and CB2 receptors with smaller effect in CB1-CB2 heteroreceptor complexes. Biochemical pharmacology. 2018; 157: 148–158. [DOI] [PubMed] [Google Scholar]
- 39.Casajuana-Martin N, Navarro G, Gonzalez A, Llinas Del Torrent C, Gómez-Autet M, Quintana García A, et al. A single point mutation blocks the entrance of ligands to the cannabinoid CB2 receptor via the lipid bilayer. Journal of Chemical Information and Modeling. 2022; 62(22): 5771–5779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nguyen T, Gamage TF, Decker AM, Finlay DB, Langston TL, Barrus D, et al. Rational design of cannabinoid type-1 receptor allosteric modulators: Org27569 and PSNCBAM-1 hybrids. Bioorg Med Chem. 2021; 41: 116215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nguyen T, Li JX, Thomas BF, Wiley JL, Kenakin TP, Zhang Y. Allosteric modulation: an alternate approach targeting the cannabinoid CB1 receptor. Medicinal Research Reviews. 2017; 37(3): 441–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ballesteros JA, Weinstein H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods in Neurosciences. 1995; 25: 366–428. [Google Scholar]
- 43.Isberg V, de Graaf C, Bortolato A, Cherezov V, Katritch V, Marshall FH, et al. Generic GPCR residue numbers - aligning topology maps while minding the gaps. Trends Pharmacol Sci. 2015; 36(1): 22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leo LM, Al-Zoubi R, Hurst DP, Stephan AP, Zhao P, Tilley DG, et al. The NPXXY motif regulates β-arrestin recruitment by the CB1 cannabinoid receptor. Cannabis Cannabinoid Res. Published online July 6, 2022. 10.1089/can.2021.0223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aderibigbe AO, Pandey P, Doerksen RJ. Negative allosteric modulators of cannabinoid receptor 1: ternary complexes including CB1, orthosteric CP55940 and allosteric ORG27569. Journal of Biomolecular Structure and Dynamics. 2022; 40(13): 5729–5747. [DOI] [PubMed] [Google Scholar]
- 46.Li X, Hua T, Vemuri K, Ho JH, Wu Y, Wu L, et al. Crystal structures of agonist-bound human cannabinoid receptor CB1. Nature. 2017; 547(7664): 468–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Srivastava A, Yano J, Hirozane Y, Kefala G, Gruswitz F, Snell G, et al. High-resolution structure of the human GPR40 receptor bound to allosteric agonist TAK-875. Nature. 2014; 513(7516): 124–127. [DOI] [PubMed] [Google Scholar]
- 48.Cheng RKY, Fiez-Vandal C, Schlenker O, Edman K, Aggeler B, Brown DG, et al. Structural insight into allosteric modulation of protease-activated receptor 2. Nature. 2017; 545(7652): 112–115. [DOI] [PubMed] [Google Scholar]
- 49.Chung H, Fierro A, Pessoa-Mahana CD. Cannabidiol binding and negative allosteric modulation at the cannabinoid type 1 receptor in the presence of delta-9-tetrahydrocannabinol: an in silico study. PLoS ONE. 2019; 14(7): e0220025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Laprairie R, Bagher A, Kelly M, Denovan-Wright E. Cannabidiol is a negative allosteric modulator of the cannabinoid CB1 receptor. British Journal of Pharmacology. 2015; 172(20): 4790–4805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jakowiecki J, Abel R, Orzeł U, Pasznik P, Preissner R, Filipek S. Allosteric modulation of the CB1 cannabinoid receptor by cannabidiol—a molecular modeling study of the N-terminal domain and the allosteric–orthosteric coupling. Molecules. 2021; 26(9): 2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vallée M, Vitiello S, Bellocchio L, Hébert-Chatelain E, Monlezun S, Martin-Garcia E, et al. Pregnenolone can protect the brain from cannabis intoxication. science. 2014; 343(6166): 94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hurst DP, Garai S, Kulkarni PM, Schaffer PC, Reggio PH, Thakur GA. Identification of CB1 receptor allosteric sites using force-biased MMC simulated annealing and validation by structure–activity relationship studies. ACS Medicinal Chemistry Letters. 2019; 10(8): 1216–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Garai S, Leo LM, Szczesniak AM, Hurst DP, Schaffer PC, Zagzoog A, et al. Discovery of a biased allosteric modulator for cannabinoid 1 receptor: preclinical anti-glaucoma efficacy. J Med Chem. 2021; 64(12): 8104–8126. [DOI] [PubMed] [Google Scholar]
- 55.Liu W, Chun E, Thompson AA, Chubukov P, Xu F, Katritch V, et al. Structural basis for allosteric regulation of GPCRs by sodium ions. Science. 2012; 337(6091): 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lu J, Byrne N, Wang J, Bricogne G, Brown FK, Chobanian HR, et al. Structural basis for the cooperative allosteric activation of the free fatty acid receptor GPR40. Nature Structural & Molecular Biology. 2017; 24(7): 570–577. [DOI] [PubMed] [Google Scholar]
- 57.Pandey P, Roy KK, Doerksen RJ. Negative allosteric modulators of cannabinoid receptor 2: protein modeling, binding site identification and molecular dynamics simulations in the presence of an orthosteric agonist. Journal of Biomolecular Structure and Dynamics. 2020; 38(1): 32–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yuan J, Jiang C, Wang J, Chen CJ, Hao Y, Zhao G, et al. In silico prediction and validation of CB2 allosteric binding sites to aid the design of allosteric modulators. Molecules. 2022; 27(2): 453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rajasekaran M. Characterization of Allosteric Modulators of CB2 Receptors as Novel Therapeutics for Inflammatory Diseases. Little Rock; University of Arkansas for Medical Sciences, 2011. [Google Scholar]
- 60.Hilger D, Kumar KK, Hu H, Pedersen MF, O’Brien ES, Giehm L, et al. Structural insights into differences in G protein activation by family A and family B GPCRs. Science. 2020; 369(6503): eaba3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Savinainen JR, Saario SM, Niemi R, Järvinen T, Laitinen JT. An optimized approach to study endocannabinoid signaling: evidence against constitutive activity of rat brain adenosine A1 and cannabinoid CB1 receptors. Br J Pharmacol. 2003; 140(8): 1451–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li X, Hua T, Vemuri K, Ho JH, Wu Y, Wu L, et al. Crystal structure of the human cannabinoid receptor CB1. Cell. 2016; 167(3): 750–762.e714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McPartland JM, Glass M, Pertwee RG. Meta-analysis of cannabinoid ligand binding affinity and receptor distribution: interspecies differences. Br J Pharmacol. 2007; 152(5): 583–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rohbeck E, Eckel J, Romacho T. Cannabinoid receptors in metabolic regulation and diabetes. Physiology (Bethesda). 2021; 36(2): 102–113. [DOI] [PubMed] [Google Scholar]
- 65.Wang J, Lu HX, Wang J. Cannabinoid receptors in osteoporosis and osteoporotic pain: a narrative update of review. J Pharm Pharmacol. 2019; 71(10): 1469–1474. [DOI] [PubMed] [Google Scholar]
- 66.Haspula D, Clark MA. Cannabinoid receptors: an update on cell signaling, pathophysiological roles and therapeutic opportunities in neurological, cardiovascular, and inflammatory diseases. Int J Mol Sci. 2020; 21(20): 7693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Puhl SL. Cannabinoid-sensitive receptors in cardiac physiology and ischaemia. Biochim Biophys Acta Mol Cell Res. 2020; 1867(3): 118462. [DOI] [PubMed] [Google Scholar]
- 68.Joshi N, Onaivi ES. Psychiatric disorders and cannabinoid receptors. Adv Exp Med Biol. 2021; 1264: 131–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Borgelt LM, Franson KL, Nussbaum AM, Wang GS. The pharmacologic and clinical effects of medical cannabis. Pharmacotherapy. 2013;33(2): 195–209. [DOI] [PubMed] [Google Scholar]
- 70.Oláh A, Szekanecz Z, Bíró T. Targeting cannabinoid signaling in the immune system: ‘high’-ly exciting questions, possibilities, and challenges. Front Immunol. 2017; 8: 1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hedderich JB, Persechino M, Becker K, Heydenreich FM, Gutermuth T, Bouvier M, et al. The pocketome of G-protein-coupled receptors reveals previously untargeted allosteric sites. Nat Commun. 2022; 13(1): 2567. [DOI] [PMC free article] [PubMed] [Google Scholar]