

Abstract
Proteolysis-targeting chimeras (PROTACs) are heterobifunctional small molecules that induce the ternary complex formation between a protein-of-interest (POI) and an E3 ligase, leading to targeted polyubiquitination and degradation of the POI. Particularly, PROTACs have the distinct advantage of targeting both canonical and noncanonical functions of epigenetic targets over traditional inhibitors, which typically target canonical functions only, resulting in greater therapeutic efficacy. In this review, we methodically analyze published PROTAC degraders of epigenetic writer, reader, and eraser proteins and their in vitro and in vivo effects. We highlight the mechanism of action of these degraders and their advantages in targeting both canonical and noncanonical functions of epigenetic targets in context of cancer treatment. Furthermore, we present the future outlook for this exciting field. Overall, pharmacological degradation of epigenetic targets has emerged as an effective and attractive strategy to thwart cancer progression and growth.
Introduction:
Epigenetics, first coined by Waddington in 1942, is characterized as the inherited changes in gene expression without any alterations in the DNA sequence.1 Over the years, seminal scientific discoveries have been made, which shed more light in the epigenetics space. Some of these discoveries include understanding the role of DNA methylation with gene suppression,2 crystal structure of nucleosome core particle with DNA,3 role of histone modification with gene activity4 and crystal structure of histone bromodomain.5 In 2000, the “histone code hypothesis”, which refers to that differential patterns of histone modifications can dictate specific biological outcomes by recruiting downstream effector proteins, was proposed.6, 7
The epigenetic effector proteins can be divided into three classes based on their activity and roles. Epigenetic ‘writers’ such as histone acetyltransferases (HATs) and histone methyltransferases (HMTs) can enact reversible epigenetic marks such as acetylation and methylation on histones and/or DNA, resulting in chromatin restructuring and alteration in gene transcription. These marks are recognized by epigenetic ‘readers’ which are primarily chromatin modulating proteins containing bromodomains, chromodomains and/or Tudor domains. Furthermore, the marks can be removed by epigenetic ‘erasers’ such as histone deacetylases (HDACs) and lysine demethylases (KDMs). Dysregulation or aberrant expression of these epigenetic modifying proteins contribute to changes in transcriptional activation or repression, leading to distinct biological effects such as apoptosis and differentiation in many diseases such as cancer, neurological disorders and inflammation.8, 9
Presently, multiple drugs targeting epigenetic proteins are under clinical investigations for cancer and several have been approved by the United States Food and Drug Administration (FDA). For example, two DNA methyltransferase (DNMT) inhibitors, 5-Azacytidine (Azacitidine) and Decitabine, have been approved for the treatment of myelodysplastic syndrome,10 and two HDAC inhibitors, Vorinostat and Romidepsin, for the treatment of cutaneous T cell lymphoma.11 Recently, Tazemetostat (EPZ-6438), an inhibitor of enhancer of zeste homolog 2 (EZH2), which is the enzymatic subunit of polycomb repressive complex 2 (PRC2) that catalyzes trimethylation of histone H3 lysine 27 (H3K27me3), has been approved as the first EZH2-targeting drug for the treatment of patients with metastatic or locally advanced epithelioid sarcoma and follicular lymphoma.12 However, it is well-established that cancer cells can acquire adaptive resistance towards inhibitors that results in cancer relapse and refractory to current therapies.13 Furthermore, in addition to canonical activity, many epigenetic targets can also possess non-canonical functions. For example, while EZH2 primarily serves as the catalytic subunit of the PRC2 repressing gene expression, it can also activate cyclin D1 transcription by binding to its promoter region in natural killer/T-cell lymphoma (NKTL) independent of its catalytic function.14 Though inhibitors of an epigenetic target, such as EZH2, could block it’s catalytic activity, they will not thwart the non-canonical activity of the protein.15 Therefore, innovative strategies with novel mechanism of action (MOA) are needed for effective cancer therapy. One of such ground-breaking and pioneering technologies is the proteolysis targeting chimeras (PROTACs) for the treatment of cancer.
PROTACs are heterobifunctional molecules, which contain two linked moieties with one ligand binding to the POI and the other ligand recruiting E3 ubiquitin ligase, offer a novel platform for chemically-induced degradation of protein of interest (POI) (Figure 1).16–18 PROTACs can effectively induce the polyubiquitination of POI and its subsequent proteasomal degradation.16 Several structural studies on ternary complex of BRD4-PROTAC-Cullin–RING E3 ligases revealed U-shaped complexes facilitating targeted ubiquitin transfer by the proximal positioning of the ubiquitin conjugated-E2 and POI (Figure 1).19, 20 One of the main advantages of PROTACs over the traditional small-molecule inhibitors is that PROTACs are catalytic in their nature and can completely degrade the target protein and be effectively recycled back, removing the need for continuous drug treatment. Another advantage of PROTACs is the possible selective degradation of target protein even though the parental inhibitor used as POI ligands lack selectivity.19, 21–23 Examples of such PROTAC selectivity is demonstrated by kinase degraders which can selectively degrade HER2 over EGFR and vice versa.24 Furthermore, DT2216, utilizing a BCL-2 and BCL-XL dual inhibitor ABT263, selectively degrades BCL-XL over BCL-2.25 Unlike traditional small molecule which binds to active site of a protein, and are occupancy-driven, PROTACs operate by event-driven MOA and can effectively degrade proteins lacking a catalytic activity such as scaffolding proteins. Thus, PROTACs could potentially overcome some point-mutation based drug resistance and can also target the non-catalytic activity of epigenetic proteins.26 Recently, PROTACs have garnered significant interest from the biomedical and drug discovery community as an emerging class of novel therapeutic approaches. Several excellent reviews on PROTACs of epigenetic targets were previously published.27, 28 Herein, we provide a systematic and comprehensive review of the published degraders of epigenetic targets including the most recent PROTACs and technologies that have not been reviewed previously, complementing the previous reviews on this subject (supplemental summary table 1). Particularly, we describe the studies underlying the advantage of degraders targeting both the canonical and non-canonical functions for epigenetic targets as well as the studies where PROTACs targeting undruggable epigenetic protein complexes. We also describe new methodologies/technologies expanding the field.
Figure 1:
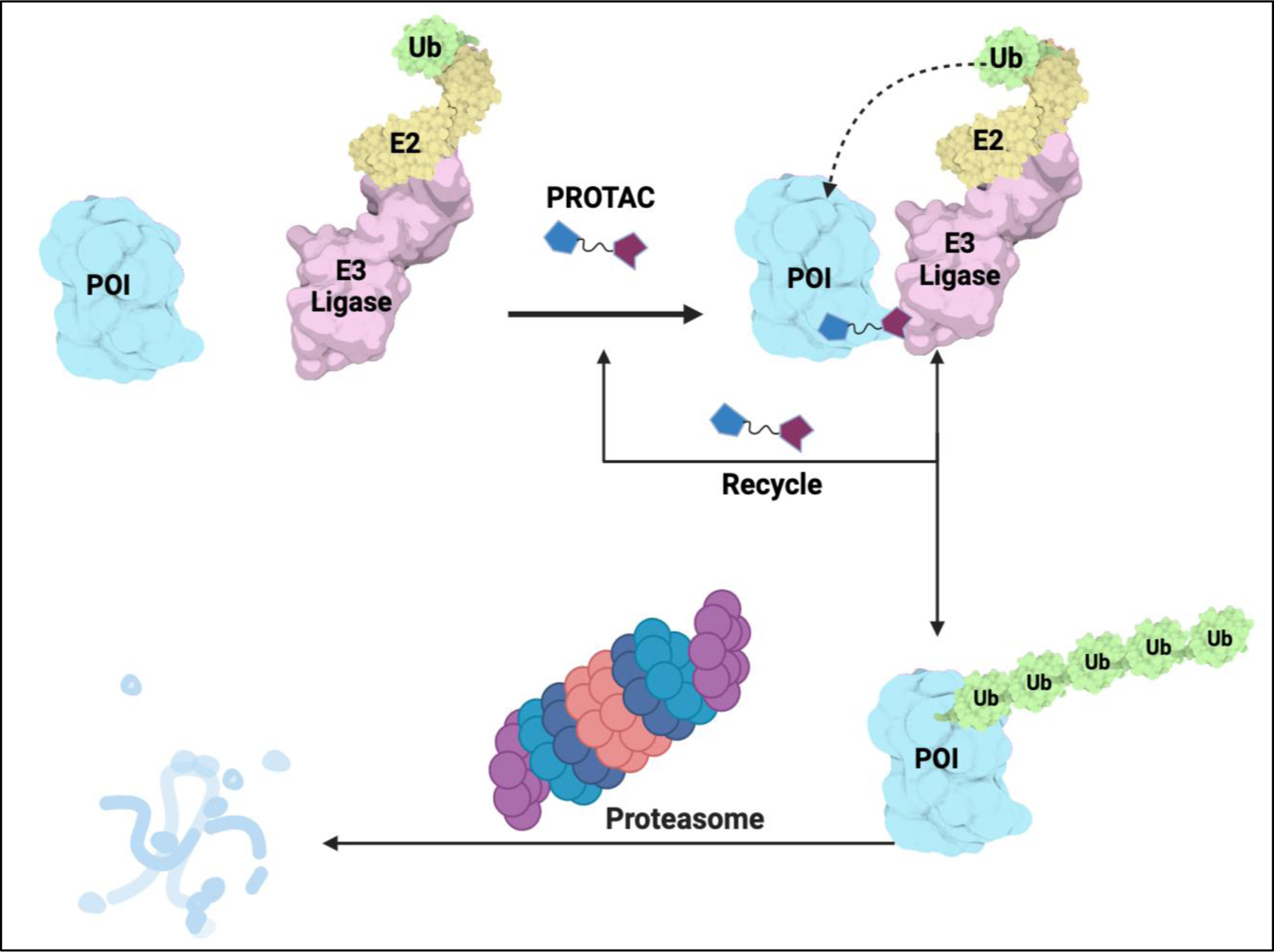
Schematic of Proteolysis targeting chimera (PROTAC). Figure generated using Biorender
1. Degraders of Epigenetic Writers:
1.1. EZH2 degraders
As one of the most important HMTs implicated in cancer, EZH2 is the main catalytic subunit of the PRC2 alongside with embryonic ectoderm development (EED) and suppressor of zeste 12 protein homolog (SUZ12) and regulate gene expression and tumorigenesis.29 PRC2 catalyzes the trimethylation of histone 3 lysine 27 residue (H3K27me3) via the EZH2 SET domain in its C-terminus and regulates gene expression, differentiation, and development. Non-canonical functions that are independent of methyltransferase activity of EZH2 have been associated with transcriptional activation such as the activation of the cMyc and cyclin D1 genes.30 It was reported that overexpression of EZH2 is correlative of poor prognosis in breast and prostate cancer as well as lymphoma and non-small cell lung cancers.31, 32 There are several S-5’-adenosyl-L-methionine (SAM)-competitive EZH2 inhibitors currently in clinical trials for the treatment of cancer. However, to date, only Tazemetostat (EPZ6438) was approved by FDA for the treatment of patients with metastatic or locally advanced epithelioid sarcoma and follicular lymphoma.33 Therefore, effective therapeutic strategies are still needed and recently degraders targeting EZH2, with the potential for limiting both the canonical and non-canonical roles of EZH2 have emerged.
Ma et al. was the first to report an EZH2 degrader, MS1943, which was synthesized by linking the solvent exposed piperazine moiety of C24, an EZH2 inhibitor,34 to a bulky adamantyl group with a short carbon linker (Figure 2).35 MS1943, a hydrophobic tagged degrader, differs from typical heterobifunctional PROTAC degraders discussed in this review. MS1943 induces EZH2 degradation most probably by prompting EZH2 misfolding or unfolding and activating the unfolded protein response. It was shown that MS1943 can effectively degrade EZH2, which led to the potent growth inhibition with a concentration for 50% of maximal inhibition (GI50) of 2.2 μM in triple negative breast cancer (TNBC) cell line MDA-MB-468. Interestingly, while the H3K27me3 mark was reduced more effectively by EZH2 inhibitors GSK126, CPI-1205 and EPZ6438 as well as C24 compared to the MS1943, the EZH2 inhibitors failed to induce growth inhibition and apoptosis. On the other hand, MS1943 was able to inhibit growth and induce apoptosis in MDA-MB-468 cells, suggesting that effects of MS1943 are largely due to its on-target EZH2 degradation effect. Furthermore, intraperitoneal (i.p.) administration of MS1943 at a dose of 150 mg/kg treatment reduced tumor growth in MDA-MB-468 breast cancer xenograft model and degraded EZH2 and lowered H3K27me3 in vivo establishing pharmacokinetic and pharmacodynamic (PK/PD) relationship with no adverse effects on mice tested. MS1943 was shown to induce the unfolded protein response (UPR), via RNA-seq experiments. Degradation of EZH2 by MS1943 suggests that pharmacological degradation of EZH2 is a potential therapeutic strategy and EZH2 degraders could be effective agents for treating TNBC.35
Figure 2:
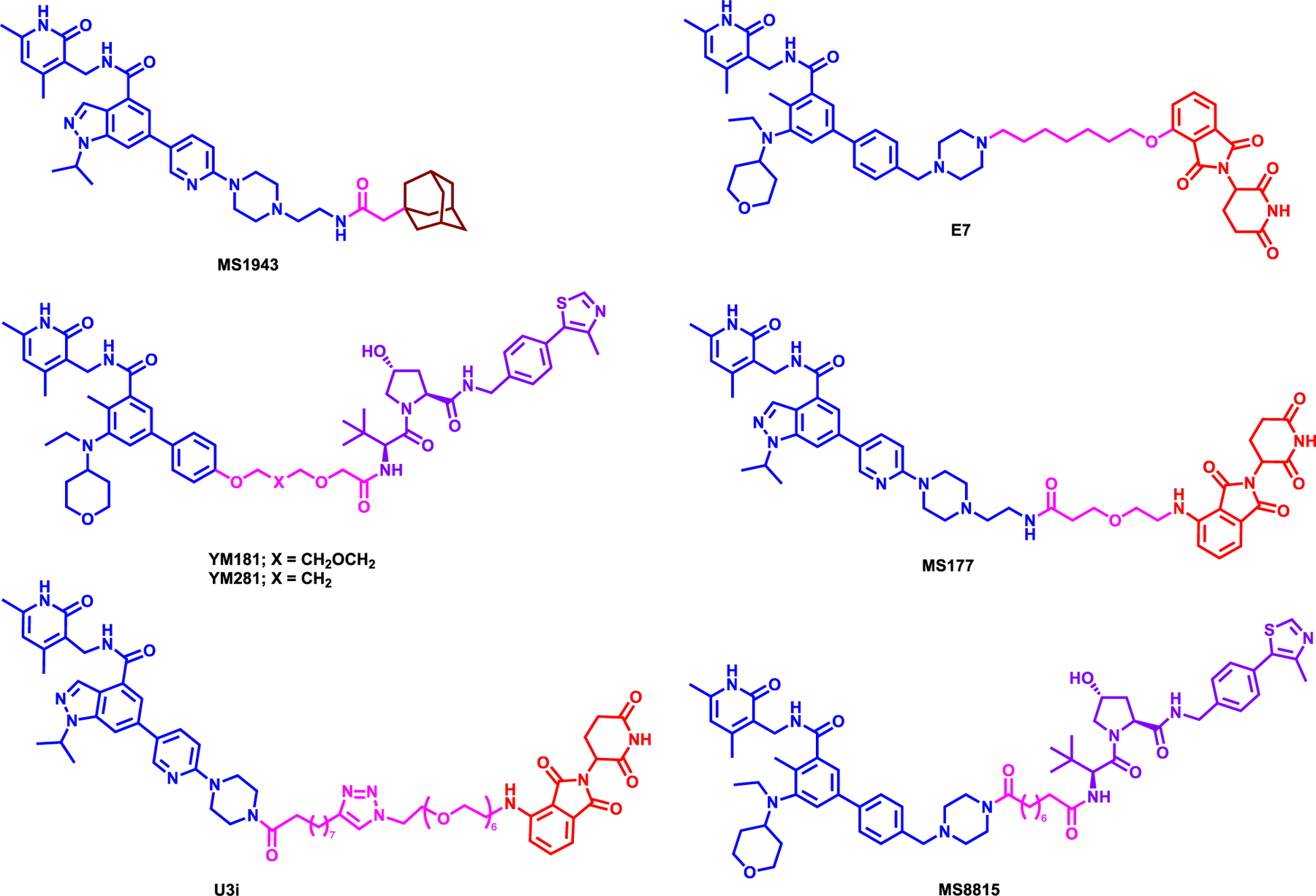
Structure of EZH2 degraders
Liu et al. was the first to report a bifunctional EZH2 PROTAC degrader E7, which utilized and modify the morpholine moiety of EPZ643836 and linked it to 4-hydroxythalidomide with a 7-carbon linker (Figure 2).37 Through a structure-activity relationship (SAR) study, the group demonstrated that GSK126 based PROTACs required longer carbon linker (8–12 carbons) to show EZH2 degradation in WSU-DLCL-2 cells. In addition, EPZ6438-based degrader with 4-carbon linker also showed PRC2 degradation, but no further studies were performed. After SAR study, Liu et al. selected E7, which showed time and dose-dependent degradation of PRC2 subunits EZH2, SUZ12, EED and RbAp48 and displayed potent reduction of H3K27me3 mark at 1 μM. Furthermore, this work demonstrated that the degradation is mediated through the Ubiquitin-Proteosome System (UPS) by inhibiting the neddylation and proteasomal degradation mechanisms. Overall, E7 was more efficacious than both GSK126 and EPZ6438 in lung and lymphoma cancer cell lines at 10 μM, establishing its superiority in anti-proliferative activity over inhibitors for the treatment of cancer. However, further studies are needed to evaluate whether E7 can degrade zing-finger containing transcription factors such as IKZF1, IKZF3, and GSPT1, well-known neo-substrates for Immunomodulatory imide Drugs- Cereblon (IMiD-CRBN) complex.38
In a separate study, Tu et. al also utilized EPZ6438 by removing the morpholine moiety to directly link E3 ligands and synthesized both VHL- and CRBN-recruiting EZH2 degraders.39 Through a series of SAR, they have discovered that VHL-recruiting YM281 and YM181 (Figure 2) which showed maximum 80% EZH2 degradation efficacy at 2 μM. However, putative degraders that linked to thalidomide to recruit CRBN did not show any PRC2 degradation at the concentrations tested. It has been shown that YM181 reduced EED and SUZ12 levels as well as EZH2 levels, in 24 hours at 2 μM concentration in both SU-DHL-2 and 22Rv1 cells via UPS. Both YM181 and YM281 showed effective in vitro efficacy in lymphoma cancer cell lines such as SU-DHL-2, −4 and −6 and furthermore, YM281 demonstrated reduction of tumor growth in vivo, in a xenograft model of DLBCL cell line SU-DHL-6 by degrading EZH2 at 80 mg/kg i.p dosage. Despite the overall efficacy of these EZH2 degraders, further studies are needed to show the degradation kinetics of the PRC2 components.39
More recently, Wang et al. reported MS177, which was synthesized by conjugating EZH2 inhibitor C24 to pomalidomide via a short PEG linker (Figure 2).15 MS177 was shown to be more potent than the parent compound C24 in EZH2 inhibition with an IC50 of 7 nM, reducing global H3K27me3 levels at 1 μM and degrading EZH2 with a half-maximal degradation concentration (DC50) of 200 nM in MV4;11 cell line as well as the other PRC2 complex core components. Particularly, it was shown that MS177 induces ubiquitination of EZH2 in dose- and time-dependent manner via UPS and highly selective for EZH2 compared to around 100 proteins including other methyltransferases and common drug targets. Furthermore, RNA sequencing (RNA-seq) profile of MS177 coincided with EZH2 knockout and gene set enrichment analysis (GSEA) revealed that MS177 stimulates the PRC2 target genes. Importantly, in this study, Wang et al. demonstrated that EZH2 interacts with cMyc via a hidden transactivation domain (TAD) and promotes oncogenesis via upregulation of TPD52 and IRF2BPL in leukemia cell lines. Particularly, MS177 can degrade cMyc through EZH2 and downregulates AML-related genes. MS177 demonstrated significant pharmacokinetic and pharmacodynamic relationships in vivo and suppressed the in vivo oncogenesis in multiple tumor cell line xenograft and PDX models and enhanced overall survival without adverse effects. MS177, by effectively suppressing both canonical and non-canonical oncogenic activities of EZH2, displayed superior growth inhibition of MLL-rearranged leukemia cells both in vitro and in vivo compared to EZH2 catalytic inhibitors. These findings clearly underscore the advantages of pharmacological degradation of EZH2 by PROTACs over the pharmacological inhibition of EZH2 for the treatment of MLL-rearranged leukemias and possibly for other EZH2-dependent diseases as a therapeutic strategy.
Wang et al. reported EZH2 degrader U3i, which was obtained by linking the piperazine moiety of EZH2 inhibitor C24 with a long PEG linker and conjugating with pomalidomide (Figure 2).40 U3i inhibited the cell growth of TNBC cell lines MDA-MB-231 and MDA-MB-468 with GI50 of 570 and 380 nM, respectively, while maintaining low toxicity in normal mammary, renal and hepatocyte cells in cell viability assays. U3i had a high binding affinity towards EZH2 (KD = 16.2 nM) and induced the degradation of EZH2, SUZ12 and EED at 1 μM. The degradation of the PRC2 complex was observed in a dose- and time-dependent manner and via the UPS. U3i displayed selectivity for PRC2 complex components in global proteomics studies. Overall, U3i displayed better anti-proliferative activity in TNBC cells compared to GSK126 and EPZ6438. However, further studies are needed to evaluate the neo-substrates degradation profile, such as for IKZF1, IKZF3, and GSPT1, by U3i.40
Dale et al. reported, EZH2 degrader MS8815 and investigated its effects in TNBC. MS8815 was synthesized by replacing the morpholinyl moiety of EPZ643836 with the piperazine moiety and linking it to VHL1 ligand through a 8-carbon linker (Figure 2).41 MS8815 induced potent EZH2 degradation at 300 nM in TNBC cell lines MDA-MB-453 and BT549. While MS8815 inhibited EZH1 and EZH2 catalytic activity with an IC50 of 62 and 8.6 nM, respectively, MS8815 did not degrade EZH1 in the MDA-MB-453 cell line. Furthermore, MS8815 degraded EZH2 with a DC50 of 140 nM and maximal degradation (Dmax) was observed at 48 h of treatment. Additional rescue experiments demonstrated that MS8815 induced EZH2 degradation via the UPS pathway. A comparison study between VHL-recruiting degraders MS8815 with YM281 revealed that MS8815 had a slightly better growth inhibition and EZH2 degradation in a series of TNBC cell lines such as BT549, SUM159, MDA-MB-453 and MDA-MB-468. However, further studies are needed to evaluate whether additional PRC2 core proteins degraded or the that target genes are regulated by the MS8815 treatment.41
These PROTACs targeting EZH2 discussed above collectively display that the pharmacological degradation of over its pharmacological inhibition is a very promising therapeutic strategy.
1.2. WDR5 degraders
WD repeat domain 5 (WDR5) is one of the important catalytic subunit of class 2 lysine methyltransferase (KTM2) comprising of the mixed-lineage leukemia family (MLL1, MLL2, MLL3, MLL4) and SET1A/SET1B enzymes.42 WDR5 forms a complex with MLL proteins alongside RBBP5, ASH2L, and DPY30 and function as the core subunit of the human MLL1–4 histone H3K4 methyltransferase complexes.43 WDR5 protein has seven WD40 repeat domains which forms a seven-bladed propeller fold, in which the WDR5 interaction (WIN) motif interacts with the SET domain of MLL H3K4 methyltransferase enzymes.44 It was shown that WDR5 overexpression is implicated in MLL-rearranged leukemia and pancreatic cancer.45, 46 There are several reported WDR5 inhibitors such as OICR-9429 or MM-401 which showed anti-tumor effects in vitro, however they failed to show potent in vivo efficacy.47, 48 Therefore, novel, and innovative therapeutic strategies are necessary for impeding WDR5 functionality in vivo.
Dölle et al. reported a first-in-class WDR5 degrader, compound 8g which induced robust degradation of WDR5 by using a modified version of WDR5 inhibitor OICR-942949 and linking it to VHL1 via 4-carbon aliphatic linker (Figure 3).50 An extensive SAR campaign was performed to better understand the effect of various linker size and E3 ligases, VHL, CRBN and MDM2. Using a WDR5-HiBiT stable MV4;11 cell line, the DC50 of compound 8g was determined to be 56 nM with a Dmax of 58%. In the same study a pyrroloimidazole-based degrader compound 17b which featured 2 PEG linker, designed to have a different linker exit position than that of 8g. However, compound 17b (Figure 4) showed a less effective degradation of WDR5 with DC50 of 1.24 μM and a Dmax of 53%. Western blot analysis to check the endogenously expressed and untagged WDR5 displayed similar degradation effect compared to HiBit-tagged WDR5 for both compounds. Furthermore, the selective degradation of WDR5 by both compounds were shown via mass-spectrometry based proteomics. However, either degrader 8g or 17b did not induce potent cell growth inhibition in MV4–11 cells. A computational docking study of WDR5 and VHL with compound 8g suggested that WDR5 and VHL do not form a dominant ternary complex and can exist in different ternary complex configurations in solution, but the hypothesis was not validated with an actual crystal structure.
Figure 3:
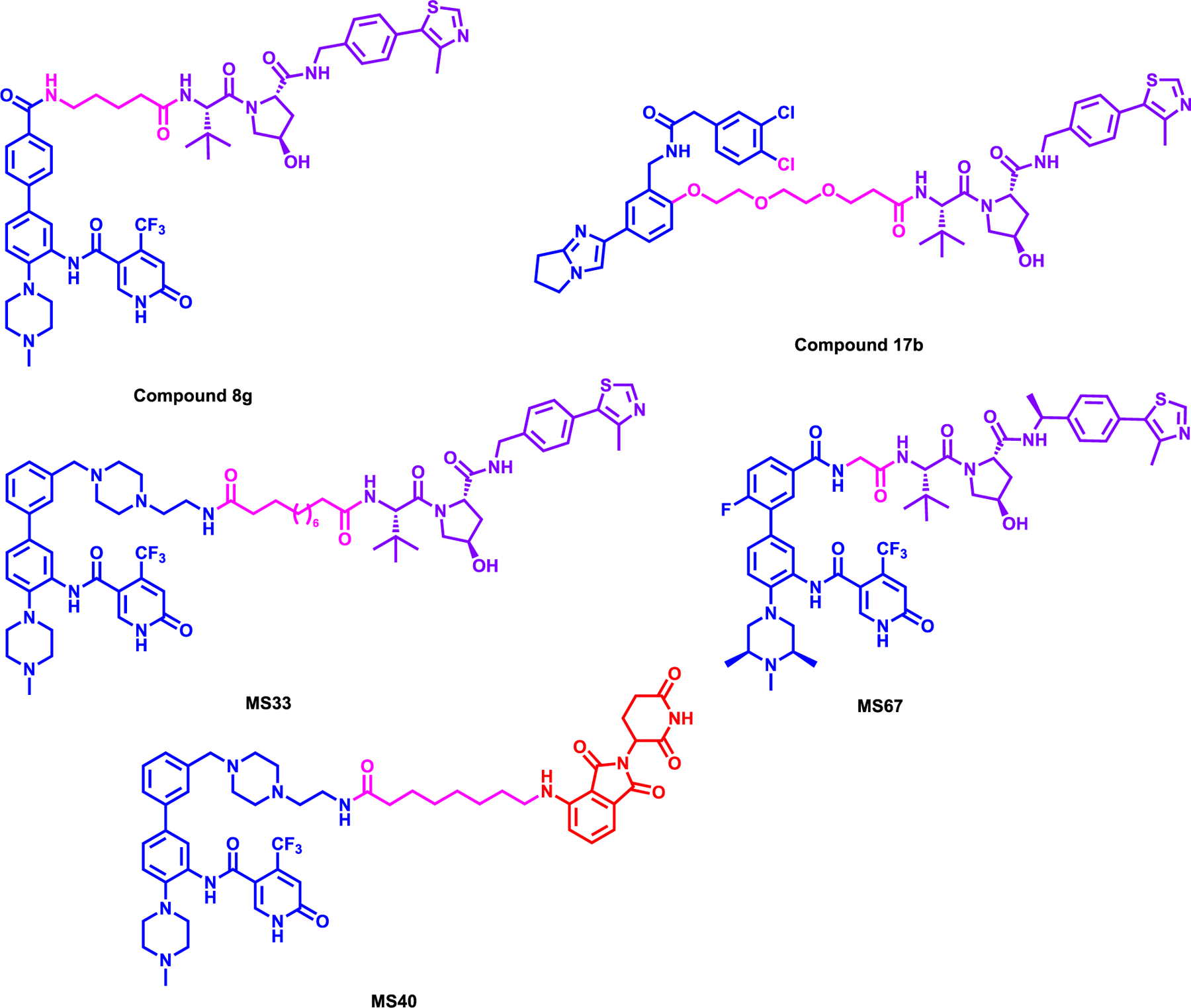
Structures of WDR5 degraders
Figure 4:
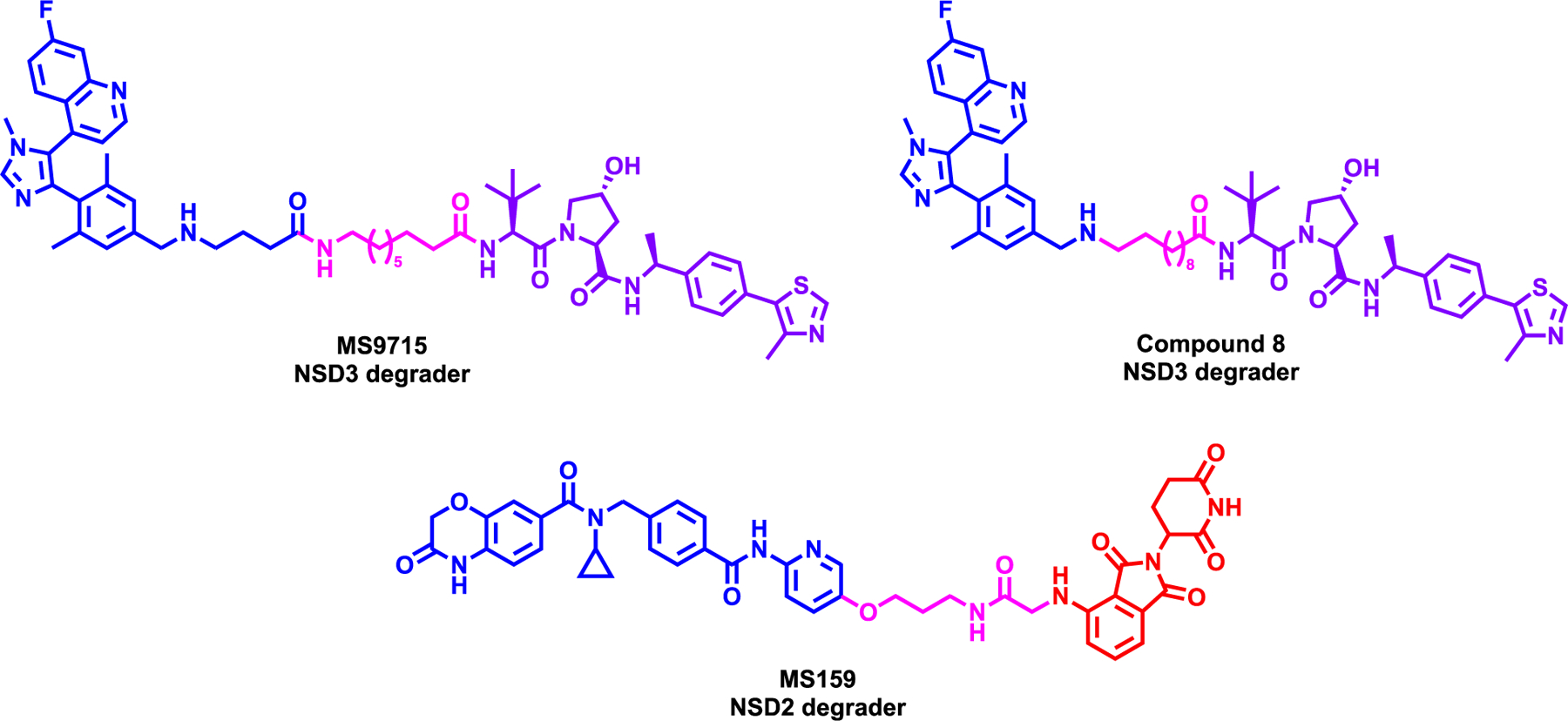
Structure of NSD3 degraders MS9715 & compound 8 (top) and NSD2 degrader MS159 (bottom)
Next, Yu et al. reported a WDR5 degrader, MS33 which was synthesized by replacing the morpholine group with piperazine of OICR-9429 and linking it to VHL ligand using a relatively long carbon linker (Figure 3).51 It was shown that MS33 induced WDR5 degradation with a DC50 of 260 nM with a maximum degradation 71% in MV4–11 cells. Further biophysical characterization led them to solve the WDR5-MS33-VHL-ElonginB-ElonginC (VBC) ternary complex structure (PDB: 7JTO) and determined that the long linker in MS33 resulted in a minimum protein-protein interface and with very limited “cross” protein-ligand interactions. Taking advantage this ternary complex structure, via a structure based PROTAC design, a new degrader MS67 was synthesized featuring a much shorter linker without piperazinyl group of MS33. In addition, further modifications were made to WDR5 and VHL binding ligands to potentially increase the binding affinities to WDR5 and VHL, respectively. The solved ternary complex structure of WDR5-MS67-VCB (PDB: 7JTP) clearly showed the more extensive VHL-WDR5 interface and the cross protein-ligand interactions which are resulted in increased cooperativity of the WDR5-MS67-VCB complex. MS67 had a DC50 of 3.7 nM and achieved a Dmax of 94% in MV4;11 cells. Selective WDR5 degradation was also demonstrated by a mass-spectrometry based proteomics study as well as selectivity against other methyltransferases and broad panel of common drug targets. Additionally, MS67 showed potent growth inhibition in a pane of MLL-rearranged cell lines and pancreatic ductal adenocarcinoma (PDAC) cells in vitro suppressed transcription of WDR5-regulated genes. MS67 effectively reduced the growth of primary cancer cells from patients with AML and suppressed tumor growth in vivo in MLL-r AML xenograft and PDX models and was well tolerated. MS67 also prolonged the survival of mice bearing MLL-r AML PDX while the parent inhibitor OICR-9429 failed to do so. However, it was shown that MS67 was insensitive towards RS4;11 and THP-1 cell lines and exhibited variation in its sensitivity to MLL-r AML cell lines and patient samples therefore, further studies are needed to examine these variations. This study underlined the power of ternary structures to facilitate a rational, structure based PROTAC design.
Li et al. from the same group that published MS33 and MS67, reported a first-in-class dual WDR5 and Ikaros degrader, MS40. This degrader was synthesized by conjugating the modified morpholine moiety of OICR-9429 with pomalidomide via a long carbon chain linker and differed from the above mentioned WDR5 degraders by recruiting CRBN rather than VHL (Figure 3).52 In a large panel of cell lines covering MLL-r AML and breast cancers MS40 robustly induced the degradation of WDR5 and IKZF1/3 in a dose- and time-dependent manner. Degradation of WDR5 and well known neosubstrates of IMiD;CRBN, IKZF1/3 was confirmed via unbiased MS-based quantitative proteomics studies. MS40 displayed GI50 of 180, 510 and 540 nM in MV4;11, EOL1, and RS4;11 cell lines, respectively, whereas it was insensitive in K562 cells, a non-MLL-r leukemia line suggesting the lack of general toxicity. MS40 also reduced the global H3K4me2 mark and reduced the chromatin bound MLL complex components. MS40 simultaneously, reduced WDR5 and CRBN target genes such as RUNX3 and SMAD3 Finally, it also effectively reduced tumor growth in vivo in PDX models at a dose of 100 mg/kg via i.p. treatment. Overall, MS40 can be utilized as a useful chemical probe to further investigate dual WDR5 and IKZF1/3 degradation as a therapeutic strategy in cancer.
1.3. NSD2 & NSD3 degraders
The Nuclear receptor binding SET domain protein (NSD) family of methyltransferases, catalyzing the mono- and di-methylation of lysine 36 on histone H3 (H3K36me1/me2), is primarily composed of three members, NSD1, NSD2 (also known as MMSET orWHSC1, and NSD3 (also known as KMT3F or WHSC1L1).53 NSD3 can exist in two isoforms, NSD3-short form (NSD3S) and NSD3-long form (NSD3L), the difference being the short isoform lacks the C-terminus SET domain.53 It was shown that NSD3S interacts with the extra terminal (ET) domain of the bromodomain-containing protein 4 (BRD4).54 Particularly, fusion between NSD3 and NUP98 is observed in AML and myelodysplastic syndrome, which induces the expression of proto-oncogenes such as HoxA7 and HoxA9.55 NSD3 overexpression and hyperactivity has also been associated with squamous cell lung cancer and breast cancer and was shown to inhibit PRC2 activity by elevating H3K36me2 and re-expressing MYC and Notch.56, 57 NSD2 is associated in the development of multiple myeloma (MM), predominantly in patients carrying a t(4,14) translocation that leads to aberrant upregulation of this gene.58 Furthermore, overexpression of NSD2 can spread the H3K36me2 mark to intragenic bodies and promote oncogenesis.59, 60 Therefore, both NSD2 and NSD3 are viable targets to inhibit the growth of cancer and garnered continuing interest as therapeutic targets. However, only limited progress has been made in developing selective inhibitors and thus, more effective therapeutic strategies are needed.
Xu et al. reported a first-in-class NSD3 degrader, MS9715, which utilized the selective antagonist of NDS3 H3K36me3/2-binding domain (PWWP1), BI-9321,61 by modifying the primary amino group with a spacer as a handle to attach VHL ligand, VHL1-Me (Figure 4, top left).62 PROTAC strategy for NSD3 was pursued because, while BI-9321 blocks the NSD3 PWWP1 domain, it does not target other functions of NSD3, and its inhibitory effect on the chromatin-reading function of the NSD3 PWWP1 domain does not result in effective killing of cancer cells. MS9715 induced the degradation of NSD3 in MOML13 cell line with a DC50 of 4.9 μM with Dmax > 80% and induced degradation at 5 μM in EOL-1 and MM1.S cell line via the UPS. Global proteomic profiling revealed the selectivity of MS9715 for NSD3, RNA-seq-based transcriptome profiling in EOL-1 and MM1.s cell lines demonstrated the downregulation of oncogenes such as CBLB, IFITM1, and MAP7 in agreement with NSD3 knockout experiment. Moreover, MS9715 effectively reduced the cell growth of several hematological cancer cell lines namely, EOL-1, RSV4;11 and MM1.S cells with a EC50 of 2.3, 3.5 and 3.3 μM, respectively, whereas the parent PWWP1 binder did not have any inhibitory activity. Furthermore, MS9715 was also shown to degrade cMyc at 2.5 μM in EOL-1 and MM1.S cell lines in a VHL dependent manner, again highlighting the advantage of degrader over parent inhibitor in targeting non-canonical functions of target protein.62
Next, Sun et al. reported a NSD3 degrader, compound 8, which was synthesized using the same NSD3 PWWP1 domain ligand BI-9321 and the same VHL ligand and linking them together directly from the terminal amino group of NSD3 binder with a 10-carbon alkyl chain (Figure 4, top right).63 Compound 8 had a DC50 of 1.43 μM and 0.94 μM in NCI-H1703 and A549 lung cell lines, respectively. Further rescue experiment with proteosome inhibitor supported the mechanism of degradation by the UPS pathway and compound 8 was shown to be selective towards NSD3 while sparing NSD2 and NSD1. Compound 8 reduced H3K36me3 levels in NCI-H1703 at 7.5 μM and in A549 at 10 μM treatment. Additional RNA-seq and western blotting studies have confirmed that compound 8 downregulated NSD3-associated genes such as CDC25A and Cyclin-D1. It also degraded NSD3 in A549 xenograft model with 100 mg/kg via single i.p. injection in 36 hours but tumor regression data was not reported. Additional proteomics and ChIP-seq studies would be desirable to validate the mechanism of action.
Meng et al. reported a first-in-class NSD2 degrader, MS159, which was synthesized by utilizing and modifying the solvent exposed pyrimidine ring of the NSD2-PWWP1 antagonist, UNC693464 and linking with CRBN ligand, pomalidomide (Figure 4, bottom).65 MS159 displayed potent NSD2 degradation at 10 M in 293FT cell line with a DC50 of 5.2 M and a DMax of 82%. The degradation of NSD2 starts at 48h while maximum degradation is observed at 72h. Further characterization revealed that the NSD2 degradation is mediated by UPS pathway, supported by no observed degradation in CRBN knockout 293FT cell line. Furthermore, MS159 also degraded IZKF1 and IZKF3 neo-substrates, however did not degrade GSPT1, which is one of the most concerning off-target of CRBN-based degrader.66 Furthermore, selectivity of MS159 against panel of 20 protein lysine and arginine methyltransferases was assessed and no inhibition was observed including NSD1/2/3 at 10 M. Finally, it was shown that MS159 has a more potent anti-proliferative activity than the parent compound UNC6934 in multiple myeloma cell lines KMS11 and H929 cell lines, consistent with NSD2 knockout in the same cell lines. This degrader was also bioavailable in mice without any adverse effect and could be used for in vivo studies in the future.
1.4. PRMT5 degraders
Protein arginine methyltransferase 5 (PRMT5) is a type II PRMT which catalyzes mono- and symmetric di-methylation by transferring the methyl groups from S-adenosyl-L-methionine (SAM) co-factor to a terminal nitrogen of protein arginine residue.67 PRMT5 forms a complex with methylosome protein 50 (MEP50) in the TIM barrel domain, followed by middle Rossman fold which aids in binding to SAM and for methyltransferase catalytic activity.68 PRMT5 overexpression has been observed in several solid tumors such as colon, breast, and prostate cancers as well as in hematologic malignancies.69 PRMT5 was shown to directly interacts with tripartite motif-containing protein 21 (TRIM21), prompting NFκB signaling in multiple myeloma.70 Overall, high expression of PRMT5 is correlated with poor cancer prognosis and has been explored as a potential therapeutic target.
Shen et al. reported the first-in-class PRMT5 degrader, MS4322, by exploiting the solvent-exposed oxetane moiety of a selective PRMT5 inhibitor, EPZ01566671 and connecting it to VHL ligand with modifying it to azetidine ring (Figure 5).72 MS4322 induced potent degradation of PRMT5 in a dose-, time-, and VHL-dependent manner with a DC50 of 1.1 μM and a Dmax of 74%. Moreover, MS4322 was slightly more effective in inhibiting the global symmetric arginine dimethylation (SDMA) than the parent compound, EPZ015666. MS4322 treatment led to potent MCF-7 cell growth inhibition as effective as the parent inhibitor. Finally, MS4322 was shown by global proteomic profiling to selectively degrade PRMT5 and was shown to be bioavailable and well-tolerated in in vivo pharmacokinetic (PK) study. However, MS4322 displayed similar potency with the parent inhibitor in anti-proliferation assay in other lung, cervical and leukemia cell lines. While this was the first example of an arginine methyl transferase PROTAC further optimization is warranted for discovery of more potent and effective degrader.
Figure 5:
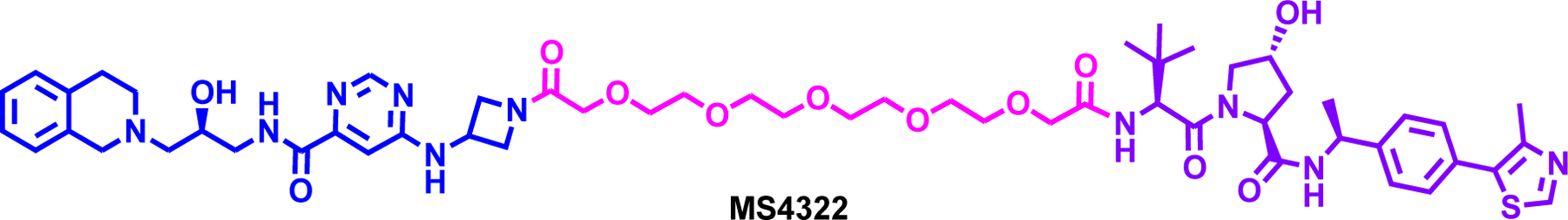
Structure of the PRMT5 degrader MS4322
1.1. KAT degraders
There are several classes of lysine acetyltransferases (KATs) that are known to acetylate lysine in histones and non-histone proteins which includes GNAT family (GCN5 and PCAF), p300/CBP, and the MYST family.73, 74 Particularly p300 and CBP (p300/CBP) shares a high sequence homology and include a bromodomain, catalytic HAT domain, followed by ZZ zinc finger, and TAZ2 domain.74, 75 On the other hand, GCN5/PCAF does not have ZZ zinc finger, TAZ2 and tandem plant homeodomain (PHD) domain.76 It was also shown that alteration in expression and activity of p300/CBP can contribute the progression of solid tumors and hematological malignancies by inducing histone 3 lysine acetylation (H3K27ac) and active recruitment of BRD4.77, 78 On the other hand, PCAF was shown to acetylate E2F1 which led to increased DNA-binding activity and protein stability.79 While selective inhibitors of several lysine acetyltransferase enzymes such as p300/CBP and PCAF/GCN5 family have been developed, inhibitors or chemical probes of PCAF/GCN5 family are still lacking. Therefore, better therapeutic approaches targeting KATs are needed to prevent cancer progression.73
Bassi et al. reported the first PCAF/GCN5 degrader, GSK699 (Figure 6), which was synthesized by utilizing inhibitor GSK4027 which effectively binds to the bromodomain of PCAF and GCN5.80 LPS-induced IL- 6 and TNF production was significantly reduced in PCAF knockout (KO) versus wild type (WT) cells. In addition, no significant effect for IL-12p70, IL-10, and IL-1β in KO macrophages validating the role for PCAF in mediating LPS-induced production of cytokines. However, inhibition of the PCAF/GCN5 bromodomains by GSK4027 was not able to phenocopy the PCAF KO to recapitulate immunomodulatory function of these proteins. Based on the co-crystal structure of GSK4027 bound to human GCN5 bromodomain, the 4-position of the pendant phenyl ring was utilized for linker attachment to recruit CRBN.81 GSK983, the cis-racemic mixture of GSK699, across the piperidine ring (Figure 6) degraded PCAF and GCN5 in THP-1 cell line with a DC50 of 1.5 and 3 nM respectively, with a “hook effect” at higher concentrations. On the other hand, GSK699, which is the cis-(R,R)-enantiomer (Figure 6) showed even better degradation efficiency at 10 nM in degrading PCAF, while GSK702, cis-(S,S)-enantiomer (Figure 6) only degraded PCAF about 35% at concentrations of 100 nM or higher. These results showed that most of the degradation activity was due to cis-(R,R)-enantiomer, GSK699 and the cis-(S,S)-enantiomer, GSK702 was used as a control for potential effects mediated by neosubstrates of CRBN. Furthermore, GSK699 induced a potent reduction of cytokine levels such as IL-6, CXCL1 and TNF in monocyte-derived dendritic cells and macrophages, recapitulating an anti-inflammatory KO phenotype. However, global proteomics and ChIP-seq were not performed to better understand the modulation of gene expression after degrader treatment. This study was another example where the epigenetic inhibitor was not effective inducing biological effects while PROTAC strategy was successful.
Figure 6:
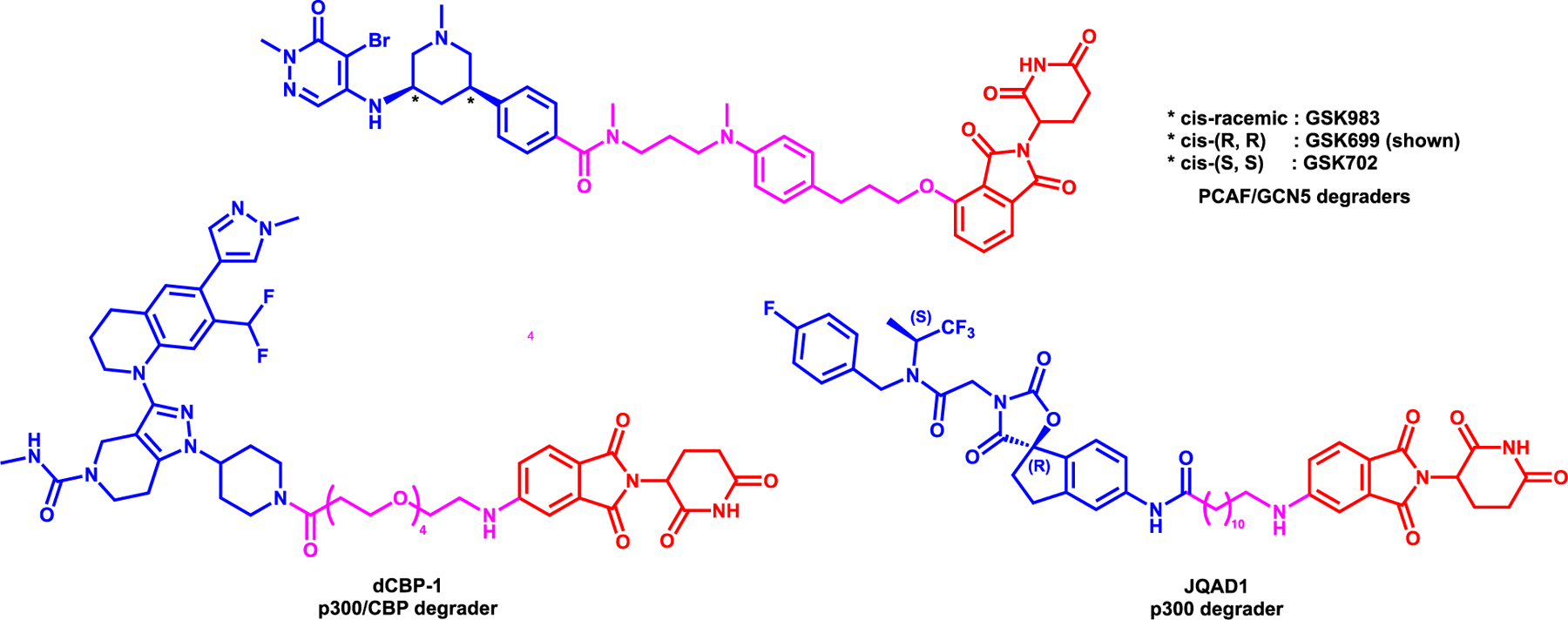
Structures of PCAF/GCN5 degraders (top) and p300 degraders (bottom)
Vannam et al. reported the p300/CBP degrader, dCBP-1, which was synthesized utilizing GNE-781, a selective and potent p300/CBP bromodomain inhibitor.82 The bromodomain inhibitor, the linker attachment point as well as the linker were decided based on a in silico study and dCBP was designed and synthesized by replacing the tetrahydropyran ring with a piperidine and linking it to thalidomide with a 4-PEG linker (Figure 6).83 The degrader, dCBP-1 showed robust degradation of both CBP and p300 at 10 nM at 6 hours treatment and induced complete degradation within an hour with 250 nM treatment via the UPS in HAP1 haploid cells. The global proteomic study in HAP1 cells showed that both p300 and CBP were among the most downregulated proteins after 3 hours of 250 nM of dCBP-1 treatment and importantly no appreciable loss of GSPT1, one of the most concerning neosubstrates of IMiD-CRBN complex, was observed. Furthermore, dCBP-1 showed potent anti-proliferative effect on multiple-myeloma cell lines compared to the CBP/p300 inhibitors A-485 and GNE-781 as well as pomalidomide. In addition, transcriptomic and proteomic profile revealed that dCBP-1 induced the reduction of Myc oncogene gene expression and protein levels. Further ChIP-seq analysis proved that dCBP-1 indeed abolished the H3K27ac mark levels while inhibitors slightly reduced acetylation level. Finally, by Assay for Transposase-Accessible Chromatin with high-throughput sequencing (ATAC-seq), it was shown that dCBP-1 induced degradation of CBP and p300 led to substantial loss of accessible chromatin at the regulatory site of the Myc promoter, confirming the complete removal of enhancer activity.
Furthermore, a novel p300 degrader, JQAD1, which was synthesized by using A-485, a selective inhibitor of p300/CBP84 and conjugating with pomalidomide with a 12-carbon linker (Figure 6), was reported by Durbin et al.85 JQAD1 induced potent p300 degradation and reduction of H3K27ac mark dose dependently at 24-hour treatment. On the other hand, CBP levels were not significantly affected by JQAD1 at the same time point and the same concentration compared to p300. The selectivity of JQAD1 for p300 degradation over CBP in Kelly neuroblastoma (NB) cell line, were corroborated by pull down assays with biotynlated-JQAD1, as well as stable isotope labeling of amino acids in cell culture (SILAC) analysis. Furthermore, it was shown that the efficacy of JQAD1 is CRBN-mediated by CRISPR knockout studies and competition assays with CRBN, p300 ligands and proteosome inhibitor. ChIP-seq studies have shown that JQAD1 reduced global H3K27ac mark in typical and super enhancer regions. JQAD1 was bioavailable and 40 mg/kg daily i.p. injection effectively reduced Kelly NB growth in vivo. Interestingly, it was shown by immunoprecipitation studies that p300, not CBP, interacts with cMyc to repress apoptosis and JQAD1 rapidly reduces the MYCN expression level compared to normal inhibition of p300 alone. Therefore, JQAD1 displayed a potent antiproliferative effect in MYCN amplified cancer cells and reduced Myc target genes in vivo. However, further studies are needed to fully describe the degradation mechanism and kinetics of p300 and MYCN. Moreover, this study is another example where the selectivity between highly homologous sub-family members of proteins could be achieved via PROTAC approach.
2. Degraders of Epigenetic Readers:
2.1. BET Family degraders
The Bromodomain and Extra-Terminal Domain (BET) family contains two tandem bromodomains (BD1 and BD2) in the N-terminus and an extra-terminal domain (ET) in the C-terminus, which helps with the recognition of the acetylated lysine.86, 87 There are four BET proteins, BRD2, BRD3, BRD4 and BRDT that has conserved amino-acid sequence forming 4 α-helices, which aids in binding the acetylated lysine residues in histones.86 Aside from histone recognizing functions, BRD-containing proteins also play a role in gene regulation and transcription modulators as part of chromatin remodeling complex.88 Mutations, dysregulation and chromosome translocation of BRD-containing proteins and genes have been discovered in several cancers such as hepatocellular carcinoma, T cell lymphoma, squamous cell lung cancer and colorectal cancer.89–91 Particularly, BRD4 was shown to form a complex with nuclear protein in testis (NUT) (BRD4-NUT) and induce pro-survival gene expression in testis (NUT) midline carcinoma.92 Therefore, inhibition of BET proteins showed initial success in NUT midline carcinomas.93 Presently, there are several BET inhibitors such as CPI-0610, OTX015 and TEN010 undergoing clinical trials either as a monotherapy or combination therapy for various cancers.94 However, resistance to BET inhibition results in reactivation of MYC in leukemia and TNBC by altering the WNT signaling pathways.95, 96 Therefore, novel therapeutic strategies are needed to overcome BET inhibitors resistance and thwart cancer growth.
BET proteins are one of the most targeted family utilizing PROTACs. There are comprehensive, recent reviews of BET protein degraders in literature.97, 98 In this review, we will only give a non-comprehensive highlights of BET protein degraders and we sub-grouped the degraders as the VHL or CRBN-recruiting PROTACs. The VHL-based BRD4 degraders include but not limited to, MZ199, ARV-771100, AT119, and MZP-54101 (Figure 7). The first VHL-based BET degrader, MZ1, reported by the Zengerle et al., was synthesized by linking the t-butyl ester moiety of (+)-JQ1 with VHL1 ligand via 3-PEG linker (Figure 7).99 MZ1 induced almost complete BRD4 (both short and long isomers) degradation at 100 nM after 24 hours of treatment in HeLa cells while near complete degradation for BRD2/3 was achieved only at 1 μM. Complete BRD4 degradation by MZ1 was observed as early as 4-hour with 1 μM treatment. Finally, a combination therapy approach of targeting SOX2-BRD4 complex using MZ1 alongside Smoothened (SMO) inhibitor MRT-92 showed in vivo efficacy in a subset of melanoma xenograft models.102 Importantly, in 2017, the same group disclosed the crystal structure of the BRD4BD2-MZ1-VHL:ElonginC:ElonginB (VCB) (PDB: 5T35), the first ternary complex structure of a PROTAC.19 Gadd et al. also reported a BRD4-selective degrader, AT1, which was designed based on the ternary structure of BRD4BD2-MZ1-VCB (PDB: 5T35) and features (+)-JQ1 linked to tert-Leu group of VH032 with a relatively shorter carbon linker.19 Comparison of the binary and ternary complexes by ITC revealed that, among the BET bromodomains, BRD4BD2 formed the most cooperative (α = 7) ternary complex with AT1. After 24 hours treatment, AT1 at 1–3 μM, selectively degraded BRD4 long and short isoforms while sparing BRD2 and BRD3 in HeLa cells. This was the first study reporting a ternary complex structure of a PROTAC bound to both the target protein and E3 ligase and measuring cooperativities of ternary-complex formation in solution using ITC. Next, Raina et al. reported, ARV-771, which again featured (+)-JQ1 linking it to VHL1-Me ligand, shown to degrade BRD2, 3, and 4 with a DC50 of less than 5 nM in various castration-resistant prostate cancer (CRPC) cell lines such as 22Rv1, VCaP and LnCaP.100 Furthermore, ARV-771 was shown to induce apoptosis in CRPC by attenuating both full length androgen-receptor (AR) (FL-AR) and AR variant AR-V7 levels. In addition, ARV-771 showed in vivo efficacy by degrading BRD4 and suppressing cMyc in 22Rv1 and VCaP tumor xenograft model. Another BET degrader, MZP-54 which was synthesized by using 3-PEG linker to connect VHL1 with the tetrahydroquinoline-based BET inhibitor I-BET726 was reported by Chan et al. differing from previously published JQ1 based VHL-recruiting degraders.101 MZP-54 induced potent BRD4 short and long-isoform degradation alongside BRD3 while sparing BRD2. MZP-54 showed anti-proliferative activity in AML cell lines and suppressed cMyc expression. However, exhibiting lower cooperativity of ternary complex formation, MZP-54 induced less effective depletion of both BRD4 and cMyc than MZ1 in MV4–11 cells.
Figure 7:
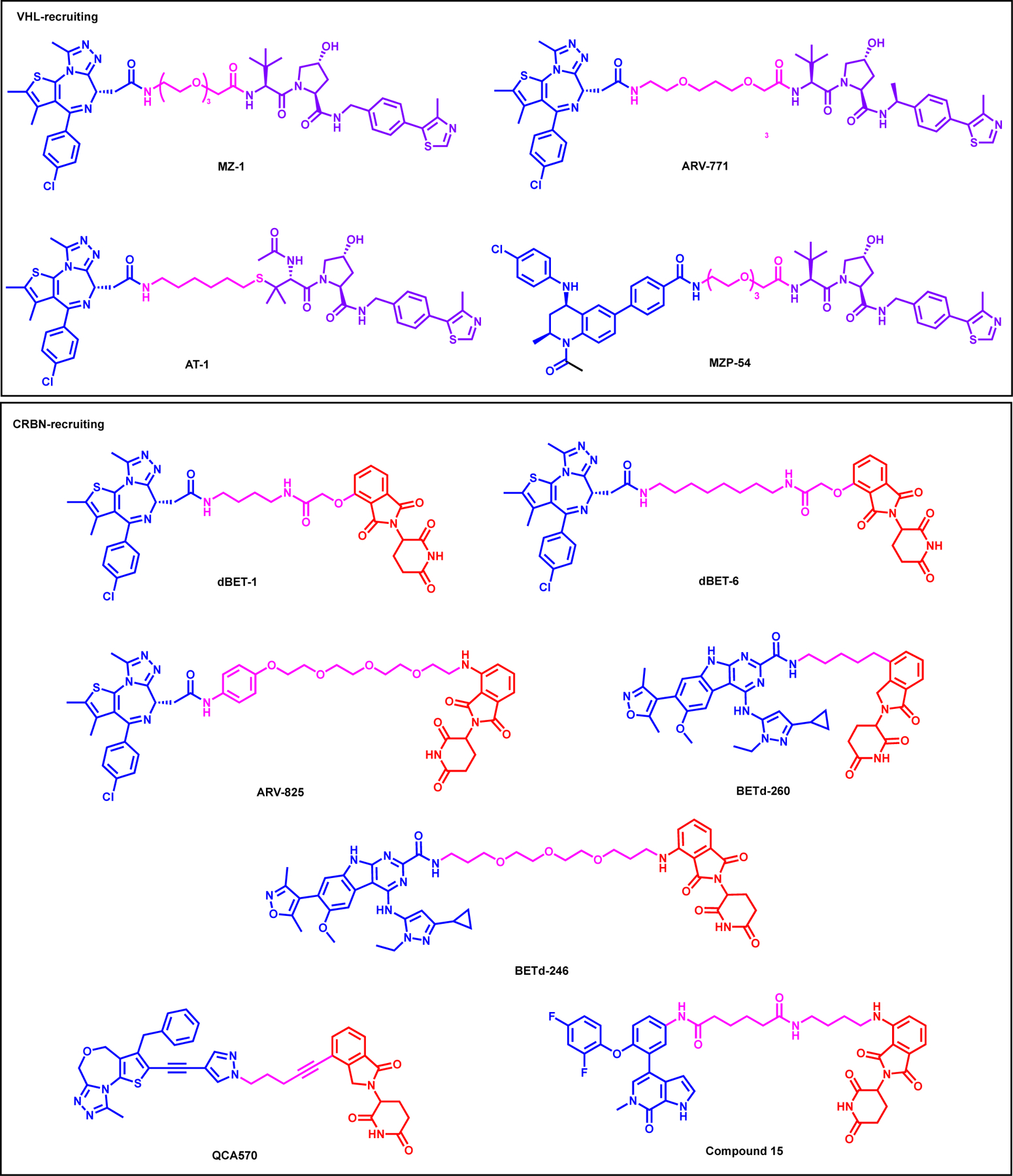
Structures of selected VHL- (top box) and CRBN-recruiting (bottom box) BET family degraders
There are several CRBN-based BET protein degraders reported including but not limited to: dBET1103, ARV-825104, dBET6105, BETd-260106, BETd-246107, QCA570108, Compound 15109 (Figure 7, bottom). Winter et al. reported the first CRBN-recruiting BET protein degrader, dBET1, which was synthesized by linking (+)-JQ1 with thalidomide showing potent BRD4 degradation in a dose- and time-dependent manner (Figure 7).103 dBET1 induced degradation of BRD2 and BRD3 as well as suppressing MYC and PIM1 transcript levels, further confirmed by proteomics study. Finally, dBET1 showed effective tumor reduction in MV4;11 xenograft model and decreased leukemic burden in bone marrow. Winter et al. introduced the second generation degrader, dBET6, which was synthesized by linking (+)-JQ1 with thalidomide similar to dBET1 with 8-carbon linker instead of 4-carbon linker of dBET1.105 dBET6 demonstrated potent BET protein degradation confirmed by global proteomics study in T cell acute lymphoblastic leukemia (T-ALL). dBET6 showed superior in vitro anti-tumor efficacy in T-ALL cell lines compared to dBET1 and further demonstrated tumor regression in disseminated SUPT11 xenograft model and survival benefit in an aggressive disseminated MOLT4 xenograft model over period of 14 days. Next, ARV-825 (Figure 7), which was synthesized by linking the phenol oxygen of OTX015 to pomalidomide via PEG linker, was reported by Lu et al.104 ARV-825 showed potent and long lasting BRD4 degradation in Burkitt’s lymphoma (BL) cell lines in a dose (DC50 < 1 nM) and time-dependent manner, mediated by the UPS system. ARV-825 showed potent BL cells anti-proliferative activity compared to BET inhibitors OTX015 and JQ1. ARV-825 also suppressed cMyc expression ARV-825 also reported to be potent in inhibiting growth in cholangiocarcinoma (CCA) cell lines by effectively degrading BRD4.110
Next, Zhou et al. reported BETd-260106 (Figure 7), which utilized close analog of the potent azacarbazole-based BET inhibitors discovered by the same research group.111 BETd-260 was synthesized by linking the 2-carboxamide group attached to the tricyclic ring system of compound 8 (HJB97) to lenalidomide via 5-carbon linker.106 BETd-260 induced potent BRD2/3/4 degradation at concentrations as low as 30 pM in RS4;11 cells while also suppressing cMyc levels which resulted in cell cycle arrest in AML cell lines. Furthermore, BETd-260 treatment led to complete tumor regression of RS4;11 xenograft model by IV dosing without any adverse toxicity. In addition, Bai et al. reported BETd-246 (Figure 7), which featured the same BET inhibitor linked to pomalidomide via longer PEG containing linker and showed potent BRD2/3/4 degradation in TNBC MDA-MB-468 cell line, confirmed by mass-spectrometry proteomics study.107 Further RNA-seq studies revealed that BETd-246 prompted downregulation of MCL1. Indeed BETd-246 led to time and dose-dependent degradation of MCL1 and played a key role in the potent apoptosis induction in a panel of TNBC cell lines. While BETd-246 demonstrated anti-tumor efficacy in “Washington Human in Mouse (WHIM)” 24 (WHIM24) PDX and MDA-MB-453 xenograft model, it did not show any activity in MDA-MB-231 or MDA-MB-468 xenograft model, partly due to poor drug exposure in the tumor tissue. Qin et al. reported QCA570 (Figure 7), which was synthesized by linking the 1-methyl-1H-pyrazole moiety of BET inhibitor 22 (QCA-276) with lenalidomide.108 QCA570 induced potent BRD2/3/4 degradation in leukemia cell lines with a picomolar efficiency and suppressed cMyc level, resulting in superior cell growth inhibition. Furthermore, QCA570 showed efficient tumor regression in RS4;11 and MV4;11 xenograft model with IV dosing.
More recently, Xiang et al. reported Compound 15 (Figure 7), which was synthesized by removing the ethane-sulfonyl moiety of pan BET inhibitor, ABBV-075 and directly linking it to thalidomide via relatively long linker.109 Compound 15 induced robust BRD4 degradation in MV4;11 and RS4;11 cell lines, in as short as 3-hour post-treatment. Compound 15 also suppressed cMyc expression and induced apoptosis in AML cell lines.
2.2. EED degraders
EED, a crucial non-catalytic component of the PRC2 complex, contains a tryptophan-aspartic acid (WD)-40-repeat domain that folds into a seven-bladed β-propeller and provides a scaffold for EZH2 and SUZ12 interaction.112 Particularly, the N-terminus of the EZH2 binds to the bottom of the β-propeller of EED, while EED recognizes the H3K27me3 and other epigenetic marks via the top of its β-propeller domain further enhancing EZH2 activity allosterically.113 Similar to EZH2, overexpression of EED also leads to poor prognosis in various cancers including breast, prostate and colorectal cancer.114 Presently, MAK-683 is a clinical-stage potent inhibitor of EED, which showed efficacy in EZH2 mutant diffuse large B-cell lymphoma.115 Importantly, EED-based degraders offer an alternative strategy to inhibit PCR2 for the treatment of cancer, particularly for EZH2 mutant tumors.
Potjewyd et al. report a EED degrader, UNC6852 which is obtained by modifying the methyl sulfone moiety of parent inhibitor, EED226116 to carboxylic acid to attach the linker, adjoining the VHL ligand (Figure 8).117 UNC6852 had a DC50 of 247 nM and 80% degradation at 5 μM compound treatment for 24 hours in HeLa cell line. Interestingly, UNC6852, in a dose-dependent manner, induced clear EED and EZH2 degradation, and to lesser extent SUZ12 degradation. Moreover, UNC6852 facilitated the PRC2 degradation via UPS and in a VHL dependent manner. A global proteomics analysis indicated that EED and EZH2 were significantly decreased after 24 hours with SUZ12 being less significantly reduced. Furthermore, UNC6852 degraded EZH2 SET domain mutant (Y641N) in DB cells. However, UNC6852 did not inhibited DB cells or Pfeiffer cells growth more potently compared to parent compound, EED226.
Figure 8:
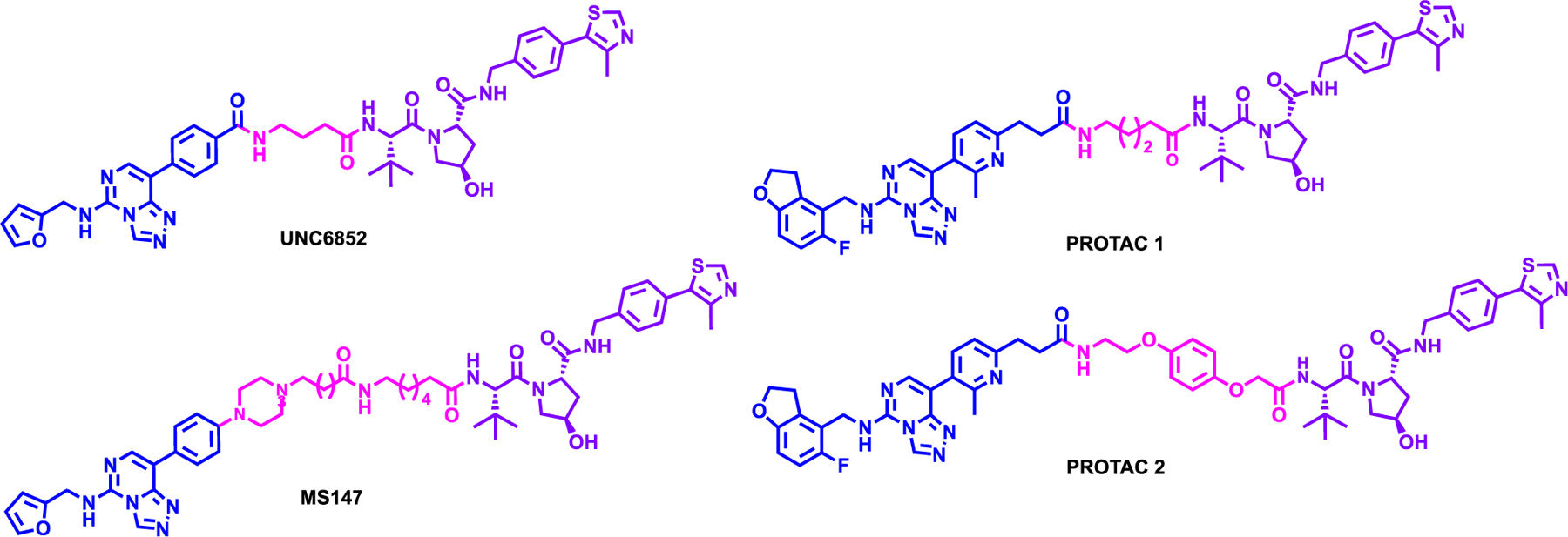
Structures of EED-based degraders
At the same time, Hsu et al. also developed EED degraders based on an analog of published EED inhibitor compound 43,118 (patent US20160176882A1) and connecting it to VHL ligand (Figure 8).119 Two highlighted degraders, PROTAC 1 and PROTAC 2 showed potent EED and EZH2 degradation even at 120 nM via the UPS. Like UNC6852, PROTAC 2 showed a preferential degradation of EED over EZH2 and SUZ12, which were the only proteins significantly decreased as displayed by mass-spectrometry based proteomics EED being the most significantly reduced followed by EZH2 and SUZ12. In addition, both PROTAC 1 and 2 showed potent growth inhibition in Karpas422 lymphoma cell lines with a GI50 of 57 and 45 nM, respectively. However, whether both EZH2 and SUZ12 is polyubiquitinated by PROTAC 2 or is degraded due to the destabilization effect of EED degradation on PCR2 needs to be further investigated.
While EED primarily known to play a crucial role in the PRC2 complex, EED has also been reported to interact with PRC1 core components BMI1 and RING1B.120, 121 Mounting evidence suggest that both enzymatic and nonenzymatic functions of PRC1 core components, particularly BMI1 and RING1B, are critical the development and progression of various cancers.122, 123 While there are inhibitors targeting PRC1 complex, either the direct interaction of the inhibitor with any PRC1 components has not been clearly shown or it displayed limited efficacy in killing various types of cancer cells.124, 125 Therefore, a novel approach such as use of PROTAC is warranted in targeting PRC1 complex.
Park et al. developed MS147, an EED-binding, VHL-recruiting PROTAC, which degrades PRC1 core components, BMI1 and RING1B, preferentially over EED.126 Very recently, Xiong et al. from the same research group reported the concept of Bridged PROTAC in which the first-in-class degrader of Cyclin D1, MS28 utilized CDK4/6-binding PROTAC to preferentially degrade Cyclin D1 taking advantage of CDK4/6-Cyclin D1 interaction.127 It is important to note that Cylin D1 does not have a known small molecule binder or inhibitor rendering it undruggable but the Bridge PROTAC, MS28 allowed it to be targeted and degraded. The first PRC1 degrader, MS147, utilizing the same bridge PROTAC concept achieved degradation of PRC1 core components exploiting the EED’s interaction with partner proteins BMI1 and RING1B. MS147 degraded BMI1 and RING1B preferentially in a dose and time-dependent manner over EED in K562 and KARPASS-422 cell lines without altering the mRNA levels. In addition, MS147 induces a complex formation between EED and BMI1/RING1B as shown by in vitro pull-down experiments. MS147 degraded PRC1 complex in EED-, VHL- and UPS dependent manner. Importantly, MS147 selectively degrades PRC1 core components more potently over PRC2 components and reduce the H2AK119ub levels while having no effect in H3K27me3 levels. Finally, MS147 can degrade PRC1 components in triple negative breast cancer cell line (MDA-MB-468) and non-small cell lung cancer cell line (NCI-H1299) and has a superior antiproliferative activity compared to PROTAC 2.119 As we discussed throughout this review, several epigenetic protein complex degraders which are mediated through the traditional PROTAC mechanism such as UNC6852 and PROTAC2 have been developed. However, it is important to point out that none of these traditional protein complex PROTACs preferentially degraded partner proteins over the protein they directly bind. MS147, on the other hand, as a bridge PROTAC preferentially degrades interacting partner proteins over the protein that it directly binds.
2.3. TRIM24 degraders
Transcription intermediary factor 1-alpha (TIF1α), also known as TRIM24, is a multi-domain protein which contains a bromodomain which recognizes the acetylated H3K23 (H3K23ac) and a PHD domain which interacts with lysine 4 residue of histone H3 tail (H3K4).128 TRIM24 also has a RING domain which was shown to function as a E3 ligase to bind and degrade p53.129 It was shown that TRIM24 is capable of binding to chromatin in presence of ligand, estrogen, to activate estrogen-dependent genes such as GREB1 and pS2/TFF1.128 Overexpression of TRIM24 is associated with poor prognosis in breast cancer and glioblastoma multiforme (GBM) which promotes cell survival by the PI3K/AKT pathway.130 There are several reported benzimidazolone-based TRIM24 bromodomain inhibitors such as IACS-9571 and Compound 34 which were shown to inhibit cell growth, however more effective therapeutics are needed for cancer treatment.131, 132
Gechijian et al. reported the first-in-class TRIM24 degrader, dTRIM24, which was synthesized by linking the sulfonamide tail of a close analog of IACS-7131 to VHL1 with a 3-PEG linker (Figure 9, top).133 dTRIM24 induced TRIM24 degradation at 5 μM and within 24 h treatment in 293FT cell line in dose- and UPS-dependent manner. dTRIM24 degraded TRIM24 in MCF-7 cell lines, however, both parent inhibitor and degrader did not impact cell viability, showing a contradiction to the dependency of TRIM24 in breast cancer. However, dTRIM24 had more potent anti-proliferative activity in MOLM-13 cell lines by degrading TRIM24, confirmed by global proteomics study. Furthermore, treatment of dTRIM24 reduced TRIM24 chromatin association and upregulated tumor suppressor genes such as BCOR, ERV3, and MZF1. However, dTRIM24 exhibited an “hook effect” in both 293FT and MOLM-13 cell lines and in vivo efficacy study was not performed.
Figure 9:
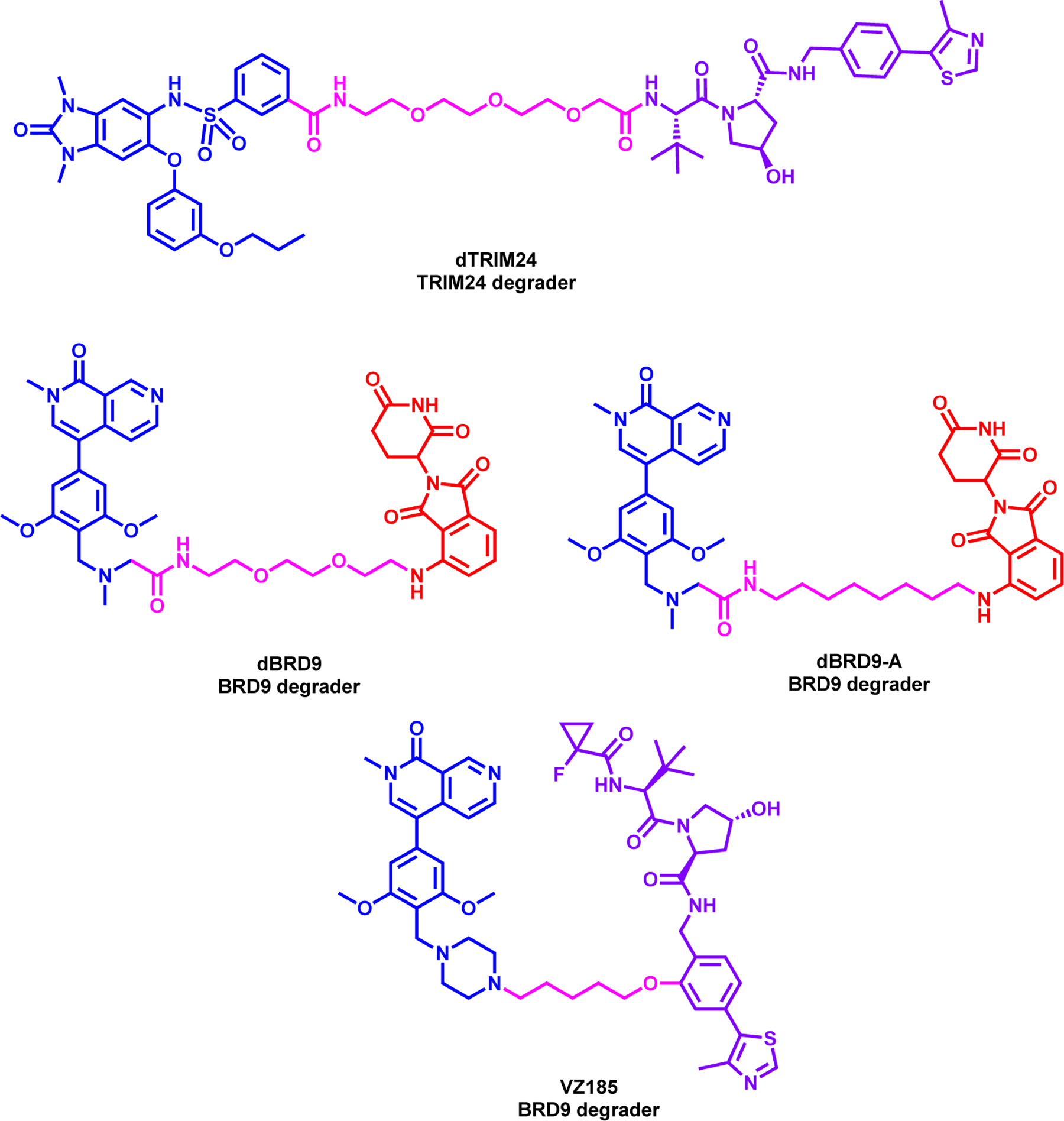
Structures of dTRIM24 (top) and BRD9 degraders (middle and bottom)
2.4. BRD9 degraders
BRD9, a type IV BRD-containing protein subfamily, contains a bromodomain and a DUF3512 domain, and importantly identified as a subunit of switch/sucrose non-fermenting (SWI/SNF) (BAF) chromatin remodeling complexes.134, 135 In particular, BRD9 is a component of BAF (BRG-/BRM-associated factor) complex, while its close homologue BRD7 is a component of PBAF (polybromo-associated BAF) complex.135, 136 It was also shown that the DUF3512 domain is critical for the interaction between BRD9 and BAF complex subunits and has an important role in transcriptional regulation.135 BRD9 was shown to enhance the transcription of MYC by binding to the promoter region and further activating STAT5 which lead to the AML cell survival and growth.137, 138 BRD9 overexpression is also documented in non-small cell lung cancer, pediatric rhabdoid tumors, prostate and cervical cancer.135, 139–141
Remillard et al. reported the first BRD9 degrader, dBRD9, which was synthesized by connecting the terminal dimethylamine moiety of BRD9 inhibitor, BI-7273142 to attach pomalidomide via 2-PEG linker (Figure 9, middle left).143 dBRD9 showed potent dose dependent degradation of BRD9 with significant degradation at 50 nM at 4-h. dBRD9 was selectively degraded BRD9 over BRD4 and BRD7, which was confirmed by global proteomics study in MOLM-13 cell line. Furthermore, dBRD9 also showed potent anti-proliferative activity in leukemia cell lines such as EOL-1 and MOLM-13, while the parent inhibitor was only effective at highest concentrations. dBRD9 also displayed activity against IKZF1/3 but its activity on GSPT1 was not reported. Brien et al. from the same group next reported dBRD9-A, which is a close analogue to dBRD9, but utilized a 8-carbon linker instead of a PEG linker (Figure 9, middle right).144 It has been shown in this study that synovial sarcoma cells were more sensitive to BRD9 bromodomain inhibition compared to other pediatric sarcomas albeit with modest effects. ChIP-seq experiments with BI-7273 treated cell showed that while reduced, BRD9 association with chromatin persists indicating that BRD9 does not rely exclusively on bromodomain function to associate with chromatin. These data, therefore, suggest that BRD9, as part of the BAF complex, can associate with chromatin in a bromodomain independent manner setting the stage for PROTAC strategy as an alternative approach. dBRD9-A induced potent BRD9 degradation in synovial sarcoma HSSYII and SYO1 cell lines at 100 nM at a time- and UPS-dependent manner. As expected, dBRD9-A treatment led to decreased BRD9 association with chromatin which led to increased anti-proliferative activity. Furthermore, dBRD9-A treatment demonstrated that BRD9 is a critical component of the SS18-SSX fusion protein containing BAF complex which induces G1 cell cycle growth arrest. dBRD9-A also inhibited tumor proregression in synovial sarcoma xenograft model because of reducing SS18-SSX1 binding at the super enhancer elements.
Next, Zoppi et al. reported a dual BRD7 & 9 degrader, VZ185, which was synthesized by replacing the dimethylamine of BI-7273142 with a piperazine moiety and linking it to VHL ligand, via 4-carbon linker (Figure 9, bottom).145 VZ185 induced potent degradation of both BRD7 and BRD9 in a dose- and time-and proteosome-dependent manner. VZ185 has a DC50 of 4.5 nM and 1.76 nM for BRD7 and BRD9, respectively, by WB analysis in diffuse large B cell lymphoma RI-1 cells at 8 hours treatment with an observed hook effect at 1 μM. It also displayed DC50 of 34.5 nM and 4.0 nM for BRD7 and BRD9, respectively, by HiBit assay in HEK293 cells. These results are correlative with higher ubiquitination of BRD9 shown by the NanoBRET experiment and corroborated by global proteomics study. VZ185 showed superior anti-proliferative activity in EOL-1 and A-204 cell lines compared to the parent inhibitor, however no in vivo efficacy studies were performed in this study.
2.5. SMARCA2/4 degraders
The ATP-dependent catalytic activity of SWI/SNF complex results in an open chromatin which is mediated through the core subunit: SMARCA4 (SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin, subfamily A, member 4) or its homologue SMARCA2.87 Aside from a ATPase domain, SMARCA2/4 also contains a bromodomain in the C-terminus which is capable of recognizing acetyl lysine 8/16 on histone 4 and acetyl lysine 9/14 on histone 3.146 While SMARCA4 induces osteoblast differentiation, SMARCA2 can suppress differentiation by binding to the repressor promoter alongside histone deacetylase 1 (HDAC1), showing distinct antagonistic relationship between SMARCA2 and SMARCA4.147 SMARCA4 was shown to have tumor suppressor activity, however it is frequently inactivated or mutated in various cancers such as non-small cell lung cancer, medulloblastoma and pancreatic cancer.148 In SMARCA4 mutated cell lines, it was demonstrated that SMARCA2 inhibition can attenuate cancer cell survival.149, 150
Farnaby et al. reported the first-in-class dual SMARCA2/4 degrader, PROTAC 1, which was synthesized by utilizing reported 2-(6-aminopyridazin-3-yl)phenols based SMARCA bromodomain inhibitor. The piperazine ring moiety of SMARCA bromodomain inhibitor was connected to VHL ligand, via a PEG linker (Figure 10).151 The ternary complex structure of SMARCA2BD:PROTAC 1:VCB (PDB: 6HAY) was solved revealing that most of the de novo protein-protein interactions were around the fluorocyclopropyl amide moiety of VHL ligand and all hydrogen bond donor groups in PROTAC 1 were already utilized in its interactions with the proteins. Structure based design then was focused on the linker region and the piperazine moiety of the SMARCA ligand was extended with a benzylic group to gain additional interactions. These structure-based modifications resulted in more potent degrader, ACBl1 (Figure 10), which degraded SMARCA2, SMARCA4 and PBRM1 in MV4;11 cells with DC50 of 6, 11 and 32 nM, respectively. Depletion of these proteins by ACBI1 was subsequently confirmed by global proteomics study. ACBl1 exhibited potent anti-proliferative activity and apoptosis in leukemic MV4;11 cell line and SMARCA4-deficient SK-MEL-5 cell line, while no activity in SMARCA2/4-deficient NCI-H1703 cell line was observed. This study demonstrated the co-dependency of SMARCA2 and SMARCA4 in cancer and exemplified the power of structure based design utilizing a ternary structure.
Figure 10:
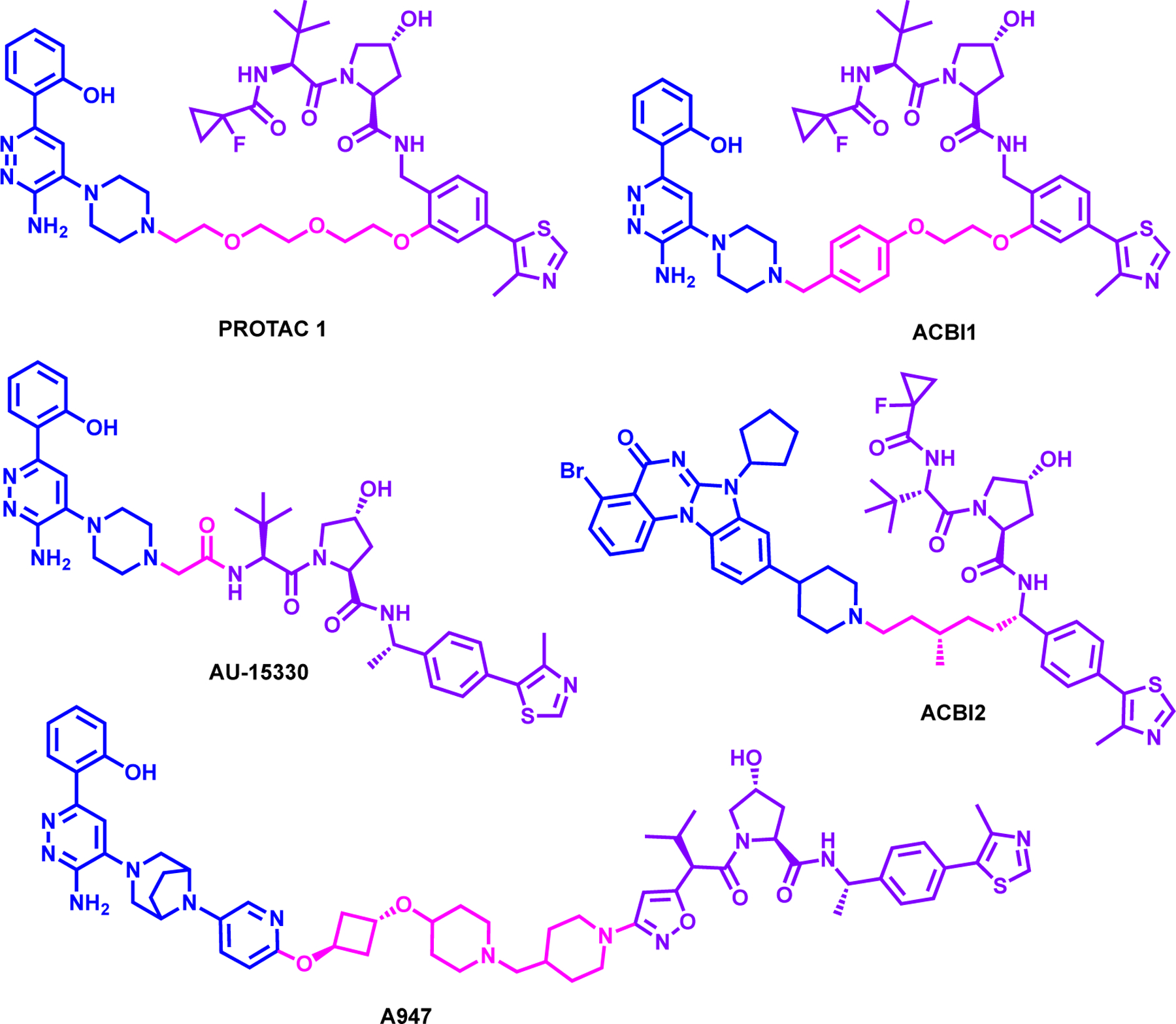
Structures of SMARCA2/4 degraders
Xiao et al. reported AU-15330, which was synthesized by using the same SMARCA bromodomain ligand described above linking it to VHL1-Me via very short linker (Figure 10).152 AU-15330 degraded SMARCA2 and 4 effectively at 1 μM in HEK293 and HeLa cells. Mass spectrometry-based proteomics analysis have confirmed the selectivity of AU-15330 in degrading SMARCA2, SMARCA4 and PBRM1 while sparing other SWI/SNF protein partners. In addition, it was shown that 1μM treatment of AU-15330 can lead to potent SMARCA2, SMARCA4 and PBRM1 degradation within 1 hour in RWPE, LNCaP and VCaP cell lines. It showed anti-proliferative effect of multiple prostate cancer cell lines with an IC50 of 19.5, 9.97 and 4.35 nM in LNCaP, VCaP, and NCI-H929, respectively. In addition, the degradation can be rescued with VHL ligand, MLN4924 and Bortezomib, confirming the proteosome dependent mechanism of degradation. Consequent assay for transposase-accessible chromatin using sequencing (ATAC-seq) studies have showed AU-15330 reduced chromatin accessibility and further reduced several oncogenes such as FOXA1 and Androgen receptor (AR). Finally, the efficacy of AU-15330 was determined in a VCaP castration-resistant prostate cancer (CRPC) in vivo model and it was shown that the combination treatment of enzalutamide with AU-15330 displayed more potent anti-tumor growth in C4–2B-derived CRPC xenografts and castration-resistant MDA-PCa-146–12 PDX model system compared to either monotherapy.
Kofink et al. reported ACBI2 (Figure 10), by using compound 3, which was designed inspired by a quinazoline based PB1 bromodomain inhibitor153 and conjugating it to VHL ligand.154 ACBI2 selectively degraded SMARCA2 over SMARCA4 with a DC50 of 1 nM and 32 nM, respectively in RKO cell lines. Furthermore, Kofink et al. using the ternary complex structure, rationalized the selectivity of degraders with linker elongation and branching towards SMARCA2 over SMARCA4. Interestingly, the addition of a branched methyl group on linker region also improved the oral bioavailability for ACBI2 to 22% from 3% of the non-branched analog. Finally, ACBl2 exhibited potent SMARCA2 degradation in NCI-H1568 and A549 xenograft mouse model and preferentially degraded SMARCA2 over SMARCA4 in different cancer cell lines. This study made great use of ternary complex structure to guide the design of a selective and orally bioavailable VHL recruiting degrader.
At the same time, Cantley et al. also reported A947 (Figure 10), which degraded SMARCA2 more potently compared to SMARCA4 despite having comparable binding affinity with a DC50 of 39 pM and 1.1 nM, respectively in SW1573 cells.155 Further ubiquitinome analysis demonstrated the majority of lysine ubiquitination occurred on the ATPase region of both SMARCA2/4 and global proteomics validated the selectivity of A947. Moreover, SMARCA4mut lung cancer cell lines such as NCI-H12944, HCC515 and NCI-H1793 were more susceptible to growth inhibition and SMARCA2 degradation by A947 treatment compared to SMARCA4WT lung cancer cell lines. The in-vitro potency of A947 was recapitulated in an HCC515 and HCC2302 xenograft models where A947 treatment led to 95% reduction in SMARCA2 protein levels and suppressed KRT80 transcript in SMARCA4mut models. Finally, A947 treatment sensitized SMARCA4mut lung cancer to MCL1 inhibitors, however further studies are needed to evaluate the combination therapy approach to treat SMARCA4mut solid cancers. This study highlights the ability of PROTACs to convert a non-selective binding ligand into a selective and efficacious in vivo PROTAC and offers a potential new therapeutic approach for tumors containing SMARCA4 mutations.
2.6. ENL Degraders
The YEATS domain family including Yaf9, ENL, AF9, Taf14, Sas5, were discovered as a family of chromatin readers that can recognize acetylated lysine residues.156 ENL (also known as MLLT1), has a disordered C-terminus which is responsible for protein interaction and the YEATS domain in the N-terminus which allows for the binding of the acetylated lysine and co-localizes with H3K9ac and H3K27ac on promoter regions.157 Furthermore, the YEATS domain was also shown to have a preference towards lysine crotonylation on histone and can contribute to transcription regulation.158 ENL was also shown to be part of the super elongation complex (SEC) regulating transcription elongation and in addition, form a complex with DOT1L to drive AML cell growth.159, 160 Fusion of ENL and AF9 also frequented in acute myeloid leukemia, which led the emergence of ENL as an attractive target to thwart against leukemia.161 While, several ENL-YEATS chemical probes were reported such as SGC-iMLLT162, XL-13m163 and XS018661164, there is still a need for novel strategies for studying the underlying role of ENL in disease progression.
Erb et al. first reported degradation of ENL degrader, using dTAG. The dTAG system platform allowing FKBP12 F36V fused target proteins to be degraded with the treatment of a hetero-bifunctional dTAG molecule that connects FKBP12F36V ligand and CRBN ligand via linker.165, 166 dTAG-13 (structure not shown), which connects FKBP12F36V ligand with CRBN very potently degraded ENL that was expressed as FKBP12F36V fusion in MV4;11-Cas9 cells.165 Confirmed by global proteomics study, dTAG-13 treatment also led to reduction of active transcripts and potent anti-proliferative activity in MV4;11 cell line. Furthermore, dTAG-13 resulted in a reduction of asymmetric localization of ENL and suppression of leukemia driver genes such as MYC, HOXA9/10, and MEIS1. While this was the first example of dTAG system and clearly validated ENL as a viable and important target for degradation. However, a degrader that can directly interact with endogenous ENL in non-engineered cancer cell lines is quite desirable as a therapeutic strategy.
Subsequently, Garnar-Wortzel et al. reported a first-in-class ENL degrader, SR-1114, which was synthesized by utilizing an imidazopyridine-based inhibitor scaffolds that was disclosed in the same study and linking it to thalidomide (Figure 11).167 SR-1114, mediated by CRBN, effectively degraded ENL in various AML cell lines such as MV4;11, MOLM-13 and OCI/AML-2 and further proteomics study confirmed the degradation of ENL alongside neo-substrate IKZF1. Several ENL target genes such as MYC, MYB, and HOXA10 was also shown to be reduced as a consequence of SR-1114 treatment. However, no further cell-based or in vivo studies to better understand the role of ENL degradation for AML cancer growth were performed with SR-1114.Next, Li et al. reported compound 1, which was synthesized by linking indazole nitrogen of YEATS inhibitor SGC-iMLLT with pomalidomide via 8-carbon alkyl linker (Figure 13).168 Briefly, compound 1 displayed DC50 of 37 nM and 44 nM in MV4;11 and MOLM13 cell lines, respectively. Furthermore, the degradation of ENL by compound 1 is mediated through the UPS pathway and can be rescued with either the parent inhibitor itself and thalidomide and proteosome inhibitor bortezomib. Compound 1 only degraded ENL and did not downregulate other SEC complex proteins such as AF9, AFF4 and cyclin-T1. It has been shown that compound 1 is more potent than the parent inhibitor in anti-proliferative activity in MLL1‑r leukemia cell lines such as Kasumi-1, U266 and RPMI-8226 cell line while no apparent activity in solid HeLa or PANC1 cancer cell lines were observed. Finally, compound 1 is bioavailable and in a mouse model of MLL1-r leukemia showed antitumor activity.
Figure 11:

Structures of ENL degraders
Figure 13:
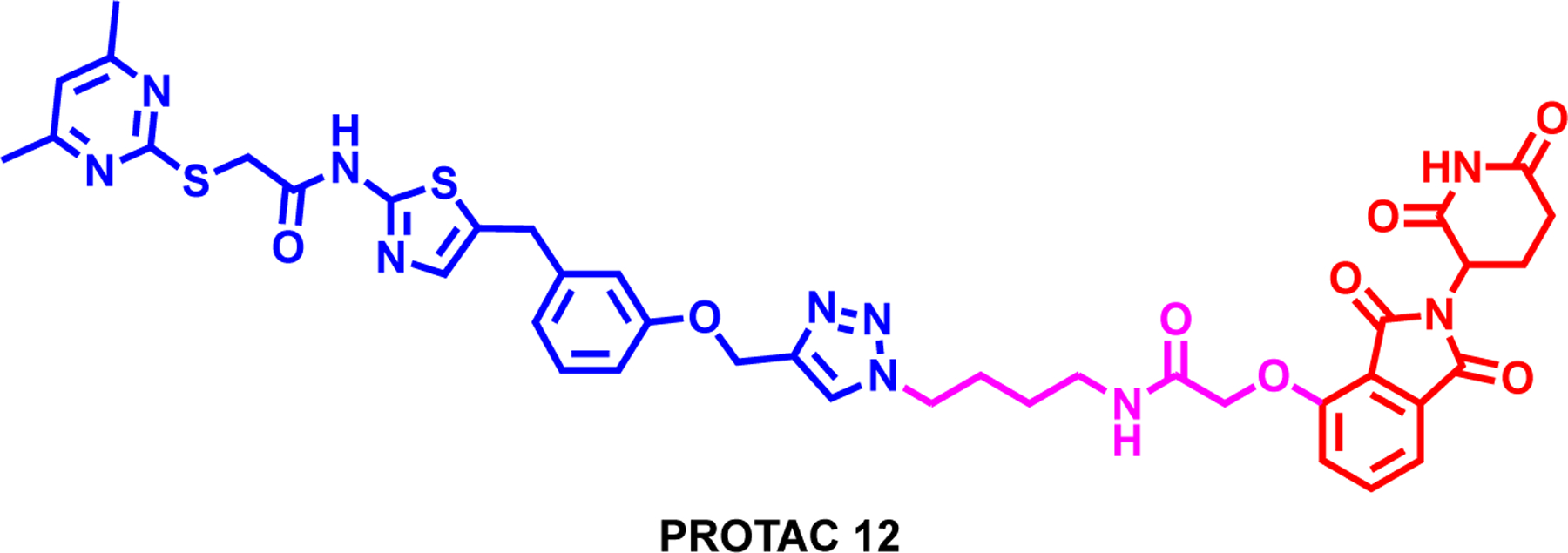
Structure of PROTAC 12, a SIRT2 degrader
3. Degraders of Epigenetic Erasers:
3.1. Zinc-dependent HDAC Degraders
Histone deacetylases (HDACs) are a group of enzymes that catalyzes the removal of acetyl groups from ε-amino lysine from histone and non-histone proteins via zinc-ion dependent hydrolysis leading to transcriptional repressive state.169 HDACs can be further divided into two major families: zinc-dependent and NAD+-dependent with four different classes.169 Zinc-mediated HDAC classes include: class I (HDAC1, HDAC2, HDAC3, HDAC8), class II (HDAC4, HDAC5, HDAC6, HDAC7, HDAC9, HDAC10) and class IV (HDAC11).170, 171 It was shown that HDACs can regulate cell-cycle progression by binding directly the p21 and p57 promoter region and also control DNA-damage response and apoptosis.172, 173 Particularly, dysregulation of class I, II, IV HDACs has been implicated in both solid tumors such as neuroblastoma, lung and gastric cancer and as well as hematological cancers such as AML and myeloma.173 Presently, there are several FDA-approved class I, II, IV HDACs inhibitors such as Vorinostat, Panobinostat and Belinostat, with many inhibitors ongoing clinical trials.173, 174
HDAC 1/2/3 Degrader:
Smalley et al. reported the first HDACs class I degrader, Compound 4, which was synthesized by linking the acetamide moiety of HDAC inhibitor, CI-994175, to VHL ligand via 10-carbon linker (Figure 12, top left).176 In a biochemical enzymatic assay, it was shown that Compound 4 had a poor target inhibition efficiency compared to shorter alkyl linker degrader and parent compound. However, in E14 mouse embryonic stem cells, it was shown that Compound 4 significantly increased the acetylation of lysine 56 of histone 3 (H3K56ac), which is a direct substrate of HDAC1. Furthermore, Compound 4 induced the potent degradation of HDAC1, to a lesser extent HDAC2 and HDAC3 in HCT116 colon cancer cell line a dose-dependent manner as well as significantly increased the acetylation of lysine 9 of histone 3 (H3K9ac). Nevertheless, Compound 4 was not more effective than the parent compound, CI-994 in the cell viability assay in HCT116 cells and further studies are needed to confirm the mode of action of the degrader and optimize to obtain more effective degrader.
Figure 12:
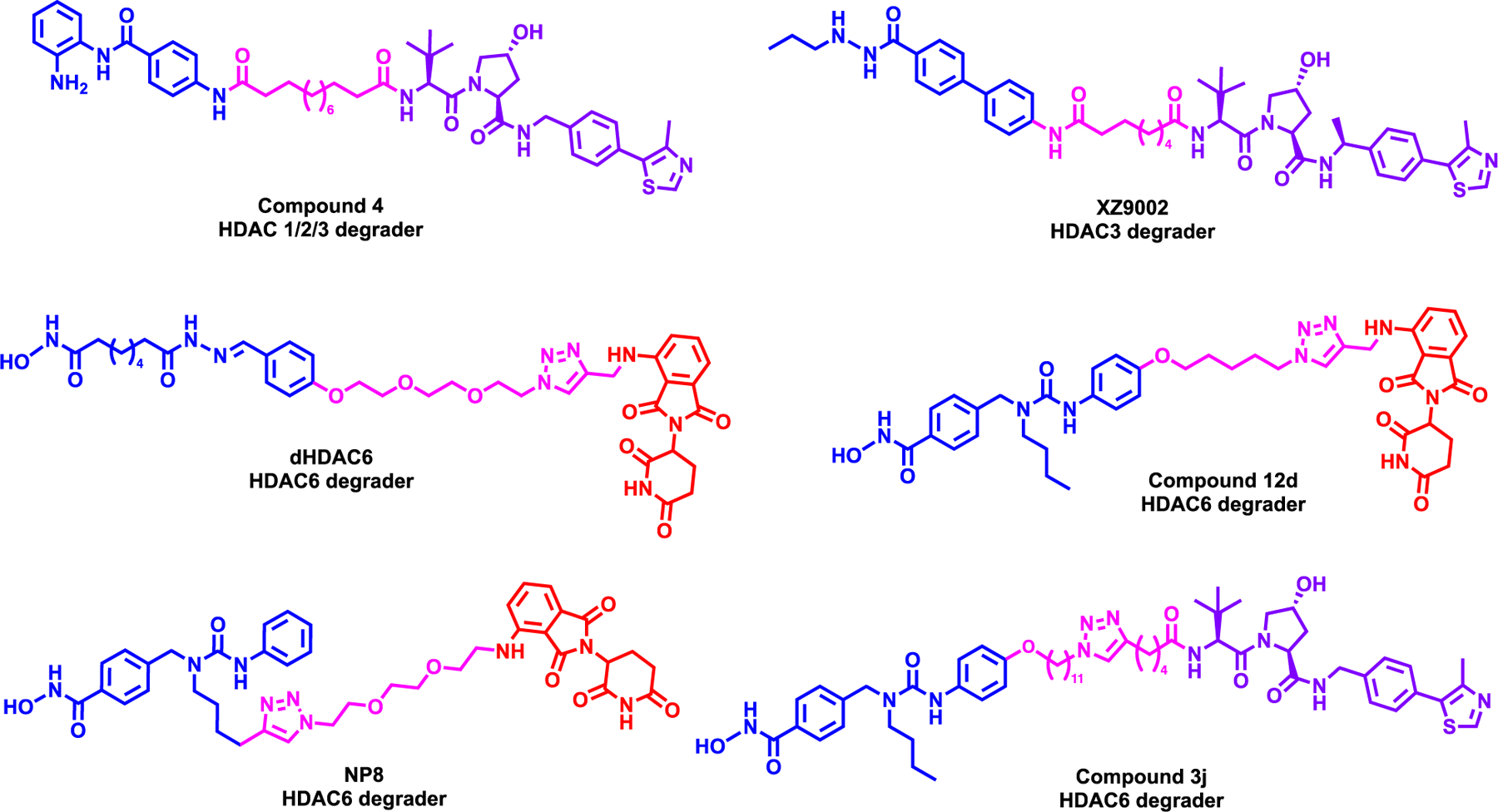
Structures of zinc-dependent HDAC degraders
HDAC3 Degrader:
Xiao et al. reported first-in-class HDAC3 selective degrader, XZ9002, which was synthesized by linking the terminal phenyl moiety of HDAC inhibitor, SR-3558177, to VHL ligand VHL1-Me (Figure 12, top right).178 XZ9002 induced potent HDAC3 degradation in MDA-MB-468 breast cancer cell line in a dose and time-dependent manner while sparing HDAC1 and HDAC2. Furthermore, XZ9002 increased the acetylation of histone 3 lysine 27 (H3K27ac) and overall acetylated lysine levels. Further mechanistic studies revealed that XZ9002-mediated HDAC3 degradation is orchestrated by the UPS system. In addition, XZ9002 induced potent clonogenic growth inhibition in multiple breast cancer cell lines such as T47D, MDA-MB-468 and BT549 while the negative control did not. However, global proteomics studies and gene expression profile were not performed to better understand the mechanism of action of the degrader.
HDAC6 Degraders:
Yang et al. was the first-in-class HDAC6 degrader, dHDAC6, which was synthesized by linking the non-selective HDAC inhibitor, AB3, to pomalidomide (Figure 12, middle left).179 dHDAC6 induced potent HDAC6 degradation with a DC50 of 34 nM with 70% Dmax observed in MCF-7 breast cancer cell line while HDAC1, HDAC2 and HDAC4 levels were not affected. Furthermore, dHDAC6 enhanced the acetylation of α-tubulin in a dose-dependent manner and mediated the degradation of HDAC6 via the UPS system. However, the anti-proliferative effect of dHDAC6 was not performed to assess its efficiency.
Wu et al. next reported second generation HDAC6 degrader, Compound 12d which was synthesized by linking HDAC6 selective inhibitor Nexturastat A180 to pomalidomide (Figure 12, middle right).181 Initial screening campaign revealed that Compound 12d achieved potent HDAC6 degradation at both 10 and 100nM concentration in MM1S multiple myeloma cell line. Compound 12d induced HDAC6 degradation with a DC50 of 1.6 nM and DMax of 86% while increasing α-tubulin acetylation levels. Further mechanistic studies exhibited that Compound 12d utilized the UPS system to degrade HDAC6, however CRBN neo-substrate, IKZF1 and IKZF3 was also degraded. The possible degradation of GSPT1 as a neosubstrate was not checked. In addition, Compound 12d had potent anti-proliferative activity in MM.1S cell line compared to the parent inhibitor, as the experiments show that the IKZF1/3 degradation contributed to this activity. Further, studies would be needed to design a PROTAC with no off-target effects.
Next, An et al reported a HDAC6 degrader, NP8, which also links Nexturastat A to pomalidomide, but by using a different linker attachment and a linker (Figure 12, bottom left).182 NP8 selectively induced potent HDAC6 degradation a DC50 of 3.8 nM without HDAC1, HDAC2 and HDAC4 degradation. Furthermore, it was shown that NP8, but not the negative control of NP8, induced degradation in a time-dependent manner. While NP8 enhanced α-tubulin acetylation, there was no significant difference in anti-proliferative efficacy in MM.1S cell line compared to the parent compound. Direct visualization of EGFP-HDAC6 in HeLa cells also confirmed the degradation of HDAC6 in-situ by NP8, however further studies are needed to understand target engagement alongside non-canonical partner proteins.
Finally, Yang et al. reported a first-in-class VHL recruiting HDAC6 degrader, compound 3j which was synthesized by linking Nexturastat A to VHL1 (Figure 12, bottom right).183 Compound 3j induced robust HDAC6 degradation in MM.1S cell line a DC50 of 7.1 nM and increased α-tubulin acetylation. While Compound 3j functioned via the UPS system to degrade HDAC6, it did not degrade HDAC1, HDAC2, HDAC3, HDAC4, HDAC7, and HDAC8. However, a hook effect was observed at higher concentrations of Compound 3j treatment, and the anti-proliferative activity was not determined.
Pan-HDAC degraders:
Xiong et al. employed a family-centric top-down approach rather than selecting a specific POI binder and screening different linkers and E3 ligases.23 In this top-down approach several pan-HDAC degraders such as Suberoylanilide hydroxamic acid (SAHA) and Dacinostat, targeting class I and IIb HDACs 1, 2, 3, 6, 8, and 10 as well as TMP269 and NVS-HD1 targeting class IIa HDACs 4, 5, 7, and 9 were used and linked to CRBN, VHL and inhibitor of apoptosis proteins (IAP) ligands via various linkers.A library of 48 pan-HDAC degraders were designed and synthesized and then degradation profiles of each degrader were assessed via quantitative proteomics measuring the change in relative abundance of protein expression in a neuroblastoma cell line, KELLY cells at 1 μM for 5 h. This interesting study exploring four pan-HDAC inhibitors as the target binding ligands and different linkers and linker attachment points as well as different E3 ligase ligands created a compound library that shed light into the degradability of the HDAC family proteins. The proteomics data across the 101 independent treatments included in this study, identified HDAC6, 3, and 8 as the most frequently degraded HDACs. Furthermore, multiple degraders for HDAC5, 7, and 10, which are for novel degradation targets, was discovered These compounds are ideal initial starting points for the further optimization and development of more selective degraders of these novel targets. While HDAC1, 2, and 4 identified as rarely degraded targets, HDAC9 was not degraded at all by any of the compounds in the library all the while the library of these degraders showed broad inhibition profiles for HDAC1–10. HDAC11 was not quantified in this study very likely due to low expression levels in the tested cell lines.
3.2. NAD+-dependent HDAC Degraders
The class III of HDACs are part of the silent information regulator 2 (Sir2) regulator family which contains a NAD/FAD-binding domain.171 In humans, there are seven Sir2-like proteins which are SIRT1 through SIRT7 which utilizes NAD+ as the cofactor to catalyze the removal acetyl marks from lysine residues.184 While SIRT3, SIRT4 and SIRT5 are localized in the mitochondria, the other SIRT deacetylases have a variety of histone lysine substrate which plays a role in DNA repair and chromatin organization.171 Particularly, overexpression of SIRT2 has been established in liver, gastric and lung cancer and can promote tumorigenesis by regulating cell cycle and metabolism.185 However, there are no effective therapeutics targets class IV HDACs inhibitors or degraders undergoing clinical trials.
SIRT2 Degrader:
Schiedel et al. reported the first-in-class SIRT2 degrader, PROTAC 12, which was synthesized by linking the triazole moiety of selective SIRT2 ligand, SirReal 3b, to thalidomide (Figure 13).186 SirReal-based PROTAC 12 showed potent and selective inhibition towards SIRT2 over SIRT1 and SIRT3 in an in-vitro fluorescence-based assay. Furthermore, PROTAC 12 showed potent SIRT2 degradation over SIRT1 in a dose and time-dependent manner which is mediated by the UPS system. In addition, the SIRT2 degradation was monitored in-situ in HeLa cells upon the PROTAC 12 treatment and moreover, increased acetylation of the microtubule network. Although, this is the first example of a SIRT HDAC eraser degrader, further studies are needed to optimize the degradation efficiency and selectivity.
4. New Methodologies and Expansion of PROTAC Toolbox:
In this section we would like to highlight some of the new methodologies and novel techniques related to PROTACs. Due to their importance as therapeutic targets, BET proteins are one of the most studied family utilizing PROTACs. Therefore, not surprisingly, owing to their well-established degradability they have been extensively utilized in the development of new methodologies and novel techniques related to PROTACs. While this is not a comprehensive list, we wanted to highlight some of these novel approaches and techniques.
4.1. Extending the E3 ligase toolbox
While VHL and CRBN-based degraders are the most established and utilized in the PROTAC field, several groups have identified ligands for other E3 ligases and expanded the scope of epigenetics degradation space. A more detailed review of E3 ligases can be found by elsewhere.187, 188 Aside from VHL and CRBN-based PROTACs, MDM2 and inhibitors of apoptosis proteins (IAPs)-based degraders garnered interest from the drug discovery community. A MDM2 based BET protein degrader, A1874, not only degraded BRD4 and cMyc with a DC50 of 32 nM and a Dmax of 98%; but A1874 also induced the expression of p53 tumor suppressor protein by stabilizing it.189 Next, SNIPER(BRD)-1, a non-genetic inhibitor of apoptosis protein (IAP)-dependent protein erasers (SNIPERs) based BRD4 degrader, not only degraded BRD4 at 100 nM, but was also able to degrade cellular inhibitor of apoptosis protein 1 (cIAP1) and an X-linked inhibitor of apoptosis protein (XIAP) through the UPS pathway.190 Other E3 ligase recruiting degraders for BRD4 has also been reported such as (1) MS83, a selective KEAP1, (also known as KLHL19)-recruiting degrader191 and CDDO-JQI, a nonselective, covalent KEAP1 recruiting degrader192 (2) DP1, a DCAF15-based degrader193 (3) CCW 28–3, a RNF4-based degrader194 (4) RNF114-recruiting degraders XH2195 and ML 2–14196 (5) KB02-JQ1, a DCAF16-based degrader197 (6) NJH-1–106, a covalent, FEM1B recruiting degrader.198 The utilization of the various E3 ligase can depend on tissue and cancer distribution.18 However, there is still a lack of knowledge regarding nuclear vs cytoplasmic degradation and a further assessment of all BRD4 degraders in the same context.
Very recently, several groups reported that JQ-1 structural modifications such as tethering with propargyl amine tail (compound 1a, a.k.a GNE-11)199, 200 or acrolein analog (TMX1)201, and aryl sulfonamide based compound 1202 act as monovalent degraders that enhances the interaction of BRD4 with DCAF16 to induce degradation of BRD4. Hsia et al. via a ternary complex structure demonstrated that compound 1 acts as a bivalent, molecular glue in which the JQ1 moiety binds to BD2 of BRD4 and the aryl sulfonamide warhead binds to BD1 of BRD4 while sulfonamide warhead does not interact with DCAF16 directly in isolation.202 These data indicates that compound 1 binds the tandem bromodomains bivalently while facilitating a multivalent gluing interaction between these two bromodomains and DCAF16 and in turn, resulting in degradation of BRD4. These recently reported BRD4 molecular glue approaches open new venues to expand the existing E3 toolbox.
4.2. Light-induced/controlled Degradation of BET Family Proteins
With the rapid advancements in the PROTAC field, additional strategies to improve their protein specificity and cell selectivity have also been explored. One of these strategies to regulate targeted protein degradation emerged as light induced/controlled degradation via PROTACs. Two major strategies were exploited for light induced/controlled PROTACs are (1) use of a photocleavable protecting group which could also be described as caging or photo-caging; and (2) use of photo switches taking advantage of reversible conformations of PROTACs, where while one confirmation is active the other one is inactive. Below examples all use BRD4 degradation by using JQ1 based degraders to showcase the light-induces/controlled strategy.
Xue et al. reported pc-PROTAC1, by introducing a photocleavable 4,5-dimethoxy-2‐nitrobenzyl (DMNB) group in dBET1 linker which showed weaker BRD4 binding affinity BRD4 degradation.203 In addition, pc-PROTAC2 was obtained by attaching the DMNB group to the imide nitrogen of the thalidomide rendering the molecule inactive in degrading BRD4. However, upon irradiation at 365 nm, pc-PROTAC1 and pc-PROTAC2 effectively were uncaged by cleavage of DMNB to release dBET1 achieving BRD4 degradation.203 It has been shown that the imide NH group of thalidomide analogs is critical for binding to CRBN.38 Taking advantage of this Naro et al. connected a 6-nitropiperonyloxymethyl (NPOM) group onto the glutarimide nitrogen to generate the caged BRD4 PROTAC 4 which is not capable of degrading BRD4.204 The NPOM caging group while is stable under aqueous conditions, undergoes photolysis when treated with 365 nm light to release PROTAC 4 to effectively degrade BRD4. Using similar strategy, Liu et al. designed opto-dBET1 featuring a photo-cleavable nitro-veratryloxycarbonyl group (NVOC) incorporated into the glutarimide nitrogen of dBET1 to effectively block CRBN recruitment. However, following exposure to UVA irradiation (365 nm), opto-dBET1 released dBET1 and effectively degraded BRD3 and BRD4.205 Similarly, Li et al. synthesized N2 by using DMNB group for caging of dBET1.206
Pfaff et al. reported a photo switchable degrader photoPROTAC-1 which was prepared by replacing the oligoether linker in ARV-771 with bis ortho-F4-azobenzene linkers.207 Efficient switching between cis- and trans-configurations upon irradiation at 530 and 415 nm, respectively was achieved. It was shown that highly stable, inactive cis-photoPROTAC-1 is isomerized with a visible light stimulus to a catalytically active trans-photoPROTAC-1 inducing degradation of BRD2. The optimal linker length of trans-photoPROTAC-1 recruiting VHL E3 ligase allowed formation of a productive ternary complex formation while cis-photoPROTAC-1 did not. Similarly, Reynders et al. reported a photo switchable PROTAC, PHOTAC-I-3, by inserting an azobenzene group into the dBET1 linker.208 Switching between trans-PHOTAC-I-3 and cis-PHOTAC-I-3 was efficiently achieved by irradiation at 390 nm (trans to cis) and 500 nm (cis to trans), respectively. In RS4–11 cells, PHOTAC-I-3 induce significant BRD4 degradation as well as BRD2 &3 c-Myc to a lesser extent when stimulated with 390 nm light conditions. In dark, PHOTAC-I-3 was inactive, and the degradation activity of cis-PHOTAC-I-3 could be blocked by irradiation with 525 nm light, showing spatiotemporal controlled as exemplified by the recovery of the BRD2 levels under these experimental conditions.
4.3. Other Novel Methodologies
Very recently, a radiotherapy inducible PROTAC strategy was developed to control PROTAC prodrug activation and protein degradation precisely and spatiotemporally, through X-ray radiation.209 This approach is quite similar in principle to the light controlled caged-PROTAC approach discussed in the previous section. Hydroxyl of VHL ligand was caged with a phenyl azide group that specifically targets BRD proteins featuring JQ1 as a ligand. While the protein degradation activity of the caged RT-PRO is blocked, upon radiation at the tumor site with X-ray, RT-PRO undergoes a cleavage chemistry reaction to release active PROTAC, to degrade BRD proteins. This work provides aims at the spatiotemporally control of protein degradation in vivo and in turn lessening of the undesired systemic toxicity of PROTACs.
Liu et al. reported a cancer cell selective delivery strategy for PROTACs via conjugation of folate group to essential hydroxyl group of VHL ligand, to achieve targeted degradation of POIs in cancer cells over noncancerous normal cells.210 The folate receptor, which is known to be overexpressed in cancer cells, is utilized for selective delivery of the Folate-PROTAC to the cancer cells. BRD4 degrader ARV-771 was used as one of the examples to create Folate-ARV-711. The active ARV-771 was released by endogenous hydrolases once in cells, effectively degrading BRD4 in cancer cells but avoiding the undesirable BRD4 degradation in normal tissues and organs.210
He et al. developed the first aptamer conjugated degrader, APR by connecting a tumor-targeting nucleolin aptamer AS1411 to VHL ligand of BRD4 degrader via a glutathione-labile ester-disulfide linker.211 This degrader designed to be specifically delivered to cancer cells overexpressing nucleolin. Once inside cancer cells, the labile disulfide bond linker is cleaved by glutathione to eventually release active BRD4 degrader resulting in effective BRD4 degradation in nucleolin-overexpressing human MCF-7 breast cancer cells. APR showed more effective cell killing of MCF-7 cells than in MCF-10A with lower nucleolin expression.
Recently it was shown that trivalent degraders can enhance the binding valency and lead to more potent degradation. Imaide et al. reported SIM1, designed based on the structural insights from ternary complexes of MZ1, a BET-degrader99 and MT1,a bivalent BRD4 binder.212 A core branched scaffold, 1,1,1-tris(hydroxymethyl)ethane was then used to attached two JQ1 molecules with 3-PEG linkers and VHL ligand, VHL1 with a short linker.213 SIM1 had a more potent BET protein degradation efficacy compared to MZ1 for BRD2, BRD3 and BRD4. Furthermore, SIM1 was shown to effectively downregulate Myc, a downstream target of BET proteins, is more potent than bivalent inhibitor, MT1 or bivalent degraders, MZ1 and ARV-771 in inducing PARP cleavage in 22Rv1 cells. Finally, it was concluded that SIM1 has favorable pharmacokinetic properties and can induce a stable ternary complex formation with augmented residence time., Huang et al. also reported a trivalent PROTAC, 1,2,5T-EG2-MZ1. 1,2,5T-EG2-MZ1 featured a 1,2,5 trisubstituted benzene ring in the center where JQ1 attached to 2-position via short linker, and VHL ligand attached to 1-position while tert-butyl ester group attached to 5-position.214 The tert-butyl ester unit on the benzene ring is designed as a handle for further functionalization to obtain trivalent degraders. 1,2,5T-EG2-MZ1 retained BRD4 degradation potency and kinetics similar to its bivalent derivative without the tert-butyl ester group. While this approach was reported as a generalizable approach to design and synthesize trivalent degraders for other applications, new linker and a functionalization site with controlled orientation optimization would be required for different targets.
In addition, several BET PROTAC-antibody conjugates have also been developed and reviewed recently elsewhere.215
Furthermore, in a proof of concept study, using a novel deep generative model, six candidate JQ1 based BRD4 PROTACs, designed, synthesized and experimentally tested and three of these degraders were validated by cell-based assays and western blot analysis.216
Future Outlook & Conclusion:
In this review, we took a systematic approach discussing the degraders developed for epigenetics writers, readers, and erasers. Significant advances have been made in this field, including the advancement of two BRD9 degraders, FHD-609 and CFT8634, to clinical development.217 We fully expect that more PROTACs targeting very important and validated epigenetic drug targets will be discovered as therapeutics or tool compounds. We also expect that PROTACs targeting less studied epigenetic proteins will be developed as chemical tools to further understand the roles of these proteins in diseases. We anticipate that the pace of the PROTAC discovery and development will accelerate.
While tremendous advancements have been made in the PROTAC field, there are still ongoing questions that remain unanswered. Such questions include: (1) whether continuous treatment of degraders can lead to resistance to PROTAC, (2) whether PROTACs towards epigenetic complex proteins can be rationally designed, and (3) which additional E3 ligases can be harnessed for PROTAC development. Some studies suggest that the resistance to BET protein degraders could be developed due to the deletion or dysregulation of the E3 ligase itself or its associated partner proteins.218, 219 Interestingly, it was recently shown that the resistance to PROTAC degraders could also occur by the introduction of hotspot mutations in the VHL or CRBN E3 ligase which alters the ternary complex formation.220 Further studies are needed to determine whether these resistance mechanisms can occur in vivo or in the clinic. The recent development of the bridged PROTAC technology to target the proteins without small-molecule binders is a quite exciting and significant advancement for targeting previously undruggable proteins. The bridged PROTAC technology could be particularly useful for targeting undruggable epigenetic proteins as some of undruggable epigenetic proteins interact with other proteins which have known small-molecule binders. As we discussed, there are continuing efforts to expand the toolbox of E3 ligase ligands that can be leveraged for PROTAC development. Further exploiting the variance of E3 ligase expression levels in tumor and normal cells to rationally develop PROTACs with reduced side effects and an improved therapeutic window is another exciting future direction. In addition, recent new methodologies to selectively deliver and activate PROTACs in cancer cells over normal cells such as light or radiation activated PROTACs, folate-caged PROTACs and Ab-conjugated PROTACs offer a potential avenue for PROTACs to be precision medicines. Furthermore, the efforts to achieve selectivity beyond what expected from parent inhibitors by PROTACs, especially against highly structural homologous proteins, have resulted in numerous successful examples. We fully expect that this additional dimension to achieve selectivity will be further exploited and lead to more success. Lastly, as discussed throughout this review, structure-based rational design of effective PROTACs, based on POI–PROTAC–E3 ternary complex structures, is a powerful approach. We expect that efforts in this area will be continually increasing and result in many more successful examples. Overall, the targeted protein degradation field has seen exciting advancements. We anticipate that more and more heterobifunctional degraders will enter clinical development, some of which will likely become effective cancer therapeutics for patients. We fully expect that amazing progress and successes will continue in targeting epigenetic proteins with PROTACs. Our hope is that this review will be a useful resource and encourage new and exciting discoveries.
Supplementary Material
Figure 14:

Structure of BRD4 degraders using E3 ligases other than VHL or CRBN
Acknowledgements:
J.J. acknowledges the support by the grants R01CA218600, R01CA230854, R01CA260666, R01CA268384 and R01CA268519 from the National Cancer Institute (NCI) at the NIH and an endowed professorship by the Icahn School of Medicine at Mount Sinai. M.K. acknowledges the support by the Training Grant in Cancer Biology (T32CA078207) from the NCI.
Conflicts of interest:
M.K., and X.Y. declare no conflicts of interest. H.U.K is a consultant for EpiCypher Inc. J.J. is a cofounder and equity shareholder in Cullgen Inc., a scientific cofounder and scientific advisory board member of Onsero Therapeutics, Inc., and a consultant for Cullgen Inc., EpiCypher Inc., Accent Therapeutics Inc., and Tavotek Biotherapeutics, Inc. The Jin laboratory received research funds from Celgene Corporation, Levo Therapeutics, Cullgen, Inc., and Cullinan Oncology.
Uncategorized References
- 1.Waddington CH, Endeavour, 1942, 1, 18–20. [Google Scholar]
- 2.Holliday R and Pugh JE, Science, 1975, 187, 226–232. [PubMed] [Google Scholar]
- 3.Luger K, Mäder AW, Richmond RK, Sargent DF and Richmond TJ, Nature, 1997, 389, 251–260. [DOI] [PubMed] [Google Scholar]
- 4.Allfrey VG, Faulkner R and Mirsky AE, Proc. Natl. Acad. Sci. U. S. A, 1964, 51, 786–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dhalluin C, Carlson JE, Zeng L, He C, Aggarwal AK and Zhou MM, Nature, 1999, 399, 491–496. [DOI] [PubMed] [Google Scholar]
- 6.Strahl BD and Allis CD, Nature, 2000, 403, 41–45. [DOI] [PubMed] [Google Scholar]
- 7.Jenuwein T and Allis CD, Science, 2001, 293, 1074–1080. [DOI] [PubMed] [Google Scholar]
- 8.Falkenberg KJ and Johnstone RW, Nat. Rev. Drug Discov, 2014, 13, 673–691. [DOI] [PubMed] [Google Scholar]
- 9.Gil J, Ramírez-Torres A and Encarnación-Guevara S, J. Proteomics, 2017, 150, 297–309. [DOI] [PubMed] [Google Scholar]
- 10.Zhou Z, Li HQ and Liu F, Curr. Top. Med. Chem, 2018, 18, 2448–2457. [DOI] [PubMed] [Google Scholar]
- 11.Chen IC, Sethy B and Liou JP, Front. Cell Dev. Biol, 2020, 8, 576391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Byrne MM, Murphy RT and Ryan AW, Front. Genet, 2014, 5, 364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holohan C, Van Schaeybroeck S, Longley DB and Johnston PG, Nat. Rev. Cancer, 2013, 13, 714–726. [DOI] [PubMed] [Google Scholar]
- 14.Yan J, Ng SB, Tay JL, Lin B, Koh TL, Tan J, Selvarajan V, Liu SC, Bi C, Wang S, Choo SN, Shimizu N, Huang G, Yu Q and Chng WJ, Blood, 2013, 121, 4512–4520. [DOI] [PubMed] [Google Scholar]
- 15.Wang J, Yu X, Gong W, Liu X, Park KS, Ma A, Tsai YH, Shen Y, Onikubo T, Pi WC, Allison DF, Liu J, Chen WY, Cai L, Roeder RG, Jin J and Wang GG, Nat. Cell Biol, 2022, 24, 384–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lai AC and Crews CM, Nat. Rev. Drug Discov, 2017, 16, 101–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schapira M, Calabrese MF, Bullock AN and Crews CM, Nat. Rev. Drug Discov, 2019, 18, 949–963. [DOI] [PubMed] [Google Scholar]
- 18.Dale B, Cheng M, Park KS, Kaniskan HÜ, Xiong Y and Jin J, Nat. Rev. Cancer, 2021, 21, 638–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gadd MS, Testa A, Lucas X, Chan KH, Chen W, Lamont DJ, Zengerle M and Ciulli A, Nat. Chem. Biol, 2017, 13, 514–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nowak RP, DeAngelo SL, Buckley D, He Z, Donovan KA, An J, Safaee N, Jedrychowski MP, Ponthier CM, Ishoey M, Zhang T, Mancias JD, Gray NS, Bradner JE and Fischer ES, Nat. Chem. Biol, 2018, 14, 706–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bondeson DP, Smith BE, Burslem GM, Buhimschi AD, Hines J, Jaime-Figueroa S, Wang J, Hamman BD, Ishchenko A and Crews CM, Cell Chem. Biol, 2018, 25, 78–87.e75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang HT, Dobrovolsky D, Paulk J, Yang G, Weisberg EL, Doctor ZM, Buckley DL, Cho JH, Ko E, Jang J, Shi K, Choi HG, Griffin JD, Li Y, Treon SP, Fischer ES, Bradner JE, Tan L and Gray NS, Cell Chem. Biol, 2018, 25, 88–99.e86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xiong Y, Donovan KA, Eleuteri NA, Kirmani N, Yue H, Razov A, Krupnick NM, Nowak RP and Fischer ES, Cell Chem. Biol, 2021, 28, 1514–1527.e1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burslem GM, Smith BE, Lai AC, Jaime-Figueroa S, McQuaid DC, Bondeson DP, Toure M, Dong H, Qian Y, Wang J, Crew AP, Hines J and Crews CM, Cell Chem. Biol, 2018, 25, 67–77.e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khan S, Zhang X, Lv D, Zhang Q, He Y, Zhang P, Liu X, Thummuri D, Yuan Y, Wiegand JS, Pei J, Zhang W, Sharma A, McCurdy CR, Kuruvilla VM, Baran N, Ferrando AA, Kim YM, Rogojina A, Houghton PJ, Huang G, Hromas R, Konopleva M, Zheng G and Zhou D, Nat. Med, 2019, 25, 1938–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paiva SL and Crews CM, Curr. Opin. Chem. Biol, 2019, 50, 111–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Webb T, Craigon C and Ciulli A, Bioorg. Med. Chem. Lett, 2022, 63, 128653. [DOI] [PubMed] [Google Scholar]
- 28.Vogelmann A, Robaa D, Sippl W and Jung M, Curr. Opin. Chem. Biol, 2020, 57, 8–16. [DOI] [PubMed] [Google Scholar]
- 29.Kim KH and Roberts CW, Nat. Med, 2016, 22, 128–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tan JZ, Yan Y, Wang XX, Jiang Y and Xu HE, Acta. Pharmacol. Sin, 2014, 35, 161–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bracken AP, Pasini D, Capra M, Prosperini E, Colli E and Helin K, EMBO J, 2003, 22, 5323–5335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bachmann IM, Halvorsen OJ, Collett K, Stefansson IM, Straume O, Haukaas SA, Salvesen HB, Otte AP and Akslen LA, J. Clin. Oncol, 2006, 24, 268–273. [DOI] [PubMed] [Google Scholar]
- 33.Duan R, Du W and Guo W, J. Hematol. Oncol, 2020, 13, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Konze KD, Ma A, Li F, Barsyte-Lovejoy D, Parton T, Macnevin CJ, Liu F, Gao C, Huang XP, Kuznetsova E, Rougie M, Jiang A, Pattenden SG, Norris JL, James LI, Roth BL, Brown PJ, Frye SV, Arrowsmith CH, Hahn KM, Wang GG, Vedadi M and Jin J, ACS Chem. Biol, 2013, 8, 1324–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ma A, Stratikopoulos E, Park KS, Wei J, Martin TC, Yang X, Schwarz M, Leshchenko V, Rialdi A, Dale B, Lagana A, Guccione E, Parekh S, Parsons R and Jin J, Nat. Chem. Biol, 2020, 16, 214–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Knutson SK, Warholic NM, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, Porter Scott M, Chesworth R, Moyer MP, Copeland RA, Richon VM, Pollock RM, Kuntz KW and Keilhack H, Proc. Natl. Acad. Sci. U. S. A, 2013, 110, 7922–7927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu Z, Hu X, Wang Q, Wu X, Zhang Q, Wei W, Su X, He H, Zhou S, Hu R, Ye T, Zhu Y, Wang N and Yu L, J. Med. Chem, 2021, 64, 2829–2848. [DOI] [PubMed] [Google Scholar]
- 38.Fischer ES, Böhm K, Lydeard JR, Yang H, Stadler MB, Cavadini S, Nagel J, Serluca F, Acker V, Lingaraju GM, Tichkule RB, Schebesta M, Forrester WC, Schirle M, Hassiepen U, Ottl J, Hild M, Beckwith RE, Harper JW, Jenkins JL and Thomä NH, Nature, 2014, 512, 49–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tu Y, Sun Y, Qiao S, Luo Y, Liu P, Jiang ZX, Hu Y, Wang Z, Huang P and Wen S, J. Med. Chem, 2021, 64, 10167–10184. [DOI] [PubMed] [Google Scholar]
- 40.Wang C, Chen X, Liu X, Lu D, Li S, Qu L, Yin F, Luo H, Zhang Y, Luo Z, Cui N, Kong L and Wang X, Eur. J. Med. Chem, 2022, 238, 114462. [DOI] [PubMed] [Google Scholar]
- 41.Dale B, Anderson C, Park KS, Kaniskan HÜ, Ma A, Shen Y, Zhang C, Xie L, Chen X, Yu X and Jin J, ACS Pharmacol. Transl. Sci, 2022, 5, 491–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang P, Lee H, Brunzelle JS and Couture JF, Nucleic Acids Res, 2012, 40, 4237–4246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rao RC and Dou Y, Nat. Rev. Cancer, 2015, 15, 334–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dharmarajan V, Lee JH, Patel A, Skalnik DG and Cosgrove MS, J. Biol. Chem, 2012, 287, 27275–27289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Krivtsov AV and Armstrong SA, Nat. Rev. Cancer, 2007, 7, 823–833. [DOI] [PubMed] [Google Scholar]
- 46.Carugo A, Genovese G, Seth S, Nezi L, Rose JL, Bossi D, Cicalese A, Shah PK, Viale A, Pettazzoni PF, Akdemir KC, Bristow CA, Robinson FS, Tepper J, Sanchez N, Gupta S, Estecio MR, Giuliani V, Dellino GI, Riva L, Yao W, Di Francesco ME, Green T, D’Alesio C, Corti D, Kang Y, Jones P, Wang H, Fleming JB, Maitra A, Pelicci PG, Chin L, DePinho RA, Lanfrancone L, Heffernan TP and Draetta GF, Cell Rep, 2016, 16, 133–147. [DOI] [PubMed] [Google Scholar]
- 47.Karatas H, Townsend EC, Cao F, Chen Y, Bernard D, Liu L, Lei M, Dou Y and Wang S, J. Am. Chem. Soc, 2013, 135, 669–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Senisterra G, Wu H, Allali-Hassani A, Wasney GA, Barsyte-Lovejoy D, Dombrovski L, Dong A, Nguyen KT, Smil D, Bolshan Y, Hajian T, He H, Seitova A, Chau I, Li F, Poda G, Couture JF, Brown PJ, Al-Awar R, Schapira M, Arrowsmith CH and Vedadi M, Biochem. J, 2013, 449, 151–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grebien F, Vedadi M, Getlik M, Giambruno R, Grover A, Avellino R, Skucha A, Vittori S, Kuznetsova E, Smil D, Barsyte-Lovejoy D, Li F, Poda G, Schapira M, Wu H, Dong A, Senisterra G, Stukalov A, Huber KVM, Schönegger A, Marcellus R, Bilban M, Bock C, Brown PJ, Zuber J, Bennett KL, Al-Awar R, Delwel R, Nerlov C, Arrowsmith CH and Superti-Furga G, Nat. Chem. Biol, 2015, 11, 571–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dölle A, Adhikari B, Krämer A, Weckesser J, Berner N, Berger LM, Diebold M, Szewczyk MM, Barsyte-Lovejoy D, Arrowsmith CH, Gebel J, Löhr F, Dötsch V, Eilers M, Heinzlmeir S, Kuster B, Sotriffer C, Wolf E and Knapp S, J. Med. Chem, 2021, 64, 10682–10710. [DOI] [PubMed] [Google Scholar]
- 51.Yu X, Li D, Kottur J, Shen Y, Kim HS, Park KS, Tsai YH, Gong W, Wang J, Suzuki K, Parker J, Herring L, Kaniskan HÜ, Cai L, Jain R, Liu J, Aggarwal AK, Wang GG and Jin J, Sci. Transl. Med, 2021, 13, eabj1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li D, Yu X, Kottur J, Gong W, Zhang Z, Storey AJ, Tsai YH, Uryu H, Shen Y, Byrum SD, Edmondson RD, Mackintosh SG, Cai L, Liu Z, Aggarwal AK, Tackett AJ, Liu J, Jin J and Wang GG, Oncogene, 2022, 41, 3328–3340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bennett RL, Swaroop A, Troche C and Licht JD, Cold Spring Harb Perspect. Med, 2017, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rahman S, Sowa ME, Ottinger M, Smith JA, Shi Y, Harper JW and Howley PM, Mol. Cell. Biol, 2011, 31, 2641–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang GG, Cai L, Pasillas MP and Kamps MP, Nat. Cell Biol, 2007, 9, 804–812. [DOI] [PubMed] [Google Scholar]
- 56.Irish JC, Mills JN, Turner-Ivey B, Wilson RC, Guest ST, Rutkovsky A, Dombkowski A, Kappler CS, Hardiman G and Ethier SP, Mol. Oncol, 2016, 10, 850–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yuan G, Flores NM, Hausmann S, Lofgren SM, Kharchenko V, Angulo-Ibanez M, Sengupta D, Lu X, Czaban I, Azhibek D, Vicent S, Fischle W, Jaremko M, Fang B, Wistuba II, Chua KF, Roth JA, Minna JD, Shao NY, Jaremko Ł, Mazur PK and Gozani O, Nature, 2021, 590, 504–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Keats JJ, Reiman T, Maxwell CA, Taylor BJ, Larratt LM, Mant MJ, Belch AR and Pilarski LM, Blood, 2003, 101, 1520–1529. [DOI] [PubMed] [Google Scholar]
- 59.Kuo AJ, Cheung P, Chen K, Zee BM, Kioi M, Lauring J, Xi Y, Park BH, Shi X, Garcia BA, Li W and Gozani O, Mol. Cell, 2011, 44, 609–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lhoumaud P, Badri S, Rodriguez-Hernaez J, Sakellaropoulos T, Sethia G, Kloetgen A, Cornwell M, Bhattacharyya S, Ay F, Bonneau R, Tsirigos A and Skok JA, Nat. Commun, 2019, 10, 4843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Böttcher J, Dilworth D, Reiser U, Neumüller RA, Schleicher M, Petronczki M, Zeeb M, Mischerikow N, Allali-Hassani A, Szewczyk MM, Li F, Kennedy S, Vedadi M, Barsyte-Lovejoy D, Brown PJ, Huber KVM, Rogers CM, Wells CI, Fedorov O, Rumpel K, Zoephel A, Mayer M, Wunberg T, Böse D, Zahn S, Arnhof H, Berger H, Reiser C, Hörmann A, Krammer T, Corcokovic M, Sharps B, Winkler S, Häring D, Cockcroft XL, Fuchs JE, Müllauer B, Weiss-Puxbaum A, Gerstberger T, Boehmelt G, Vakoc CR, Arrowsmith CH, Pearson M and McConnell DB, Nat. Chem. Biol, 2019, 15, 822–829. [DOI] [PubMed] [Google Scholar]
- 62.Xu C, Meng F, Park KS, Storey AJ, Gong W, Tsai YH, Gibson E, Byrum SD, Li D, Edmondson RD, Mackintosh SG, Vedadi M, Cai L, Tackett AJ, Kaniskan HÜ, Jin J and Wang GG, Cell Chem. Biol, 2022, 29, 386–397.e389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sun Y, Zhang Y, Chen X, Yu A, Du W, Huang Y, Wu F, Yu L, Li J, Wen C, Yang H, Shi Q, Geng M, Huang X and Xu S, Eur. J. Med. Chem, 2022, 239, 114528. [DOI] [PubMed] [Google Scholar]
- 64.Dilworth D, Hanley RP, Ferreira de Freitas R, Allali-Hassani A, Zhou M, Mehta N, Marunde MR, Ackloo S, Carvalho Machado RA, Khalili Yazdi A, Owens DDG, Vu V, Nie DY, Alqazzaz M, Marcon E, Li F, Chau I, Bolotokova A, Qin S, Lei M, Liu Y, Szewczyk MM, Dong A, Kazemzadeh S, Abramyan T, Popova IK, Hall NW, Meiners MJ, Cheek MA, Gibson E, Kireev D, Greenblatt JF, Keogh MC, Min J, Brown PJ, Vedadi M, Arrowsmith CH, Barsyte-Lovejoy D, James LI and Schapira M, Nat. Chem. Biol, 2022, 18, 56–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Meng F, Xu C, Park KS, Kaniskan HÜ, Wang GG and Jin J, J. Med. Chem, 2022, 65, 10611–10625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ishoey M, Chorn S, Singh N, Jaeger MG, Brand M, Paulk J, Bauer S, Erb MA, Parapatics K, Müller AC, Bennett KL, Ecker GF, Bradner JE and Winter GE, ACS Chem. Biol, 2018, 13, 553–560. [DOI] [PubMed] [Google Scholar]
- 67.Stopa N, Krebs JE and Shechter D, Cell Mol Life Sci, 2015, 72, 2041–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Antonysamy S, Bonday Z, Campbell RM, Doyle B, Druzina Z, Gheyi T, Han B, Jungheim LN, Qian Y, Rauch C, Russell M, Sauder JM, Wasserman SR, Weichert K, Willard FS, Zhang A and Emtage S, Proc. Natl. Acad. Sci. U. S. A, 2012, 109, 17960–17965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yang Y and Bedford MT, Nat. Rev. Cancer, 2013, 13, 37–50. [DOI] [PubMed] [Google Scholar]
- 70.Gullà A, Hideshima T, Bianchi G, Fulciniti M, Kemal Samur M, Qi J, Tai YT, Harada T, Morelli E, Amodio N, Carrasco R, Tagliaferri P, Munshi NC, Tassone P and Anderson KC, Leukemia, 2018, 32, 996–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chan-Penebre E, Kuplast KG, Majer CR, Boriack-Sjodin PA, Wigle TJ, Johnston LD, Rioux N, Munchhof MJ, Jin L, Jacques SL, West KA, Lingaraj T, Stickland K, Ribich SA, Raimondi A, Scott MP, Waters NJ, Pollock RM, Smith JJ, Barbash O, Pappalardi M, Ho TF, Nurse K, Oza KP, Gallagher KT, Kruger R, Moyer MP, Copeland RA, Chesworth R and Duncan KW, Nat. Chem. Biol, 2015, 11, 432–437. [DOI] [PubMed] [Google Scholar]
- 72.Shen Y, Gao G, Yu X, Kim H, Wang L, Xie L, Schwarz M, Chen X, Guccione E, Liu J, Bedford MT and Jin J, J. Med. Chem, 2020, 63, 9977–9989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dancy BM and Cole PA, Chem. Rev, 2015, 115, 2419–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Whedon SD and Cole PA, Curr. Opin. Chem. Biol, 2023, 72, 102255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ludlam WH, Taylor MH, Tanner KG, Denu JM, Goodman RH and Smolik SM, Mol. Cell. Biol, 2002, 22, 3832–3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liu X, Wang L, Zhao K, Thompson PR, Hwang Y, Marmorstein R and Cole PA, Nature, 2008, 451, 846–850. [DOI] [PubMed] [Google Scholar]
- 77.Sun XJ, Man N, Tan Y, Nimer SD and Wang L, Front Oncol, 2015, 5, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Attar N and Kurdistani SK, Cold Spring Harb. Perspect. Med, 2017, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Martínez-Balbás MA, Bauer UM, Nielsen SJ, Brehm A and Kouzarides T, EMBO J, 2000, 19, 662–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Humphreys PG, Bamborough P, Chung CW, Craggs PD, Gordon L, Grandi P, Hayhow TG, Hussain J, Jones KL, Lindon M, Michon AM, Renaux JF, Suckling CJ, Tough DF and Prinjha RK, J. Med. Chem, 2017, 60, 695–709. [DOI] [PubMed] [Google Scholar]
- 81.Bassi ZI, Fillmore MC, Miah AH, Chapman TD, Maller C, Roberts EJ, Davis LC, Lewis DE, Galwey NW, Waddington KE, Parravicini V, Macmillan-Jones AL, Gongora C, Humphreys PG, Churcher I, Prinjha RK and Tough DF, ACS Chem. Biol, 2018, 13, 2862–2867. [DOI] [PubMed] [Google Scholar]
- 82.Romero FA, Murray J, Lai KW, Tsui V, Albrecht BK, An L, Beresini MH, de Leon Boenig G, Bronner SM, Chan EW, Chen KX, Chen Z, Choo EF, Clagg K, Clark K, Crawford TD, Cyr P, de Almeida Nagata D, Gascoigne KE, Grogan JL, Hatzivassiliou G, Huang W, Hunsaker TL, Kaufman S, Koenig SG, Li R, Li Y, Liang X, Liao J, Liu W, Ly J, Maher J, Masui C, Merchant M, Ran Y, Taylor AM, Wai J, Wang F, Wei X, Yu D, Zhu BY, Zhu X and Magnuson S, J. Med. Chem, 2017, 60, 9162–9183. [DOI] [PubMed] [Google Scholar]
- 83.Vannam R, Sayilgan J, Ojeda S, Karakyriakou B, Hu E, Kreuzer J, Morris R, Herrera Lopez XI, Rai S, Haas W, Lawrence M and Ott CJ, Cell Chem. Biol, 2021, 28, 503–514.e512. [DOI] [PubMed] [Google Scholar]
- 84.Michaelides MR, Kluge A, Patane M, Van Drie JH, Wang C, Hansen TM, Risi RM, Mantei R, Hertel C, Karukurichi K, Nesterov A, McElligott D, de Vries P, Langston JW, Cole PA, Marmorstein R, Liu H, Lasko L, Bromberg KD, Lai A and Kesicki EA, ACS Med. Chem. Lett, 2018, 9, 28–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Durbin AD, Wang T, Wimalasena VK, Zimmerman MW, Li D, Dharia NV, Mariani L, Shendy NAM, Nance S, Patel AG, Shao Y, Mundada M, Maxham L, Park PMC, Sigua LH, Morita K, Conway AS, Robichaud AL, Perez-Atayde AR, Bikowitz MJ, Quinn TR, Wiest O, Easton J, Schönbrunn E, Bulyk ML, Abraham BJ, Stegmaier K, Look AT and Qi J, Cancer Discov, 2022, 12, 730–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zeng L and Zhou MM, FEBS Lett, 2002, 513, 124–128. [DOI] [PubMed] [Google Scholar]
- 87.Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert JP, Barsyte-Lovejoy D, Felletar I, Volkmer R, Müller S, Pawson T, Gingras AC, Arrowsmith CH and Knapp S, Cell, 2012, 149, 214–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fujisawa T and Filippakopoulos P, Nat. Rev. Mol. Cell Biol, 2017, 18, 246–262. [DOI] [PubMed] [Google Scholar]
- 89.Cleary SP, Jeck WR, Zhao X, Chen K, Selitsky SR, Savich GL, Tan TX, Wu MC, Getz G, Lawrence MS, Parker JS, Li J, Powers S, Kim H, Fischer S, Guindi M, Ghanekar A and Chiang DY, Hepatology, 2013, 58, 1693–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Odejide O, Weigert O, Lane AA, Toscano D, Lunning MA, Kopp N, Kim S, van Bodegom D, Bolla S, Schatz JH, Teruya-Feldstein J, Hochberg E, Louissaint A, Dorfman D, Stevenson K, Rodig SJ, Piccaluga PP, Jacobsen E, Pileri SA, Harris NL, Ferrero S, Inghirami G, Horwitz SM and Weinstock DM, Blood, 2014, 123, 1293–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Oh HR, An CH, Yoo NJ and Lee SH, Cell Oncol. (Dordr), 2014, 37, 455–461. [DOI] [PubMed] [Google Scholar]
- 92.Reynoird N, Schwartz BE, Delvecchio M, Sadoul K, Meyers D, Mukherjee C, Caron C, Kimura H, Rousseaux S, Cole PA, Panne D, French CA and Khochbin S, EMBO J, 2010, 29, 2943–2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Stathis A, Zucca E, Bekradda M, Gomez-Roca C, Delord JP, de La Motte Rouge T, Uro-Coste E, de Braud F, Pelosi G and French CA, Cancer Discov, 2016, 6, 492–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shorstova T, Foulkes WD and Witcher M, Br J Cancer, 2021, 124, 1478–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rathert P, Roth M, Neumann T, Muerdter F, Roe JS, Muhar M, Deswal S, Cerny-Reiterer S, Peter B, Jude J, Hoffmann T, Boryń Ł M, Axelsson E, Schweifer N, Tontsch-Grunt U, Dow LE, Gianni D, Pearson M, Valent P, Stark A, Kraut N, Vakoc CR and Zuber J, Nature, 2015, 525, 543–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shu S, Lin CY, He HH, Witwicki RM, Tabassum DP, Roberts JM, Janiszewska M, Huh SJ, Liang Y, Ryan J, Doherty E, Mohammed H, Guo H, Stover DG, Ekram MB, Brown J, D’Santos C, Krop IE, Dillon D, McKeown M, Ott C, Qi J, Ni M, Rao PK, Duarte M, Wu SY, Chiang CM, Anders L, Young RA, Winer E, Letai A, Barry WT, Carroll JS, Long H, Brown M, Liu XS, Meyer CA, Bradner JE and Polyak K, Nature, 2016, 529, 413–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yang CY, Qin C, Bai L and Wang S, Drug Discov. Today Technol, 2019, 31, 43–51. [DOI] [PubMed] [Google Scholar]
- 98.Zhou XL, Zhao F, Xu YT, Guan YY, Yu T, Zhang YZ, Duan YC and Zhao Y, Bioorg. Med. Chem, 2022, 73, 117033. [DOI] [PubMed] [Google Scholar]
- 99.Zengerle M, Chan KH and Ciulli A, ACS Chem Biol, 2015, 10, 1770–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Raina K, Lu J, Qian Y, Altieri M, Gordon D, Rossi AM, Wang J, Chen X, Dong H, Siu K, Winkler JD, Crew AP, Crews CM and Coleman KG, Proc. Natl. Acad. Sci. U. S. A, 2016, 113, 7124–7129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chan KH, Zengerle M, Testa A and Ciulli A, J. Med. Chem, 2018, 61, 504–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pietrobono S, Gaudio E, Gagliardi S, Zitani M, Carrassa L, Migliorini F, Petricci E, Manetti F, Makukhin N, Bond AG, Paradise BD, Ciulli A, Fernandez-Zapico ME, Bertoni F and Stecca B, Oncogene, 2021, 40, 3799–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S and Bradner JE, Science, 2015, 348, 1376–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lu J, Qian Y, Altieri M, Dong H, Wang J, Raina K, Hines J, Winkler JD, Crew AP, Coleman K and Crews CM, Chem. Biol, 2015, 22, 755–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Winter GE, Mayer A, Buckley DL, Erb MA, Roderick JE, Vittori S, Reyes JM, di Iulio J, Souza A, Ott CJ, Roberts JM, Zeid R, Scott TG, Paulk J, Lachance K, Olson CM, Dastjerdi S, Bauer S, Lin CY, Gray NS, Kelliher MA, Churchman LS and Bradner JE, Mol. Cell, 2017, 67, 5–18.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhou B, Hu J, Xu F, Chen Z, Bai L, Fernandez-Salas E, Lin M, Liu L, Yang CY, Zhao Y, McEachern D, Przybranowski S, Wen B, Sun D and Wang S, J. Med. Chem, 2018, 61, 462–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bai L, Zhou B, Yang CY, Ji J, McEachern D, Przybranowski S, Jiang H, Hu J, Xu F, Zhao Y, Liu L, Fernandez-Salas E, Xu J, Dou Y, Wen B, Sun D, Meagher J, Stuckey J, Hayes DF, Li S, Ellis MJ and Wang S, Cancer Res, 2017, 77, 2476–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Qin C, Hu Y, Zhou B, Fernandez-Salas E, Yang CY, Liu L, McEachern D, Przybranowski S, Wang M, Stuckey J, Meagher J, Bai L, Chen Z, Lin M, Yang J, Ziazadeh DN, Xu F, Hu J, Xiang W, Huang L, Li S, Wen B, Sun D and Wang S, J. Med. Chem, 2018, 61, 6685–6704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Xiang W, Wang Q, Ran K, Ren J, Shi Y and Yu L, Bioorg. Chem, 2021, 115, 105238. [DOI] [PubMed] [Google Scholar]
- 110.Lu Q, Ding X, Huang T, Zhang S, Li Y, Xu L, Chen G, Ying Y, Wang Y, Feng Z, Wang L and Zou X, Am. J. Transl. Res, 2019, 11, 5728–5739. [PMC free article] [PubMed] [Google Scholar]
- 111.Ran X, Zhao Y, Liu L, Bai L, Yang CY, Zhou B, Meagher JL, Chinnaswamy K, Stuckey JA and Wang S, J. Med. Chem, 2015, 58, 4927–4939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Han Z, Xing X, Hu M, Zhang Y, Liu P and Chai J, Structure, 2007, 15, 1306–1315. [DOI] [PubMed] [Google Scholar]
- 113.Margueron R, Justin N, Ohno K, Sharpe ML, Son J, Drury WJ 3rd, Voigt P, Martin SR, Taylor WR, De Marco V, Pirrotta V, Reinberg D and Gamblin SJ, Nature, 2009, 461, 762–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Liu YL, Gao X, Jiang Y, Zhang G, Sun ZC, Cui BB and Yang YM, J. Cancer Res. Clin. Oncol, 2015, 141, 661–669. [DOI] [PubMed] [Google Scholar]
- 115.Zhang JYJ, Zhang J, Kiffe M, Walles M, Jin Y, Blanz J, Dayer J, Sanchez A, Zhang C, Zhang L, Huang Y and Oyang C, Xenobiotica, 2022, 52, 65–78. [DOI] [PubMed] [Google Scholar]
- 116.Qi W, Zhao K, Gu J, Huang Y, Wang Y, Zhang H, Zhang M, Zhang J, Yu Z, Li L, Teng L, Chuai S, Zhang C, Zhao M, Chan H, Chen Z, Fang D, Fei Q, Feng L, Feng L, Gao Y, Ge H, Ge X, Li G, Lingel A, Lin Y, Liu Y, Luo F, Shi M, Wang L, Wang Z, Yu Y, Zeng J, Zeng C, Zhang L, Zhang Q, Zhou S, Oyang C, Atadja P and Li E, Nat. Chem. Biol, 2017, 13, 381–388. [DOI] [PubMed] [Google Scholar]
- 117.Potjewyd F, Turner AW, Beri J, Rectenwald JM, Norris-Drouin JL, Cholensky SH, Margolis DM, Pearce KH, Herring LE and James LI, Cell Chem. Biol, 2020, 27, 47–56.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Huang Y, Zhang J, Yu Z, Zhang H, Wang Y, Lingel A, Qi W, Gu J, Zhao K, Shultz MD, Wang L, Fu X, Sun Y, Zhang Q, Jiang X, Zhang J, Zhang C, Li L, Zeng J, Feng L, Zhang C, Liu Y, Zhang M, Zhang L, Zhao M, Gao Z, Liu X, Fang D, Guo H, Mi Y, Gabriel T, Dillon MP, Atadja P and Oyang C, J. Med. Chem, 2017, 60, 2215–2226. [DOI] [PubMed] [Google Scholar]
- 119.Hsu JH, Rasmusson T, Robinson J, Pachl F, Read J, Kawatkar S, DH OD, Bagal S, Code E, Rawlins P, Argyrou A, Tomlinson R, Gao N, Zhu X, Chiarparin E, Jacques K, Shen M, Woods H, Bednarski E, Wilson DM, Drew L, Castaldi MP, Fawell S and Bloecher A, Cell Chem. Biol, 2020, 27, 41–46.e17. [DOI] [PubMed] [Google Scholar]
- 120.Cao Q, Wang X, Zhao M, Yang R, Malik R, Qiao Y, Poliakov A, Yocum AK, Li Y, Chen W, Cao X, Jiang X, Dahiya A, Harris C, Feng FY, Kalantry S, Qin ZS, Dhanasekaran SM and Chinnaiyan AM, Nat. Commun, 2014, 5, 3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Stuckey JI, Dickson BM, Cheng N, Liu Y, Norris JL, Cholensky SH, Tempel W, Qin S, Huber KG, Sagum C, Black K, Li F, Huang XP, Roth BL, Baughman BM, Senisterra G, Pattenden SG, Vedadi M, Brown PJ, Bedford MT, Min J, Arrowsmith CH, James LI and Frye SV, Nat. Chem. Biol, 2016, 12, 180–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Siddique HR and Saleem M, Stem Cells, 2012, 30, 372–378. [DOI] [PubMed] [Google Scholar]
- 123.van den Boom V, Maat H, Geugien M, Rodríguez López A, Sotoca AM, Jaques J, Brouwers-Vos AZ, Fusetti F, Groen RW, Yuan H, Martens AC, Stunnenberg HG, Vellenga E, Martens JH and Schuringa JJ, Cell Rep, 2016, 14, 332–346. [DOI] [PubMed] [Google Scholar]
- 124.Kreso A, van Galen P, Pedley NM, Lima-Fernandes E, Frelin C, Davis T, Cao L, Baiazitov R, Du W, Sydorenko N, Moon YC, Gibson L, Wang Y, Leung C, Iscove NN, Arrowsmith CH, Szentgyorgyi E, Gallinger S, Dick JE and O’Brien CA, Nat. Med, 2014, 20, 29–36. [DOI] [PubMed] [Google Scholar]
- 125.Shukla S, Ying W, Gray F, Yao Y, Simes ML, Zhao Q, Miao H, Cho HJ, González-Alonso P, Winkler A, Lund G, Purohit T, Kim E, Zhang X, Ray JM, He S, Nikolaidis C, Ndoj J, Wang J, Jaremko Ł, Jaremko M, Ryan RJH, Guzman ML, Grembecka J and Cierpicki T, Nat. Chem. Biol, 2021, 17, 784–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Park KS, Qin L, Kabir M, Luo K, Dale B, Zhong Y, Kim A, Wang GG, Kaniskan HÜ and Jin J, Adv. Sci. (Weinh), 2023, 10, e2205573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Xiong Y, Zhong Y, Yim H, Yang X, Park KS, Xie L, Poulikakos PI, Han X, Xiong Y, Chen X, Liu J and Jin J, J. Am. Chem. Soc, 2022, 144, 22622–22632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Tsai WW, Wang Z, Yiu TT, Akdemir KC, Xia W, Winter S, Tsai CY, Shi X, Schwarzer D, Plunkett W, Aronow B, Gozani O, Fischle W, Hung MC, Patel DJ and Barton MC, Nature, 2010, 468, 927–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Allton K, Jain AK, Herz HM, Tsai WW, Jung SY, Qin J, Bergmann A, Johnson RL and Barton MC, Proc. Natl. Acad. Sci. U. S. A, 2009, 106, 11612–11616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zhang LH, Yin AA, Cheng JX, Huang HY, Li XM, Zhang YQ, Han N and Zhang X, Oncogene, 2015, 34, 600–610. [DOI] [PubMed] [Google Scholar]
- 131.Palmer WS, Poncet-Montange G, Liu G, Petrocchi A, Reyna N, Subramanian G, Theroff J, Yau A, Kost-Alimova M, Bardenhagen JP, Leo E, Shepard HE, Tieu TN, Shi X, Zhan Y, Zhao S, Barton MC, Draetta G, Toniatti C, Jones P, Geck Do M and Andersen JN, J. Med. Chem, 2016, 59, 1440–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Bennett J, Fedorov O, Tallant C, Monteiro O, Meier J, Gamble V, Savitsky P, Nunez-Alonso GA, Haendler B, Rogers C, Brennan PE, Müller S and Knapp S, J. Med. Chem, 2016, 59, 1642–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Gechijian LN, Buckley DL, Lawlor MA, Reyes JM, Paulk J, Ott CJ, Winter GE, Erb MA, Scott TG, Xu M, Seo HS, Dhe-Paganon S, Kwiatkowski NP, Perry JA, Qi J, Gray NS and Bradner JE, Nat. Chem. Biol, 2018, 14, 405–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J and Crabtree GR, Nat. Genet, 2013, 45, 592–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wang X, Wang S, Troisi EC, Howard TP, Haswell JR, Wolf BK, Hawk WH, Ramos P, Oberlick EM, Tzvetkov EP, Ross A, Vazquez F, Hahn WC, Park PJ and Roberts CWM, Nat. Commun, 2019, 10, 1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kaeser MD, Aslanian A, Dong MQ, Yates JR 3rd and Emerson BM, J. Biol. Chem, 2008, 283, 32254–32263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hohmann AF, Martin LJ, Minder JL, Roe JS, Shi J, Steurer S, Bader G, McConnell D, Pearson M, Gerstberger T, Gottschamel T, Thompson D, Suzuki Y, Koegl M and Vakoc CR, Nat. Chem. Biol, 2016, 12, 672–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Del Gaudio N, Di Costanzo A, Liu NQ, Conte L, Migliaccio A, Vermeulen M, Martens JHA, Stunnenberg HG, Nebbioso A and Altucci L, Cell Death Dis, 2019, 10, 338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Scotto L, Narayan G, Nandula SV, Subramaniyam S, Kaufmann AM, Wright JD, Pothuri B, Mansukhani M, Schneider A, Arias-Pulido H and Murty VV, Mol. Cancer, 2008, 7, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Huang H, Wang Y, Li Q, Fei X, Ma H and Hu R, Cancer Lett, 2019, 446, 81–89. [DOI] [PubMed] [Google Scholar]
- 141.Alpsoy A, Utturkar SM, Carter BC, Dhiman A, Torregrosa-Allen SE, Currie MP, Elzey BD and Dykhuizen EC, Cancer Res, 2021, 81, 820–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Martin LJ, Koegl M, Bader G, Cockcroft XL, Fedorov O, Fiegen D, Gerstberger T, Hofmann MH, Hohmann AF, Kessler D, Knapp S, Knesl P, Kornigg S, Müller S, Nar H, Rogers C, Rumpel K, Schaaf O, Steurer S, Tallant C, Vakoc CR, Zeeb M, Zoephel A, Pearson M, Boehmelt G and McConnell D, J. Med. Chem, 2016, 59, 4462–4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Remillard D, Buckley DL, Paulk J, Brien GL, Sonnett M, Seo HS, Dastjerdi S, Wühr M, Dhe-Paganon S, Armstrong SA and Bradner JE, Angew. Chem. Int. Ed. Engl, 2017, 56, 5738–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Brien GL, Remillard D, Shi J, Hemming ML, Chabon J, Wynne K, Dillon ET, Cagney G, Van Mierlo G, Baltissen MP, Vermeulen M, Qi J, Fröhling S, Gray NS, Bradner JE, Vakoc CR and Armstrong SA, Elife, 2018, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Zoppi V, Hughes SJ, Maniaci C, Testa A, Gmaschitz T, Wieshofer C, Koegl M, Riching KM, Daniels DL, Spallarossa A and Ciulli A, J. Med. Chem, 2019, 62, 699–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Singh M, Popowicz GM, Krajewski M and Holak TA, Chembiochem, 2007, 8, 1308–1316. [DOI] [PubMed] [Google Scholar]
- 147.Flowers S, Nagl NG Jr., Beck GR Jr. and Moran E, J. Biol. Chem, 2009, 284, 10067–10075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wilson BG and Roberts CW, Nat. Rev. Cancer, 2011, 11, 481–492. [DOI] [PubMed] [Google Scholar]
- 149.Wilson BG, Helming KC, Wang X, Kim Y, Vazquez F, Jagani Z, Hahn WC and Roberts CW, Mol. Cell. Biol, 2014, 34, 1136–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Hoffman GR, Rahal R, Buxton F, Xiang K, McAllister G, Frias E, Bagdasarian L, Huber J, Lindeman A, Chen D, Romero R, Ramadan N, Phadke T, Haas K, Jaskelioff M, Wilson BG, Meyer MJ, Saenz-Vash V, Zhai H, Myer VE, Porter JA, Keen N, McLaughlin ME, Mickanin C, Roberts CW, Stegmeier F and Jagani Z, Proc. Natl. Acad. Sci. U. S. A, 2014, 111, 3128–3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Farnaby W, Koegl M, Roy MJ, Whitworth C, Diers E, Trainor N, Zollman D, Steurer S, Karolyi-Oezguer J, Riedmueller C, Gmaschitz T, Wachter J, Dank C, Galant M, Sharps B, Rumpel K, Traxler E, Gerstberger T, Schnitzer R, Petermann O, Greb P, Weinstabl H, Bader G, Zoephel A, Weiss-Puxbaum A, Ehrenhöfer-Wölfer K, Wöhrle S, Boehmelt G, Rinnenthal J, Arnhof H, Wiechens N, Wu MY, Owen-Hughes T, Ettmayer P, Pearson M, McConnell DB and Ciulli A, Nat. Chem. Biol, 2019, 15, 672–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Xiao L, Parolia A, Qiao Y, Bawa P, Eyunni S, Mannan R, Carson SE, Chang Y, Wang X, Zhang Y, Vo JN, Kregel S, Simko SA, Delekta AD, Jaber M, Zheng H, Apel IJ, McMurry L, Su F, Wang R, Zelenka-Wang S, Sasmal S, Khare L, Mukherjee S, Abbineni C, Aithal K, Bhakta MS, Ghurye J, Cao X, Navone NM, Nesvizhskii AI, Mehra R, Vaishampayan U, Blanchette M, Wang Y, Samajdar S, Ramachandra M and Chinnaiyan AM, Nature, 2022, 601, 434–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Sutherell CL, Tallant C, Monteiro OP, Yapp C, Fuchs JE, Fedorov O, Siejka P, Müller S, Knapp S, Brenton JD, Brennan PE and Ley SV, J. Med. Chem, 2016, 59, 5095–5101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kofink C, Trainor N, Mair B, Wöhrle S, Wurm M, Mischerikow N, Roy MJ, Bader G, Greb P, Garavel G, Diers E, McLennan R, Whitworth C, Vetma V, Rumpel K, Scharnweber M, Fuchs JE, Gerstberger T, Cui Y, Gremel G, Chetta P, Hopf S, Budano N, Rinnenthal J, Gmaschitz G, Mayer M, Koegl M, Ciulli A, Weinstabl H and Farnaby W, Nat. Commun, 2022, 13, 5969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Cantley J, Ye X, Rousseau E, Januario T, Hamman BD, Rose CM, Cheung TK, Hinkle T, Soto L, Quinn C, Harbin A, Bortolon E, Chen X, Haskell R, Lin E, Yu SF, Del Rosario G, Chan E, Dunlap D, Koeppen H, Martin S, Merchant M, Grimmer M, Broccatelli F, Wang J, Pizzano J, Dragovich PS, Berlin M and Yauch RL, Nat. Commun, 2022, 13, 6814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Schulze JM, Wang AY and Kobor MS, Biochem Cell Biol, 2009, 87, 65–75. [DOI] [PubMed] [Google Scholar]
- 157.Wan L, Wen H, Li Y, Lyu J, Xi Y, Hoshii T, Joseph JK, Wang X, Loh YE, Erb MA, Souza AL, Bradner JE, Shen L, Li W, Li H, Allis CD, Armstrong SA and Shi X, Nature, 2017, 543, 265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Andrews FH, Shinsky SA, Shanle EK, Bridgers JB, Gest A, Tsun IK, Krajewski K, Shi X, Strahl BD and Kutateladze TG, Nat. Chem. Biol, 2016, 12, 396–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Mueller D, Bach C, Zeisig D, Garcia-Cuellar MP, Monroe S, Sreekumar A, Zhou R, Nesvizhskii A, Chinnaiyan A, Hess JL and Slany RK, Blood, 2007, 110, 4445–4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.He N, Chan CK, Sobhian B, Chou S, Xue Y, Liu M, Alber T, Benkirane M and Zhou Q, Proc. Natl. Acad. Sci. U. S. A, 2011, 108, E636–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Daser A and Rabbitts TH, Genes Dev, 2004, 18, 965–974. [DOI] [PubMed] [Google Scholar]
- 162.Moustakim M, Christott T, Monteiro OP, Bennett J, Giroud C, Ward J, Rogers CM, Smith P, Panagakou I, Díaz-Sáez L, Felce SL, Gamble V, Gileadi C, Halidi N, Heidenreich D, Chaikuad A, Knapp S, Huber KVM, Farnie G, Heer J, Manevski N, Poda G, Al-Awar R, Dixon DJ, Brennan PE and Fedorov O, Angew. Chem. Int. Ed. Engl, 2018, 57, 16302–16307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Li X, Li XM, Jiang Y, Liu Z, Cui Y, Fung KY, van der Beelen SHE, Tian G, Wan L, Shi X, Allis CD, Li H, Li Y and Li XD, Nat. Chem. Biol, 2018, 14, 1140–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Christott T, Bennett J, Coxon C, Monteiro O, Giroud C, Beke V, Felce SL, Gamble V, Gileadi C, Poda G, Al-Awar R, Farnie G and Fedorov O, SLAS Discov, 2019, 24, 133–141. [DOI] [PubMed] [Google Scholar]
- 165.Erb MA, Scott TG, Li BE, Xie H, Paulk J, Seo HS, Souza A, Roberts JM, Dastjerdi S, Buckley DL, Sanjana NE, Shalem O, Nabet B, Zeid R, Offei-Addo NK, Dhe-Paganon S, Zhang F, Orkin SH, Winter GE and Bradner JE, Nature, 2017, 543, 270–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Nabet B, Roberts JM, Buckley DL, Paulk J, Dastjerdi S, Yang A, Leggett AL, Erb MA, Lawlor MA, Souza A, Scott TG, Vittori S, Perry JA, Qi J, Winter GE, Wong KK, Gray NS and Bradner JE, Nat. Chem. Biol, 2018, 14, 431–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Garnar-Wortzel L, Bishop TR, Kitamura S, Milosevich N, Asiaban JN, Zhang X, Zheng Q, Chen E, Ramos AR, Ackerman CJ, Hampton EN, Chatterjee AK, Young TS, Hull MV, Sharpless KB, Cravatt BF, Wolan DW and Erb MA, ACS Cent. Sci, 2021, 7, 815–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Li X, Yao Y, Wu F and Song Y, J. Hematol. Oncol, 2022, 15, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Khochbin S, Verdel A, Lemercier C and Seigneurin-Berny D, Curr. Opin. Genet. Dev, 2001, 11, 162–166. [DOI] [PubMed] [Google Scholar]
- 170.Leipe DD and Landsman D, Nucleic Acids Res, 1997, 25, 3693–3697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Seto E and Yoshida M, Cold Spring Harb. Perspect. Biol, 2014, 6, a018713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Yamaguchi T, Cubizolles F, Zhang Y, Reichert N, Kohler H, Seiser C and Matthias P, Genes Dev, 2010, 24, 455–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Li Y and Seto E, Cold Spring Harb. Perspect. Med, 2016, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Ho TCS, Chan AHY and Ganesan A, J. Med. Chem, 2020, 63, 12460–12484. [DOI] [PubMed] [Google Scholar]
- 175.Beckers T, Burkhardt C, Wieland H, Gimmnich P, Ciossek T, Maier T and Sanders K, Int. J. Cancer, 2007, 121, 1138–1148. [DOI] [PubMed] [Google Scholar]
- 176.Smalley JP, Adams GE, Millard CJ, Song Y, Norris JKS, Schwabe JWR, Cowley SM and Hodgkinson JT, Chem. Commun. (Camb), 2020, 56, 4476–4479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Wang Y, Stowe RL, Pinello CE, Tian G, Madoux F, Li D, Zhao LY, Li JL, Wang Y, Wang Y, Ma H, Hodder P, Roush WR and Liao D, Chem. Biol, 2015, 22, 273–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Xiao Y, Wang J, Zhao LY, Chen X, Zheng G, Zhang X and Liao D, Chem. Commun. (Camb), 2020, 56, 9866–9869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Yang K, Song Y, Xie H, Wu H, Wu YT, Leisten ED and Tang W, Bioorg. Med. Chem. Lett, 2018, 28, 2493–2497. [DOI] [PubMed] [Google Scholar]
- 180.Bergman JA, Woan K, Perez-Villarroel P, Villagra A, Sotomayor EM and Kozikowski AP, J. Med. Chem, 2012, 55, 9891–9899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Wu H, Yang K, Zhang Z, Leisten ED, Li Z, Xie H, Liu J, Smith KA, Novakova Z, Barinka C and Tang W, J. Med. Chem, 2019, 62, 7042–7057. [DOI] [PubMed] [Google Scholar]
- 182.An Z, Lv W, Su S, Wu W and Rao Y, Protein & Cell, 2019, 10, 606–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Yang K, Wu H, Zhang Z, Leisten ED, Nie X, Liu B, Wen Z, Zhang J, Cunningham MD and Tang W, ACS Med. Chem. Lett, 2020, 11, 575–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Frye RA, Biochem Biophys. Res. Commun, 2000, 273, 793–798. [DOI] [PubMed] [Google Scholar]
- 185.Chen G, Huang P and Hu C, Int. J. Cancer, 2020, 147, 3297–3304. [DOI] [PubMed] [Google Scholar]
- 186.Schiedel M, Herp D, Hammelmann S, Swyter S, Lehotzky A, Robaa D, Oláh J, Ovádi J, Sippl W and Jung M, J. Med. Chem, 2018, 61, 482–491. [DOI] [PubMed] [Google Scholar]
- 187.Iconomou M and Saunders DN, Biochem. J, 2016, 473, 4083–4101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Ishida T and Ciulli A, SLAS Discov, 2021, 26, 484–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Hines J, Lartigue S, Dong H, Qian Y and Crews CM, Cancer Res, 2019, 79, 251–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Ohoka N, Ujikawa O, Shimokawa K, Sameshima T, Shibata N, Hattori T, Nara H, Cho N and Naito M, Chem. Pharm. Bull. (Tokyo), 2019, 67, 203–209. [DOI] [PubMed] [Google Scholar]
- 191.Wei J, Meng F, Park KS, Yim H, Velez J, Kumar P, Wang L, Xie L, Chen H, Shen Y, Teichman E, Li D, Wang GG, Chen X, Kaniskan HÜ and Jin J, J. Am. Chem. Soc, 2021, 143, 15073–15083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Tong B, Luo M, Xie Y, Spradlin JN, Tallarico JA, McKenna JM, Schirle M, Maimone TJ and Nomura DK, Sci. Rep, 2020, 10, 15543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Li L, Mi D, Pei H, Duan Q, Wang X, Zhou W, Jin J, Li D, Liu M and Chen Y, Signal Transduct. Target Ther, 2020, 5, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Ward CC, Kleinman JI, Brittain SM, Lee PS, Chung CYS, Kim K, Petri Y, Thomas JR, Tallarico JA, McKenna JM, Schirle M and Nomura DK, ACS Chem. Biol, 2019, 14, 2430–2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Spradlin JN, Hu X, Ward CC, Brittain SM, Jones MD, Ou L, To M, Proudfoot A, Ornelas E, Woldegiorgis M, Olzmann JA, Bussiere DE, Thomas JR, Tallarico JA, McKenna JM, Schirle M, Maimone TJ and Nomura DK, Nat. Chem. Biol, 2019, 15, 747–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Luo M, Spradlin JN, Boike L, Tong B, Brittain SM, McKenna JM, Tallarico JA, Schirle M, Maimone TJ and Nomura DK, Cell Chem. Biol, 2021, 28, 559–566.e515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Zhang X, Crowley VM, Wucherpfennig TG, Dix MM and Cravatt BF, Nat. Chem. Biol, 2019, 15, 737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Henning NJ, Manford AG, Spradlin JN, Brittain SM, Zhang E, McKenna JM, Tallarico JA, Schirle M, Rape M and Nomura DK, J. Am. Chem. Soc, 2022, 144, 701–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Shergalis AG, Marin VL, Rhee DY, Senaweera S, McCloud RL, Ronau JA, Hutchins CW, McLoughlin S, Woller KR, Warder SE, Vasudevan A and Reitsma JM, ACS Chem. Biol, 2023, 18, 331–339. [DOI] [PubMed] [Google Scholar]
- 200.Dragovich P, Thomas P, Blake RA & Wertz I Chemical Inducers of Degradation and Methods of Use. WO2020086858, 2020.
- 201.Li YD, Ma MW, Hassan MM, Hunkeler M, Teng M, Puvar K, Lumpkin R, Sandoval B, Jin CY, Ficarro SB, Wang MY, Xu S, Groendyke BJ, Sigua LH, Tavares I, Zou C, Tsai JM, Park PMC, Yoon H, Majewski FC, Marto JA, Qi J, Nowak RP, Donovan KA, Słabicki M, Gray NS, Fischer ES and Ebert BL, bioRxiv, 2023, DOI: 10.1101/2023.02.14.528208. [DOI] [Google Scholar]
- 202.Hsia O, Hinterndorfer M, Cowan AD, Iso K, Ishida T, Sundaramoorthy R, Nakasone MA, Rukavina A, Husnjak K, Wegner M, Correa-Sáez A, Craigon C, Maniaci C, Testa A, Kaulich M, Dikic I, Winter GE and Ciulli A, bioRxiv, 2023, DOI: 10.1101/2023.02.14.528511. [DOI] [Google Scholar]
- 203.Xue G, Wang K, Zhou D, Zhong H and Pan Z, J. Am. Chem. Soc, 2019, 141, 18370–18374. [DOI] [PubMed] [Google Scholar]
- 204.Naro Y, Darrah K and Deiters A, J. Am. Chem. Soc, 2020, 142, 2193–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Liu J, Chen H, Ma L, He Z, Wang D, Liu Y, Lin Q, Zhang T, Gray N, Kaniskan HÜ, Jin J and Wei W, Sci. Adv, 2020, 6, eaay5154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Li Z, Ma S, Yang X, Zhang L, Liang D, Dong G, Du L, Lv Z and Li M, Eur. J. Med. Chem, 2021, 222, 113608. [DOI] [PubMed] [Google Scholar]
- 207.Pfaff P, Samarasinghe KTG, Crews CM and Carreira EM, ACS Cent. Sci, 2019, 5, 1682–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Reynders M, Matsuura BS, Bérouti M, Simoneschi D, Marzio A, Pagano M and Trauner D, Sci. Adv, 2020, 6, eaay5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Yang C, Yang Y, Li Y, Ni Q and Li J, J Am Chem Soc, 2023, 145, 385–391. [DOI] [PubMed] [Google Scholar]
- 210.Liu J, Chen H, Liu Y, Shen Y, Meng F, Kaniskan HÜ, Jin J and Wei W, J. Am. Chem. Soc, 2021, 143, 7380–7387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.He S, Gao F, Ma J, Ma H, Dong G and Sheng C, Angew. Chem. Int. Ed. Engl, 2021, 60, 23299–23305. [DOI] [PubMed] [Google Scholar]
- 212.Tanaka M, Roberts JM, Seo HS, Souza A, Paulk J, Scott TG, DeAngelo SL, Dhe-Paganon S and Bradner JE, Nat. Chem. Biol, 2016, 12, 1089–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Imaide S, Riching KM, Makukhin N, Vetma V, Whitworth C, Hughes SJ, Trainor N, Mahan SD, Murphy N, Cowan AD, Chan KH, Craigon C, Testa A, Maniaci C, Urh M, Daniels DL and Ciulli A, Nat. Chem. Biol, 2021, 17, 1157–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Huang Y, Yokoe H, Kaiho-Soma A, Takahashi K, Hirasawa Y, Morita H, Ohtake F and Kanoh N, Bioconjug. Chem, 2022, 33, 142–151. [DOI] [PubMed] [Google Scholar]
- 215.Dragovich PS, Chem. Soc. Rev, 2022, 51, 3886–3897. [DOI] [PubMed] [Google Scholar]
- 216.Zheng S, Tan Y, Wang Z, Li C, Zhang Z, Sang X, Chen H and Yang Y, Nat. Mach. Intell, 2022, 4, 739–748. [Google Scholar]
- 217.Békés M, Langley DR and Crews CM, Nat. Rev. Drug Discov, 2022, 21, 181–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Zhang L, Riley-Gillis B, Vijay P and Shen Y, Mol. Cancer Ther, 2019, 18, 1302–1311. [DOI] [PubMed] [Google Scholar]
- 219.Shirasaki R, Matthews GM, Gandolfi S, de Matos Simoes R, Buckley DL, Raja Vora J, Sievers QL, Brüggenthies JB, Dashevsky O, Poarch H, Tang H, Bariteau MA, Sheffer M, Hu Y, Downey-Kopyscinski SL, Hengeveld PJ, Glassner BJ, Dhimolea E, Ott CJ, Zhang T, Kwiatkowski NP, Laubach JP, Schlossman RL, Richardson PG, Culhane AC, Groen RWJ, Fischer ES, Vazquez F, Tsherniak A, Hahn WC, Levy J, Auclair D, Licht JD, Keats JJ, Boise LH, Ebert BL, Bradner JE, Gray NS and Mitsiades CS, Cell Rep, 2021, 34, 108532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Hanzl A, Casement R, Imrichova H, Hughes SJ, Barone E, Testa A, Bauer S, Wright J, Brand M, Ciulli A and Winter GE, Nat. Chem. Biol, 2023, 19, 323–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.