

Abstract
Ex vivo gene editing in hematopoietic stem and progenitor cells (HSPCs) represents a promising curative treatment strategy for monogenic blood disorders. Gene editing using the homology-directed repair (HDR) pathway enables precise genetic modifications ranging from single base pair correction to replacement or insertion of large DNA segments. Hence, HDR-based gene editing could facilitate broad application of gene editing across monogenic disorders, but the technology still faces challenges for clinical translation. Among these, recent studies demonstrate induction of a DNA damage response (DDR) and p53 activation caused by DNA double-strand breaks and exposure to recombinant adeno-associated virus vector repair templates, resulting in reduced proliferation, engraftment, and clonogenic capacity of edited HSPCs. While different mitigation strategies can reduce this DDR, more research is needed on this phenomenon to ensure safe and efficient implementation of HDR-based gene editing in the clinic.
Keywords: p53, p21, GSE56, DDR, DNA damage response, CRISPR-Cas, ZFNs, TALENs, gene editing, Cas9, sgRNA, HDR, NHEJ, homology-directed repair, HR, homologous recombination
Graphical abstract
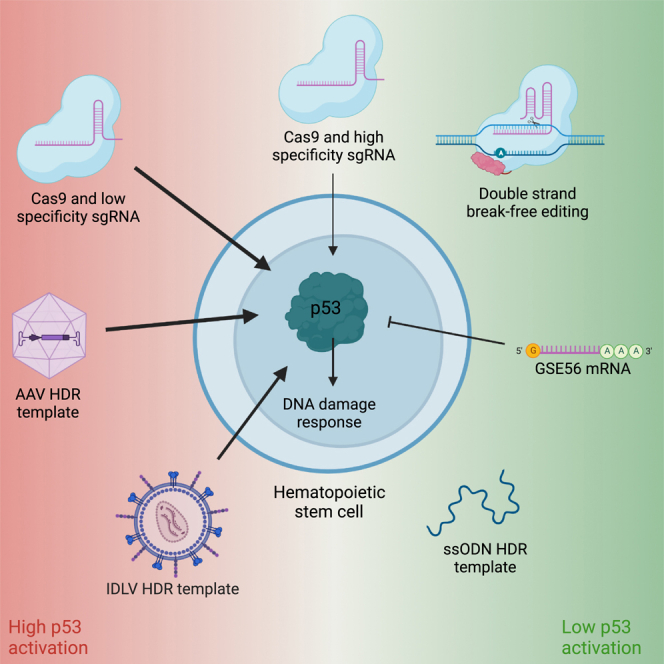
Dorset and Bak review the challenge of the p53-induced DNA damage response (DDR) following CRISPR-Cas9-based gene editing of hematopoietic stem and progenitor cells (HSPCs). They describe strategies for mitigation of the DDR and provide suggestions for additional pre-clinical evaluation of HSPC functionality upon gene editing for ex vivo gene therapies.
Gene editing in HSPCs with the CRISPR-Cas9 system
Ex vivo genome editing of HSPCs with programmable nucleases is a promising platform for development of curative treatments for monogenic blood disorders, with remarkable results from the first clinical trials in sickle cell disease (SCD) and β-thalassemia.1,2,3 CRISPR-Cas gene editing tools were developed from adaptive defense systems of bacteria and archaea and have advanced to become the most utilized methodology for genome engineering. The conventional CRISPR-Cas system is comprised of the endonuclease Cas9 and a single guide RNA (sgRNA) that guides Cas9 to a specific chromosomal location, where Cas9 introduces a DNA double-strand break (DSB), which is repaired mainly through one of two pathways: non-homologous end joining (NHEJ) or homology-directed repair (HDR). Whereas NHEJ generates small insertions or deletions (indels) of various types, the HDR pathway can be leveraged for precise gene editing by supplying a homologous DNA repair template carrying specific genetic changes. CRISPR-Cas9 gene editing in hematopoietic stem and progenitor cells (HSPCs) can be performed by electroporation of cells with precomplexed ribonucleoprotein (RNP) consisting of recombinant Cas9 protein and a synthetic sgRNA, followed by delivery of the repair template by transduction of the cells with recombinant adeno-associated virus 6 (rAAV6) vectors or integration-defective lentiviral vectors (IDLVs).4,5,6 Alternatively, the repair template can be co-delivered during electroporation as single- or double-stranded DNA.7,8,9
Blood disorders that can be treated with an NHEJ-based gene editing strategy constitute the “low-hanging fruit” for clinical translation and commercialization because of the highly efficient NHEJ pathway and no requirement for exposing cells to exogenous DNA. Thus, this strategy has already proven efficient in β-globinopathies, where induced indels in the erythroid enhancer of the BCL11a gene reactivates γ-globin, which compensates for the lack of functional β-globin. This strategy has proven highly efficient and safe, with the first treated patients showing no adverse effects more than 3 years post treatment.1,2,3 On the other hand, HDR has been shown in numerous preclinical studies to be relatively inefficient in long-term repopulating hematopoietic stem cells (LT-HSCs), and a reduction in repopulation capacity of treated HSPCs has also been widely observed.5,9,10 One of the challenges of HDR-based gene editing in LT-HSCs is quiescence or slow cycling of the cells because the HDR machinery is mostly active in the S/G2 phases of the cell cycle. Opposite to that, the NHEJ repair pathway is active throughout the cell cycle and, hence, is the main repair mechanism in LT-HSCs.11,12 Therefore, stimulating the cells into cycling using cytokine-rich medium is essential to achieve HDR. The competition between HDR and NHEJ constitutes another challenge for HDR-based gene editing because the indels produced by NHEJ, when within the coding region of a gene, can result in frameshift mutations and, thus, non-functional gene products from these alleles.
HSPC exposure to stress stimuli, such as reactive oxygen species and DNA damage, can result in loss of self-renewal capacity or apoptosis; for example, through increased expression of cell cycle inhibitors such as p16INK4a,13 activation of p38 mitogen-activated protein kinase14 or inhibitor of DNA binding 1,15 or by the unfolded protein response, which is a focal point of stress stimuli.16
The p53-induced DNA damage response
Studies in non-HSPCs have reported that Cas9-mediated DSBs induce a DNA damage response (DDR) and activation of the tumor suppressor p53.17,18,19,20,21 p53 is a transcription factor with an essential role in mediating a DDR because it upregulates the expression of genes involved in DNA repair, cell-cycle arrest, and apoptosis.22 In addition, p53 can induce apoptosis through a transcription-independent mechanism involving Bcl-2-family proteins.23 Accordingly, Cas9-mediated p53 activation results in cytotoxicity and cell-cycle arrest of gene-edited cells.17,18,20 Additionally, studies in human and mouse cell lines have shown that gene editing can result in enrichment of p53-deficient cells with a correlation between the magnitude of the DDR and the degree of enrichment of p53-deficient cells.17,19,21 Because the p53-encoding gene, TP53, is the most frequently mutated gene in cancer, enrichment of p53-deficient cells could predispose to cancer development upon transplantation of edited cells. However, Cromer et al.24 recently used ultra-deep sequencing of ex vivo-expanded edited HSPCs and did not identify any enrichment of tumorigenic gene variants among 523 known tumor suppressors and oncogenes, including TP53. Even without enrichment for p53-deficient cells, there might be an increased risk of developing malignancies following autologous transplantation of ex vivo gene-edited HSPCs. Myeloid malignancies caused by clonal expansion of preexisting TP53 mutant cells have been observed in some SCD patients following allogeneic transplantation, but only in patients who did not successfully engraft.25 These patients had undergone non-myeloablative conditioning prior to the transplantation, whereas myeloablative conditioning has been used so far in autologous transplantation of ex vivo gene-edited HSPCs.1,2,3 In a large study of 910 SCD patients who received transplants, none of the 478 patients who underwent myeloablative conditioning developed malignant neoplasms, while 6 of the 432 patients who underwent non-myeloablative or reduced intensity conditioning regimens did.26 In another study using autologous transplantation of ex vivo lentivirally transduced HSPCs, 2 of 47 participants developed therapy-related myeloid neoplasm, of which one might be ascribed to the myeloablative conditioning. It was also hypothesized that the stress of switching from homeostatic to regenerative hematopoiesis, possibly combined with exposure of the HSCs to chronic inflammation and hypoxemia in SCD patients, might have promoted malignant transformation of dormant pre-malignant clones.27,28
p53 activation upon gene editing in HSPCs
Transient DDR and p53 activation caused by Cas9-mediated DSBs have also been observed in HSPCs, resulting in cell proliferation delay, cell-cycle arrest, and reduced engraftment of edited cells (Figure 1).9 The number of DSBs correlates with the amplitude of the DDR and the impact on HSPC colony-forming unit (CFU) capacity. Schiroli et al.9 found that electroporation of HSPCs with RNP complexes with a low-specificity sgRNA causing multiple DSBs resulted in a greater than 20-fold upregulation of the p53 target gene CDKN1A (hereafter referred to as p21), a greater than 25-fold reduction in cell number 7 days post electroporation, as well as a 2-fold reduction in colony formation. In contrast, editing with a highly specific sgRNA only caused a 3-fold increase in p21 levels and did not adversely affect clonogenic output or HSPC apoptosis. While this study did not investigate any engraftment differences between control and NHEJ-edited HSPCs, other studies in mice and non-human primates (NHPs) show varying results.29,30,31,32,33 Some studies show marked reductions in engraftment between control cells and edited cells as well as decreased indel frequencies in LT-HSCs compared with transplanted HSPCs,29,32,33 while other studies show minimal impact on both parameters.30,31 These discrepancies might be ascribed to differences in sgRNA specificity and number of transplanted HSPCs, inefficient myeloablation in the autologous NHP setting, and optimization of editing protocols. Furthermore, NHPs may not serve as a good model to monitor effects of p53 activation on HSPCs because p53 upregulation has been shown to last longer in NHP HSPCs compared with human HSPCs using the same sgRNA.29 Although this sgRNA was found to be highly specific, this difference might also be caused by different off-target activity in the two species. Because the number of DSBs correlates with p53 activation, future studies may analyze the distinct contribution of this parameter to evaluate clinical gene editing strategies. For example, γ-globin reactivation is also pursued by targeting the promoter region of the two γ-globin genes that are juxtaposed in the globin gene cluster. With a total of four potential DSBs, excluding any off-target DSBs, and deletions being frequently introduced between the DSBs, this strategy might exacerbate the DDR compared with single-target-site strategies. sgRNA selection for clinical editing approaches involves bioinformatic sgRNA design and off-target analyses and may be complemented with quantitative studies of DDR; for example, by high-throughput imaging, as recently presented by Allen et al.34 Notwithstanding the pre-clinical results, clinical trials have proven that NHEJ-based gene editing can be safe and efficient. Here, indel frequencies have been maintained at 60% or greater for more than 1 year of follow-up in 7 SCD patients1 and in 15 of 16 β-thalassemia patients,2,3 and while nothing is reported on hematopoietic clonality in two of the studies, the third study shows consistent and broad indel distribution in edited cells of two patients over the 18-month follow-up period, indicating no substantial clonal expansion and no adverse impact on hematopoietic recovery following transplantation.3 This suggests that use of highly specific sgRNAs suffices to keep the p53-mediated DDR at low levels with minimal impact on HSC function and repopulation capacity. However, longer follow-up and inclusion of more patients is essential to validate NHEJ-based gene editing as a viable clinical strategy.
Figure 1.

DDR upon gene editing of HSPCs
Cas9-induced DSBs activate a DDR mediated by p53 activation in HSPCs, with the DDR magnitude dependent on the number of DSBs. The response is exacerbated by rAAV6 transduction because of p53 activation caused by the rAAV vector DNA in the nuclei of the cells. The DDR causes decreased proliferation, cell-cycle arrest, and apoptosis, ultimately leading to impaired engraftment and reduced clonality of the human graft upon xenotransplantation in mice. The DDR can be partially mitigated through p53 inhibition using a dominant-negative p53 mutant (GSE56), resulting in higher clonality and engraftment of gene-edited HSPCs.
On the other hand, gene editing relying on rAAV6 to facilitate HDR was found to induce additional p53 activation and delay cell proliferation, with cumulative responses observed for treatment with both RNP complexes and rAAV6.6,9 Schiroli et al.9 showed that rAAV6 transduction alone led to an almost 10-fold upregulation of p21 expression, whereas RNP (high-specificity sgRNA) + rAAV6 resulted in almost 15-fold upregulation. Repopulation studies in immunodeficient NSG mice showed a drop from 40% chimerism to around 15% in HDR-edited HSPCs compared with control cells, a decrease that is in line with other studies.35,36 Ferrari et al.6 observed similar accumulating adverse effects of RNP + rAAV6 inducing 25- to 30-fold p21 upregulation across three different genomic targets as well as rAAV6 dose-dependent decreases in CFU output and engraftment in immunodeficient mice (Figure 1). Clonal tracking studies of transplanted HSPCs using genetic barcodes in the rAAV6 repair templates can validate successful engraftment of edited LT-HSCs with multilineage potential and investigate the graft clonality.37,38 Such studies reveal that human grafts in mice generally have an oligoclonal composition.37,38 In fact, Ferrari et al.37 found that the human grafts originated from as few as 6 dominant edited clones 18 weeks after transplantation of all cells originating from 1–3 × 105 HSPCs transplanted 4 days post seeding with editing efficiencies of ∼65%. Aside from prolonging hematopoietic recovery time and reducing graft size, oligoclonal composition of the graft might compromise the long-term stability and safety of the graft because of replicative stress.39 Ferrari et al.6 found that the p53-mediated DDR from rAAV transduction was mainly caused by the AAV vector genome because empty AAV particles without DNA cargo induced 3-fold lower p53 activation and an almost 2-fold increase in engraftment up to 20 weeks after transplantation in immunodeficient mice. In addition, nuclear entry of the AAV vector genome was necessary for p53 activation, and the response was exacerbated for self-complementary rAAV6 compared with single-stranded rAAV6.6 The authors suggest that the inverted terminal repeats of the vector genome are the main source of DDR activation caused by sensing by the MRE11-RAD50-NBS1 (MRN) complex. However, such responses can be difficult to distinguish from other innate immune responses. Nevertheless, these findings pose a clear challenge for clinical application of CRISPR-Cas9- and rAAV6-based ex vivo HSPCs gene editing, with major concerns related to loss of LT-HSCs and risk of graft failure and/or decreased editing rates in the LT-HSC pool.
Mitigation of the p53-mediated DNA damage response
The aggravating effect of rAAV6 donor delivery on p53 responses and engraftment potential has been mitigated using the dominant-negative p53 mutant GSE56. Co-delivery of GSE56-encoding mRNA reduced p53 transcriptional responses 2-fold upon RNP + rAAV6 gene editing (Figure 1).9,37 Accordingly, GSE56 treatment partially rescued the proliferation delay following RNP + rAAV6 treatment, and CFU numbers and mouse engraftment increased ∼2-fold and ∼5-fold, respectively.9,37 This was accompanied by an increase in edited cells, with up to 50% of the human graft consisting of HDR-edited cells.9,37 Importantly, GSE56 treatment did not lead to compromised chromosomal stability, nor did it significantly increase mutations in any of 151 cancer-associated genes examined by deep sequencing.9
Another approach for reducing the p53-induced DDR is by decreasing the AAV6 dose needed for gene editing. Studies clearly show that increasing the nuclear concentration of the repair template can push the repair outcome to favor HDR.40 Hence, lowering the AAV6 dose would favor NHEJ, but this can be circumvented by transient suppression of NHEJ; e.g., by inhibiting p53-binding protein 1 (53BP1), which promotes NHEJ-repair of DSBs. This can increase HDR in HSPCs up to 2- to 3-fold without affecting the viability of the cells.41,42,43 Consequently, an 8-fold reduction of the AAV6 dose could be used without reducing HDR rates, which consequently reduced p21 induction.43 Small-molecule NHEJ inhibitors have also been used to enhance HDR rates up to 2.5-fold across 22 genes in primary T cells,8 and recently, a Cas9 fusion protein that promotes HDR and inhibits NHEJ has been shown to increase the HDR/NHEJ ratio of the induced edit 1.3- to 1.8-fold in HSPCs compared with Cas9.44 A positive derived effect of NHEJ inhibition is reduction in indels and, therefore, increased purity of the gene edit in the HSPC population. Finally, forcing cell cycle progression using the adenovirus 5 E4orf6/7 protein can also promote HDR.37 To avoid pushing HSCs into differentiation, induction of quiescence after a short, controlled period of cycling to maintain the stemness of the HSPC population after editing has been shown to increase the HDR/NHEJ ratio by 6-fold.12
Because rAAV transduction led to p53 activation even without the presence of RNP complexes, Ferrari et al.6 examined whether they could dampen the DDR by changing to an IDLV for template delivery instead of rAAV6. They found that RNP + IDLV treatment resulted in less efficient HDR than when using rAAV, but IDLV without GSE56 co-delivery did not activate p53 responses to the same degree as AAV with co-delivery of GSE56 mRNA. In addition, use of IDLV rather than rAAV6 resulted in a higher clonogenic capacity with a 2-fold increase in colony number as well as an ∼50% larger and more stable HDR-edited population in xenografts up to 14 weeks after transplantation.6 Because previous studies show that IDLVs efficiently escape innate immune sensing,45 this might be a contributing factor and subject of investigation in rAAV6 template delivery.
Another platform for template delivery in HSPC gene editing is single- and double-stranded DNA templates. These are preferable because they are non-viral and only induce DDRs similar to that of RNP complexes.9 Whereas long double- and single-stranded DNA templates induce only less than 20% HDR in vitro,8,9 short single-stranded deoxynucleotides (ssODNs) can lead to the same HDR efficiency as rAAV6 transduction.9 However, the capacity of ssODNs is limited to a few hundred bases; thus, ssODNs can only be used for correction of small mutations.
In recent years, novel CRISPR-Cas technologies, including base editing46 and prime editing,47 have been developed. Base editing is based on a fusion protein of Cas9 nickase and a deaminase enzyme programmed with an sgRNA and can be used for introduction of the four transition mutations.46,48 Prime editing is also based on a Cas9-nickase fusion protein but with an engineered reverse transcriptase and is programmed using a prime editing guide RNA (pegRNA), which, aside from defining the target site, also encodes the desired edit. Even though prime editing is template based, it can only be used for relatively small genomic changes of less than 100 bp.47 These technologies do not introduce DSBs and, hence, have been found to induce minimal indel formation and genome products of high purity. In addition, they should not induce p53-mediated DDR caused by DSBs. However, while adenine base editors have not been found to induce p53 activation over that observed for mock electroporated cells, cytosine base editors have been found to induce p53 activation comparable with Cas9 in HSPCs.49,50 In addition, transient p53 inhibition by co-delivery of a plasmid encoding a dominant-negative fragment of p53 has been found to increase the efficiency of cytosine base editing and prime editing 2- to 3-fold in a human pluripotent stem cell line.51 Base editing can reach greater than 80% efficiency in HSPCs, and base-edited cells have shown engraftment similar to mock-treated cells, with no decrease in editing 16 weeks after xenotransplantation in mice.50,52,53,54 Recently, prime editing efficiencies of 15%–40% in HSPCs has been reported without a decrease 17 weeks after xenotransplantation in mice.55 Furthermore, base editing and prime editing have shown promising results for in vivo genome editing. Approximately 60% adenine base editing and ∼40% prime editing have been achieved in murine peripheral blood mononuclear cells (PBMCs) and bone marrow mononuclear cells after a single intravenous vector injection and following O6-benzylguanine and carmustine selection for edited cells. This selection was necessary because editing could not be detected before selection.56,57 Because neither of the technologies are dependent on delivery of an exogenous DNA template, they may be more cost effective compared with HDR-based gene editing of small indels. However, they cannot substitute HDR-based gene editing for insertion of large transgenes.
One promising technique for ex vivo precise large gene insertion in HSPCs is an extension of the prime editing technology called programmable addition via site-specific targeting elements (PASTE). The PASTE system is based on a fusion protein comprised of Cas9 nickase, reverse transcriptase, and a serine integrase and first installs a landing pad sequence using prime editing, followed by targeted insertion into the landing pad by the serine integrase. This technology can be used for programmable genome integration of up to 36-kb DNA sequences. Like prime editing, PASTE is not dependent on cell cycling of the target cells, and, in addition, it does not introduce DSBs and, therefore, induces minimal indel formation.58 However, the efficiency of PASTE in primary cells is still low, and editing in HSPCs has not yet been reported.
Clinical translation of ex vivo HSPC gene editing therapies
Ex vivo HSPC gene editing therapies based on HDR are starting to emerge in clinical trials. Recently, the first clinical trial for SCD using HDR by rAAV6 repair template delivery (ClinicalTrials.gov: NCT04819841) was initiated by Graphite Bio. Even though the pre-clinical experiments showed median gene editing of 30% in HSPCs from SCD patients as well as multilineage engraftment 16 weeks after transplantation in mice, a 2-fold decrease in gene editing and engraftment was observed compared with control treated cells. At clinical-scale manufacturing, gene-edited HSPCs from healthy donors showed a 6-fold decrease in engraftment in the bone marrow 20 weeks after transplantation compared with unedited cells.35 The trial was voluntarily discontinued after treatment of the first patient because of a severe adverse event of pancytopenia reported in January 2023. Graphite Bio recently announced discontinuation of this gene therapy, and the community awaits further analysis of the adverse event. The reduced engraftment of the gene-edited HSPCs observed in the preclinical experiments leads one to speculate whether loss of stemness of the gene-edited HSCs could have been the cause of the observed adverse event. A similar trial led by UCSF Benioff Children’s Hospital Oakland for SCD will be using ssODNs as HDR repair templates (ClinicalTrials.gov: NCT04774536). This academic trial is approved and is currently enrolling patients. Results from this trial will hopefully provide more vital knowledge about clinical translation of ex vivo HDR-based gene editing of HSPCs.
Still, stringent preclinical evaluation of the state and functionality of gene-edited HSPCs products is warranted. Limiting-dilution transplantation studies of HSPCs and hematopoietic clonality studies optimally using barcoded DNA donors37,38,59 or analyzing the indel spectrum over time,3,6 albeit with reduced clonal resolution, would be tools to consider implementing routinely. Engraftment studies in NHPs would be optimal because these larger animals better recapitulate regenerative hematopoiesis in humans and allow prolonged examination of long-term engraftment but also because engraftment might be established by different HSPC subpopulations in mice and NHPs.60 In addition to these pre-clinical studies, optimization of the manufacturing protocols for HDR-based ex vivo gene-edited HSPC products could include transient inhibition of p53 as well as minimizing p53-induced DDR through template optimization or template delivery choice.
The serious consequences of DDR and p53 activation in edited HSPCs highlight the importance of HSPC state and viability at the time of transplantation. Mitigation of these responses to increase the size and clonal diversity of the graft seems to be needed to realize the therapeutic potential of ex vivo HSPC gene editing by HDR. While such therapies are slowly emerging in the clinical setting, the previously described DNA break-free editing modalities that do not require DNA template delivery have matured. For example, Beam Therapeutics enrolled the first patient for SCD base editing in November 2022 (ClinicalTrials.gov: NCT05456880). Even so, HDR still constitutes the most flexible gene editing modality in terms of editing outcome possibilities and the ability to make sizable changes to the genome. Future insights into the impact of HDR gene editing on HSPC function and scrutiny of the first clinical data may deliver mitigation strategies that solve these toxicity issues. These strategies could, for example, entail general improvements to genome editing protocols, modifications to template delivery strategies, and manipulation of cellular response pathways, which can hopefully pave the way for successful and broad implementation of gene editing in blood disorders and beyond.
Acknowledgments
Funding in the Bak Lab is supported by a grant from the Danish health authorities (SST) (4-1612-391/1), the EU Commission in the form of an ERC starting grant (project 101041231, Horizon Europe Pillar I) and a grant from the Horizon Research and Innovation Actions (project 101057438, Horizon Europe Pillar II), a Lundbeck Foundation fellowship (R238-2016-3349), the Independent Research Fund Denmark (0134-00113B, 0242-00009B, and 9144-00001B), an AIAS-COFUND (Marie Curie) fellowship from the Aarhus Institute of Advanced Studies (AIAS) co-funded by Aarhus University’s Research Foundation and the European Union’s Seventh Framework Program under grant agreement 609033, the Novo Nordisk Foundation (NNF19OC0058238 and NNF17OC0028894), Innovation Fund Denmark (8056-00010B), the Carlsberg Foundation (CF20-0424 and CF17-0129), Slagtermester Max Wørzner og Hustru Inger Wørzners Mindelegat, the A.P. Møller Foundation, the Riisfort Foundation, the Agnes and Poul Friis’ Foundation, and a Genome Engineer Innovation Grant from Synthego. The figure was created with BioRender.
Author contributions
S.R.D. took the lead in writing the manuscript with assistance from R.O.B. S.R.D. drafted the figures with input from R.O.B. Both authors reviewed, edited, and approved the final manuscript.
Declaration of interests
R.O.B. is a co-inventor of patents on HDR in HSPCs, holds equity in Graphite Bio and UNIKUM Tx, and is a part-time employee of UNIKUM Therapeutics.
References
- 1.Frangoul H., Locatelli F., Bhatia M., Mapara M.Y., Molinari L., Sharma A., Lobitz S., de Montalembert M., Rondelli D., Steinberg M., et al. Efficacy and Safety of a Single Dose of Exagamglogene Autotemcel for Severe Sickle Cell Disease. Blood. 2022;140:29–31. doi: 10.1182/blood-2022-162353. [DOI] [Google Scholar]
- 2.Locatelli F., Lang P., Li A., Corbacioglu S., de la Fuente J., Wall D.A., Liem R., Meisel R., Mapara M.Y., Shah A.J., et al. Efficacy and Safety of a Single Dose of Exagamglogene Autotemcel for Transfusion-Dependent β-Thalassemia. Blood. 2022;140:4899–4901. doi: 10.1182/blood-2022-166881. [DOI] [Google Scholar]
- 3.Fu B., Liao J., Chen S., Li W., Wang Q., Hu J., Yang F., Hsiao S., Jiang Y., Wang L., et al. CRISPR-Cas9-mediated gene editing of the BCL11A enhancer for pediatric beta(0)/beta(0) transfusion-dependent beta-thalassemia. Nat. Med. 2022;28:1573–1580. doi: 10.1038/s41591-022-01906-z. [DOI] [PubMed] [Google Scholar]
- 4.Bak R.O., Dever D.P., Porteus M.H. CRISPR/Cas9 genome editing in human hematopoietic stem cells. Nat. Protoc. 2018;13:358–376. doi: 10.1038/nprot.2017.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cromer M.K., Camarena J., Martin R.M., Lesch B.J., Vakulskas C.A., Bode N.M., Kurgan G., Collingwood M.A., Rettig G.R., Behlke M.A., et al. Gene replacement of α-globin with β-globin restores hemoglobin balance in β-thalassemia-derived hematopoietic stem and progenitor cells. Nat. Med. 2021;27:677–687. doi: 10.1038/s41591-021-01284-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ferrari S., Jacob A., Cesana D., Laugel M., Beretta S., Varesi A., Unali G., Conti A., Canarutto D., Albano L., et al. Choice of template delivery mitigates the genotoxic risk and adverse impact of editing in human hematopoietic stem cells. Cell Stem Cell. 2022;29:1428–1444.e9. doi: 10.1016/j.stem.2022.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DeWitt M.A., Magis W., Bray N.L., Wang T., Berman J.R., Urbinati F., Heo S.J., Mitros T., Muñoz D.P., Boffelli D., et al. Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Sci. Transl. Med. 2016;8:360ra134. doi: 10.1126/scitranslmed.aaf9336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shy B.R., Vykunta V.S., Ha A., Talbot A., Roth T.L., Nguyen D.N., Pfeifer W.G., Chen Y.Y., Blaeschke F., Shifrut E., et al. High-yield genome engineering in primary cells using a hybrid ssDNA repair template and small-molecule cocktails. Nat. Biotechnol. 2023;41:521–531. doi: 10.1038/s41587-022-01418-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schiroli G., Conti A., Ferrari S., Della Volpe L., Jacob A., Albano L., Beretta S., Calabria A., Vavassori V., Gasparini P., et al. Precise Gene Editing Preserves Hematopoietic Stem Cell Function following Transient p53-Mediated DNA Damage Response. Cell Stem Cell. 2019;24:551–565.e8. doi: 10.1016/j.stem.2019.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dever D.P., Bak R.O., Reinisch A., Camarena J., Washington G., Nicolas C.E., Pavel-Dinu M., Saxena N., Wilkens A.B., Mantri S., et al. CRISPR/Cas9 beta-globin gene targeting in human haematopoietic stem cells. Nature. 2016;539:384–389. doi: 10.1038/nature20134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Genovese P., Schiroli G., Escobar G., Tomaso T.D., Firrito C., Calabria A., Moi D., Mazzieri R., Bonini C., Holmes M.C., et al. Targeted genome editing in human repopulating haematopoietic stem cells. Nature. 2014;510:235–240. doi: 10.1038/nature13420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shin J.J., Schröder M.S., Caiado F., Wyman S.K., Bray N.L., Bordi M., Dewitt M.A., Vu J.T., Kim W.T., Hockemeyer D., et al. Controlled Cycling and Quiescence Enables Efficient HDR in Engraftment-Enriched Adult Hematopoietic Stem and Progenitor Cells. Cell Rep. 2020;32 doi: 10.1016/j.celrep.2020.108093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yahata T., Takanashi T., Muguruma Y., Ibrahim A.A., Matsuzawa H., Uno T., Sheng Y., Onizuka M., Ito M., Kato S., Ando K. Accumulation of oxidative DNA damage restricts the self-renewal capacity of human hematopoietic stem cells. Blood. 2011;118:2941–2950. doi: 10.1182/blood-2011-01-330050. [DOI] [PubMed] [Google Scholar]
- 14.Ito K., Hirao A., Arai F., Takubo K., Matsuoka S., Miyamoto K., Ohmura M., Naka K., Hosokawa K., Ikeda Y., Suda T. Correction: Corrigendum: Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat. Med. 2010;16:129. doi: 10.1038/nm0110-129a. [DOI] [PubMed] [Google Scholar]
- 15.Singh S.K., Singh S., Gadomski S., Sun L., Pfannenstein A., Magidson V., Chen X., Kozlov S., Tessarollo L., Klarmann K.D., Keller J.R. Id1 Ablation Protects Hematopoietic Stem Cells from Stress-Induced Exhaustion and Aging. Cell Stem Cell. 2018;23:252–265.e8. doi: 10.1016/j.stem.2018.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van Galen P., Kreso A., Mbong N., Kent D.G., Fitzmaurice T., Chambers J.E., Xie S., Laurenti E., Hermans K., Eppert K., et al. The unfolded protein response governs integrity of the haematopoietic stem-cell pool during stress. Nature. 2014;510:268–272. doi: 10.1038/nature13228. [DOI] [PubMed] [Google Scholar]
- 17.Haapaniemi E., Botla S., Persson J., Schmierer B., Taipale J. CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 2018;24:927–930. doi: 10.1038/s41591-018-0049-z. [DOI] [PubMed] [Google Scholar]
- 18.Ihry R.J., Worringer K.A., Salick M.R., Frias E., Ho D., Theriault K., Kommineni S., Chen J., Sondey M., Ye C., et al. p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat. Med. 2018;24:939–946. doi: 10.1038/s41591-018-0050-6. [DOI] [PubMed] [Google Scholar]
- 19.Enache O.M., Rendo V., Abdusamad M., Lam D., Davison D., Pal S., Currimjee N., Hess J., Pantel S., Nag A., et al. Cas9 activates the p53 pathway and selects for p53-inactivating mutations. Nat. Genet. 2020;52:662–668. doi: 10.1038/s41588-020-0623-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Geisinger J.M., Stearns T. CRISPR/Cas9 treatment causes extended TP53-dependent cell cycle arrest in human cells. Nucleic Acids Res. 2020;48:9067–9081. doi: 10.1093/nar/gkaa603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang L., Ingelshed K., Shen Y., Boddul S.V., Iyer V.S., Kasza Z., Sedimbi S., Lane D.P., Wermeling F. CRISPR/Cas9-Induced DNA Damage Enriches for Mutations in a p53-Linked Interactome: Implications for CRISPR-Based Therapies. Cancer Res. 2022;82:36–45. doi: 10.1158/0008-5472.Can-21-1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hafner A., Bulyk M.L., Jambhekar A., Lahav G. The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Biol. 2019;20:199–210. doi: 10.1038/s41580-019-0110-x. [DOI] [PubMed] [Google Scholar]
- 23.Chipuk J.E., Kuwana T., Bouchier-Hayes L., Droin N.M., Newmeyer D.D., Schuler M., Green D.R. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science. 2004;303:1010–1014. doi: 10.1126/science.1092734. [DOI] [PubMed] [Google Scholar]
- 24.Cromer M.K., Barsan V.V., Jaeger E., Wang M., Hampton J.P., Chen F., Kennedy D., Xiao J., Khrebtukova I., Granat A., et al. Ultra-deep sequencing validates safety of CRISPR/Cas9 genome editing in human hematopoietic stem and progenitor cells. Nat. Commun. 2022;13:4724. doi: 10.1038/s41467-022-32233-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ghannam J.Y., Xu X., Maric I., Dillon L., Li Y., Hsieh M.M., Hourigan C.S., Fitzhugh C.D. Baseline TP53 mutations in adults with SCD developing myeloid malignancy following hematopoietic cell transplantation. Blood. 2020;135:1185–1188. doi: 10.1182/blood.2019004001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eapen M., Brazauskas R., Walters M.C., Bernaudin F., Bo-Subait K., Fitzhugh C.D., Hankins J.S., Kanter J., Meerpohl J.J., Bolaños-Meade J., et al. Effect of donor type and conditioning regimen intensity on allogeneic transplantation outcomes in patients with sickle cell disease: a retrospective multicentre, cohort study. Lancet. Haematol. 2019;6:e585–e596. doi: 10.1016/s2352-3026(19)30154-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hsieh M.M., Bonner M., Pierciey F.J., Uchida N., Rottman J., Demopoulos L., Schmidt M., Kanter J., Walters M.C., Thompson A.A., et al. Myelodysplastic syndrome unrelated to lentiviral vector in a patient treated with gene therapy for sickle cell disease. Blood Adv. 2020;4:2058–2063. doi: 10.1182/bloodadvances.2019001330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jones R.J., DeBaun M.R. Leukemia after gene therapy for sickle cell disease: insertional mutagenesis, busulfan, both, or neither. Blood. 2021;138:942–947. doi: 10.1182/blood.2021011488. [DOI] [PubMed] [Google Scholar]
- 29.Humbert O., Radtke S., Samuelson C., Carrillo R.R., Perez A.M., Reddy S.S., Lux C., Pattabhi S., Schefter L.E., Negre O., et al. Therapeutically relevant engraftment of a CRISPR-Cas9-edited HSC-enriched population with HbF reactivation in nonhuman primates. Sci. Transl. Med. 2019;11 doi: 10.1126/scitranslmed.aaw3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Métais J.Y., Doerfler P.A., Mayuranathan T., Bauer D.E., Fowler S.C., Hsieh M.M., Katta V., Keriwala S., Lazzarotto C.R., Luk K., et al. Genome editing of HBG1 and HBG2 to induce fetal hemoglobin. Blood Adv. 2019;3:3379–3392. doi: 10.1182/bloodadvances.2019000820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu Y., Zeng J., Roscoe B.P., Liu P., Yao Q., Lazzarotto C.R., Clement K., Cole M.A., Luk K., Baricordi C., et al. Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nat. Med. 2019;25:776–783. doi: 10.1038/s41591-019-0401-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Demirci S., Zeng J., Wu Y., Uchida N., Shen A.H., Pellin D., Gamer J., Yapundich M., Drysdale C., Bonanno J., et al. BCL11A enhancer-edited hematopoietic stem cells persist in rhesus monkeys without toxicity. J. Clin. Invest. 2020;130:6677–6687. doi: 10.1172/JCI140189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weber L., Frati G., Felix T., Hardouin G., Casini A., Wollenschlaeger C., Meneghini V., Masson C., De Cian A., Chalumeau A., et al. Editing a gamma-globin repressor binding site restores fetal hemoglobin synthesis and corrects the sickle cell disease phenotype. Sci. Adv. 2020;6 doi: 10.1126/sciadv.aay9392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Allen D., Weiss L.E., Saguy A., Rosenberg M., Iancu O., Matalon O., Lee C., Beider K., Nagler A., Shechtman Y., et al. High-Throughput Imaging of CRISPR- and Recombinant Adeno-Associated Virus-Induced DNA Damage Response in Human Hematopoietic Stem and Progenitor Cells. Crispr j. 2022;5:80–94. doi: 10.1089/crispr.2021.0128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lattanzi A., Camarena J., Lahiri P., Segal H., Srifa W., Vakulskas C.A., Frock R.L., Kenrick J., Lee C., Talbott N., et al. Development of beta-globin gene correction in human hematopoietic stem cells as a potential durable treatment for sickle cell disease. Sci. Transl. Med. 2021;13 doi: 10.1126/scitranslmed.abf2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Romero Z., Lomova A., Said S., Miggelbrink A., Kuo C.Y., Campo-Fernandez B., Hoban M.D., Masiuk K.E., Clark D.N., Long J., et al. Editing the Sickle Cell Disease Mutation in Human Hematopoietic Stem Cells: Comparison of Endonucleases and Homologous Donor Templates. Mol. Ther. 2019;27:1389–1406. doi: 10.1016/j.ymthe.2019.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ferrari S., Jacob A., Beretta S., Unali G., Albano L., Vavassori V., Cittaro D., Lazarevic D., Brombin C., Cugnata F., et al. Efficient gene editing of human long-term hematopoietic stem cells validated by clonal tracking. Nat. Biotechnol. 2020;38:1298–1308. doi: 10.1038/s41587-020-0551-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sharma R., Dever D.P., Lee C.M., Azizi A., Pan Y., Camarena J., Köhnke T., Bao G., Porteus M.H., Majeti R. The TRACE-Seq method tracks recombination alleles and identifies clonal reconstitution dynamics of gene targeted human hematopoietic stem cells. Nat. Commun. 2021;12:472. doi: 10.1038/s41467-020-20792-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schiroli G., Ferrari S., Conway A., Jacob A., Capo V., Albano L., Plati T., Castiello M.C., Sanvito F., Gennery A.R., et al. Preclinical modeling highlights the therapeutic potential of hematopoietic stem cell gene editing for correction of SCID-X1. Sci. Transl. Med. 2017;9 doi: 10.1126/scitranslmed.aan0820. [DOI] [PubMed] [Google Scholar]
- 40.Hendel A., Kildebeck E.J., Fine E.J., Clark J., Punjya N., Sebastiano V., Bao G., Porteus M.H. Quantifying genome-editing outcomes at endogenous loci with SMRT sequencing. Cell Rep. 2014;7:293–305. doi: 10.1016/j.celrep.2014.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.De Ravin S.S., Brault J., Meis R.J., Liu S., Li L., Pavel-Dinu M., Lazzarotto C.R., Liu T., Koontz S.M., Choi U., et al. Enhanced homology-directed repair for highly efficient gene editing in hematopoietic stem/progenitor cells. Blood. 2021;137:2598–2608. doi: 10.1182/blood.2020008503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sweeney C.L., Pavel-Dinu M., Choi U., Brault J., Liu T., Koontz S., Li L., Theobald N., Lee J., Bello E.A., et al. Correction of X-CGD patient HSPCs by targeted CYBB cDNA insertion using CRISPR/Cas9 with 53BP1 inhibition for enhanced homology-directed repair. Gene Ther. 2021;28:373–390. doi: 10.1038/s41434-021-00251-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baik R., Cromer M., Glenn S., Vakulskas C., Dudek A., Feist W., Shipp S., Dever D., Porteus M. Transient Inhibition of 53BP1 Increases the Frequency of Targeted Integration in Human Hematopoietic Stem and Progenitor Cells. Research Square. 2023 doi: 10.21203/rs.3.rs-2621625/v1. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carusillo A., Haider S., Schäfer R., Rhiel M., Türk D., Chmielewski K.O., Klermund J., Mosti L., Andrieux G., Schäfer R., et al. A novel Cas9 fusion protein promotes targeted genome editing with reduced mutational burden in primary human cells. Nucleic Acids Res. 2023;51:4660–4673. doi: 10.1093/nar/gkad255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Piras F., Riba M., Petrillo C., Lazarevic D., Cuccovillo I., Bartolaccini S., Stupka E., Gentner B., Cittaro D., Naldini L., Kajaste-Rudnitski A. Lentiviral vectors escape innate sensing but trigger p53 in human hematopoietic stem and progenitor cells. EMBO Mol. Med. 2017;9:1198–1211. doi: 10.15252/emmm.201707922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Komor A.C., Kim Y.B., Packer M.S., Zuris J.A., Liu D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533:420–424. doi: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Anzalone A.V., Randolph P.B., Davis J.R., Sousa A.A., Koblan L.W., Levy J.M., Chen P.J., Wilson C., Newby G.A., Raguram A., Liu D.R. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576:149–157. doi: 10.1038/s41586-019-1711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gaudelli N.M., Komor A.C., Rees H.A., Packer M.S., Badran A.H., Bryson D.I., Liu D.R. Programmable base editing of A·T to G·C in genomic DNA without DNA cleavage. Nature. 2017;551:464–471. doi: 10.1038/nature24644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Antoniou P., Hardouin G., Martinucci P., Frati G., Felix T., Chalumeau A., Fontana L., Martin J., Masson C., Brusson M., et al. Base-editing-mediated dissection of a γ-globin cis-regulatory element for the therapeutic reactivation of fetal hemoglobin expression. Nat. Commun. 2022;13:6618. doi: 10.1038/s41467-022-34493-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Newby G.A., Yen J.S., Woodard K.J., Mayuranathan T., Lazzarotto C.R., Li Y., Sheppard-Tillman H., Porter S.N., Yao Y., Mayberry K., et al. Base editing of haematopoietic stem cells rescues sickle cell disease in mice. Nature. 2021;595:295–302. doi: 10.1038/s41586-021-03609-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li M., Zhong A., Wu Y., Sidharta M., Beaury M., Zhao X., Studer L., Zhou T. Transient inhibition of p53 enhances prime editing and cytosine base-editing efficiencies in human pluripotent stem cells. Nat. Commun. 2022;13:6354. doi: 10.1038/s41467-022-34045-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hardouin G., Antoniou P., Martinucci P., Felix T., Manceau S., Joseph L., Masson C., Scaramuzza S., Ferrari G., Cavazzana M., Miccio A. Adenine base editor-mediated correction of the common and severe IVS1-110 (G>A) β-thalassemia mutation. Blood. 2023;141:1169–1179. doi: 10.1182/blood.2022016629. [DOI] [PubMed] [Google Scholar]
- 53.Liao J., Chen S., Hsiao S., Jiang Y., Yang Y., Zhang Y., Wang X., Lai Y., Bauer D.E., Wu Y. Therapeutic adenine base editing of human hematopoietic stem cells. Nat. Commun. 2023;14:207. doi: 10.1038/s41467-022-35508-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Siegner S.M., Ugalde L., Clemens A., Garcia-Garcia L., Bueren J.A., Rio P., Karasu M.E., Corn J.E. Adenine base editing efficiently restores the function of Fanconi anemia hematopoietic stem and progenitor cells. Nat. Commun. 2022;13:6900. doi: 10.1038/s41467-022-34479-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Everette K.A., Newby G.A., Levine R.M., Mayberry K., Jang Y., Mayuranathan T., Nimmagadda N., Dempsey E., Li Y., Bhoopalan S.V., et al. Ex vivo prime editing of patient haematopoietic stem cells rescues sickle-cell disease phenotypes after engraftment in mice. Nat. Biomed. Eng. 2023;7:616–628. doi: 10.1038/s41551-023-01026-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li C., Georgakopoulou A., Newby G.A., Chen P.J., Everette K.A., Paschoudi K., Vlachaki E., Gil S., Anderson A.K., Koob T., et al. In vivo HSC prime editing rescues Sickle Cell Disease in a mouse model. Blood. 2023;141:2085–2099. doi: 10.1182/blood.2022018252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li C., Georgakopoulou A., Newby G.A., Everette K.A., Nizamis E., Paschoudi K., Vlachaki E., Gil S., Anderson A.K., Koob T., et al. In vivo base editing by a single i.v. vector injection for treatment of hemoglobinopathies. JCI Insight. 2022;7 doi: 10.1172/jci.insight.162939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yarnall M.T.N., Ioannidi E.I., Schmitt-Ulms C., Krajeski R.N., Lim J., Villiger L., Zhou W., Jiang K., Garushyants S.K., Roberts N., et al. Drag-and-drop genome insertion of large sequences without double-strand DNA cleavage using CRISPR-directed integrases. Nat. Biotechnol. 2023;41:500–512. doi: 10.1038/s41587-022-01527-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Adair J.E., Enstrom M.R., Haworth K.G., Schefter L.E., Shahbazi R., Humphrys D.R., Porter S., Tam K., Porteus M.H., Kiem H.P. DNA Barcoding in Nonhuman Primates Reveals Important Limitations in Retrovirus Integration Site Analysis. Mol. Ther. Methods Clin. Dev. 2020;17:796–809. doi: 10.1016/j.omtm.2020.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Horn P.A., Thomasson B.M., Wood B.L., Andrews R.G., Morris J.C., Kiem H.P. Distinct hematopoietic stem/progenitor cell populations are responsible for repopulating NOD/SCID mice compared with nonhuman primates. Blood. 2003;102:4329–4335. doi: 10.1182/blood-2003-01-0082. [DOI] [PubMed] [Google Scholar]