


Abstract
A novel method for detection of any mutation located within a PCR-amplified DNA sequence was demonstrated. The method is based on the inhibition of spontaneous DNA branch migration. Partial duplexes produced by PCR amplification of a test and a reference genomic DNA sample anneal to form four-stranded cruciform structures. Spontaneous DNA branch migration results in dissociation of these structures when the test and reference sequences are identical. Any base substitution, deletion or insertion inhibits branch migration and produces stable cruciform structures. When suitable ligands are attached to the PCR primers, the cruciform structures can be detected by standard immunochemical methods. This approach was tested using several commonly occurring mutations within the human cystic fibrosis gene. New methods for increasing the specificity of PCR amplifications are described that were used for successful mutation analysis.
INTRODUCTION
There is a need for simple, universal, high throughput methods for mutation detection. A variety of methods for determination of sequence abnormalities that avoid complete sequencing of PCR amplicons have been devised (1). Many of these methods are laborious, require electrophoretic separation of the products and are capable of detecting only specific sequence abnormalities. A method that is independent of the exact type of mutation would be particularly valuable in the diagnosis and risk assessment of genetic diseases when any of several mutations within a given sequence can often give rise to the disease. Such a method would also be useful for verification of sequence homology as related to various applications in molecular biology, molecular medicine and population genetics. A particularly useful application could be in the emerging field of pharmacogenomics, which is focused on identification of genetic variations in the form of single nucleotide polymorphisms (SNPs).
It has been shown that strand exchange between two homologous DNA duplexes by spontaneous branch migration is inhibited by a single base pair difference between the duplexes (2–4). This phenomenon would provide an opportunity for detecting differences in related DNA sequences if four-stranded cruciform DNA structures could be easily formed from a test and a reference DNA sample. We describe here a PCR-based procedure for generation and detection of such structures.
MATERIALS AND METHODS
Samples and reagents
Human genomic DNA samples were purchased from the Coriell Institute for Medical Research (Camden, NJ) or were provided by Dr D. O’Kane (Mayo Foundation, Rochester, MN). PCR primers were purchased from Oligos Etc. (Wilsonville, OR). Cloned Pfu DNA polymerase was from Stratagene (San Diego, CA).
Oligonucleotides
Exon 10, forward primer 1, 5′-CTCAGTTTTCCTGGATTATGCCɛAɛAA-3′; exon 10, reverse primer 1, 5′-ACCATGC-TCGAGATTACGAGCTAACCGATTGAATATGGAGCCɛAɛAG-3′; exon 10, reverse primer 2, 5′-GATCCTAGGCCT-CACGTATTCTAACCGATTGAATATGGAGCCɛAɛAG-3′; exon 10, reverse primer 3, 5′-CTAACCGATTGAATATGG-AGCC-3′; exon 10, reverse primer 4, 5′-AGCCTAATCGTC-CACGATGTATAAATATATAATTTGGGTAGTGT-3′; exon 11, forward primer 1, 5′-TAGAAGGAAGATGTGCCTTTC-AɛAɛAA-3′; exon 11, forward primer 2, 5′-GCCTTTCAAATTC-AGATTGAGCɛAɛAG-3′; exon 11, reverse primer 1, 5′-AC-CATGCTCGAGATTACGAGTTCTTAACCCACTAGCCA-TAAAAɛAɛAA-3′; exon 11, reverse primer 2, 5′-GATC-CTAGGCCTCACGTATTTTCTTAACCCACTAGCCATA-AAAɛAɛAA-3′; exon 11, reverse primer 3, 5′-ACCATGC-TCGAGATTACGAGGACATTTACAGCAAATGCTTGCɛAɛAG-3′; exon 11, reverse primer 4, 5′-GATCC-TAGGCCTC-ACGTATTGACATTTACAGCAAATGCTTGCɛAɛAG-3′.
The underlined ‘tail’ portions of the reverse primers and the ɛAɛAN 3′-termini are not complementary to the target DNA. The same pair of tail sequences are used in both exon 10 reverse primers 1 and 2 and exon 11 reverse primers 1–4. These are the same tail sequences used in the synthetic branch migration substrates by Panyutin and Hsieh (5). ɛA is ethenoadenosine.
PCR amplification
PCR amplifications were carried out using a TRIO thermocycler (Biometra Inc., Tampa, FL). A hot start was used with AmpliWax PCR gems according to the instructions provided by the manufacturer (PE Bioscience, Foster City, CA). Forty PCR cycles were performed with 30 s denaturation at 94°C, 1 min re-annealing at 64°C and 1 min extension at 72°C, except where otherwise indicated. The reaction mixtures contained 40 ng genomic DNA, 250 nM forward primer (50% 5′-end-labeled with biotin and 50% 5′-end-labeled with digoxigenin), 125 nM each of two differently tailed reverse primers, 0.5 U Pfu polymerase and 200 µM each dNTP in 40 µl of 10 mM Tris–HCl, pH 8.3, 50 mM KCl, 4 mM MgCl2 and 200 µg/ml BSA.
Branch migration
For detection of heterozygotes, PCR-amplified samples were heat denatured for 2 min at 94°C and then allowed to re-anneal and undergo strand exchange for 30 min at 65°C. Unresolved cruciform structures from heterozygous samples were detected by their ability to cause association of biotin with digoxigenin using the luminescent oxygen channeling immunoassay (LOCI™) (6) or enzyme immunoassay (ELISA). For LOCI, 2 µl aliquots of amplified and re-annealed samples were added to suspensions of 1.25 µg of streptavidin-coated latex beads loaded with a photosensitizer and 0.625 µg of anti-digoxigenin monoclonal antibody-coated latex beads loaded with a chemiluminescent olefin in 50 µl of the above PCR buffer. The mixtures were incubated for 30 min at 37°C and then irradiated for 1 s at 680 nm. The ensuing chemiluminescent emission intensity at 560–630 nm provided a measure of the number of bound bead pairs and thus a measure of the number of cruciform structures. Alternatively, digoxigenin–biotin association was measured using a standard ELISA protocol with streptavidin-coated microtiter plates and a horseradish peroxidase–anti-digoxigenin monoclonal antibody conjugate.
For simultaneous detection of both heterozygotes and mutant homozygotes the amplified samples were mixed with equal volumes of a reference PCR-amplified wild-type sample. The solutions were then heat denatured for 2 min at 94°C and incubated for 30 min at 65°C to re-anneal and undergo strand exchange. Detection of unresolved cruciform structures was carried out as above.
RESULTS AND DISCUSSION
Assay concept
The principle of the method is illustrated in Figure 1. A reference sample containing the fully base-paired DNA duplex sequence, Aa, is modified by appending to one end non-complementary tails, D and e, to give the partial duplex I. The non-complementary tails are allowed to hybridize to a similar partial duplex II that has non-complementary tails, d and E, appended to one end. The fully base-paired sequence A*a* in duplex II differs from Aa by a possible mutation. When the reference and test sequences, Aa and A*a*, are identical, the four-stranded cruciform structure III that is formed can spontaneously dissociate into fully complementary duplexes IV and V. This process occurs by repetitive dissociation of base pairs within one pair of arms of the cruciform structure and re-association of the base pairs within the other pair of arms. Branch migration can only take place in one direction because of the non-identity of Dd and Ee. Once IV and V have formed the process is irreversible. When there is a sequence difference between the reference and test sequences, i.e. Aa and A*a* are different, branch migration past the base pair difference is energetically unfavorable because it would lead to the creation of two base-pair mismatches (2,4). Branch migration in both directions is then inhibited and structure III is stabilized. If biotin and digoxigenin labels are attached to the 5′-ends of the positive strands of I and II, respectively, the association of these labels in III will signal the presence of a mutation.
Figure 1.

Principle of mutation detection by BMI. A pair of partial duplexes, I and II, can assemble into a four-stranded cruciform structure III that contains two ligands, biotin and digoxigenin. Resolution into fully hybridized duplexes IV and V occurs when III has a pair of identical opposite arms. Provided sequences Dd and Ee are different, a single base mutation in the test sequence II (indicated by the square) is sufficient to inhibit resolution. The association of the two ligands in III permits detection by standard immunochemical methods.
A PCR strategy for preparing partial duplexes I and II is illustrated in Figure 2. The genomic test sample containing the sequence A*a* and wild-type reference sample containing the sequence Aa are each amplified by PCR. The same forward primer is used in both amplifications but is 5′-end-labeled with biotin for amplification of the test sample and 5′-end-labeled with digoxigenin for amplification of the reference sample. The reverse primer in both amplifications is an equimolar mixture of two oligonucleotides. Both have identical 3′-priming sequences but one has a 20 nt 5′-tail sequence, d, and the other a 20 nt 5′-tail sequence, e. Termination of each PCR amplification with a final primer extension yields the two fully base-paired amplicons, AD:ad and AE:ae. The individual strands formed from the reference sequence are 5′-digoxigenyl-AD, 5′-digoxigenyl-AE and unlabeled ad and ae. Similarly, the individual strands formed from the test sequence include 5′-biotinyl-A*D, 5′-biotinyl-A*E and unlabeled a*d and a*e.
Figure 2.

Preparation of partial duplexes for BMI analysis. One forward and two tailed reverse primers are used to amplify test and reference DNA sequences. The forward primers are labeled with biotin and digoxigenin. Combination of the amplicons followed by denaturation and re-annealing produces partial duplexes that assemble into four-stranded cruciform structures.
This scheme can be simplified by including all the reagents in a single PCR reaction mixture. However, in the present work the test and reference samples were amplified separately and a mixture of both the 5′-biotinylated and 5′-digoxigenin-labeled forward primers was used in each amplification. Single amplifications were avoided because of the potential difficulty of delivering similar quantities of genomic DNA to the reaction mixtures.
Branch migration was initiated by combining the PCR mixtures, heating to denature the double strands and then incubating. The maximum mutant:wild-type signal ratio was attained within ~20 min at 65°C, although an incubation period of 30 min at 65°C was routinely used to assure complete resolution of the cruciform structures. At 60°C the resolution was much slower and it was necessary to incubate for ~2 h. At 75°C single base mutations failed to inhibit branch migration and discrimination between mutant and wild-type samples was lost.
During incubation the single-stranded PCR products, A*D, A*E, AD and AE, can complex with any of the corresponding complementary strands, a*d, a*e, ad and ae, to form eight fully base-paired duplexes and eight partial duplexes in which the two tails are not hybridized. These can combine to form 16 different cruciform structures, only two of which will have the two different fully homologous Aa and A*a* strands needed to inhibit branch migration (2). Hence, only a sixteenth of the PCR products become incorporated into detectable cruciform structures. Inclusion of both biotinylated and digoxigenin-labeled forward primers in each PCR reaction produces twice as many PCR products, but only half of the potentially detectable cruciform structures are labeled with both biotin and digoxigenin. Accordingly, the yield of detectable cruciform structures is only 1/32 of the total amplified material. Despite this low theoretical yield, these structures can readily be detected by methods that sense association of the two labels, such as ELISA or LOCI.
Detection of mutations in the CFTR gene
For detection of both mutant homozygotes and heterozygotes the samples were amplified with 1:1 mixtures of the biotinylated and digoxigenin-labeled forward primer and 1:1 mixtures of the two reverse primers. The solutions were then combined with a similarly amplified wild-type reference prior to denaturation and re-annealing. For detection of heterozygous samples no reference was added. Tables 1 and 2 illustrate detection of the most frequently occurring mutations within the human cystic fibrosis gene (CFTR) using this method.
Table 1. Detection of mutations in exon 10 of the human cystic fibrosis gene.
| Genotype | LOCI without reference DNA | LOCI with reference DNA | ELISA without reference DNA |
|---|---|---|---|
| Wild-type homozygotes | |||
| wt/wt | 2.5 (2.1) | 2.3 | 9.4 |
| wt/wt | 2.3 (3.2) | 2.9 | 7.4 |
| wt/wt | 2.4 (2.4) | 3.7 | 8.2 |
| wt/wt | 2.1 (2.7) | 3.1 | 7.6 |
| Heterozygotes | |||
| ΔF508/wt 3 bp deletion | 61 (100) | 53 | 63 |
| ΔF508/wt 3 bp deletion | 105 (109) | 102 | 102 |
| ΔF508/wt 3 bp deletion | 110 (114) | 94 | 108 |
| ΔF508/wt 3 bp deletion | 138 (147) | 119 | 136 |
| ΔI508/wt 3 bp deletion | 107 (76) | 96 | 116 |
| F580C/wt T→G | 95 (57) | 87 | 92 |
| ΔF508/F508C 3 bp deletion/T→G | 93 (85) | 139 | 97 |
| ΔF508/F508C 3 bp deletion/T→G | 90 (113) | 111 | 86 |
| Mutant homozygotes | |||
| ΔF508/ΔF508 3 bp deletion | 2.6 (3.2) | 149 | 6.7 |
| ΔF508/ΔF508 3 bp deletion | 2.0 (3.2) | 184 | 7.4 |
| ΔF508/ΔF508 3 bp deletion | 2.4 (2.1) | 181 | 7.8 |
| ΔF508/ΔF508 3 bp deletion | 2.2 (3.0) | 212 | 7.9 |
| Blank (no target DNA) | 1.2 | 6.7 |
Genomic DNA samples were analyzed for the presence of mutations in CFTR exon 10 using LOCI and ELISA for detection. Amplifications of genomic DNA samples were carried out with a hot start. The primers were a 1:1 mixture of biotin- and digoxigenin-labeled exon 10 forward primer 1 and a 1:1 mixture of exon 10 reverse primers 1 and 2, each of which had the same 22-nt priming sequence plus a 3′-terminal ɛAɛAG but different 20-nt 5′-tails. The amplicon length was 220 bp. A homozygous wild-type sample was used as reference. The assay responses were normalized with the average heterozygote signal taken as 100. Analysis results from a duplicate PCR amplification are given in parentheses.
Table 2. Detection of mutations in exon 11 of the human cystic fibrosis gene.
| Genotype | Without reference DNA | With reference DNA |
|---|---|---|
| Wild-type homozygotes | ||
| wt/wt | 1.4 (1.2) | 2.4 |
| wt/wt | 1.6 (1.3) | 2.1 |
| wt/wt | 2.0 (1.5) | 2.4 |
| wt/wt | 2.1 (1.4) | 2.0 |
| wt/wt | 1.6 (1.2) | 3.2 |
| wt/wt | 1.7 (1.4) | 1.5 |
| Heterozygotes | ||
| G542X/wt G→T | 41 (42) | 38 |
| G542X/wt G→T | 73 (90) | 66 |
| G551D/wt G→A | 83 (84) | 93 |
| G551D/wt G→A | 99 (69) | 76 |
| R553X/wt C→T | 92 (84) | 57 |
| R553X/wt C→T | 105 (95) | 61 |
| R560T/wt G→C | 130 (123) | 70 |
| R560T/wt G→C | 109 (135) | 111 |
| G551D/R553X G→A/C→T | 134 (134) | 193 |
| G551D/R553X G→A/C→T | 134 (144) | 235 |
| Mutant homozygotes | ||
| G542X/G542X G→T | 1.5 (1.4) | 174 |
| G542X/G542X G→T | 1.4 (1.5) | 133 |
| Blank (no target DNA) | 1.6 |
Genomic DNA samples were analyzed for the presence of mutations in CFTR exon 11 as in Table 1 using LOCI detection. Exon 11 forward primer 1 was used with exon 11 reverse primers 1 and 2, each of which had the same 23-nt priming sequence plus a 3′-terminal ɛAɛAA and a different 20-nt 5′-tail. The amplicon length was 333 bp.
The results obtained using ELISA and LOCI were similar and correlated well (r2 = 0.996), although the latter provided a 2.5-fold higher positive signal:negative background ratio. The signal from double mutants and mutant homozygotes averaged >80% higher than from heterozygotes. However, signal intensity alone was a poor measure of heterozygosity since the signals from heterozygotes varied by up to 3-fold. Differences in the yields of amplified DNA are probably responsible for most of the variances, although differences in assay response when the same amplified samples were re-analyzed suggest that incompletely controlled assay conditions may play a role.
Because a failed PCR amplification might cause a mutant sample to be missed, in subsequent testing each amplified negative sample was confirmed by combining it with an unrelated amplicon prepared using the same reverse primer tails. With a successful amplification a stable cruciform structure will be formed because it will have different sequences in each arm. Failure to produce a signal could therefore be attributed to a failed PCR amplification. In principle, the signal intensities obtained in this way could also be used to normalize and quantitate positive branch migration inhibition (BMI) assay responses if the irrelevant sample was labeled with a capture ligand different from biotin or digoxigenin. The method may even provide sufficient quantitation to distinguish heterozygous and homozygous mutants.
Alternatively, amplification can be verified by including 0.5 µg/ml ethidium bromide in the PCR reaction mixture and measuring its increased fluorescence in the presence of double-stranded DNA using a microplate fluorometer or simply by placing PCR tubes on a UV transilluminator (7).
A study using BMI to detect mutations in CFTR exons 10 and 11 was carried out with 50 clinical samples kindly provided by Dennis O’Kane (Mayo Foundation). The results are illustrated in Figure 3. Heterozygous samples were amplified and identified without adding a reference sample (heterozygote screen) and the negatives then combined with a reference wild-type sample and re-measured to identify homozygous mutants (homozygote screen). The remaining negatives were checked for PCR failures. BMI analysis identified all samples correctly. One heterozygous sample failed to amplify but could be detected following re-amplification.
Figure 3.
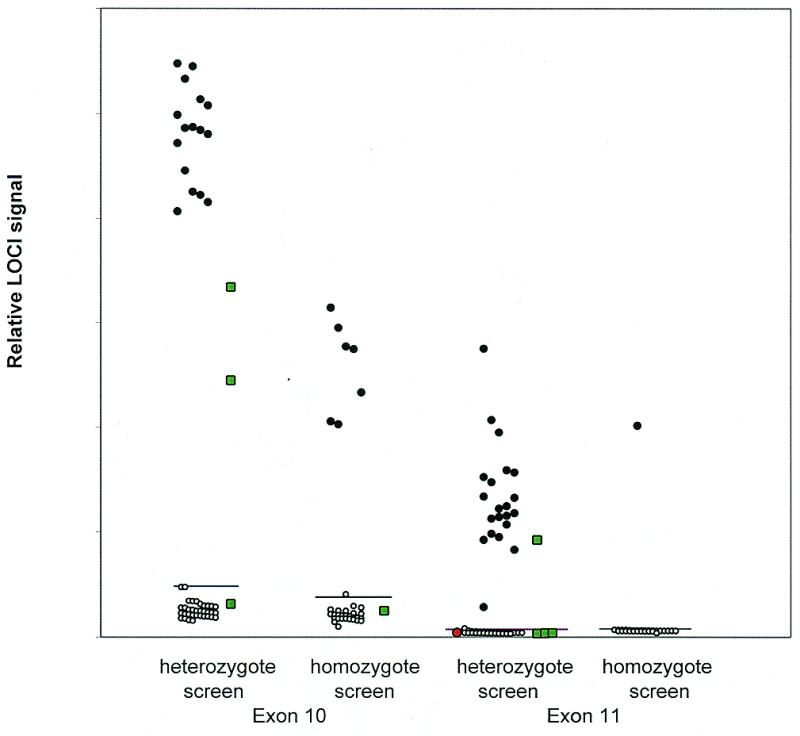
BMI assay of clinical samples for CFTR exons 10 and 11. Fifty samples consisting of 16 exon 10 and 22 exon 11 heterozygotes, seven exon 10 and one exon 11 homozygotes and eight wild-type homozygotes plus controls (green squares) were assayed by LOCI following 35 PCR cycles with hot start. Filled circles are positive and open circles are negative by sequencing. Forward primer 1 and reverse primers 1 and 2 with (exon 11) and without (exon 10) the 3′-terminal ɛAɛAN sequences were used. Heterozygous positives were identified without an added reference sample. The only false negative (red circle) was due to failed amplification. Horizontal lines are drawn at three standard deviations above the mean negative signal.
BMI assay limitations
Non-specific priming. Non-specific PCR amplification leads to increased background signals and reduced BMI sensitivity. Amplicons that are formed by non-specific priming become labeled and tailed in exactly the same manner as the specific amplicon. Upon denaturing and re-annealing, they form partial duplexes that interact with partial duplexes from the specific amplicon to yield four-stranded DNA structures. These structures are incapable of branch migration and produce an increased background signal. Primer selection and hybridization stringency are therefore critically important.
A high annealing temperature together with a ‘hot start’ (8) was useful but did not suppress background sufficiently in all cases. For this reason two ethenoadenosine (ɛA) residues (9) were introduced at the 3′-terminus of each primer. Since these bases are incapable of hybridizing to natural DNA, the primers are not initially extended. Instead, active primer is gradually released due to the 3′-exonuclease activity of the Pfu polymerase. Reduction in the concentration of active primers during the initial stages of amplification increases stringency during the critical early amplification steps. The method greatly increased selectivity (Table 3) and was used together with hot start for most studies.
Table 3. Effect of 3′-termination of primers with ɛAɛAG.
| Genotype | Without ɛAɛAG | With ɛAɛAG |
|---|---|---|
| wt/wt | 37 | 1.4 |
| wt/wt | 39 | 1.4 |
| wt/wt | 41 | 1.7 |
| wt/wt | 41 | 1.5 |
| G542X/wt | 121 | 126 |
| G551D/wt | 100 | 93 |
| R553X/wt | 82 | 85 |
| R560T/wt | 97 | 96 |
Thirty-five cycle PCR amplification of exon 11 was carried out with hot start as in Table 2, followed by LOCI detection. Exon 11 forward primer 2 and reverse primers 3 and 4 were used with and without a 3′-terminating ɛAɛAG sequence. The amplicon length was 203 bp. Data are normalized with the average mutant response taken as 100.
A PCR modification similar to the use of nested primers provided a powerful alternative method for reducing background signals. This method utilizes the same forward primer as in Figure 2 but two different reverse priming sequences (Fig. 4). The outer reverse primer, f, is a standard primer without a 5′-tail. The inner reverse primer has a nested priming sequence that binds adjacent to the outer primer and a 5′-tail sequence, g, which is unrelated to the outer primer. Each reverse primer therefore produces a similar length amplicon but with different termini that correspond to f and g, respectively. As already illustrated in Figure 2, denaturation and re-annealing of such amplicons provides duplexes with unhybridized tails.
Figure 4.
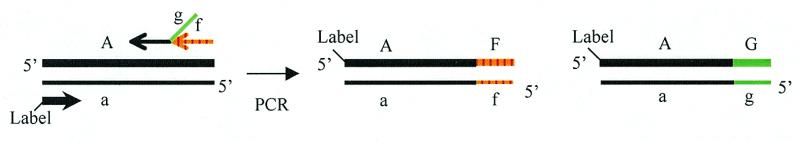
Alternative primer scheme for preparation of partial duplexes. Two reverse primers anneal to adjacent sites in target DNA. The inner primer has a tail that is unrelated to the outer primer. Partial duplexes produced upon denaturation and re-annealing of resulting amplicons assemble into four-stranded cruciform structures as shown in Figure 2. The forward primer label is biotin or digoxigenin.
Table 4 illustrates reduction in background signal in detection of a heterozygous exon 10 deletion mutation using this method. To demonstrate the power of the method the results are compared with results obtained using the original procedure with a sub-optimal PCR re-annealing temperature. The background signal was reduced about 13-fold and even with the use of a hot start the method afforded a 3-fold suppression of the background. The ɛAɛA terminal sequences were not used in these experiments and two separate amplifications were carried out, each using one of the reverse primers. Similar results could be obtained when the amplifications were combined, but lengthy optimization was needed to identify suitable concentration ratios of the reverse primers to permit equal participation in the amplification.
Table 4. Effect of hot start and nested reverse primers.
| Genotype | Without hot start | With hot start | ||
|---|---|---|---|---|
| Scheme 1 | Scheme 2 | Scheme 1 | Scheme 2 | |
| wt/wt | 61 | 4.2 | 15.1 | 2.8 |
| wt/wt | 56 | 5.2 | 8.2 | 3.4 |
| ΔF508/wt | 98 | 94 | 101 | 99 |
| ΔF508/wt | 101 | 106 | 99 | 101 |
Thirty-five cycle PCR amplification of CFTR exon 10 was carried out with cycle temperatures of 94, 54 and 72°C, followed by LOCI detection. A reference DNA sample was not used. Scheme 1: the procedure of Table 1 was followed using the same primers but without the 3′-terminal ɛAɛAG sequence. The amplicon length was 220 bp. Scheme 2: separate PCR reactions were carried out. Each used one of the exon 10 reverse primers, 3 and 4, together with the exon 10 forward primer 1 without its 3′-terminal ɛAɛAA sequence. The PCR mixtures were combined prior to annealing and analysis. Reverse primer 3 had the same 22-nt priming sequence as reverse primers 1 and 2 without a 5′-tail sequence. Reverse primer 4 contained a 22-nt nested primer sequence that binds adjacent to the 3′-end of reverse primer 3 and had a 22-nt 5′-tail. The amplicon lengths were each 200 bp. For each primer scheme, the amplifications were performed either with or without hot start.
Although a second reverse primer might be expected to increase the number of false priming products, the number of irrelevant products that were detectable by BMI was actually reduced. This is because formation of cruciform structures from partial duplexes can only occur if the two pairs of unhybridized tails are complementary to each other. Such partial duplexes can arise in error only if the two reverse primers randomly prime two contiguous sequences in the genomic DNA, which is a highly unlikely event.
Amplification fidelity. Another source of background signal in BMI is infidelity of amplification. This was exemplified by comparing BMI analyses using Pfu and Taq polymerases, respectively. The latter has 6- to 12-fold lower fidelity than Pfu (10,11). Thus amplification of wild-type exon 11 with Taq produced an 8-fold higher normalized background signal than with Pfu (316 bp amplicon). Increasing the number of PCR cycles also decreases amplification accuracy (12), but was less troublesome. Less than a 3-fold increase was observed in the wild-type signal from amplified exon 10 (220 bp amplicon) upon increasing number of cycles from 24 to 45, whereas the mutant signal grew by two orders of magnitude.
Amplicon length. The effect of amplicon length on mutation detection by BMI is not fully understood. In general, longer sequences produce reduced specific signal:background ratios. In the present work with the CFTR gene (exons 11 and 7) these ratios dropped with amplicon length from a high of about 50:1 to below 15:1 at ~400 bp. However, in a similar study of a mutant glucose 6-phosphate dehydrogenase from Leuconostoc mesenteroides the effect was less dramatic. A 15:1 ratio was not reached until an amplicon length of ~750 bp and sequences as long as 900 bp could be successfully analyzed. Similarly, amplicons of up to 500–600 bp in length of the rpoB and pncA genes of Mycobacterium tuberculosis could be assayed with signal:background ratios ranging from 12 to 90.
At least part of the reduced signal:background ratio with longer amplicons is due simply to lower PCR yields. Modifications that improved yield, such as elimination of potassium chloride from the buffer, were found to increase the lengths that could be analyzed. Thus, undetectable signal:background ratios of 1:1 obtained with a 445 bp amplicon from exon 11 and a 475 bp amplicon from exon 7 were increased to 5:1 and 8:1, respectively.
The limitation of BMI to relatively short amplicons is not an impedement in the application of, for example, scanning the genome for SNPs that occur approximately every 1000 bp along the human genome (13).
CONCLUSION
BMI is suited for rapid screening of a large number of samples for sequence differences within a PCR amplicon. This method makes it possible to avoid sequencing normal samples that will often be in the vast majority. Only the BMI-positive samples need to be the subject of further analysis by sequencing. Two related methods for generating PCR amplicons capable of branch migration have been used. The method least sensitive to mispriming is more labor intensive, requiring two separate amplifications with different nested reverse primers, one of which has a 5′-tail. The other method, using a pair of tailed reverse primers that bind to the same site, is more convenient. Only a single amplification is required and there is no need to optimize the primer ratios. However, stringent control of non-specific amplification such as using ethenoadenosine-terminated primers is sometimes required. Preliminary studies (unpublished) suggest that AmpliTaq Gold™ (PE Bioscience) can be used in place of ethenoadenosine termination, but the lower fidelity of this polymerase may prove troublesome.
Detection of stable cruciform structures by LOCI gives excellent sensitivity using a protocol that does not require separation. Alternatively, more readily accessible ELISA reagents can provide adequate signal:background ratios. Because of the relatively simple protocols BMI offers a good opportunity for automation. The primary limitation of the method is that sequence lengths suitable for analysis are currently limited to a maximum of 400–1000 bp depending on the sequence of the target DNA and primers that are used.
Acknowledgments
ACKNOWLEDGEMENTS
The authors wish to thank Yen Ping Liu for data on M.tuberculosis and the gift of LOCI beads, Marc Taylor for applying BMI to other cystic fibrosis mutations and Sam Rose and Alan Dafforn for critical reading of the manuscript.
REFERENCES
- 1.Cotton R.G. (1997) Trends Genet., 13, 43–46. [DOI] [PubMed] [Google Scholar]
- 2.Panyutin I.G. and Hsieh,P. (1993) J. Mol. Biol., 230, 413–424. [DOI] [PubMed] [Google Scholar]
- 3.Hsieh P. and Panyutin,I.G. (1995) In Eckstein,F. and Lilley,D.M.J. (eds), Nucleic Acids and Molecular Biology. Springer-Verlag, Berlin, Germany, Vol. 9, pp. 42–65.
- 4.Biswas I., Yamamoto,A. and Hsieh,P. (1998) J. Mol. Biol., 279, 795–806. [DOI] [PubMed] [Google Scholar]
- 5.Panyutin I.G. and Hsieh,P. (1994) Proc. Natl Acad. Sci. USA, 91, 2021–2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ullman E.F., Kirakossian,H., Singh,S., Wu,Z.P., Irvin,B.R., Pease,J.S., Switchenko,A.C., Irvine,J.D., Dafforn,A., Skold,C.N. and Wagner,D.B. (1994) Proc. Natl Acad. Sci. USA, 91, 5426–5430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Higuchi R., Dollinger,G., Walsh,P.S. and Griffith,R. (1992) Biotechnology, 10, 413–417. [DOI] [PubMed] [Google Scholar]
- 8.Chou Q., Russel,M., Birch,D.E., Raymond,J. and Bloch,W. (1992) Nucleic Acids Res., 20, 1717–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Litinski V., Chenna,A., Sagi,J. and Singer,B. (1997) Carcinogenesis, 18, 1609–1615. [DOI] [PubMed] [Google Scholar]
- 10.Lundberg K.S., Shoemaker,D.D., Adams,M.W.W., Short,J.M., Sorge,J.A. and Mathur,E.J. (1991) Gene, 180, 1–6. [DOI] [PubMed] [Google Scholar]
- 11.Cline J., Braman,J.C. and Hogrefe,H.H. (1996) Nucleic Acids Res., 24, 3546–3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reiss J., Krawczak,M., Schloesser,M., Wagner,M. and Cooper,D.N. (1990) Nucleic Acids Res., 18, 973–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang D.G., Fan,J.-B., Siao,C.-J., Berno,A., Young,P., Sapolsky,R., Ghandour,G., Perkins,N., Winchester,E., Spencer,J. et al. (1998) Science, 280, 1077–1082. [DOI] [PubMed] [Google Scholar]