

Abstract
A cardiac glycoside epoxide, (−)-cryptanoside A (1), was isolated from the stems of Cryptolepis dubia collected in Laos, for which the complete structure was confirmed by analysis of its spectroscopic and single-crystal X-ray diffraction data, using copper radiation at a low temperature. This cardiac glycoside epoxide exhibited potent cytotoxicity against several human cancer cell lines tested, including HT-29 colon, MDA-MB-231 breast, OVCAR3 and OVCAR5 ovarian cancer, and MDA-MB-435 melanoma cells, with the IC50 values found to be in the range 0.1–0.5 μM, which is comparable with that observed for digoxin. However, it exhibited less potent activity (IC50 1.1 μM) against FT194 benign/non-malignant human fallopian tube secretory epithelial cells when compared with digoxin (IC50 0.16 μM), indicating its more selective activity toward human cancer versus benign/non-malignant cells. (−)-Cryptanoside A (1) also inhibited Na+/K+-ATPase activity and increased the expression of Akt and the p65 subunit of NF-κB but did not show any effects on the expression of PI3K. A molecular docking profile showed that (−)-cryptanoside A (1) binds to Na+/K+-ATPase, and thus 1 may directly target Na+/K+-ATPase to mediate its cancer cell cytotoxicity.
Graphical Abstract
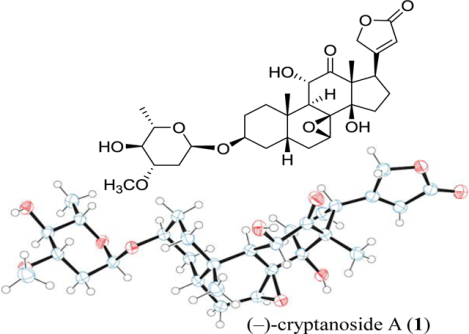
Cryptolepis dubia (Burm.f.) M.R. Almeida (Apocynaceae) is an evergreen liana used widely in folk medicine in Southeast Asia for the treatment of infections and other diseases.1,2 This species has several synonyms, including Chonemorpha reticulata G. Don, Cryptolepis buchananii Roem. & Schult. and Cryptolepis reticulata (Roth) Wall. ex Steud., Echites cuspidatus B. Heyne ex Hook.f. and Echites reticulatus (Roxb.) Roth, Nerium reticulatum Roxb., Periploca dubia Burm.f. and Periploca viridiflora Kostel., and Trachelospermum cavaleriei H. Lév. (all belonging to the Apocynaceae family).3,4 A biological investigation showed that both the CHCl3 and EtOH extracts of the leaves, roots, seeds, and stems of C. dubia exhibited activity against the bacteria Staphylococcus citreus and Shigella flexneri,5 and another study demonstrated that the MeOH extract of its stems showed analgesic, anti-inflammatory, and chondroprotective effects.1 In addition, the EtOH extract of the roots of C. dubia exhibited potent in vivo immunostimulant activity while its aqueous root extract showed hepatoprotective activity in mice.6,7 Also, silver nanoparticles prepared from the water extract of the leaves of C. dubia showed cytotoxicity against human HeLa cervical and MCF-7 breast cancer cells.8
Thus far, several cardiac glycoside epoxides have been identified from C. dubia, of which the structure of cryptosin isolated from the leaves of C. dubia has been determined by analysis of its single-crystal X-ray diffraction data.9–11 Similar to other cardiac glycosides, cryptosin was found to show cardiotonic action,10 and it also binds to Na+/K+-ATPase (NKA) and inhibits its activity.12,13 Both cryptanoside A (also named as buchanin, sarverogenin 3β-O-α-L-oleandroside, and 7,8β-epoxysinogenin-3β-L-oleandropyranoside) and cryptanoside C were isolated from the roots of C. dubia while cryptanosides A and B were identified from the leaves of this species.14,15 The structure of 7,8β-epoxysinogenin-3β-L-oleandropyranoside was confirmed by analysis of its single-crystal X-ray diffraction data, following which the NMR data measured in different solvents were reported, but the configuration of the entire molecule was inconclusive.16,17
No isolation work on any cardiac glycosides from the stems of C. dubia has been documented, even though the presence of cryptanosides A and C in the stems of C. dubia has been discussed in an early short report.5 Several other types of natural products have been isolated from this plant part, including pyridine alkaloids, a coumarin, and lignans.18–20 In addition, no cancer cell cytotoxicity data have been reported for any of these cardiac glycoside expoxides isolated from Cryptolepis species thus far.
Cardiac glycosides have attracted wide interest due to their therapeutic use in treating congestive heart disease, as well as their potential anticancer activity, for which several compounds have been evaluated in cancer clinical trials, including digoxin and oleandrin.21 Digoxin was found to mediate its antitumor activity by targeting directly HIF-1α, NKA, and NF-κB,22,23 and its mono-glycoside analogue, strebloside, exhibited potent cancer cell cytotoxicity by targeting potentially NKA, NF-κB, PI3K, and p53.24–26 In a continuing search for anticancer agents from higher plants,27 a CHCl3-soluble extract of the mature thickened stems of C. dubia collected in Laos was found to be cytotoxic toward several human cancer cell lines. Fractionation guided by bioassay against these cells yielded the cardiac glycoside epoxide, (–)-cryptanoside A (1), which showed cytotoxicity toward the human HT-29 colon, MDA-MB-231 breast, and OVCAR3 and OVCAR5 ovarian cancer and MDA-MB-435 melanoma cell lines. Also, the molecular targets were investigated by western blotting and molecular modeling for this compound.
RESULTS AND DISCUSSION
A sample of the stems of C. dubia (1.9 kg) collected in Laos was extracted with MeOH, and the MeOH extract was then partitioned with n-hexane and CH2Cl2. When the cytotoxic CH2Cl2 partition (6.8 g, IC50 <20.0 μg/mL) was subjected to chromatographic separation guided by inhibitory activity against human HT-29 colon and OVCAR3 ovarian cancer and MDA-MB-435 melanoma cells, three active fractions (650 mg, IC50 around 2 μg/mL) were obtained. Further purification of these combined fractions afforded a cytotoxic compound, (–)-cryptanoside A (1, 35.0 mg, IC50 0.1–0.5 μM).
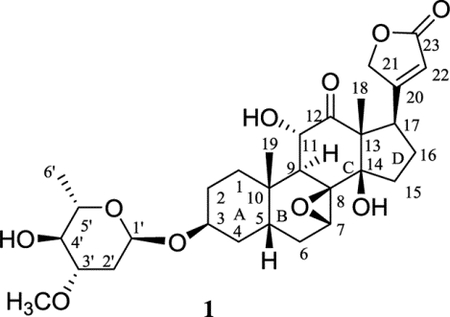
Compound 1 was purified as colorless rod-like crystals with a molecular formula of C30H42O10, as determined by the HRESIMS in conjunction with the 13C NMR spectroscopic data. The 1H and 13C NMR data (Table S1 and Figure S1)28 exhibited the characteristic resonances for a cardiac glycoside.24 Comparison of the NMR spectroscopic data of 1 with those reported for cryptanoside A indicated that these compounds are the same. This was substantiated by the almost identical mp and specific rotation values observed for 1 and those published previously for cryptanoside A, for which the mp 230–231 °C and [α]20D−28 values of 1 were comparable with those reported (mp 230 °C and [α]20D−30) in the literature.15
The aglycone of (–)-cryptanoside A, sarverogenin, is a hydrolyzed product of sarveroside that was isolated from the seeds of Strophanthus sarmentosus DC.29 A 7,15α-oxide unit was assigned in the original structure of sarverogenin through a series of chemical reactions,30 but such an assignment was found to be ambiguous when investigating the NMR spectroscopic data of sarverogenin acetate.31 It was hence proposed as 7,8β-epoxy-3β,11α,17β-trihydroxy-12-oxo-5β-card-20(22)-enolide by analysis of the IR, NMR, and mass spectra of this cardenolide,32 as supported by degradation of sarverogenin to several known bile-acid derivatives.33
Thus far, (–)-cryptanoside A (1) has been identified from different plant parts of C. dubia and assigned with several names. Its structure was proposed by analysis of its spectroscopic data and confirmed using its hydrolyzed products sarverogenin and L-oleandrose, but its NMR spectroscopic data were not assigned correctly, and its absolute configuration was not determined unambiguously by any of these previous investigations.14–16 Even though the crystal structure of 1 was reported previously, the authors did not provide detailed analytical data while the cif file was not presented properly, and the absolute configuration was not determined completely.16 Thus, in the present investigation, the analytical data of 1 are reported, and its 1H and 13C NMR spectroscopic data have been assigned, aided by analysis of its 1D and 2D NMR spectra (Table S1), while the absolute configuration has been determined unambiguously by analysis of its single-crystal X-ray diffraction data.
The X-ray diffraction data of 1 were collected at 100(2) K on a Bruker Kappa Photon II CPAD diffractometer, using Cu Kα radiation (Tables S2–S7), which showed that the crystal structure of 1 contains two independent molecules in the asymmetric unit (Figure 1 and Figure S2). The A and C rings are in a chair conformation, and the B ring is in a half-chair conformation while the D ring is in an envelope conformation. The lactone unit is relatively planar, and the saccharide ring is in a chair conformation (Figure 1). The A, B, C, D rings are fused in a cis-trans-cis fashion, and a (3S, 5S, 7S, 8R, 9S, 10S, 11S, 13R, 14R, 17R, 1’R, 3’S, 4’S, 5’S) absolute configuration was determined for 1 by anomalous dispersion using Parson’s method, with a Flack parameter of 0.034(40) being observed (Table S2).
Figure 1.
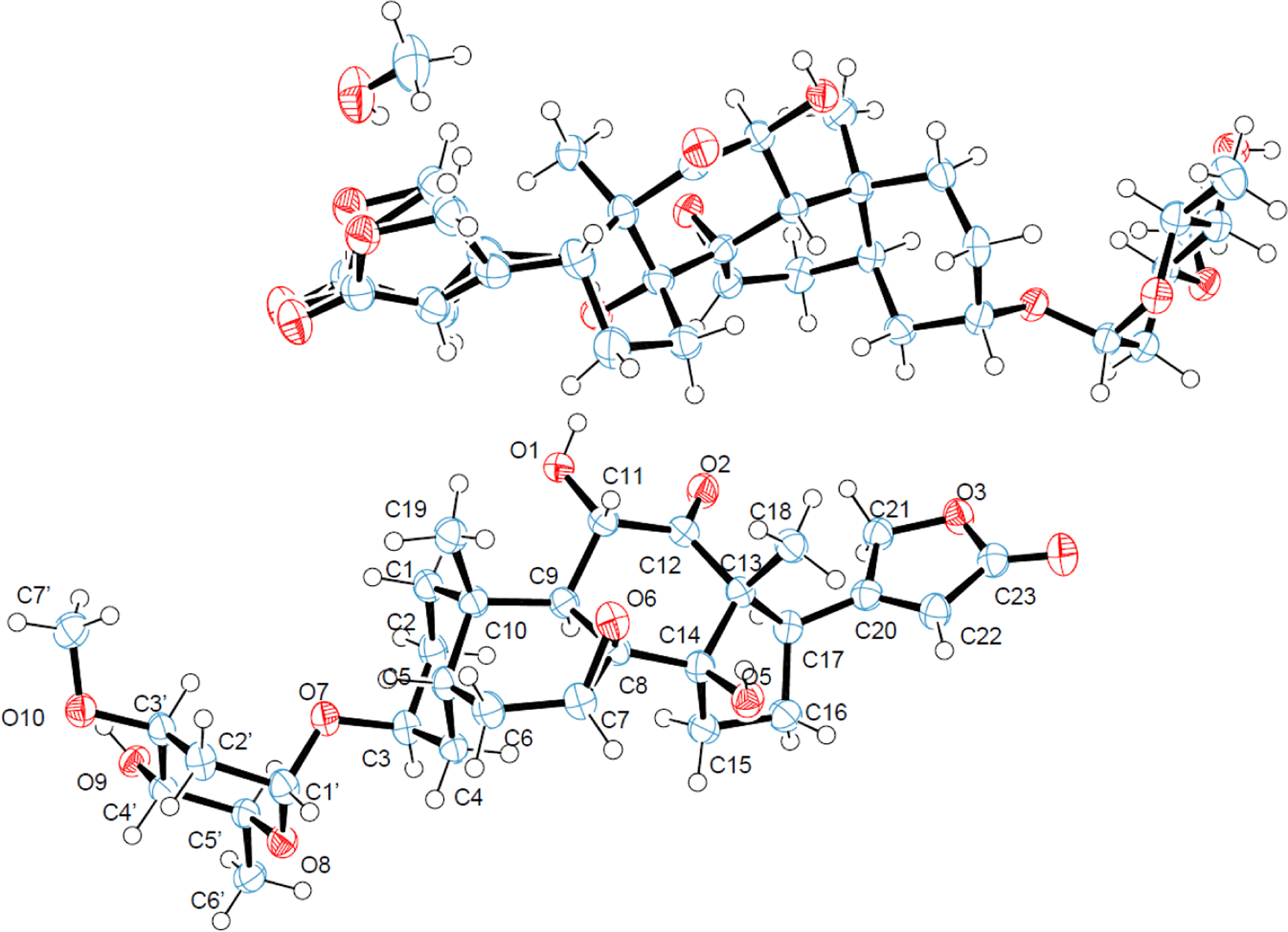
ORTEP plots for the molecular structure of (–)-cryptanoside A (1) (two independent molecules in the asymmetric unit) drawn with 50% probability displacement ellipsoids (oxygen atoms are red, carbon atoms are blue, and the small white circles represent hydrogen atoms, which are drawn with an artificial radius).
To support the future determination of the absolute configuration of (–)-cryptanoside A-like cardiac glycosides, the ECD spectrum of 1 was recorded, from which three positive Cotton effects (CE) around 280, 241, and 203 nm and a negative CE around 224 nm were observed (Figure S3). Of these, the positive CE around 241 nm, along with the negative CE around 224 nm, could correlate to the presence of a 17β lactone unit, as supported by the UV absorption maximum at ca. 215 nm (Figure S3), which indicates that these CEs are indicative of exciton coupling arising from the α,β-unsaturated carbonyl chromophore.22 The other two positive CEs around 280 and 203 nm may result from the 7,8β-epoxy group and the 11α-hydroxy-12-oxo substitution and their resultant conformational effects on the ECD spectrum of 1.22,34
Compound 1 was evaluated for its cytotoxicity against human HT-29 colon, MDA-MB-231 breast, and OVCAR3 and OVCAR5 ovarian cancer, and MDA-MB-435 melanoma cells, using a protocol reported previously, with paclitaxel as the positive control (Figure S4).25 This compound exhibited potent cytotoxicity toward all cancer cell lines tested, with IC50 values in the range 0.1‒0.5 μM, which is comparable with those observed previously for digitoxin, digoxin, and strebloside (Table 1).22,24,25 Comparison of the amount (35 mg) of this compound isolated and its cytotoxic potency (IC50 0.1–0.5 μM) with those of the active fractions (650 mg, IC50 around 2 μg/mL) separated from the cytotoxic CH2Cl2 extract (6.8 g, IC50 <20.0 μg/mL) suggests that (–)-cryptanoside A (1) is the major cytotoxic component of the stems of C. dubia. The stems of C. dubia are used medicinally, and its extracts were found to show antibacterial, analgesic, anti-inflammatory, and chondroprotective activities.1,5 However, the major active components that represent the bioactivities have not been characterized for this plant part in any previous investigations. Thus, identification of the cytotoxic (–)-cryptanoside A (1) from the stems of C. dubia in the present study could provide supportive information for the medicinal use of this plant.
Table 1.
Cytotoxicity of (–)-cryptanoside A (1)a
| compound | HT-29 | MDA-MB-231 | OVCAR3 | OVCAR5 | MDA-MB-435 | FT-194 |
|---|---|---|---|---|---|---|
| 1 | 0.22 ± 0.23 | 0.50 ± 0.05 | 0.14 ± 0.05 | 0.44 ± 0.08 | 0.20 ± 0.12 | 1.1 ± 0.17 |
| digoxin | 0.28 ± 0.03b | 0.31 ± 0.03b | 0.10 ± 0.01b | NT c | 0.17 ± 0.02b | 0.16 ± 0.15 |
| paclitaxeld | 3.4 ± 0.1 | 3.8 ± 0.1 | 4.9 ± 0.1 | 7.3 ± 0.1 | 1.9 ± 0.1 | NT c |
IC50 values are the concentration (μM) required for 50% inhibition of viability of the human HT-29 colon, MDA-MB-231 breast, OVCAR3 and OVCAR5 ovarian cancer and MDA-MB-435 melanoma and the FT194 benign/non-malignant human fallopian tube secretory epithelial cell lines for a given test compound with a 72 h treatment and were calculated using nonlinear regression analysis with measurements performed in triplicate and representative of three independent experiments.
Data reported previously.22
NT: Not tested.
Positive control (IC50 nM).
Interestingly, when tested against the FT194 benign/non-malignant human fallopian tube secretory epithelial cell line, compound 1 exhibited less potent activity (IC50 1.1 μM) when compared with its cancer cell cytotoxicity and with the value observed for digoxin (IC50 0.16 μM) (Table 1 and Figure S4), indicating that 1 shows somewhat selective activity against cancer versus benign/non-malignant cells.
Cardiac glycosides are well-known Na+/K+-ATPase (NKA) inhibitors, and both digoxin and strebloside were reported previously for their potent inhibitory activity against this enzyme, with the substituents at C-10, C-12, and C-17 found to be important for mediating the activity.22,35 The C. dubia-derived cardiac glycoside epoxide, cryptosin, a closely similar analogue of 1, was found previously to bind to NKA to inhibit its activity.12,13 Following a previous procedure,22 the cellular enzyme adenosine 5′-triphosphatase from the porcine cerebral cortex was treated in the present study with various concentrations of 1 and digitoxin, and both compounds were found to inhibit NKA activity in a concentration-dependent manner, with IC50 values of 1.2 μM and 0.16 μM being observed for 1 and digitoxin, respectively (Figure 2).
Figure 2.
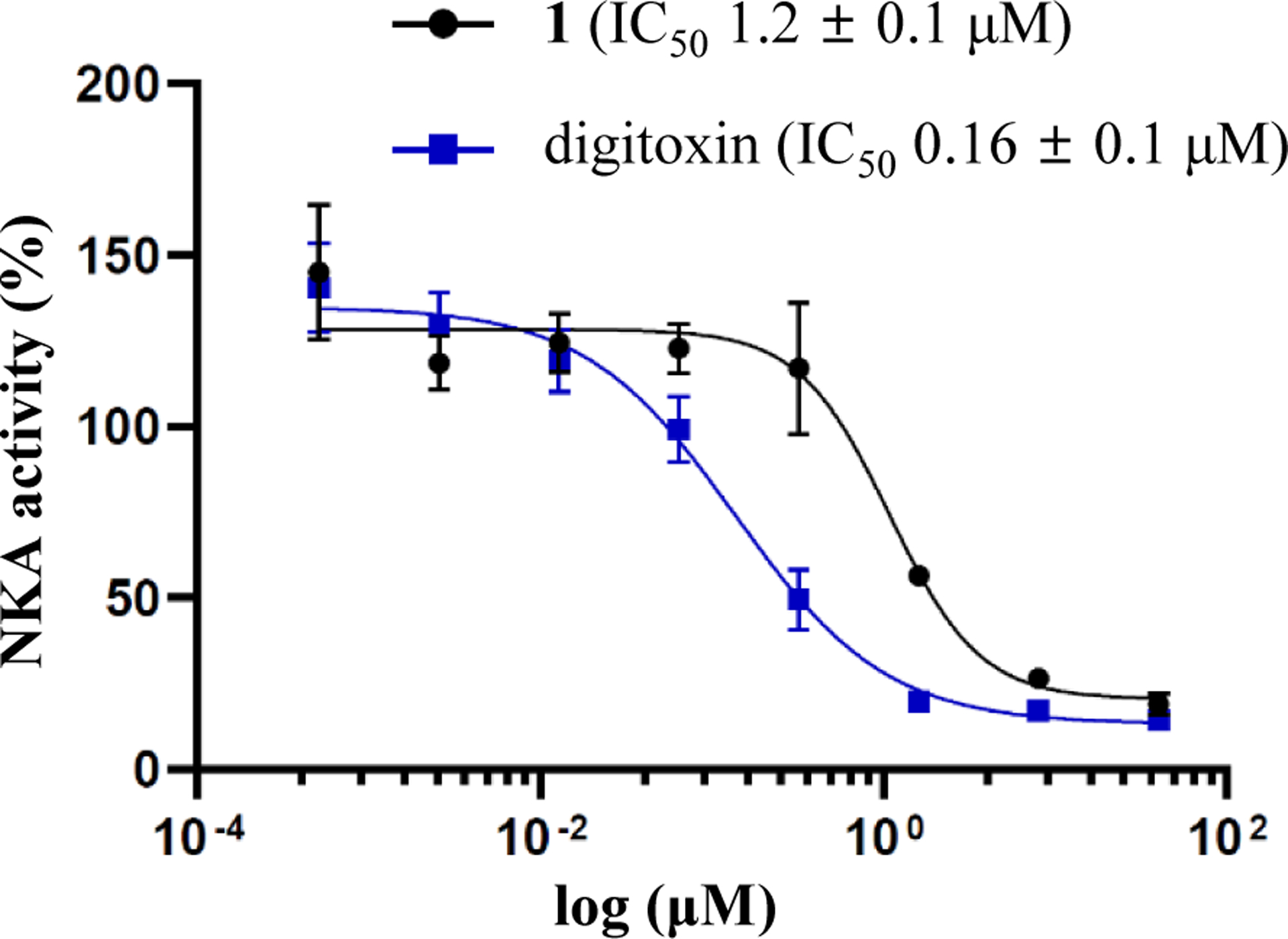
Inhibition of Na+/K+-ATPase by (–)-cryptanoside A (1) and digitoxin.
To support these observations, the docking profile for 1 and NKA has been investigated by AutoDock Vina, using a previously published procedure, and compared with analogous information reported previously for digoxin.23,26 This profile showed that the lactone unit of 1 can insert into the NKA binding pocket, with the binding pose being similar to that observed for digoxin. Hydrogen bonds are formed between the C-14 hydroxy and C-12 carbonyl groups of 1 and the Thr797 and Asn122 residues of NKA, respectively, and between the saccharide moiety of 1 and several polar residues at the entrance of the cavity, including Arg880, Glu312. Also, hydrophobic interactions were observed between 1 and the Phe783 and Ile315 residues of NKA (Figure 3). Both Thr797 and Asn122 are important residues of NKA, and thus these interactions could facilitate the binding between 1 and NKA, indicating that 1 does bind to NKA, as supported by its docking score of −9.2 kcal/mol (minimal, PDB:4RET). However, the 7,8-epoxy moiety restricts the interaction of 1 and Asp121 and Phe783 and hence prevents its docking deeply into the pocket, while digoxin can interact with Glu327 at the bottom to fit the pocket more completely. Thus, the binding between 1 and NKA is not as strong as that observed for digoxin, which showed docking scores of −12.0 kcal/mol (minimal, PDB:4RET).23 The binding and the docking score observed for 1 and digoxin are consistent with their NKA inhibitory activity, indicating that these two compounds could directly bind to NKA to inhibit its activity. However, the overview binding pose of 1 and digoxin is different when they dock the active binding pocket of NKA (Figure 3), which may contribute to the different cytotoxic potency observed for these compounds.
Figure 3.
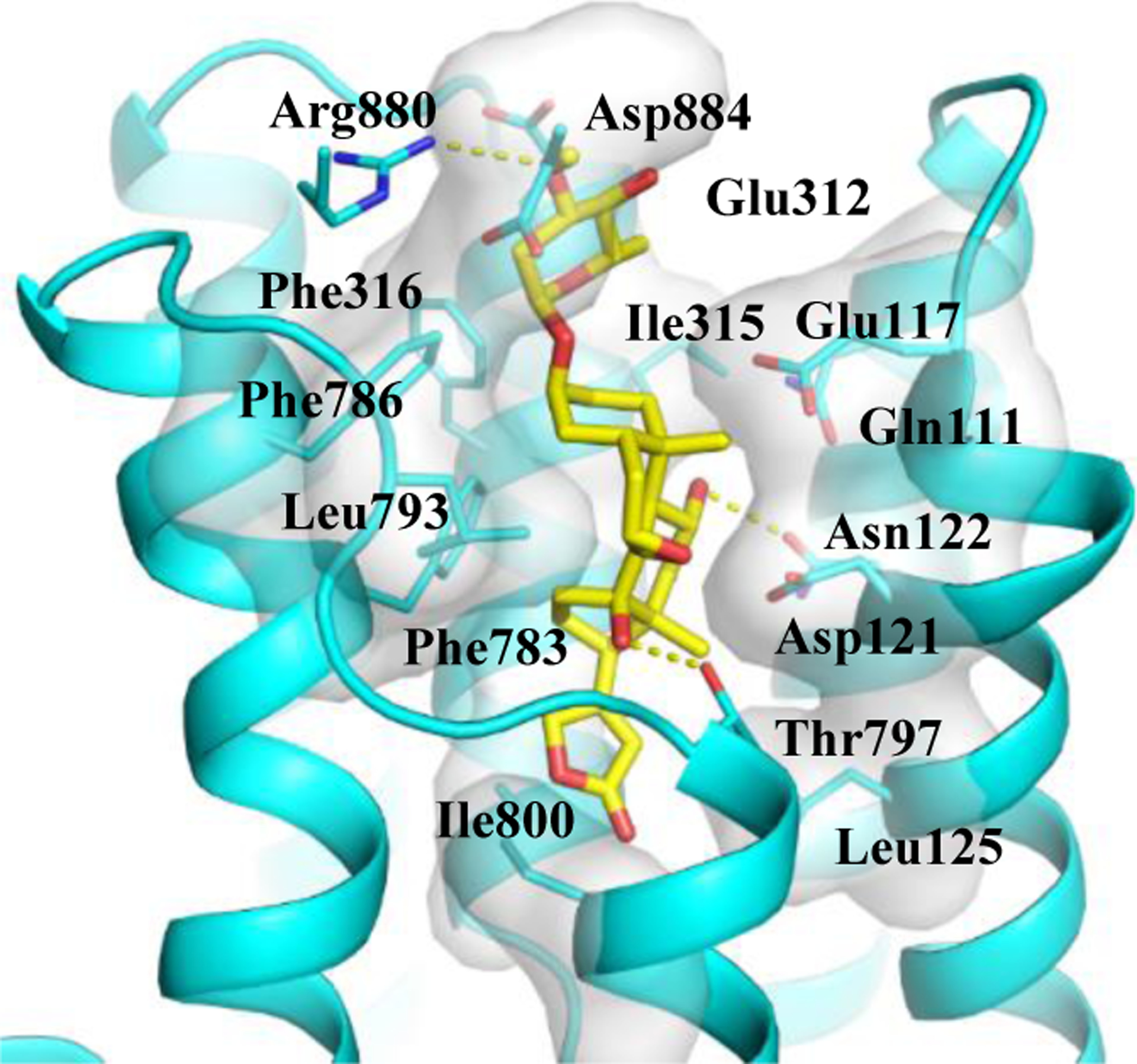
Docking profile for (–)-cryptanoside A (1) (yellow) and Na+/K+-ATPase (NKA).
Previously, (–)-cryptanoside A (1) was found to inhibit the growth of the bacterium Staphylococcus citreus,5 and thus it possesses some anti-infective potential. Inflammation is an immediate body response to tissue injury caused by infection or other noxious stimuli, and one of the mechanisms of inflammation-induced oncogenesis is the induction of ROS generation, which can trigger Akt, and this, in turn, activates NF-κB.36 NF-κB, a transcriptional activator of many inflammatory and tumor-promoting genes, has been proposed as an important link between inflammation and cancer, and it is a protein also involved in immunoregulation.36 Several cardiac glycosides have been reported to inhibit NF-κB activation to exhibit antitumor activity,37 and their major molecular target, NKA and its signalosome complex, has been demonstrated to play a critical role in the immune modulation of these compounds.38 In addition, both NF-κB and PI3K correlate to increased levels of reactive oxygen species, and inhibition of NKA can activate PI3K,39 which may then mediate its downstream pathways, including Akt signaling.40,41 Thus, regulation of the expression of p-Akt and the NF-κB p50 by 1 and digoxin was tested by western blotting herein.42,43 After 1 (0.5 μM) or digoxin (0.48 μM) was co-cultured with MDA-MB-231 cells for 5 h, the expression of phosphor-Akt (p-Akt) was found to be increased, but no significant changes were observed for the NF-κB p50 subunit for these compounds (Figure 4).
Figure 4.
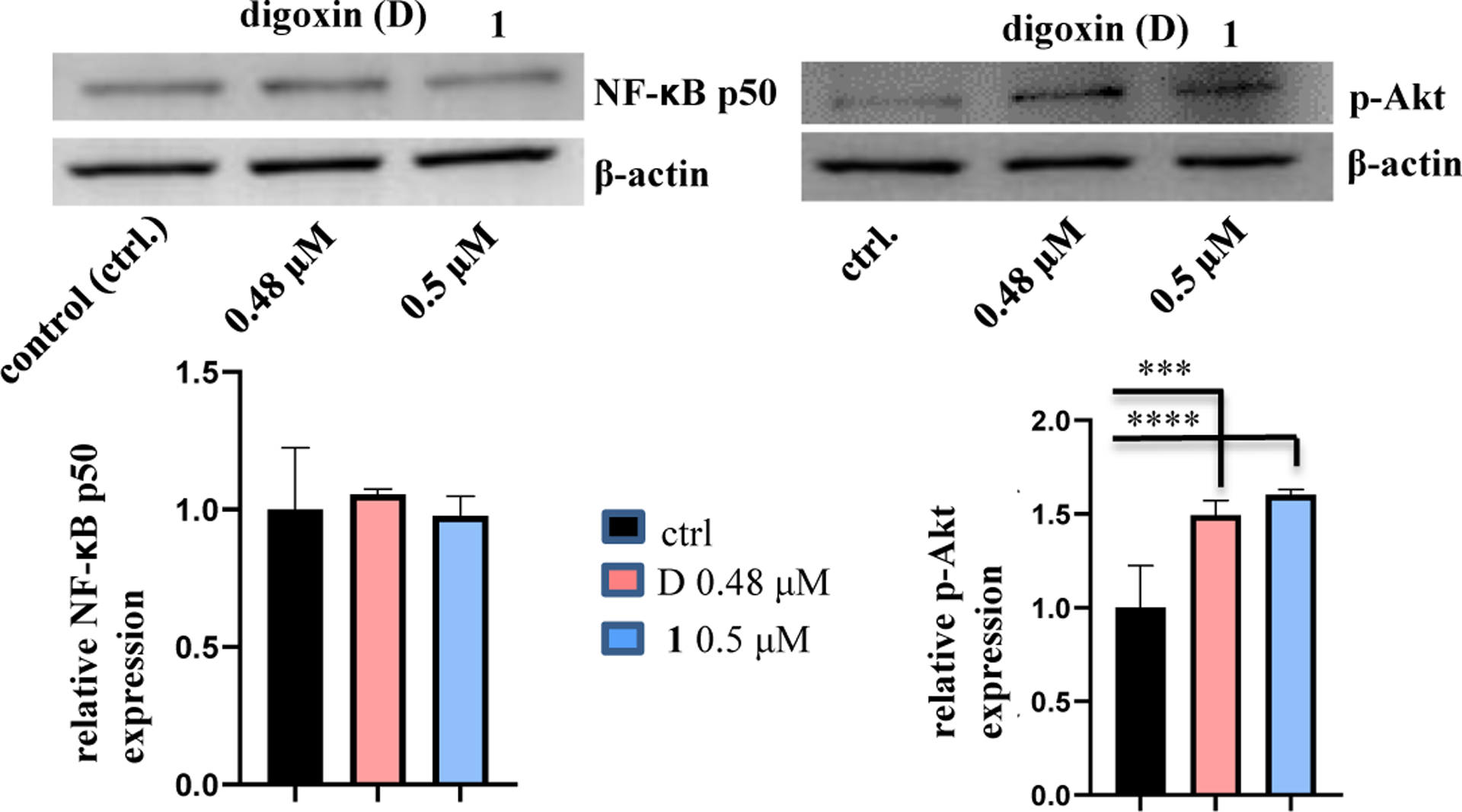
Regulation of the expression of p-Akt and the NF-κB p50 subunit of (–)-cryptanoside A (1). MDA-MB-231 cells were incubated with 1 or digoxin for 5 h, at concentrations of 0.5 μM and 0.48 μM, respectively. Expression of p-Akt and the NF-κB p50 subunit was determined by western blotting using rabbit monoclonal cleaved antibodies, and data for the targeted protein were normalized by the corresponding β-actin results of the same samples. A representative blot is shown from three independent experiments with similar results being shown (Experimental Section) [columns, mean in each group (n ≥ 3); bars, SE; *** p ≤ 0.05 and **** p ≤ 0.01 when compared to the control group (ctrl.)].
In addition, after 1 or digoxin was co-cultured with MDA-MB-231 cells at concentrations of 1, 5, or 10 μM for 72 h, and the cells still viable under these conditions were tested for western blotting. The expression of Akt and the NF-κB p65 subunit was found to be increased while that of the NF-κB p50 subunit was decreased, but no significant changes were observed for PI3K and the NF-κB p52 subunit, when compared with the control group. However, at concentrations of 5.0 or 10.0 μM, 1 seems to down-regulate significantly the expression of the NF-κB p52 subunit when compared with its 1.0 μM-treatment and with digoxin at these same concentrations used (Figure 5), suggesting that the activity of 1 and digoxin on the NF-κB p52 subunit may be different.
Figure 5.
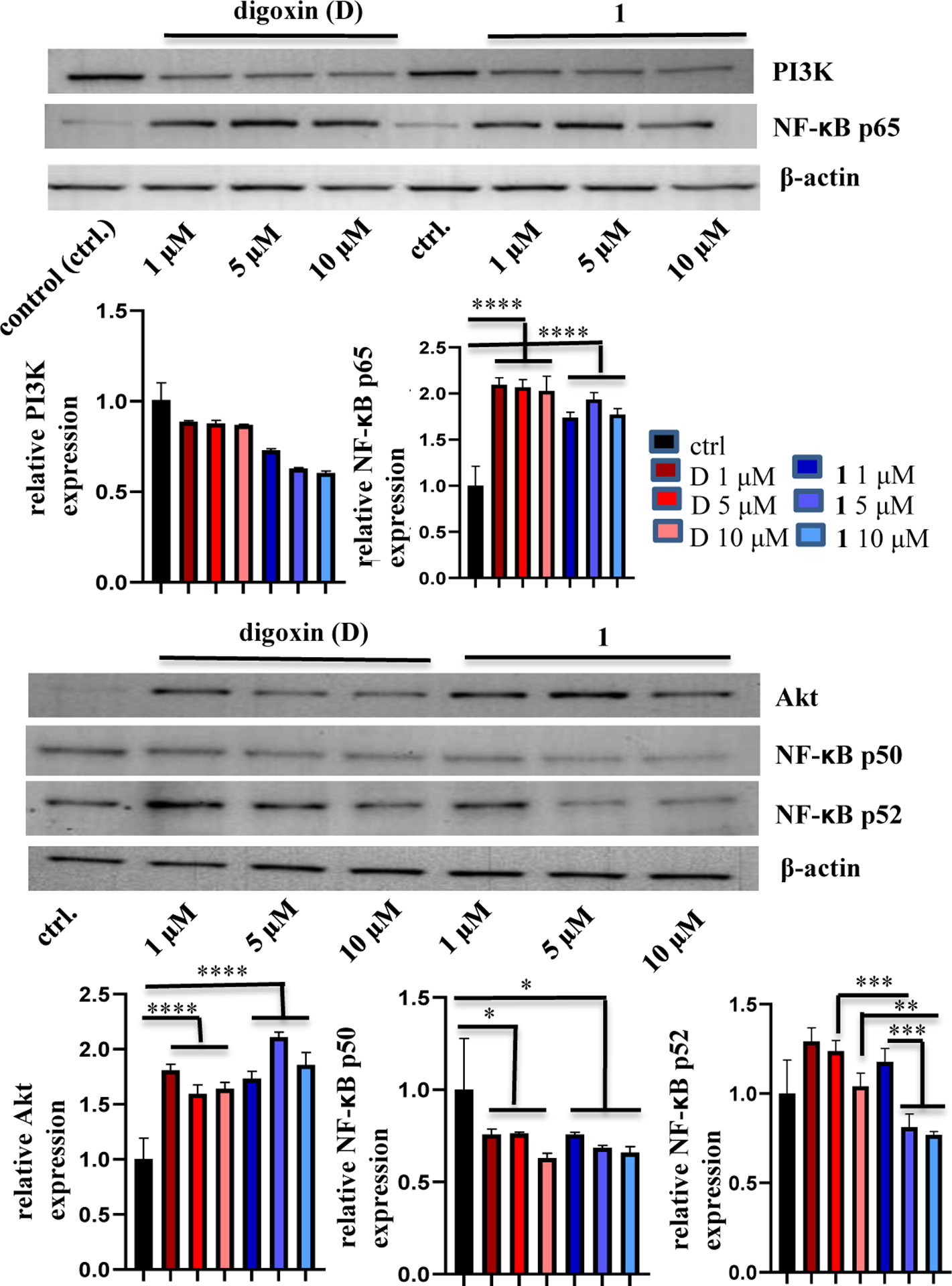
Regulation of the expression of Akt, the NF-κB p50, p52, and p65 subunits, and PI3K of 1. MDA-MB-231 cells were incubated with (–)-cryptanoside A (1) or digoxin for 72 h at different concentrations. Expression of the proteins tested was determined by western blotting using rabbit monoclonal cleaved antibodies, and data for the targeted proteins were normalized by the corresponding β-actin results of the same samples. A representative blot is shown from three independent experiments with similar results being shown (Experimental Section) [columns, mean in each group (n ≥ 3); bars, SE; * p ≤ 0.5, ** p ≤ 0.1, *** p ≤ 0.05, and **** p ≤ 0.01 when compared to the control group (ctrl.) or digoxin].
Na+/K+-ATPase (NKA) is a ubiquitous electrogenic pump located in the cell membrane to transport potassium ions in and sodium ions out to keep an equilibrium Na+/K+ ratio in cells.44,45 Inhibition of this pump could result in a change of the Na+/K+ ratio followed by a resultant higher concentration of cytoplasmic Ca2+, which increases calcium uptake into the endoplasmic reticulum to lead to tumor cell apoptosis.44 Thus, NKA has been defined as a promising target for the development of novel anticancer agents.46 In the present investigation, (–)-cryptanoside A (1) was found to bind to NKA to inhibit its activity, and one of its close analogues, cryptosin, was reported previously to inhibit NKA and to reduce significantly the Ca2+ channel and the β-adrenoceptor densities, of which reduction of the calcium channel density may result from enhanced Ca2+ levels.12,13,47,48 Thus, (–)-cryptanoside A (1) could mediate its cancer cell cytotoxicity by directly targeting NKA to induce cancer cell apoptosis.
Akt, also named as protein kinase B (PKB), is a set of three serine/threonine-specific protein kinases (Akt1, Akt2, and Akt3) that play a key role in cell survival, cell cycle, metabolism, autophagy, and angiogenesis. Akt regulates many downstream proteins, including NF-κB, Bcl-2, TFEB, MDM2, and Wnt and thus becomes a dependent effector for cell survival, and activation of Akt can promote tumor growth.49 Also, Akt is a major protein in PI3K/Akt/mTOR signaling, and several Akt inhibitors have been thus far evaluated in clinical trials for the development of new anticancer agents,50 even though some adverse effects such as hyperglycemia and hyperinsulinemia have been observed for them.51 Compound 1 was found to inhibit NKA, which would lead to cancer cells being stressed. As a result, these stressed cells activate Akt to enhance their survival, and thus the expression of p-Akt was increased after MDA-MB-231 cells were treated by 1 at 0.5 μM for 5 h. Consistently, the expression of Akt was also increased after cells were treated by 1 at high concentrations for 72 h, from which a small proportion of cells survived (Figures 4 and 5).
Activated Akt can result in activation of NF-κB,49 which controls DNA transcription, cytokine production, and cell survival, and connects infection, cancer, and cancer immunity, and thus promotes tumorigenesis and modulates the cancer response to therapy.52,53 NF-κB supports cancer cell proliferation and tumor growth through the transcription of antiapoptotic proteins, and thus inhibition of NF-κB can be of benefit to the cancer treatment.52 However, the p50 subunit of NF-κB has been found to act as both a tumor-driver and a tumor-suppressor to promote and inhibit tumor growth, respectively, and thus this subunit seems to contribute to cancer growth with a complex role.54 Therefore, at high concentrations, both 1 and digoxin were found to increase the expression of the NF-κB p65 subunit while they decreased significantly the expression of the NF-κB p50 subunit with a 72 h treatment (Figure 5). However, these compounds did not show any effects on the NF-κB p50 subunit at a low concentration (less than 1.0 μM) with a 5 h treatment (Figure 4). These could result from activity of 1 and digoxin on all of Akt, NKA, and NF-κB, and thus the role of NF-κB in the mediation of the cytotoxicity of these compounds is complex, involving suppression and promotion of cancer cell growth, of which the p50, p52 and p65 subunits function differently.
Even though it has been demonstrated that inhibition of NKA could activate PI3K,39 no significant effects on PI3K were observed for 1 and digoxin (Figure 5), indicating that the expression of PI3K would not be changed by their activity on Akt, NF-κB, and NKA.
In the class of cardiac glycosides, cardiac glycoside epoxides are a small group. In general, these compounds contain an epoxy group at the C-7 and C-8 positions and a 11-hydroxy and 12-oxo group substitution, and compound 1 is the first member to be reported for its cancer cell cytotoxicity. This compound was also found to bind to NKA to inhibit its activity, with the potency being less than that observed for digoxin, which could result from the different binding poses observed. Thus, compound 1 could mediate its cancer cell cytotoxicity by inhibiting NKA, which results in activation of Akt and then the NF-κB p65 subunit to support cancer cell survival.
EXPERIMENTAL SECTION
General Experimental Procedures.
The melting point was measured using a Fisher Scientific apparatus that is uncorrected. The specific rotation was measured at room temperature on an Anton Paar MCP 150 polarimeter (Anton Paar). The ultraviolet (UV) spectrum was recorded on a Hitachi U2910 ultraviolet spectrophotometer. Electronic circular dichroism (ECD) measurement was performed using a JASCO J-810 spectropolarimeter. The infrared (IR) spectrum was recorded on a Nicolet 6700 FT-IR spectrometer (Thermo Scientific). 1H and 13C, DEPT 90, DEPT 135, COSY, HSQC, HMBC, and NOESY nuclear magnetic resonance (NMR) spectra were recorded at room temperature on a Bruker Avance III 400 or a Bruker Avance III HD 700 MHz NMR spectrometer. The 1H NMR and 13C NMR spectroscopic data (δ) measured in CDCl3 were referenced to the solvent residual peaks at δ 7.26 and δ 77.16, respectively, while those measured in DMSO-d6 were referenced to the solvent residual peaks at δ 2.50 and δ 39.52, respectively.28 ESIMS and HRESIMS data were collected on a Bruker Maxis 4G Q-TOF mass spectrometer in the positive-ion mode. A Dionex/Thermo UltiMate 3000 HPLC System was used to evaluate the purity of 1. Column chromatography was conducted using silica gel (65 × 250 or 230 × 400 mesh, Sorbent Technologies). Analytical TLC was performed on precoated silica gel 60 F254 plates (Sorbent Technologies). Sephadex LH-20 was purchased from Amersham Biosciences. For visualization of TLC plates, H2SO4 was used as a spray reagent. All procedures were carried out using solvents purchased from commercial sources and employed without further purification. Digoxin, digitoxin, and paclitaxel were obtained from Sigma-Aldrich (purity ≥ 98%).
Plant Material.
A sample of the stems of Cryptolepis dubia (Burm.f.) M.R. Almeida (Apocynaceae) (acquisition number AAA07194) was identified by D.D.S. and collected in November 2018 by D.D.S., K.S., and M.X. (voucher specimen: DDS 15365), from the mature thickened stems base of a liana, bearing greenish yellow to pale yellow flowers and exuding a milky white latex when the young stems were cut, at the Kham Medicinal Biodiversity Preserve (19° 42.637’ N; 103° 35.350’ E), Kham District, Xieng Khouang Province, Laos. A voucher herbarium specimen has been deposited at the John G. Searle Herbarium of the Field Museum of Natural History, Chicago, IL, USA, under the accession number FM-2326945. This plant sample was collected under a collaborative arrangement between the University of Illinois at Chicago (USA) and the Institute of Traditional Medicine, Ministry of Health, Vientiane, Laos.
Extraction and Isolation.
The milled air-dried stems of Cryptolepis dubia (sample AAA07194, 1935 g) were extracted with MeOH (7 L × 5) at room temperature (rt). The solvent was evaporated in vacuo, and the dried MeOH extract (138.5 g, 7.2%) was resuspended in 10% H2O in MeOH (700 mL) and partitioned with n-hexane (500, 300, and 200 mL), to yield an n-hexane-soluble residue (26.0 g, 1.3%). Then, 100 mL of H2O were added to the aqueous MeOH layer, and this was partitioned with CH2Cl2 (400, 300, and 200 mL). The combined CH2Cl2 partition was washed with a 1% aqueous solution of NaCl, to partially remove plant phenolic components, and the solvent was evaporated to afford a CH2Cl2-soluble extract (7.0 g, 0.36%). This extract exhibited cytotoxicity toward the human HT-29 colon and OVCAR3 ovarian cancer and MDA-MB-435 melanoma cell lines (IC50 < 20.0 μg/mL), but both the n-hexane- and water-soluble extracts were inactive against all these cancer cells tested (IC50 >20.0 μg/mL) in the bioassay system used. The CH2Cl2-soluble extract (6.8 g) was subjected to silica gel column chromatography (5.0 × 25 cm) and eluted with a gradient of CH2Cl2-MeOH. Eluates were pooled by TLC analysis to give 32 combined fractions (D2F1−D2F32), of which fractions D2F9–D2F11 (650 mg) were deemed active against all the cancer cells tested, with the IC50 value being around 2 μg/mL. These active fractions were combined and chromatographed over a silica gel column (4.5 × 40 cm), eluted with a gradient of CH2Cl2-MeOH, followed by separation over a Sephadex LH-20 column eluted with CH2Cl2-MeOH (1:1) and crystallization in MeOH, affording (–)-cryptanoside A [1, (–)-7,8β-epoxysinogenin-3β-L-oleandropyranoside, 35.0 mg (purity > 98%)].
(–)-cryptanoside A (1): Colorless rod-like crystals (MeOH), mp 230–231 °C; [α]20D −28 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 215 (4.04) nm; ECD (0.053 μM, MeOH) λmax (Δε) 203 (+3.58), 224 (−1.38), 241 (+1.07), 280 (+3.01) nm; IR (dried film) νmax 3460, 2925, 1744, 1732, 1633, 1442, 1378, 1343, 1298, 1260, 1197, 1077, 990, 896 cm−1; 1H and 13C NMR data, Table S1; positive-ion HRESIMS m/z 585.2656 [M + Na]+ (calcd for C30H42O10Na+, 585.2670).
X-ray Crystal Structure Analysis of 1.
Intensity data for a small colorless rod; molecular formula C30H42O10 with 0.0785 CH3OH, MW = 565.16, MeOH, space group P212121, a = 10.0990(6) Å, b = 15.4768(9) Å, c = 35.817(2) Å, V = 5598.2(6) Å3, Z = 8, density (calculated) = 1.341 mg/m3, size 0.226 × 0.172 × 0.067 mm3] from 1 were collected at 100(2) K on a Bruker Kappa Photon II CPAD diffractometer equipped with Cu Kα radiation (λ = 1.54178 Å). The crystal was mounted on a Cryoloop with Paratone 24EX oil, and data were collected in a nitrogen gas stream at 100(2) K using φ and ω scans. The crystal-to-detector distance was 40 mm using variable exposure times (20–60 s) depending on θ with a scan width of 1.0°. Data collection was 99.7% complete to 68.00° in θ (0.83 Å). A total of 59910 reflections were collected covering the indices, −12 ≤ h ≤ 12, −19 ≤ k ≤ 18, −44 ≤ l ≤ 40, and 11286 reflections were found to be symmetry independent, with a Rint of 0.0443. Indexing and unit cell refinement indicated a primitive, orthorhombic lattice, and the space group was found to be P212121. The data were integrated using the Bruker SAINT software program and scaled using the SADABS software program, and solution by direct methods (SHELXT) produced a complete phasing model for refinement.55 Full-matrix least-squares refinements based on F2 were performed in SHELXL-2014,56 and all non-hydrogen atoms were refined anisotropically by full-matrix least-squares while all hydrogen atoms were placed using a riding model, with their positions constrained relative to their parent atom using the appropriate HFIX command in SHELXL-2014. The absolute configuration of the molecule was established by anomalous dispersion using Parson’s method with a Flack parameter of 0.034(40). The final refinement cycle was based on 11286 intensities, 111 restraints, and 768 variables and resulted in agreement factors of R1(F) = 0.0405 and wR2(F2) = 0.0947. For the subset of data with I > 2* sigma(I), the R1(F) value is 0.0394. The final difference electron density map contains maximum and minimum peak heights of 0.207 and −0.190 e/Å3. Neutral atom scattering factors were used and include terms for anomalous dispersion.57 The CIF file of the X-ray data of 1 has been deposited in the Cambridge Crystallographic Data Centre (deposition no.: CCDC 2238464).
Cytotoxicity Assays.
All cell lines were purchased from the American Type Culture Collection (ATCC) and cultured at 37 °C in 5% CO2. The human HT-29 colon, MDA-MB-231 breast, and OVCAR3 and OVCAR5 ovarian cancer and MDA-MB-435 melanoma and the FT194 benign/non-malignant human fallopian tube epithelial cell lines were cultured in RPMI 1640 medium, and supplemented with fetal bovine serum (FBS) (10%), penicillin (100 units/mL), and streptomycin (100 μg/mL).The cytotoxicity of 1 was screened against these cell lines, using a procedure reported previously,24,25 with the vehicle and paclitaxel used as the negative and positive control, respectively. Briefly, after log-phase-growth, cells were seeded in 96-well clear flat-bottomed plates (Microtest 96, Falcon) (5,000 cells per well) and incubated overnight. The cells were treated with the test sample or paclitaxel (both dissolved in DMSO and diluted to the different concentrations required) or the vehicle (DMSO) for 72 h. The viability of cells was evaluated using a commercial absorbance assay (CellTiter-Blue Cell Viability Assay, Promega Corp.), with the IC50 values calculated from the vehicle control.
Na+/K+-ATPase Activity Assay.
Na+/K+-ATPase (NKA) activity was assessed using a luminescent ADP detection assay (ADP-Glo Max Assay; Promega) that measures enzymatic activity by quantitating the ADP produced during the enzymatic first half-reaction.35 Specifically, 10 μL of 1 × NKA reaction assay buffer containing adenosine 5′-triphosphatase (ATP) from porcine cerebral cortex (Sigma) were added to the wells of a 96-well plate followed by adding 10 μL of DMSO or the test compound dissolved in DMSO at different concentrations. Then, 5.0 μL of 5 mM ATP were added to each well followed by a 15 min incubation at 37 °C and subsequently adding 25 μL of ADP-Glo Reagent. After a 40 min incubation at rt, 50 μL of kinase detection reagent were added to each well followed by a 60 min incubation at rt. ATP was then measured versus a luciferin/luciferase reaction using a Synergy Mx microplate reader (BioTek) to assess luminescence.
Western Blot Analysis.
MDA-MB-231 human breast cancer cells were propagated at 37 °C in 5% CO2 in DMEM medium, supplemented with fetal bovine serum (10%), penicillin (100 units/mL), and streptomycin (100 μg/mL). Cells in the log phase of growth were harvested by trypsinization followed by two washings to remove all traces of enzyme. A total of 5,000 cells were seeded per well of 6-well clear flat-bottom plates (Microtest 96, Falcon) and incubated overnight (37 °C in 5% CO2). For the higher doses, compound 1 or digoxin dissolved in DMSO was diluted to 1.0 μM, 5.0 μM, and 10.0 μM for each and added to the appropriate wells containing MDA-MB-231 cells, which were incubated in the presence of the test samples for 72 h at 37 °C. For the lower dose, compound 1 (0.5 μM) or digoxin (0.48) μM was dissolved in DMSO and added to the appropriate wells containing MDA-MB-231 cells, which were incubated in the presence of the test samples for 5 h at 37 °C. The cells were then harvested and washed once with ice-cold PBS, and lysed (108 cells/mL lysis buffer) in RIPA (Thermo Fisher) with 1 mM EDTA and protease inhibitor cocktail tablet (Roche Applied Science). Cell lysates containing equal amounts of protein for the test samples and control, adding 4 or 2× Laemmli buffer (Bio-Rad) by supplementing with 2.5% β-mercaptoethanol to give 1× SDS sample buffer, were boiled for 10 min and subjected to western blot analysis, as described previously.42,43 To determine protein concentrations, protein levels were assessed using a BCA protein assay kit (Thermo Fisher), standardized with BSA. Protein samples were resolved on 4–15% SDS-PAGE (Bio-Rad) and western blot analysis was performed using Abs against the indicated signaling molecules, and data for the targeted protein were normalized by the corresponding β-actin results of the same samples. The antibodies used were rabbit/mouse monoclonal Akt, p-Akt, the NF-κB p50, p52, and p65 subunits (Cell Signaling Technology), PI3K (Thermo Fisher), and mouse polyclonal β-actin (Proteintech).
Molecular Modeling.
Following previous procedures,22,23,26 the crystal structure of Sus scrofa NKA (sNKA) was obtained from the Protein Data Bank (PDB, 4RET) and used as the receptor, and the conformations of 1 generated by LigPrep from Schrödinger Suite 2018–2 [Schrödinger Suite 2018–2 Protein Preparation Wizard] were used in molecular docking against the receptor by AutoDock Vina.58,59 Geometric optimization was performed using the OPLS3 force field with all possible ionization states at pH 7.4 ± 0.1 created by Epik.59 AutoDock Vina was used to generate the docking profiles, which were analyzed by PyMol.60,61
Statistical Analysis.
The in vitro measurements were performed in triplicate and are representative of three independent experiments. The dose response curve was calculated for IC50 determinations using non-linear regression analysis (Table Curve2DV4; AISN Software Inc.). A two-sided Student’s t test was used to compare two independent conditions. Differences among samples were assessed by one-way ANOVA followed by Tukey-Kramerʼs test. Statistical software GraphPad, R.3.6.3 and SAS 9.4 were used for the analysis, and the significance level was set at p < 0.5, p < 0.1, p < 0.05, or p < 0.01.
Supplementary Material
ACKNOWLEDGMENTS
This investigation was supported by grants P01 CA125066 and K12 GM139186 funded to A. D. Kinghorn by the National Cancer Institute and to E. M. Kaweesa by National Institute of General Medical Sciences, respectively, NIH, Bethesda. MD, USA. Scientists from The Ohio State University, including Drs. C. Yuan, A. Somogy, G. Wu, and N. M. Kleinholz, Campus Chemical Instrument Center, for assistance with NMR and MS data collection, Drs. D. Uchenik, D. Krishnan, and J. C. Gallucci, College of Pharmacy, for access to the instrumentation used and helpful information about the crystal structure of compound presented, are acknowledged.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website, including mass and NMR spectra, the single-crystal X-ray diffraction data and cif file, and the raw data for bioassay testing of 1. The raw NMR data have been uploaded to the NP-MRD website, with a deposition ID of NPd000000072 being assigned.
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Hanprasertpong N; Teekachunhatean S; Chaiwongsa R; Ongchai S; Kunanusorn P; Sangdee C; Panthong A; Bunteang S; Nathasaen N; Reutrakul V BioMed Res. Int 2014, 978582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Bisht Priyanka, Juyal AS, D. Pharm. Innov. J 2018, 7, 39–41. [Google Scholar]
- (3). http://worldfloraonline.org/taxon/wfo-0000628338;jsessionid=3B2F2A335C837A3F7FAFCA3F3CF97F63.
- (4). https://www.iapt-taxon.org/nomen/main.php;https://jagiroadcollegelive.co.in/attendence/classnotes/files/1587290512.pdf.
- (5).Vasanth S; Gopal RH; Rao RB Fitoterapia 1997, 68, 463–464. [Google Scholar]
- (6).Kaul A; Bani S; Zutshi U; Suri KA; Satti NK; Suri OP Phytother. Res 2003, 17, 1140–1144. [DOI] [PubMed] [Google Scholar]
- (7).Rajeshwari HM; Lakshman K; Ranjan Kumar M; Nandish C IOSR J. Pharm 2021, 11, 45–58. [Google Scholar]
- (8).Panja S; Patra A; Khanra K; Choudhuri I; Pati BR; Bhattacharyya N Nanomed. Res. J 2020, 5, 369–377. [Google Scholar]
- (9).Narendra N; Viswamitra MA; Venkateswara R; Rao KS; Vaidyanathan CS Acta Cryst. 1987, C43, 1562–1564. [Google Scholar]
- (10).Venkateswara R; Rao KS; Vaidyanathan CS Plant Cell Rep. 1987, 6, 291–293. [DOI] [PubMed] [Google Scholar]
- (11).Venkateswara R; Narendra N; Viswamitra MA; Vaidyanathan CS Phytochemistry 1989, 28, 1203–1205. [Google Scholar]
- (12).Rao VR; Dasgupta D; Vaidyanathan CS Biochem. Pharmacol 1989, 38, 2039–2041. [DOI] [PubMed] [Google Scholar]
- (13).Venkateswara RR; Dasgupta D; Banning JW; Vaidyanathan CS Biochem. Int 1990, 22, 287–294. [PubMed] [Google Scholar]
- (14).Khare MP; Shah BB J. Nep. Chem. Soc 1983, 3, 21–30. [Google Scholar]
- (15).Purushothaman KK; Vasanth S; Connolly JD; Rycroft DS Rev. Latinoamer. Quím 1988, 19, 28–31. [Google Scholar]
- (16).Nethaji M; Pattabhi V; Gabe EJ Acta Cryst. 1988, C44, 1066–1070. [DOI] [PubMed] [Google Scholar]
- (17).Pan L; Wang M-K; Peng S-L; Zhang X-F; Ding L-S Nat. Prod. Res. Dev 2002, 14, 29–31. [Google Scholar]
- (18).Dutta SK; Sharma BN; Sharma PV Phytochemistry 1978, 17, 2047–2048. [Google Scholar]
- (19).Dutta SK; Sharma BN; Sharma PV Phytochemistry 1980, 19, 1278. [Google Scholar]
- (20).Li J-L; Zhao Y-L; Qin X-J; Liu Y-P; Luo X-D Chin. Trad. Herb. Drugs 2014, 45, 1677–1681. [Google Scholar]
- (21).Reddy D; Kumavath R; Barh D; Azevedo V; Ghosh P Molecules 2020, 25, 3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Ren Y; Ribas HT; Heath K; Wu S; Ren J; Shriwas P; Chen X; Johnson ME; Cheng X; Burdette JE; Kinghorn AD J. Nat. Prod 2020, 83, 638–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Ren Y; Wu S; Burdette JE; Cheng X; Kinghorn AD Molecules 2021, 26, 3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Ren Y; Chen W-L; Lantvit DD; Sass EJ; Shriwas P; Ninh TN; Chai H-B; Zhang X; Soejarto DD; Chen X; Lucas DM; Swanson SM; Burdette JE; Kinghorn AD J. Nat. Prod 2017, 80, 648‒658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Chen W-L; Ren Y; Ren J; Erxleben C; Johnson ME; Gentile S; Kinghorn AD; Swanson SM; Burdette JE J. Nat. Prod 2017, 80, 659–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Ren Y; Wu S; Chen S; Burdette JE; Cheng X; Kinghorn AD Molecules 2021, 26, 5675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Aldrich LN; Burdette JE; Carcache de Blanco E; Coss CC; Eustaquio AS; Fuchs JR; Kinghorn AD; MacFarlane A; Mize BK; Oberlies NH; Orjala J; Pearce CJ; Phelps MA; Rakotondraibe LH; Ren Y; Soejarto DD; Stockwell BR; Yalowich JC; Zhang XJ Nat. Prod 2022, 85, 702–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Gottlieb HE; Kotlyar V; Nudelman AJ Org. Chem 1997, 62, 7512‒7515. [DOI] [PubMed] [Google Scholar]
- (29).Buzas A; Euw JV; Reichstein T Helv. Chim. Acta 1950, 33, 465–485. [Google Scholar]
- (30).Taylor DAH Chem. Ind 1953, 62–63. [Google Scholar]
- (31).Taylor DAH J. Chem. Soc. (C) 1966, 790–791. [Google Scholar]
- (32).Fuhrer H; Züercher RF; Reichstein T Helv. Chim. Acta 1969, 52, 616–621. [Google Scholar]
- (33).Lardon A; Reichstein T Helv. Chim. Acta 1987, 70, 894–928. [Google Scholar]
- (34).Padula D; Pescitelli G Molecules 2018, 23, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Ren Y; Tan Q; Heath K; Wu S; Wilson JR; Ren J; Shriwas P; Yuan C; Ninh TN; Chai H-B; Chen X; Soejarto DD; Johnson ME; Cheng X; Burdette JE; Kinghorn AD Bioorg. Med. Chem 2020, 28, 115301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Kundu JK; Surh Y-J Free Rad. Biol. Med 2012, 52, 2013‒2037. [DOI] [PubMed] [Google Scholar]
- (37).Slingerland M; Cerella C; Guchelaar HJ; Diederich M; Gelderblom H Invest. New Drugs 2013, 31, 1087‒1094. [DOI] [PubMed] [Google Scholar]
- (38).Škubník J; Pavlíčková V; Rimpelová S Biomolecules 2021, 11, 659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Zhou X; Jiang G; Zhao A; Bondeva T; Hirszel P; Balla T Biochem. Biophys. Res. Commun 2001, 285, 46–51. [DOI] [PubMed] [Google Scholar]
- (40).Zhang J; Wang X; Vikash V; Ye Q; Wu D; Liu Y; Dong W Oxidat. Med. Cell. Longev 2016, 4350965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Khan H; Singh A; Thapa K; Garg N; Grewal AK Singh TG Brain Res. 2021, 1761, 147399. [DOI] [PubMed] [Google Scholar]
- (42).Xu B; Tian L; Chen J; Wang J; Ma R; Dong W; Li A; Zhang J; Chiocca EA; Kaur B; Feng M; Caligiuri MA; Yu J Nat. Commun 2021, 12, 5908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Tian L; Xu B; Chen Y; Li Z; Wang J; Zhang J; Ma R; Cao S; Hu W; Chiocca EA; Kaur B; Caligiuri MA; Yu J Nat. Cancer 2022, 3, 1318–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Patel S Biomed. Pharmacother 2016, 84, 1036–1041. [DOI] [PubMed] [Google Scholar]
- (45).Manoj KM; Gideon DA; Bazhin NM.; Tamagawa H; Nirusimhan V; Kavdia M; Jaeken L J. Cell. Physiol 2023, 238, 109–136. [DOI] [PubMed] [Google Scholar]
- (46).Themistocleous SC; Yiallouris A; Tsioutis C; Zaravinos A; Johnson EO; Patrikios I Oncol. Lett 2021, 22, 658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Rao VR; Banning JW Life Sci. 1990, 47, 1667–1676. [DOI] [PubMed] [Google Scholar]
- (48).Rao VR; Banning JW Drug Chem. Toxicol 1990, 13, 173–194. [DOI] [PubMed] [Google Scholar]
- (49).Tsai P-J; Lai Y-H; Manne RK; Tsai Y-S; Sarbassov D; Lin H-KJ Biomed. Sci 2022, 29, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Toson B; Fortes IS; Roesler R; Andrade SF Pharmacol. Res 2022, 183, 106403. [DOI] [PubMed] [Google Scholar]
- (51).Wang Q; Chen X; Hay N Br. J. Cancer 2017, 117, 159–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Escárcega RO; Fuentes-Alexandro S; García-Carrasco M; Gatica A; Zamora A Clin. Oncol 2007, 19, 154–161. [DOI] [PubMed] [Google Scholar]
- (53).Taniguchi K; Karin M Nat. Rev. Immunol 2018, 18, 309–324. [DOI] [PubMed] [Google Scholar]
- (54).Concetti J; Wilson CL Cells 2018, 7, 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).APEX2 v2010.3.0 and SAINT v7.60A data collection and data processing programs, respectively. Bruker Analytical X-ray Instruments, Inc., Madison, WI; SADABS v2008/1, Semi-empirical absorption and beam correction program Sheldrick GM, University of Göttingen, Germany. [Google Scholar]
- (56).Sheldrick GM Acta Cryst 2015, C71, 3–8. [Google Scholar]
- (57).Prince E International Tables for Crystallography Volume C; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1992. [Google Scholar]
- (58).Verdonk ML; Cole JC; Hartshorn MJ; Murray CW; Taylor RD Proteins Struct. Funct. Gene 2003, 52, 609–623. [DOI] [PubMed] [Google Scholar]
- (59).Shelley JC; Cholleti A; Frye LL; Greenwood JR; Timlin MR; Uchimaya MJ Comp. Aided Mol. Des 2007, 21, 681–691. [DOI] [PubMed] [Google Scholar]
- (60).Trott O; Olson AJ J. Comp. Chem 2010, 31, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Seeliger D; de Groot BL J. Comp. Aided Mol. Des 2010, 24, 417–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.