

ABSTRACT
The gut microbiota plays a pivotal role in health and disease. The use of probiotics as microbiota-targeted therapies is a promising strategy to improve host health. However, the molecular mechanisms involved in such therapies are often not well understood, particularly when targeting the small intestinal microbiota. In this study, we investigated the effects of a probiotic formula (Ecologic®825) on the adult human small intestinal ileostoma microbiota. The results showed that supplementation with the probiotic formula led to a reduction in the growth of pathobionts, such as Enterococcaceae and Enterobacteriaceae, and a decrease in ethanol production. These changes were associated with significant alterations in nutrient utilization and resistance to perturbations. These probiotic mediated alterations which coincided with an initial increase in lactate production and decrease in pH were followed by a sharp increase in the levels of butyrate and propionate. Moreover, the probiotic formula increased the production of multiple N-acyl amino acids in the stoma samples. The study demonstrates the utility of network theory in identifying novel microbiota-targeted therapies and improving existing ones. Overall, the findings provide insights into the dynamic molecular mechanisms underlying probiotic therapies, which can aid in the development of more effective treatments for a range of conditions.
KEYWORDS: Probiotics, gut microbiota, small intestine, dynamic correlation-based networks, metabolites, sequencing
Introduction
The human gastrointestinal tract is home to a diverse microbial community comprising hundreds of microbial species.1 The stability, resilience, and resistance to perturbations of this community depend on the composition of the microbiota and the interactions between the microorganisms as well as with the host.2,3 These interactions are often mediated by microbiota-produced metabolites, which form metabolic interaction networks.4,5 Any disruption to these networks, whether caused by medication, disease, or dietary interventions, may affect the community’s function and ultimately impact the host’s health, which may result in disorders such as anxiety, depression or type 2 diabetes.6–8 For example, Sung et al. used a network approach combining metagenomic sequencing data with experimentally validated metabolic reactions to construct and compare a metabolic interaction network of healthy controls and type 2 diabetic patients. The authors demonstrated changes in the produced metabolites as well as in the species with the most significant influence on the network.9 Metabolic network approaches have also been utilized to explore the role of the microbiota in diseases such as inflammatory bowel disease, obesity, and Parkinson’s disease.10,11 Therefore, network-based approaches can serve as a knowledge-driven method to identify new microbiota-targeted therapies.12 For example, Steinway et al. employed a dynamic microbial interaction network approach to identify Barnesiella intestinehominis as a potential candidate for the treatment of Clostridium difficile infection.13
Probiotics, which are defined as live microorganisms that confer a health benefit on the host when administered in adequate amounts, are currently being used for the prevention and treatment of several disorders.14–16 The potential impact of multi-strain probiotic formulas, as opposed to single-strain probiotics, on the host is greater because the strains within the formula can complement each other.17 Previous studies have shown that the oral administration of a probiotic formula, Ecologic® 825, to healthy volunteers did not result in an overall shift in fecal community composition of the microbiota. However, the LEfSe analyses identified two features belonging to a Bacteroides sp. and Alistipes sp., which were positively associated with probiotic supplementation.18 Additionally, the administration of this probiotic formula to patients with irritable bowel syndrome resulted in an increase in fecal levels of acetate and butyrate, accompanied by a reduction in fecal zonulin and symptom severity.19 When this probiotic formula was administered together with a prebiotic (Ecologic® 825/FOS P6) to healthy volunteers, it increased stool frequency, but did not affect zonulin levels and gastrointestinal symptoms.20 These reported effects of the supplemented probiotics may be due to their interaction with the resident microbiota. However, a better understanding of the metabolic interactions between the different strains within a probiotic formula as well as between the probiotic formula and the residing microbiota is needed to elucidate the molecular mechanisms underlying these effects.
The investigation of the small intestine is critical for understanding the efficacy of probiotics, as this region represents a key target for their activity.21 Yet, sampling the small intestine in humans is challenging, which limits our ability to comprehensively explore the microbiota, diet, and host interactions as well as small intestinal disorders.22 Additionally, the small intestine is a highly dynamic environment with varying oxygen, pH, and nutrient gradients, making it difficult to investigate probiotic therapies that target this region.23 However, previous studies have demonstrated that stoma effluent from ileostomy subjects can serve as a surrogate for the in-vivo small intestine, providing a useful tool for investigating the effects of probiotics in this region.24–26
In this study, we employed a dynamic correlation-based metabolic network approach and multivariate analysis, incorporating experimental data obtained from proton-nuclear magnetic resonance (1H-NMR), liquid chromatography-mass spectrometry (LC-MS), shallow shotgun sequencing and flow cytometry performed on ileostoma samples grown using ex vivo SIFR® technology27 to investigate the alterations caused by the supplementation of a 9-species probiotic formula.
Material and methods
Product supplementation to the ileostoma effluent
Ecologic® 825 (Winclove probiotics, The Netherlands), a probiotic formula, consisting of 9 different strains; Bifidobacterium bifidum W23, Bifidobacterium lactis W51, Bifidobacterium lactis W52, Lactobacillus acidophilus W22, Lactobacillus casei W56, Lactobacillus paracasei W20, Lactobacillus plantarum W62 Lactobacillus salivarius W24 and Lactococcus lactis W19, was tested at a dose of 107 CFU/mL. Prior to the introduction of the formula in the bioreactors, the microbial cells were washed by centrifugation and resuspension in anaerobic PBS.
Fermentation experiment using SIFR® technology
Individual bioreactors were processed in parallel in a bioreactor management device (Cryptobiotix, Ghent, Belgium). Each bioreactor contained 5 mL of nutritional medium (M0014, Cryptobiotix, Ghent, Belgium) – microbial inoculum blend with or without supplementation of the probiotic formula (at 107 CFU probiotic/mL), then sealed individually, before being rendered anaerobic. At the start of the incubation, oxygen was introduced at pO2 = 15 mmHg, thus providing relevant intraluminal oxygen levels for the proximal ileum.28 After preparation, bioreactors were incubated under continuous agitation (140 rpm) at 37°C for 24 h (MaxQ 6000, Thermo Scientific, Thermo Fisher Scientific, Merelbeke, Belgium). Upon gas pressure measurement in the headspace, liquid samples were collected for subsequent analysis. Both the blank and probiotic treatment were tested for 6 different test subjects; 4 male and 2 female with an age range between 55–86 years old. For each test subject, multiple technical replicates were performed and harvested at either 0 h (only blank), 3 h, 6 h, 9 h or 24 h.
Ileostomy samples were collected from healthy middle aged-elderly subjects after the participants signed an informed consent in which they donated their sample (procedure approved by Ethics Committee of the University Hospital Ghent; reference number BC-09977).
Absolute microbial community analysis
Quantitative insights were obtained by correcting proportional data (%; shallow shotgun sequencing) with total cell counts for each sample (cells/mL; flow cytometry), resulting in estimated cell counts/mL.
DNA extraction was performed as previously described.29 From the cell pellets obtained by centrifuging 1 mL culture for 1 min at 15.000 RPM. Details are depicted in the supplementary methods.
DNA libraries were prepared using the Nextera XT DNA Library Preparation Kit (Illumina) and IDT Unique Dual Indexes with total DNA input of 1 ng. Libraries were then sequenced on an Illumina Nextseq 2000 platform 2 × 150bp. Unassembled sequencing reads were directly analyzed by CosmosID-HUB Microbiome Platform (CosmosID Inc., Germantown, MD) described elsewhere30–33 for multi-kingdom microbiome analysis and profiling of antibiotic resistance and virulence genes and quantification of organisms’ relative abundance. Cleaned reads were assembled using metaSpades in default configuration.34 Genes were predicted using Prodigal in metagenomics mode and subsequently functions were assigned by EggnogMapper.35,36 Details regarding library preparation, sequencing and bioinformatics analysis are depicted in the supplementary methods.
For total cell count analysis, liquid samples were diluted in anaerobic phosphate-buffered saline, after which cells were stained with SYTO 16 at a final concentration of 1 µM and counted via a BD FACS Verse flow cytometer (BD, Erembodegem, Belgium). Data was analyzed using FlowJo, version 10.8.1.
Extraction and LC-MS/MS
Metabolites for LC-MS/MS analysis were extracted from 350 µL culture supernatant using ethyl acetate. LC-MS/MS acquisition was performed using Shimadzu Nexera X2 UHPLC system, with attached PDA, coupled to Shimadzu 9030 QTOF mass spectrometer, equipped with a standard ESI source unit, in which a calibrant delivery system (CDS) is installed. All the samples were analyzed in positive and negative polarity, using data dependent acquisition mode. Full scan MS spectra (m/z 100–1700, scan rate 10 Hz, ID enabled) were followed by two data dependent MS/MS spectra (m/z 100–1700, scan rate 10 Hz, ID disabled) for the two most intense ions per scan. Details regarding the metabolite extraction and LC-MS/MS are depicted in the supplementary methods.
1 H NMR spectroscopy and data processing
1 mL culture was centrifuged for 1 min at 21,130 rcf, 250 µL of the supernatant was added to 400 µL NMR buffer (200 mM Na2HPO4, 44 mM NaH2PO4, 1 mM TSP, 3 mM NaN3 and 20% (v/v) D2O), centrifuged at 4°C, 21130 rcf for 20 min and 550 µL was transferred to a 5 mm NMR tube.
All 1H-NMR spectra were recorded using a Bruker 600 MHz AVANCE II spectrometer equipped with a 5 mm triple resonance inverse cryoprobe and a z-gradient system. One-dimensional (1D) 1H-NMR spectra were recorded using the first increment of a NOESY pulse sequence with pre-saturation (γB1 = 50 Hz) for water suppression during a relaxation delay of 4 s and a mixing time of 10 ms. A 256 scans of 65,536 points covering 13,658 Hz were recorded and zero filled to 65,536 complex points prior to Fourier transformation, an exponential window function was applied with a line-broadening factor of 1.0 Hz. The spectra were phase and baseline corrected and referenced to the internal standard (TSP; δ 0.0 ppm), using the MestReNova software (v.12.0.0–20080, Mesterlab Research). The annotation of the bins was performed with the Chenomx Profiler software (Chenomx NMR Suite 8.6 and Chenomx 600 MHz, version 11) and the HMDB database 5.0 (http://www.hmdb.ca). Details regarding spectral imaging and data processing are depicted in the supplementary methods.
Comparative metabolomics
Raw data obtained from the LC-MS analysis were converted to mzXML centroid files using Shimadzu LabSolutions Postrun Analysis. The files were imported into Mzmine 2.53 for data processing.37 The resulting quantification tables were uploaded to MetaboAnalyst and subjected to RM two-way ANOVA.38 The exported quantification table for GNPS and the MS2 spectra were uploaded to Feature Based Molecular Networking on the GNPS platform39,40 using the default settings. The resulting network was visualized in Cytoscape 3.4.0. Details regarding data processing are depicted in the supplementary methods.
Statistical and network analysis
All analysis of variance (ANOVA) were performed using GraphPad Prism 7.0. When a comparison resulted in a statistically significant difference (P < 0.05), multiple comparisons testing was performed by controlling the false discovery rate (FDR) according to the Benjamini-Hochberg (BH) method (α < 0.05). The R package MixOmics was used for ordination and multivariate statistical analysis of the 1H-NMR spectra.41 The dynamic profile comparison using Kendall’s τ correlation with the BH (α < 0.05) was performed using the R package psych. In CytoScape 3.9.1 the plugin CoNet42 was used for network construction. The network properties were obtained using the network analyzer tool in Cytoscape.
Results
Supplementation of Ecologic®825 to the ileostoma microbiota alters the microbiota composition and function
To investigate the impact of probiotics supplementation on the small intestinal microbial community, healthy middle aged-elderly test subjects (n = 6) who provided ileostoma effluent as a noninvasive access route to the small intestine.26 Ileostoma samples were inoculated in an ex vivo SIFR fermenter platform consisting of 54 bioreactors. Each ileostoma sample was incubated with or without a probiotic formula Ecologic® 825, which consists of 9 probiotic species, in separate bioreactors, at 37°C with an initial pO2 of 15 mmHg, to simulate intraluminal oxygen levels of the proximal ileum28 (Figure 1a).
Figure 1.
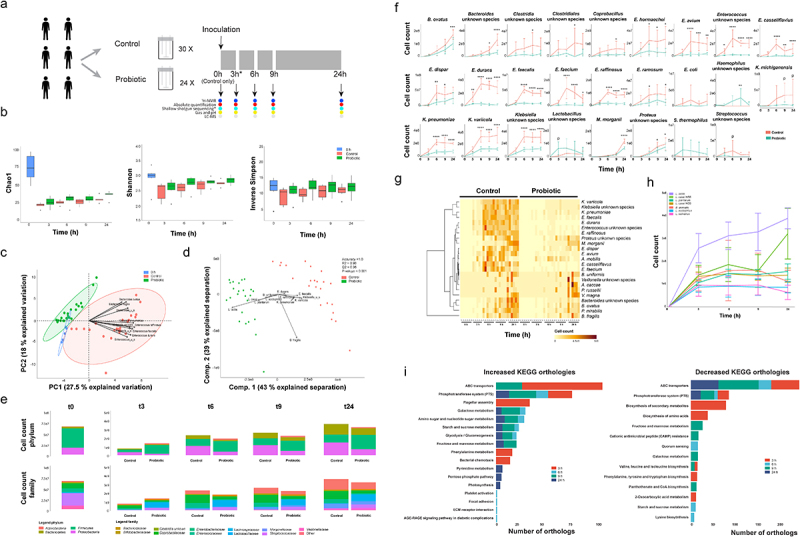
A probiotic formula reduces the cell counts of Enterococcus and Klebsiella species in compositional data obtained from ileostomy effluent.
(a) Schematic overview of the experimental procedure. Ileostomy effluent of 6 donors is collected and divided over 54 bioreactors. 24/54 bioreactors are supplemented with 107 CFU/mL of the probiotic formula. The other 30/54 are controls without supplementation. The black arrows indicate sampling timepoints, where the contents of 6 control and 6 supplemented bioreactors are subjected to shallow shotgun metagenomic, flow cytometry, 1H-NMR, LC-MS and fermentation parameter analysis. The contents of 6 control bioreactors are subjected to the same analysis before the start of the experiment. The asterisk depicts the DNA sample obtained from donor 5 at timepoint 3 h, which got destroyed during sample processing. Therefore this sample could not be used for the shallow shotgun sequencing analysis. (b) The mean and standard deviation of the microbial richness as assessed by the Chao1 index (ordinary two-way ANOVA; F(1,49) = 2.1, P = .15 and F(4,49) = 0.15, P = .96) and the microbial diversity as assessed by Shannon’s H (ordinary two-way ANOVA; F(1,49) = 0.08, P = .77 and F(4,49) = 0.40, P = .81)and the inverse Simpson index (ordinary two-way ANOVA; F(1,49) = 3.0, P = .088 and F(4,49) = 0.24, P = .91). Differences are calculated using an ordinary two-way ANOVA. (c) Principal component analysis of the relative abundance as obtained via shallow shotgun sequencing multiplied by the absolute cell count as obtained via flow cytometry. The ellipses represent the 95% confidence interval. The taxa contributing the most to the separation are indicated with arrows. (d) Partial least square – discriminant analyses of the relative abundance as obtained via shallow shotgun sequencing multiplied by the absolute cell count as obtained via flow cytometry. The taxa contributing the most to the separation are depicted with arrows. The accuracy, R2 and Q2 values are statistical parameters which estimate the predictive ability of the model and are calculated via cross validation. The P-value is the results of a permutation test performed with the separation distance statistic and 1000 permutations. All parameters are calculated using MetaboAnalyst 5.0. (e) Depiction of the average cell counts timepoint at the phylum and family levels as calculated by multiplying the relative abundance data obtained from the shallow shotgun sequencing and the absolute cell counts as obtained via flow cytometry. (f) Dynamic cell count profiles of significantly different taxa between the control and probiotics-supplemented ileostoma microbiotas according to the factor condition as calculated by an ordinary two-way ANOVA with multiple comparisons testing by controlling the false discovery rate according to the Benjamini-Hochberg procedure; *: q < 0.05, **: q < 0.01, ***: q < 0.001, ****: q < 0.0001, P: P < .05, q > 0.05. (g) Heatmap showing the significantly different taxa between the control and probiotics-supplemented ileostoma microbiota according to the factor time as calculated by an ordinary two-way ANOVA with multiple comparisons testing by controlling the false discovery rate according to the Benjamini-Hochberg procedure. (h) Dynamic cell count profiles of 7/9 species present in the probiotic formula. Bifidobacterium animalis W51 and W52 could not be distinguished and Bifidobacterium bifidum W23 could not be identified in the shallow shotgun sequencing data. (i) Differences in KEGG orthologs within the microbiota of the control vs probiotics-supplemented community. The differences are obtained by comparing the orthologs of the control and probiotics-supplemented community per timepoint using a linear modeling approach.
Shallow shotgun metagenomic sequencing combined with flow cytometry was used to investigate the dynamic changes in the ileostoma microbiota composition upon probiotics supplementation at different time points (0, 3, 6, 9, 24 h) after incubation with or without the probiotic formula (Figure 1a).
Comparing microbial richness and diversity did not result in significant differences between control and probiotics-supplemented samples (Figure 1b). However, the addition of probiotic cells decreased the absolute number of cells in the probiotics-supplemented samples compared to the control, except after 3 h of growth (Supplementary figure S1), indicating an altered microbiota composition.
The absolute abundance of the ileostoma microbiota was investigated by multiplying the relative shallow shotgun data by the absolute cell count data per sample (Supplementary data 1). Principal component analysis (PCA) and partial least square – discriminant analysis (PLS-DA) showed distinct clustering of the probiotics-supplemented and control samples, mainly due to variations in the absolute abundance of multiple species of Bacteroides, Klebsiella, and Enterococcus (Figure 1c,d).
Next, compositional changes over time were compared to identify which taxa were affected by the supplementation of the probiotic formula.
The abundance of Firmicutes (ordinary one-way ANOVA; F(4,24) = 20.5, P < .0001), Proteobacteria (ordinary one-way ANOVA; F(4,24) = 3.8, P = .016) and Bacteroidetes (ordinary one-way ANOVA; F(4,24) = 11.4, P < .0001) were significantly altered throughout the experiment, particularly at 3 h and 24 h of the fermentation in the control samples (Figure 1e, Supplementary Figure S2). Only the abundance of Actinobacteria (ordinary one-way ANOVA; F(4,25) = 6.1, P = .001) was altered at 3 h of the experiment in the probiotics-supplemented samples, and the changes in abundance of Firmicutes (ordinary one-way ANOVA; F(4,25) = 5.6, P = .002) and Bacteroidetes (ordinary one-way ANOVA; F(4,25) = 13.0, P < .0001) were most notable after 24 h (Figure 1e, Supplementary Figure S2).
In contrast to the control samples, the levels of Proteobacteria did not change throughout the experiment in the probiotics-supplemented samples (ordinary one-way ANOVA; F(4,25) = 2.5, P = .07) (Figure 1e, Supplementary Figure S2). The difference in the levels of Actinobacteria and the differences in the levels of Firmicutes between control and probiotics-supplemented samples are likely due to the composition of the probiotic formula, which contains seven Firmicutes and two Actinobacteria species. The abundance of Enterobacteriaceae was significantly altered in the control samples, but not in the probiotics-supplemented samples.
Enterobacteriaceae (ordinary one-way ANOVA; F(4,24) = 6.3, P = .001), Bacteroidaceae (ordinary one-way ANOVA; F(4,24) = 12.9, P < .0001), and Enterococcaceae (ordinary one-way ANOVA; F(4,24) = 13.1, P < .0001) abundances were altered throughout the experiment on the family level, with the most prominent changes after 3 h and 24 h of growth in the control samples. The abundance of Enterobacteriaceae did not significantly change throughout the experiment in the probiotics-supplemented samples. In the presence of probiotics, the abundance of Bacteroidaceae (ordinary one-way ANOVA; F(4,25) = 14.6, P < .0001) and Enterococcaceae (ordinary one-way ANOVA; F(4,25) = 58.6, P < .0001) were significantly altered throughout the experiment (Figure 1e, Supplementary Figure S2). On the species level, only 3 out of 26 taxa were higher in the probiotics-supplemented samples (Figure 1f), while 23 taxa, mainly Enterobacteriaceae, Bacteroidaceae, and Enterococcaceae, were significantly lower (Figure 1f)), even though most species increased in number over time in both the control and probiotics-supplemented samples (Figure 1g), confirming the ordination analysis (Figure 1c,d). On the probiotics level, only L. casei W56 (ordinary one-way ANOVA; F(3,20) = 6.5, P = .003) and L. lactis W19 (ordinary one-way ANOVA; F(3,20) = 4.2, P = .018) showed an increase in cell counts over time (Figure 1h).
Metagenomic functional profiling was conducted to identify changes in metabolic pathways associated with the addition of probiotics to the ileostoma samples. Results showed that 17 KEGG orthologs (KO) increased while 14 decreased throughout the experiment. The abundance of KO related to flagellar assembly, bacterial chemotaxis, and phenylalanine metabolism increased in the probiotics-supplemented samples after 3 h of incubation, while the biosynthesis of secondary metabolites, amino acids, and 2-oxocarboxylic acid metabolism decreased. Notably, phosphotransferase system (PTS) and ABC transporters were the most affected KO at every sampling point, indicating that bacteria capable of utilizing available metabolites could survive (Figure 1i).
Altogether, while the microbial richness and diversity were not significantly different, the addition of probiotics altered the human stoma composition, with distinct clustering of probiotics-supplemented and control samples mainly due to a significant decreased representation of multiple species of Bacteroides, Klebsiella and Enterococcus over time.
The metabolic activity of the ileostoma microbiota is altered in response to the probiotic formula supplementation
Alterations in the microbiota composition and function are likely accompanied by alterations in the metabolic environment.43
To investigate the metabolic changes associated with the probiotic formula supplementation, we measured fermentation parameters, pH, and gas production over time in the ileostoma samples (Figure 1a). Both conditions showed a decrease in the measured parameters over time, but the reduction was significantly lower in the probiotics-supplemented samples compared to the control samples, indicating an altered metabolism (Figure 2a,b).
Figure 2.
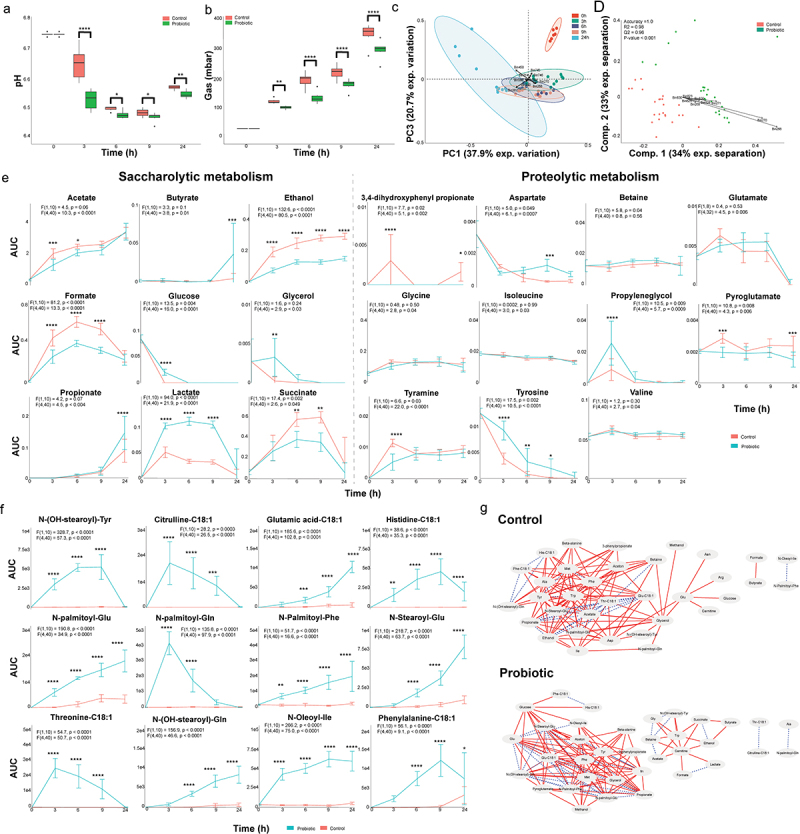
A probiotic formula alters the metabolic environment when supplemented to ileostomy effluent.
(a) The mean and standard deviation of the pH measured at each timepoint. (b) The mean and standard deviation of the gas production at each timepoint. For panel B and C: Significance is obtained via a repeated measure two-way ANOVA with multiple comparison testing by controlling the false discovery rate according to the Benjamini-Hochberg procedure; *: q < 0.05, **: q < 0.01, ***: q < 0.001, ****: q < 0.0001. (c) Principal component analysis performed with the binned spectra as input. The ellipses represent the 95% confidence interval. The 10 bins contributing the most to the separation are indicated with arrows and belong to propionate (bin 214), acetate (bin 387 and 388), lactate (bin 268 and 270), acetone (bin 450), ethanol (bin 240) and an unidentified compound (bin 744, 745 and 746). (d) Partial least square – discriminant analyses performed with the binned spectra as input. The 9 bins contributing the most to the separation are indicated with arrows and belong to lactate (bin 268, 269, 270 and 271) and ethanol (bin 823, 825, 826, 828 and 830). The accuracy, R2 and Q2 values are statistical parameters which estimate the predictive ability of the model and are calculated via cross validation. The P-value is the results of a permutation test performed with the separation distance statistic and 1000 permutations. All parameters are calculated using MetaboAnalyst 5.0. (e) Dynamic profiles of the significantly different saccharolytic and proteolytic metabolites as obtained by measuring the area under the curve of a representative peak per metabolite. The error bars indicate the standard deviation. Significant differences are obtained via repeated measure two-way ANOVA. The F statistic and P value for the factor condition and for the interaction between time and condition are indicated for each metabolite. The result of multiple comparison testing by controlling the false discovery rate according to the Benjamini-Hochberg procedure is indicated with asterisks; *: q < 0.05, **: q < 0.01, ***: q < 0.001, ****: q < 0.0001. (f) Dynamic profiles of the significantly different and identified metabolites as measured the area under the curve obtained by LC-MS. The error bars indicate the standard deviation. Significant differences are obtained via repeated measure two-way ANOVA. The F statistic and P value for the factor condition and for the interaction between time and condition are indicated for each metabolite. The result of multiple comparison testing by controlling the false discovery rate according to the Benjamini-Hochberg procedure is indicated with asterisks; *: q < 0.05, **: q < 0.01, ***: q < 0.001, ****: q < 0.0001. (g) Dynamic correlation based networks of all metabolites measured in the control and probiotics-supplemented community. Nodes represent metabolites. Edges represents correlations between the two connected nodes and are obtained when 2/3 methods give a positive results; Kendall’s -0.8 > τ > 0.8, Spearman’s -0.8 > ρ > 0.8 and Brown’s randomization method with 1000 iterations has a Benjamini-Hochberg corrected P-value <0.05. The calculation of the edges is performed using the Cytoscape plugin CoNet. Red solid edges represent negative correlations, blue dashed edges represent positive correlations.
The samples were subjected to 1H-NMR and LC-MS analysis to further investigate the metabolic changes associated with the addition of the probiotic formula (Figure 1a). Ordination analysis of binned and processed 1H-NMR spectra showed a time-dependent clustering of the spectra, driven by several metabolites including acetate, propionate, lactate, acetone, ethanol, and an unidentified compound (Figure 2c). PLS-DA revealed distinct separation between the control and probiotics-supplemented samples, primarily due to differences in lactate and ethanol (Figure 2d). We also detected a separation between the control and probiotics-supplemented samples as early as 3 h of community growth, with lactate as the biggest contributor to the separation (Supplementary Figure S3s), without donor specific separation (Supplementary Figure S3b). Repeated measure (RM) two-way ANOVA identified altered levels of several saccharolytic metabolites over time such as butyrate, propionate, lactate, acetate, ethanol as well as proteolytic metabolites including tyrosine and tyramine (Figure 2e). Notably, 3,4-dihydroxyphenylpropionic acid was exclusively detected in the control samples (Figure 2e). However, several dynamically altered metabolites were similar between control and probiotics-supplemented samples (Supplementary Figure S4).
In addition to 1H-NMR, LC-MS was used to investigate metabolites that were not detectable by 1H-NMR. Similar to the 1H-NMR data, RM two-way ANOVA analysis of the LC-MS data showed significant differences in 64 and 98 features in negative and positive mode, respectively, between control and probiotics-supplemented samples (Supplementary data 2). The interaction between time and probiotics supplementation revealed significant differences in 62 and 66 LC-MS features in negative and positive mode, respectively (Supplementary data 2) (Supplementary Figures S5–S7). Importantly, all the identified metabolites were N-acyl-amino acid conjugates (Figure 2f).
To assess the global impact of probiotics on the microbial community and the coordinated behavior of metabolites, we compared whole network topology features such as the clustering coefficient (indicative of resistance to perturbations) and network density (indicative of nutrient utilization)44 using dynamic correlation-based networks constructed via the Cytoscape plugin CoNet.42 The nodes in these networks represented the 31 identified 1H-NMR and 12 identified LC-MS metabolites, while the edges represented correlations between dynamic metabolic profiles per condition, as obtained via 1H-NMR and LC-MS data. The control network consisted of 34 out of 43 nodes with 132 edges (clustering coefficient = 0.413, network density = 0.299) (Figure 2g). Although probiotics supplementation only slightly increased the number of nodes (35/43) and edges (135), the clustering coefficient (0.658) and network density (0.529) increased significantly (Figure 2g). This indicates that the ileostoma microbiota, in conjunction with the tested probiotic formula, is more resistant to external perturbations and is better suited to utilize the available metabolites compared to the ileostoma microbiota without the supplemented probiotics. To identify the nodes with the largest impact on the global organization of the networks, we compared node parameters such as the node degree (number of edges per node) and node betweenness centrality (measure of the number of shortest paths between any two nodes that pass through the node)45,46 (Supplementary data 3). N-palmitoyl-phenylalanine (13), glucose (10) and glutamate (9) showed the largest increase in node degree due to probiotics supplementation, while alanine (11), acetate (11) and threonine-C18:1 (13) showed the largest decrease. Moreover, probiotics supplementation altered the node betweenness centrality, with the largest increase observed for carnitine and glucose, and the largest decrease observed for glycerol and glutamate (Supplementary data 3). Together, these results indicate that the altered metabolic activity in the tested probiotics-supplemented ileostoma microbiota generated a dynamic network which makes the community more resistant to external perturbations and more capable to utilize nutrients.
Increased lactate and N-acyl amino acid production by the probiotic formula correlates with reduced growth of Enterobacteriaceae, Enterococcaceae and ethanol production
To examine whether changes in microbiota composition were responsible for alterations in community metabolism following probiotic supplementation, Kendall’s τ correlation analysis was conducted (Figure 3, Supplementary data 4). The results showed positive correlations between probiotic species and lactate, acetate, and propionate (Figure 2e). Negative correlations were found between lactate and other lactic acid bacteria such as Streptococcaceae and Enterococcaceae.47
Figure 3.
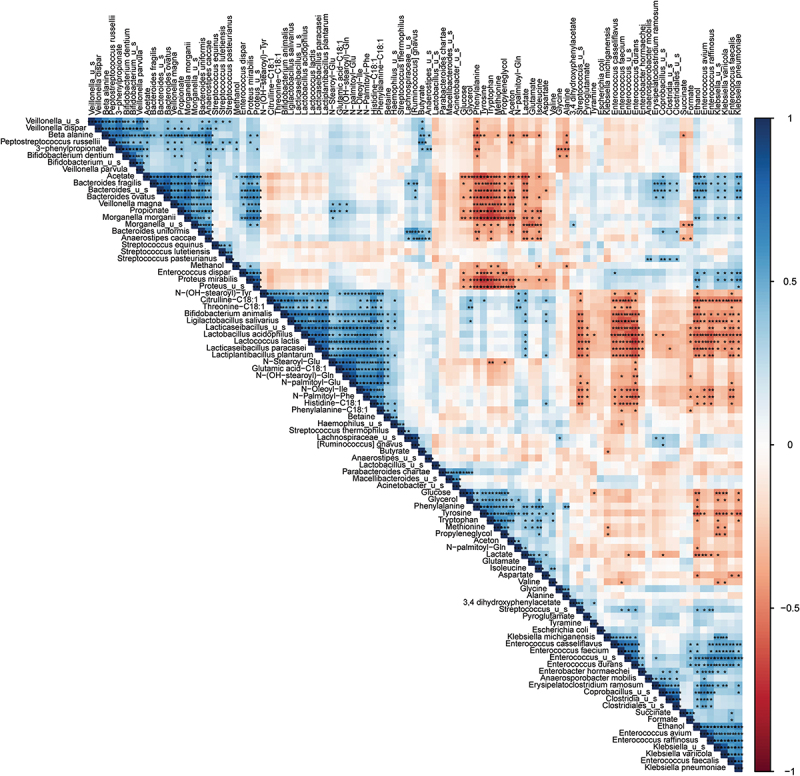
Dynamic correlations within the microbiota show a different dynamic metabolic fingerprint of the community with and without probiotic supplementation.
Kendall’s τ correlation matrix of all metabolites and bacterial species present. Positive correlations are indicated in blue and negative correlations are indicated in red according the legend on the right. Significance is calculated by controlling the false discovery rate according to the Benjamini-Hochberg correction method and indicated with asterisks; *: q < 0.05, **: q < 0.01, ***: q < 0.001.
Acetate and propionate showed positive correlations with Bacteroides, Morganella, and Proteus species, which were significantly reduced in the probiotic-supplemented samples. Additionally, N-acyl amino acids were negatively correlated with Enterococcaceae but positively correlated with the probiotic species. Our findings suggest that the observed changes in the adult human small intestinal stoma microbiota following probiotic supplementation were linked to alterations in the metabolic environment. The analysis also revealed positive correlations between certain metabolites and probiotic species, while negative correlations were observed between these metabolites and certain bacterial species such as Bacteroides, Morganella, and Klebsiella (Figure 2e, g). These findings suggest that the observed changes in the adult human small intestinal stoma microbiota following probiotic supplementation were linked to alterations in the metabolic environment.
Discussion
This study investigated the impact of the supplementation of a 9-species probiotic formula (Ecologic®825) on the dynamic metabolic interaction network of the microbiota of ileostoma samples obtained from healthy middle aged-elderly subjects. The network topology features showed that the supplementation of the tested probiotic formula increased the resistance to external perturbations and the nutrient utilization in the adult human small intestinal stoma microbiota (Figure 2g). This action appears to be mediated via two mechanisms; pH mediated competitive exclusion and alteration of the metabolic interactions.
PH mediated competitive exclusion within the microbial community occurred via increased production of lactate and the subsequent increase in acidity, which is the most prevalent probiotic mechanism of action.48–50 This mechanism has been linked to eradication of several pathogens, including C. difficile, Escherichia coli and Klebsiella pneumonia, increased production of short chain fatty acids51,52 and reduced production of succinate and formate.53,54 In line with the previous reports, our data showed an exclusion of Enterococcaceae, Bacteroidaceae and Enterobacteriaceae from the ileostoma community (Figure 1f), higher levels of butyrate and propionate after 24 h of the addition of the probiotics and lower levels of succinate and formate over the course of the experiment (Figure 2e).
The increased acidity of the microbial environment resulted in activation of bacterial acid stress mechanisms to maintain their intracellular pH.55 Enterococcaceae, one of the most dominant families in the small intestine (Figure 1e),26 tolerate acid stress via activation of a tyrosine decarboxylase56, an enzyme which converts tyrosine to tyramine. The present data infers higher tyrosine decarboxylase activity in the control ileostoma samples because the levels of tyramine and tyrosine significantly decreased and increased, respectively, compared to the samples supplemented with the probiotic formula (Figure 2a,e), which is opposite to our hypothesis. However, these results could be explained by the significant decrease in the absolute abundance of Enterococcus species upon the addition of probiotics (Figure 1f). This scenario is further supported by the negative correlations detected between multiple Enterococcus species and the probiotic species as well as the lower abundance of Enterococcus species upon probiotics supplementation (Figure 3a). Analogously, Fernandez et al. found a reduction of Enterococcus durans growth with a lower pH.57
The significant reduction in Enterococcus species could be related to the reduced levels of ethanol detected in the probiotic supplemented samples (Figures 1f and 2e). Indeed, our correlation analysis shows negative correlations between ethanol and the probiotic species, and positive correlations between ethanol and multiple Enterococcus species (Figure 3a), which is in agreement with previous observations showing a positive association between Enterococcus levels and increased ethanol levels and pH in the small intestine, which promotes alcoholic liver disease.58 The reduced levels of ethanol observed in the present study may be related to the lower abundance of K. pneumoniae upon probiotics supplementation (Figure 1f). K. pneumoniae, which has been detected in the normal healthy human microbiota,26,59,60 can produce high levels of ethanol, endogenously in humans, from glucose and glycerol via the 2,3-butanediol fermentation pathway, and this has been linked to nonalcoholic fatty liver disease.61,62 However, the growth of K. pneumoniae is reduced at acidic pH.63,64 This is in agreement with our correlation analysis, which showed a positive correlation between ethanol and Klebsiella species, including K. pneumoniae and negative correlations between ethanol and glucose and glycerol, as well as a significant reduction of glycerol in the control community only after 3 h of incubation (Figures 1f, 2e and 3a), coinciding with the drop in the pH (Figure 1a). The observed restricted growth of Enterobacteriaceae species in our data could explain the increase in absolute abundance of Clostridiales species (depicted in our data as Anaerostipes) and higher levels of butyrate production (Figures 1g and 2e). In fact, it has been shown that the interaction between Lactobacillus and Clostridiales species caused restriction in the growth of Enterobacteriaceae species and the authors hypothesized that Lactobacillus supplementation could promote the Clostridiales growth.65 Together, the probiotic formula used in this study may have elicited the increased resistance to perturbations and the metabolic usage in the stoma community by lowering the pH, reducing ethanol production via reduction of K. pneumonia growth, consequently lowering Enterococcus growth. Subsequently, the increased lactate levels, most likely, resulted in higher production of propionate and butyrate by Anaerostipes and Veillonella species (Figure 4).
Figure 4.
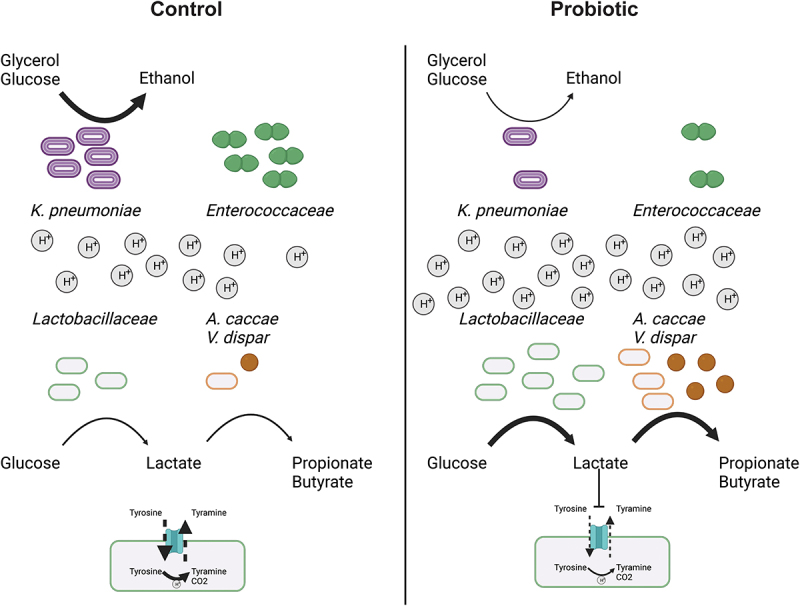
The probiotics supplementation to the ileostoma microbiota alters the metabolic environment and microbiota composition in a pH dependent manner.
Supplementation of the probiotic formula increase the level of Lactobacillaceae, which reduces the pH of the metabolic environment by increasing the production of lactate, which also increases the production of propionate and butyrate by Anaestipes cacae and Veillonella dispar. Furthermore, the cells counts of Enterococcaceae are reduced due to lactate since lactate inhibits the tyrosine decarboxylase of Enterococcoceae, which is their main pH stress regulator. Additionally, the levels of Klebsiella pneumonia are reduced. Subsequently, reducing the conversion of glucose and glycerol into ethanol by K. pneumoniae.
Another potential mechanism through which the probiotic formula may have influenced the bacterial community in the ileostoma is via the production of lantibiotics, although this aspect was not investigated in the present study. Lantibiotics are a class of metabolites produced by microorganisms that can shape the composition of the microbiota.66 Notably, lactic acid bacteria such as L. lactis and L. plantarum, which are members of the Ecologic®825 probiotic formula, are well-known producers of lantibiotics.67
Although the higher bacterial cell count had a direct impact on the levels of the produced metabolites, the interaction of specific bacterial strains within the probiotic formula with the surrounding microbial community may be equally important in determining the effect of the supplementation of probiotics. Among the 9 probiotic strains that constitute the probiotic formula, our data revealed an increase in the cell count of only 2/9 species (Figure 1h), inferring that the metabolic interaction with the surrounding bacteria rather than the growth of the probiotic strains was the determinant factor of the measured effects of the probiotics. To understand these metabolic interactions, we constructed metabolic networks based on the dynamic profile of the identified metabolites. The increase in network density and the clustering coefficient of the metabolic networks (Figure 2g) further support the importance of the metabolic interactions within the community. Our observations are in agreement with Jeong et al. who compared the network diameter among metabolic networks of individual organisms and compared these networks with non-biological networks. The network diameter is the largest number of edges between two nodes in the network and is a measure of connectivity within the network, comparable to the network density. The authors showed that the addition of a new node to a non-biological network increased the network diameter, thereby reducing the connectivity. In contrast, the addition of nodes to biological networks did not alter the network diameter, implying a large resistance to external perturbations and tolerance to node removal,68 which is supported by the low number of essential genes in bacteria69,70 Closer inspection of the changed network showed that N-palmitoyl-phenylalanine and threonine-C18:1 were among the metabolites with the largest differences in network impact. Moreover, these two metabolites positively correlated with the probiotic species and N-palmitoyl-phenylalanine positively correlated with the other identified N-acyl-amino acids (Figure 3). Additionally, all N-acyl-amino acids were all produced in higher amounts in the probiotics-supplemented samples (Figure 2f), however, this increase does not necessarily mean that they are directly produced by the probiotic strains but due to the changes in the microbiota composition that followed the supplementation. Indeed, gut bacteria, notably Clostridia, can produce fatty acid conjugates,71 and these metabolites can be used by the gut microbiota as signaling molecules, and have been found to act as antimicrobials.71,72 Although the precise biological role of the N-acyl-amino acids in the gut is largely unknown,73 these molecules have been categorized, together with the endocannabinoid system, as the endocannabinoidome74 and alterations in the endocannabinoidome have been implicated in a variety of gut related disorders.75,76 Further research is needed to elucidate the precise role of the N-acyl-amino acids within the microbiota and between microbiota and host. The findings of this study should be interpreted in the context of certain limitations. The observed dynamic effects on the human small intestinal ileostoma microbiota were a result of the supplementation with the probiotic formula Ecologic®825, but it cannot be definitively concluded whether these alterations are specific to this particular probiotic formula or if they could also be observed with other probiotic formulations or individual species. Furthermore, the proposed resistance to perturbations, which is inferred from the observed increase in network parameters, has not been experimentally validated in this study. Challenging a control community and a probiotic-supplemented community with an invading pathogen, as an example, would contribute to a deeper understanding of the probiotic-mediated alterations of the metabolic network and its implications for resilience in the face of external disturbances.
Our study did not include experimental testing of the effects of Ecologic®825 supplementation on the host. The next translatable step would be to conduct human intervention studies to further investigate these effects. One promising approach would be to utilize the recently developed ingestible device, which allows for sample collection from the human small intestinal tract during normal digestion.77 By co-culturing the collected digesta with organoids derived from the same subject, it would be possible to provide valuable insights into the precise mechanisms and impacts of Ecologic®825 supplementation on the host. Overall, the present study shows that combining metabolic network construction with compositional analysis of adult human small intestinal stoma microbiota supplemented with the tested mixture of probiotics revealed a dynamic increase in the resistance of the community to perturbations and nutrient utilization. These alterations were the consequences of a pH related competitive exclusion of Enterobacteriaceae and Enterococcaceae. These findings highlight the potential of using probiotics as a therapeutic intervention to improve the health and functionality of the human microbiota. Further studies are needed to evaluate the long-term effects of probiotic supplementation on human health and to identify the optimal probiotic formulations for specific clinical applications. The results of this study may contribute to the advancement of our knowledge on the mechanisms behind the positive impacts of probiotic supplementation on the host, as well as aid in the identification of novel microbiota-targeted therapies. In particular, the findings suggest that the small intestine, a crucial site for probiotic activity, may be a promising target for improving current treatments or developing new ones. Further studies are needed to place the current findings in the context of potential health benefits. Further research is needed to contextualize the current findings with respect to the long-term effects of probiotic supplementation on human health.78
Supplementary Material
Funding Statement
SEA has received research funding support from Winclove, B.V. This support neither influenced nor constrained the contents of this article.
Author’s contributions
JJ, SEA: Conceptualization, investigation, writing-original draft, and editing; JJ, MS, PvA, NuT: data analysis; JJ, ACC, NuT, SsES, PvA: methodology; SvH, GpvW: Conceptualization, writing review; SEA: funding acquisition. All authors have read and agreed to the published version of the manuscript.
Disclosure statement
S. van Hemert is employee in Winclove (Winclove manufactures and markets probiotics). The content of this study was neither influenced nor constrained by this fact. The other authors have no conflicts of interest to declare.
Data availability statement
The raw LC-MS/MS data was deposited in the MASSIVE database, accession ID: MSV000091160.
The shallow shotgun sequencing data were deposited under BioProject number PRJNA928311.
The raw 1H-NMR data was deposited in the Metabolights public repository under the accession number: MTBLS6987
Ethics approval and consent to participate
Ileostomy samples were collected from healthy subjects after the participants signed an informed consent in which they donated their sample (procedure approved by Ethics Committee of the University Hospital Ghent; reference number BC-09977).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19490976.2023.2232506
References
- 1.Faith JJ, Guruge JL, Charbonneau M, Subramanian S, Seedorf H, Goodman AL, Clemente JC, Knight R, Heath AC, Leibel RL, et al. The long-term stability of the human gut microbiota. Science (1979). 2013;341(6141). doi: 10.1126/science.1237439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fassarella M, Blaak EE, Penders J, Nauta A, Smidt H, Zoetendal EG.. Gut microbiome stability and resilience: elucidating the response to perturbations in order to modulate gut health. Gut. 2021;70:595–17. doi: 10.1136/gutjnl-2020-321747. [DOI] [PubMed] [Google Scholar]
- 3.Kern L, Abdeen SK, Kolodziejczyk AA, Elinav E. Commensal inter-bacterial interactions shaping the microbiota. Curr Opin Microbiol. 2021;63:158–171. doi: 10.1016/j.mib.2021.07.011. [DOI] [PubMed] [Google Scholar]
- 4.Weiss AS, Burrichter AG, Durai Raj AC, von Strempel A, Meng C, Kleigrewe K, Münch PC, Rössler L, Huber C, Eisenreich W, et al. In vitro interaction network of a synthetic gut bacterial community. Isme J. 2022;16:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Layeghifard M, Hwang DM, Guttman DS. Disentangling interactions in the microbiome: a network perspective. Trends Microbiol. 2017;25(3):217–228. doi: 10.1016/j.tim.2016.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barabási AL, Gulbahce N, Loscalzo J. Network medicine: a network-based approach to human disease. Nat Rev Genet. 2011;12:56–68. doi: 10.1038/nrg2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rathour D, Shah S, Khan S, Singh PK, Srivastava S, Singh SB, Khatri DK. Role of gut microbiota in depression: Understanding molecular pathways, recent research, and future direction. Behav Brain Res. 2023;436:114081. doi: 10.1016/j.bbr.2022.114081. [DOI] [PubMed] [Google Scholar]
- 8.Yang B, Wei J, Ju P, Chen J. Effects of regulating intestinal microbiota on anxiety symptoms: A systematic review. Gen Psychiatr. 2019;32(2):100056. doi: 10.1136/gpsych-2019-100056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sung J, Kim S, Cabatbat JJT, Jang S, Jin YS, Jung GY, Chia N, Kim P-J. Global metabolic interaction network of the human gut microbiota for context-specific community-scale analysis. Nat Commun. 2017;8(1):1–12. doi: 10.1038/ncomms15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Greenblum S, Turnbaugh PJ, Borenstein E. Metagenomic systems biology of the human gut microbiome reveals topological shifts associated with obesity and inflammatory bowel disease. Proc Natl Acad Sci USA. 2012;109(2):594–599. doi: 10.1073/pnas.1116053109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rosario D, Bidkhori G, Lee S, Bedarf J, Hildebrand F, le Chatelier E, Uhlen M, Ehrlich SD, Proctor G, Wüllner U, et al. Systematic analysis of gut microbiome reveals the role of bacterial folate and homocysteine metabolism in Parkinson’s disease. Cell Rep. 2021;34:108807. doi: 10.1016/j.celrep.2021.108807. [DOI] [PubMed] [Google Scholar]
- 12.Dohlman AB, Shen X. Mapping the microbial interactome: Statistical and experimental approaches for microbiome network inference. Exp Biol Med (Maywood). 2019;244(6):445. doi: 10.1177/1535370219836771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Steinway SN, Biggs MB, Loughran TP, Papin JA, Albert R, Maranas CD. Inference of network dynamics and metabolic interactions in the gut microbiome. PLoS Comput Biol. 2015;11(6):e1004338. doi: 10.1371/journal.pcbi.1004338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hill C, Guarner F, Reid G, Gibson GR, Merenstein DJ, Pot B, Morelli L, Canani RB, Flint HJ, Salminen S, et al. The international scientific association for probiotics and prebiotics consensus statement on the scope and appropriate use of the term probiotic. Nat Rev Gastro Hepat. 2014;11(8):506–514. doi: 10.1038/nrgastro.2014.66. [DOI] [PubMed] [Google Scholar]
- 15.Zucko J, Starcevic A, Diminic J, Oros D, Mortazavian AM, Putnik P. Probiotic – friend or foe? Curr Opin Food Sci. 2020;32:45–49. doi: 10.1016/j.cofs.2020.01.007. [DOI] [Google Scholar]
- 16.Ritchie ML, Romanuk TN, Heimesaat MM. A meta-analysis of probiotic efficacy for gastrointestinal diseases. PLoS One. 2012;7(4):e34938. doi: 10.1371/journal.pone.0034938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Puvanasundram P, Chong CM, Sabri S, Yusoff MSM, Lim KC, Karim M. Efficacy of single and multi-strain probiotics on in vitro strain compatibility, pathogen inhibition, biofilm formation capability, and stress tolerance. Biology (Basel). 2022;11:1644. doi: 10.3390/biology11111644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bagga D, Reichert JL, Koschutnig K, Aigner CS, Holzer P, Koskinen K, Moissl-Eichinger C, Schöpf V. Probiotics drive gut microbiome triggering emotional brain signatures. Gut Microbes. 2018;9:1–11. doi: 10.1080/19490976.2018.1460015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moser AM, Spindelboeck W, Halwachs B, Strohmaier H, Kump P, Gorkiewicz G, Högenauer C. Effects of an oral synbiotic on the gastrointestinal immune system and microbiota in patients with diarrhea-predominant irritable bowel syndrome. Eur J Nutr. 2019;58:2767–2778. doi: 10.1007/s00394-018-1826-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wilms E, Gerritsen J, Smidt H, Besseling-Van Der Van Vaart I, Rijkers GT, Fuentes ARG, Masclee AAM, Troost FJ. Effects of supplementation of the synbiotic Ecologic® 825/FOS P6 on intestinal barrier function in healthy humans: A randomized controlled trial. PLoS One. 2016;11(12):e0167775. doi: 10.1371/journal.pone.0167775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martinez-Guryn K, Hubert N, Frazier K, Urlass S, Musch MW, Ojeda P, Pierre JF, Miyoshi J, Sontag TJ, Cham CM, et al. Small intestine microbiota regulate host digestive and absorptive adaptive responses to dietary lipids. Cell Host & Microbe. 2018;23(4):458. doi: 10.1016/j.chom.2018.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rios-Morales M, van Trijp MPH, Rösch C, An R, Boer T, Gerding A, de Ruiter N, Koehorst M, Heiner-Fokkema MR, Schols HA, et al. A toolbox for the comprehensive analysis of small volume human intestinal samples that can be used with gastrointestinal sampling capsules. Sci Rep. 2021;11(1):1–14. doi: 10.1038/s41598-021-86980-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kastl AJ, Terry NA, Wu GD, Albenberg LG. The structure and function of the human small intestinal microbiota: current understanding and future directions. Cell Mol Gastroenterol Hepatol. 2020;9:33–45. doi: 10.1016/j.jcmgh.2019.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matsuzawa H, Munakata S, Kawai M, Sugimoto K, Kamiyama H, Takahashi M, KOJIMA Y, SAKAMOTO K. Analysis of ileostomy stool samples reveals dysbiosis in patients with high-output stomas. Biosci Microbiota Food Health. 2021;40(3):135. doi: 10.12938/bmfh.2020-062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Colom J, Freitas D, Simon A, Brodkorb A, Buckley M, Deaton J, Winger AM. Presence and germination of the probiotic bacillus subtilis DE111® in the human small intestinal tract: a randomized, crossover, double-blind, and placebo-controlled study. Front Microbiol. 2021;12:2189. doi: 10.3389/fmicb.2021.715863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yilmaz B, Fuhrer T, Morgenthaler D, Krupka N, Wang D, Spari D, Candinas D, Misselwitz B, Beldi G, Sauer U, et al. Plasticity of the adult human small intestinal stoma microbiota. Cell Host & Microbe. 2022;30(12):1773–1787.e6. doi: 10.1016/j.chom.2022.10.002. [DOI] [PubMed] [Google Scholar]
- 27.van den Abbeele P, Deyaert S, Thabuis C, Perreau C, Bajic D, Wintergerst E, Joossens M, Firrman J, Walsh D, Baudot A. Bridging preclinical and clinical gut microbiota research using the ex vivo SIFR® technology. Front Microbiol. 2023. doi: 10.3389/fmicb.2023.1131662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Vos WM, Tilg H, van Hul M, Cani PD, de Vos WM. Gut microbiome and health: mechanistic insights. Gut. 2022;71(5):1020–1032. doi: 10.1136/gutjnl-2021-326789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu Z, Morrison M. Improved extraction of PCR-quality community DNA from digesta and fecal samples. Biotechniques. 2004;36(5):808–812. doi: 10.2144/04365ST04. [DOI] [PubMed] [Google Scholar]
- 30.Lax S, Smith DP, Hampton-Marcell J, Owens SM, Handley KM, Scott NM, Gibbons SM, Larsen P, Shogan BD, Weiss S, et al. Longitudinal analysis of microbial interaction between humans and the indoor environment. Science (1979). 2014;345(6200):1048–1052. doi: 10.1126/science.1254529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hasan NA, Young BA, Minard-Smith AT, Saeed K, Li H, Heizer EM, McMillan NJ, Isom R, Abdullah AS, Bornman DM, et al. Microbial community profiling of human saliva using shotgun metagenomic sequencing. PLoS One. 2014;9(5):e97699. doi: 10.1371/journal.pone.0097699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ponnusamy D, Ev K, Sha J, Erova TE, Azar SR, Fitts EC, Kirtley ML, Tiner BL, Andersson JA, Grim CJ, et al. Cross-talk among flesh-eating Aeromonas hydrophila strains in mixed infection leading to necrotizing fasciitis. Proc Natl Acad Sci. 2016;113(3):722–727. doi: 10.1073/pnas.1523817113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ottesen A, Ramachandran P, Reed E, White JR, Hasan N, Subramanian P, Ryan G, Jarvis K, Grim C, Daquiqan N, et al. Enrichment dynamics of Listeria monocytogenes and the associated microbiome from naturally contaminated ice cream linked to a listeriosis outbreak. BMC Microbiol. 2016;16(1). doi: 10.1186/s12866-016-0894-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nurk S, Meleshko D, Korobeynikov A, Pevzner PA. MetaSPAdes: A new versatile metagenomic assembler. Genome Res. 2017;27(5):824–834. doi: 10.1101/gr.213959.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cantalapiedra CP, Hern̗andez-Plaza A, Letunic I, Bork P, Huerta-Cepas J, Tamura K. eggNOG-mapper v2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol Biol Evol. 2021;38:5825–5829. doi: 10.1093/molbev/msab293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huerta-Cepas J, Szklarczyk D, Heller D, Hernández-Plaza A, Forslund SK, Cook H, Mende DR, Letunic I, Rattei T, Jensen L, et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019;47(D1):D309–D314. doi: 10.1093/nar/gky1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pluskal T, Castillo S, Villar-Briones A, Orešič M. Mzmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinform. 2010;11(1):1–11. doi: 10.1186/1471-2105-11-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xia J, Psychogios N, Young N, Wishart DS. MetaboAnalyst: a web server for metabolomic data analysis and interpretation. Nucleic Acids Res. 2009;37(Web Server):W652. doi: 10.1093/nar/gkp356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang M, Carver JJ, Vv P, Sanchez LM, Garg N, Peng Y, Nguyen DD, Watrous J, Kapono CA, Luzzatto-Knaan T, et al. Sharing and community curation of mass spectrometry data with global natural products social molecular networking. Nat Biotechnol. 2016;34(8):828–837. doi: 10.1038/nbt.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nothias LF, Petras D, Schmid R, Dührkop K, Rainer J, Sarvepalli A, Protsyuk I, Ernst M, Tsugawa H, Fleischauer M, et al. Feature-based molecular networking in the GNPS analysis environment. Nat Methods. 2020;1717(9):905–908. doi: 10.1038/s41592-020-0933-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rohart F, Gautier B, Singh A, Lê Cao KA, Schneidman D. mixOmics: An R package for ‘omics feature selection and multiple data integration. PLoS Comput Biol. 2017;13(11):e1005752. doi: 10.1371/journal.pcbi.1005752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Faust K, Raes J, Wilmes P, Heintz-Buschart A, Eiler A. CoNet app: inference of biological association networks using cytoscape. F1000 Res. 2016;5:1519. doi: 10.12688/f1000research.9050.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cao Y, Liu Y, Dong Q, Wang T, Niu C. Alterations in the gut microbiome and metabolic profile in rats acclimated to high environmental temperature. Microb Biotechnol. 2022;15(1):276–288. doi: 10.1111/1751-7915.13772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Batushansky A, Matsuzaki S, Newhardt MF, West MS, Griffin TM, Humphries KM. GC-MS metabolic profiling reveals Fructose-2,6-bisphosphate regulates branched chain amino acid metabolism in the heart during fasting. Metabolomics. 2019;15:18. doi: 10.1007/s11306-019-1478-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vernocchi P, Gili T, Conte F, Del Chierico F, Conta G, Miccheli A, Botticelli A, Paci P, Caldarelli G, Nuti M, et al. Network analysis of gut microbiome and metabolome to discover microbiota-linked biomarkers in patients affected by non-small cell lung cancer. Int J Mol Sci. 2020;21(22):8730. doi: 10.3390/ijms21228730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tipton L, Müller CL, Kurtz ZD, Huang L, Kleerup E, Morris A, Bonneau R, Ghedin E. Fungi stabilize connectivity in the lung and skin microbial ecosystems. Microbiome. 2018;6(1):1–14. doi: 10.1186/s40168-017-0393-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Y, Wu J, Lv M, Shao Z, Hungwe M, Wang J, Bai X, Xie J, Wang Y, Geng W. Metabolism characteristics of lactic acid bacteria and the expanding applications in food industry. Front Bioeng Biotechnol. 2021;9. doi: 10.3389/fbioe.2021.612285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mohan R, Koebnick C, Schildt J, Mueller M, Radke M, Blaut M. Effects of bifidobacterium lactis Bb12 Supplementation on body weight, fecal pH, acetate, lactate, calprotectin, and iga in preterm infants. Pediatr Res. 2008;64(4):418–422. doi: 10.1203/PDR.0b013e318181b7fa. [DOI] [PubMed] [Google Scholar]
- 49.Kim HK, Rutten NBMM, Besseling-van der Vaart I, Niers LEM, Choi YH, Rijkers GT, van Hemert S. Probiotic supplementation influences faecal short chain fatty acids in infants at high risk for eczema. Benef Microbes. 2015;6:783–790. doi: 10.3920/BM2015.0056. [DOI] [PubMed] [Google Scholar]
- 50.van Thu T, Foo HL, Loh TC, Bejo MH. Inhibitory activity and organic acid concentrations of metabolite combinations produced by various strains of Lactobacillus plantarum. Afr J Biotechnol. 2013;10:1359–1363. [Google Scholar]
- 51.Sorbara MT, Dubin K, Littmann ER, Moody TU, Fontana E, Seok R, Leiner IM, Taur Y, Peled JU, van den Brink MRM, et al. Inhibiting antibiotic-resistant Enterobacteriaceae by microbiota-mediated intracellular acidification. J Exp Med. 2019;216(1):84–98. doi: 10.1084/jem.20181639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kolling GL, Wu M, Warren CA, Durmaz E, Klaenhammer TR, Guerrant RL. Lactic acid production by Streptococcus thermophilus alters Clostridium difficile infection and in vitro Toxin a production. Gut Microbes. 2012;3(6):523. doi: 10.4161/gmic.21757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ternes D, Tsenkova M, VI P, Meyers M, Koncina E, Atatri S, Schmitz M, Karta J, Schmoetten M, Heinken A, et al. The gut microbial metabolite formate exacerbates colorectal cancer progression. Nat Metabolism. 2022;4(4):458–475. doi: 10.1038/s42255-022-00558-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Serena C, Ceperuelo-Mallafré V, Keiran N, Queipo-Ortuño MI, Bernal R, Gomez-Huelgas R, Urpi-Sarda M, Sabater M, Pérez-Brocal V, Andrés-Lacueva C, et al. Elevated circulating levels of succinate in human obesity are linked to specific gut microbiota. Isme J. 2018;12(7):1642–1657. doi: 10.1038/s41396-018-0068-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guan N, Liu L. Microbial response to acid stress: mechanisms and applications. Appl Microbiol Biotechnol. 2020;104(1):51–65. doi: 10.1007/s00253-019-10226-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Perez M, Calles-Enríquez M, Nes I, Martin MC, Fernandez M, Ladero V, Alvarez MA. Tyramine biosynthesis is transcriptionally induced at low pH and improves the fitness of Enterococcus faecalis in acidic environments. Appl Microbiol Biotechnol. 2015;99(8):3547–3558. doi: 10.1007/s00253-014-6301-7. [DOI] [PubMed] [Google Scholar]
- 57.Fernández M, Linares DM, Rodríguez A, Alvarez MA. Factors affecting tyramine production in Enterococcus durans IPLA 655. Appl Microbiol Biotechnol. 2007;73(6):1400–1406. doi: 10.1007/s00253-006-0596-y. [DOI] [PubMed] [Google Scholar]
- 58.Llorente C, Jepsen P, Inamine T, Wang L, Bluemel S, Wang HJ, Loomba R, Bajaj JS, Schubert ML, Sikaroodi M, et al. Gastric acid suppression promotes alcoholic liver disease by inducing overgrowth of intestinal Enterococcus. Nat Commun. 2017;8(1):1–15. doi: 10.1038/s41467-017-00796-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Conlan S, Kong HH, Segre JA, Highlander SK. Species-level analysis of DNA sequence data from the NIH human microbiome project. PLoS One. 2012;7(10):e47075. doi: 10.1371/journal.pone.0047075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zoetendal EG, Raes J, van den Bogert B, Arumugam M, Booijink CC, Troost FJ, Bork P, Wels M, de Vos WM, Kleerebezem M. The human small intestinal microbiota is driven by rapid uptake and conversion of simple carbohydrates. Isme J. 2012;6(7):1415–1426. doi: 10.1038/ismej.2011.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li NN, Li W, Feng JX, Zhang WW, Zhang R, Du SH, Liu S-Y, Xue G-H, Yan C, Cui J-H, et al. High alcohol-producing Klebsiella pneumoniae causes fatty liver disease through 2,3-butanediol fermentation pathway in vivo. Gut Microbes. 2021;13(1). doi: 10.1080/19490976.2021.1979883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yuan J, Chen C, Cui J, Lu J, Yan C, Wei X, Zhao X, Li N, Li S, Xue G, et al. Fatty liver disease caused by high-alcohol-producing Klebsiella pneumoniae. Cell Metab. 2019;30(4):675–688.e7. doi: 10.1016/j.cmet.2019.08.018. [DOI] [PubMed] [Google Scholar]
- 63.Abbas SZ, Riaz M, Ramzan N, Zahid MT, Shakoori FR, Rafatullah M. Isolation and characterization of arsenic resistant bacteria from wastewater. Braz J Microbiol. 2014;45(4):1309. doi: 10.1590/S1517-83822014000400022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mitrea L, Vodnar DC. Klebsiella pneumoniae—a useful pathogenic strain for biotechnological purposes: diols biosynthesis under controlled and uncontrolled pH levels. Pathogens. 2019;8(4):293. doi: 10.3390/pathogens8040293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Djukovic A, Garzón MJ, Canlet C, Cabral V, Lalaoui R, García-Garcerá M, Rechenberger J, Tremblay-Franco M, Peñaranda I, Puchades-Carrasco L, et al. Lactobacillus supports Clostridiales to restrict gut colonization by multidrug-resistant Enterobacteriaceae. Nat Commun. 2022;13(1):1–18. doi: 10.1038/s41467-022-33313-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Field D, Cotter PD, Hill C, Ross RP. Bioengineering lantibiotics for therapeutic success. Front Microbiol. 2015;6:1363. doi: 10.3389/fmicb.2015.01363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Twomey D, Ross RP, Ryan M, Meaney B, Hill C. Lantibiotics produced by lactic acid bacteria: Structure, function and applications. Antonie van Leeuwenhoek, Int J Gen Mol Microbiol. 2002;82(1/4):165–185. doi: 10.1023/A:1020660321724. [DOI] [PubMed] [Google Scholar]
- 68.Jeong H, Tombor B, Albert R, Oltval ZN, Barabásl AL. The large-scale organization of metabolic networks. Nature. 2000;407(6804):651–654. doi: 10.1038/35036627. [DOI] [PubMed] [Google Scholar]
- 69.Kobayashi K, Ehrlich SD, Albertini A, Amati G, Andersen KK, Arnaud M, Asai K, Ashikaga S, Aymerich S, Bessieres P, et al. Essential Bacillus subtilis genes. Proc Natl Acad Sci USA. 2003;100(8):4678–4683. doi: 10.1073/pnas.0730515100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gerdes SY, Scholle MD, Campbell JW, Balázsi GBalazsi G, Ravasz E, Daugherty MD, Somera AL, Kyrpides NC, Anderson I, Gelfand MS, et al. Experimental determination and system level analysis of essential genes in Escherichia coli MG1655. J Bacteriol. 2003;185(19):5673. 10.1128/JB.185.19.5673-5684.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chang FY, Siuti P, Laurent S, Williams T, Glassey E, Sailer AW, Gordon DB, Hemmerle H, Voigt CA. Gut-inhabiting Clostridia build human GPCR ligands by conjugating neurotransmitters with diet- and human-derived fatty acids. Nat microbiol. 2021;6(6):792–805. doi: 10.1038/s41564-021-00887-y. [DOI] [PubMed] [Google Scholar]
- 72.Brady SF, Clardy J. Long-chain N -Acyl amino acid antibiotics isolated from heterologously expressed environmental DNA. J Am Chem Soc. 2000;122(51):12903–12904. doi: 10.1021/ja002990u. [DOI] [Google Scholar]
- 73.Battista N, Bari M, Bisogno T. N-Acyl amino acids: metabolism, molecular targets, and role in biological processes. Biomolecules. 2019;9:822. doi: 10.3390/biom9120822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.di Marzo V. The endocannabinoidome as a substrate for noneuphoric phytocannabinoid action and gut microbiome dysfunction in neuropsychiatric disorders. Dialogues Clin Neurosci. 2020;22(3):259. doi: 10.31887/DCNS.2020.22.3/vdimarzo. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lian J, Casari I, Falasca M. Modulatory role of the endocannabinoidome in the pathophysiology of the gastrointestinal tract. Pharmacol Res. 2022;175:106025. doi: 10.1016/j.phrs.2021.106025. [DOI] [PubMed] [Google Scholar]
- 76.Kim JT, Terrell SM, Li VL, Wei W, Fischer CR, Long JZ. Cooperative enzymatic control of N-ACYL amino acids by PM20D1 and FAAH. Elife. 2020;9. doi: 10.7554/eLife.55211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shalon D, Culver RN, Grembi JA, Folz J, Treit PV, Shi H, Rosenberger FA, Dethlefsen L, Meng X, Yaffe E, et al. Profiling the human intestinal environment under physiological conditions. Nature. 2023;617(7961):581–591. doi: 10.1038/s41586-023-05989-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Haug K, Salek RM, Conesa P, Hastings J, de Matos P, Rijnbeek M, Mahendraker T, Williams M, Neumann S, Rocca-Serra P, et al. MetaboLights—an open-access general-purpose repository for metabolomics studies and associated meta-data. Nucleic Acids Res. 2013;41(D1):D781–D786. doi: 10.1093/nar/gks1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw LC-MS/MS data was deposited in the MASSIVE database, accession ID: MSV000091160.
The shallow shotgun sequencing data were deposited under BioProject number PRJNA928311.
The raw 1H-NMR data was deposited in the Metabolights public repository under the accession number: MTBLS6987